
Abstract
The Wnt/beta-catenin/TCF4 pathway plays critical roles in the maintenance of small intestinal epithelium; however, downstream targets of the beta-catenin/TCF4 complex are not extensively characterized. We identified miR-30e as an immediate target activated by the beta-catenin/TCF4 complex. miR-30e was detected in the peri-nuclear region of the intestinal crypt IEC-6 cells. Bioinformatics analysis revealed clustered beta-catenin/TCF4 binding sites within the miR-30e promoter region. This promoter region was cloned into pGL3-control luciferase reporter vector, with the enhancer region removed. Transfection of pCMV-SPORT6-beta-catenin expression vector dose-dependently increased luciferase activity, and co-transfection of pCMV-SPORT6-TCF4 expression vector further enhanced the promoter activity. Dexamethasone-induced IEC-6 cells differentiation caused a 2.5-fold increase in miR-30e expression, and upon beta-catenin siRNA transfection, miR-30e increased 1.3-fold. Electrophoretic mobility shift assay and chromatin immunoprecipitation assay confirmed the binding between beta-catenin/TCF4 complexes from IEC-6 nuclear extracts and the putative sequences in the miR-30e promoter. These results demonstrate that beta-catenin/TCF4 transactivates miR-30e during intestinal cell differentiation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The small intestine has two functionally distinct compartments, villi and crypts of Lieberkühn (crypts). Villi are finger-like protrusions extending to the intestinal lumen, and contain three types of terminally differentiated cells: (1) enterocytes are the only absorptive cells, whose function is to absorb nutrients from food sources, and comprise the majority of the villi; (2) enteroendocrine cells are local endocrine cells that secret gastrointestinal hormones, such as somatostatin, cholecystokinin and glucagon; and (3) goblet cells which have the sole function to secret mucus, protecting the intestinal barrier from mechanical damage. The crypts contain two types of cells: (1) Paneth cells are also terminally differentiated, and secrete anti-microbial peptides in response to pathogens; and (2) undifferentiated stem cells which reside within crypts, and undergo self-renewal, proliferation, and eventually differentiate into all the above-mentioned terminally differentiated cells [1–3]. The structural and functional integrity of the small intestine is largely dependent upon a continuous and well-organized flow of fully differentiated cell lineages migrating from the crypts. However, the molecular mechanisms by which the individual cell lineages are derived from the crypts await full characterization.
A number of cellular signaling pathways have been suggested to be involved in small intestinal cell proliferation and differentiation, among which the Delta-Notch pathway and Wnt/beta-catenin/T cell factor 4 (TCF4) pathway have drawn most attention [3–5]. Activation of the highly conserved Delta-Notch signaling pathway is usually considered to maintain the proliferative state of the intestinal progenitor cells, and lead to columnar cell lineages rather than secretory cells [6–10]; The Wnt/beta-catenin/TCF4 signaling pathway instead plays a key role in maintaining progenitor cells in the proliferative state and making them select towards secretory cells [11–15]. Decreased Wnt signaling decreases the size of crypts [12], and aberrant upregulation of the Wnt signaling is associated with local tumorigenesis [16, 17]. However, the molecular determination of individual terminally differentiated cell lineages does not rely solely on either Notch or Wnt signaling but on the coordinate interaction of the two [3, 18, 19].
Wnt signal activates three independent pathways, the canonical Wnt/beta-catenin cascade, the noncanonical planar cell polarity pathway, and the Wnt/Ca2+ pathway. The intestinal crypts express members of the canonical Wnt/beta-catenin pathway [20]. The locally expressed Wnt molecule binds to the surface receptor frizzled, and stabilizes cytosolic transcriptional factor beta-catenin from degradation. Subsequently, downstream functional outcomes are elicited by the translocation of beta-catenin to the nucleus, where it forms a nuclear binding complex with TCF4, a member of T-cell factors and specifically expressed in the small intestine, and the complex subsequently regulates transcription of a variety of genes.
The end effect of Wnt signaling is a result of gene transactivation by the nuclear beta-catenin/TCF4 complex. To date, genes in the intestine that have been found to be downstream targets of beta-catenin/TCF4 transactivation include PKC [21], p16(INK4a) [22], S100A4 [23], Rad6B [24], c-myc [25], cyclin D1 [26], MMP-7 [27], PPARδ [28], and cohesion SMC3 [29], which are mostly involved in tumorigenesis. The downstream targets of beta-catenin/TCF4 binding complex responsible for normal intestinal homeostasis, however, remain an open question.
microRNAs (miRNAs), typically ~22 nucleotides (nt) in size, are a large family of non-coding RNA molecules that can repress gene expression post-transcriptionally [30–32]. miRNAs regulate genes in a variety of biological processes, and have been shown to mediate differentiation of specific cell types, including B-cells and T-cells [33], cardiac and somatic muscle progenitors [34], osteoblasts [35], adipocytes [36], neurons [37], and embryonic stem cells [38]. In particular, miR-194 was recently found to interact with the transcription factor HNF1-alpha during intestinal epithelial cell differentiation in a Caco-2 cell model [39].
The objective of this study was to determine whether specific miRNA(s) could be the downstream target of the beta-catenin/TCF4 complex and is/are involved in intestinal cell differentiation.
Materials and methods
Confocal microscopy
IEC-6 cells were seeded onto sterile glass coverslips at 85% confluency per 24-well culture plates the day prior to each experiment, and cells were fixed with 4% paraformaldehyde in DEPC-treated 1× phosphate-buffered saline (PBS) for 30 min and permeabilized with 0.2% Triton X-100 in DEPC-treated 1× PBS for 5 min. Endogenous peroxidase activity was blocked in 1% H2O2 for 3 min. Non-specific hybridization was blocked with hybridization buffer (50% formamide, 0.5% SDS, 5× SSPE, 5× Denhardt’s solution, and 20 μg/ml sheared, denatured, salmon sperm DNA) at 42°C for 2 h. miR-30e LNA probe 5′-labeled with Digoxigenin (Exiqon) was diluted to 2.5 nM in hybridization buffer and incubated with the cells for 16 h at 42°C. Stringency wash was performed with 2× SSC, 0.1% SDS at 42°C three times for 10 min each, and 0.1× SSC, 0.1% SDS at 65°C three times for 10 min each. The cells were incubated with 5% BSA in TNT buffer (0.1 M Tris–HCl, pH 7.5, 0.15 M NaCl, and 0.05% Tween 20) for 30 min, and incubated with anti-DIG-HRP (Roche) for 30 min. Subsequently, fluorescence was detected with the TSA Cy5 system (Perkin Elmer). Non-specific staining was determined by using scrambled-miR detection probe (Exiqon), RNase A (Invitrogen) treatment of the cells before hybridization, incubating the sections without probe, and incubating with TSA Cy5 system alone after hybridization. Nuclei were stained with 0.3 μg/ml DAPI (Invitrogen) in 1× PBS for 5 min. Coverslips were mounted in ProLong Gold (Invitrogen) and sealed with nail polish. Fluorescent signals were captured under the Olympus FV1000 confocal microscope at the Microscopy and Imaging facility of the section of Molecular and Cell Biology at UCDavis.
Bioinformatic sequence analysis
The mature miR-30e sequences of human, mouse, and rat were retrieved from the miRBase Sequence Database (http://microrna.sanger.ac.uk/sequences/) and the sequences were found to be identical and conserved between the three species.
The sequence of the miR-30e promoter was retrieved from the Table Browser (http://genome.ucsc.edu/cgi-bin/hgTables) at UCSC (University of California, Santa Cruz) Genome Bioinformatics (http://genome.ucsc.edu). The BAC containing the miR-30e promoter region was identified from the Genome Browser (http://genome.ucsc.edu/cgi-bin/hgGateway) using human Mar. 2006 assembly.
Transcriptional factor binding sites were predicted by the Transcription Element Search System (http://www.cbil.upenn.edu/cgi-bin/tess/tess) and the TFSEARCH system (http://www.cbrc.jp/research/db/TFSEARCH.html).
Plasmid vector construction
A 2,000-bp fragment of the miR-30e promoter region was PCR amplified from a BAC CTD-3060E5 (Open Biosystems). The primer sequences used for PCR amplification were promoter forward, 5′CGCAGATCTCTTAACTATACATAATATGTTGGG3′, reverse, 5′CGCAAGCTTGTAGCAAAGACTGCCCAGAAAG3′. The PCR product was cloned into pBluescript SK(+) vector (Stratagene). The enhancer region of the pGL3-control luciferase reporter vector (Promega) was removed via BglII and HindIII sites. Subsequently, the promoter sequence was excised from pBluescript SK (+) and cloned into pGL3-control luciferase reporter vector without the enhancer region; the resulting vector was named miR30epromoter-pGL3 vector.
The pBluescriptR-TCF4 vector containing TCF4 full-length cDNA was from Open Biosystems. The TCF4 cDNA sequences containing the coding sequence (CDS) and the PCMV-SPORT6 vector backbone were retrieved from pBluescriptR-TCF4 vector and PCMV-SPORT6-beta-catenin vector (Open Biosystems) by ApaI and EcoRI, respectively. Subsequently, the TCF4 cDNA CDS was cloned into the PCMV-SPORT6 vector backbone.
Site-directed mutagenesis was performed to silence the putative TCF4 binding site within the miR-30e promoter region. The mutation was introduced by PCR using the following primers: 30emut1 forward, 5′-GTAAAAATCTTTTAATTTTATTATTCAAAGAAAGGAAG-3′, reverse 5′-CTTCCTTTCTTTGAATAATAAAATTAAAAGATTTTTAC-3′; 30emut2 forward, 5′-GTATCTCTTTCTTGCGCAACGATGGTTGCCCACCAG-3′, reverse 5′-CTGGTGGGCAACCATCGTTGCGCAAGAAAGAGATAC-3′ (mutation underlined). Mutagenesis was carried out using the Quickchange site-directed mutagenesis kit from Stratagene. The mutated vector was named mut-pGL3-miR30e.
Fidelity and orientation of the cloning were confirmed by DNA sequencing (UCDavis, Division of Biological Sciences Automated DNA Sequencing Facility).
Cell culture and transfections
HEK293 cells (ATCC# CRL-1573) were maintained in Dulbecco’s modified Eagle’s medium (DMEM) with 1.5 g/l sodium bicarbonate, 10% FBS (Sigma), 100 U/ml penicillin, 100 mg/ml streptomycin (Sigma), and incubated at 37°C, 5% CO2.
IEC-6 cells (ATCC# CRL-1592) were maintained in DMEM with 1.5 g/l sodium bicarbonate, 10% FBS (Sigma), 100 U/ml penicillin, and 100 mg/ml streptomycin (Sigma), 0.1 U/ml bovine insulin (Sigma) and incubated at 37°C, 5% CO2. Cells were passaged when approximately 90% confluent at 1:4, and cell stocks were not used after 7 passages upon arrival from ATCC.
All transfections were carried out with Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions.
For luciferase assays, HEK293 cells were seeded at 70% confluency per 96-well plates the day prior to transfection with 3.5 μg/ml luciferase expression construct, pCMV-SPORT6-beta-catenin expression vector (Open Biosystems), and pCMV-SPORT6-TCF4 expression vector. Mock transfected cells were transfected with luciferase expression construct alone. PGL4.73[hRluc/SV40] vector (Promega) served as an internal control for transfection efficiency. Luciferase activity was measured 24 h after transfection using the Dual-Glo Luciferase Assay System (Promega).
For dexamethasone treatment, IEC-6 cells were seeded at 60% confluency per 6-well plates the day prior to treatment with 100 nM cell culture grade dexamethasone (Sigma), the control group was treated with the vehicle ethanol alone, and the cells were collected 48 h after treatment for real-time Quantitative-Polymerase Chain Reaction (Q-PCR) and immunoblotting analysis.
For beta-catenin siRNA and TCF4 siRNA, IEC-6 cells were seeded at 70% confluency per 6-well plates the day prior to transfection with 75 nM beta-catenin siRNA (siRNA ID: 190086), or 100 nM TCF4 siRNA (siRNA ID: 195500). Mock transfected cells were transfected with transfection reagent alone, and the cells were collected 48 h after transfection for real-time Q-PCR and immunoblotting analysis.
Immunoblotting analysis
IEC-6 cells were homogenized in Tris buffer [50 mM Tris·HCl pH 7.4, 150 mM NaCl, 1% Triton X-100, 0.1% SDS, 1 mM EDTA (ethylenediamine tetraacetic acid), and 0.5% sodium deoxycholate] containing 1× complete EDTA-free protease inhibitor (Roche), and 40 μg proteins were electrophoresed through 10% polyacrylamide gel, transferred onto nitrocellulose membrane at 350 mA for 60 min, and blocked overnight in 1× PBS/0.1% Tween-20 (PBST) with 5% non-fat milk at 4°C.
Antibodies against beta-catenin and TCF4 were purchased from Santa Cruz Biotechnology. The blots were incubated with anti-rat beta-catenin (1:500 in PBST with 5% non-fat milk) and anti-rat-TCF4 (1:200 in PBST with 5% non-fat milk) at room temperature for 1 h, washed for 15 min with PBST, and incubated with rabbit anti-mouse IgG horseradish peroxidase linked secondary antibody (DakoCytomation) (1:2,000 in PBST with 5% non-fat milk). Bands were detected using Super Signal Femto chemiluminescent reagent (Pierce) and quantified using the Chemi-doc gel quantification system (Bio-Rad). All data were normalized to beta-actin.
Real-time Q-PCR
Total RNA from IEC-6 cells were isolated using mirVana microRNA isolation kit (Ambion) and diluted to 2 μg/μl in DEPC-treated water (Ambion).
For miR-30e assay, cDNA was generated from 2 μg RNA using TaqMan® MicroRNA Reverse Transcription Kit (Applied Biosystems) according to the manufacturer’s protocol. The miR-30e gene specific stem-loop RT primer for reverse transcription was designed according to miRNAs sequences listed in the Sanger miRBase (http://microrna.sanger.ac.uk/sequences/), and the reaction was performed at 16°C for 30 min, followed by 42°C for 30 min and 85°C for 5 min. Relative expression of miR-30e and U87 (internal standard) were determined by real-time Q-PCR using a FAM detection system (Applied Biosystems). The reaction was performed in a 15-μl reaction mixture using the ABI 7900 HT real-time thermocycler (Applied Biosystems) with the following protocol: 50°C for 2 min, 95°C for 10 min, 40 cycles of 95°C for 15 s, 60°C for 1 min.
Each sample was analyzed in triplicate and normalized to U87 for miR-30e, using the following equation: ΔCtGENE = CtGENE − CtU87. The fold change, relative to the control group which was used as the internal control, was calculated using the following equation: 2(ΔΔCtGENE) where ΔΔCtGENE = ΔCtGENE of U87 − ΔCtGENE of each well.
Electrophoretic mobility shift assay (EMSA)
Nuclear proteins from IEC-6 cells treated with dexamethasone were extracted by NE-PER nuclear extraction kit (Pierce). Biotin labeled probes were synthesized at Bioneer. The following oligos and their anti-sense oligos were synthesized and annealed: 5′CTGCTTGCTTTGTTATATAAAAC3′, corresponding to the −272/−250 bp region of miR-30e promoter; 5′CCGCTCTTCTTTGTTACAGAGAAATG3′, corresponding to the −1,239/−1,214 region of miR-30e promoter. EMSA reactions (20 μl) were assembled using LightShift Chemiluminescent EMSA Kit (Pierce), and the reaction contained 2 nM of biotin-labeled ds oligonucleotide, 1× binding buffer, 1 μl 1 μg/μl Poly (dI•dC), 1 μl 50% glycerol, 1 μl 1% NP-40, 1 μl 1 M KCl, 1 μl 100 mM MgCl2, and/or 3 μl NE-PER nuclear extracts, 4 pmol unlabeled probe. Mixtures of protein and DNA were incubated for 30 min at room temperature. Following addition of 5 μl loading buffer, bound and free DNA were resolved by fractionation on 5% native polyacrylamide TBE gel (Bio-Rad) in 0.5× TBE. Gels were pre-run for 1 h at 100 V at room temperature using the Miniprotean II system (Bio-Rad), and run with samples under the same conditions for 45 min. The DNA/protein complex was transferred onto Biodyne® B Nylon Membrane (Pierce) at 400 mA for 35 min. The transferred DNA was cross-linked to the membrane at 120 mJ/cm2 for 60 s using a GS Gene Linker (Bio-Rad) equipped with a 254 nm bulb. The biotin-labeled DNA was detected by means of the Chemiluminescent Nucleic Acid Detection Module (Pierce).
Chromatin immunoprecipitation (ChIP)
IEC-6 cells in 6-well culture plates were treated with dexamethasone, and fixed with 1% formaldehyde, washed with ice-cold 1× PBS, harvested, lysed and sonicated. The soluble chromatin containing supernatant was then subjected to immunoprecipitation using anti-beta-catenin and anti-TCF4 antibodies (Santa Cruz Biotechnology) at 4°C overnight, followed by adding protein A agarose and salmon sperm DNA. Unrelated immunoglobulin G (IgG) was used as a negative control for immunoprecipitation. Precipitated DNA was recovered via phenol/chloroform extraction and amplified by RT-PCR for 40 cycles using specific primer sets for the indicated specific promoter regions of miR-30e. The PCR primers for ChIP assays are: region1 (−1348/−1103) forward, 5′CAGCTGTAGACTTGCCCGTAG3′, reverse 5′CTGCAATCCCTTCACAGCTC3′; region2 (−386/−149) forward, 5′AATAGACCTCTGTCCTTTAGAGC3′, reverse, CTCTGACTGGTTATTAATACAGGC3′; region3 (−1535/−1328) forward, 5′CTGCTAATTGAGATTACTAGGTTTG3′, reverse, CTACGGGCAAGTCTACAGCTG3′; region4 (−1991/−1787) forward, 5′CTTAACTATACATAATATGTT3′, reverse 5′CATAGCTTGTGAGAATTCTGATAT3′. The DNA sequence corresponding to the +927/+1,168 region served as negative control: control forward, 5′GAGGCCACTGCTAAAGTTGA3′, reverse, 5′TTTAGTGGGGCAGAAGTCAG3′.
Data analysis
All data were analyzed by Prism (Prism GraphPad Software). The effect of beta-catenin/TCF4 overexpression on the luciferase activity in HEK293 cells and beta-catenin/TCF4 siRNAs on miR-30e expression in IEC-6 cells were tested by Student’s t test. Data are shown as means ± SEM of two replicates and are representative of three independent experiments. Differences were considered significant when P < 0.05.
Results
miR-30e is present in the IEC-6 crypt cell line
To investigate if miR-30e is present in small intestinal crypt cells, we used confocal microscopy and determined that, in IEC-6 cells, there is an intense staining at the peri-nuclear region for miR-30e, with low levels of diffusion in the cytosol and nucleus (Fig. 1a). No staining was observed by incubating with scrambled miRNA detection probe, with RNase A treatment before hybridization, or with TSA Cy5 system alone.

Subcellular localization of miR-30e in IEC-6 cells. miR-30e distribution in IEC-6 cells was examined by confocal microscopy. Paraformaldehyde-fixed cells were probed with LNA DIG-miR-30e probe. Reactivities were visualized by TSA Cy5 system (red a), and nuclei were stained with DAPI nucleic acid dye (blue b). c The peri-nuclear localization of miR-30e. Magnification of ×60 under oil. Scale bar 10 μm
The miR-30e promoter region contains clustered binding sites for beta-catenin/TCF4
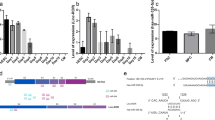
To characterize the transcriptional factor binding sites within the miR-30e promoter, miRBase Sequences Database was used to identify the mature miR-30e sequences from human, mouse, and rat, and they were found to share the identical sequence of 5′UGUAAACAUCCUUGACUGGAAG3′. Furthermore, the promoter sequences were examined for the presence of putative transcriptional factor binding sites for beta-catenin/TCF4 in TESS and TFSEARCH databases. Within a 2,000-bp promoter region, all three sequences contained two canonical beta-catenin/TCF4 binding sites (5′CTTTGTT3′) (black arrowhead in Fig. 2); moreover, the mouse and rat promoter harbored one additional beta-catenin/TCF4 binding site, 5′AACAAAG3′ (empty arrowhead in Fig. 2). The human, mouse, and rat promoter further contained two, three, and four TCF4 binding sequences (5′TTTTGTT3′) (sketched arrowhead in Fig. 2).

Comparative sequence analysis of miR-30e gene promoter from human, mouse, and rat. The promoter sequences were retrieved from miRBase Sequences Database, and the analysis was performed at TESS and TFSEARCH. The binding domains for two types of beta-catenin/TCF4 complex are shown with black and empty arrowheads, and for TCF4 with hatched arrowheads
Overexpression of beta-catenin and TCF4 enhances miR-30e promoter activity
To assess the potential role of beta-catenin and TCF4 in regulation of the miR-30e gene promoter, we determined if the promoter activity was affected by the level of intracellular beta-catenin and TCF4 using a luciferase reporter assay. A dose-dependent increase in the luciferase activity was observed with increasing concentrations of beta-catenin expression vector ranging from 0.15 to 1.5 μg/ml (P < 0.05) (Fig. 3a), indicating increased luciferase gene transactivation through the promoter. Beta-catenin at concentrations higher than 2 μg/ml decreased cell survival when examined 24 h post-transfection (data not shown). Co-transfection of 1.2 μg/ml TCF4 expression vector further potentiated the enhancing effect of beta-catenin (P < 0.01) (Fig. 3b), in line with the fact that TCF4 facilitates translocalization of beta-catenin to the nucleus and binding to target genes. When the TTTTGTT sites were mutated, TCF4 overexpression did not result in further enhancement of the luciferase activity, as indicated by similar luciferase activity shown in Fig. 3c and d. This provides functional evidence that TCF4 directly regulates beta-catenin activity through TTTTGTT sites within the miR30e promoter region.

Effect of beta-catenin/TCF4 overexpression on the miR-30e gene promoter activity. IEC-6 cells were co-transfected with 3.5 μg/ml luciferase expression vector containing wild-type and mutated miR-30e promoter and increasing concentrations of pCMV-SPORT6-beta-catenin expression vector without (a,c) or with (b,d) 1.2 μg/ml pCMV-SPORT6-TCF4 expression vector. Luciferase activity was corrected for transfection efficiency based on pGL4.73[hRluc/SV40] vector transfection. Beta-catenin overexpression dose-dependently increased the luciferase activity (P < 0.05) (a) and overexpresison of TCF4 further enhanced the effect from beta-catenin (P < 0.05) (b). When the TTTTGTT sites were mutated, overexpression of TCF4 did not further enhance the effects of beta-catenin (c,d). Values are means ± SEM run in triplicates; asterisks indicate significant differences between treatment and control groups
miR-30e is associated with beta-catenin during dexamethasone induced cell differentiation
To determine the functional relationship between miR-30e and beta-catenin, IEC-6 cells were treated with 100 nM dexamethasone for 48 h to induce cell differentiation. In differentiated cells, miR-30e expression was upregulated compared to undifferentiated cells (1.00 ± 0.20 vs 2.58 ± 0.26; P < 0.01), while 75 nM beta-catenin siRNA knocked-down beta-catenin protein expression by 50%, and miR-30e expression increased to 1.3-fold compared to undifferentiated cells (1.00 ± 0.20 vs 1.29 ± 0.32; P = 0.49), which is significantly different from cells treated with dexamethasone alone (1.29 ± 0.32 vs 2.58 ± 0.26; P < 0.05) (Fig. 4a); In addition, 100 nM TCF4 siRNA knocked-down TCF4 protein expression by 50%, and miR-30e expression increased 0.4-fold compared to undifferentiated cells (1.00 ± 0.04 vs 1.42 ± 0.16; P = 0.1148), which is also significantly different from cells treated with dexamethasone alone (1.42 ± 0.16 vs 1.86 ± 0.17, P < 0.05) (Fig. 4b).

Effects of beta-catenin and TCF4 knock-down on miR-30e expression during dexamethasone induced cell differentiation. IEC-6 cells were treated with 100 nM dexamethasone for 48 h with or without 75 nM beta-catenin siRNA, 100 nM TCF4 siRNA. Total RNA was extracted and analyzed for miR-30 expression by real-time Q-PCR. miR-30e significantly increased upon dexamethasone treatment (P < 0.05), beta-catenin siRNA and TCF4 siRNA both blocked the upregulation. Values are means ± SEM run in triplicates, asterisk indicates significant differences between dexamethasone treatment alone and control group, letters indicate significant differences between dexamethasone treatments without (a) and with (b) siRNA treatment
Putative binding sequences within the miR-30e promoter actively bind beta-catenin/TCF4
The binding properties of the beta-catenin were confirmed by performing EMSA and supershift assay. DNA probes of 23–26 bp long, containing the putative beta-catenin/TCF4 core binding sequences (−265/−259, −1,231/−1,225) and their flanking region at both sides within the human miR-30e promoter, were tested. Incubation of the biotin-labeled probe −1,239/−1,214 with IEC-6 cell nuclear extracts caused a major retarded band as a result of the binding between the labeled probe and the beta-catenin/TCF4 transactivation complex (Fig. 5a, lane 1). An excessive amount of unlabeled probe prevented the labeled probe from binding with the transactivation complex, as indicated by no retarded band (Fig. 5a, lane 2). Incubation with beta-catenin antibody, but not an irrelevant antibody (alkaline phosphatase), further shifted the band indicating the presence of beta-catenin in the binding complex (Fig. 5a, lanes 3 and 4). Identical data were obtained for the other probe corresponding to −272/−250 bp within the human miR-30e promoter (data not shown).

Identification of beta-catenin/TCF4 binding sites within the 2,000-bp miR-30e promoter region. a 3 μl of IEC-6 cell NE-PER nuclear extracts were incubated with 2 nM biotin-labeled DNA probe corresponding to the human miR-30e promoter sequence −1,239/−1,214 bp (lane 1). For binding competetion, 50-fold excess of unlabeled DNA probe was included in the reaction (lane 2). For supershift assay, 0.5 μg beta-catenin antibody (lane 3) or an irrelevant antibody (antibody for alkaline phosphatase, lane 4) were included. Supershift was also observed for probe corresponding to −272/−250 bp region of the miR-30e promoter (data not shown). b IEC-6 cells treated with dexamethasone, the precipitated DNA, along with the DNA isolated before immunoprecipitation (Input), were analyzed by PCR amplication with specific primers corresponding to the region1 (−1,348/−1,103), region2 (−386/−149), region3 (−1,535 to −1,328), region4 (−1,991 to −1,787) and negative control (+927/+1,168)
The binding properties of beta-catenin/TCF4 to the miR-30e promoter were also observed by ChIP assay. Antibody against beta-catenin/TCF4 was used to immunoprecipitate the protein–chromatin complex. As depicted in Fig. 5b, specific binding of beta-catenin/TCF4 to the miR-30e promoter region (−1,348/−1,103, Fig. 5b region 1 panel; −386/−149, Fig. 5b region 2 panel), and specific binding of TCF4 to miR-30e promoter region (−1,535/−1,328, Fig. 5b region 3 panel; −1,991/−1,787, Fig. 5b region 4 panel), but not to a negative control region (+927/+1,168, Fig. 5b bottom panel) was observed.
Discussion
miRNAs are non-protein coding RNA molecules, classified as small regulatory RNAs. Although miRNAs have been suggested to mediate the differentiation of neurogenesis, myogenesis, and lymphogenesis, there have been very few reports demonstrating a role for miRNAs during small intestinal cell differentiation. However, a recent report of Hino et al. [39] found miR-194 to be the downstream regulator of transcriptional factor HNF-1α, which has been suggested to be required in the lineage selection toward enteroendocrine cells [40] and enterocytes [41]. The roles of other miRNAs in the classical signaling pathways of small intestinal stem cell differentiation remain to be determined. During intestinal cell differentiation, a large number of kinases and phosphatases are activated/inactivated, which all require regulatory molecules. As one of the characteristics of the miRNA pathway is their multi-target mechanism in coordinately fine-tuning biological networks, and the Wnt/beta-catenin/TCF4 signaling has the potential of transactivating different downstream genes by DNA binding, we hypothesized that, in addition to targeting protein-coding genes, beta-catenin/TCF4 targets miRNA during intestinal cell differentiation.
We initiated this project by finding candidate miRNAs in the miRBase sequence database by using Transcriptional Factor Prediction tools. These analyses pinpointed miRNAs of interest and, as a result of these bioinformatics analyses, miR-30e was identified to have high frequency clustered beta-catenin/TCF4 binding sites at the gene promoter. So far, there are limited reports specifically addressing the function of miR-30e. Wu et al. [54] found that miR-30e regulates Ubc9 expression in cancer cells, and ectopic expression of miR-30e was found to suppress cell proliferation. Our results support miR-30e as a mediator in the cell differentiation process, which suggests a role for miR-30 in the maintenance of cellular development.
Furthermore, the localization of two beta-catenin/TCF4 binding sites is conserved across the three species; one is a canonical beta-catenin/TCF4 binding site (5′CTTTGTT3′), located at −265/−259 and −1,231/−1,225 upstream of the transcription start site; and the other is a TCF4 binding site (5′TTTTGTT3′), located at −1,947/−1,941 and −1,412/−1,406 from the transcription start site. Remarkably, in the luciferase reporter assay, with pGL3 vector containing both CTTTGTT and TTTTGTT binding sites, TCF4 co-transfection further enhanced the beta-catenin up-regulated luciferase activities by 1.62-, 1.38-, 1.27-, 1.12-, and 1.15-fold at beta-catenin concentrations 0.15, 0.3, 0.6, 1.2, and 1.5 μg/ml, respectively (Fig. 3b); mutation of the two TTTTGTT sites abolished the ability of the TCF4 expression vector to further enhance the up-regulation of luciferase activity (Fig. 3c, d), supporting the argument for TTTTGTT being one of the TCF4 binding sites which directly regulates beta-catenin activity.
Interestingly, putative binding sites for some other transcriptional factors involved in intestinal cell differentiation were also identified within the miR-30e promoter region (TESS/TFSEARCH). miR-30e promoter of human, mouse, and rat all contain two CDX-1 binding sites. CDX-1 is an intestine-specific transcriptional factor expressed in the lower part of the crypts, and overexpression of CDX-1 by stable transfection in IEC-6 cells induced enterocyte differentiation; morphology of the top layer also showed characteristics of epithelial cells after transfection [42, 43]. The miR-30e promoter also contained more than 15 binding sites for nuclear factor AP-1 (human 19, mouse 18, and rat 19), and AP-1 binding activity was increased in Caco-2 cells at post-confluency during spontaneous differentiation into the enterocyte phenotype [44]. Lastly, there is a presence of conserved HES1 binding sites (human 5, mouse 4, rat 2). HES1 is a transcriptional gene repressor: HES1 knock-out mice have an increased number of lysozyme-positive and cryptdin-4-positive cells, indicating their inhibitory role in Paneth cell differentiation [45]. The presence of binding domains for other differentiation-related transcriptional factors further supports the concept that miR-30e targets intestinal cell differentiation.
IEC-6 cells were originally established from intestinal crypt cells, and bear the characteristics of undifferentiated intestinal cells morphologically and immunologically [46]. Cortisol and thyroid hormones are essential in the maturation of fetal organs, tissues and molecular signaling, and glucocorticoids have been well characterized to stimulate intestinal maturation by influencing morphological, cytological, and functional differentiation [47, 48]. In our study, dexamethasone was used to elicit differentiation events in IEC-6 cells, and the treatment was able to upregulate gene markers for absorptive enterocytes (ALP) [49, 50], Paneth cells (Lyzs, Pglyrp1 and spink4) [15, 51], goblet cells (trefoil F3), and enteroendocrine cells (CCK and Collagen IV) [52, 53] (data not shown). Normally, beta-catenin in the cells is constantly expressed and degraded, and in the Wnt signaling pathway, the presence of extracellular Wnt signal stabilizes beta-catenin, which in turn associates with TCF family transcription factors, resulting in beta-catenin/TCF complex formation and subsequent translocation to the nucleus. There is a balanced expression level of beta-catenin in the cytoplasm, and it is the translocation to the nucleus that matters to transduce the Wnt signal. In the present study, dexamethasone did not significantly affect total beta-catenin or TCF4 protein, but as can be seen from the EMSA and ChIP data, beta-catenin and TCF4 were both found to be translocated to the nucleus, where they cause the change in miR-30e expression.
According to the cellular model suggested by Crosnier et al. [3], the Wnt/beta-catenin/TCF4 pathway and Delta-Notch pathway jointly maintain and commit the intestinal progenitor cells: (Wnt+, Notch−) cells become secretory cell types; (Wnt+, Notch+) cells continue to divide without differentiation; thereafter, cells which lose Wnt activation differentiate into absorptive cells if Notch remains activated in them and differentiate into secretory cells if Notch signaling is also inactivated. From the observations of this study, we suggest that dexamethasone treatment elicited dynamic responses in IEC-6 cells in regard to the control of Wnt and Notch signaling, and as a result, the undifferentiated crypt cells were able to give rise to terminally differentiated intestinal cell types, further showing that the IEC-6 cell line is a good model to study Wnt/Notch signaling during intestinal cell differentiation. In the present study, the in situ hybridization (Fig. 2) experiment confirmed the cytolocalization of miR-30e in IEC-6 cells, and down-regulation of miR-30e after beta-catenin or TCF4 knock-down during dexamethasone-induced cell differentiation (off-target effect was excluded by using siRNA targeting a different region of the gene). Furthermore, in luciferase reporter assays, not only did the beta-catenin transactivation cause an increase in miR-30e promoter activity but also co-expression of TCF4 enhanced the effect of beta-catenin on miR-30e promoter activity, mimicking in vivo events, where TCF4 enters the nucleus in response to Wnt signal and forms stimulatory complexes with beta-catenin. These observations all directly argue the presence of a dexamethasone–beta-catenin/TCF4-miR-30e axis.
In conclusion, in intestinal crypt cells, beta-catenin/TCF4, an essential component in Wnt signalling, directly binds to miR-30e, and this binding is directly involved in dexamethasone-induced cell differentiation, suggesting that miR-30e is an immediate downstream target for the beta-catenin/TCF4 transactivation pathway during intestinal cell differentiation.
References
Barker N, van de Wetering M, Clevers H (2008) The intestinal stem cell. Genes Dev 22:1856–1864
Sancho E, Batlle E, Clevers H (2003) Live and let die in the intestinal epithelium. Curr Opin Cell Biol 15:763–770
Crosnier C, Stamataki D, Lewis J (2006) Organizing cell renewal in the intestine: stem cells, signals and combinatorial control. Nat Rev Genet 7:349–359
Scoville DH, Sato T, He XC, Li L (2008) Current view: intestinal stem cells and signaling. Gastroenterology 134:849–864
Walters JR (2005) Recent findings in the cell and molecular biology of the small intestine. Curr Opin Gastroenterol 21:135–140
Riccio O, van Gijn ME, Bezdek AC, Pellegrinet L, van Es JH, Zimber-Strobl U, Strobl LJ, Honjo T, Clevers H, Radtke F (2008) Loss of intestinal crypt progenitor cells owing to inactivation of both Notch1 and Notch2 is accompanied by derepression of CDK inhibitors p27Kip1 and p57Kip2. EMBO Rep 9:377–383
Schroder N, Gossler A (2002) Expression of Notch pathway components in fetal and adult mouse small intestine. Gene Expr Patterns 2:247–250
Jensen J, Pedersen EE, Galante P, Hald J, Heller RS, Ishibashi M, Kageyama R, Guillemot F, Serup P, Madsen OD (2000) Control of endodermal endocrine development by Hes-1. Nat Genet 24:36–44
Crosnier C, Vargesson N, Gschmeissner S, Ariza-McNaughton L, Morrison A, Lewis J (2005) Delta-Notch signalling controls commitment to a secretory fate in the zebrafish intestine. Development 132:1093–1104
Yang Q, Bermingham NA, Finegold MJ, Zoghbi HY (2001) Requirement of Math1 for secretory cell lineage commitment in the mouse intestine. Science 294:2155–2158
van Es JH, Jay P, Gregorieff A, van Gijn ME, Jonkheer S, Hatzis P, Thiele A, van den Born M, Begthel H, Brabletz T, Taketo MM, Clevers H (2005) Wnt signalling induces maturation of Paneth cells in intestinal crypts. Nat Cell Biol 7:381–386
Pinto D, Gregorieff A, Begthel H, Clevers H (2003) Canonical Wnt signals are essential for homeostasis of the intestinal epithelium. Genes Dev 17:1709–1713
Korinek V, Barker N, Moerer P, van Donselaar E, Huls G, Peters PJ, Clevers H (1998) Depletion of epithelial stem-cell compartments in the small intestine of mice lacking Tcf-4. Nat Genet 19:379–383
Andreu P, Colnot S, Godard C, Gad S, Chafey P, Niwa-Kawakita M, Laurent-Puig P, Kahn A, Robine S, Perret C, Romagnolo B (2005) Crypt-restricted proliferation and commitment to the Paneth cell lineage following Apc loss in the mouse intestine. Development 132:1443–1451
Andreu P, Peignon G, Slomianny C, Taketo MM, Colnot S, Robine S, Lamarque D, Laurent-Puig P, Perret C, Romagnolo B (2008) A genetic study of the role of the Wnt/beta-catenin signalling in Paneth cell differentiation. Dev Biol 15:288–296
Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler KW, Vogelstein B, Clevers H (1997) Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC-/- colon carcinoma. Science 275:1784–1787
Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW (1997) Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science 275:1787–1790
Nakamura T, Tsuchiya K, Watanabe M (2007) Crosstalk between Wnt and Notch signaling in intestinal epithelial cell fate decision. J Gastroenterol 42:705–710
Katoh M (2007) WNT antagonist, DKK2, is a Notch signaling target in intestinal stem cells: augmentation of a negative regulation system for canonical WNT signaling pathway by the Notch-DKK2 signaling loop in primates. Int J Mol Med 19:197–201
Clevers H (2006) Wnt/beta-catenin signaling in development and disease. Cell 127:469–480
Raab MS, Breitkreutz I, Tonon G, Zhang J, Hayden PJ, Nguyen T, Fruehauf JH, Lin BK, Chauhan D, Hideshima T, Munshi NC, Anderson KC, Podar K (2009) Targeting PKC: a novel role for beta-catenin in ER stress and apoptotic signaling. Blood 113:1513–1521
Wassermann S, Scheel SK, Hiendlmeyer E, Palmqvist R, Horst D, Hlubek F, Haynl A, Kriegl L, Reu S, Merkel S, Brabletz T, Kirchner T, Jung A (2009) p16(INK4a) is a beta-catenin target gene and indicates low survival in human colorectal tumors. Gastroenterology 136:196–205.e2
Stein U, Arlt F, Walther W, Smith J, Waldman T, Harris ED, Mertins SD, Heizmann CW, Allard D, Birchmeier W, Schlag PM, Shoemaker RH (2006) The metastasis-associated gene S100A4 is a novel target of beta-catenin/T-cell factor signaling in colon cancer. Gastroenterology 131:1486–1500
Shekhar MP, Tait L, Gerard B (2006) Essential role of T-cell factor/beta-catenin in regulation of Rad6B: a potential mechanism for Rad6B overexpression in breast cancer cells. Mol Cancer Res 4:729–745
Yochum GS, Cleland R, Goodman RH (2008) A genome-wide screen for {beta}-catenin binding sites identifies a downstream enhancer element that controls c-Myc gene expression. Mol Cell Biol 28:7368–7379
Tetsu O, McCormick F (1999) Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 398:422–426
Brabletz T, Jung A, Dag S, Hlubek F, Kirchner T (1999) beta-catenin regulates the expression of the matrix metalloproteinase-7 in human colorectal cancer. Am J Pathol 155:1033–1038
He TC, Chan TA, Vogelstein B, Kinzler KW (1999) PPARdelta is an APC-regulated target of nonsteroidal anti-inflammatory drugs. Cell 99:335–345
Ghiselli G, Coffee N, Munnery CE, Koratkar R, Siracusa LD (2003) The cohesin SMC3 is a target the for beta-catenin/TCF4 transactivation pathway. J Biol Chem 278:20259–20267
Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116:281–297
Bushati N, Cohen SM (2007) microRNA functions. Annu Rev Cell Dev Biol 23:175–205
Rana TM (2007) Illuminating the silence: understanding the structure and function of small RNAs. Nat Rev Mol Cell Biol 8:23–36
Turner M, Vigorito E (2008) Regulation of B- and T-cell differentiation by a single microRNA. Biochem Soc Trans 36:531–533
Kwon C, Han Z, Olson EN, Srivastava D (2005) MicroRNA1 influences cardiac differentiation in Drosophila and regulates Notch signaling. Proc Natl Acad Sci USA 102:18986–18991
Mizuno Y, Yagi K, Tokuzawa Y, Kanesaki-Yatsuka Y, Suda T, Katagiri T, Fukuda T, Maruyama M, Okuda A, Amemiya T, Kondoh Y, Tashiro H, Okazaki Y (2008) miR-125b inhibits osteoblastic differentiation by down-regulation of cell proliferation. Biochem Biophys Res Commun 368:267–272
Wang Q, Li YC, Wang J, Kong J, Qi Y, Quigg RJ, Li X (2008) miR-17–92 cluster accelerates adipocyte differentiation by negatively regulating tumor-suppressor Rb2/p130. Proc Natl Acad Sci USA 105:2889–2894
Schratt GM, Tuebing F, Nigh EA, Kane CG, Sabatini ME, Kiebler M, Greenberg ME (2006) A brain-specific microRNA regulates dendritic spine development. Nature 439:283–289
Murchison EP, Partridge JF, Tam OH, Cheloufi S, Hannon GJ (2005) Characterization of Dicer-deficient murine embryonic stem cells. Proc Natl Acad Sci USA 102:12135–12140
Hino K, Tsuchiya K, Fukao T, Kiga K, Okamoto R, Kanai T, Watanabe M (2008) Inducible expression of microRNA-194 is regulated by HNF-1alpha during intestinal epithelial cell differentiation. RNA 14:1433–1442
Serfas MS, Tyner AL (1993) HNF-1 alpha and HNF-1 beta expression in mouse intestinal crypts. Am J Physiol 265:G506–G513
Traber PG, Wu GD, Wang W (1992) Novel DNA-binding proteins regulate intestine-specific transcription of the sucrase-isomaltase gene. Mol Cell Biol 12:3614–3627
Moucadel V, Soubeyran P, Vasseur S, Dusetti NJ, Dagorn JC, Iovanna JL (2001) Cdx1 promotes cellular growth of epithelial intestinal cells through induction of the secretory protein PAP I. Eur J Cell Biol 80:156–163
Soubeyran P, Andre F, Lissitzky JC, Mallo GV, Moucadel V, Roccabianca M, Rechreche H, Marvaldi J, Dikic I, Dagorn JC, Iovanna JL (1999) Cdx1 promotes differentiation in a rat intestinal epithelial cell line. Gastroenterology 117:1326–1338
Ding Q, Dong Z, Evers BM (1999) Enterocyte-like differentiation of the Caco-2 intestinal cell line is associated with increases in AP-1 protein binding. Life Sci 64:175–182
Suzuki K, Fukui H, Kayahara T, Sawada M, Seno H, Hiai H, Kageyama R, Okano H, Chiba T (2005) Hes1-deficient mice show precocious differentiation of Paneth cells in the small intestine. Biochem Biophys Res Commun 328:348–352
Quaroni A, Wands J, Trelstad RL, Isselbacher KJ (1979) Epithelioid cell cultures from rat small intestine. Characterization by morphologic and immunologic criteria. J Cell Biol 80:248–265
Blake HH, Henning SJ (1983) Weaning in the rat: a study of hormonal influences. Am J Physiol 244:R537–R543
Trahair JF, Sangild PT (1997) Systemic and luminal influences on the perinatal development of the gut. Equine Vet J Suppl 24:40–50
Hinnebusch BF, Siddique A, Henderson JW, Malo MS, Zhang W, Athaide CP, Abedrapo MA, Chen X, Yang VW, Hodin RA (2004) Enterocyte differentiation marker intestinal alkaline phosphatase is a target gene of the gut-enriched Kruppel-like factor. Am J Physiol Gastrointest Liver Physiol 286:G23–G30
Hodin RA, Shei A, Meng S (1997) Transcriptional activation of the human villin gene during enterocyte differentiation. J Gastrointest Surg 1:433–438 (discussion 438)
Renes IB, Verburg M, Bulsing NP, Ferdinandusse S, Buller HA, Dekker J, Einerhand AW (2002) Protection of the Peyer’s patch-associated crypt and villus epithelium against methotrexate-induced damage is based on its distinct regulation of proliferation. J Pathol 198:60–68
Gregorieff A, Clevers H (2005) Wnt signaling in the intestinal epithelium: from endoderm to cancer. Genes Dev 19:877–890
Kirkland SC, Henderson K (2001) Collagen IV synthesis is restricted to the enteroendocrine pathway during multilineage differentiation of human colorectal epithelial stem cells. J Cell Sci 114:2055–2064
Wu F, Zhu S, Ding Y, Beck WT, Mo Y-Y (2009) MicroRNA-mediated regulation of Ubc9 expression in cancer cells. Clin Cancer Res 15:1550–1557
Acknowledgments
The authors would like to thank Dr. Michael Paddy for assistance with confocal microscopy and the support of the UCDavis Clinical Nutrition Research Unit, NIH DK 35747 for real-time Q-PCR analyses.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Liao, Y., Lönnerdal, B. Beta-catenin/TCF4 transactivates miR-30e during intestinal cell differentiation. Cell. Mol. Life Sci. 67, 2969–2978 (2010). https://doi.org/10.1007/s00018-010-0366-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-010-0366-y