
Abstract
A considerable number of genes that code for AU-rich mRNAs including cytokines, growth factors, transcriptional factors, and certain receptors are involved in both chronic inflammation and cancer. Overexpression of these genes is affected by aberrations or by prolonged activation of several signaling pathways. AU-rich elements (ARE) are important cis-acting short sequences in the 3′UTR that mediate recognition of an array of RNA-binding proteins and affect mRNA stability and translation. This review addresses the cellular and molecular mechanisms that are common between inflammation and cancer and that also govern ARE-mediated post-transcriptional control. The first part examines the role of the ARE-genes in inflammation and cancer and sequence characteristics of AU-rich elements. The second part addresses the common signaling pathways in inflammation and cancer that regulate the ARE-mediated pathways and how their deregulations affect ARE-gene regulation and disease outcome.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cancer and chronic inflammatory conditions including autoimmune diseases are among the leading causes of morbidity. Knowledge about the signaling pathways that are common between inflammation and cancer can lead to deeper insights into the underlying molecular mechanisms of disease and improved therapeutic approaches. A large body of literature supports the notion that common signaling pathways are deregulated in chronic inflammation and cancer (Table 1). In normal cells or cells that are not undergoing inflammatory response, many signaling pathways are dormant, but become transiently activated during immune response and cellular growth. In chronic inflammation and cancer, continued activation of these signaling pathways are distinct features in the biology of these two processes. These same signaling pathways also lead to increased stabilization of a class of mRNAs contributing to the increased expression of these gene products that are involved in inflammation and cancer. Cancer, as a disease, spans alterations in many biological processes and is thus involved not only in oncogenesis but also in maintenance, invasion, hypoxia/nutrient deprivation response, angiogenesis, and metastasis. Certain genes of pro-inflammatory nature contribute to one or more of these processes (Fig. 1).

ARE-genes in inflammation-cancer crosstalk. Infectious agents or their products, such as endotoxin, and chronic irritations stimulate inflammatory and immune system cells such as the macrophages to release pro-inflammatory cytokines. Many of these cytokines are encoded by AU-rich mRNAs. In a chronic inflammation setting, several signaling pathways that lead to stabilization of ARE-mRNAs are continually activated leading to sustained production of the cytokines. In a cancer-associated microenvironment, these cytokines can promote the several processes that underlie cancer, including tumor growth, resistance to apoptosis, angiogenesis, invasion and metastasis. In turn, tumor cells themselves can release chemokines and other mediators that amplify the inflammatory response by attracting further inflammatory cells and stimulating further ARE-gene expression of cytokines and growth factors. Conditions in the tumor microenvironment such as hypoxia can also trigger the expression of ARE-mRNAs of pro-inflammatory cytokines, chemokines, and growth factors
Several key proteins that share common functions in inflammation and cancer process are coded by AU-rich mRNAs (Table 2). The AU-rich mRNAs are a class of mRNAs that bear AU-rich elements (ARE) in their 3′ untranslated regions (3′UTR). Estimated now to be in the vicinity of 10–15% of all transcripts [1], the ARE-mRNAs comprise a functionally diverse group including inflammatory and immune response, transcription, cellular proliferation, RNA metabolism, development, and signaling [2]. This functional enrichment of ARE-genes correlates with the rapid mRNA decay patterns, particularly with those involved in transcription and signaling [3]. The mRNA stability is a product of not only the cis-acting sequences such as the ARE but also transacting factors, e.g., RNA-binding proteins, that bind directly or indirectly to the cis-acting elements and promote the deadenylation and degradation of the mRNA. The most studied roles of ARE are in mRNA stability and translation. These are modulated during cellular stimulation by inactivation of RNA decay-promoting proteins such as zinc-finger protein, tristetraprolin (TTP) [4], K-homology splicing-regulatory protein (KSRP) [5], butyrate response factor 1 and 2 [6], and certain gene products of the AU-rich element (ARE)/poly(U)-binding/degradation factor 1 (AUF1) [7]. The mRNA stability can be enhanced by the activity of mRNA stabilization-promoting proteins notably the human antigen R (HuR). ARE-mediated changes in mRNA stability are important in processes that require transient responses such as cellular growth, defense to pathogens, cardiovascular toning, and external stress-mediated pathways. Stabilization of the ARE-mRNAs can cause prolonged responses that subsequently may lead to undesirable, e.g., diseased, states including chronic inflammatory diseases and cancer.
ARE-genes in inflammation and cancer
A number of important ARE-genes are commonly involved in inflammation and cancer (Table 2). Different functional categories and sequence classes of ARE genes are represented in these genes which regulate processes underlying inflammation and cancer. These genes include cytokines, chemokines, growth factors, transcriptional factors, RNA-binding proteins, and others. The following is an appraisal of notable examples of ARE-genes that have been widely studied for their involvement in inflammation and cancer.
Tumor necrosis factor in chronic inflammatory conditions and cancer
Tumor necrosis factor (TNF-α) is an AU-rich mRNA coding gene (ARE-gene) that has well-validated ARE. TNF-α is a pro-inflammatory cytokine that triggers the expression of many inflammatory mediators including chemokines. In addition to inducing transcription of a number of ARE-genes, such as interleukin-8 (IL-8), IFN-γ induced IP-10 (CXCL-10), and colony stimulating factors, TNF-α also mediates post-transcriptional regulation by increasing the stability of their mRNAs [8, 9]. Notable evidence on the role of ARE in TNF-α pro-inflammatory action comes from studies of mice with targeted deletion of the ARE in the TNF-α gene [8]. Here, chronic inflammatory arthritis and Crohn’s-like inflammatory bowel disease were observed. Further indirect evidence of the role of ARE in TNF-α production and associated inflammatory pathology is from studies on mice with targeted deletion of ZFP36 that code for the ARE-mRNA destabilizing protein, TTP; the mice exhibit severe inflammatory syndrome, characterized by severe polyarticular arthritis, myeloid hyperplasia, autoimmunity, dermatitis, conjunctivitis, glomerular mesangial thickening, and cachexia [9]. These pathologies are largely the result of increased stability of the mRNAs for TNF-α and GM-CSF and thereby increased secretion of these cytokines [8–10].
Despite the notion of its name as tumor necrosis protein, TNF-α is in fact a pro-tumor molecule in a number of cancers (reviewed in [11]). Blockage of TNF-α in mice models results in: failure of hepatitis to develop hepatocellular carcinoma [12], reduction of colorectal carcinogenesis associated with chronic colitis [13], inhibition of pancreatic tumor growth and metastasis [14], and promotion of epithelial ovarian cancer [15]. Other possibilities of TNF-α as a cancer promoter are also shown with other types of cancer [11]. It can act as a growth factor in certain leukemias and lymphomas [16]. Thus, TNF-α is involved both in inflammation and cancer and provides one of the links between chronic inflammation and, in cancer, at least certain tumor types.
Colitis-associated cancer is a progression from inflammatory bowel disease, and thus it is one of the best examples of inflammation-induced cancer situations. It has been demonstrated that TNF-α can induce the epithelial-to-mesenchymal transition of colon carcinoma [17]. Drug-induced colitis that leads to tumors is associated with increased TNF-α secretion; in TNF-Receptor p55-knockout mice, reduced mucosal damage, reduced infiltration of macrophages and neutrophils, and attenuated subsequent tumor formation are clearly seen [13]. In addition, monocyte chemoattractant protein-1 (CCL2) plays a role in colitis-associated colon tumors, and this could be mediated by the expression of cyclooxygenase (COX-2) [18].
Involvement of COX-2 and VEGF in inflammation and cancer
COX-2, a prostaglandin synthase enzyme, is an important AU-rich mRNA which codes for Class 2 ARE (Table 2). COX-2 has two isoforms, both of which catalyze the committed step in the prostaglandin production pathway; COX-1 is present under basal conditions while COX-2 is an inducible form by many inflammatory and growth promoters that result in prostaglandin synthesis associated with inflammation and cancer. Nonsteroidal antiinflammatory inhibitor drugs (NSAIDs) such as aspirin and indomethacin inhibit both COX-1 and COX-2 while newer generations of NSAIDs selectively inhibit COX-2.
The increased activity of COX-2 is associated with colon and other cancers, and promotes cellular proliferation, resistance to apoptosis, angiogenesis, and metastasis [19–21]. The inflammatory cells in the tumor microenvironment contribute further to COX-2-mediated tumorgenesis via induction of factors and cytokines that increase COX-2 gene expression which involves ARE-mediated mechanisms. Other pro-inflammatory ARE-genes such as TNF-α, IL-1, platelet derived factor (PDGF), vascular endothelial growth factor (VEGF), and others can upregulate COX-2, and, thus, inflammation amplifies cancer through the cytokine induction of COX-2 (Fig. 1).
Another important ARE gene that mediates both inflammation and cancer is VEGF. VEGF is upregulated during inflammatory response to increase blood supply, and, therefore, increases in oxygen, nutrients, and infiltrating leukocytes, to the site of inflammation for lesion repair. The source of VEGF is the leukocytes themselves and endothelial tissues, and both can promote angiogenesis of tumors. VEGF can promote inflammatory intestinal angiogenesis and leukocyte adhesion during inflammatory bowel syndrome [22] which can lead to colon cancer. VEGF is potently induced by the hypoxia-induced factor, HIF1α, a transcriptional factor and also an ARE-gene. Hypoxia is a prominent feature of malignant tumors that is characterized by angiogenesis and vascular hyper permeability. HIF-1α is constitutively overexpressed in several cancers as a result of hypoxia and induces VEGF mRNA stability [23]. Pro-inflammatory cytokines, in the tumor microenvironment, such as TNF-α and IL-1, activate HIF1α. HIF1α, as result of hypoxia during tumor expansion, can act as an inflammatory mediator, and has been shown to have a role in leukocyte migration and in stromal inflammatory cells [24, 25].
Vascular endothelial growth factor triggers the expression of urokinase activator, uPA, a serine protease, and its receptor, uPAR, which is also an ARE-gene; uPA converts plasminogen to plasmin that leads to the degradation of the extra-cellular matrix, an important component of the invasion and metastasis. The increased activity of uPA and its receptor has been associated with invasiveness in a number of tumors [26]. Another related angiogenesis-promoting protein is IL-8, a member of the inflammatory chemokines, a group of cytokines that are involved in chemotaxis of leukocytes to inflamed regions. IL-8 can promote angiogenesis and metastasis in a number of cancers [27, 28]. Thus, in general, chemokines can amplify cancer processes in which tumor-associated leukocytes can release chemokines and growth factors that further maintain or promote tumor growth and survival. Certain chemokines can act as growth factors and some of these chemokines are coded by ARE-genes such as IL-8 and the GRO family which play a role in melanoma growth and metastasis [29].
Other pro-inflammatory cytokines and mediators such as IL-1, IL-6, and inducible nitric oxide synthase (iNOS) play roles in cancer (Table 2). Overexpression or prolongation of iNOS due to chronic inflammation can cause cellular injury in arthritis or colitis-promoting tumors by inducing DNA damage and cellular proliferation [30]. Pro-inflammatory cytokines, many of which are coded by ARE-genes, are released from the tumor micro-environment favoring the initiation and progression of cancer. These happen when these cytokine ARE-genes themselves tend to exert further ARE-mediated post-transcriptional effects that lead to production of the growth factors in neighboring cells (Fig. 1). Having described the role of ARE-genes in chronic inflammation and cancer, the next section addresses the ARE and their sequence structure–function relationships.
AU-rich elements and their sequence characteristics
Sequence characteristics of ARE classes
The ARE are a heterogeneous class of adenylate uridylate-rich sequences that range from loosely defined U-rich or AU-rich sequences to well-defined repeats of overleaping pentamers of AUUUA or the nonamer UUAUUUAUU. The latter motif or its variation UUAUUUAWW is considered the minimally functional ARE sequence; a stronger effect on RNA stability is obtained with two overlapping copies of this nonamer [31–35]. The pentamer AUUUA in non-AU context is not functional, and, as a matter of fact, it is ubiquitous in many mRNAs, regardless of the mRNA region. With computational biology analysis of labile mRNAs, WWWUAUUUAUWW is the most consensus motif in labile AU-rich mRNAs and is highly specific to the 3′UTR when compared to other regions of the mRNA, e.g., 5′UTR and coding regions [36]. This motif, including variations around the core pentamer, is the basis for building up the ARE-mRNA database (ARED). The ARED motif is closely related to the Class 2 ARE based on the Chen et al. classification where Class 1 ARE have discontinuous nonamers and Class 2 ARE have at least two overlapping copies of the nonamer [31]. Both class II ARE and ARED motifs are found preferentially in labile transcripts in different cell types that are stimulated with different ligands including fibroblasts, hepatocytes, and immune system-type cells such as α-CD3- or CD28-activated T cells and endotoxin-induced monocytes; thus, in general, genome-wide studies confirm the association between these ARE and rapid mRNA decay [3, 37–39].
ARE recognition
Further definition and minimal function of the ARE motif was sought by interaction studies of synthetic RNA molecules and RNA-binding proteins. It appears that from the various studies that functional ARE can exist as either linear non-structured RNA molecules or as structural RNA, i.e., stem-loop structures [40–42], as in the case of short ARE or certain non-ARE mRNA decay elements. In the first case, the minimally functional linear ARE, UUAUUUAUU, or the 13-bp ARED motif is required. The RNA motif NNUUNNUUU for HuR binding is also functional as linear conformation or as a linear segment within a secondary structure; the latter conformation promotes further recognition by the RNA-binding protein [40–43]. This RNA motif is still within the 13-bp ARED motif WWWUAUUUAUWWW, and also within the linear ARE segment within a structured RNA of HuR targets that contains UUNNUU(G/U)UUWW [43]. Similarly, work with the neuronal RNA-binding protein HuD shows that the three RNA-binding domains interact with a minimal 13-bp motif that includes UU(AUUU)NAUU [44]. Further evidence of ARE functionality is from work on interactions of synthetic RNA molecules with TTP also showing that as small as UAUUUAU and UUAUUUAUU can bind TTP and that the 13-bp motif ARE has a strong affinity for TTP [45, 46]. The adenylate residues in the pentamer (AUUUA)or hexamer (AUUUUA) core are important for TTP binding but not necessarily for Hu proteins, which unlike TTP, can bind a U-rich tract [44–46]. Large-scale analysis of ARE based on reporter live cell-based assays are still needed to correlate many of these findings with actual functional effects of ARE variations on RNA and protein levels. A different approach to delineate the ARE and other mRNA destabilization sequence elements has been adopted in recent years in which target mRNAs physically associated with RNA-binding protein are profiled using microarrays followed by RNA motif derivation bioinformatics [43, 47–49]. These motifs are still characterized by short U-rich dinucleotides and clusters but may inherit authenticity of the RNA-binding protein association with its target transcripts since not all the target mRNAs harbor the identified sequence signature.
Length of the 3′UTR and the ARE
Although the ARE can exist in other regions of the mRNA, though to a much lesser extent, such as 5′UTR and coding regions, the 3′UTR are the main source of ARE [36]. The 5′UTR are well known for their translation regulatory role in cellular and viral mRNA including cap-dependent and internal ribosomal entry site (IRES)-mediated processes [50] but can have a function in mRNA decay, for example iron response element [49, 50]. Few coding regions have been reported to bear mRNA instability motifs, though not typical of ARE, such as those in interferon-β and CD38 coding regions [51, 52].
The 3′UTR length can be a determinant factor in RNA stability. It appears that the 3′UTR of ARE-mRNA is longer than non-ARE mRNAs such as those of housekeeping genes [53–55]. Longer 3′UTR tend to have a higher proportion of miRNA targets, indicating the higher order of post-transcriptional regulation complexity. Certain miRNAs may synergize with ARE on the same 3′UTR by reducing RNA stability and/or translation [56]. Alternative polyadenylation that results in differential expression of short and long 3’UTR is widespread in the transcriptome. This length alteration can reflect functional responses towards miRNAs, ARE involvement, and RNA-binding proteins. Short 3′UTRs escape at least a part of miRNA-mediated suppression and ARE-mediated decay [57, 58]. Indeed, this 3′UTR length-mediated control has a functional outcome on protein levels and activity including cellular growth [57, 58].
The length of ARE itself, e.g., the number of the overlapping pentamer, AUUUA, may contribute to the mRNA half-life. Experimentally, it has been shown that the number of ARE pentamers correlate with the half-life of ARE-mRNAs [59]. Bioinformatics clustering of ARED resulted in five groups, I–V, that include mRNAs whose 3′UTRs have five, four, three, and two consecutive AUUUA pentamers, and single AUUUA pentamer in flanking AU-context, respectively. The number of these transcripts in these groups increased dramatically from group I to group V. The last group is heterogeneous and large and not necessarily functional while the key characteristic of other groups’ transcripts is the presence of the WWAUUUAUUUAWW sequence which likely RNA-binding proteins such as TTP could bind to, thus resulting in a large number of potential TTP and other RNA-binding protein mRNA targets [60]. In a detailed analysis of ARE-mRNA half-lives in endotoxin-stimulated monocytes and activated T cells, longer ARE are associated with shorter half-lives [37, 38].
The ARE are conserved among different species [1]. However, orthologs may bear different ARE arrangements such as the number of overlapping pentamers in AU-rich clusters. Although a significant proportion (>25%) of human genes differ in their ARE patterns from mouse and rat transcripts [1]. This suggests that the ARE-mediated post-transcriptional control for a given gene may be functional in one organism but not in another. The heterogeneity may also reflect diversity of the AU-rich binding proteins and their protein–protein interactions. In general, the ARE are the most studied and validated mRNA destabilization elements. However, other mRNA destabilization elements do exist [3]. The existence of the GU-rich element motif, UGUUUGUUUGU, which may mediate mRNA decay, in a number of labile mRNAs has been recently shown [61]. Altogether, it is clear that there are several mRNA decay elements in the 3′UTR which may explain the repertoire of RNA-binding proteins that are thought to exist in thousands.
Regulation of ARE-gene expression: RNA-binding proteins
The RNA-binding proteins play key roles in each of the processes in which the ARE are involved including RNA processing, export, localization, degradation, and translation. At default, the ARE maintain the instability behavior of many ARE-mRNAs and have an inhibitory action on translation [62, 63]. In certain conditions, the ARE can switch into a translation activation signal, such as in the case of TNF-α mRNA translation during serum starvation that leads to cell cycle arrest [64]. Not all the RNA-binding proteins that affect post-transcriptional regulation of genes are discussed here. The HuR and TTP proteins are the most studied RNA-binding proteins in the context of chronic inflammatory conditions and cancer and regulate ARE-genes involved in these diseases. Thus, more emphasis on these two proteins is given throughout this review.
The tristetraprolin and HuR RNA-binding proteins
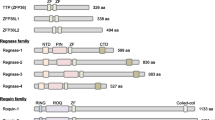
TTP is a part of the zinc-finger family proteins consisting of conserved Cys-Cys-Cys-His domain and comprising TTP (also known as ZFP36, TIS11, G0S24 and NUP475), ZFP36L1 (TIS11b, cMG1, ERF1, BRF1 and Berg36) and ZFP36L2 (TIS11d, ERF2 and BRF2) [65]. Mutation of either of the two zinc-fingers of TTP by a single-point mutation (Cys to Arg), e.g., C124R mutant, fails to bind TNF-α 3′UTR and its ARE [66]. Well-established mRNA targets for TTP binding are involved in inflammation include the mRNAs of TNF-α, GM-CSF and IL-3 [10, 67–69]. Furthermore, two targets of TTP that are related to both inflammation and cancer have been identified, namely COX-2 [70, 71] and VEGF [72]. A polyadenylation ARE-mRNA transcript variant of HuR has recently been uncovered and is responsive to HuR auto-regulation and to wild-type TTP mRNA decay action but not to the zinc-finger mutant (C124R)[73]. Transcriptome analysis using RNA from TTP wild-type and knockout cells identified further mRNA targets [55, 74].
Human antigen R is a member of the mammalian homologues of embryonic lethal abnormal vision (ELAV) proteins comprising a group of RNA-binding proteins first described in Drosophila [75]. It is predominantly nuclear, but translocates to the cytoplasm upon cellular activation, binds selected mRNAs and causes their stabilization [76]. HuR and other members of ELAV proteins possess three RNA-recognition motifs through which they bind with high affinity to specific mRNAs bearing AU- and U-rich sequences affecting their stability and/or translation [77–81]. The array of HuR target mRNAs is large, as analyzed by profiling of mRNAs that are bound to HuR protein which include many cytokines, growth factors, cell-cycle regulators, and many others [43, 80, 82]. Recent work, particularly with conditional deletion of HuR in mice, reveals complex and essential roles of HuR in development including thymic T cell maturation, placental branching morphogenesis, spleen ontogeny, intestinal integrity, and hematopoietic progenitor cell survival [83–85].
Tristetraprolin and several other RNA-binding proteins such as BRF1, KSRP, and AUF1 appear to function by targeting AU-rich mRNA to the exosome, a multiprotein complex with 3′–5′ exoribonuclease activity representing a major degradation scheme in eukaryotes [86]. When phosphorylated, these RNA-binding proteins are complexed with 14-3-3 proteins that influence their sub-cellular localization and mRNA decay activity [6, 87, 88]. The 14-3-3 proteins also protect RNA-binding proteins such as TTP from de-phosphorylation by the protein phosphatase 2α40, resulting in further stabilization of ARE-mRNA [89]. ARE binding proteins can also function by recruitment of degradation factors other than the exosome [90, 91]. Another pathway of 5′–3′ decay can also occur during degradation of ARE-mRNAs in the cytoplasmic RNA foci called processing bodies [92].
In tumor cells or during the cell cycle, and also in response to inflammatory stimuli, HuR may stabilize certain mRNAs by the displacement or inhibition of factors such as TTP, KSRP or AUF1 that specifically cleave or deadenylate these mRNAs. Associations of KSRP and AUF1 p45, or KSRP and HuR, allow fine tuning of ARE-mRNA stability [93, 94]. A specific example is that siRNA-mediated knockdown of HuR led to larger AUF1 association with cyclin D and p21 mRNAs [80]. Ectopic expression of HuR overcomes TTP-mediated destabilization of IL-3 mRNA and HuR mRNA in an ARE-dependent manner [69, 73]. CUGBP2 can override HuR and suppresses COX-2 mRNA translation [95]. Pro-inflammatory cytokines can reduce KSRP binding and enhance HuR binding to iNOS 3′UTR-ARE leading to its stabilization [96]. Thus, it appears that HuR antagonize ARE-mRNA decay-promoting proteins during chronic inflammation and cancer.
Other RNA-binding proteins
AUF1 is another characterized RNA-binding protein with two quasi-RRM domains that consists of four protein isomers of 37, 40, 42, and 45 kDa differing in their terminal domains and their RNA-binding activity [97]. AUF1 binds to and destabilizes both inflammation and cancer-associated ARE-mRNAs such as c-myc, TNF-α, GM-CSF and others [98, 99]. Like TTP, AUF1 acts as another negative feedback mediator of the innate response to inflammatory stimuli such as endotoxin; LPS challenge in AUF1-knockout mice displays symptoms of severe endotoxic shock due to overproduction of TNF-α and IL-1β [99]. However, in certain conditions, certain isoforms of AUF1 or during effects on certain ARE classes, AUF1 can stabilize mRNAs, for example during heat shock, and in certain cancer situations such as in the case of cyclin D1 and c-fos mRNA [98, 100]. The fragile X mental retardation, autosomal homolog 1 (FXR1), is a K–H type RNA-binding protein that interacts with TNF-α ARE to either repress or upregulate mRNA translation during serum growth or starvation conditions, respectively [64, 101]. FXR1 knockout and conditional knockout mice exhibit muscle wasting that may be attributed to increased TNF-α mRNA translation [101]. A recent review describes in detail the role of the miRNP (microribonucleoprotein)-associated proteins FXR1 and Argonaute 2 as effector molecules that bind the ARE to activate TNF-α mRNA translation [102].
These examples demonstrate that there is a complex interplay of RNA-binding protein in the 3′UTR and ARE regions that determines the outcome of the mRNA half-life as affected by external stimuli. Further details on the function of the RNA-binding proteins are given in the next section. What are the common signaling pathways that operate during chronic inflammation and cancer and how do they affect RNA-binding proteins interactions with ARE?
Common ARE-mediated signaling pathways in inflammation and cancer
A significant link between inflammation and cancer arise from the molecular and physiological observations of the effect of NSAIDs; attenuation of chronic inflammation by NSAIDs corresponds to lower incidence of cancer, notably colon cancer. Thus, one may conclude that signaling pathways or at least part of them—that are targeted by NSAIDs—are operative in both inflammation and cancer. One of these is the NF-κB pathway (recently reviewed in [103, 104]). Many of the cancer-promoting proteins are ARE-genes in which their gene transcriptions are triggered by NF-κB (Fig. 1). Stimulation of these genes by pro-inflammatory cytokines themselves or ligands of toll-like receptor initiates signaling that diverges into transcriptional (NF-κB) and post-transcriptional regulatory pathways [105, 106]. The latter pathways constitute the mitogen-activated protein kinase (MAPK), phosphatidylinositol 3-kinase (PI3 K)-AKT, Wnt/β-catenin pathways, and others [6, 37, 87, 107]. Since signaling pathways are deregulated in cancer and common with those occurring during chronic inflammatory conditions, it is likely these continually active signaling pathways lead to changes in the expression and localization of RNA-binding proteins that favor tumorgenesis and chronic inflammation. Most of the studies on signaling pathways have been performed with transcriptional induction in contrast to post-transcriptional modulation. However, the p38 MAP kinase and PI3 kinase pathways are the most studied signaling pathways regulating ARE-mediated post-transcriptional events (Fig. 2). That does not mean the other pathways are not involved since there are indeed scattered but non-extensive reports on their involvements (Table 1).

Common signaling pathways in inflammation and cancer including those regulating ARE-gene expression and mRNA stability. Microbial stimuli, inflammatory and tumor inducers, and ARE-gene products themselves such as certain growth factors and pro-inflammatory cytokines trigger a number of signaling pathways. One of those is the p38 MAPK pathway that leads to phosphorylation and inactivation of the activity of the RNA decay-promoting proteins such as TTP and KSRP. This results in stabilization of ARE-mRNAs that are involved in chronic inflammation and cancer. The RNA-binding stabilizing protein, HuR, is able to compete with the RNA-binding proteins such as TTP and AUF1 for the same mRNA targets; however, it can also act independently on mRNA targets for promoting mRNA stabilization
The mitogen-activated protein kinase pathways
Inflammatory stimuli activate the three MAPK major pathways: extracellular signal-regulated kinase (ERK), c-jun N-terminal kinase (JNK), and p38 MAPK pathways. Abnormalities in signaling pathways of MAPK, particularly the ERK pathway, are associated with cancers in mice and humans (reviewed in [108]). Constitutive activation of ERK can occur with activation of Raf and Ras oncogenes that are commonly deregulated in cancer. The JNK pathway can be activated in certain cancer types [109] and has also been demonstrated in the stabilization of VEGF and iNOS [110]. Downstream of MAPK are other kinases such as the MAPK-activated protein kinase 2 (MAPKAPK2 or MK2) which regulate both transcriptional and post-transcriptional gene expression. The p38 MAPK pathway activation in inflammation is one of the most common pathways that regulate mRNA stability in multiple cell types. This pathway is central to the stabilization of many ARE-mRNA involved in inflammation and cancer, including COX-2, TNF-α, IL-3, IL-6, IL-8, macrophage inhibitory protein 1 (MIP1), GM-CSF, VEGF, c-fos, and uPA [69, 111–116]. The pro-inflammatory cytokines, IL-1α and TNF-α, have been found to stabilize a significant number of cytokines and chemokines including CXCL-10 (IP-10), GRO proteins, and fractalkine (SCYD1), and several genes involved in regulation of inflammatory responses such as TNFAIP3, NFκBαI, and mannose-binding lectin (MBL2) [117–119]. These cytokines and many others are regulated by the p38 MAPK signaling and its target, MK2, to trigger the ARE-mRNA stabilization. Not all ARE-mRNAs, regardless of the ARE class, are targets for the MAPK p38 pathway [37]. Thus, the p38 MAPK pathway is not essential for all ARE-mRNAs and appears to be also dependent on the stimulus. Cooperative action of p38 MAPK and ERK pathways can be necessary for optimal ARE-mRNA stabilization, particularly in the case of inhibition of TTP [120]. Another p38 MAPK downstream target with structural and functional similarity to MK2 is MK3; it is thought to cooperate with MK2 in TNF-α mRNA stabilization [121].
Effect of p38 MAPK on TTP and HuR functions
Activation of the p38 MAPK pathway leads to MK2-mediated phosphorylation of TTP and KSRP leading to loss of function of these RNA-binding proteins, e.g., reduced affinity to ARE regions, and subsequently ARE-mRNA stabilization (Fig. 2) [122–124]. A detailed role for MK2 is demonstrated, for example, in the stability of IL-8 mRNA since mutants of MK2 interfere with IL-8 mRNA stability [116]. Selective activation of the p38 MAPK pathway by MAPK kinase 6 induces mRNA stabilization of IL-8 [112]. MK2 can regulate IL-6 at the levels of mRNA stability, and of TNF-α mainly through TNF-ARE-dependent translational control [125]. In vitro evidence shows that TTP can be directly phosphorylated by either p38 or MK2 potentially modifying mRNA destabilizing activity of TTP [121, 122]. Activation of p38 MAPK pathway by pro-inflammatory cytokines such as IL-1α can phosphorylate KSRP leading to loss of its binding activity to ARE and subsequently mRNA stabilization [93, 124]. The MK2-knockout mice with induced collagen arthritis exhibit decreased disease incidence and severity, associated with reduced TNF-α and IL-6 serum levels [123]. Inhibition of p38 using the pharmacological inhibitor SB203580 (SB), or by expressing dominant negative MK2, abolishes endotoxin-induced COX-2 mRNA stabilization and leads to rapid degradation of COX-2 mRNA [124–126]. Recent evidence shows that p38 regulates stability of several ARE-mRNAs, e.g., COX-2, IL-1α, IL-6, and IL-10 exclusively through TTP phosphorylation such as in murine bone marrow-derived macrophages [126].These above studies and others offer strong evidence for the p38 MAPK regulation of ARE-mRNA stability and the association with loss of TTP function.
The p38 MAPK activation further results in hosting of the RNA stabilizing proteins such as HuR in place of the displaced RNA decay-promoting proteins. The p38 MAPK may also phosphorylate ARE-stabilizing proteins such as HuR which could compete with the destabilizing proteins such as TTP as suggested in studies on the regulation of IL-3 mRNA decay [127]. The p38 MAPK regulates HuR localization and subsequently HuR mRNA targets that are involved in chronic inflammation and cancer such as uPA and COX-2 [128–130]. Certain signaling pathways converge or cooperate with MAPK pathways in the ARE-mediated post-transcriptional control such as the PI3-AKT and β-catenin pathways (Fig. 2); both of these and others are discussed in the next section. For further details on MAPK and post-transcriptional gene regulation, several excellent reviews have appeared in the literature [131, 132].
The PI3-AKT pathway
The PI3 kinase pathway comprises lipid and protein kinases that regulate numerous biological processes, including cell growth, differentiation, survival, chemotaxis, and metabolism. As a result of ligand engagement of a number of receptors for integrin, tyrosine kinases, cytokines, and growth factors, activation of the PI3 kinase catalyzes the addition of a phosphate group to the phosphatidylinositol lipids (PIP2), generating different 3′ phosphorylated products (PIP3) that act as second messengers. PIP3 leads to membrane recruitment of the cystolic AKT3 to the membrane and its activation (Fig. 2). Activated AKT3 (also called protein kinase B) is a central serine-threonine kinase effector that participates in the various biological processes such as the cell cycle progression and survival (e.g., reduced apoptosis). Deregulation of the PI3 K signaling pathway has been foundto be frequently associated with cancer, and simple overexpression of wild-type PI3 Kα can be sufficient to induce an oncogenic phenotype in cultured cells [133]. Mutations in the PI3 kinase components, genetic alterations in tyrosine kinase receptors, or loss of PI3 kinase negative regulator, PTEN, can be detected in a large number of human tumors. Several tyrosine kinase growth factor oncoproteins activate the PI3-kinase pathway including EGFR (an ARE-gene), PDGFR, VEGFR, stem-cell factor (kit, an ARE-gene) and hepatocyte growth factor (met). As a result, increased PI3 kinase activity in cancer cells and subsequently increased growth and survival are observed (reviewed in [134]). In inflammation, the PI3 Kinase pathway drives certain inflammatory processes such as neutrophil chemotaxis, mast cell and eosinophil migration, and degranulation. Inflammatory cytokines such as TNF-α can activate the PI3-AKT pathway which subsequently leads to activation of the NF-κB pathway, and this appears to be the case with certain cell types [135]. The PI3 kinase pathway has been shown to be involved in the mRNA stabilization of several reporter genes linked to the ARE of IL-3, GM-CSF, TNF-α, IL-2, and IL-6 [127, 136]. Hence, PI3 kinase activation further amplifies the processes underlying chronic inflammation and cancer partially by post-transcriptional augmentation of cytokines and tyrosine growth factors.
Other signaling pathways
Other signaling pathways which operate both in chronic inflammation and cancer and also regulate ARE-dependent post-transcriptional control of gene expression exist (Table 1). For example, protein kinase C (PKC) activation has been shown to increase COX-2 mRNA stability, and this effect appears to be mediated by crosstalk with the MAPK pathway [137, 138]. The Wnt β-catenin pathway activation can stabilize ARE-mRNAs. For example, β-catenin binds to the ARE region of the COX-2 mRNA and increases its stability and interacts with HuR [139]. Further amplification of the Wnt pathway can arise from the action of pro-inflammatory prostaglandins, in particular PGE2, produced by COX-2 [140]. The PI3-AKT pathway can increase the stability of the β-catenin pathway by inactivating the RNA decay properties of KSRP through AKT-mediated phosphorylation at a unique serine residue in KSRP, that results in association with the multifunctional protein 14-3-3 and prevents KSRP interaction with the exoribonucleolytic complex exosome [94].
Signaling pathways such as the NF-κB, STAT-JAK, and β-catenin pathways activate transcription of RNA-binding proteins or ARE-genes that are involved in chronic inflammation and cancer which lead to post-transcriptional effects. Increased transcription of HuR may be mediated by a putative NF-κB binding site in the HuR promoter [141]. Thus, increased NF-κB activity during chronic inflammation and cancer may be associated with elevated HuR transcription, and, subsequently, higher HuR-mediated stabilizing effects on target mRNAs. Because of the apparent heterogeneity of ARE sequences and interactions with their binding proteins, it is expected that the heterogeneity also extends to the signaling pathways regulating ARE-mediated events.
How do deregulated pathways in inflammation and cancer affect ARE-mediated processes?
The various signaling pathways discussed above are dysregulated in chronic inflammation and cancer, either by prolonged activation and/or by loss of negative feedback control of mRNA expression (Figs. 1 and 2). One outcome is alterations in the behavior of ARE-binding proteins due to the signaling pathway aberrations; these events cause overproduction of pro-inflammatory molecules and cancer-promoting gene products. Further, defects in ARE-mediated processes may arise by different mechanisms including mutations in the ARE themselves, chromosomal deletions of ARE regions, promoter silencing of RNA-binding proteins, differential expression of miRNA targeting ARE-genes or RNA-binding proteins, and differential abundance of ARE-deficient alternative mRNA forms. The following summarizes these potential mechanisms that affect ARE-mediated post-transcriptional control, and the associated diseases of inflammation origin and cancer.
Deregulation in expression and localization of RNA-binding proteins
Overexpression of RNA-binding protein that tends to stabilize AU-rich mRNAs such as HuR and certain isoforms of AUF1 is observed in certain cancers. Also, under-expression of RNA destabilizing proteins such as TTP can lead to increase in AU-rich mRNAs that are involved in chronic inflammation and cancer. Changes in localization of the RNA-binding proteins towards a compartment that facilitates their action comprise a possible deregulated mechanism that leads to overexpression of the proteins involved in chronic inflammation and cancer crosstalk. Many RNA-binding proteins are nuclear-cytoplasmic shuttling proteins that can translocate during activation. During disease, an RNA-binding protein may be locked into one compartment that becomes either active or non-active, depending on the protein, and thus influences the outcome of RNA turnover.
Alternations in the cellular localization and expression of HuR
Human antigen R is predominantly nuclear, but during cellular growth and activation, it translocates to the cytoplasm where it binds AU-rich mRNAs, including those involved in inflammation and cancer such as VEGF, COX-2, and IL-8, IL-6, and TNF- α, and promotes their mRNA stabilization. In particular, HuR has the capacity to upregulate many cell-cycle and pro-growth genes by binding and stabilizing their mRNAs such as cyclins, growth factors, and transcriptional factors. Several independent lines of evidence strongly support the role of HuR in promoting cellular growth. HuR knockdown of HuR by siRNA or antisense vectors decreases cellular growth [142, 143]. Knockdown of HuR also leads to decreased cellular growth and VEGF expression in colorectal cancer cells [144]. miR-519 represses HuR translation, in turn reducing HuR-regulated gene expression and cell division [145]. Further indirect evidence is that RNase L suppression of cellular growth is associated with reduction in HuR mRNA expression [146].
Several studies have pointed out that HuR is overexpressed in tumor cells when compared to their normal counterparts, with increased localization of HuR in cytoplasmic compartments. Breast, colon, gastric, prostate, and ovarian cancers have been linked to overexpression of HuR and increased cytoplasmic localization, when compared to normal tissues [143, 147–150]. HuR is particularly seen in higher amounts with advanced versus early tumors such as in case of gastric tumors, glioblastomas, breast cancer, and lung cancer [149, 151]. Cytoplasmic expression of HuR is increased in urethane-induced neoplasia and in butylated hydroxytoluene-induced compensatory hyperplasia in mouse lung tissue [152]. However, certain studies have shown that HuR overexpression may be limited to a small sub-set of tumors [153]. Cytoplasmic but not nuclear HuR expression is significantly associated with tumor grade, poor differentiation, and lymph node metastasis in non-small cell lung cancer with increased levels of VEGF-C [154].
In an epithelial to mesenchymal transition colon cancer rat cellular model that undergoes invasive phenotype in response to Ras and TGF-β, a synergistic effect is observed with the stability of several ARE-mRNA including COX-2 and VEGF [155]. The VEGF mRNA is a target for HuR binding and effect. It is associated with increased HuR expression in different types of cancers; for example, major HuR expression is detected in perinecrotic areas in which VEGF, COX-2, and IL-8, all angiogenesis factors, are up-regulated [151]. HuR overexpression and cytoplasmic localization leads to binding to, and upregulation of, COX-2 mRNA expression in a number of cancers [71, 156–159]. The highly invasive breast cancer cell line, MDA-MB-231, which has increased constitutive levels of uPA, is due to impairment in the ARE-mediated decay of uPA mRNA; this is correlated with HuR overexpression and p38 MAP kinase induction [113]. Thus, these many studies clearly show consistent correlation of cytoplasmic localization of HuR with tumor grade, tumor size, and patient survival in many types of cancer tissues.
How is cytoplasmic localization of HuR promoted in tumor cells? Certain signaling aberrations in cancer may promote cytoplasmic HuR localization such as p38 MAPK and PKC pathways. The overexpression of constitutively active MK2 results in increased HuR cytoplasmic localization in the MDA-MB-231 invasive breast cancer line [129]. HuR phosphorylation by p38 MAPK, cAMP kinase, PKC, and cell-cycle G2-phase kinase Cdk1 can regulate HuR shuttling and thus affect the mRNA stabilization outcome [128, 160, 161]. Aberrations in the expression or activity of these kinases in cancer can lead to changes in HuR phosphorylation that favor cytoplasmic localization of HuR. A detailed review on the role of phosphorylation in HuR translocation, see [161].
Loss of TTP expression and function in cancer and chronic inflammation
Continued secretion of cytokines and growth factors during chronic inflammation and cancer contribute to a prolonged state of activated pathways such as the MAPK pathway that maintains mRNA stabilization by inactivation of RNA-binding proteins that participate in mRNA decay such as TTP and KSRP (Fig. 2). These events allow the prevalence of HuR stabilizing action. Elevated HuR expression occurs with the loss of TTP expression at an early stage of colorectal tumorigenesis that is associated with increased COX-2 expression [71]. TTP mRNA and protein levels are significantly decreased in tumors of the thyroid, lung, ovary, uterus, and breast compared to non-transformed tissues [153]. Loss of TTP is observed in the highly metastatic breast tumor line, MDA-MB-231, that is functionally associated with increased invasion and elevated expression of uPA and uPAR mRNAs due to their stabilization [162]. It is not necessarily that TTP levels are reduced in order to observe reduced TTP-induced mRNA decay, since phosphorylation of TTP due to aberration in signaling pathways, such as overactivated p38 MAPK, is a defect that is observed in several cancers. An example is demonstrated with malignant gliomas in which TTP is hyperphosphorylated due most likely to p38 MAPK activation [163]. Here, loss of TTP can be linked to overexpression of TTP mRNA targets that are involved in inflammation and cancer such as VEGF [163]. Since the ERK pathway is overactivated in cancer, it is thought that ERK activation results in phosphorylation of TTP, leading to an increase in TTP mRNA targets such as VEGF [72]. Loss of TTP in hepatocellular carcinoma is thought to lead to an increase in HIF1α, a key player in angiogenesis [164].
A possible mechanism of TTP deficiency in tumor cell lines and tumors is silencing of TTP promoter due to epigenetic alterations as a result of changes in DNA methylation or altered chromatin [71]. Possible loss of TTP and probably other ARE-mRNA decay-promoting proteins is overexpression of miRNAs that target the mRNAs of these proteins; the miR-29a which may be overexpressed in breast cancer tissues can suppress TTP [165]. Thus, the loss of post-transcriptional regulation due to decreased expression of mRNA decay proteins such as TTP can occur with different mechanisms and allow enhanced expression of common inflammation and cancer genes.
An indirect involvement of TTP dysfunction in chronic inflammatory conditions may be deduced from the observation that the anti-inflammatory cytokine, IL-10, increases TTP expression and subsequently reduces the pro-inflammatory cytokines, TNF-α and IL-1 [166]. Certain IL-10 promoter polymorphisms that result in reduced IL-10 production may be associated with a number of autoimmune and inflammatory chronic diseases such as inflammatory bowel disease [167]. Thus, it is possible that dysfunction in IL-10 production would lead to reduced TTP levels and subsequently elevated pro-inflammatory cytokines. Thus, the strong anti-inflammatory properties of TTP can be explained by both direct targeting of pro-inflammatory mRNAs for degradation and by helping the potent anti-inflammatory cytokine, IL-10, to exert its immunosuppressive effects. IL-10-deficient mice develop a range of auto-immune diseases, colitis and colorectal cancer similar to the inflammatory bowel disease-associated cancer in humans [168, 169]. The role of IL-10 in either promoting or inhibiting tumor growth in humans may still be controversial and would require larger patient data [170]. Since IL-0 mRNA itself is subject to decay promotion by TTP [126], this suggests that ARE-mediated decay is important for immune homeostasis.
Changes in alternative splicing and polyadenylation
Certain ARE-genes code for alternative polyadenylation variants that have multiple poly(A) sites that may be differentially used during inflammation and cancer. The loss of the ARE in the 3′UTRs due to changes in alternative splicing or polyadenylation pattern may lead to overexpression of an undesirable pathway triggered by the alternative transcript. The existence of two transcripts generated by the use of alternative polyadenylation sequences that differ in their 3′UTRs and the differential presence of ARE is found in a number of genes. Notably, these are COX-2, cyclin D1, VEGF, fibroblast growth factor 2, and, recently, HuR [70, 73, 171–173]. In the latter case, the different alternative variants have differential response to HuR-induced stabilization and destabilization to TTP and other mRNA decay players [73, 146]. Similarly, alternative splicing that result in ARE and non-ARE form can also be differentially regulated.
The COX-2 gene codes for two alternative polyadenylation variants that differ in their 3′UTR length with different patterns of post-transcriptional regulation [70, 171]. Both the major transcripts of COX-2 gene, 2.8- and 4.6-kb mRNAs, contain a highly conserved ARE region (cluster III ARE, Class 2) in the proximal part of the 3′UTR. However, the longer transcript contains an additional conserved ARE region (Class 1 ARE). These different transcripts respond differentially to TTP-mediated RNA decay [70, 171]. In certain cancer types, the abundance of expression of the shorter transcript is higher, which may contribute to the overexpression of COX-2 in cancer and during inflammatory activation [70, 171–173]. In some primary tumors and in cell lines, cyclin D1 mRNAs of smaller size—due to chromosomal alterations that lead to deletion of ARE-3′UTR in CCND1 gene—can be detected and substantially increased in mantle cell lymphoma cells [173]. Cyclin D1 overexpression is known to perturb the G1-S transition of the cell cycle and thereby contributes to tumor development. HuR mRNA also has several alternative polyadenylation variants, with the least abundant being a long 3′UTR transcript that harbors an ARE and is responsive to HuR auto-regulatory process [73]. Short stable FGF-2 mRNAs are present in transformed cells, whereas normal fibroblasts contain mostly long ARE containing unstable mRNAs [172]. The effect of alternative polyadenylation on ARE-mediated mRNA stability was also demonstrated with VEGF mRNA [174].
Effect of hypoxia and angiogenesis factors on ARE-mediated pathways
Hypoxia in the tumor environment or in an inflammatory tumor microenvironment triggers the expression of the ARE-mRNA product, HIFα, which in turn induces a range of HIF-responsive genes including those encoding other ARE-mRNAs, notably VEGF (Fig. 1). In addition to transcriptional induction, both VEGF and HIF expression is modulated by ARE-mediated post-transcriptional effects in response to hypoxia. HuR binds to a U-rich region in VEGF 3′UTR and mediates increased mRNA stability [175]. A number of RNA-binding proteins, e.g., HuR and PTB (polypyrimidine tract-binding protein), bind to U-rich/AU-rich region of VEGF and HIF 3′UTRs [176]. HIF1α also has a VEGF-independent role in the regulation of glycolytic capacity in myeloid cells contributing to the survival of macrophages and other cells in the tumor microenvironment [25]. The loss of Von Hippel-Lindau (VHL) tumor suppressor action leads to increased VEGF expression at a post-transcriptional level contributing further to hypoxia-induced angiogenesis in certain tumor types such as renal carcinomas [177]. This effect may be mediated by TTP down-regulation since VHL decreases TTP expression [178]. Another protein affected in renal cancer by VHL gene loss is the survival factor, parathyroid hormone-related protein (PTHrP), in which its mRNA stability is altered by HuR [179]. As HIF-1α is constitutively overexpressed in several cancers as a result of hypoxia, it also increases ARE-mRNA stability of VEGF [23] in a manner that is associated with HuR expression and cytoplasmic localization. Thus, both overexpression and cytoplasmic localization of HuR during tumorgenesis further promote the processes of cancer such as angiogenesis (e.g., VEGF) and invasion/metastasis (uPA). Additionally, loss of TTP in cancer increases HIF1α [164], and promotes angiogenesis-related processes.
Other mechanisms for impairment of ARE-mediated decay
The ARE binding proteins T cell internal antigen-1 (TIA-1) and TIA-1 related protein (TIAR) are translational silencers for TNF-α and certain Class II ARE mRNA translations. They bind ARE in TNF-α mRNA molecules without affecting the mRNA stability itself. As a result, mRNAs are diverted from the polysomes to untranslated messenger ribonucleoproteins in a form of stress granules, cytoplasmic domains into which mRNAs are sorted dynamically [180, 181]. The role of TIA in inflammation is observed with TIA knockdown mice which develop mild arthritis [182]. In cancer, loss of ARE-mediated translational repression by TIA due to defective binding to the ARE promotes COX-2 expression as observed with certain colon cancer cell lines [183].
Removal of the ARE stretch correlates with the increased oncogenicity of proto-oncogene fos [184]. Bcl-2 mRNA is an ARE-mRNA in which its increased stability may lead to overproduction of the antiapoptotic BCL-2 protein likely responsible for neoplastic transformation of follicular B cell lymphoma [185, 186]. The mRNA of the transcription factor CHOP, which is involved in cell division and apoptosis in response to stress, is regulated by ARE [187]. Increased constitutive expression of an ARE-mRNA isoform for 6-phosphofructoso-2-kinase, which is a key enzyme in the Warbug effect of the oxygen-dependent enhanced glycolysis in cancer cells, has been shown to be required for tumor growth in vitro and in vivo [188]. In the same context of enhanced glucose metabolism in cancer cells, the stability of glucose transporter Glut1 mRNA has been shown to be regulated by ARE and ARE binding proteins and correlates with certain tumors including gliomas [189]. Increased production of hematopoietic growth factors, e.g., GM-CSF, acting as autocrine growth factor due to defects in ARE-mediated stability may contribute to leukemia pathogenesis [190].
Conclusions and final remarks
The sequence heterogeneity of ARE, the high number ARE-genes, and the multiple signaling pathways that change the behavior of so many RNA-binding proteins are indicative of the high complexity and repertoire of ARE interactome. Changes in any of these various players and processes may disturb ARE-mediated post-transcriptional pathways leading to specific abnormalities seen in chronic inflammation and cancer. Overexpression and cytoplasmic localization of stabilizing ARE binding proteins, notably HuR, with commitment functional loss of RNA decay-promoting proteins such as TTP can be associated with chronic inflammation and cancer. Continued production of pro-inflammatory and pro-cancer ARE-gene products can then result from aberrations in ARE-mediated processes that affect mRNA stability and translation (Table 3). The ARE-gene family is close to 15% of the human transcriptome, which contains functionally diverse proteins, thus one can speculate on the involvement of many of the deregulated ARE-mRNAs in chronic inflammation and any of the cancer processes. Therapeutic interventions that aim to control aberrant signaling pathways, both in chronic inflammation and cancer, might also lead to the control of sustained ARE-mediated post-transcriptional gene expression events. Knowledge on the interplay and redundancy of RNA-protein interactions in inflammation and cancer may result in the design of new therapeutic approaches that aim at chronic inflammatory diseases and cancer, and their linked pathology.
References
Halees AS, El-Badrawi R, Khabar KS (2008) ARED organism: expansion of ARED reveals AU-rich element cluster variations between human and mouse. Nucleic Acids Res 36:D137–D140
Bakheet T, Williams BR, Khabar KS (2006) ARED 3.0: the large and diverse AU-rich transcriptome. Nucleic Acids Res 34:D111–D114
Yang E, Van Nimwegen E, Zavolan M, Rajewsky N, Schroeder M, Magnasco M, Darnell JE Jr (2003) Decay rates of human mRNAs: correlation with functional characteristics and sequence attributes. Genome Res 13:1863–1872
Blackshear PJ (2002) Tristetraprolin and other CCCH tandem zinc-finger proteins in the regulation of mRNA turnover. Biochem Soc Trans 30:945–952
Chou CF, Mulky A, Maitra S, Lin WJ, Gherzi R, Kappes J, Chen CY (2006) Tethering KSRP, a decay-promoting AU-rich element-binding protein, to mRNAs elicits mRNA decay. Mol Cell Biol 26:3695–3706
Schmidlin M, Lu M, Leuenberger SA, Stoecklin G, Mallaun M, Gross B, Gherzi R, Hess D, Hemmings BA, Moroni C (2004) The ARE-dependent mRNA-destabilizing activity of BRF1 is regulated by protein kinase B. EMBO J 23:4760–4769
Barreau C, Paillard L, Osborne HB (2005) AU-rich elements and associated factors: are there unifying principles? Nucleic Acids Res 33:7138–7150
Kontoyiannis D, Pasparakis M, Pizarro TT, Cominelli F, Kollias G (1999) Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU- rich elements: implications for joint and gut-associated immunopathologies. Immunity 10:387–398
Taylor GA, Carballo E, Lee DM, Lai WS, Thompson MJ, Patel DD, Schenkman DI, Gilkeson GS, Broxmeyer HE, Haynes BF, Blackshear PJ (1996) A pathogenetic role for TNF alpha in the syndrome of cachexia, arthritis, and autoimmunity resulting from tristetraprolin (TTP) deficiency. Immunity 4:445–454
Carballo E, Lai WS, Blackshear PJ (1998) Feedback inhibition of macrophage tumor necrosis factor-alpha production by tristetraprolin. Science 281:1001–1005
Balkwill F (2009) Tumour necrosis factor and cancer. Nat Rev Cancer 9:361–371
Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, Gutkovich-Pyest E, Urieli-Shoval S, Galun E, Ben-Neriah Y (2004) NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature 431:461–466
Popivanova BK, Kitamura K, Wu Y, Kondo T, Kagaya T, Kaneko S, Oshima M, Fujii C, Mukaida N (2008) Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. J Clin Invest 118:560–570
Egberts JH, Cloosters V, Noack A, Schniewind B, Thon L, Klose S, Kettler B, von Forstner C, Kneitz C, Tepel J, Adam D, Wajant H, Kalthoff H, Trauzold A (2008) Anti-tumor necrosis factor therapy inhibits pancreatic tumor growth and metastasis. Cancer Res 68:1443–1450
Kulbe H, Thompson R, Wilson JL, Robinson S, Hagemann T, Fatah R, Gould D, Ayhan A, Balkwill F (2007) The inflammatory cytokine tumor necrosis factor-alpha generates an autocrine tumor-promoting network in epithelial ovarian cancer cells. Cancer Res 67:585–592
Liu RY, Fan C, Liu G, Olashaw NE, Zuckerman KS (2000) Activation of p38 mitogen-activated protein kinase is required for tumor necrosis factor-alpha -supported proliferation of leukemia and lymphoma cell lines. J Biol Chem 275:21086–21093
Bates RC, Mercurio AM (2003) Tumor necrosis factor-alpha stimulates the epithelial-to-mesenchymal transition of human colonic organoids. Mol Biol Cell 14:1790–1800
Popivanova BK, Kostadinova FI, Furuichi K, Shamekh MM, Kondo T, Wada T, Egashira K, Mukaida N (2009) Blockade of a chemokine, CCL2, reduces chronic colitis-associated carcinogenesis in mice. Cancer Res 69:7884–7892
Tsujii M, Kawano S, Tsuji S, Sawaoka H, Hori M, DuBois RN (1998) Cyclooxygenase regulates angiogenesis induced by colon cancer cells. Cell 93:705–716
Sawaoka H, Tsuji S, Tsujii M, Gunawan ES, Sasaki Y, Kawano S, Hori M (1999) Cyclooxygenase inhibitors suppress angiogenesis and reduce tumor growth in vivo. Lab Invest 79:1469–1477
Li G, Yang T, Yan J (2002) Cyclooxygenase-2 increased the angiogenic and metastatic potential of tumor cells. Biochem Biophys Res Commun 299:886–890
Franco S, Stefania V, Miquel S, Vincenzo A, Giuseppe S, Egidio S, Alessandro R, Andreas S, Alberto M, Julian P, Seppo YH, Claudio F, Silvio D (2009) VEGF-A links angiogenesis and inflammation in inflammatory bowel disease pathogenesis. Gastroenterology 136:585–595, e585
Liu LX, Lu H, Luo Y, Date T, Belanger AJ, Vincent KA, Akita GY, Goldberg M, Cheng SH, Gregory RJ, Jiang C (2002) Stabilization of vascular endothelial growth factor mRNA by hypoxia-inducible factor 1. Biochem Biophys Res Commun 291:908–914
Scortegagna M, Cataisson C, Martin RJ, Hicklin DJ, Schreiber RD, Yuspa SH, Arbeit JM (2008) HIF-1{alpha} regulates epithelial inflammation by cell autonomous NF{kappa}B activation and paracrine stromal remodeling. Blood 111:3343–3354
Cramer T, Yamanishi Y, Clausen BE, Forster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V, Firestein GS, Gerber HP, Ferrara N, Johnson RS (2003) HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell 112:645–657
Andreasen PA, Egelund R, Petersen HH (2000) The plasminogen activation system in tumor growth, invasion, and metastasis. Cell Mol Life Sci 57:25–40
Shi Q, Abbruzzese JL, Huang S, Fidler IJ, Xiong Q, Xie K (1999) Constitutive and inducible interleukin 8 expression by hypoxia and acidosis renders human pancreatic cancer cells more tumorigenic and metastatic. Clin Cancer Res 5:3711–3721
Inoue K, Slaton JW, Eve BY, Kim SJ, Perrotte P, Balbay MD, Yano S, Bar-Eli M, Radinsky R, Pettaway CA, Dinney CP (2000) Interleukin 8 expression regulates tumorigenicity and metastases in androgen-independent prostate cancer. Clin Cancer Res 6:2104–2119
Haghnegahdar H, Du J, Wang D, Strieter RM, Burdick MD, Nanney LB, Cardwell N, Luan J, Shattuck-Brandt R, Richmond A (2000) The tumorigenic and angiogenic effects of MGSA/GRO proteins in melanoma. J Leukoc Biol 67:53–62
Hussain SP, He P, Subleski J, Hofseth LJ, Trivers GE, Mechanic L, Hofseth AB, Bernard M, Schwank J, Nguyen G, Mathe E, Djurickovic D, Haines D, Weiss J, Back T, Gruys E, Laubach VE, Wiltrout RH, Harris CC (2008) Nitric oxide is a key component in inflammation-accelerated tumorigenesis. Cancer Res 68:7130–7136
Chen CY, Shyu AB (1995) AU-rich elements: characterization and importance in mRNA degradation. Trends Biochem Sci 20:465–470
Lagnado CA, Brown CY, Goodall GJ (1994) AUUUA is not sufficient to promote poly(A) shortening and degradation of an mRNA: the functional sequence within AU-rich elements may be UUAUUUA(U/A)(U/A). Mol Cell Biol 14:7984–7995
Lewis T, Gueydan C, Huez G, Toulme JJ, Kruys V (1998) Mapping of a minimal AU-rich sequence required for lipopolysaccharide- induced binding of a 55-kDa protein on tumor necrosis factor-alpha mRNA. J Biol Chem 273:13781–13786
Zubiaga AM, Belasco JG, Greenberg ME (1995) The nonamer UUAUUUAUU is the key AU-rich sequence motif that mediates mRNA degradation. Mol Cell Biol 15:2219–2230
Lai WS, Carrick DM, Blackshear PJ (2005) Influence of nonameric AU-rich tristetraprolin-binding sites on mRNA deadenylation and turnover. J Biol Chem 280:34365–34377
Bakheet T, Frevel M, Williams BR, Greer W, Khabar KS (2001) ARED: human AU-rich element-containing mRNA database reveals an unexpectedly diverse functional repertoire of encoded proteins. Nucleic Acids Res 29:246–254
Frevel MA, Bakheet T, Silva AM, Hissong JG, Khabar KS, Williams BR (2003) p38 Mitogen-activated protein kinase-dependent and -independent signaling of mRNA stability of AU-rich element-containing transcripts. Mol Cell Biol 23:425–436
Raghavan A, Dhalla M, Bakheet T, Ogilvie RL, Vlasova IA, Khabar KS, Williams BR, Bohjanen PR (2004) Patterns of coordinate down-regulation of ARE-containing transcripts following immune cell activation. Genomics 84:1002–1013
Sharova LV, Sharov AA, Nedorezov T, Piao Y, Shaik N, Ko MS (2009) Database for mRNA half-life of 19 977 genes obtained by DNA microarray analysis of pluripotent and differentiating mouse embryonic stem cells. DNA Res 16:45–58
Ray D, Kazan H, Chan ET, Castillo LP, Chaudhry S, Talukder S, Blencowe BJ, Morris Q, Hughes TR (2009) Rapid and systematic analysis of the RNA recognition specificities of RNA-binding proteins. Nat Biotechnol 27:667–670
Meisner NC, Hackermuller J, Uhl V, Aszodi A, Jaritz M, Auer M (2004) mRNA openers and closers: modulating AU-rich element-controlled mRNA stability by a molecular switch in mRNA secondary structure. Chembiochem 5:1432–1447
Hackermuller J, Meisner NC, Auer M, Jaritz M, Stadler PF (2005) The effect of RNA secondary structures on RNA-ligand binding and the modifier RNA mechanism: a quantitative model. Gene 345:3–12
Lopez de Silanes I, Zhan M, Lal A, Yang X, Gorospe M (2004) Identification of a target RNA motif for RNA-binding protein HuR. Proc Natl Acad Sci USA 101:2987–2992
Park-Lee S, Kim S, Laird-Offringa IA (2003) Characterization of the interaction between neuronal RNA-binding protein HuD and AU-rich RNA. J Biol Chem 278:39801–39808
Brewer BY, Malicka J, Blackshear PJ, Wilson GM (2004) RNA sequence elements required for high affinity binding by the zinc-finger domain of tristetraprolin: conformational changes coupled to the bipartite nature of Au-rich MRNA-destabilizing motifs. J Biol Chem 279:27870–27877
Worthington MT, Pelo JW, Sachedina MA, Applegate JL, Arseneau KO, Pizarro TT (2002) RNA-binding properties of the AU-rich element-binding recombinant Nup475/TIS11/tristetraprolin protein. J Biol Chem 277:48558–48564
Mazan-Mamczarz K, Kuwano Y, Zhan M, White EJ, Martindale JL, Lal A, Gorospe M (2009) Identification of a signature motif in target mRNAs of RNA-binding protein AUF1. Nucleic Acids Res 37:204–214
Lopez de Silanes I, Galban S, Martindale JL, Yang X, Mazan-Mamczarz K, Indig FE, Falco G, Zhan M, Gorospe M (2005) Identification and functional outcome of mRNAs associated with RNA-binding protein TIA-1. Mol Cell Biol 25:9520–9531
Andersen JB, Mazan-Mamczarz K, Zhan M, Gorospe M, Hassel BA (2009) Ribosomal protein mRNAs are primary targets of regulation in RNase-L-induced senescence. RNA Biol 6:305–315
Forte GI, Scola L, Bellavia D, Vaccarino L, Sanacore M, Sisino G, Scazzone C, Caruso C, Barbieri R, Lio D (2009) Characterization of two alternative Interleukin(IL)-10 5’UTR mRNA sequences, induced by lipopolysaccharide (LPS) stimulation of peripheral blood mononuclear cells. Mol Immunol 46:2161–2166
Paste M, Huez G, Kruys V (2003) Deadenylation of interferon-beta mRNA is mediated by both the AU-rich element in the 3’-untranslated region and an instability sequence in the coding region. Eur J Biochem 270:1590–1597
Prechtel AT, Chemnitz J, Schirmer S, Ehlers C, Langbein-Detsch I, Stulke J, Dabauvalle MC, Kehlenbach RH, Hauber J (2006) Expression of CD83 is regulated by HuR via a novel cis-active coding region RNA element. J Biol Chem 281:10912–10925
Al-Zoghaibi F, Ashour T, Al-Ahmadi W, Abulleef H, Demirkaya O, Khabar KS (2007) Bioinformatics and experimental derivation of an efficient hybrid 3’ untranslated region and use in expression active linear DNA with minimum poly(A) region. Gene 391:130–139
Mukherjee N, Lager PJ, Friedersdorf MB, Thompson MA, Keene JD (2009) Coordinated posttranscriptional mRNA population dynamics during T-cell activation. Mol Syst Biol 5:288
Stoecklin G, Tenenbaum SA, Mayo T, Chittur SV, George AD, Baroni TE, Blackshear PJ, Anderson P (2008) Genome-wide analysis identifies interleukin-10 mRNA as target of tristetraprolin. J Biol Chem 283:11689–11699
Jing Q, Huang S, Guth S, Zarubin T, Motoyama A, Chen J, Di Padova F, Lin SC, Gram H, Han J (2005) Involvement of microRNA in AU-rich element-mediated mRNA instability. Cell 120:623–634
Mayr C, Bartel DP (2009) Widespread shortening of 3’UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell 138:673–684
Sandberg R, Neilson JR, Sarma A, Sharp PA, Burge CB (2008) Proliferating cells express mRNAs with shortened 3’ untranslated regions and fewer microRNA target sites. Science 320:1643–1647
Akashi M, Shaw G, Hachiya M, Elstner E, Suzuki G, Koeffler P (1994) Number and location of AUUUA motifs: role in regulating transiently expressed RNAs. Blood 83:3182–3187
Blackshear PJ, Lai WS, Kennington EA, Brewer G, Wilson GM, Guan X, Zhou P (2003) Characteristics of the interaction of a synthetic human tristetraprolin tandem zinc-finger peptide with AU-rich element-containing RNA substrates. J Biol Chem 278:19947–19955
Vlasova IA, Tahoe NM, Fan D, Larsson O, Rattenbacher B, Sternjohn JR, Vasdewani J, Karypis G, Reilly CS, Bitterman PB, Bohjanen PR (2008) Conserved GU-rich elements mediate mRNA decay by binding to CUG-binding protein 1. Mol Cell 29:263–270
Kruys V, Wathelet M, Poupart P, Contreras R, Fiers W, Content J, Huez G (1987) The 3’ untranslated region of the human interferon-beta mRNA has an inhibitory effect on translation. Proc Natl Acad Sci USA 84:6030–6034
Kruys V, Marinx O, Shaw G, Deschamps J, Huez G (1989) Translational blockade imposed by cytokine-derived UA-rich sequences. Science 245:852–855
Vasudevan S, Steitz JA (2007) AU-rich-element-mediated upregulation of translation by FXR1 and Argonaute 2. Cell 128:1105–1118
Cao H, Deterding LJ, Blackshear PJ (2007) Phosphorylation site analysis of the anti-inflammatory and mRNA-destabilizing protein tristetraprolin. Expert Rev Proteomics 4:711–726
Lai WS, Carballo E, Strum JR, Kennington EA, Phillips RS, Blackshear PJ (1999) Evidence that tristetraprolin binds to AU-Rich elements and promotes the deadenylation and destabilization of tumor necrosis factor alpha mRNA. Mol Cell Biol 19:4311–4323
Taylor GA, Thompson MJ, Lai WS, Blackshear PJ (1996) Mitogens stimulate the rapid nuclear to cytosolic translocation of tristetraprolin, a potential zinc-finger transcription factor. Mol Endocrinol 10:140–146
Carballo E, Lai WS, Blackshear PJ (2000) Evidence that tristetraprolin is a physiological regulator of granulocyte-macrophage colony stimulating factor messenger RNA deadenylation and stability. Blood 95:1891–1899
Stoecklin G, Ming X-F, Looser R, Moroni C (2000) Somatic mRNA turnover mutants implicate tristetraprolin in the interleukin-3 mRNA degradation pathway. Mol Cell Biol 20:3753–3763
Sawaoka H, Dixon DA, Oates JA, Boutaud O (2003) Tristetraprolin binds to the 3’-Untranslated Region of cyclooxygenase-2 mRNA. A polyadenylation variant in a cancer cell line lacks the binding site. J Biol Chem 278:13928–13935
Young LE, Sanduja S, Bemis-Standoli K, Pena EA, Price RL, Dixon DA (2009) The mRNA-binding proteins HuR and tristetraprolin regulate cyclooxygenase 2 expression during colon carcinogenesis. Gastroenterology 136:1669–1679
Essafi-Benkhadir K, Onesto C, Stebe E, Moroni C, Pages G (2007) Tristetraprolin inhibits ras-dependent tumor vascularization by inducing vascular endothelial growth factor mRNA degradation. Mol Biol Cell 18:4648–4658
Al-Ahmadi W, Al-Ghamdi M, Al-Haj L, Al-Saif M, Khabar KS (2009) Alternative polyadenylation variants of the RNA-binding protein, HuR: abundance, role of AU-rich elements and auto-Regulation. Nucleic Acids Res 37:3612–3624
Lai WS, Parker JS, Grissom SF, Stumpo DJ, Blackshear PJ (2006) Novel mRNA targets for tristetraprolin (ttp) identified by global analysis of stabilized transcripts in TTP-deficient fibroblasts. Mol Cell Biol 26:9196–9208
Ma WJ, Cheng S, Campbell C, Wright A, Furneaux H (1996) Cloning and characterization of HuR, a ubiquitously expressed Elav-like protein. J Biol Chem 271:8144–8151
Atasoy U, Watson J, Patel D, Keene JD (1998) ELAV protein HuA (HuR) can redistribute between nucleus and cytoplasm and is upregulated during serum stimulation and T cell activation. J Cell Sci 111:3145–3156
Levine TD, Gao F, King PH, Andrews LG, Keene JD (1993) Hel-N1: an autoimmune RNA-binding protein with specificity for 3’ uridylate-rich untranslated regions of growth factor mRNAs. Mol Cell Biol 13:3494–3504
Keene JD (1999) Why is Hu where? Shuttling of early-response-gene messenger RNA subsets. Proc Natl Acad Sci USA 96:5–7
Wang W, Furneaux H, Cheng H, Caldwell MC, Hutter D, Liu Y, Holbrook N, Gorospe M (2000) HuR regulates p21 mRNA stabilization by UV light. Mol Cell Biol 20:760–769
Lal A, Mazan-Mamczarz K, Kawai T, Yang X, Martindale JL, Gorospe M (2004) Concurrent versus individual binding of HuR and AUF1 to common labile target mRNAs. EMBO J 23:3092–3102
Casolaro V, Fang X, Tancowny B, Fan J, Wu F, Srikantan S, Asaki SY, De Fanis U, Huang SK, Gorospe M, Atasoy UX, Stellato C (2008) Posttranscriptional regulation of IL-13 in T cells: role of the RNA-binding protein HuR. J Allergy Clin Immunol 121:853–859,e854
Lopez de Silanes I, Fan J, Galban CJ, Spencer RG, Becker KG, Gorospe M (2004) Global analysis of HuR-regulated gene expression in colon cancer systems of reducing complexity. Gene Expr 12:49–59
Papadaki O, Milatos S, Grammenoudi S, Mukherjee N, Keene JD, Kontoyiannis DL (2009) Control of thymic T cell maturation, deletion and egress by the RNA-binding protein HuR. J Immunol 182:6779–6788
Katsanou V, Milatos S, Yiakouvaki A, Sgantzis N, Kotsoni A, Alexiou M, Harokopos V, Aidinis V, Hemberger M, Kontoyiannis DL (2009) The RNA-binding protein Elavl1/HuR is essential for placental branching morphogenesis and embryonic development. Mol Cell Biol 29:2762–2776
Ghosh M, Aguila HL, Michaud J, Ai Y, Wu MT, Hemmes A, Ristimaki A, Guo C, Furneaux H, Hla T (2009) Essential role of the RNA-binding protein HuR in progenitor cell survival in mice. J Clin Invest 119:3530–3543
Houseley J, LaCava J, Tollervey D (2006) RNA-quality control by the exosome. Nat Rev Mol Cell Biol 7:529–539
Gherzi R, Trabucchi M, Ponassi M, Ruggiero T, Corte G, Moroni C, Chen CY, Khabar KS, Andersen JS, Briata P (2006) The RNA-binding protein KSRP promotes decay of beta-Catenin mRNA and is inactivated by PI3 K-AKT signaling. PLoS Biol 5:e5
He C, Schneider R (2006) 14–3-3sigma is a p37 AUF1-binding protein that facilitates AUF1 transport and AU-rich mRNA decay. EMBO J 25:3823–3831
Sun L, Stoecklin G, Van Way S, Hinkovska-Galcheva V, Guo RF, Anderson P, Shanley TP (2007) Tristetraprolin (TTP)-14–3-3 complex formation protects TTP from dephosphorylation by protein phosphatase 2a and stabilizes tumor necrosis factor-alpha mRNA. J Biol Chem 282:3766–3777
Fenger-Gron M, Fillman C, Norrild B, Lykke-Andersen J (2005) Multiple processing body factors and the ARE binding protein TTP activate mRNA decapping. Mol Cell 20:905–915
Lykke-Andersen J, Wagner E (2005) Recruitment and activation of mRNA decay enzymes by two ARE-mediated decay activation domains in the proteins TTP and BRF-1. Genes Dev 19:351–361
Stoecklin G, Mayo T, Anderson P (2006) ARE-mRNA degradation requires the 5’–3’ decay pathway. EMBO Rep 7:72–77
Briata P, Forcales SV, Ponassi M, Corte G, Chen CY, Karin M, Puri PL, Gherzi R (2005) p38-dependent phosphorylation of the mRNA decay-promoting factor KSRP controls the stability of select myogenic transcripts. Mol Cell 20:891–903
Briata P, Ilengo C, Corte G, Moroni C, Rosenfeld MG, Chen CY, Gherzi R (2003) The Wnt/beta-catenin– > Pitx2 pathway controls the turnover of Pitx2 and other unstable mRNAs. Mol Cell 12:1201–1211
Sureban SM, Murmu N, Rodriguez P, May R, Maheshwari R, Dieckgraefe BK, Houchen CW, Anant S (2007) Functional antagonism between RNA-binding proteins HuR and CUGBP2 determines the fate of COX-2 mRNA translation. Gastroenterology 132:1055–1065
Linker K, Pautz A, Fechir M, Hubrich T, Greeve J, Kleinert H (2005) Involvement of KSRP in the post-transcriptional regulation of human iNOS expression-complex interplay of KSRP with TTP and HuR. Nucleic Acids Res 33:4813–4827
Wagner BJ, DeMaria CT, Sun Y, Wilson GM, Brewer G (1998) Structure and genomic organization of the human AUF1 gene: alternative pre-mRNA splicing generates four protein isoforms. Genomics 48:195–202
Gouble A, Grazide S, Meggetto F, Mercier P, Delsol G, Morello D (2002) A new player in oncogenesis: AUF1/hnRNPD overexpression leads to tumorigenesis in transgenic mice. Cancer Res 62:1489–1495
Lu JY, Sadri N, Schneider RJ (2006) Endotoxic shock in AUF1 knockout mice mediated by failure to degrade proinflammatory cytokine mRNAs. Genes Dev 20:3174–3184
Laroia G, Cuesta R, Brewer G, Schneider RJ (1999) Control of mRNA decay by heat shock-ubiquitin-proteasome pathway. Science 284:499–502
Garnon J, Lachance C, Di Marco S, Hel Z, Marion D, Ruiz MC, Newkirk MM, Khandjian EW, Radzioch D (2005) Fragile X-related protein FXR1P regulates proinflammatory cytokine tumor necrosis factor expression at the post-transcriptional level. J Biol Chem 280:5750–5763
Steitz JA, Vasudevan S (2009) miRNPs: versatile regulators of gene expression in vertebrate cells. Biochem Soc Trans 37:931–935
Porta C, Larghi P, Rimoldi M, Totaro MG, Allavena P, Mantovani A, Sica A (2009) Cellular and molecular pathways linking inflammation and cancer. Immunobiology 214:761–777
Lin Y, Bai L, Chen W, Xu S (2010) The NF-kappaB activation pathways, emerging molecular targets for cancer prevention and therapy. Expert Opin Ther Targets 14:45–55
Hartupee J, Li X, Hamilton T (2008) Interleukin 1alpha-induced NFkappaB activation and chemokine mRNA stabilization diverge at IRAK1. J Biol Chem 283:15689–15693
Hoffmann E, Dittrich-Breiholz O, Holtmann H, Kracht M (2002) Multiple control of interleukin-8 gene expression. J Leukoc Biol 72:847–855
Gaestel M (2006) MAPKAP kinases—MKs—two’s company, three’s a crowd. Nat Rev Mol Cell Biol 7:120–130
Dhillon AS, Hagan S, Rath O, Kolch W (2007) MAP kinase signalling pathways in cancer. Oncogene 26:3279–3290
Vivanco I, Palaskas N, Tran C, Finn SP, Getz G, Kennedy NJ, Jiao J, Rose J, Xie W, Loda M, Golub T, Mellinghoff IK, Davis RJ, Wu H, Sawyers CL (2007) Identification of the JNK signaling pathway as a functional target of the tumor suppressor PTEN. Cancer Cell 11:555–569
Korhonen R, Linker K, Pautz A, Forstermann U, Moilanen E, Kleinert H (2007) Post-transcriptional regulation of human inducible nitric-oxide synthase expression by the Jun N-terminal kinase. Mol Pharmacol 71:1427–1434
Brook M, Sully G, Clark AR, Saklatvala J (2000) Regulation of tumour necrosis factor alpha mRNA stability by the mitogen-activated protein kinase p38 signalling cascade. FEBS Lett 483:57–61
Holtmann H, Winzen R, Holland P, Eickemeier S, Hoffmann E, Wallach D, Malinin NL, Cooper JA, Resch K, Kracht M (1999) Induction of interleukin-8 synthesis integrates effects on transcription and mRNA degradation from at least three different cytokine- or stress-activated signal transduction pathways. Mol Cell Biol 19:6742–6753
Montero L, Nagamine Y (1999) Regulation by p38 mitogen-activated protein kinase of adenylate- and uridylate-rich element-mediated urokinase-type plasminogen activator (uPA) messenger RNA stability and uPA-dependent in vitro cell invasion. Cancer Res 59:5286–5293
Pages G, Berra E, Milanini J, Levy AP, Pouyssegur J (2000) Stress-activated protein kinases (JNK and p38/HOG) are essential for vascular endothelial growth factor mRNA stability [In Process Citation]. J Biol Chem 275:26484–26491
Wang SW, Pawlowski J, Wathen ST, Kinney SD, Lichenstein HS, Manthey CL (1999) Cytokine mRNA decay is accelerated by an inhibitor of p38-mitogen-activated protein kinase. Inflamm Res 48:533–538
Winzen R, Kracht M, Ritter B, Wilhelm A, Chen CY, Shyu AB MM, Gaestel M, Resch K, Holtmann H (1999) The p38 MAP kinase pathway signals for cytokine-induced mRNA stabilization via MAP kinase-activated protein kinase 2 and an AU-rich region-targeted mechanism. EMBO J 18:4969–4980
Green J, Khabar KS, Koo BC, Williams BR, Polyak SJ (2006) Stability of CXCL-8 and related AU-rich mRNAs in the context of hepatitis C virus replication in vitro. J Infect Dis 193:802–811
Datta S, Novotny M, Li X, Tebo J, Hamilton TA (2004) Toll IL-1 receptors differ in their ability to promote the stabilization of adenosine and uridine-rich elements containing mRNA. J Immunol 173:2755–2761
Novotny M, Datta S, Biswas R, Hamilton T (2005) Functionally independent AU-rich sequence motifs regulate KC (CXCL1) mRNA. J Biol Chem 280:30166–30174
Deleault KM, Skinner SJ, Brooks SA (2008) Tristetraprolin regulates TNF TNF-[alpha] mRNA stability via a proteasome dependent mechanism involving the combined action of the ERK and p38 pathways. Mol Immunol 45:13–24
Ronkina N, Kotlyarov A, Dittrich-Breiholz O, Kracht M, Hitti E, Milarski K, Askew R, Marusic S, Lin LL, Gaestel M, Telliez JB (2007) The mitogen-activated protein kinase (MAPK)-activated protein kinases MK2 and MK3 cooperate in stimulation of tumor necrosis factor biosynthesis and stabilization of p38 MAPK. Mol Cell Biol 27:170–181
Stoecklin G, Stubbs T, Kedersha N, Wax S, Rigby WF, Blackwell TK, Anderson P (2004) MK2-induced tristetraprolin:14–3-3 complexes prevent stress granule association and ARE-mRNA decay. EMBO J 23:1313–1324
Hitti E, Iakovleva T, Brook M, Deppenmeier S, Gruber AD, Radzioch D, Clark AR, Blackshear PJ, Kotlyarov A, Gaestel M (2006) Mitogen-activated protein kinase-activated protein kinase 2 regulates tumor necrosis factor mRNA stability and translation mainly by altering tristetraprolin expression, stability, and binding to adenine/uridine-rich element. Mol Cell Biol 26:2399–2407
Winzen R, Thakur BK, Dittrich-Breiholz O, Shah M, Redich N, Dhamija S, Kracht M, Holtmann H (2007) Functional analysis of KSRP interaction with the AU-rich element of interleukin-8 and identification of inflammatory mRNA targets. Mol Cell Biol 27:8388–8400
Neininger A, Kontoyiannis D, Kotlyarov A, Winzen R, Eckert R, Volk HD, Holtmann H, Kollias G, Gaestel M (2002) MK2 targets AU-rich elements and regulates biosynthesis of tumor necrosis factor and interleukin-6 independently at different post-transcriptional levels. J Biol Chem 277:3065–3068
Tudor C, Marchese FP, Hitti E, Aubareda A, Rawlinson L, Gaestel M, Blackshear PJ, Clark AR, Saklatvala J, Dean JL (2009) The p38 MAPK pathway inhibits tristetraprolin-directed decay of interleukin-10 and pro-inflammatory mediator mRNAs in murine macrophages. FEBS Lett 583:1933–1938
Ming XF, Stoecklin G, Lu M, Looser R, Moroni C (2001) Parallel and independent regulation of interleukin-3 mRNA turnover by phosphatidylinositol 3-kinase and p38 mitogen-activated protein kinase. Mol Cell Biol 21:5778–5789
Lafarga V, Cuadrado A, Lopez de Silanes I, Bengoechea R, Fernandez-Capetillo O, Nebreda AR (2009) p38 Mitogen-activated protein kinase- and HuR-dependent stabilization of p21Cip1 mRNA mediates the G1/S checkpoint. Mol Cell Biol 29:4341–4351
Tran H, Maurer F, Nagamine Y (2003) Stabilization of urokinase and urokinase receptor mRNAs by HuR is linked to its cytoplasmic accumulation induced by activated mitogen-activated protein kinase-activated protein kinase 2. Mol Cell Biol 23:7177–7188
Dixon DA, Tolley ND, Bemis-Standoli K, Martinez ML, Weyrich AS, Morrow JD, Prescott SM, Zimmerman GA (2006) Expression of COX-2 in platelet-monocyte interactions occurs via combinatorial regulation involving adhesion and cytokine signaling. J Clin Invest 116:2727–2738
Clark A, Dean J, Tudor C, Saklatvala J (2009) Post-transcriptional gene regulation by MAP kinases via AU-rich elements. Front Biosci 14:847–871
Sandler H, Stoecklin G (2008) Control of mRNA decay by phosphorylation of tristetraprolin. Biochem Soc Trans 36:491–496
Kang S, Denley A, Vanhaesebroeck B, Vogt PK (2006) Oncogenic transformation induced by the p110beta, -gamma, and -delta isoforms of class I phosphoinositide 3-kinase. Proc Natl Acad Sci USA 103:1289–1294
Hirsch E, Ciraolo E, Ghigo A, Costa C (2008) Taming the PI3 K team to hold inflammation and cancer at bay. Pharmacol Ther 118:192–205
Madge LA, Pober JS (2000) A Phosphatidylinositol 3-kinase/akt pathway, activated by tumor necrosis factor or interleukin-1, inhibits apoptosis but does not activate NFκB in human endothelial cells. J Biol Chem 275:15458–15465
Stoecklin G, Stoeckle P, Lu M, Muehlemann O, Moroni C (2001) Cellular mutants define a common mRNA degradation pathway targeting cytokine AU-rich elements. RNA 7:1578–1588
Yu W, Murray NR, Weems C, Chen L, Guo H, Ethridge R, Ceci JD, Evers BM, Thompson EA, Fields AP (2003) Role of cyclooxygenase 2 in protein kinase C beta II-mediated colon carcinogenesis. J Biol Chem 278:11167–11174
Subbaramaiah K, Marmao TP, Dixon DA, Dannenberg AJ (2003) Regulation of cyclooxygenase-2 mRNA stability by taxanes. Evidence for involvement of p38, MAPKAPK-2 and HuR. J Biol Chem 25:25
Lee HK, Jeong S (2006) Beta-catenin stabilizes cyclooxygenase-2 mRNA by interacting with AU-rich elements of 3’-UTR. Nucleic Acids Res 34:5705–5714
Castellone MD, Teramoto H, Williams BO, Druey KM, Gutkind JS (2005) Prostaglandin E2 promotes colon cancer cell growth through a Gs-axin-beta-catenin signaling axis. Science 310:1504–1510
Kang MJ, Ryu BK, Lee MG, Han J, Lee JH, Ha TK, Byun DS, Chae KS, Lee BH, Chun HS, Lee KY, Kim HJ, Chi SG (2008) NF-kappaB activates transcription of the RNA-binding factor HuR, via PI3 K-AKT signaling, to promote gastric tumorigenesis. Gastroenterology 135:2030–2042, 2042 e2031–2033
Dormoy-Raclet V, Menard I, Clair E, Kurban G, Mazroui R, Di Marco S, von Roretz C, Pause A, Gallouzi IE (2007) The RNA-binding protein HuR promotes cell migration and cell invasion by stabilizing the {beta}-actin mRNA in a U-rich-element-dependent manner. Mol Cell Biol 27:5365–5380
Lopez de Silanes I, Fan J, Yang X, Zonderman AB, Potapova O, Pizer ES, Gorospe M (2003) Role of the RNA-binding protein HuR in colon carcinogenesis. Oncogene 22:7146–7154
Yoo PS, Mulkeen AL, Silva T, Schmitz J, Tai N, Uchio EM, Chu E, Cha CH (2006) RNA-binding protein HUR regulates VEGF expression in human colorectal cancer cells. J Surg Res 130:219–220
Abdelmohsen K, Srikantan S, Kuwano Y, Gorospe M (2008) miR-519 reduces cell proliferation by lowering RNA-binding protein HuR levels. Proc Natl Acad Sci USA 105:20297–20302
Al-Ahmadi W, Al-Haj L, Al-Mohanna FA, Silverman RH, Khabar KS (2009) RNase L downmodulation of the RNA-binding protein, HuR, and cellular growth. Oncogene 28:1782–1791
Denkert C, Weichert W, Pest S, Koch I, Licht D, Kobel M, Reles A, Sehouli J, Dietel M, Hauptmann S (2004) Overexpression of the embryonic-lethal abnormal vision-like protein HuR in ovarian carcinoma is a prognostic factor and is associated with increased cyclooxygenase 2 expression. Cancer Res 64:189–195
Mrena J, Wiksten JP, Thiel A, Kokkola A, Pohjola L, Lundin J, Nordling S, Ristimaki A, Haglund C (2005) Cyclooxygenase-2 is an independent prognostic factor in gastric cancer and its expression is regulated by the messenger RNA stability factor HuR. Clin Cancer Res 11:7362–7368
Heinonen M, Bono P, Narko K, Chang SH, Lundin J, Joensuu H, Furneaux H, Hla T, Haglund C, Ristimaki A (2005) Cytoplasmic HuR expression is a prognostic factor in invasive ductal breast carcinoma. Cancer Res 65:2157–2161
Niesporek S, Kristiansen G, Thoma A, Weichert W, Noske A, Buckendahl AC, Jung K, Stephan C, Dietel M, Denkert C (2008) Expression of the ELAV-like protein HuR in human prostate carcinoma is an indicator of disease relapse and linked to COX-2 expression. Int J Oncol 32:341–347
Nabors LB, Gillespie GY, Harkins L, King PH (2001) HuR, a RNA stability factor, is expressed in malignant brain tumors and binds to adenine- and uridine-rich elements within the 3’ untranslated regions of cytokine and angiogenic factor mRNAs. Cancer Res 61:2154–2161
Blaxall BC, Dwyer-Nield LD, Bauer AK, Bohlmeyer TJ, Malkinson AM, Port JD (2000) Differential expression and localization of the mRNA-binding proteins, AU-rich element mRNA-binding protein (AUF1) and Hu antigen R (HuR), in neoplastic lung tissue. Mol Carcinog 28:76–83
Brennan SE, Kuwano Y, Alkharouf N, Blackshear PJ, Gorospe M, Wilson GM (2009) The mRNA-destabilizing protein tristetraprolin is suppressed in many cancers, altering tumorigenic phenotypes and patient prognosis. Cancer Res 69:5168–5176
Wang J, Zhao W, Guo Y, Zhang B, Xie Q, Xiang D, Gao J, Wang B, Chen Z (2009) The expression of RNA-binding protein HuR in non-small cell lung cancer correlates with vascular endothelial growth factor-C expression and lymph node metastasis. Oncology 76:420–429
Kanies CL, Smith JJ, Kis C, Schmidt C, Levy S, Khabar KS, Morrow J, Deane N, Dixon DA, Beauchamp RD (2008) Oncogenic Ras and transforming growth factor-beta synergistically regulate AU-rich element-containing mRNAs during epithelial to mesenchymal transition. Mol Cancer Res 6:1124–1136
Dixon DA, Tolley ND, King PH, Nabors LB, McIntyre TM, Zimmerman GA, Prescott SM (2001) Altered expression of the mRNA stability factor HuR promotes cyclooxygenase-2 expression in colon cancer cells. J Clin Invest 108:1657–1665
Oyesanya RA, Lee ZP, Wu J, Chen J, Song Y, Mukherjee A, Dent P, Kordula T, Zhou H, Fang X (2008) Transcriptional and post-transcriptional mechanisms for lysophosphatidic acid-induced cyclooxygenase-2 expression in ovarian cancer cells. FASEB J 22:2639–2651
Lim SJ, Kim HJ, Kim JY, Park K, Lee CM (2007) Expression of HuR is associated with increased cyclooxygenase-2 expression in uterine cervical carcinoma. Int J Gynecol Pathol 26:229–234
Denkert C, Koch I, von Keyserlingk N, Noske A, Niesporek S, Dietel M, Weichert W (2006) Expression of the ELAV-like protein HuR in human colon cancer: association with tumor stage and cyclooxygenase-2. Mod Pathol 19:1261–1269
Farooq F, Balabanian S, Liu X, Holcik M, MacKenzie A (2009) p38 Mitogen-activated protein kinase stabilizes SMN mRNA through RNA-binding protein HuR. Hum Mol Genet 18:4035–4045
Doller A, Pfeilschifter J, Eberhardt W (2008) Signalling pathways regulating nucleo-cytoplasmic shuttling of the mRNA-binding protein HuR. Cell Signal 20:2165–2173
Al-Souhibani N, Al-Ahmadi W, Hesketh J, Blackshear P, Khabar K (2010) The RNA-binding zinc-finger protein tristetraprolin regulates AU-rich mRNAs involved in breast cancer-related processes. Oncogene (in press)
Suswam E, Li Y, Zhang X, Gillespie GY, Li X, Shacka JJ, Lu L, Zheng L, King PH (2008) Tristetraprolin down-regulates interleukin-8 and vascular endothelial growth factor in malignant glioma cells. Cancer Res 68:674–682
Kim TW, Yim S, Choi BJ, Jang Y, Lee JJ, Sohn BH, Yoo H-S, Yeom YI, Park KC (2010) Tristetraprolin regulates the stability of HIF-1[alpha] mRNA during prolonged hypoxia. Biochem Biophys Res Commun 391:963–968
Gebeshuber CA, Zatloukal K, Martinez J (2009) miR-29a suppresses tristetraprolin, which is a regulator of epithelial polarity and metastasis. EMBO Rep 10:400–405
Schaljo B, Kratochvill F, Gratz N, Sadzak I, Sauer I, Hammer M, Vogl C, Strobl B, Muller M, Blackshear PJ, Poli V, Lang R, Murray PJ, Kovarik P (2009) Tristetraprolin is required for full anti-inflammatory response of murine macrophages to IL-10. J Immunol 183:1197–1206
Correa I, Veny M, Esteller M, Pique JM, Yague J, Panes J, Salas A (2009) Defective IL-10 production in severe phenotypes of Crohn’s disease. J Leukoc Biol 85:896–903
Sturlan S, Oberhuber G, Beinhauer BG, Tichy B, Kappel S, Wang J, Rogy MA (2001) Interleukin-10-deficient mice and inflammatory bowel disease associated cancer development. Carcinogenesis 22:665–671
Mosser DM, Zhang X (2008) Interleukin-10: new perspectives on an old cytokine. Immunol Rev 226:205–218
Howell WM, Rose-Zerilli MJ (2007) Cytokine gene polymorphisms, cancer susceptibility, and prognosis. J Nutr 137:194S–199S
Ristimaki A, Narko K, Hla T (1996) Down-regulation of cytokine-induced cyclo-oxygenase-2 transcript isoforms by dexamethasone: evidence for post-transcriptional regulation. Biochem J 318:325–331
Touriol C, Morillon A, Gensac MC, Prats H, Prats AC (1999) Expression of human fibroblast growth factor 2 mRNA is post- transcriptionally controlled by a unique destabilizing element present in the 3’-untranslated region between alternative polyadenylation sites. J Biol Chem 274:21402–21408
Rimokh R, Berger F, Bastard C, Klein B, French M, Archimbaud E, Rouault JP, Santa Lucia B, Duret L, Vuillaume M et al (1994) Rearrangement of CCND1 (BCL1/PRAD1) 3’ untranslated region in mantle-cell lymphomas and t(11q13)-associated leukemias. Blood 83:3689–3696
Yoo PS, Mulkeen AL, Cha CH (2006) Post-transcriptional regulation of vascular endothelial growth factor: implications for tumor angiogenesis. World J Gastroenterol 12:4937–4942
Lejbkowicz F, Goldberg-Cohen I, Levy AP (2005) New horizons for VEGF. Is there a role for nuclear localization? Acta Histochem 106:405–411
Claffey KP, Shih SC, Mullen A, Dziennis S, Cusick JL, Abrams KR, Lee SW, Detmar M (1998) Identification of a human VPF/VEGF 3’ untranslated region mediating hypoxia-induced mRNA stability. Mol Biol Cell 9:469–481
Gnarra JR, Zhou S, Merrill MJ, Wagner JR, Krumm A, Papavassiliou E, Oldfield EH, Klausner RD, Linehan WM (1996) Post-transcriptional regulation of vascular endothelial growth factor mRNA by the product of the VHL tumor suppressor gene. Proc Natl Acad Sci USA 93:10589–10594
Sinha S, Dutta S, Datta K, Ghosh AK, Mukhopadhyay D (2009) Von Hippel-Lindau gene product modulates TIS11B expression in renal cell carcinoma: impact on vascular endothelial growth factor expression in hypoxia. J Biol Chem 284:32610–32618
Danilin S, Sourbier C, Thomas L, Rothhut S, Lindner V, Helwig JJ, Jacqmin D, Lang H, Massfelder T (2009) von Hippel-Lindau tumor suppressor gene-dependent mRNA stabilization of the survival factor parathyroid hormone-related protein in human renal cell carcinoma by the RNA-binding protein HuR. Carcinogenesis 30:387–396
Piecyk M, Wax S, Beck AR, Kedersha N, Gupta M, Maritim B, Chen S, Gueydan C, Kruys V, Streuli M, Anderson P (2000) TIA-1 is a translational silencer that selectively regulates the expression of TNF-alpha. EMBO J 19:4154–4163
Gueydan C, Droogmans L, Chalon P, Huez G, Caput D, Kruys V (1999) Identification of TIAR as a protein binding to the translational regulatory AU-rich element of tumor necrosis factor alpha mRNA. J Biol Chem 274:2322–2326
Phillips K, Kedersha N, Shen L, Blackshear PJ, Anderson P (2004) Arthritis suppressor genes TIA-1 and TTP dampen the expression of tumor necrosis factor alpha, cyclooxygenase 2, and inflammatory arthritis. Proc Natl Acad Sci USA 101:2011–2016
Dixon DA, Balch GC, Kedersha N, Anderson P, Zimmerman GA, Beauchamp RD, Prescott SM (2003) Regulation of cyclooxygenase-2 expression by the translational silencer TIA-1. J Exp Med 198:475–481
Raymond V, Atwater JA, Verma IM (1989) Removal of an mRNA destabilizing element correlates with the increased oncogenicity of proto-oncogene fos. Oncogene Res 5:1–12
Capaccioli S, Quattrone A, Schiavone N, Calastretti A, Copreni E, Bevilacqua A, Canti G, Gong L, Morelli S, Nicolin A (1996) A bcl-2/IgH antisense transcript deregulates bcl-2 gene expression in human follicular lymphoma t(14;18) cell lines. Oncogene 13:105–115
Schiavone N, Rosini P, Quattrone A, Donnini M, Lapucci A, Citti L, Bevilacqua A, Nicolin A, Capaccioli S (2000) A conserved AU-rich element in the 3’ untranslated region of bcl-2 mRNA is endowed with a destabilizing function that is involved in bcl-2 down- regulation during apoptosis. FASEB J 14:174–184
Ubeda M, Schmitt-Ney M, Ferrer J, Habener JF (1999) CHOP/GADD153 and methionyl-tRNA synthetase (MetRS) genes overlap in a conserved region that controls mRNA stability. Biochem Biophys Res Commun 262:31–38
Chesney J, Mitchell R, Benigni F, Bacher M, Spiegel L, Al-Abed Y, Han JH, Metz C, Bucala R (1999) An inducible gene product for 6-phosphofructo-2-kinase with an AU-rich instability element: role in tumor cell glycolysis and the Warburg effect. Proc Natl Acad Sci USA 96:3047–3052
Hamilton BJ, Nichols RC, Tsukamoto H, Boado RJ, Pardridge WM, Rigby WF (1999) hnRnp A2 and hnRnp L bind the 3’UTR of glucose transporter 1 mRna and exist as a complex in vivo. Biochem Biophys Res Commun 261:646–651
Hoyle PE, Steelman LS, McCubrey JA (1997) Autocrine transformation of human hematopoietic cells after transfection with an activated granulocyte/macrophage colony stimulating factor gene. Cytokines Cell Mol Ther 3:159–168
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Khabar, K.S.A. Post-transcriptional control during chronic inflammation and cancer: a focus on AU-rich elements. Cell. Mol. Life Sci. 67, 2937–2955 (2010). https://doi.org/10.1007/s00018-010-0383-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-010-0383-x