
Abstract
The central nervous system (CNS) is capable of gathering information on the body’s nutritional state and it implements appropriate behavioral and metabolic responses to changes in fuel availability. This feedback signaling of peripheral tissues ensures the maintenance of energy homeostasis. The hypothalamus is a primary site of convergence and integration for these nutrient-related feedback signals, which include central and peripheral neuronal inputs as well as hormonal signals. Increasing evidence indicates that glucose and lipids are detected by specialized fuel-sensing neurons that are integrated in these hypothalamic neuronal circuits. The purpose of this review is to outline the current understanding of fuel-sensing mechanisms in the hypothalamus, to integrate the recent findings in this field, and to address the potential role of dysregulation in these pathways in the development of obesity and type 2 diabetes mellitus.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Precise control of energy intake, storage, and expenditure is indispensable in keeping body weight and blood glucose concentrations within physiological ranges. Dysregulation of these homeostatic processes is associated with the development of obesity and type 2 diabetes mellitus; however, the exact underlying mechanisms leading to these diseases are still not fully elucidated.
Communication of the body’s nutritional state by feedback signaling of peripheral organs to the CNS and the appropriate behavioral and metabolic responses initiated by the brain are pivotal processes in maintaining energy homeostasis. The hypothalamus, and in particular the arcuate nucleus (the most external hypothalamic nucleus), are located close to the median eminence, a site characterized by a discontinuous blood–brain barrier, allowing for direct access of circulating hormones and nutrients to the CNS [1]. Thus, the hypothalamus is a potential primary site of convergence and integration for nutrient-related signals, which include central and peripheral neuronal inputs as well as hormonal signals [2]. Besides fuel-communicating hormones such as insulin and leptin, increasing evidence indicates that glucose and lipids are detected by specialized fuel-sensing neurons that are incorporated in critical hypothalamic neuronal circuits. Hence, circulating nutrients cooperate with hormones, such as insulin, leptin, and ghrelin, to regulate the activity of distinct neuron populations that control food intake, energy expenditure, and glucose homeostasis. Thus, this review aims to provide an updated view of the pathways of glucose and lipid sensing in the hypothalamus and to delineate the contribution of impaired neuronal nutrient sensing to the etiology of obesity and type 2 diabetes mellitus.
Central glucose sensing
Glucose sensing in the hypothalamus
The earliest demonstration that the brain is involved in the control of blood glucose levels was provided by Claude Bernard, who showed that hypothalamic lesions induce hyperglycemia in dogs [3]. In 1953, John Mayer proposed that cells located in the hypothalamus could be specialized to monitor variations in blood glucose concentrations and further postulated that these cells translate these variations in glucose concentrations in electrical or chemical signals that control feeding behavior [4]. About 10 years later, two groups indeed demonstrated the presence of such specialized glucose-sensing neurons by electrophysiological analysis of hypothalamic slides. Anand et al. and Oomura et al. independently identified hypothalamic neurons that are able to modulate their firing activity in response to changes in extracellular glucose concentrations [5, 6].
Essentially two different types of glucose-responsive neurons can monitor changes in blood glucose levels: glucose-excited (GE) neurons, whose firing rate is increased by elevation of extracellular glucose concentrations, and glucose-inhibited (GI) neurons, which are activated when glucose concentrations decrease [7]. Both types of neurons are widely distributed throughout the brain but highly represented in hypothalamic nuclei, which are involved in the control of energy homeostasis. GE neurons are most abundant in the ventromedial nucleus (VMN), the arcuate nucleus (ARC), and the paraventricular nucleus (PVN), whereas GI neurons are mostly located in the lateral hypothalamus (LH), the median ARC, and the PVN [8]. In the ARC, the presence of GE and GI neurons responsive to glucose over either a low range (0–5 mM) or a high range (5–20 mM) of glucose concentrations has been described, the latter are referred to as HGE (high glucose excited) or HGI (high glucose inhibited) neurons, respectively [9, 10].
GE and GI neurons are also present in the brain stem, in particular in the area postrema (AP), the nucleus of solitary tract (NTS), and the dorsal motor nucleus of the vagus (DMNX) [11]. The NTS represents a critical node of convergence that integrates various signals from the periphery and relays them to the hypothalamus. Neurons in the NTS are sensitive to small variations in blood glucose concentrations and may regulate the activity of hypothalamic neurons since they project widely into hypothalamic nuclei implicated in the control of blood glucose levels and food intake [12].
Neuronal circuits of the ARC are among the best-studied systems in the central regulation of energy homeostasis. Key players are two functionally opposing neuron populations, the agouti-related peptide/neuropeptide Y (AgRP/NPY)-expressing and the proopiomelanocortin and cocaine-and amphetamine-related transcript (POMC/CART)-expressing neurons [13, 14]. The anorectic POMC/CART neurons express POMC as a precursor peptide, which, dependent on the cell-type specific expression pattern of prohormone convertases, is processed to different bioactive products [15]. Among these are the melanocyte-stimulating hormones (α-, β-, and γ-MSH). α-MSH and β-MSH reduce food intake and increase energy expenditure both in animals and in humans [16–18]. α-MSH and β-MSH act on melanocortin receptor (MC-R) types 3 and 4, which are expressed in the ARC, the PVN, LH, VMN, and dorsomedial hypothalamus [19, 20]. The second key neuron population in the ARC is formed by the orexigenic AgRP/NPY neurons. NPY is a potent stimulator of food intake and it reduces energy expenditure [21, 22]. AgRP acts as an inverse agonist of the MC3/4-R and prevents the anorectic effect of α-MSH [23]. Besides their regulation by hormones, such as insulin, leptin, and ghrelin, these both types of neurons represent prototypic glucose-sensing neurons. In particular, through electrophysiological recordings of identified, genetically marked neuron populations, it has been demonstrated that increasing extracellular glucose levels inhibit AgRP/NPY neurons and excite POMC neurons [24–27].
AgRP/NPY and POMC neurons extend broad projections to various brain regions including the LH that harbors two other populations of glucose-sensing neurons, the orexin-expressing and the melanin-concentrating hormone (MCH) neurons. Orexin neurons are inhibited and MCH neurons are excited by glucose, in addition both populations receive inputs from AgRP/NPY and POMC neurons [28–30].
Molecular mechanisms of glucose sensing
Since GE neurons increase their firing activity when extracellular glucose rises, they share similarity to pancreatic β-cells [31–33]. Glucose signaling in β-cells requires glucose uptake by the low-affinity glucose transporter type 2 (GLUT2), glucose phosphorylation by glucokinase, the rate-limiting enzyme of glycolysis, and subsequent metabolism of glucose to increase intracellular ATP concentration [34]. This in turn leads to closure of ATP-sensitive potassium (KATP) channels [35], membrane depolarization, and the entry of Ca2+, which triggers insulin secretion. Thus, many studies have evaluated the role of GLUT2, glucokinase, and the KATP channel subunits SUR1, SUR2, and Kir6.2 in central glucose sensing.
GLUT2 is expressed in hypothalamic nuclei where glucose-sensitive neurons are present [36–39]. In transgenic mice, central GLUT2 has been shown to be involved in the counter-regulatory response to hypoglycemia [40].
In the pancreatic β-cell, glucokinase is the critical regulator of glycolytic production of ATP and KATP channel activity [41]. The pancreatic form of glucokinase is also present in brain areas involved in glucose sensing and in about 70% of GE neurons glucokinase mRNA is detectable [37, 42–44]. Indeed, glucokinase has been shown to regulate the ability of GE neurons to sense glucose [32, 37, 42, 45]. In addition, the selective down-regulation of glucokinase in primary VMH neuronal cultures led to selective loss of glucose sensing [46].
KATP channels play a fundamental role since they link changes in glucose metabolism to electrical activity [47] and expression has been demonstrated throughout the brain, including hypothalamic regions involved in glucose sensing [48–51]. Using single-cell RT-PCR to analyze glucose-sensing neurons, investigators have shown the expression of the KATP channel subunits SUR1 and Kir6.2 in the hypothalamus [37]. In addition, electrophysiological studies have demonstrated that pharmacological inhibition or activation of KATP channels can alter the response of GE neurons to changes in ambient glucose in vitro and in vivo [32, 48, 52–57]. In line with a role in glucose sensing, the expression of a mutated version of the KATP subunit Kir6.2 (resulting in dramatically reduced ATP sensitivity) specifically in POMC neurons blunted glucose sensing. Interestingly, obesity-induced POMC glucose insensitivity is mediated by the mitochondrial protein uncoupling protein 2 (UCP2), which impairs glucose-stimulated ATP production and thus KATP channel closure, while its genetic deletion or pharmacological inhibition reversed the phenotype [58].
Taken together, there is considerable evidence that similar to pancreatic β-cells, an increase in extracellular glucose raises ATP levels in GE neurons, which leads to the closure of KATP channels [49, 53, 57, 59, 60], and thus plasma membrane depolarization. Subsequent Ca2+ entry through voltage-gated channels finally increases neuronal activity and neurotransmitter secretion [61, 62] (Fig. 1a).

Potential mechanisms of glucose sensing in hypothalamic neurons. a An increase in extracellular glucose and/or lactate raises the ATP/ADP ratio in GE neurons, which leads to the closure of KATP channels and thus plasma membrane depolarization. Subsequent Ca2+ entry through voltage-gated channels finally increases neuronal activity and neurotransmitter secretion. b In GI neurons, suppression of neurotransmitter secretion at elevated glucose might be caused by an ATP-mediated increase in Na+–K+-ATPase activity and/or inhibition of AMPK, which in turn opens Cl− or other ion channels. In other GI neurons, glucose-induced hyperpolarization might be due to K+ channels opened by an ATP- and AMPK-independent mechanism
There is however also indication that neuronal glucose sensing could be independent of KATP channels [9], glucokinase [63], or GLUT2 [37]. For instance, the activation of an ARC GE neuron population by glucose seems to depend on the glucose-regulated activity of transient response potential (TRP) channels [9]. Moreover, a recent study identified the heterodimeric G-protein-coupled sweet receptor T1R2/T1R3 as a candidate membrane-bound brain glucose sensor that allows neurons to change firing rates independently of intracellular glucose metabolism [64].
In GI neurons, the mechanism linking a decrease in glucose concentrations to increased firing activity is less clear (Fig. 1b). An early study proposed that suppression of firing activity at elevated glucose levels might be controlled by the increase in the ATP-to-ADP ratio, which leads to an increase in Na+–K+-ATPase activity [8, 65]. A further study indicated that the opening of plasma membrane Cl− channels at elevated glucose concentrations leads to cell hyperpolarization and inhibition of VMN and ARC GI neurons [27, 54, 63]. For GI orexin neurons, it has been shown that tandem-pore K+ channels could mediate their inhibition by glucose [66], however a recent study demonstrated that glucose inhibition persists in orexin neurons lacking tandem-pore K+ channels [67]. Orexin neurons further exhibit carbohydrate selectivity and can operate independently of glucose metabolism [68]. Moreover, a distinct group of orexin neurons exhibits only transient inhibitory responses to sustained rises in glucose levels due to time-dependent recovery from inhibition via adaptive closure of leak-like K+ channels, presumably the same channels as those activated by glucose [69]. This adaptive glucose sensing allows orexin cell firing to maintain sensitivity to small fluctuations in glucose levels despite large background levels of glucose.
Changes in AMP-activated protein kinase (AMPK) activity are likely to mediate some effects of glucose on VMN and ARC GI neurons [26, 70]. AMPK is activated in response to a rise in intracellular AMP levels, which increase under conditions of cellular stress or energy deficiency, such as hypoxia, ischemia, and glucose deprivation [71, 72]. Activated AMPK acts to switch off energy-consuming anabolic processes and switch on energy producing catabolic processes [73] and has therefore been suggested as an important glucose sensor. As shown in cell lines and ex vivo hypothalamic tissue, AMPK activity is stimulated by low glucose levels and AMPK activation increases orexigenic AgRP expression [74]. This study also demonstrated that small changes in hypothalamic glucose levels in the physiological range of 1–5 mM altered cellular ATP levels sufficiently to induce AMPK activation and gene expression. Recently, the cAMP response element binding protein (CREB) co-activator CRTC2 was identified as a new hypothalamic AMPK target, linking hypothalamic glucose sensing with cAMP response element (CRE) gene expression [75]. Mechanistically, it is possible that AMPK activation in GI neurons leads to the phosphorylation and thereby inactivation of plasma membrane Cl− and/or other ion channels [54], leading to depolarization, and activation of the neuron. Consistently, studies of Murphy et al. [76] suggest that in VMH GI neurons activated AMPK phosphorylates neuronal nitric oxide synthase (nNOS) leading to nitric oxide production, which in turn amplifies AMPK activation and thereby closure of the Cl− channel cystric fibrosis transmembrane regulator (CFTR).
Since glucokinase mRNA is expressed in about 40% of GI neurons it may also hold a regulatory role in these neurons [42, 46], although it seems that AMPK activity plays a predominant role in GI neuronal glucose sensing.
Glucose-sensing neurons do not perform their function in isolation. They are surrounded and provided with metabolic fuels by glia cells. Astrocytes readily take up and store glucose as glycogen, which is hydrolyzed to release lactate into the extracellular space when glucose is scarce. Extracellular lactate is taken up by neurons and converted to pyruvate, which is then oxidized in mitochondria to provide ATP [37, 77, 78]. By this mechanism, lactate is able to trigger KATP channel closure [79, 80] and thereby reverse the inhibition of GE activity, which occurs at low ambient glucose levels [63]. Importantly, levels of the lactate/monocarboxylate transporters, which are very low in pancreatic β-cells [81, 82], appear to be relatively high in the hypothalamus [78, 79]. The role of astrocytes in central glucose sensing is further supported by the already-mentioned finding that re-expression of GLUT2 in astrocytes was sufficient to rescue the phenotype of mice lacking central GLUT2 [40]. However, it is conceivable that depending on substrate availability, one or more glucose-sensing pathways are used by different neuronal populations.
Physiological functions regulated by central glucose sensing
Food intake
The role of glucose in the regulation of feeding has early been postulated [4, 83]. It has been shown that initiation of feeding is preceded by a drop in blood glucose concentration [84] and if this is prevented by glucose infusion, then initiation of feeding is suppressed [85]. In addition, intrahypothalamic infusion of glucose leads to a decrease in food intake and body weight [86–88]. Induction of cellular glucoprivation by central administration of 2-deoxy-d-glucose (2-DG), a glucose analogue able to inhibit glucose metabolism, however, induces food intake [89, 90]. In particular, brain stem areas such as the DMNX and NTS appear to be the primary site of glucoprivation detection, since direct injections of 5-thioglucose (5-TG), a 2-DG analogue, into these areas stimulated food intake [89, 91] whereas direct injection of 2-DG into the VMH or LH failed to activate feeding.
As already mentioned, glucose-sensitive neurons of the brain stem project widely into the hypothalamus, in particular the PVN and the ARC. Destruction of these neuronal projections by immunotoxins suppressed the effect of peripheral 2-DG administration on food intake [92] and on the regulation of NPY and AgRP expression [93]. The pivotal role of AgRP/NPY-expressing neurons in the acute regulation of feeding in adult mammals has been impressively demonstrated by two independent studies using diphtheria-toxin-mediated ablation of this neuron population [13, 94]. However, the extent to which these neurons contribute to glucose-induced inhibition of feeding is still unclear. When Luquet et al. [95] destroyed AgRP/NPY neurons in neonatal mice, they found that feeding responses to glucoprivation were unaffected. The same group previously showed that in contrast to starvation induced upon elimination in adult mice, neonatal destruction of these neurons has minimal effects on feeding in general, which illustrates the impressive plasticity of this circuitry [94]. Thus, it is still possible that glucose-induced inhibition of AgRP/NPY neurons has profound effects on feeding when the brain is allowed to develop normally.
On a molecular level, many of the critical components of glucose sensing have been demonstrated to be involved in the regulation of food intake. For instance, central GLUT2 expression has been linked to feeding regulation by knockdown experiments using antisense oligonucleotides as well as targeted disruption of the Glut2 gene. Injection of antisense oligonucleotides in the ARC decreased food intake and led to resistance towards the effects of 2-DG on feeding behavior [96]. In the GLUT2-null mice, ad libitum feeding as well as refeeding after a 24 h fast was higher than in controls [97]. Moreover, during the fast-to-refed transition, expression of the orexigenic (NPY, AgRP) and anorexigenic (POMC, CART) neuropeptides was dysregulated in the absence of GLUT2. This was directly attributed to the loss of central glucose sensing, as intracerebroventricular (ICV) glucose in fasted GLUT2-null mice failed to reduce NPY and to increase POMC expression. In parallel in GLUT2 knockdown animals, feeding responses to ICV glucose or 2-DG were also lost. In humans, a genetic variation in GLUT2 is associated with a higher daily intake of sugars [98].
These data indicate that GLUT2 is required for physiological control of feeding by glucose. However, attempts to localize GLUT2 in the brain so far failed to show its expression in POMC or NPY neurons, although it was found to be associated with other hypothalamic and brain stem structures [36–38, 99, 100] and it was further shown to be expressed in astrocytes, endothelial cells, and tanycytes of the third ventricle [101–103]. Thus, the regulation of orexigenic and anorexigenic peptide expressions by GLUT2-dependent sensors may be controlled indirectly, probably by glucose-sensitive neurons located in other brain regions, such as the brain stem.
Haploinsufficiency for glucokinase in mice gave rise to effects similar to those observed during hypoglycemia, i.e., increased food intake and a hypothalamic gene expression profile similar to fasted mice (increased hypothalamic NPY mRNA and reduced hypothalamic POMC mRNA), indicating impaired glucose sensing in these animals [104].
Evidence for a role of KATP channels in the control of feeding comes from the study of mice with inactivation of the Kir6.2 gene as well as mice with constitutive active KATP channels exclusively in POMC neurons. Whereas, Kir6.2-null mice display a smaller feeding response to intraperitoneal 2-DG administration than control mice [53], mice expressing a constitutively active Kir6.2 subunit in POMC cells exhibited normal feeding behavior despite the complete unresponsiveness of these neurons to elevations in extra-cellular glucose concentration [58]. However, studies in POMC cells with constitutive active phosphatidylinositol 3,4,5-triphosphate (PIP3) formation, mimicking insulin-stimulated activation of the PI3 kinase pathway (see below), demonstrated an effect of constitutive active KATP channels on feeding behavior [105]. In the hypothalamus, PIP3 has been shown to regulate cell excitability by binding to the regulatory subunit of KATP channels and modulating their activity by at least three different mechanisms: (i) by increasing the probability that KATP channels are open, which indirectly lowers the ability of ATP to inhibit the channels, (ii) by directly decreasing ATP binding to the channel, and (iii) by modulating the local cytoskeleton in the vicinity of the channels [106–109]. We demonstrated that augmented PIP3-dependent KATP channel activation leads to electrical silencing of POMC neurons and consequently to hyperphagia [105].
The role of the metabolic sensor AMPK in feeding control was addressed by generating mice lacking AMPKα2 in either POMC (POMCα2KO) or AgRP/NPY (AgRPα2KO) neurons [110]. In both neuron populations, inactivation of AMPK suppressed the glucose-mediated regulation of firing activity, indicating that AMPK function is necessary for glucose sensing in these neuron populations. Absence of this enzyme from POMC neurons led to increased feeding in response to fasting, an imbalance in the expression of hypothalamic orexigenic and anorexigenic peptides, favoring an orexigenic output and body weight gain. Inactivation of AMPK in AgRP/NPY neurons led to a modest reduction of body weight, which developed after 3 months of age. However, this was not associated with detectable differences in feeding or in orexigenic and anorexigenic neuropeptide expressions. The molecular mechanism how AMPK allows POMC and AGRP/NPY neurons to sense glucose and thus alters their neuronal excitation is currently unknown.
Energy expenditure
It has been known for several years that infusions of glucose can both increase general sympathetic activity (as evidenced by increased plasma norepinephrine levels) [111, 112] and produce an increase in thermogenesis, which is partly due to such sympathetic activation [113]. These effects of glucose were independent of insulin’s well-known function as activator of sympathetic activity, since glucose evoked these effects in insulin-deficient animals [112] as well as after direct infusion into the forebrain via the carotid artery (without altering plasma insulin levels) [114, 115]. Such forebrain infusions activate neurons in several hypothalamic areas known to contain glucose-sensing neurons such as those in the PVN [51, 116]. The thermogenic effects of glucose are probably also mediated by hypothalamic glucose-sensing neurons, since intracarotid and direct injections of glucose into the VMH and PVN produce increased activity in the sympathetic efferents to brown adipose tissue [117, 118], and mice lacking AMPK in POMC neurons developed obesity partially caused by reduced resting metabolic rate with concomitant downregulation of brown adipose tissue thermogenic genes [110].
Hepatic glucose production
Similar to what is seen in the central regulation of energy intake and expenditure, the brain also directly responds to changes in fuel availability to maintain glucose homeostasis. In the regulation of blood glucose levels, the liver plays a pivotal role. During times of starvation, breakdown of hepatic glycogen stores (glycogenolysis) and hepatic de novo synthesis of glucose (gluconeogenesis) from precursors such as lactate, gluconeogenic amino acids, and glycerol provide the organism with glucose. On the other hand, when food (and consequently glucose) are available, hepatic glucose production (HGP) needs to be suppressed. Besides being subjected to tight hormonal control [119], these processes are also directly regulated by glucose. In response to changing concentrations of hypothalamic glucose, gluconeogenesis, glycogenolysis, and with it total HGP are significantly reduced [80]. The fact that besides glucose also lactate is able to inhibit glucose production when administered directly into the ARC suggests that mitochondrial oxidation is necessary for the CNS to respond to glucose. The finding that pharmacologically blocking lactate dehydrogenase inhibited both lactate- and glucose-induced decrease in HGP, further supported this notion. In contrast, the activation of pyruvate dehydrogenase mimicked the inhibition of HGP [80]. These findings suggest that the neuronal tricarboxylic acid cycle may function as a biochemical sensor for carbohydrate availability, which in turn modulates HGP. The effects of glucose and lactate seem to depend critically on the activation of KATP channels. In fact, the suppressive effect of intra-ARC infusions of glucose or lactate on HGP is blocked by a concomitant infusion of glibenclamide, a KATP channel blocker [80, 120]. Thus, besides feeding behavior, glucose homeostasis is also linked with KATP channel function in glucose-sensing neurons. In a more recent study, Kokorovic et al. demonstrated that hypothalamic sensing of circulating lactate levels also regulates glucose production [121]. In the presence of physiologically relevant increases in the levels of plasma lactate (intravenous injection), inhibition of central lactate-sensing mechanisms by lactate dehydrogenase inhibitor oxamate or KATP channel blocker glibenclamide increased glucose production. Furthermore, direct administration of oxamate into the mediobasal hypothalamus increased glucose production in the presence of a similar elevation of circulating lactate.
Interestingly, central glucose and lactate sensing seems to also affect hepatic lipid metabolism. ICV administration of glucose or lactate was found to lower circulating lipid concentrations and, more importantly, hepatic lipid production [122]. Again, pharmacologically blocking either lactate dehydrogenase or KATP channel function blunted these effects [122].
Counter-regulatory processes
Glucose sensing in the brain is also implicated in the counter-regulatory response to hypoglycemia. In particular, this involves the activation of glucagon secretion from pancreatic α-cells and catecholamines from adrenal glands [123–125].
Experimentally, counter-regulation is activated either by insulin-induced hypoglycemia or by injection of 2-DG or 5-TG to induce cellular glucoprivation. In both conditions, afferent neurons located in the abdominal regions relay the information to the brain stem and the hypothalamus, which can also be directly activated by hypoglycemia or the glucoprivic signal. The role of central glucose sensing in the control of counter-regulation can be evidenced by intracarotid glucose infusion, which blocks hypoglycemia-induced secretion of counter-regulatory hormones [126, 127], or by ICV injection of 2-DG, which stimulates glucagon and catecholamine secretion [128]. The involvement of hypothalamic nuclei, in particular the VMH, has been assessed in lesion studies and by pharmacological or genetic interference with glucose detection systems [127]. For instance, glucagon secretion can be induced by direct injection of 2-DG in the VMH [128], or, in contrast, hypoglycemia-induced glucagon secretion can be suppressed by direct VMH injection of glucose [129]. In a series of studies, Chan et al. [130] demonstrated that modulation of the γ-aminobutyric acid (GABA) inhibitory tone in the VMH modifies the magnitude of glucagon and sympathoadrenal responses to hypoglycemia. They could show that during hypoglycemia, KATP channels are implicated in the regulation of GABA release in this area of the hypothalamus [131] and that an increased GABAergic tone in the VMH contributes to suppression of counter-regulatory responses after recurrent hypoglycemia [132]. In addition to GABA, the excitatory neurotransmitter glutamate released by VMH neurons has also been linked to the prevention of hypoglycemia. Mice lacking synaptic vesicular transporters for glutamate (VGLUT2) selectively in SF1 neurons (a major subset of VMH neurons) exhibit defective counter-regulatory responses during fasting as well as to insulin- or 2-DG-induced hypoglycemia [133].
Recently, hyperinsulinemic clamp studies in humans could show that the hypothalamus is sensitive to small decrements in systemic glucose levels and that hypothalamic blood flow and presumably neuronal activity, precedes the rise in counter-regulatory hormones seen during hypoglycemia [134].
There is also strong evidence that brain stem nuclei play an important role in the control of glucagon secretion. For instance, when the cerebral aqueduct, which allows circulation of cerebrospinal fluid between the third and fourth ventricle, is obstructed, 5-TG induces a glucoregulatory response only when injected in the fourth but not in the third ventricle [135]. In addition, whereas 5-TG injections in different nuclei of the hypothalamus fail to induce a glucoregulatory response, its injections into the NTS, which projects to different sites of the hypothalamus, induce a strong response [91–93]. Moreover, c-fos immunostaining revealed that the activated neurons are present in the NTS [136].
Thus, central glucose-sensing units involved in the physiological control of counter-regulation are present at the brain stem and the hypothalamus. These sites are synaptically connected to form a glucose-monitoring network. The gathered information is integrated to control the counter-regulatory response by activating efferent autonomic nerves that stimulate pancreatic glucagon secretion and the secretion of epinephrine by the adrenals, and at the same time block insulin secretion.
The role of GLUT2 and the KATP channel in the control of glucagon secretion has been investigated in several studies. Mice with inactivation of the Glut2 gene, while expressing a transgenic glucose transporter (GLUT1) in their pancreatic β-cells to preserve normal glucose-stimulated insulin secretion (ripglut1::glut2-null-mice) [137] display an abnormal increase in random fed plasma glucagon levels. More importantly, ICV 2-DG injections failed to stimulate glucagon secretion in these mutant mice attributing a role of GLUT2-mediated central glucose sensing also in the control of counter-regulatory processes [40]. Furthermore, the analysis of c-fos-positive cells in response to intraperitoneal 2-DG injections indicated a decreased number of activated cells in the brain stem, in particular in the NTS and the DMNX in the Glut2-null mice. At the level of the hypothalamus, a reduced stimulation of c-fos-positive cells was observed in the PVN but not in the VMH, indicating that the glucose detection mechanisms used to control glucagon secretion might vary between several hypothalamic nuclei.
The involvement of KATP channel-mediated hypothalamic glucose sensing in the central regulation of glucagon secretion has also been demonstrated. For instance, the counter-regulatory response in a hypoglycemic clamp is blocked upon ICV or direct VMH injection of the KATP channel inhibitor glibenclamide [55]. Genetic inactivation of KATP channel by deletion of Kir6.2 gene also leads to impaired glucagon response, correlated with suppressed glucose-regulated firing activity of VMH neurons [53]. In contrast, the activation of KATP channels in the VMH amplifies counter-regulatory hormone responses to recurrent hypoglycemia [56].
The above-summarized findings clearly indicate that central glucose sensing is implicated in a variety of important physiological functions. In particular the coordinated crosstalk between different sites of glucose detection allows for the precise control of energy homeostasis and normoglycemia. These studies further demonstrate that there is more than one mechanism to sense either an increase or decrease in blood glucose levels, and that the β-cell model of glucose sensing may have many variations.
Central lipid sensing
Cerebral lipids represent 50% of the brain dry weight, which is the highest organ lipid content after adipose tissue. Since neurons do not use fatty acids as a fuel, it was thought for a long time that they are not able to cross the blood–brain barrier. In recent years, however, it has been demonstrated that cerebral lipids arise from both local synthesis as well as plasma origin. Fatty acids cross the blood–brain barrier mainly by simple diffusion in the unbound form, so that access of circulating free fatty acids to the CNS is proportional to their plasma concentration [138, 139]. However, a small proportion of fatty acid uptake into the brain may also occur through direct uptake of lipoprotein particles mediated by lipoprotein receptors [140, 141]. Upon entry into the cell, fatty acids are rapidly esterified to fatty acyl-coenzyme A (acyl-CoAs) in a process catalyzed by the enzyme acyl-CoA synthetase (ACS). Once integrated to the intracellular acyl-CoA pool, fatty acid fate is diverse and depends on the respective fatty acid. For example, Rapoport et al. [141] reported that 50% of the palmitate taken up by the brain is oxidized, whereas 80% of the arachidonate is incorporated into phospholipids. Furthermore increasing evidence shows that fatty acids are used in the CNS as cellular messengers that act like glucose to communicate the body’s energy status and thus are involved in the control of feeding behavior, HGP, and insulin secretion. Especially the hypothalamus is able to detect and respond to changes in circulating fatty acid concentrations through the involvement of lipid-sensing neurons. Indeed, these neurons use fatty acids and their metabolites as signaling molecules to regulate their membrane potential and action potential frequency in a concentration-dependent manner.
Lipid sensing in the hypothalamus
In 1975, Oomura et al. [142] demonstrated that fatty acids activate neurons of the LH, indicating a role for fatty acids as cellular messengers. Accordingly, a study using c-fos immunoreactivity as marker for neuronal activity indicated an activating effect of lipids on neurons located in the LH, whereas neurons in the ARC, DMH, VMH, and PVN were inhibited in response to lipid infusion [143]. Wang et al. [144] demonstrated that distinct neuron populations in the ARC modify their neuronal firing rate in response to fatty acids and that these effects depend on ambient glucose levels.
The molecular mechanisms involved in neuronal lipid sensing are far from being elucidated. However, a body of literature indicates that, in general, fatty acids regulate the conductance of a wide variety of ion channels, which include Cl−, GABAA [145], ClC-2 [146], K+, K+–Ca2+, KATP [147], and Ca2+ channels [148]. Additionally, fatty acids inhibit the Na+–K+-ATPase [149]. Addressing hypothalamic lipid-sensing mechanisms, Lam et al. and Pocai et al. evidenced that the central effects of oleic acid on liver metabolism are abolished by genetic or pharmacological inhibition of the KATP channel [120, 150]. Patch-clamp recordings of oleic acid-inhibited neurons in the ARC further supported a role for KATP channels in hypothalamic lipid sensing [151]. In contrast, neuronal excitation by oleic acid seems to be mediated by the closure of Cl− channels [144].
The physiological relevance of lipid sensing is supported by various studies. For instance, intravenous infusion of a lipid emulsion is sufficient to suppress food intake in baboons [152]. This signal is independent of measurable changes in plasma insulin and does not require gastrointestinal nutrient absorption [152–155]. Consistent with the hypothesis that circulating lipids act on hypothalamic energy centers to generate a signal of nutrient surfeit, the group of Rossetti and colleagues demonstrated that ICV administration of the long-chain fatty acid (LCFA) oleic acid inhibits food intake and leads to a decrease in HGP [156, 157]. Interestingly, using the same protocol octanoic acid, a medium-chain fatty acid had no effect, suggesting that fatty acid action is related to the respective chain length. The authors attributed the anorectic effect of oleic acid to its action on orexigenic ARC neuropeptide expression, i.e., reduction of AgRP and NPY mRNA expression, whereas POMC expression was not affected by oleic acid treatment. In the liver, ICV oleic acid inhibited the expression of glucose-6-phosphatase, the rate-limiting gluconeogenic enzyme [156, 157].
A decrease in food intake and HGP is also obtained by inhibiting hypothalamic carnitine palmitoyltransferase (CPT) 1 [158]. These effects on food intake and HGP were proposed to be caused by the accumulation of intracellular LCFA-CoA due to the inhibition of CPT1-mediated mitochondrial import of LCFA-CoA for β-oxidation. This finding suggests that an increase in intracellular LCFA-CoA concentrations, rather than fatty acids themselves, is the "final" satiety signal that activates neural pathways designed to regulate the input of nutrients in the circulation from either exogenous (food intake) or endogenous sources (HGP) [158–160]. Indeed, a sustained two-to-threefold elevation in circulating long-chain fatty acid levels is sufficient to double LCFA-CoA concentrations within the mediobasal hypothalamus. Furthermore, this increase is prevented by intrahypothalamic infusion of triascin C, a pharmacological inhibitor of LCFA-CoA synthetase [150]. The inhibition of hypothalamic LCFA-CoA synthetase activity also abolishes the ability of circulating LCFA to suppress HGP, indicating that hypothalamic esterification of LCFA to LCFA-CoA is required for the observed effects of LCFA on liver metabolism [150]. However, it has to be noted that inhibition of CPT1 leads to increased levels of LCFA-CoA in the ARC, but not in other hypothalamic nuclei [158], and inhibition of β-oxidation in the LH does not alter food intake [161].
The role of hypothalamic fatty acid metabolism
Fatty acid metabolism seems crucial to relay the central action of fatty acids on energy homeostasis. Enzymes involved in fatty acid metabolism, such as acetyl-CoA carboxylase (ACC), fatty acid synthase (FAS), malonyl-CoA decarboxylase (MCD), and CPT1 are expressed in various brain cell types including hypothalamic neuron populations in the ARC, VMH, and dorsomedial hypothalamus [162]. Furthermore, double-labeling studies have shown that FAS colocalizes with NPY (and thus AgRP) in ARC neurons [163], indicating that modulation of fatty acid synthesis may act on hypothalamic pathways regulating feeding. Feeding increases the concentration of cytoplasmic malonyl-CoA, which is suggested to be one of the main energy sensors in the hypothalamus. Accumulation of malonyl-CoA derives from an increased flux of glucose into the lipogenic pathway during the fed state, i.e., when nutrients are abundant. The steady-state level of malonyl-CoA is determined by its rate of synthesis catalyzed by ACC relative to its rate of turnover catalyzed by FAS. The synthesis of malonyl-CoA is the first step of fatty acid synthesis and ACC is the major site of regulation. In addition, malonyl-CoA is a potent inhibitor of CPT1 and thereby β-oxidation. Thus, in times of ample energy supply, when there is a decreased need for fatty acid oxidation, increased malonyl-CoA levels may inhibit β-oxidation and increase fatty acid synthesis and esterification to triglycerides.
Consistent with a role for neuronal FAS in the modulation of feeding, central administration of C75, a potent inhibitor of FAS, increases malonyl-CoA concentrations in the hypothalamus, suppresses food intake and leads to profound weight loss [164, 165]. In mice deprived of food, central administration of C75 at anorexic doses also significantly increases hypothalamic malonyl-CoA levels [166]. Moreover, pharmacological inhibition of ACC, and thereby malonyl-CoA synthesis, largely reverses the effect of C75 on feeding and malonyl-CoA accumulation [164, 166], supporting the idea that malonyl-CoA availability is essential for the hypothalamic control of food intake. In vitro and in vivo studies demonstrate that at least parts of C75’s effects are mediated through AMPK [167, 168]. Indeed, ICV administration of 5-aminoimidazole-4-carboxamide-ribonfuranoside (AICAR), an AMPK activator, rapidly lowers hypothalamic malonyl-CoA concentrations, increases food intake, and also attenuates C75-induced anorexia [167, 169]. These effects correlate closely with the phosphorylation and thus inactivation of ACC, an established AMPK target.
The anorectic action of FAS inhibition is linked to decreased expression of orexigenic and increased expression of anorexigenic neuropeptides in the ARC, as central administration of C75 blocks fasting-induced up-regulation of AgRP/NPY and down-regulation of POMC/CART expression [164–166, 170]. The molecular mechanisms underlying these effects of FAS inhibition are not completely understood. It has been reported that changes in hypothalamic malonyl-CoA levels correlate with C75’s effects on neuropeptide expression [166]. However, it is so far unclear how accumulation of malonyl-CoA is linked to changes in neuropeptide gene expression and whether its effects are direct or indirect via inhibition of CPT1 and the ensuing accumulation of LCFA-CoA. The latter is supported by the anorectic action of both hypothalamic inhibition of CPT1 [158] and central administration of LCFA [156, 157].
Interrelationship between glucose and lipid sensing
It is well known from pancreatic islets that glucose and lipid sensing are interrelated, since elevated fatty acid concentrations are able to modify the secretory response to glucose [171, 172]. This has been attributed to the intracellular accumulation of various lipid metabolites that interfere with the function of ion channels or other proteins regulating the exocytosis of insulin granules [173]. Moreover, free fatty acids have been demonstrated to decrease the expression of key components of the glucose-signaling pathway such as GLUT2 and glucokinase [174]. There is now also increasing evidence that elevated concentrations of fatty acids may induce ER stress that, under chronic conditions, exerts deleterious effects on pancreatic islet cells [175].
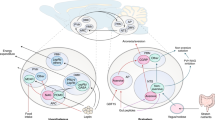
The observation that anorexia resulting from FAS inhibition is dependent on changes in central glucose uptake and/or metabolism [176] supported the idea that nutrient-sensing pathways are also interconnected in the brain. High glucose levels are known to elevate intracellular malonyl-CoA, which in turn inhibits CPT1 and thereby β-oxidation. Increased levels of malonyl-CoA may in turn induce an increase in glucose oxidation as a consequence of decreasing fatty acid oxidation. Thus, the CNS may use the relative ratio of glucose to fatty acid utilization as an index of the overall energy status of the body [176]. Further points of interaction between glucose and lipid metabolism are KATP channels and AMPK (Fig. 2). Increased glucose oxidation raises intracellular ATP levels, which leads to the closure of KATP channels. These channels also seem to be affected by intracellular accumulation of LCFA-CoA [120, 150]. Finally, a decrease in glucose concentration activates AMPK, which phosphorylates and thereby inhibits ACC, thus reducing malonyl-CoA formation and increasing β-oxidation.
Electrophysiological evidence for a direct functional interaction of central lipid and glucose sensing in the hypothalamus has been provided by measuring the firing rate of ARC and VMH neurons in response to oleic acid in the presence of different ambient glucose concentrations [144, 177]. These data revealed an interaction between glucose and fatty acids to regulate oleic acid sensing in neurons of these hypothalamic nuclei, implying that subtypes of lipid-sensing neurons are activated or inhibited, depending on the hypo-, normo- or hyperglycemic status. Even more interesting in this context is the proposed role for reactive oxygen species (ROS) signaling in glucose and lipid sensing by AgRP/NPY and POMC neurons [178–180]. ROS production naturally occurs during mitochondrial respiration of products resulting from both glucose and fatty acid metabolism and thus may represent an additional interconnection of neuronal nutrient sensing. This notion is further supported by an increasing number of studies indicating that ROS production is not simply a byproduct of substrate oxidation but, instead, is implicated in the regulation of neuronal function in a substrate-dependent manner [179–182]. Andrews et al. observed that during positive energy balance, glucose-excited POMC neurons accumulate ROS due to increased glucose utilization. Contrariwise, during negative energy balance, when low glucose levels activate AgRP/NPY neurons, elevated fatty acid oxidation in these cells does not increase intracellular ROS levels. Endogenous buffering of ROS production seems to be indispensable for the function of AgRP/NPY neurons, since uncontrolled ROS formation impairs neuronal firing of these cells. Thus, persistent high ROS levels in active POMC neurons seem to favor satiety during energy surplus, whereas in active AgRP/NPY neurons ROS production needs to be buffered to allow for the appropriate orexigenic responses to energy deficiency [180, 183].
Hormonal modulation of glucose and lipid sensing
Apart from influencing each other, hypothalamic glucose and lipid sensing is controlled by peripheral hormones, such as insulin, leptin, and ghrelin. The central actions of these hormones comprise several sites of convergence with nutrient sensing, as, for instance, in the ARC, they regulate neuronal activity of glucose-excited POMC as well as glucose-inhibited AgRP/NPY neurons [2, 14, 184].
The finding that central applied insulin decreases food intake and body weight led to the proposal that insulin serves as an adiposity signal in the brain, decreasing food intake and increasing energy expenditure [185–187]. Along these lines, mice with brain-specific insulin receptor deletion are mildly obese and display impaired fertility, assigning insulin receptor signaling in the brain a role in regulation of fuel metabolism and reproduction [188]. Importantly, insulin action in the brain not only controls body weight [185, 188–190] but also peripheral glucose and fat metabolism [191–193]. Insulin’s central action in controlling food intake and hepatic gluconeogenesis is mediated by the activation of the PI3 kinase and subsequent regulation of KATP channel activity by its product PIP3 [105, 192, 194]. Thus, KATP channel activation might represent a mechanism allowing for integration of hormonal and nutrient (glucose)-dependent modulation of glucose and lipid sensing in the hypothalamus (Fig. 2). This notion is supported by the recent finding that insulin attenuates the ability of VMN GE neurons to sense decreased glucose via the activation of PI3 kinase and KATP channels [195]. These data are consistent with the role of insulin as satiety factor, as in the presence of insulin glucose levels must decline to a greater extend before GE neurons respond.

Crosstalk and hormonal regulation of glucose and lipid sensing. This schematic representation of the interrelationship between glucose and lipid-sensing mechanisms and their modulation by hormones underlines the convergence of these signals on three key players: AMPK, malonyl-CoA, and KATP channels. LCFA-CoA long-chain fatty acyl-CoA, ACC acetyl-CoA carboxylase, FAS fatty acid synthase, CPT1 carnitine palmitoyltransferase 1
Similarly, the adipocyte-derived hormone leptin, which is secreted in proportion to the body fat content [196, 197], has provided the prototypic adiposity signal in the control of energy homeostasis [198]. Mice carrying a mutation in the leptin gene (ob) exhibit dramatic weight gain (ob/ob) [198], and a naturally occurring loss-of-function mutation of the leptin receptor in the CNS and periphery underlies the phenotype of obese and diabetic mice (db/db) [199]. Importantly, the phenotype of mice with a neuron-specific deletion of the leptin receptor resembles that of the db/db mouse [200], and neuron-specific reconstitution of the leptin receptor is sufficient to reverse the obese phenotype of db/db mice [201], indicating that indeed the major role of leptin action to control energy homeostasis is accounted for by its signaling in the CNS. Consistent with this, central application of leptin decreases food intake and body weight in various species, underlining the pivotal role of leptin in the regulation of energy homeostasis via its action in the CNS [202]. Moreover, leptin, via activation of the CNS PI3 kinase pathway, is crucial for the regulation of glucose metabolism and peripheral insulin sensitivity [105, 203–207]. Leptin has been shown to inhibit hypothalamic AMPK activity [208]. Furthermore, expression of a constitutively active AMPK mutant in the hypothalamus blunts leptin’s effects on food intake, thus indicating that AMPK serves as a principal mediator of leptin’s effects on food intake and fatty-acid metabolism [26, 209, 210]. Since AMPK is also inhibited by insulin [208], regulation of hypothalamic AMPK activity represents a further site of convergence between hormonal and nutritional control of neuron excitability (Fig. 2).
However, studies in mice lacking a functional AMPKα2 subunit specifically in either AgRP or POMC neurons suggest differential mechanisms for glucose sensing and leptin-/insulin-stimulated pathways, since these neurons were still responsive to both insulin and leptin but not glucose [110]. Conversely, Murphy et al. [211] recently demonstrated that leptin-induced changes in glucose sensing of NPY-expressing GI neurons are mediated by AMPK and concluded that during energy sufficiency, leptin reduces the ability of AgRP/NPY neurons to sense decreased glucose concentrations.
AMPK activity is further regulated by ghrelin, an orexigenic gut hormone, which is released to the circulation upon fasting [212, 213]. In the arcuate nucleus, ghrelin activates AgRP/NPY neurons, thereby mediating its orexigenic effect [214]. In addition to its stimulatory effect on food intake, centrally applied ghrelin, presumably via control of sympathetic innervation, influences adipocyte metabolism [215]. Central ghrelin induces AMPK activity via the activation of Ca2+ signaling and subsequent activation of CamKK2 in NPY neurons [216–218]. The following chain of intracellular events seems to contribute to ghrelin’s effect on AgRP/NPY neurons. AMPK inhibits ACC activity, thereby eliminating the inhibitory effect of malonyl-CoA on CPT1 activity. CPT1 activation enhances mitochondrial β-oxidation of long-chain fatty acids, which ultimately promotes generation of reactive oxygen species (ROS). Fatty acids and ROS promote UCP2 transcription and activity, which aims to neutralize ROS, thereby enabling continuous CPT1-promoted fatty acid β-oxidation. Moreover, UCP2 can increase NPY/AgRP neuronal activity, thereby increasing inhibitory GABA-ergic input onto POMC neurons, ultimately stimulating food intake [180, 219].
Taken together, these findings clearly point towards a convergence between hypothalamic nutrient sensing and hormone signaling in order to maintain energy homeostasis via the integration of multiple signals.
Fuel sensing in obesity and type 2 diabetes mellitus
Since peripheral nutrient sensing and its dysregulation represent key components of diet-induced obesity and diabetes mellitus, impaired fuel sensing in the CNS, especially the hypothalamus, may also be involved in the development of high-fat-diet-induced obesity and peripheral insulin resistance. In peripheral tissues, sustained exposure to a supply of nutrients that exceeds energy requirements induces insulin resistance via multiple, convergent mechanisms [220]. Given the diversity of tissues affected by nutrient excess, it is not surprising that resistance to the actions of both insulin and leptin in the CNS represents a hallmark during the development of high-caloric-diet-induced weight gain and peripheral insulin resistance [190]. However, this response has the potential to cause obesity, rather than simply being its consequence, because insulin and leptin are crucial hormonal signals that convey adiposity negative feedback information to the hypothalamus regarding the amount of body fuel stored in the form of fat. When input from these hormones is reduced, this triggers an adaptive increase of food intake that favors body weight gain. Consistently, high-fat-diet-induced obesity reduces the ability of intracerebroventricularly applied insulin, leptin, and LCFA to reduce food intake and glucose production [157, 221–224]. These effects are also present in rats fed a high-fat diet that are calorically and body weight matched to rats on a low-fat diet [222]. Moreover, exposing the animals to a normal chow diet can restore high-fat-feeding-induced leptin resistance in mice, even before body weight has been normalized [225]. Taken together, these data indicate that diet-induced leptin and insulin resistance in the CNS may be caused directly by food components, rather than obesity per se.
Indeed, saturated fatty acids, such as palmitate, which is also increased in the hypothalamus upon high-fat feeding [226], acutely causes leptin resistance in the CNS [227]. Saturated fatty acids represent important candidates for causing diet-induced leptin and insulin resistance, they act as ligands for Toll-like receptor 4 (TLR4) [228], thus activating JNK- and IKK-signaling cascades, both of which inhibit insulin action in peripheral tissues such as skeletal muscle, liver, and adipose tissue [229] as well as in the hypothalamus [230–232]. In the hypothalamus, pro-inflammatory signaling via TLRs not only inhibits neuronal insulin action but can also inhibit leptin signal transduction to further aggravate obesity [227, 233–235]. Consistently, the ability of a high-fat diet to induce obesity and its associated metabolic derangements depends upon neuronal expression of MyD88, a key intracellular mediator of inflammatory signaling [227].
Obesity also correlates with altered hypothalamic glucose sensing, as obese Zucker rats display central glucose hypersensitivity, which has recently been linked to mitochondrial dysfunction leading to changes in redox signaling [236].
A better understanding of the mechanisms by which high-caloric diets and and/or the resulting obesity reduce central sensitivity to peripheral hormonal and nutritional signals may allow for the development of potential novel pharmacological targets for the treatment of obesity and type 2 diabetes mellitus. Nevertheless, many aspects regarding the extra- and intracellular mechanisms that contribute to CNS nutrient and hormonal resistance are still elusive and further investigations will have to unravel the complex network of the CNS in sensing nutrients and transmitting their effects.
Concluding remarks
The global prevalence of obesity and diabetes mellitus is increasing at an alarming rate and dysregulation of the central control of energy homeostasis has been posited as a causative mechanism of these metabolic diseases. Besides the capability of selective brain regions, especially the hypothalamus, to integrate a variety of neuronal and hormonal signals, increasing evidence indicates that fuel sensing also plays a pivotal role in the central regulation of food intake, energy expenditure, and glucose homeostasis. Recent progress has confirmed the presence of distinct hypothalamic neuron populations that respond to changes in circulating nutrient concentrations by either activating or inhibiting their firing activity. At the molecular level, some proteins required for neuronal fuel sensing have already been identified. Among them, KATP channels and the well-known fuel sensor AMPK represent critical points of interaction between hypothalamic glucose and lipid sensing. A wide range of data further implicates the modulation of neuronal fuel sensing by a variety of hormones such as insulin, leptin, and ghrelin, leading to an increased degree of complexity in the response of these neurons to changes in whole-body energy level. Beyond the scope of this review is another relative new concept in central nutrient sensing involving the role of amino acids in the regulation of energy homeostasis. Studies have demonstrated that increasing or decreasing the dietary intake of leucine, a branched-chain amino acid, as well as ICV administration of this amino acid affects glucose metabolism and food intake [237, 238]. The latter was attributed to changes in hypothalamic AgRP expression and mechanistically linked to mammalian target of rapamycine (mTOR) [237, 239].
A better understanding of the complex interaction of nutrients, hormones, and neuronal circuitries in the modulation of central behavioral and metabolic output signals might point to novel interventions to prevent or treat obesity and type 2 diabetes mellitus.
References
Banks WA (2006) Blood–brain barrier and energy balance. Obesity (Silver Spring) 14(Suppl 5):234S–237S
Sanchez-Lasheras C, Christine Konner A, Bruning JC (2009) Integrative neurobiology of energy homeostasis-neurocircuits, signals and mediators. Front Neuroendocrinol 31:4–15
Bernard C (1855) Leçons de physiologie experimentale appliquée à la medecine. Baillère et Fils Paris 2:296–313
Mayer J (1953) Glucostatic mechanism of regulation of food intake. N Engl J Med 249:13–16
Oomura Y, Ono T, Ooyama H, Wayner MJ (1969) Glucose and osmosensitive neurones of the rat hypothalamus. Nature 222:282–284
Anand BK, Chhina GS, Sharma KN, Dua S, Singh B (1964) Activity of single neurons in the hypothalamic feeding centers: effect of glucose. Am J Physiol 207:1146–1154
Oomura Y, Yoshimatsu H (1984) Neural network of glucose monitoring system. J Auton Nerv Syst 10:359–372
Silver IA, Erecinska M (1998) Glucose-induced intracellular ion changes in sugar-sensitive hypothalamic neurons. J Neurophysiol 79:1733–1745
Fioramonti X, Lorsignol A, Taupignon A, Penicaud L (2004) A new ATP-sensitive K+ channel-independent mechanism is involved in glucose-excited neurons of mouse arcuate nucleus. Diabetes 53:2767–2775
Penicaud L, Fioramonti X, Leloup C, Lorsignol A (2006) Glucose sensors and food intake. J Annu Diabetol Hotel Dieu 2006:1–11
Marty N, Dallaporta M, Thorens B (2007) Brain glucose sensing, counterregulation, and energy homeostasis. Physiology (Bethesda) 22:241–251
Norgren R (1978) Projections from the nucleus of the solitary tract in the rat. Neuroscience 3:207–218
Gropp E, Shanabrough M, Borok E, Xu AW, Janoschek R, Buch T, Plum L, Balthasar N, Hampel B, Waisman A, Barsh GS, Horvath TL, Bruning JC (2005) Agouti-related peptide-expressing neurons are mandatory for feeding. Nat Neurosci 8:1289–1291
Konner AC, Klockener T, Bruning JC (2009) Control of energy homeostasis by insulin and leptin: targeting the arcuate nucleus and beyond. Physiol Behav 97:632–638
Zhou A, Bloomquist BT, Mains RE (1993) The prohormone convertases PC1 and PC2 mediate distinct endoproteolytic cleavages in a strict temporal order during proopiomelanocortin biosynthetic processing. J Biol Chem 268:1763–1769
Fan W, Boston BA, Kesterson RA, Hruby VJ, Cone RD (1997) Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature 385:165–168
Biebermann H, Castaneda TR, van Landeghem F, von Deimling A, Escher F, Brabant G, Hebebrand J, Hinney A, Tschop MH, Gruters A, Krude H (2006) A role for beta-melanocyte-stimulating hormone in human body-weight regulation. Cell Metab 3:141–146
Lee YS, Challis BG, Thompson DA, Yeo GS, Keogh JM, Madonna ME, Wraight V, Sims M, Vatin V, Meyre D, Shield J, Burren C, Ibrahim Z, Cheetham T, Swift P, Blackwood A, Hung CC, Wareham NJ, Froguel P, Millhauser GL, O’Rahilly S, Farooqi IS (2006) A POMC variant implicates beta-melanocyte-stimulating hormone in the control of human energy balance. Cell Metab 3:135–140
Adan RA, Cone RD, Burbach JP, Gispen WH (1994) Differential effects of melanocortin peptides on neural melanocortin receptors. Mol Pharmacol 46:1182–1190
Mountjoy KG (1994) The human melanocyte stimulating hormone receptor has evolved to become “super-sensitive” to melanocortin peptides. Mol Cell Endocrinol 102:R7–R11
Stanley BG, Leibowitz SF (1984) Neuropeptide Y: stimulation of feeding and drinking by injection into the paraventricular nucleus. Life Sci 35:2635–2642
Clark JT, Kalra PS, Crowley WR, Kalra SP (1984) Neuropeptide Y and human pancreatic polypeptide stimulate feeding behavior in rats. Endocrinology 115:427–429
Ollmann MM, Wilson BD, Yang YK, Kerns JA, Chen Y, Gantz I, Barsh GS (1997) Antagonism of central melanocortin receptors in vitro and in vivo by agouti-related protein. Science 278:135–138
Ibrahim N, Bosch MA, Smart JL, Qiu J, Rubinstein M, Ronnekleiv OK, Low MJ, Kelly MJ (2003) Hypothalamic proopiomelanocortin neurons are glucose responsive and express K(ATP) channels. Endocrinology 144:1331–1340
Muroya S, Yada T, Shioda S, Takigawa M (1999) Glucose-sensitive neurons in the rat arcuate nucleus contain neuropeptide Y. Neurosci Lett 264:113–116
Mountjoy PD, Bailey SJ, Rutter GA (2007) Inhibition by glucose or leptin of hypothalamic neurons expressing neuropeptide Y requires changes in AMP-activated protein kinase activity. Diabetologia 50:168–177
Fioramonti X, Contie S, Song Z, Routh VH, Lorsignol A, Penicaud L (2007) Characterization of glucosensing neuron subpopulations in the arcuate nucleus: integration in neuropeptide Y and pro-opio melanocortin networks? Diabetes 56:1219–1227
Burdakov D, Luckman SM, Verkhratsky A (2005) Glucose-sensing neurons of the hypothalamus. Phil Trans R Soc Lond B Biol Sci 360:2227–2235
Elias CF, Saper CB, Maratos-Flier E, Tritos NA, Lee C, Kelly J, Tatro JB, Hoffman GE, Ollmann MM, Barsh GS, Sakurai T, Yanagisawa M, Elmquist JK (1998) Chemically defined projections linking the mediobasal hypothalamus and the lateral hypothalamic area. J Comp Neurol 402:442–459
Broberger C, De Lecea L, Sutcliffe JG, Hokfelt T (1998) Hypocretin/orexin- and melanin-concentrating hormone-expressing cells form distinct populations in the rodent lateral hypothalamus: relationship to the neuropeptide Y and agouti gene-related protein systems. J Comp Neurol 402:460–474
Schuit FC, Huypens P, Heimberg H, Pipeleers DG (2001) Glucose sensing in pancreatic beta-cells: a model for the study of other glucose-regulated cells in gut, pancreas, and hypothalamus. Diabetes 50:1–11
Yang XJ, Kow LM, Funabashi T, Mobbs CV (1999) Hypothalamic glucose sensor: similarities to and differences from pancreatic beta-cell mechanisms. Diabetes 48:1763–1772
Mountjoy PD, Rutter GA (2007) Glucose sensing by hypothalamic neurones and pancreatic islet cells: AMPle evidence for common mechanisms? Exp Physiol 92:311–319
Panten U, Christians J, von Kriegstein E, Poser W, Hasselblatt A (1973) Effect of carbohydrates upon fluorescence of reduced pyridine nucleotides from perifused isolated pancreatic islets. Diabetologia 9:477–482
Ashcroft FM, Harrison DE, Ashcroft SJ (1984) Glucose induces closure of single potassium channels in isolated rat pancreatic beta-cells. Nature 312:446–448
Arluison M, Quignon M, Nguyen P, Thorens B, Leloup C, Penicaud L (2004) Distribution and anatomical localization of the glucose transporter 2 (GLUT2) in the adult rat brain—an immunohistochemical study. J Chem Neuroanat 28:117–136
Kang L, Routh VH, Kuzhikandathil EV, Gaspers LD, Levin BE (2004) Physiological and molecular characteristics of rat hypothalamic ventromedial nucleus glucosensing neurons. Diabetes 53:549–559
Leloup C, Arluison M, Lepetit N, Cartier N, Marfaing-Jallat P, Ferre P, Penicaud L (1994) Glucose transporter 2 (GLUT 2): expression in specific brain nuclei. Brain Res 638:221–226
Navarro M, Rodriquez de Fonseca F, Alvarez E, Chowen JA, Zueco JA, Gomez R, Eng J, Blazquez E (1996) Colocalization of glucagon-like peptide-1 (GLP-1) receptors, glucose transporter GLUT-2, and glucokinase mRNAs in rat hypothalamic cells: evidence for a role of GLP-1 receptor agonists as an inhibitory signal for food and water intake. J Neurochem 67:1982–1991
Marty N, Dallaporta M, Foretz M, Emery M, Tarussio D, Bady I, Binnert C, Beermann F, Thorens B (2005) Regulation of glucagon secretion by glucose transporter type 2 (glut2) and astrocyte-dependent glucose sensors. J Clin Invest 115:3545–3553
Matschinsky FM (1996) Banting Lecture 1995. A lesson in metabolic regulation inspired by the glucokinase glucose sensor paradigm. Diabetes 45:223–241
Dunn-Meynell AA, Routh VH, Kang L, Gaspers L, Levin BE (2002) Glucokinase is the likely mediator of glucosensing in both glucose-excited and glucose-inhibited central neurons. Diabetes 51:2056–2065
Jetton TL, Liang Y, Pettepher CC, Zimmerman EC, Cox FG, Horvath K, Matschinsky FM, Magnuson MA (1994) Analysis of upstream glucokinase promoter activity in transgenic mice and identification of glucokinase in rare neuroendocrine cells in the brain and gut. J Biol Chem 269:3641–3654
Lynch RM, Tompkins LS, Brooks HL, Dunn-Meynell AA, Levin BE (2000) Localization of glucokinase gene expression in the rat brain. Diabetes 49:693–700
Wang R, Liu X, Hentges ST, Dunn-Meynell AA, Levin BE, Wang W, Routh VH (2004) The regulation of glucose-excited neurons in the hypothalamic arcuate nucleus by glucose and feeding-relevant peptides. Diabetes 53:1959–1965
Kang L, Dunn-Meynell AA, Routh VH, Gaspers LD, Nagata Y, Nishimura T, Eiki J, Zhang BB, Levin BE (2006) Glucokinase is a critical regulator of ventromedial hypothalamic neuronal glucosensing. Diabetes 55:412–420
Ashcroft FM, Gribble FM (1999) ATP-sensitive K+ channels and insulin secretion: their role in health and disease. Diabetologia 42:903–919
Ashford ML, Boden PR, Treherne JM (1990) Tolbutamide excites rat glucoreceptive ventromedial hypothalamic neurones by indirect inhibition of ATP-K+ channels. Br J Pharmacol 101:531–540
Ashford ML, Boden PR, Treherne JM (1990) Glucose-induced excitation of hypothalamic neurones is mediated by ATP-sensitive K+ channels. Pflugers Arch 415:479–483
Dunn-Meynell AA, Rawson NE, Levin BE (1998) Distribution and phenotype of neurons containing the ATP-sensitive K+ channel in rat brain. Brain Res 814:41–54
Dunn-Meynell AA, Levin BE (1997) Histological markers of neuronal, axonal and astrocytic changes after lateral rigid impact traumatic brain injury. Brain Res 761:25–41
Spanswick D, Smith MA, Groppi VE, Logan SD, Ashford ML (1997) Leptin inhibits hypothalamic neurons by activation of ATP-sensitive potassium channels. Nature 390:521–525
Miki T, Liss B, Minami K, Shiuchi T, Saraya A, Kashima Y, Horiuchi M, Ashcroft F, Minokoshi Y, Roeper J, Seino S (2001) ATP-sensitive K+ channels in the hypothalamus are essential for the maintenance of glucose homeostasis. Nat Neurosci 4:507–512
Song Z, Levin BE, McArdle JJ, Bakhos N, Routh VH (2001) Convergence of pre- and postsynaptic influences on glucosensing neurons in the ventromedial hypothalamic nucleus. Diabetes 50:2673–2681
Evans ML, McCrimmon RJ, Flanagan DE, Keshavarz T, Fan X, McNay EC, Jacob RJ, Sherwin RS (2004) Hypothalamic ATP-sensitive K+ channels play a key role in sensing hypoglycemia and triggering counterregulatory epinephrine and glucagon responses. Diabetes 53:2542–2551
McCrimmon RJ, Evans ML, Fan X, McNay EC, Chan O, Ding Y, Zhu W, Gram DX, Sherwin RS (2005) Activation of ATP-sensitive K+ channels in the ventromedial hypothalamus amplifies counterregulatory hormone responses to hypoglycemia in normal and recurrently hypoglycemic rats. Diabetes 54:3169–3174
Dallaporta M, Perrin J, Orsini JC (2000) Involvement of adenosine triphosphate-sensitive K+ channels in glucose-sensing in the rat solitary tract nucleus. Neurosci Lett 278:77–80
Parton LE, Ye CP, Coppari R, Enriori PJ, Choi B, Zhang CY, Xu C, Vianna CR, Balthasar N, Lee CE, Elmquist JK, Cowley MA, Lowell BB (2007) Glucose sensing by POMC neurons regulates glucose homeostasis and is impaired in obesity. Nature 449:228–232
van den Top M, Lyons DJ, Lee K, Coderre E, Renaud LP, Spanswick D (2007) Pharmacological and molecular characterization of ATP-sensitive K(+) conductances in CART and NPY/AgRP expressing neurons of the hypothalamic arcuate nucleus. Neuroscience 144:815–824
Lee K, Dixon AK, Richardson PJ, Pinnock RD (1999) Glucose-receptive neurones in the rat ventromedial hypothalamus express KATP channels composed of Kir6.1 and SUR1 subunits. J Physiol 515(Pt 2):439–452
Amoroso S, Schmid-Antomarchi H, Fosset M, Lazdunski M (1990) Glucose, sulfonylureas, and neurotransmitter release: role of ATP-sensitive K+ channels. Science 247:852–854
Moriyama R, Tsukamura H, Kinoshita M, Okazaki H, Kato Y, Maeda K (2004) In vitro increase in intracellular calcium concentrations induced by low or high extracellular glucose levels in ependymocytes and serotonergic neurons of the rat lower brainstem. Endocrinology 145:2507–2515
Song Z, Routh VH (2005) Differential effects of glucose and lactate on glucosensing neurons in the ventromedial hypothalamic nucleus. Diabetes 54:15–22
Ren X, Zhou L, Terwilliger R, Newton SS, de Araujo IE (2009) Sweet taste signaling functions as a hypothalamic glucose sensor. Front Integr Neurosci 3:12
Oomura Y, Ooyama H, Sugimori M, Nakamura T, Yamada Y (1974) Glucose inhibition of the glucose-sensitive neurone in the rat lateral hypothalamus. Nature 247:284–286
Burdakov D, Jensen LT, Alexopoulos H, Williams RH, Fearon IM, O’Kelly I, Gerasimenko O, Fugger L, Verkhratsky A (2006) Tandem-pore K+ channels mediate inhibition of orexin neurons by glucose. Neuron 50:711–722
Guyon A, Tardy MP, Rovere C, Nahon JL, Barhanin J, Lesage F (2009) Glucose inhibition persists in hypothalamic neurons lacking tandem-pore K+ channels. J Neurosci 29:2528–2533
Gonzalez JA, Jensen LT, Fugger L, Burdakov D (2008) Metabolism-independent sugar sensing in central orexin neurons. Diabetes 57:2569–2576
Williams RH, Burdakov D (2008) Hypothalamic orexins/hypocretins as regulators of breathing. Expert Rev Mol Med 10:e28
Canabal DD, Potian JG, Duran RG, McArdle JJ, Routh VH (2007) Hyperglycemia impairs glucose and insulin regulation of nitric oxide production in glucose-inhibited neurons in the ventromedial hypothalamus. Am J Physiol Regul Integr Comp Physiol 293:R592–R600
Hardie DG, Hawley SA, Scott JW (2006) AMP-activated protein kinase—development of the energy sensor concept. J Physiol 574:7–15
Kahn BB, Alquier T, Carling D, Hardie DG (2005) AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab 1:15–25
Hardie DG, Carling D (1997) The AMP-activated protein kinase—fuel gauge of the mammalian cell? Eur J Biochem 246:259–273
Kitamura T, Feng Y, Ido Kitamura Y, Chua SC, Xu AW, Barsh GS, Rossetti L, Accili D (2006) Forkhead protein FoxO1 mediates Agrp-dependent effects of leptin on food intake. Nat Med 12(5):534–540
Lerner RG, Depatie C, Rutter GA, Screaton RA, Balthasar N (2009) A role for the CREB co-activator CRTC2 in the hypothalamic mechanisms linking glucose sensing with gene regulation. EMBO Rep 10:1175–1181
Murphy BA, Fakira KA, Song Z, Beuve A, Routh VH (2009) AMP-activated protein kinase and nitric oxide regulate the glucose sensitivity of ventromedial hypothalamic glucose-inhibited neurons. Am J Physiol Cell Physiol 297:C750–C758
Magistretti PJ, Pellerin L, Rothman DL, Shulman RG (1999) Energy on demand. Science 283:496–497
Pierre K, Pellerin L, Debernardi R, Riederer BM, Magistretti PJ (2000) Cell-specific localization of monocarboxylate transporters, MCT1 and MCT2, in the adult mouse brain revealed by double immunohistochemical labeling and confocal microscopy. Neuroscience 100:617–627
Ainscow EK, Mirshamsi S, Tang T, Ashford ML, Rutter GA (2002) Dynamic imaging of free cytosolic ATP concentration during fuel sensing by rat hypothalamic neurones: evidence for ATP-independent control of ATP-sensitive K(+) channels. J Physiol 544:429–445
Lam TK, Gutierrez-Juarez R, Pocai A, Rossetti L (2005) Regulation of blood glucose by hypothalamic pyruvate metabolism. Science 309:943–947
Sekine N, Cirulli V, Regazzi R, Brown LJ, Gine E, Tamarit-Rodriguez J, Girotti M, Marie S, MacDonald MJ, Wollheim CB (1994) Low lactate dehydrogenase and high mitochondrial glycerol phosphate dehydrogenase in pancreatic beta-cells. Potential role in nutrient sensing. J Biol Chem 269:4895–4902
Ishihara H, Wang H, Drewes LR, Wollheim CB (1999) Overexpression of monocarboxylate transporter and lactate dehydrogenase alters insulin secretory responses to pyruvate and lactate in beta cells. J Clin Invest 104:1621–1629
Mayer J (1953) Genetic, traumatic and environmental factors in the etiology of obesity. Physiol Rev 33:472–508
Louis-Sylvestre J, Le Magnen J (1980) Fall in blood glucose level precedes meal onset in free-feeding rats. Neurosci Biobehav Rev 4(Suppl 1):13–15
Campfield LA, Brandon P, Smith FJ (1985) On-line continuous measurement of blood glucose and meal pattern in free-feeding rats: the role of glucose in meal initiation. Brain Res Bull 14:605–616
Davis JD, Wirtshafter D, Asin KE, Brief D (1981) Sustained intracerebroventricular infusion of brain fuels reduces body weight and food intake in rats. Science 212:81–83
Panksepp J, Rossi J 3rd (1981) D-glucose infusions into the basal ventromedial hypothalamus and feeding. Behav Brain Res 3:381–392
Kurata K, Fujimoto K, Sakata T, Etou H, Fukagawa K (1986) D-glucose suppression of eating after intra-third ventricle infusion in rat. Physiol Behav 37:615–620
Berthoud HR, Mogenson GJ (1977) Ingestive behavior after intracerebral and intracerebroventricular infusions of glucose and 2-deoxy-D-glucose. Am J Physiol 233:R127–R133
Miselis RR, Epstein AN (1975) Feeding induced by intracerebroventricular 2-deoxy-D-glucose in the rat. Am J Physiol 229:1438–1447
Ritter S, Dinh TT, Zhang Y (2000) Localization of hindbrain glucoreceptive sites controlling food intake and blood glucose. Brain Res 856:37–47
Ritter S, Bugarith K, Dinh TT (2001) Immunotoxic destruction of distinct catecholamine subgroups produces selective impairment of glucoregulatory responses and neuronal activation. J Comp Neurol 432:197–216
Fraley GS, Ritter S (2003) Immunolesion of norepinephrine and epinephrine afferents to medial hypothalamus alters basal and 2-deoxy-D-glucose-induced neuropeptide Y and agouti gene-related protein messenger ribonucleic acid expression in the arcuate nucleus. Endocrinology 144:75–83
Luquet S, Perez FA, Hnasko TS, Palmiter RD (2005) NPY/AgRP neurons are essential for feeding in adult mice but can be ablated in neonates. Science 310:683–685
Luquet S, Phillips CT, Palmiter RD (2007) NPY/AgRP neurons are not essential for feeding responses to glucoprivation. Peptides 28:214–225
Wan HZ, Hulsey MG, Martin RJ (1998) Intracerebroventricular administration of antisense oligodeoxynucleotide against GLUT2 glucose transporter mRNA reduces food intake, body weight change and glucoprivic feeding response in rats. J Nutr 128:287–291
Bady I, Marty N, Dallaporta M, Emery M, Gyger J, Tarussio D, Foretz M, Thorens B (2006) Evidence from glut2-null mice that glucose is a critical physiological regulator of feeding. Diabetes 55:988–995
Eny KM, Wolever TM, Fontaine-Bisson B, El-Sohemy A (2008) Genetic variant in the glucose transporter type 2 is associated with higher intakes of sugars in two distinct populations. Physiol Genomics 33:355–360
Arluison M, Quignon M, Thorens B, Leloup C, Penicaud L (2004) Immunocytochemical localization of the glucose transporter 2 (GLUT2) in the adult rat brain. II. Electron microscopic study. J Chem Neuroanat 28:137–146
Li B, Xi X, Roane DS, Ryan DH, Martin RJ (2003) Distribution of glucokinase, glucose transporter GLUT2, sulfonylurea receptor-1, glucagon-like peptide-1 receptor and neuropeptide Y messenger RNAs in rat brain by quantitative real-time RT-PCR. Brain Res Mol Brain Res 113:139–142
Garcia MA, Millan C, Balmaceda-Aguilera C, Castro T, Pastor P, Montecinos H, Reinicke K, Zuniga F, Vera JC, Onate SA, Nualart F (2003) Hypothalamic ependymal-glial cells express the glucose transporter GLUT2, a protein involved in glucose sensing. J Neurochem 86:709–724
Maekawa F, Toyoda Y, Torii N, Miwa I, Thompson RC, Foster DL, Tsukahara S, Tsukamura H, Maeda K (2000) Localization of glucokinase-like immunoreactivity in the rat lower brain stem: for possible location of brain glucose-sensing mechanisms. Endocrinology 141:375–384
Ngarmukos C, Baur EL, Kumagai AK (2001) Co-localization of GLUT1 and GLUT4 in the blood–brain barrier of the rat ventromedial hypothalamus. Brain Res 900:1–8
Yang XJ, Mastaitis J, Mizuno T, Mobbs CV (2007) Glucokinase regulates reproductive function, glucocorticoid secretion, food intake, and hypothalamic gene expression. Endocrinology 148:1928–1932
Plum L, Ma X, Hampel B, Balthasar N, Coppari R, Munzberg H, Shanabrough M, Burdakov D, Rother E, Janoschek R, Alber J, Belgardt BF, Koch L, Seibler J, Schwenk F, Fekete C, Suzuki A, Mak TW, Krone W, Horvath TL, Ashcroft FM, Bruning JC (2006) Enhanced PIP3 signaling in POMC neurons causes KATP channel activation and leads to diet-sensitive obesity. J Clin Invest 116:1886–1901
Baukrowitz T, Schulte U, Oliver D, Herlitze S, Krauter T, Tucker SJ, Ruppersberg JP, Fakler B (1998) PIP2 and PIP as determinants for ATP inhibition of KATP channels. Science 282:1141–1144
MacGregor GG, Dong K, Vanoye CG, Tang L, Giebisch G, Hebert SC (2002) Nucleotides and phospholipids compete for binding to the C terminus of KATP channels. Proc Natl Acad Sci USA 99:2726–2731
Shyng SL, Nichols CG (1998) Membrane phospholipid control of nucleotide sensitivity of KATP channels. Science 282:1138–1141
Mirshamsi S, Laidlaw HA, Ning K, Anderson E, Burgess LA, Gray A, Sutherland C, Ashford ML (2004) Leptin and insulin stimulation of signalling pathways in arcuate nucleus neurones: PI3K-dependent actin reorganization and KATP channel activation. BMC Neurosci 5:54
Claret M, Smith MA, Batterham RL, Selman C, Choudhury AI, Fryer LG, Clements M, Al-Qassab H, Heffron H, Xu AW, Speakman JR, Barsh GS, Viollet B, Vaulont S, Ashford ML, Carling D, Withers DJ (2007) AMPK is essential for energy homeostasis regulation and glucose sensing by POMC and AgRP neurons. J Clin Invest 117:2325–2336
Levin BE, Sullivan AC (1987) Glucose-induced norepinephrine levels and obesity resistance. Am J Physiol 253:R475–R481
Levin BE, Sullivan AC (1987) Glucose, insulin and sympathoadrenal activation. J Auton Nerv Syst 20:233–242
Acheson KJ, Ravussin E, Wahren J, Jequier E (1984) Thermic effect of glucose in man. Obligatory and facultative thermogenesis. J Clin Invest 74:1572–1580
Levin BE, Sullivan AC (1989) Glucose-induced sympathetic activation in obesity-prone and resistant rats. Int J Obes 13:235–246
Levin BE (1991) Glucose increases rat plasma norepinephrine levels by direct action on the brain. Am J Physiol 261:R1351–R1357
Levin BE, Govek EK, Dunn-Meynell AA (1998) Reduced glucose-induced neuronal activation in the hypothalamus of diet-induced obese rats. Brain Res 808:317–319
Sakaguchi T, Bray GA (1987) The effect of intrahypothalamic injections of glucose on sympathetic efferent firing rate. Brain Res Bull 18:591–595
Sakaguchi T, Bray GA (1988) Sympathetic activity following paraventricular injections of glucose and insulin. Brain Res Bull 21:25–29
Rother E, Konner AC, Bruning JC (2008) Neurocircuits integrating hormone and nutrient signaling in control of glucose metabolism. Am J Physiol Endocrinol Metab 294:E810–E816
Pocai A, Lam TK, Gutierrez-Juarez R, Obici S, Schwartz GJ, Bryan J, Aguilar-Bryan L, Rossetti L (2005) Hypothalamic K(ATP) channels control hepatic glucose production. Nature 434:1026–1031
Kokorovic A, Cheung GW, Rossetti L, Lam TK (2008) Hypothalamic sensing of circulating lactate regulates glucose production. J Cell Mol Med 57:594–605
Lam TK, Gutierrez-Juarez R, Pocai A, Bhanot S, Tso P, Schwartz GJ, Rossetti L (2007) Brain glucose metabolism controls the hepatic secretion of triglyceride-rich lipoproteins. Nat Med 13:171–180
Cryer PE (2004) Diverse causes of hypoglycemia-associated autonomic failure in diabetes. N Engl J Med 350:2272–2279
Mitrakou A, Ryan C, Veneman T, Mokan M, Jenssen T, Kiss I, Durrant J, Cryer P, Gerich J (1991) Hierarchy of glycemic thresholds for counterregulatory hormone secretion, symptoms, and cerebral dysfunction. Am J Physiol 260:E67–E74
Taborsky GJ Jr, Ahren B, Havel PJ (1998) Autonomic mediation of glucagon secretion during hypoglycemia: implications for impaired alpha-cell responses in type 1 diabetes. Diabetes 47:995–1005
Biggers DW, Myers SR, Neal D, Stinson R, Cooper NB, Jaspan JB, Williams PE, Cherrington AD, Frizzell RT (1989) Role of brain in counterregulation of insulin-induced hypoglycemia in dogs. Diabetes 38:7–16
Frizzell RT, Jones EM, Davis SN, Biggers DW, Myers SR, Connolly CC, Neal DW, Jaspan JB, Cherrington AD (1993) Counterregulation during hypoglycemia is directed by widespread brain regions. Diabetes 42:1253–1261
Borg WP, Sherwin RS, During MJ, Borg MA, Shulman GI (1995) Local ventromedial hypothalamus glucopenia triggers counterregulatory hormone release. Diabetes 44:180–184
Borg MA, Sherwin RS, Borg WP, Tamborlane WV, Shulman GI (1997) Local ventromedial hypothalamus glucose perfusion blocks counterregulation during systemic hypoglycemia in awake rats. J Clin Invest 99:361–365
Chan O, Zhu W, Ding Y, McCrimmon RJ, Sherwin RS (2006) Blockade of GABA(A) receptors in the ventromedial hypothalamus further stimulates glucagon and sympathoadrenal but not the hypothalamo-pituitary-adrenal response to hypoglycemia. Diabetes 55:1080–1087
Chan O, Lawson M, Zhu W, Beverly JL, Sherwin RS (2007) ATP-sensitive K(+) channels regulate the release of GABA in the ventromedial hypothalamus during hypoglycemia. Diabetes 56:1120–1126
Chan O, Cheng H, Herzog R, Czyzyk D, Zhu W, Wang A, McCrimmon RJ, Seashore MR, Sherwin RS (2008) Increased GABAergic tone in the ventromedial hypothalamus contributes to suppression of counterregulatory responses after antecedent hypoglycemia. Diabetes 57:1363–1370
Tong Q, Ye C, McCrimmon RJ, Dhillon H, Choi B, Kramer MD, Yu J, Yang Z, Christiansen LM, Lee CE, Choi CS, Zigman JM, Shulman GI, Sherwin RS, Elmquist JK, Lowell BB (2007) Synaptic glutamate release by ventromedial hypothalamic neurons is part of the neurocircuitry that prevents hypoglycemia. Cell Metab 5:383–393
Page KA, Arora J, Qiu M, Relwani R, Constable RT, Sherwin RS (2009) Small decrements in systemic glucose provoke increases in hypothalamic blood flow prior to the release of counterregulatory hormones. Diabetes 58:448–452
Ritter RC, Slusser PG, Stone S (1981) Glucoreceptors controlling feeding and blood glucose: location in the hindbrain. Science 213:451–452
Ritter S, Llewellyn-Smith I, Dinh TT (1998) Subgroups of hindbrain catecholamine neurons are selectively activated by 2-deoxy-D-glucose induced metabolic challenge. Brain Res 805:41–54
Thorens B, Guillam MT, Beermann F, Burcelin R, Jaquet M (2000) Transgenic reexpression of GLUT1 or GLUT2 in pancreatic beta cells rescues GLUT2-null mice from early death and restores normal glucose-stimulated insulin secretion. J Biol Chem 275:23751–23758
Rapoport SI (1996) In vivo labeling of brain phospholipids by long-chain fatty acids: relation to turnover and function. Lipids 31(Suppl):S97–S101
Miller JC, Gnaedinger JM, Rapoport SI (1987) Utilization of plasma fatty acid in rat brain: distribution of [14C]palmitate between oxidative and synthetic pathways. J Neurochem 49:1507–1514
Qi K, Hall M, Deckelbaum RJ (2002) Long-chain polyunsaturated fatty acid accretion in brain. Curr Opin Clin Nutr Metab Care 5:133–138
Rapoport SI (2001) In vivo fatty acid incorporation into brain phosholipids in relation to plasma availability, signal transduction and membrane remodeling. J Mol Neurosci 16:243–261 discussion 279–284
Oomura Y, Nakamura T, Sugimori M, Yamada Y (1975) Effect of free fatty acid on the rat lateral hypothalamic neurons. Physiol Behav 14:483–486
Cruciani-Guglielmacci C, Hervalet A, Douared L, Sanders NM, Levin BE, Ktorza A, Magnan C (2004) Beta oxidation in the brain is required for the effects of non-esterified fatty acids on glucose-induced insulin secretion in rats. Diabetologia 47:2032–2038
Wang R, Cruciani-Guglielmacci C, Migrenne S, Magnan C, Cotero VE, Routh VH (2006) Effects of oleic acid on distinct populations of neurons in the hypothalamic arcuate nucleus are dependent on extracellular glucose levels. J Neurophysiol 95:1491–1498
Witt MR, Nielsen M (1994) Characterization of the influence of unsaturated free fatty acids on brain GABA/benzodiazepine receptor binding in vitro. J Neurochem 62:1432–1439
Tewari KP, Malinowska DH, Sherry AM, Cuppoletti J (2000) PKA and arachidonic acid activation of human recombinant ClC-2 chloride channels. Am J Physiol Cell Physiol 279:C40–C50
Zheng HF, Li XL, Jin ZY, Sun JB, Li ZL, Xu WX (2005) Effects of unsaturated fatty acids on calcium-activated potassium current in gastric myocytes of guinea pigs. World J Gastroenterol 11:672–675
Honen BN, Saint DA, Laver DR (2003) Suppression of calcium sparks in rat ventricular myocytes and direct inhibition of sheep cardiac RyR channels by EPA, DHA and oleic acid. J Membr Biol 196:95–103
Oishi K, Zheng B, Kuo JF (1990) Inhibition of Na, K-ATPase and sodium pump by protein kinase C regulators sphingosine, lysophosphatidylcholine, and oleic acid. J Biol Chem 265:70–75
Lam TK, Pocai A, Gutierrez-Juarez R, Obici S, Bryan J, Aguilar-Bryan L, Schwartz GJ, Rossetti L (2005) Hypothalamic sensing of circulating fatty acids is required for glucose homeostasis. Nat Med 11:320–327
Migrenne S, Magnan C, Cruciani-Guglielmacci C (2007) Fatty acid sensing and nervous control of energy homeostasis. Diabetes Metab 33:177–182
Woods SC, Stein LJ, McKay LD, Porte D Jr (1984) Suppression of food intake by intravenous nutrients and insulin in the baboon. Am J Physiol 247:R393–R401
Monnikes H, Lauer G, Bauer C, Tebbe J, Zittel TT, Arnold R (1997) Pathways of Fos expression in locus ceruleus, dorsal vagal complex, and PVN in response to intestinal lipid. Am J Physiol 273:R2059–R2071
Matzinger D, Degen L, Drewe J, Meuli J, Duebendorfer R, Ruckstuhl N, D’Amato M, Rovati L, Beglinger C (2000) The role of long-chain fatty acids in regulating food intake and cholecystokinin release in humans. Gut 46:688–693
Schwartz GJ, Salorio CF, Skoglund C, Moran TH (1999) Gut vagal afferent lesions increase meal size but do not block gastric preload-induced feeding suppression. Am J Physiol 276:R1623–R1629
Obici S, Feng Z, Morgan K, Stein D, Karkanias G, Rossetti L (2002) Central administration of oleic acid inhibits glucose production and food intake. Diabetes 51:271–275
Morgan K, Obici S, Rossetti L (2004) Hypothalamic responses to long-chain fatty acids are nutritionally regulated. J Biol Chem 279:31139–31148
Obici S, Feng Z, Arduini A, Conti R, Rossetti L (2003) Inhibition of hypothalamic carnitine palmitoyltransferase-1 decreases food intake and glucose production. Nat Med 9:756–761
He W, Lam TK, Obici S, Rossetti L (2006) Molecular disruption of hypothalamic nutrient sensing induces obesity. Nat Neurosci 9:227–233
Lam TK, Schwartz GJ, Rossetti L (2005) Hypothalamic sensing of fatty acids. Nat Neurosci 8:579–584
Beverly JL, Martin RJ (1991) Influence of fatty acid oxidation in lateral hypothalamus on food intake and body composition. Am J Physiol 261:R339–R343
Sorensen A, Travers MT, Vernon RG, Price NT, Barber MC (2002) Localization of messenger RNAs encoding enzymes associated with malonyl-CoA metabolism in mouse brain. Brain Res Gene Expr Patterns 1:167–173
Kim EK, Miller I, Landree LE, Borisy-Rudin FF, Brown P, Tihan T, Townsend CA, Witters LA, Moran TH, Kuhajda FP, Ronnett GV (2002) Expression of FAS within hypothalamic neurons: a model for decreased food intake after C75 treatment. Am J Physiol Endocrinol Metab 283:E867–E879
Loftus TM, Jaworsky DE, Frehywot GL, Townsend CA, Ronnett GV, Lane MD, Kuhajda FP (2000) Reduced food intake and body weight in mice treated with fatty acid synthase inhibitors. Science 288:2379–2381
Shimokawa T, Kumar MV, Lane MD (2002) Effect of a fatty acid synthase inhibitor on food intake and expression of hypothalamic neuropeptides. Proc Natl Acad Sci USA 99:66–71
Hu Z, Cha SH, Chohnan S, Lane MD (2003) Hypothalamic malonyl-CoA as a mediator of feeding behavior. Proc Natl Acad Sci USA 100:12624–12629
Hu Z, Dai Y, Prentki M, Chohnan S, Lane MD (2005) A role for hypothalamic malonyl-CoA in the control of food intake. J Biol Chem 280:39681–39683
Ronnett GV, Kim EK, Landree LE, Tu Y (2005) Fatty acid metabolism as a target for obesity treatment. Physiol Behav 85:25–35
Kim EK, Miller I, Aja S, Landree LE, Pinn M, McFadden J, Kuhajda FP, Moran TH, Ronnett GV (2004) C75, a fatty acid synthase inhibitor, reduces food intake via hypothalamic AMP-activated protein kinase. J Biol Chem 279:19970–19976
Gao S, Lane MD (2003) Effect of the anorectic fatty acid synthase inhibitor C75 on neuronal activity in the hypothalamus and brainstem. Proc Natl Acad Sci USA 100:5628–5633
Sako Y, Grill VE (1990) A 48-hour lipid infusion in the rat time-dependently inhibits glucose-induced insulin secretion and B cell oxidation through a process likely coupled to fatty acid oxidation. Endocrinology 127:1580–1589
Zhou YP, Grill VE (1994) Long-term exposure of rat pancreatic islets to fatty acids inhibits glucose-induced insulin secretion and biosynthesis through a glucose fatty acid cycle. J Clin Invest 93:870–876
Prentki M, Nolan CJ (2006) Islet beta cell failure in type 2 diabetes. J Clin Invest 116:1802–1812
Gremlich S, Bonny C, Waeber G, Thorens B (1997) Fatty acids decrease IDX-1 expression in rat pancreatic islets and reduce GLUT2, glucokinase, insulin, and somatostatin levels. J Biol Chem 272:30261–30269
Eizirik DL, Cardozo AK, Cnop M (2008) The role for endoplasmic reticulum stress in diabetes mellitus. Endocr Rev 29:42–61
Wortman MD, Clegg DJ, D’Alessio D, Woods SC, Seeley RJ (2003) C75 inhibits food intake by increasing CNS glucose metabolism. Nat Med 9:483–485
Le Foll C, Irani BG, Magnan C, Dunn-Meynell A, Levin BE (2009) Effects of maternal genotype and diet on offspring glucose and fatty acid-sensing ventromedial hypothalamic nucleus neurons. Am J Physiol Regul Integr Comp Physiol 297:R1351–R1357
Leloup C, Magnan C, Benani A, Bonnet E, Alquier T, Offer G, Carriere A, Periquet A, Fernandez Y, Ktorza A, Casteilla L, Penicaud L (2006) Mitochondrial reactive oxygen species are required for hypothalamic glucose sensing. Diabetes 55:2084–2090
Benani A, Troy S, Carmona MC, Fioramonti X, Lorsignol A, Leloup C, Casteilla L, Penicaud L (2007) Role for mitochondrial reactive oxygen species in brain lipid sensing: redox regulation of food intake. Diabetes 56:152–160
Andrews ZB, Liu ZW, Walllingford N, Erion DM, Borok E, Friedman JM, Tschop MH, Shanabrough M, Cline G, Shulman GI, Coppola A, Gao XB, Horvath TL, Diano S (2008) UCP2 mediates ghrelin’s action on NPY/AgRP neurons by lowering free radicals. Nature 454:846–851
Auerbach JM, Segal M (1997) Peroxide modulation of slow onset potentiation in rat hippocampus. J Neurosci 17:8695–8701
Avshalumov MV, Bao L, Patel JC, Rice ME (2007) H2O2 signaling in the nigrostriatal dopamine pathway via ATP-sensitive potassium channels: issues and answers. Antioxid Redox Signal 9:219–231
Horvath TL, Andrews ZB, Diano S (2009) Fuel utilization by hypothalamic neurons: roles for ROS. Trends Endocrinol Metab 20:78–87
Belgardt BF, Okamura T, Bruning JC (2009) Hormone and glucose signalling in POMC and AgRP neurons. J Physiol 587:5305–5314
Woods SC, Lotter EC, McKay LD, Porte D Jr (1979) Chronic intracerebroventricular infusion of insulin reduces food intake and body weight of baboons. Nature 282:503–505
McGowan MK, Andrews KM, Fenner D, Grossman SP (1993) Chronic intrahypothalamic insulin infusion in the rat: behavioral specificity. Physiol Behav 54:1031–1034
Porte D Jr, Woods SC (1981) Regulation of food intake and body weight in insulin. Diabetologia 20(Suppl):274–280
Bruning JC, Gautam D, Burks DJ, Gillette J, Schubert M, Orban PC, Klein R, Krone W, Muller-Wieland D, Kahn CR (2000) Role of brain insulin receptor in control of body weight and reproduction. Science 289:2122–2125
Schwartz MW, Woods SC, Porte D Jr, Seeley RJ, Baskin DG (2000) Central nervous system control of food intake. Nature 404:661–671
Schwartz MW, Porte D Jr (2005) Diabetes, obesity, and the brain. Science 307:375–379
Koch L, Wunderlich FT, Seibler J, Konner AC, Hampel B, Irlenbusch S, Brabant G, Kahn CR, Schwenk F, Bruning JC (2008) Central insulin action regulates peripheral glucose and fat metabolism in mice. J Clin Invest 118:2132–2147
Konner AC, Janoschek R, Plum L, Jordan SD, Rother E, Ma X, Xu C, Enriori P, Hampel B, Barsh GS, Kahn CR, Cowley MA, Ashcroft FM, Bruning JC (2007) Insulin action in AgRP-expressing neurons is required for suppression of hepatic glucose production. Cell Metab 5:438–449
Obici S, Zhang BB, Karkanias G, Rossetti L (2002) Hypothalamic insulin signaling is required for inhibition of glucose production. Nat Med 8:1376–1382
Niswender KD, Morrison CD, Clegg DJ, Olson R, Baskin DG, Myers MG Jr, Seeley RJ, Schwartz MW (2003) Insulin activation of phosphatidylinositol 3-kinase in the hypothalamic arcuate nucleus: a key mediator of insulin-induced anorexia. Diabetes 52:227–231
Cotero VE, Routh VH (2009) Insulin blunts the response of glucose-excited neurons in the ventrolateral-ventromedial hypothalamic nucleus to decreased glucose. Am J Physiol Endocrinol Metab 296:E1101–E1109
Maffei M, Halaas J, Ravussin E, Pratley RE, Lee GH, Zhang Y, Fei H, Kim S, Lallone R, Ranganathan S, Kern PA, Friedman JM (1995) Leptin levels in human and rodent: measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat Med 1:1155–1161
Frederich RC, Hamann A, Anderson S, Lollmann B, Lowell BB, Flier JS (1995) Leptin levels reflect body lipid content in mice: evidence for diet-induced resistance to leptin action. Nat Med 1:1311–1314
Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM (1994) Positional cloning of the mouse obese gene and its human homologue. Nature 372:425–432
Chen H, Charlat O, Tartaglia LA, Woolf EA, Weng X, Ellis SJ, Lakey ND, Culpepper J, Moore KJ, Breitbart RE, Duyk GM, Tepper RI, Morgenstern JP (1996) Evidence that the diabetes gene encodes the leptin receptor: identification of a mutation in the leptin receptor gene in db/db mice. Cell 84:491–495
Cohen P, Zhao C, Cai X, Montez JM, Rohani SC, Feinstein P, Mombaerts P, Friedman JM (2001) Selective deletion of leptin receptor in neurons leads to obesity. J Clin Invest 108:1113–1121
de Luca C, Kowalski TJ, Zhang Y, Elmquist JK, Lee C, Kilimann MW, Ludwig T, Liu SM, Chua SC Jr (2005) Complete rescue of obesity, diabetes, and infertility in db/db mice by neuron-specific LEPR-B transgenes. J Clin Invest 115:3484–3493
Schwartz MW, Seeley RJ, Campfield LA, Burn P, Baskin DG (1996) Identification of targets of leptin action in rat hypothalamus. J Clin Invest 98:1101–1106
Plum L, Belgardt BF, Bruning JC (2006) Central insulin action in energy and glucose homeostasis. J Clin Invest 116:1761–1766
Hill JW, Williams KW, Ye C, Luo J, Balthasar N, Coppari R, Cowley MA, Cantley LC, Lowell BB, Elmquist JK (2008) Acute effects of leptin require PI3K signaling in hypothalamic proopiomelanocortin neurons in mice. J Clin Invest 118:1796–1805
Niswender KD, Morton GJ, Stearns WH, Rhodes CJ, Myers MG Jr, Schwartz MW (2001) Intracellular signalling. Key enzyme in leptin-induced anorexia. Nature 413:794–795
Belgardt BF, Husch A, Rother E, Ernst MB, Wunderlich FT, Hampel B, Klockener T, Alessi D, Kloppenburg P, Bruning JC (2008) PDK1 deficiency in POMC-expressing cells reveals FOXO1-dependent and -independent pathways in control of energy homeostasis and stress response. Cell Metab 7:291–301
Plum L, Schubert M, Bruning JC (2005) The role of insulin receptor signaling in the brain. Trends Endocrinol Metab 16:59–65
Minokoshi Y, Alquier T, Furukawa N, Kim YB, Lee A, Xue B, Mu J, Foufelle F, Ferre P, Birnbaum MJ, Stuck BJ, Kahn BB (2004) AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature 428:569–574
Andersson U, Filipsson K, Abbott CR, Woods A, Smith K, Bloom SR, Carling D, Small CJ (2004) AMP-activated protein kinase plays a role in the control of food intake. J Biol Chem 279:12005–12008
Minokoshi Y, Kim YB, Peroni OD, Fryer LG, Muller C, Carling D, Kahn BB (2002) Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature 415:339–343
Murphy BA, Fioramonti X, Jochnowitz N, Fakira K, Gagen K, Contie S, Lorsignol A, Penicaud L, Martin WJ, Routh VH (2009) Fasting enhances the response of arcuate neuropeptide Y-glucose-inhibited neurons to decreased extracellular glucose. Am J Physiol Cell Physiol 296:C746–C756
Cummings DE, Purnell JQ, Frayo RS, Schmidova K, Wisse BE, Weigle DS (2001) A preprandial rise in plasma ghrelin levels suggests a role in meal initiation in humans. Diabetes 50:1714–1719
Date Y, Kojima M, Hosoda H, Sawaguchi A, Mondal MS, Suganuma T, Matsukura S, Kangawa K, Nakazato M (2000) Ghrelin, a novel growth hormone-releasing acylated peptide, is synthesized in a distinct endocrine cell type in the gastrointestinal tracts of rats and humans. Endocrinology 141:4255–4261
Cowley MA, Smith RG, Diano S, Tschop M, Pronchuk N, Grove KL, Strasburger CJ, Bidlingmaier M, Esterman M, Heiman ML, Garcia-Segura LM, Nillni EA, Mendez P, Low MJ, Sotonyi P, Friedman JM, Liu H, Pinto S, Colmers WF, Cone RD, Horvath TL (2003) The distribution and mechanism of action of ghrelin in the CNS demonstrates a novel hypothalamic circuit regulating energy homeostasis. Neuron 37:649–661
Theander-Carrillo C, Wiedmer P, Cettour-Rose P, Nogueiras R, Perez-Tilve D, Pfluger P, Castaneda TR, Muzzin P, Schurmann A, Szanto I, Tschop MH, Rohner-Jeanrenaud F (2006) Ghrelin action in the brain controls adipocyte metabolism. J Clin Invest 116:1983–1993
Kohno D, Sone H, Minokoshi Y, Yada T (2008) Ghrelin raises [Ca2 +]i via AMPK in hypothalamic arcuate nucleus NPY neurons. Biochem Biophys Res Commun 366:388–392
Anderson KA, Ribar TJ, Lin F, Noeldner PK, Green MF, Muehlbauer MJ, Witters LA, Kemp BE, Means AR (2008) Hypothalamic CaMKK2 contributes to the regulation of energy balance. Cell Metab 7:377–388
Sleeman MW, Latres E (2008) The CAMplexities of central ghrelin. Cell Metab 7:361–362
Lopez M, Lage R, Saha AK, Perez-Tilve D, Vazquez MJ, Varela L, Sangiao-Alvarellos S, Tovar S, Raghay K, Rodriguez-Cuenca S, Deoliveira RM, Castaneda T, Datta R, Dong JZ, Culler M, Sleeman MW, Alvarez CV, Gallego R, Lelliott CJ, Carling D, Tschop MH, Dieguez C, Vidal-Puig A (2008) Hypothalamic fatty acid metabolism mediates the orexigenic action of ghrelin. Cell Metab 7:389–399
Muoio DM, Newgard CB (2008) Mechanisms of disease: molecular and metabolic mechanisms of insulin resistance and beta-cell failure in type 2 diabetes. Nat Rev Mol Cell Biol 9:193–205
Wang J, Obici S, Morgan K, Barzilai N, Feng Z, Rossetti L (2001) Overfeeding rapidly induces leptin and insulin resistance. Diabetes 50:2786–2791
Woods SC, D’Alessio DA, Tso P, Rushing PA, Clegg DJ, Benoit SC, Gotoh K, Liu M, Seeley RJ (2004) Consumption of a high-fat diet alters the homeostatic regulation of energy balance. Physiol Behav 83:573–578
El-Haschimi K, Pierroz DD, Hileman SM, Bjorbaek C, Flier JS (2000) Two defects contribute to hypothalamic leptin resistance in mice with diet-induced obesity. J Clin Invest 105:1827–1832
Arase K, Fisler JS, Shargill NS, York DA, Bray GA (1988) Intracerebroventricular infusions of 3-OHB and insulin in a rat model of dietary obesity. Am J Physiol 255:R974–R981
Enriori PJ, Evans AE, Sinnayah P, Jobst EE, Tonelli-Lemos L, Billes SK, Glavas MM, Grayson BE, Perello M, Nillni EA, Grove KL, Cowley MA (2007) Diet-induced obesity causes severe but reversible leptin resistance in arcuate melanocortin neurons. Cell Metab 5:181–194
Posey K, Clegg DJ, Printz RL, Byun J, Morton GJ, Vivekanandan-Giri A, Pennathur S, Baskin DG, Heinecke JW, Woods SC, Schwartz MW, Niswender KD (2008) Hypothalamic proinflammatory lipid accumulation, inflammation, and insulin resistance in rats fed a high-fat diet. Am J Physiol Endocrinol Metab 296(5):E1003–E1012
Kleinridders A, Schenten D, Konner AC, Belgardt BF, Mauer J, Okamura T, Wunderlich FT, Medzhitov R, Bruning JC (2009) MyD88 signaling in the CNS is required for development of fatty acid-induced leptin resistance and diet-induced obesity. Cell Metab 10:249–259
Lee JY, Sohn KH, Rhee SH, Hwang D (2001) Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through Toll-like receptor 4. J Biol Chem 276:16683–16689
Wellen KE, Hotamisligil GS (2003) Obesity-induced inflammatory changes in adipose tissue. J Clin Invest 112:1785–1788
De Souza CT, Araujo EP, Bordin S, Ashimine R, Zollner RL, Boschero AC, Saad MJ, Velloso LA (2005) Consumption of a fat-rich diet activates a proinflammatory response and induces insulin resistance in the hypothalamus. Endocrinology 146:4192–4199
Posey KA, Clegg DJ, Printz RL, Byun J, Morton GJ, Vivekanandan-Giri A, Pennathur S, Baskin DG, Heinecke JW, Woods SC, Schwartz MW, Niswender KD (2009) Hypothalamic proinflammatory lipid accumulation, inflammation, and insulin resistance in rats fed a high-fat diet. Am J Physiol Endocrinol Metab 296:E1003–E1012
Belgardt BF, Mauer J, Wunderlich FT, Ernst MB, Pal M, Spohn G, Bronneke HS, Brodesser S, Hampel B, Schauss AC, Bruning JC (2010) Hypothalamic and pituitary c-Jun N-terminal kinase 1 signaling coordinately regulates glucose metabolism. Proc Natl Acad Sci USA 107(13):6028–6033
Milanski M, Degasperi G, Coope A, Morari J, Denis R, Cintra DE, Tsukumo DM, Anhe G, Amaral ME, Takahashi HK, Curi R, Oliveira HC, Carvalheira JB, Bordin S, Saad MJ, Velloso LA (2009) Saturated fatty acids produce an inflammatory response predominantly through the activation of TLR4 signaling in hypothalamus: implications for the pathogenesis of obesity. J Neurosci 29:359–370
Ozcan L, Ergin AS, Lu A, Chung J, Sarkar S, Nie D, Myers MG Jr, Ozcan U (2009) Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab 9:35–51
Zhang X, Zhang G, Zhang H, Karin M, Bai H, Cai D (2008) Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell 135:61–73
Colombani AL, Carneiro L, Benani A, Galinier A, Jaillard T, Duparc T, Offer G, Lorsignol A, Magnan C, Casteilla L, Penicaud L, Leloup C (2009) Enhanced hypothalamic glucose sensing in obesity: alteration of redox signaling. Diabetes 58:2189–2197
Cota D, Proulx K, Smith KA, Kozma SC, Thomas G, Woods SC, Seeley RJ (2006) Hypothalamic mTOR signaling regulates food intake. Science 312:927–930
Zhang Y, Guo K, LeBlanc RE, Loh D, Schwartz GJ, Yu YH (2007) Increasing dietary leucine intake reduces diet-induced obesity and improves glucose and cholesterol metabolism in mice via multimechanisms. Diabetes 56:1647–1654
Morrison CD, Xi X, White CL, Ye J, Martin RJ (2007) Amino acids inhibit Agrp gene expression via an mTOR-dependent mechanism. Am J Physiol Endocrinol Metab 293:E165–E171
Acknowledgments
We apologize to all colleagues whose important contributions could not be cited due to space limitations. We thank G. Schmall and T. Rayle for excellent secretarial assistance. This work was supported by grants from the CMMC (TV2), the EU (LSHM-CT-2003-503041) and the DFG (Br. 1492/7-1) to J.C.B.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Jordan, S.D., Könner, A.C. & Brüning, J.C. Sensing the fuels: glucose and lipid signaling in the CNS controlling energy homeostasis. Cell. Mol. Life Sci. 67, 3255–3273 (2010). https://doi.org/10.1007/s00018-010-0414-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-010-0414-7