
Abstract
Introduction:
Proteus penneri lipopolysaccharide (LPS) core regions are characterized by a greater structural variability than that observed in other Enterobacteriaceae. This fact and the small amount of published data concerning the serological activity of this part of P. penneri LPS prompted an examination of which fragment might determine cross-reactions with antibodies. To date, such epitopes have been found in the LPS core regions of P. mirabilis and P. vulgaris strains.
Materials and Methods:
Proteus sp. LPSs were tested with unabsorbed rabbit antisera by enzyme-linked immunosorbent assay (ELISA), sodium dodecyl sulfate polyacrylamide gel electrophoresis and Western blot, and once again by ELISA or passive immunohemolysis after the absorption of these antisera with selected LPSs.
Results:
The serological studies of P. penneri 8 LPS demonstrated antibodies in the tested antisera recognizing a common epitope located in the core regions of six of the LPSs, i.e. P. penneri 8, 34, 133, 7, 14, and 15. Additionally, another type of antibody directed against some fragment of P. penneri 13 and the core regions of other LPSs investigated was observed in one antiserum.
Conclusions:
A distal, trisaccharide fragment of the P. penneri 8 LPS core region is suggested to determine the cross-reactions of the tested antisera with the six P. penneri LPSs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Previous chemical studies allowed elucidating the structures of the LPS core regions of twenty-one P. penneri strains [11–15]. Those data showed a greater structural variability in the LPS core regions than that observed in other Enterobacteriaceae [3]. It is worth mentioning that even one P. penneri strain can be characterized by possessing a few structural variants of the LPS core region [15]. Such a huge structural heterogeneity of the LPS core region of P. penneri strains, its immunogenic properties, and the small amount of published data concerning the serological activity of this part of LPS encouraged us to find out which fragment of the LPS core region of P. penneri may be responsible for its reaction with antibodies. The results of immunochemical studies conducted before enabled the identification of two fragments in the core region of other Proteus species which react as epitopes: the amide of galacturonic acid with l-lysine located in the core region of the P. mirabilis O28 and R14 LPSs and N-(l-alanylamino)-4,6-dideoxy-β-d-glucose in the core part of the LPSs of P. mirabilis O6 and P. vulgaris TG103 [5, 8]. These fragments were determined by use of polyclonal antisera containing antibodies against both the O-polysaccharide and the core region. The existence of O-polysaccharide-specific antibodies in such antisera can influence the cross-reactivity of core-specific antibodies and may not demonstrate the real serological activity of the core region of the lipopolysaccharide (LPS). In this paper we report on serological studies performed with two different kinds of sera against P. penneri 8 containing only core-specific antibodies. The application of this material allowed a more precise determination of which fragment of the P. penneri 8 LPS core region may react with specific antibodies.
Materials And Methods
P. penneri 8 (O67) was a clinical isolate from the urine of a patient with bacteriuria in Missouri (USA). This strain and 26 others, i.e. P. penneri 1 (O72a), 2 (O66), 4 (O72a,b), 7 (O61), 11 (O58), 12 (O58), 13 (R form), 14 (O59), 15 (O52), 16 (O17), 17 (undefined O-serogroup), 18 (O17), 19 (O64a,b,c), 22 (O63), 25 (O69), 26 (O31a), 27 (O64a,b,c), 28 (O31a,b), 31 (O19a,b), 34 (O65), 37 (R form), 39 (O64a,b,d), 40 (O64a,b,d), 41 (O62), 42 (O71), and 44 (R form), were kindly provided by Prof. D. J. Brenner, Centers for Disease Control and Prevention in Atlanta (USA). P. penneri 60 (O70), 63 (O68), and 75 (O73a,c) were isolated from the urine of patients with bacteriuria in a hospital in Łódź and, along with P. penneri 71 (O64a,c,e) and the R110 mutant of P. mirabilis S1959, they belong to the collection of the Department of General Microbiology, University of Łódź, Poland. The other strains and their contributors were: P. penneri 47 (O59) by Dr. E. Falsen, Department of Clinical Bacteriology, Göteborg (Sweden); P. penneri 77 (O65) and 93 (O67) by Dr. B. Senior, Department of Medical Microbiology in Dundee (UK); P. penneri 103 (O73a,b), 104 (O61), 107 (O8), 112 (O8) and P. penneri 124 (R form) by Dr. B. Holmes, Central Public Health Laboratory in London (UK); and P. penneri 133 (O61) by Dr. G. Giammanco, Institute of Hygiene and Prevention Medicine in Catania (Italy).
Cultivation of bacteria, isolation, and saponification of the LPSs
The bacteria were grown under aerobic conditions in liquid nutrient broth containing 1% glucose (BTL, Łódź, Poland), then killed with phenol, centrifuged, washed twice with water, and lyophilized in order to obtain dried bacterial cells. Isolation of the LPS from the dried bacterial cells of the smooth or rough strains and saponification of the LPS were performed according to procedures described in detail elsewhere [2, 5, 16].
Degradation of LPS
The LPS was degraded by treatment with 1.5% acetic acid at 100°C for 1 h to obtain a precipitate of lipid A. The reaction mixture was centrifuged (13,000×g, 20 min, 4°C) to separate lipid A from the water-soluble carbohydrate portion, which was then fractionated on Bio-Gel P-10 column (1.6×100 cm, equilibrated with 0.05 M pyridine/acetic acid buffer, pH 5.6) [9]. The eluates were monitored with a Knauer differential refractometer. The fractions were eluted, freeze-dried, and checked by matrix-assisted laser-desorption/ionization time-of-flight mass spectrometry (MALDI-TOF) run on a Kratos Kompact-SEQ instrument [7].
Preparation of core oligosaccharide conjugate with diphtheria toxoid
The conjugation was performed by the method of Jennings and Ługowski [4] based on reductive amination. Fractions containing the conjugate were concentrated by ultrafiltration up to 2 mg/ml of the product.
Immunization procedure and sources of the sera
Sera against the whole bacterial cells of P. penneri 8 and 34 were provided by the Department of General Microbiology, University of Łódź The anti-conjugate serum was obtained by immunization of New Zealand white rabbits in the foot pads with 50 µg of the conjugate suspended in Freund’s complete adjuvant at days 0 and 21. Two weeks after administration of the last dose of vaccine, the rabbits were bled [6].
The LPS samples were tested with rabbit antisera by sodium dodecyl sulfate polyacrylamide gel electrophoresis and Western blot, enzyme-linked immunosorbent assay (ELISA), or passive immunohemolysis (PIH) with antisera absorbed with selected LPSs and unabsorbed sera. All assays were carried out as previously described [8]. In PIH, sheep red blood cells were sensitized with alkali-treated LPSs (64–128 µg/0.2 ml of sheep red blood cells). Using the alkali-treatment process for the preparation of antigens used in PIH or absorption procedures is necessary because only LPSs deprived of ester-bound fatty acids as a result of saponification with 0.25 M NaOH (2 h, 56°C) can easily sensitize sheep red blood cells. The polystyrene microtiter plates used in ELISA were coated with 200 ng LPS per well. Antibody titers (the highest dilution of antiserum giving optical density (OD)405≥0.2) were determined three times for each system. In Western blot the antisera were diluted 1:100 in a dot-blot 10% skimmed milk buffer.
Antiserum was diluted with veronal buffer (1:50) and incubated with sheep red blood cells (0.2 ml) sensitized with the respective alkali-treated LPSs (200 µg) for 30 min in ice. Antibodies that bound to LPS were removed from the antiserum by centrifugation. The reactivities of the 2-fold serially diluted antisera were tested using ELISA or PIH. In the latter test the antiserum titers were established as the last dilution of antiserum resulting in 50% hemolysis [8].
Results And Discussion
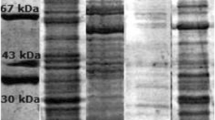
In this paper the serological and epitope characterization of the P. penneri 8 LPS core region is presented. In the first stage of this study, serum against whole cells of the P. penneri 8 strain was absorbed with alkali-treated LPS of P. penneri 93, which has the same O-polysaccharide structure as the P. penneri 8 antigen but their core regions are serologically different [1]. The aim of this procedure was to eliminate antibodies against O-polysaccharide from the tested antiserum and leave only those directed against the core region of the LPS. The absorption efficiency was checked by the Western blot technique. The lack of ladder-like patterns referring to the O-polysaccharide of P. penneri 93 LPS showed that all the O-polysaccharide-specific antibodies were successfully removed from the tested antiserum by the absorption procedure (Fig. 1). In the further part of the study this antiserum is referred to as anti-core serum. The other antiserum used in this study was directed against the core oligosaccharide of the P. penneri 8-diphtheria toxoid conjugate (anti-conjugate serum).

Western blot of P. penneri LPSs with serum against the P. penneri 8 LPS core region.
Apart from the antisera, Proteus LPS samples were used here as antigens. They were chosen on the basis of results of serological studies performed earlier at the Department of General Microbiology. Those investigations resulted in the preliminary division of 133 P. penneri strains into nineteen groups differing in the serological activities of their LPS core regions [unpublished data]. One to six representatives of each group were selected for the isolation of LPSs to create a set of 40 P. penneri antigens used in this study. Additionally, LPS from mutant R110 of P. mirabilis S1959 was added to the set, which thus contained 41 antigens.
The reactivities of the anti-conjugate and anti-core sera were tested by ELISA with all the mentioned LPS preparations. In this assay, both antisera reacted with six LPSs: P. penneri 8, 34, 133, 7, 14, and 15 (Table 1). Some differences in reactivity of both antisera were observed. The anti-core serum reacted with equal strength with P. penneri 8, 34, and 133 LPSs and the reactions were only a little weaker with the P. penneri 7, 14, and 15 LPS samples. On the other hand, the anti-conjugate serum reacted strongly with the LPSs of the first group, but much more weakly with the second group of LPS preparations. Remarkably, only the anti-core serum reacted weakly with the LPS of the rough P. penneri 13.
The dissimilarities in the reactivities of the two antisera could be explained by the different compositions of the immunoglobulin classes presented in these antisera. The core oligosaccharide linked to the diphtheria toxoid is a T-dependent antigen and induces the production of anti-conjugate immunoglobulin G (IgG) but not the production of immunoglobulin M (IgM). The use in ELISA of Fc fragment-specific goat anti-IgG and anti-IgM as the second antibodies enabled confirmation of the proposed theory by detecting only IgG in the anticonjugate serum (antiserum titer in the homologous system: 1:8000). In contrast, the application of the mentioned antibodies showed the presence of both immunoglobulin classes (higher level of IgG: 1:8000 and a lower level of IgM: 1:2000) in the anti-core serum. This may explain the stronger reactions of this antiserum with P. penneri 7, 14, and 15 LPSs and the presence of antibodies cross-reacting with P. penneri 13 which were not found in the anti-conjugate serum.
In Western blot, both the anti-conjugate and the anti-core sera bound to low-molecular-mass LPS species consisting of the core-lipid A moieties of P. penneri 8, 34, 133, 7, 14, and 15 LPS samples (Figs. 1, 2). As in ELISA, only the anti-core serum reacted with P. penneri 13 LPS. It showed only a weak banding pattern.

Western blot of P. penneri LPSs with serum against the core oligosaccharide of P. penneri 8-diphtheria toxoid conjugate.
The absorption of both tested antisera with the respective P. penneri 8, 34, 133, 7, 14, and 15 LPSs followed by ELISA completely abolished their reactivity with these antigens. This suggests the presence of a common epitope in the core region of all the mentioned LPSs. As the anti-core serum was the one that reacted with the LPS of P. penneri 13, only this antiserum was absorbed with the antigen in question. This absorption decreased the anticore serum titers with the LPSs of P. penneri 8, 34, 133 (from 16,000 to 4000) and 7, 14, and 15 (from 8000 to 2000), which suggested the presence of another type of antibody in this antiserum cross-reacting with some fragment of P. penneri 13 and other LPSs core regions used.
The data reported earlier by other authors [10] also suggested the presence of a common fragment in the P. penneri 8 and 34 core regions reacting as an epitope. The identical serological activities of these LPSs observed in the present serological study may also demonstrate the structural identity of their LPS core regions. This probably also refers to the P. penneri 133 LPS core region, which reacted identically to the P. penneri 8 and P. penneri 34 antigens in all the assays. To verify these assumptions, an experiment with the serum against whole cells of P. penneri 34 was conducted. At first this antiserum was absorbed with the alkali-treated LPS of P. penneri 78 strain, which has the same structure of the O-polysaccharide chains as the homologous antigen although the core regions of both LPSs are seriologically different [1]. This absorption resulted in an antiserum containing only core-specific antibodies.
According to the serological data obtained in this study, this antiserum reacted in the same way in ELISA with the LPSs of P. penneri 8, 34, and 133 (antiserum titer 1:8000; data not shown). As expected, in Western blot this antiserum recognized fast-migrating bands (restricted to the core-lipid A molecule) of the three tested antigens. The LPS of P. penneri 78 was used here as a control of O-polysaccharide-specific antibody elimination from the antiserum (Fig. 3).

Western blot of P. penneri LPSs with serum against the P. penneri 34 LPS core region.
The absorption of the core-antiserum against P. penneri 34 with the LPSs of P. penneri 8, 34, and 133 followed by PIH completely abolished its reactivity with these antigens, and the results may confirm the previous suggestions of the identity of the P. penneri 8, 34, and probably 133 core regions (not shown data).
The main aim of the present investigation was to discover which fragment of the P. penneri 8 LPS core region may be recognized by the antibodies and react as an epitope as well. The results of the serological studies carried out using anti-core and anti-conjugate sera against P. penneri 8 referred to the known chemical structures of the LPS core regions of P. penneri 8, 7, 14, 15, and 13 (Fig. 4A–E) indicated that this epitope may be associated with the fragment α-GalNAc-(1→2)-α-dd-Hep-(1→6)-α-GlcN. It is located in the distal part of the LPS core region of P. penneri 8, 7, 14, and 15. The only strain that does not have this fragment in the LPS core region is the rough P. penneri 13 strain (Fig. 4E). The remaining part of the core region of this LPS that could react with both tested antisera was identical in all the tested antigens. Accordingly, the lack of an anti-conjugate serum reaction with the P. penneri 13 LPS both in ELISA and Western blot suggests the existence of antibodies in this antiserum which recognize only a common epitope (Table 1, Fig. 2). Similarly, the antibodies remaining in the anti-core serum after its absorption with the LPS of P. penneri 13 probably reacted only with this common fragment.

General structures of the core-lipid A region of P. penneri 8 (A), 7 (B), 14 (C), 15 (D), and 13 (E) LPSs. The putative common epitope in the P. penneri 8 (A), 7 (B), 14 (C), and 15 (D) core regions is indicated by an arc. Arap4N — 4-amino-4-deoxy-L-arabinopyranose, GalA — galacturonic acid, GalNA — amides of GalA with aliphatic polyamines, GlcNGly — 2-deoxy-2-glycylamino-d-glucose, Hep — l-glycero-d-manno-heptose, dd-Hep — d-glycero-d-manno-heptose, Kdo — 3-deoxy-d-manno-oct-2-ulosonic acid, PEtn — 2-aminoethyl phosphate.
The identical reactivity of the tested antisera with the P. penneri 8, 34, and 133 LPSs in all the serological assays also suggests the existence of the same epitope in the core regions of the P. penneri 34 and 133 LPSs of undefined core structure.
Comparison of the results of the serological studies with the well-defined core structures of P. penneri 8, 7, 14, 15 (Fig. 4A–D) also revealed that the glucose residue linked to the epitope located in the core region of the LPSs of P. penneri 7, 14, and 15 (Fig. 4B–D) and absent from this part of the LPS of P. penneri 8 (and, presumably, P. penneri 34, 133) (Fig. 4A) might weaken the binding of antibodies by this common fragment. Therefore, in ELISA both antisera (P. penneri 8 anti-conjugate and anti-core serum) reacted with the homologous LPSs and P. penneri 34 and 133 antigens more strongly than with the second group of LPS preparations (P. penneri 7, 14, 15) having the glucose residue in their distal part of the LPS core region (Table 1).
Moreover, the structural and serological data suggest that the glycine residue attached to α-GlcN of the epitope located in the P. penneri 8 (and probably P. penneri 34 and 133), 7, 14 LPS core regions (Fig. 4A–C) might not influence the serological specificity of the antigens in question. This is more likely because of the fact that in the LPS core region of the mentioned antigens, the amino group of the GlcN residue may be acylated by 10–20% by a glycine residue [15]. This amino-acid residue was not found in the P. penneri 15 LPS core region (Fig. 4D). In spite of this small difference between the core region structures of the P. penneri 15 and P. penneri 8, 7, 14 (and probably P. penneri 34 and 133) LPSs, the P. penneri 8 anti-conjugate and anti-core sera absorbed with P. penneri 15 LPS did not react with any of the tested antigens, both those possessing and those not possessing the glycine residue in the core region. This finding might confirm that the common fragment of the LPS core region responsible for the cross-reactivity of P. penneri 8 antisera is α-GalNAc-(1→2)-α-dd-Hep-(1→6)-α-GlcN without a glycine residue. Establishing more numbers of similar epitopes may be helpful in choosing appropriate R antigens for the preparation of a vaccine protecting against infections caused by P. penneri 8 and other Proteus cross-reacting strains.
Abbreviations
- LPS:
-
lipopolysaccharide
- Glc:
-
glucose
- GlcN:
-
glucosamine
- IgG:
-
immunoglobulin G
- IgM:
-
immunoglobulin M
- ELISA:
-
enzyme-linked immunosorbent assay
- PIH:
-
passive immunohemolysis
References
Drzewiecka D., Zych K. and Sidorczyk Z. (2004): Characterization and serological classification of a collection of Proteus penneri clinical strains. Arch. Immunol. Ther. Exp., 52, 121–128.
Galanos C., Lüderitz O. and Westphal O. (1969): A new method for the extraction of R lipopolysaccharides. Eur. J. Biochem., 9, 245–249.
Holst O. (2002): Chemical structure of the core region of lipopolysaccharides — an update. Trends Glycosci. Glycotech., 14, 87–103.
Jennings H.J. and Ługowski C. (1981): Immunochemistry of groups A, B, and C meningococcal polysaccharide-tetanus toxoid conjugates. J. Immunol., 127, 1011–1018.
Ko3odziejska K., Perepelov A.V., Zabłotni A., Drzewiecka D., Senchenkova S.N., Zych K., Shashkov A.S., Knirel Y.A. and Sidorczyk Z. (2006): Structure of the glycerol phosphate-containing O-polysaccharides and serological studies of the lipopolysaccharides of Proteus mirabilis CCUG 10704 (OE) and Proteus vulgaris TG 103 classified into a new Proteus serogroup, O54. FEMS Immunol. Med. Microbiol., 47, 267–274.
Ługowski C., Jachymek W., Niedziela T. and Rowiński S. (1996): Serological characterization of anti-endotoxin sera directed against the conjugates of oligosaccharide core of Escherichia coli type R1, R2, R3, J5 and Salmonella Ra with tetanus toxoid. FEMS Immunol. Med. Microbiol., 16, 21–30.
Łukasiewicz J., Jachymek W., Niedziela T., Malik-Gębicka M., Dzieciątkowska M. and Ługowski C. (2002): Comparison of serological specificity of anti-endotoxin sera directed against whole bacterial cells and core oligosaccharide of Escherichia coli J5-tetanus toxoid conjugate. Acta Biochim. Pol., 49, 721–734.
Sidorczyk Z., Zych K., Toukach F.V., Arbatsky N.P., Zabłotni A., Shashkov A.S. and Knirel Y.A. (2002): Structure of the O-polysaccharide and classification of Proteus mirabilis strain G1 in Proteus serogroup O3. Eur. J. Biochem., 269, 1406–1412.
Petersson C., Niedziela T., Jachymek W., Kenne L., Zarzecki P. and Ługowski C. (1997): Structural studies of the O-specific polysaccharide of Hafnia alvei strain PCM 1206 lipopolysacchride containing D-allothreonine. Eur. J. Biochem., 244, 580–586.
Toukach F.V., Arbatsky N.P., Shashkov A.S., Knirel Y.A., Zych K. and Sidorczyk Z. (1998): Structure of a new neutral O-specific polysaccharide of Proteus penneri 34. Carbohydr. Res., 312, 97–101.
Vinogradov E. and Sidorczyk Z. (2000): The structure of the core part of Proteus penneri strain 16 lipopolysaccharide. Carbohydr. Res., 326, 185–193.
Vinogradov E. and Sidorczyk Z. (2001): The structure of the carbohydrate backbone of the core-lipid A region of the lipoplysaccharide from Proteus penneri strain 40: new Proteus strains containing open-chain acetal-linked N-acetylgalactosamine in the core part of the LPS. Carbohydr. Res., 330, 537–540.
Vinogradov E. and Sidorczyk Z. (2002): The structure of the carbohydrate backbone of the rough type lipopolysaccharides from Proteus penneri strains 12, 13, 37 and 44. Carbohydr. Res., 337, 835–840.
Vinogradov E., Sidorczyk Z. and Knirel Y.A. (2002): Structure of the core part of the lipopolysaccharides from Proteus penneri strains 7, 8, 14, 15, and 21. Carbohydr. Res., 337, 643–649.
Vinogradov E., Sidorczyk Z. and Knirel Y.A. (2002): Structure of the lipopolysaccharide core region of the bacteria of the genus Proteus. Aust. J. Chem., 55, 61–67.
Westphal O. and Jann K. (1965): Bacterial lipopolysaccharides. Extraction with phenol-water and further applications of the procedure. Methods Carbohydr. Chem., 5, 83–91.
Acknowledgment
This work was supported by the Ministry of Science and Higher Education (Poland, grant no. N 401 001 31/0009).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Palusiak, A., Dzieciątkowska, M. & Sidorczyk, Z. Application of two different kinds of sera against the Proteus penneri lipopolysaccharide core region in search of epitopes determining cross-reactions with antibodies. Arch. Immunol. Ther. Exp. 56, 135–140 (2008). https://doi.org/10.1007/s00005-008-0012-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00005-008-0012-7