
Abstract
Objective and design
TNF-α neutralization is associated with increased mortality in mouse cecal ligation puncture (CLP) models. AZD9773 is an ovine polyclonal human TNF-α immune Fab, with pharmacological properties that differ from previously studied anti-TNF-α agents. We explored the safety and efficacy of therapeutically administered AZD9773 in mouse CLP sepsis.
Methods
A moderate/severe-grade CLP model resulting in 20–30 % 5-day survival and a mild-grade CLP model resulting in ~70 % 5-day survival were established in human TNF-α transgene/murine TNF null (Tg1278/−/−) mice.
Treatment
Mice received saline resuscitation and imipenem administration every 12 h (0–72 h post-CLP). AZD9773 (or DigiFab control) was dosed 24, 36, 48 and 60 h post-CLP.
Results
Therapeutic dosing of AZD9773 in moderate/severe-grade CLP resulted in significantly increased survival (>70 %) compared with DigiFab (27 %, P < 0.05). Therapeutic dosing of AZD9773 in mild-grade CLP did not significantly affect survival outcome compared with DigiFab or imipenem alone (~60–70 % survival).
Conclusions
These data demonstrate that TNF-α neutralization can improve survival in moderate/severe CLP sepsis. TNF-α suppression in mild-grade models was not associated with survival benefit and did not increase 5-day mortality. These findings suggest that therapeutic benefit following TNF-α attenuation in models of sepsis may depend on model severity.
Similar content being viewed by others


Introduction
Sepsis is the primary cause of death in non-cardiac intensive care units (ICUs) and leads to substantially decreased quality of life in surviving patients [1, 2]. Current management of sepsis includes eradication of the infection source and/or use of appropriate antimicrobial treatment, together with aggressive supportive care such as fluid resuscitation and restoration of tissue perfusion, or pulmonary therapy using supplemental oxygen and mechanical ventilation [3, 4]. The release of cytokines into the circulation is key in the initial development and perpetuation of sepsis [5]. Animal septicemia models have demonstrated that the pro-inflammatory cytokine tumor necrosis factor-alpha (TNF-α) plays a significant role in activating the cytokine cascade [6–8]. TNF-α is one of the first cytokines to be released by macrophages in response to infection, stimulating the production of downstream cytokines such as interleukin-6 (IL-6) and IL-8 [5]. TNF-α acts in association with other cytokines to produce the clinical signs of the systemic inflammatory response syndrome, and their synergistic effects are probably responsible for the hypotension and resultant organ dysfunction seen early in the course of severe sepsis [9]. Despite evidence of a key role for TNF-α in driving adverse clinical effects during sepsis, in vivo experiments employing live infectious agents also reveal that TNF-α is a critical factor in promoting host defense during both localized and invasive bacterial infection [10].
Preclinical cecal ligation and puncture (CLP) models involve perforation of the gastrointestinal tract and can be used to assess the impact of immunomodulatory agents on both injury-related inflammation and host defense and repair. Depending on variations in the CLP model and other factors, anti-TNF-α therapy has been shown to result in increased or unaltered mortality [11–13]. However, in contrast to data obtained following pharmacological modulation of TNF-α, mortality following CLP in transgenic mice lacking TNF receptor 1 (TNFR1) and TNFR2 is markedly inhibited [14]. In the clinic, trials designed to investigate the effect of neutralizing TNF-α with either monoclonal antibodies (mAbs) or TNF-α receptor fusion proteins in severe sepsis/septic shock have largely mirrored the findings of preclinical polymicrobial sepsis models, i.e. no significant beneficial survival effect, although a consistent trend towards clinical benefit was apparent [15]. A recent retrospective analysis considered a further level of complexity that may relate to the severity of the sepsis syndrome and the consequent risk of death [16]. Literature analysis of data with anti-inflammatory agents (including anti-TNFs) and a prospective study in a rat sepsis model examining the influence of the route, type and severity of infection appeared to indicate a relationship between efficacy and risk of death: anti-inflammatory agents were beneficial at high control mortality rates but were apparently harmful (i.e. reduced survival) at lower, more moderate mortality rates [16].
AZD9773 is a polyclonal Fab preparation with distinctive pharmacokinetic properties compared with many other anti-TNF-α biopharmaceuticals, e.g. a markedly shorter plasma half-life (cleared in hours rather than days) and more rapid tissue distribution compared with anti-TNF-α mAbs and TNF receptor-Ig fusion proteins. Thus, AZD9773 may represent a highly effective agent for the rapid neutralization and elimination of TNF-α in the setting of severe sepsis. Given the differentiated pharmacological attributes of AZD9773, we re-examined the effect of TNF-α neutralization in preclinical models of sepsis. We initially validated AZD9773 effects on mouse endotoxemic cytokine response and then examined AZD9773 effects in CLP models. Two murine models of sepsis were utilized: a mild-grade (low-risk-of-death) CLP model and a severe-grade (high-risk-of-death) CLP model. Together, these models were used to determine whether the survival outcome following therapeutic treatment with AZD9773 was related to risk of death. In these models, AZD9773 was administered therapeutically, i.e. 24 h post-surgery when clinical signs of sepsis became apparent, and in combination with a broad-spectrum antibiotic.
Methods
Animals
As AZD9773 does not bind or neutralize murine TNF-α, CLP models were established in Tg1278/TNF−/− (human TNF-α transgene/mouse TNF-α null) mice. Male Tg1278/TNF−/− transgenic (Tg) mice [17] were obtained from Biomedcode (Athens, Greece) as hemizygous Tg (human TNF-α transgene), homozygous knockout (mouse TNF-α KO). They were re-derived via in-vitro fertilization using C57BL/6J oocytes, producing animals that were either hemizygous Tg or wild-type (WT) Tg (i.e. human TNF-α transgene absent) and heterozygous KO. These animals were then bred to produce hemizygous or WT Tg and homozygous, heterozygous or WT KO. Only mice that were hemizygous or WT Tg and homozygous for the KO were used for further breeding. All breeding units had the following animals: hemizygous Tg, homozygous KO crossed with WT Tg, and homozygous KO. The Tg1278/TNF−/− mice used in these experiments were backcrossed at least eight generations and can be considered to be predominantly C57BL/6.
In-vivo experiments were designed and undertaken to minimize pain and distress, and the minimum number of mice was used to achieve the scientific objectives. Experimental design, statistical justification, animal welfare and endpoints were reviewed and approved by the AstraZeneca global veterinary council. For cecal ligation and puncture studies, a detailed program of observation was implemented to enable humane endpoints as a proxy measure for mortality. Study protocols were approved by the Transpharm Animal Care and Use Committee and were conducted in a research facility approved by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC).
Lipopolysaccharide challenge
Animals were randomly allocated to dose groups and then processed according to a blocking design. Five blocks containing one animal from each dose group were sequentially processed in terms of dosing, challenge and termination to avoid the risk of ‘order’ bias. Intraperitoneal (ip) injection of AZD9773 (14–850 U/kg) or vehicle was administered 2 h before lipopolysaccharide (LPS) challenge (0.01 mg/kg) at a dose volume of 10 μL/g animal (n = 5 for AZD9773 and LPS, n = 5 for LPS alone, n = 3 for vehicle). All mice were sacrificed 2 h after LPS challenge. Blood was collected by cardiac puncture, the serum extracted, and human TNF-α levels measured by enzyme-linked immunosorbent assay (ELISA). A large panel of murine cytokines and related factors (~60 antigens) were measured using multiplexed immunoassays (mouse CytokineMAP, Rules-Based Medicine, Inc., Austin, Texas, USA), which were optimized to maximize detection of human TNF-α and murine cytokines. AZD9773 Fabs were quantitatively determined by ELISA.
Induction of sepsis with CLP
Mice were anesthetized using isoflurane and the cecum was exteriorized and 100 % ligated with sterile 9.5 mm stainless steel surgical clips below the ileocecal valve. Using an 18-, 20-, 25- or 27-gauge needle, a single through-and-through puncture midway between the ligation and the tip of the cecum was performed and a small droplet of cecal contents was expressed from both the mesenteric and antimesenteric penetration holes. The cecum was then relocated into the abdominal cavity, the peritoneum, fascia and abdominal musculature closed by applying simple running sutures, and the skin incision closed with 9 mm Autoclips. Mice were resuscitated immediately after surgery by a subcutaneous (sc) injection of 2.5 mL/100 g body weight of 37 °C saline (0.9 % w/vol). Two h post-surgery, mice received 37 °C imipenem sc (12.5 mg/kg) in 0.5 mL/100 g saline, followed by ip imipenem in 2.5 mL/100 g saline at 12 h post-surgery. At 24 h post-surgery, mice were administered single ip injections (2.5 mL/100 g body weight) of AZD9773 or DigiFab (4.53 mg/mL) in 37 °C saline containing imipenem (12.5 mg/kg); vehicle mice received imipenem only. At 36, 48 and 60 h post-surgery, mice were administered a single ip injection of AZD9773 or DigiFab (2.27 mg/mL) in 37 °C saline containing imipenem (12.5 mg/kg); vehicle mice received imipenem only. All mice received an ip injection (2.5 mL/100 g) of 37 °C saline at 72 h post-surgery. The mice were monitored every 2–3 min for approximately 30 min until they recovered. Thereafter, they were observed hourly for at least 6 h, then regularly until study termination at 108 h. At each assessment, the likelihood of a moribund mouse surviving until the next assessment was evaluated; if the probability was low, the mouse was sacrificed.
AZD9773 pharmacokinetics
Mice received ip AZD9773 85 or 850 U/kg. Serum samples were obtained at 0.5, 1, 2, 3, 4, 6, 8, 10 and 12 h post-dosing from three male and three female mice per time point. Serum samples were analyzed for AZD9773 total Fabs using ELISA. For the purpose of simulating future studies, a two-compartment model was developed using mouse AZD9773 pharmacokinetic data. It was assumed that absorption of AZD9773 into the systemic circulation was instantaneous, which is justified for modeling purposes because the data do not define an absorption phase. Parameter estimation, simulation and graphics were performed in ACSLX version 2.5 (The AEgis Technologies Group, Inc., Huntsville, Alabama, USA). Parameter estimation was based on the sum of squared residuals weighted by the model prediction. A bridging AZD9773 pharmacokinetic study was conducted in control CLP mice to determine whether sepsis-induced hemodynamic changes modified pharmacokinetic properties. AZD9773 was dosed 24 h post-CLP at 850 or 4,000 U/kg; serum AZD9773 was measured by ELISA at 2, 4 or 12 h post-dose.
CLP histopathology
Histopathology was performed on the following tissues at 24 and 48 h post-CLP: cecum, femorotibial joint, sternum, spleen, liver, lung, kidney and heart. Analysis was conducted with and without imipenem to reveal any masking effect of antibiotic on host defense phenotype. All tissues (apart from sternum and femorotibial joint) were fixed for 48 h, dehydrated in 70–100 % ethanol, cleared in xylene and then processed in paraffin wax. The sternum and femorotibial joint were decalcified in 5 % formic acid for 2 days prior to further processing. Tissues were cut on a Shandon Finesse microtome (Thermo Fisher Scientific, Loughborough, UK) at a thickness of 4 μm. Standard hematoxylin/eosin (HE) staining was performed on all tissues. Special stains were used on selected tissues as required, including Martius scarlet blue to identify intravascular thromboemboli (common sequelae of sepsis) and periodic acid-Schiff to identify glycogen.
Immunohistochemistry
Immunohistochemistry was performed on selected formalin-fixed paraffin-embedded tissue sections using the Labvision Autostainer (Thermo Fisher Scientific, Loughborough, UK). 4 μm-thick sections were mounted on glass slides, dewaxed and rehydrated. All washes were performed in Tris-buffered saline (TBS) containing 0.1 % Tween, and all antibodies were diluted in TBS with 0.1 % Tween. To allow visualization of antibodies, sections were incubated with 3,3′-diaminobenzidine (A Menarini Diagnostics, Winnersh-Wokingham, UK) and counterstained with Carazzi’s hematoxylin. Sections were then dehydrated, cleared and mounted. Antibodies used were: CD3 (Pan T-cell), a rabbit polyclonal anti-CD3 anti-serum (Zymed/Invitrogen, Paisley, UK; see Supplementary Fig. 1); Pax-5 (Pan B-cell), a rabbit polyclonal anti-PAX-5 (Neomarkers, Fremont, California, USA; see Supplementary Fig. 1); TNF-α, a polyclonal rabbit anti-TNF-α (Abcam 66579, Cambridge, UK). Numbers of cells with positive TNF-α immunohistochemistry were graded semi-quantitatively by light microscopy (1 rare, 2 slight increase, 3 moderate increase, 4 marked increase). Detailed methods are available on request.
Statistical methods
For LPS-challenge studies, antigen data analysis comprised a univariate two-stage non-parametric approach involving a Kruskal–Wallis (KW) analysis of variance (ANOVA) that, if statistically significant, was followed by a pairwise test of each dose against the combined LPS control group using Wilcoxon rank sum test and a two-sided 5 % significance level. Since anti-inflammatory agents have been reported to be beneficial at high control mortality rates but apparently harmful (i.e. reduced survival) at lower, more moderate mortality rates [16], our statistical analysis of AZD9773 in the mild-grade sepsis model was centered on demonstrating non-inferiority. This was defined as confidence (95 %) that the odds ratio (OR) of survival between control and AZD9773 was less than the critical threshold of 2.6. With regard to detection of benefit in the mild-grade sepsis model, 60 animals per group would mean an 80 % chance that a true compound-treated survival rate improvement from 70 to 90 % would be detected using a one-sided 5 % Fisher’s test. For assessment of AZD9773 efficacy in the severe-grade model with 15 animals per group, there was an 80 % chance that survival rate improvement from 30 to 90 % would be detected using a one-sided 5 % Fisher’s test.
Results
AZD9773 comprises polyclonal human TNF-α immune Fab populations and is highly specific for human TNF-α. It does not bind to, or is minimally effective in neutralizing, mouse, rat, porcine and canine TNF-α (data not shown). In order to explore the in-vivo pharmacology of AZD9773 as it pertains to humans more accurately, the mouse strain Tg1278/TNF−/− (mouse TNF-α double gene deletion:human TNF-α transgene backcross, hereafter termed ‘Tg’), which is null for mouse TNF-α but transgenic for human TNF-α under the control of the human TNF-α promoter, was used [17].
We initially examined the effect of AZD9773 on endotoxin-induced human TNF-α and measured murine IL-6 to determine the biological response to TNF-α neutralization by AZD9773 [18, 19]. We also measured a large panel of LPS-induced serum cytokines, chemokines and inflammatory biomarkers. Human TNF-α and 55 serum murine cytokine (and related factors) levels were determined 2 h post-LPS challenge in Tg mice. Forty-two serum antigens, including human TNF-α, were significantly regulated by LPS or AZD9773. Human TNF-α was induced from 31 pg/mL at baseline to >60,000 pg/mL 2 h post-LPS. Pre-treatment (LPS −2 h) with AZD9773 resulted in dose-related exposure (measured 4 h post-dose) (Fig. 1a). The highest dose of AZD9773 (850 U/kg) resulted in a statistically significant reduction in human TNF-α (~9-fold) and 29 of 55 other cytokines/immune antigens; 24 cytokines/immune antigens also showed a statistically significant reduction following 425 U/kg of AZD9773 (P < 0.05) (Fig. 1b and Supplementary Table 1). Interestingly, lower doses of AZD9773 (107 U/kg) produced a small increase in human TNF-α, while 14 or 27 U/kg doses were associated with a 1.2- to 2-fold increase in four antigens (VEGF, SCF, IL-7 and FGF9; data not shown).

AZD9773 pharmacokinetics and pharmacological effects following LPS challenge of Tg mice. AZD9773 biological activity is expressed as units; 1 unit of AZD9773 neutralizes ~12,800 international units of human TNF-α: a AZD9773 plasma concentration 4 h after ip dosing; b plasma cytokine measurements 2 h post-LPS challenge. x-axes, AZD9773 dose (U/kg) administered 2 h pre-LPS dose; y-axes, plasma cytokine concentration (pg/mL)
Taken together, these data demonstrate that LPS induces human TNF-α in the Tg mouse and that AZD9773 administration attenuates serum TNF-α, resulting in reduced biological activity; this was evidenced by reduced serum IL-6. This effect was specific to AZD9773 since an equivalent protein dose of albumin did not significantly affect LPS-induced human TNF-α or IL-6 (data not shown). We also found that AZD9773 reduced the levels of 28/54 other cytokines and related factors, indicating that TNF-α participates in the regulation of a broad panel of LPS-induced genes.
To assess the effects of AZD9773 in an experimental model of sepsis, we developed the CLP method in the Tg mouse line. In our method, CLP surgery was followed by high-volume fluid (saline) resuscitation administered immediately after surgery and then co-administered twice daily with the broad-spectrum antibiotic imipenem. Survival was broadly similar 5 days post-CLP in both control and Tg mice after CLP surgery, with outcomes broadly similar across a range of needle gauges (higher gauge number reflects a smaller diameter needle) used to modulate model severity (Fig. 2). However, these data appeared to indicate that Tg mice may be less sensitive to imipenem compared with control mice (compare Fig. 2c, d).

Development of CLP models in Tg mice. a Mild- versus severe-grade CLP model development: WT mouse 5-day survival responses following CLP conducted with a range of needle grades in the absence of imipenem co-administration. b Mild-grade CLP transgenic (no imipenem) versus C57BL/6. 27-gauge needle CLP with imipenem administration resulted in ~70 % 5-day survival. Tg1278/TNF−/− mice responded similarly to WT mice. c Mild-grade CLP (+ imipenem) (C57BL/6). A mild-grade, low-mortality CLP with imipenem treatment was obtained with a 20-gauge needle in WT mice. d Mild-grade CLP transgenic (+ imipenem) versus C57BL/6. Tg1278/TNF−/− and WT mice responded similarly
The polymicrobial sepsis induced by CLP is a complex physiological process; therefore, to ensure that the human TNF-α expressed by the Tg1278/TNF−/− mice did not induce an atypical CLP phenotype, we compared the histopathological response of Tg1278/TNF−/− mice and control C57/BL6J mice in our CLP protocol. In mice treated with and without imipenem, the cecum of WT and Tg animals following CLP was necrotic and hemorrhagic at 24 h, with increased severity of inflammation at 48 h. In the absence of imipenem, large numbers of bacterial rods were present in the necrotic and hemorrhagic cecal wall, and this was considered to be due to the lack of antibiotic intervention. Occasional TNF-α-positive cells were observed in the cecal mucosa of sham-treated animals, while increased numbers were present in the CLP animals (Fig. 3).

Mouse CLP tissue histopathology of cecum HE and cecum TNF-α Left panel a, b In sham-treated WT and Tg animals with imipenem at both time points (only 24 h time point is shown), the cecum had normal glandular mucosa (M) overlying the smooth muscle layer (SM) and serosa (S). The lumen (L) contained food material and bacteria. The cecal mucosa of WT and Tg animals following CLP in the presence of imipenem was diffusely hemorrhagic and necrotic at 24 h and the lumen contained free blood cells and necrotic sloughed epithelial cells, shown in (c) and (d). At 48 h in the presence of imipenem, there was increased inflammation in WT and Tg animals, especially in the smooth muscle and serosal layers, and occasional intramucosal thrombi, shown in (e) and (f). In the absence of imipenem and in both WT and Tg animals, large numbers of streaming bacterial rods were present as shown in (g) and (h), particularly in the cecal wall smooth muscle layer at both time points; this was considered to be due to lack of antibiotic intervention. All images were taken with HE staining at ×20 magnification. Right panel TNF-α-positive cells were infrequently observed in the mucosa and underlying submucosa (SB) of all WT and Tg sham-treated animals at both time points, as shown in (a) and (b) (24 h + imipenem only shown). In CLP animals, there were many positive cells (mostly neutrophils, indicated with arrows; shown in c–h), predominantly in the SB, serosa (S) and mesentery (ME). In CLP animals, the appearance was similar in both the presence and absence of imipenem. Brown staining of luminal contents is due to non-specific antibody staining. All images are of TNF-α immunohistochemistry and were taken at ×20 magnification
Analysis of the liver in both studies revealed that hepatocytes were smaller and darker in appearance due to glycogen depletion in most animals after 24 h, with all animals showing this change by 48 h. Intracytoplasmic microvesicular vacuolation of hepatocytes (consistent with fat accumulation) was also present at both time points, although the severity was greater when antibiotic had not been given. Further findings observed without antibiotic in both WT and Tg animals included Kupffer cell apoptosis and cytoplasmic vacuolation, periportal oval cell proliferation, capsule-mixed inflammatory cell infiltration and vessel dilatation (see Supplementary Fig. 2). Additional histopathological findings in WT and Tg animals were typical of an acute inflammatory response to gut perforation and a resultant mild sepsis. There was a decrease in bone marrow myeloid cells at 24 h, reflecting their systemic release into the circulation and to the site of trauma, while at 48 h there was rebound myeloid hyperplasia (Supplementary Fig. 3). These changes were paralleled in the spleen, where white pulp apoptosis was noted at 24 h and increased red pulp myeloid cells at 48 h. Notably, in the absence of imipenem, bone marrow and splenic myeloid hyperplasia began at 24 h. The severity of hemopoietic changes in the presence of imipenem was generally less in Tg animals; in the absence of imipenem, the converse was true. The increased inflammatory response in CLP animals compared with sham-treated animals was also supported by TNF-α immunohistochemistry in several tissues, as were the subtle differences in response between WT and Tg animals in the presence or absence of imipenem (see lung TNF-α expression in Supplementary Fig. 4).
Taken together, these histopathological data indicate a reasonably similar tissue response in WT and Tg mice following CLP, either with or without imipenem. Critically, the histopathological data from both Tg and WT mice demonstrate histological TNF-α expression at both 24 and 48 h and that imipenem reduces the host response to polymicrobial infection following CLP.
AZD9773 dose
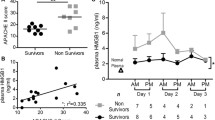
Mouse AZD9773 pharmacokinetic parameters were derived following ip dosing of AZD9773 by determining plasma AZD9773 between 0 and 12 h post-dose (Fig. 4a). Non-compartmental analysis revealed that the terminal half-life of AZD9773 in the mouse was ~3.5 h, indicating that AZD9773 was eliminated more rapidly than in humans (terminal half-life ~19 h) [20]. Therefore, simulations were conducted to define a dose that would result in acceptable pharmacological exposure. The pharmacokinetic model was shown to predict the observed AZD9773 serum concentrations after CLP (Fig. 4b) and was then used to investigate the dosing regimen in mice that would ensure sufficient pharmacological exposure between doses and match human clinical exposure to AZD9773. It was found that a 4,000 U/kg loading dose followed by 2,000 U/kg every 12 h produced an acceptable murine plasma exposure and was comparable to that in humans (Fig. 4c).

Determination of AZD9773 pharmacokinetics in mouse CLP and in comparison with AZD9773 clinical pharmacokinetics: a mean ± SE serum concentrations of total AZD9773 Fabs dosed 24 h post-CLP (27-gauge needle); b the murine PK model was used to simulate mouse exposure after various doses, until a good comparison with clinical exposure was obtained; c clinical pharmacokinetic data for AZD9773 were utilized from two AZD9773 cohorts which received 500 U/kg followed by 100 U/kg maintenance, or 750 U/kg followed by 250 U/kg maintenance [20]. The mouse pharmacokinetic model was used to simulate mouse exposure following various doses until a good comparison with clinical exposure was obtained. Since AZD9773 is eliminated more rapidly in mice than in humans, a higher dose is required, and although this leads to higher maximum serum concentrations than observed clinically, it results in a comparable duration of exposure
AZD9773 in high- and low-risk-of-death CLP sepsis
To determine whether AZD9773 had differential effects on survival (i.e. beneficial or detrimental) following CLP sepsis, we assayed the effects in mild-grade (low-risk-of-death) and severe-grade (high-risk-of-death) Tg mouse CLP models. In these models, AZD9773 was administered therapeutically, i.e. 24 h post-surgery when clinical signs of sepsis became apparent, and in combination with a broad-spectrum antibiotic.
In the first CLP model, AZD9773 at 4,000 U/kg (24 h post-CLP surgery when a sepsis response was fully evident) followed by three doses of 2,000 U/kg (36, 48 and 60 h post-CLP) was administered to Tg mice with moderate- to severe-grade sepsis induced with an 18-gauge needle. Survival was monitored for 108 h post-CLP. A statistically significant difference in survival was observed: 73 % for AZD9773 and imipenem versus 20 % for DigiFab control and imipenem (P < 0.0092; Fig. 5a). Inspection of time-of-death/euthanasia data revealed that four animals in the DigiFab group died before receiving treatment as a consequence of the severe sepsis response to CLP. Exclusion of these animals resulted in a corrected survival frequency of 27 % at 5 days post-surgery, although the difference remained statistically significant (P < 0.05; Fig. 5b). Thus, therapeutic neutralization of TNF-α by AZD9773 is effective in protecting Tg mice at high risk of death from CLP-induced sepsis.

Effect of therapeutic AZD9773 treatment on 5-day survival following severe CLP in Tg1278/TNF−/− mice: a intention-to-treat survival curves; b baseline-controlled survival curves
We next explored whether if there was a relationship between AZD9773-driven TNF-α neutralization efficacy and risk of death, and specifically whether TNF-α neutralization could be harmful (i.e. result in reduced survival) at lower, more moderate mortality rates. Since mild-grade sepsis results in a low mortality rate at 5 days post-CLP (~30 %), 60 animals per group were studied to ensure that genuine non-inferiority (i.e. control survival not detrimentally impacted as a result of AZD9773 administration) would be detected. Survival frequencies were 63 % for imipenem alone, 65 % for AZD9773 + imipenem and 69 % for DigiFab + imipenem (Fig. 6). These data translate into survival odds of 1.68, 1.86 and 2.28, respectively (Table 1). The OR of survival for AZD9773 and DigiFab compared with imipenem alone were both <1 (0.91 and 0.74, respectively). Therefore, the odds of survival in these two groups is in excess of that in the imipenem alone group, and the one-sided upper 95 % confidence limits for these ORs are both less than the critical threshold of 2.6 (1.7 and 1.4, respectively; Table 2). This indicates that AZD9773 treatment with imipenem is not inferior (in the sense of leading to a decrease in survival) to treatment with DigiFab + imipenem or imipenem alone. In addition, these data also fail to demonstrate a statistically significant benefit of AZD9773 + imipenem in the low-mortality-rate CLP model.

Survival in the mild-grade sepsis model
Discussion
The potential role of TNF-α in sepsis has been extensively investigated preclinically. Experimental animal models of infection, inflammation and sepsis (see [21] for more detailed references) have shown that TNF-α is the first cytokine that appears in the circulation following an ‘infective insult’, and that TNF-α release is proportional to the insult (e.g. experimental endotoxin load). Importantly, increased TNF-α levels are associated with mortality in models of sepsis and septic shock. However, variable results are obtained depending on the nature of the sepsis model, the anti-TNF-α treatment modalities or the presence/absence of TNF-α, TNFR1 and TNFR2 [11–14, 22–24]. Here, using a polyclonal TNF-α-neutralizing Fab preparation (AZD9773), we have explored the relationship between efficacy and risk of death in murine CLP sepsis and found that AZD9773 reduced the risk of death in high-mortality CLP but was neither efficacious nor detrimental in low-mortality CLP.
In order to characterize AZD9773, we first demonstrated that AZD9773 dose dependently suppressed LPS-induced human TNF-α and TNF-α-dependent biomarkers such as IL-6 in the Tg mouse line, indicating that AZD9773 rapidly and effectively neutralized high levels of human TNF-α in vivo. In addition, AZD9773 significantly down-regulated 29/55 other cytokines/immune antigens, confirming the broad influence of TNF-α on endotoxin-regulated cytokine networks.
To determine the effects of AZD9773 on mouse survival following CLP-induced polymicrobial sepsis, we developed and characterized a low- and high-mortality CLP protocol in Tg mice. Our mild-grade protocol, despite resulting in ~30 % mortality, was associated with relatively mild pathological responses in the liver, heart and lungs. Comparisons between Tg and WT mouse histopathological reactions revealed that the Tg mouse CLP model was typical of WT counterparts. Although histomorphological and immunochemical changes following CLP in Tg and WT mice were similar, Tg mice had a slightly slower, less robust immune response to CLP compared with WT mice in the presence of imipenem, but a slightly more robust response in its absence. The reasons for this are unclear but may be related to altered human TNF-α pharmacology versus mouse TNFR1/TNFR2 [25].
Since a prospective study in rodent sepsis models has indicated a relationship between anti-inflammatory agent efficacy and risk of death—agents were beneficial at high control mortality rates but reduced survival at lower, more moderate mortality rates [16]—we tested AZD9773 in both low- and high-mortality-rate models. Our experiments were powered to detect an increase in survival rate of 20 % or a decrease of 23 % in the low-mortality-rate model, or an increase of 50 % in the high-mortality-rate model. We found that AZD9773 dosed at exposures comparable to those that can be achieved clinically, and in combination with imipenem, significantly improved murine survival (5-day survival after CLP surgery) following severe-grade CLP from 27 % in controls to 70 %. However, there was no evidence of a benefit or adverse effect in a CLP model where the risk of death was relatively low (~30 %). Thus, this is the first report of an anti-TNF-α neutralizing agent affording a true therapeutic survival benefit following murine CLP sepsis; however, the efficacy of TNF-α neutralization by AZD9773 appears to correlate with risk of death.
CLP in rodents is a model of polymicrobial sepsis simulating human sepsis associated with perforation of the gastrointestinal tract. Depending on variations in the model and other factors such as the mode of TNF-α inhibition, anti-TNF-α therapy results in unaltered or increased mortality in the CLP model [11–13], or in the case of an anti-TNF F(ab′)2 agent, reduces mortality at intermediate doses (1 mg/kg) but not low (0.1 mg/kg) or high doses (10 mg/kg) [26]. Thus, biopharmaceutical inhibition of TNF-α appears only to protect mice from polymicrobial sepsis when TNF-α suppression is carefully titrated with anti-TNF-α antibody fragments. Thus, biopharmaceutical inhibition of TNF-α fails to protect mice from polymicrobial sepsis. Studies with genetically modified mice appear to indicate a different outcome. Ebach and co-workers [24] examined the effects of CLP in TNFR1-null or TNFR2-null mouse lines and found that in CLP sepsis, TNFR1 mediates much of the TNF-α-induced pathology, whereas TNFR2 mediates protective effects; however, mice lacking both TNFR1 and TNFR2 were markedly protected following CLP [14]. More recently, the immune and inflammatory responses in TNFR1/R2 double-deficient mice have been revisited in a polymicrobial model of abdominal infection similar to CLP. TNFR1 and TNFR2 deficiency was found to be associated with a protective effect [23].
The reasons for the divergent conclusions drawn from the genetic and TNF-neutralization CLP studies are not clear; however, Secher and co-workers [23] speculated that incomplete neutralization of TNF by the biological agents employed in previous studies may be a critical determinant, since genetic ablation of TNFR1 and TNFR2 results in complete abolition of TNF-α signaling. These data may imply that neutralization of other TNFR ligands is critical in affording survival following CLP (e.g. lymphotoxin) or that the anti-TNF-α agents employed fail to fully neutralize TNF-α in the extravascular space of tissues undergoing organ failure (as detected in our studies; Supplementary Fig. 4). The timing of anti-TNF-α therapy pre- or post-CLP may influence the responses to CLP, especially with respect to host defense [26]. Indeed, a protective role for TNF-α in sepsis has been proposed, since it appears to facilitate the formation of a local fibrous adhesion following sub-lethal mouse CLP [27, 28]. This TNF-α-driven response is probably most important in the early phase of peritonitis to control and encapsulate the spread of infection and may explain the importance of anti-TNF-α agent dose titration during this early period: intermediate anti-TNF treatment is more effective than high-dose anti-TNF-α treatment [26]. In this study, AZD9773 was administered 24 h after surgery, not prophylactically as in the Echtenacher [27] publication, which may also have contributed to our positive safety and survival data. Histologically, we noted evidence of TNF-α expression in inflammatory cells in the wall of the cecum at 24 and 48 h post-surgery; therefore, early inflammation in the smooth muscle and serosal layers of the cecum would likely have contributed to sepsis containment. Thus, early or prophylactic inhibition of TNF-α in mild-grade CLP sepsis may be detrimental [11–13] or require careful dose titration of anti-TNF-α treatment [26], while incomplete therapeutic inhibition (i.e. whole antibody anti-TNF) in severe-grade sepsis is ineffective [26].
The preclinical sepsis model efficacy of AZD9773 is at odds with the findings of a double-blind Phase IIb study comparing the efficacy and safety of two doses of AZD9773 with placebo in patients with severe sepsis and/or septic shock [29]. Treatment with AZD9773 did not show any significant improvements versus placebo with respect to the primary endpoint, ventilator-free days, or the secondary endpoints, including mortality. The overall mortality rates across treatment groups were 15.0 % for low-dose AZD9773, 27.0 % for high-dose AZD9773 and 20.0 % for placebo. The relative risk of death in the treated group versus placebo at day 29 was not statistically significant. Although the 20 % placebo mortality rate is lower than that detected historically (36 % [30]), it is comparable to that observed in severe sepsis and septic shock patients entered into recent randomized double-blind controlled trials and observational studies (20.9 %, ARISE [31]; 24 %, E5664 [32]; 33.3 %, TAK242 [33]; 24.2 %, activated protein C [34]). The general lack of immunomodulatory agent clinical efficacy in severe sepsis and septic shock is in contrast with the data presented here. The reasons for this discrepancy are unclear; one explanation may be lack of clinical relevance of the murine genomic response to acute pro-inflammatory stress associated with trauma and infection [35], while another possibility is that benefit of anti-inflammatory treatment strategies may be restricted to the setting of early severe sepsis with high risk of death. Indeed, the preclinical data presented here and those published by Eichacker and co-workers [16] support this hypothesis. In addition, while some patients may benefit from immunosupressive therapies, it is likely that others may benefit from immunostimulation so that they may more effectively eliminate the microbial challenge; thus, treatment regimens may need to be tailored to the patient’s host response to the invading pathogens (i.e. excessive hyper-inflammatory state or compromised host response) [36, 37].
In conclusion, therapeutic benefit following TNF-α attenuation in models of sepsis may be dependent on model severity. Thus, when considering the translation of preclinical findings to the clinical setting of severe sepsis, it is recommended that preclinical studies of novel immunomodulatory therapeutic agents should include both low- and high-mortality preclinical sepsis models. Ideally, these models should also attempt to account for the dynamic range of immunocompetence, the pharmacological mechanism of target modulation, the timing of the therapy and the dosing strategy employed.
References
Perl TM, Dvorak L, Hwang T, Wenzel RP. Long-term survival and function after suspected gram-negative sepsis. JAMA. 1995;274:338–45.
Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003;348:1546–54.
Sharma VK, Dellinger RP. Treatment options for severe sepsis and septic shock. Expert Rev Anti Infect Ther. 2006;4:395–403.
Schuerholz T, Marx G. Management of sepsis. Minerva Anestesiol. 2008;74:181–95.
Blackwell TS, Christman JW. Sepsis and cytokines: current status. Br J Anaesth. 1996;77:110–7.
Fong Y, Tracey KJ, Moldawer LL, Hesse DG, Manogue KB, Kenney JS, Lee AT, Kuo GC, Allison AC, Lowry SF. Antibodies to cachectin/tumor necrosis factor reduce interleukin 1 beta and interleukin 6 appearance during lethal bacteremia. J Exp Med. 1989;170:1627–33.
Hinshaw LB, Emerson TE Jr, Taylor FB Jr, Chang AC, Duerr M, Peer GT, Flournoy DJ, White GL, Kosanke SD, Murray CK. Lethal Staphylococcus aureus-induced shock in primates: prevention of death with anti-TNF antibody. J Trauma. 1992;33:568–73.
Redl H, Schlag G, Ceska M, Davies J, Buurman WA. Interleukin-8 release in baboon septicemia is partially dependent on tumor necrosis factor. J Infect Dis. 1993;167:1464–6.
Rice TW, Bernard GR. Therapeutic intervention and targets for sepsis. Annu Rev Med. 2005;56:225–48.
Lorente JA, Marshall JC. Neutralization of tumor necrosis factor in preclinical models of sepsis. Shock. 2005;24(Suppl 1):107–19.
Eskandari MK, Bolgos G, Miller C, Nguyen DT, DeForge LE, Remick DG. Anti-tumor necrosis factor antibody therapy fails to prevent lethality after cecal ligation and puncture or endotoxemia. J Immunol. 1992;148:2724–30.
Evans GF, Snyder YM, Butler LD, Zuckerman SH. Differential expression of interleukin-1 and tumor necrosis factor in murine septic shock models. Circ Shock. 1989;29:279–90.
Tracey KJ, Fong Y, Hesse DG, Manogue KR, Lee AT, Kuo GC, Lowry SF, Cerami A. Anti-cachectin/TNF monoclonal antibodies prevent septic shock during lethal bacteraemia. Nature. 1987;330:662–4.
Leon LR, White AA, Kluger MJ. Role of IL-6 and TNF in thermoregulation and survival during sepsis in mice. Am J Physiol. 1998;275:R269–77.
Qiu P, Cui X, Barochia A, Li Y, Natanson C, Eichacker PQ. The evolving experience with therapeutic TNF inhibition in sepsis: considering the potential influence of risk of death. Expert Opin Investig Drugs. 2011;20:1555–64.
Eichacker PQ, Parent C, Kalil A, Esposito C, Cui X, Banks SM, Gerstenberger EP, Fitz Y, Danner RL, Natanson C. Risk and the efficacy of antiinflammatory agents: retrospective and confirmatory studies of sepsis. Am J Respir Crit Care Med. 2002;166:1197–205.
Keffer J, Probert L, Cazlaris H, Georgopoulos S, Kaslaris E, Kioussis D, Kollias G. Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis. EMBO J. 1991;10:4025–31.
Dinarello CA. The biology of interleukin 1 and comparison to tumor necrosis factor. Immunol Lett. 1987;16:227–31.
Sironi M, Gadina M, Kankova M, Riganti F, Mantovani A, Zandalasini M, Ghezzi P. Differential sensitivity of in vivo TNF and IL-6 production to modulation by anti-inflammatory drugs in mice. Int J Immunopharmacol. 1992;14:1045–50.
Morris PE, Zeno B, Bernard AC, Huang X, Das S, Edeki T, Simonson SG, Bernard GR. A placebo-controlled, double-blind, dose-escalation study to assess the safety, tolerability, and pharmacokinetics/pharmacodynamics of single and multiple intravenous infusions of AZD9773 in patients with severe sepsis and septic shock. Crit Care. 2012;16:R31.
van der Poll T, van Deventer SJ. Cytokines and anticytokines in the pathogenesis of sepsis. Infect Dis Clin North Am. 1999;13:413–26.
Covell DG, Barbet J, Holton OD, Black CD, Parker RJ, Weinstein JN. Pharmacokinetics of monoclonal immunoglobulin G1, F(ab′)2, and Fab′ in mice. Cancer Res. 1986;46:3969–78.
Secher T, Vasseur V, Poisson DM, Mitchell JA, Cunha FQ, ves-Filho JC, Ryffel B. Crucial role of TNF receptors 1 and 2 in the control of polymicrobial sepsis. J Immunol. 2009;182:7855–64.
Ebach DR, Riehl TE, Stenson WF. Opposing effects of tumor necrosis factor receptor 1 and 2 in sepsis due to cecal ligation and puncture. Shock. 2005;23:311–8.
Lewis M, Tartaglia LA, Lee A, Bennett GL, Rice GC, Wong GH, Chen EY, Goeddel DV. Cloning and expression of cDNAs for two distinct murine tumor necrosis factor receptors demonstrate one receptor is species specific. Proc Natl Acad Sci USA. 1991;88:2830–4.
Marquez-Velasco R, Bojalil R, Buelna A, Flores-Guzman F, Estevez-Ramirez J, Laguna J, Hernandez AM, az-Quinonez A, Paniagua-Solis JF. Anti-tumor necrosis factor alpha F(ab′)2 antibody fragments protect in murine polymicrobial sepsis: concentration and early intervention are fundamental to the outcome. Inflamm Res. 2006;55:378–84.
Echtenacher B, Falk W, Mannel DN, Krammer PH. Requirement of endogenous tumor necrosis factor/cachectin for recovery from experimental peritonitis. J Immunol. 1990;145:3762–6.
Echtenacher B, Falk W, Mannel DN, Krammer PH. Survival from cecal ligation and puncture and the formation of fibrous adhesions in the peritoneal cavity depend on endogenous tumor necrosis factor. In: Faist E, Meakins JL, Schildberg FW, editors. Host Defense Dysfunction in Trauma, Shock and Sepsis. Berlin Heidelberg: Springer-Verlag; 1993. pp. 755–758.
Bernard GR, Francois B, Mira J-P, Vincent J-L, Dellinger RP, Russell JA, LaRosa SP, Laterre P-F, Levy MM, Dankner W, Schmitt N, Lindemann J, Wittebole X. Evaluating the efficacy and safety of two doses of the polyclonal anti-TNF-α fragment antibody AZD9773 in adult patients with severe sepsis and/or septic shock: randomized, double-blind, placebo-controlled Phase IIb study. Crit Care Med. 2014; in press.
Levy MM, Dellinger RP, Townsend SR, Linde-Zwirble WT, Marshall JC, Bion J, Schorr C, Artigas A, Ramsay G, Beale R, Parker MM, Gerlach H, Reinhart K, Silva E, Harvey M, Regan S, Angus DC. The Surviving Sepsis Campaign: results of an international guideline-based performance improvement program targeting severe sepsis. Crit Care Med. 2010;38:367–74.
ARISE; ANZICS APD Management Committee. The outcome of patients with sepsis and septic shock presenting to emergency departments in Australia and New Zealand. Crit Care Resusc. 2007;9:8–18.
Tidswell M, Tillis W, LaRosa SP, Lynn M, Wittek AE, Kao R, Wheeler J, Gogate J, Opal SM. Phase 2 trial of eritoran tetrasodium (E5564), a toll-like receptor 4 antagonist, in patients with severe sepsis. Crit Care Med. 2010;38:72–83.
Rice TW, Wheeler AP, Bernard GR, Vincent J-L, Angus DC, Aikawa N, Demeyer I, Sainati S, Amlot N, Cao C, Ii M, Matsuda H, Mouri K, Cohen J. A randomized, double-blind, placebo-controlled trial of TAK-242 for the treatment of severe sepsis. Crit Care Med. 2010;38:1685–94.
Ranieri VM, Thompson BT, Barie PS, Dhainaut JF, Douglas IS, Finfer S, Gårdlund B, Marshall JC, Rhodes A, Artigas A, Payen D, Tenhunen J, Al-Khalidi HR, Thompson V, Janes J, Macias WL, Vangerow B, Williams MD. Drotrecogin alfa (activated) in adults with septic shock. N Engl J Med. 2012;366:2055–64.
Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, Richards DR, Donald-Smith GP, Gao H, Hennessy L, Finnerty CC, Lopez CM, Honari S, Moore EE, Minei JP, Cuschieri J, Bankey PE, Johnson JL, Sperry J, Nathens AB, Billiar TR, West MA, Jeschke MG, Klein MB, Gamelli RL, Gibran NS, Brownstein BH, Miller-Graziano C, Calvano SE, Mason PH, Cobb JP, Rahme LG, Lowry SF, Maier RV, Moldawer LL, Herndon DN, Davis RW, Xiao W, Tompkins RG. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci USA. 2013;110:3507–12.
Hotchkiss RS, Monneret G, Payen D. Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. Lancet Infect Dis. 2013;13:260–8.
Remick DG, Call DR, Ebong SJ, Newcomb DE, Nybom P, Nemzek JA, Bolgos GE. Combination immunotherapy with soluble tumor necrosis factor receptors plus interleukin 1 receptor antagonist decreases sepsis mortality. Crit Care Med. 2001;29:473–81.
Acknowledgments
We would like to thank Mike Snaith (MedImmune) and Lisa Stone (The Jackson Labs) for their management of the Tg mouse line, Yannis Sotsios (Biomedcode) for conducting the LPS studies, and Kerry Ratcliffe (Pathology Department, AstraZeneca, UK) for sectioning and HE staining of tissue blocks. We also thank Dr. Andrew Jones from Mudskipper Business Ltd, who provided editing assistance funded by AstraZeneca.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible Editor: Michael J. Parnham.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Newham, P., Ross, D., Ceuppens, P. et al. Determination of the safety and efficacy of therapeutic neutralization of tumor necrosis factor-α (TNF-α) using AZD9773, an anti-TNF-α immune Fab, in murine CLP sepsis. Inflamm. Res. 63, 149–160 (2014). https://doi.org/10.1007/s00011-013-0683-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00011-013-0683-3