
Abstract
Background and Objective
Almorexant is the first representative of the new class of orexin receptor antagonists, which could become a new treatment option for insomnia. The present study investigated the potential interaction between almorexant and warfarin.
Methods
In this open-label, two-way crossover, drug–drug interaction study, healthy male subjects received, in a randomized fashion, almorexant 200 mg once daily for 10 days and a single dose of 25 mg warfarin co-administered on day 5 (treatment A) and a single dose of 25 mg warfarin on day 1 (treatment B). Serial blood samples for warfarin pharmacokinetics and pharmacodynamics were drawn during both treatments.
Results
Of the 14 enrolled subjects, one withdrew due to an adverse event and 13 completed the study. Almorexant had no effect on the pharmacokinetics of warfarin. The geometric mean ratios (90 % confidence interval) for the area under the plasma concentration–time curve to infinity (AUC0–∞) of S- and R-warfarin were 0.99 (0.89, 1.09) and 1.05 (0.95, 1.16), respectively, and for the maximum plasma concentration (Cmax) were 0.99 (0.86, 1.14) and 1.00 (0.88, 1.13), respectively. The main pharmacodynamic variable was the AUC for the international normalized ratio (AUCINR). Almorexant had no effect on this variable as demonstrated by a geometric mean ratio of 0.99 (0.82, 1.19). Secondary pharmacodynamic variables including maximum effect (Emax), the time to the maximum INR, and factor VII plasma concentrations were also not affected by almorexant.
Conclusion
No dose adjustment of warfarin is necessary when concomitantly administered with almorexant.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
The orexin or hypocretin system was discovered in 1998, and a number of experimental observations have suggested that this system plays an important role in the sleep–wake cycle [1–5]. These observations have spurred considerable interest in the development of orexin receptor antagonists as a potential new treatment for insomnia [6, 7]. Almorexant is the first representative of this new class of compounds, which has shown promising sleep-promoting activity in animals, healthy subjects, and patients with primary insomnia [8–10].
The pharmacokinetics of almorexant after single- and multiple-dose administration to healthy subjects have been described previously [9, 11, 12]. In brief, they are characterized by a clearance of 43 L/h, a large volume of distribution (683 L), a fast absorption (time to Cmax [tmax] ~1 h), and a rapid disposition due to a pronounced distribution phase, with concentrations decreasing to less than 20 % of maximum plasma concentration (Cmax) 8 h after administration. Following multiple-dose administration, steady-state concentrations were reached after 4–5 days of dosing, and accumulation was minimal. In vitro, almorexant has been shown to be an inhibitor (inhibitory constant approximately 2 μM) of cytochrome P450 (CYP) isoenzymes CYP2C9, CYP2D6, and CYP3A4 but not of other CYP isoenzymes (Actelion Pharmaceuticals, data on file). For this reason, a drug–drug interaction study was performed in healthy subjects investigating the effect of almorexant on midazolam and simvastatin, two model substrates of CYP3A4 [13]. This study showed that almorexant only marginally increased exposure to midazolam, but exposure to simvastatin and its hydroxyacid metabolite was increased by 3.4- and 2.8-fold, respectively [14]. The difference in sensitivity of both CYP3A4 substrates is consistent with the observation that in vitro almorexant inhibited testosterone 6β-hydroxylation but not midazolam 1′-hydroxylation (Actelion Pharmaceuticals, data on file).
The anticoagulant warfarin acts by inhibiting the regeneration of vitamin K1 epoxide, which is necessary for the post-ribosomal synthesis of vitamin K-dependent clotting factors such as factors II, VII, IX, and X. Warfarin is administered as a racemic mixture of S- and R-enantiomers. Its elimination is almost entirely by metabolism followed by urinary excretion of metabolites with minimal anticoagulation activity [15]. Warfarin is metabolized by CYP isoenzymes to inactive hydroxylated metabolites and by reductases to reduced metabolites. The S-enantiomer is primarily metabolized by CYP2C9 and less by CYP2C19 and CYP3A4, whereas the R-enantiomer is mainly metabolized by CYP1A2 with a smaller contribution of CYP3A4 [16]. Warfarin has a narrow therapeutic index, and small changes in its pharmacokinetics may lead to the need for dose adaptation.
The present study investigated further the drug–drug interaction potential of almorexant by studying its effects on the pharmacokinetics and pharmacodynamics of warfarin.
2 Methods
2.1 Study Design
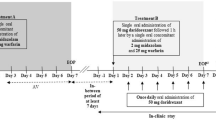
This was a single-center, open-label, randomized, two-way crossover, drug–drug interaction study. Screening of subjects took place between 21 and 3 days before first study drug administration. Enrolled subjects were randomized to treatment sequences A/B or B/A. Treatment A consisted of almorexant 200 mg once daily on day 1–10 and a single dose of 25 mg warfarin co-administered on day 5; treatment B consisted of a single dose of 25 mg warfarin on day 1. A 2-week washout period between treatments was respected. A dose of 200 mg almorexant was chosen because it was expected to be well tolerated and it was the highest dose investigated in phase III trials. Study drugs were administered in the morning to subjects in the fasted state, with breakfast served 2 h thereafter. During both treatments, subjects were confined to the study center from approximately 12 h prior to warfarin administration until 144 h thereafter. Because of the sleep-promoting properties of almorexant, subjects stayed in the clinic under supervision for approximately 5 h after its intake on days 1–4 of treatment A. After this 5-h observation period, a physician determined whether the subject was fully alert and could be allowed to go home or whether there were any residual effects that could be attributed to a sleep-promoting drug (e.g., muscular weakness, dizziness, fatigue, or somnolence). Subjects were not to drive a car or engage in activities that required operating vehicles or dangerous machinery. From screening until the end-of-study examination, which was performed 144 h after warfarin administration in the second treatment period, subjects had to refrain from excessive physical exercise and strenuous sports activities and were not allowed to consume cranberries, grapefruit, cranberry juice, or grapefruit juice. Although no effect of grapefruit juice on the pharmacodynamics of warfarin could be shown [17], cranberry juice increased the international normalized ratio (INR) [18].
This study was conducted in full conformity with the Declaration of Helsinki and its amendments. The protocol was approved by an independent ethics committee (Ethics Committee of the Medical University, Graz, Austria). Each subject provided written informed consent prior to any study procedure.
2.2 Inclusion and Exclusion Criteria
Eligible subjects were healthy males aged between 18 and 45 years who had a body mass index between 18 and 28 kg/m2 at screening. Subjects were judged to be healthy based on medical history, physical examination, ECG, vital signs, and clinical laboratory tests. Subjects were not enrolled if they had a history of hemorrhagic disease, frequent nasal, hemorrhoidal, or gingival bleeding, an activated partial thromboplastin time >40 s, an INR >1.15, a low (<150 × 109) or high (>400 × 109) platelet count, or had been treated with any medication (including over-the-counter and herbal medicines) within 2 weeks prior to screening.
2.3 Blood Sampling and Bioanalytical Methods
Blood samples for the determination of S- and R-warfarin were collected in tubes with ethylenediaminetetra-acetic acid (EDTA) as anticoagulant at the following time points: pre-dose, and 1, 2, 4, 8, 12, 24, 36, 48, 60, 72, 96, 120, and 144 h following the warfarin dose during both treatment periods. The tubes with blood were centrifuged, the plasma separated, and all plasma samples were stored in an upright position at −20 °C pending analysis. The stereoselective bioanalysis of warfarin in plasma was done using a validated high pressure liquid chromatography (HPLC) coupled to tandem mass spectrometry (MS/MS) method. In brief, 300 μL of acetonitrile containing internal standards (deuterated S- and R-warfarin) was added to 100 μL of plasma. Following protein precipitation and centrifugation, 15 μL of the supernatant was injected onto the HPLC system. The latter consisted of a C18 pre-column (5 μm, 4 × 3.0 mm; Phenomenex, Aschaffenburg, Germany), a Reprosil Chiral-NR analytical column (8 μm, 125 × 3.0 mm; Dr. Maisch GmbH, Ammerbruch, Germany), a Waters Alliance 2795 pump, degasser, and autosampler (Waters, Eschborn, Germany). The columns were eluted with a mixture of methanol:5 mM ammonium acetate pH 4.0 (70:30 v/v) for 11 min. The MS/MS analysis (Quattro LC, Micromass, Wythenshawe, UK) was performed in the positive ionization mode, and the limit of detection was 20 ng/mL for both analytes. For R-warfarin, the inter-day coefficients of variation (imprecision) were ≤11.0 %, whereas inter-day inaccuracy ranged between −1.1 and 0.6 %. For S-warfarin, imprecision was ≤10.1 %, whereas inter-day inaccuracy ranged between −2.0 and −0.4 %.
Blood samples for the determination of factor VII and INR were collected pre-dose, and 4, 8, 12, 24, 36, 48, 60, 72, 96, 120, and 144 h after dosing with warfarin during both treatment periods in tubes containing citrate as anticoagulant. These samples were put on ice and sent as soon as possible to the local clinical laboratory for analysis. The assay of factor VII was performed by a standard one-stage method on fresh plasma. The results are expressed in percent of the laboratory reference value. The prothrombin time of each sample was measured using a standard test and then standardized to yield the INR, a fraction that has no unit. In treatment A, blood samples for determination of trough almorexant plasma concentrations were collected pre-dose on days 1–10 and 24 h after the last almorexant dose on day 10 in tubes with EDTA as anticoagulant. Concentrations in plasma were measured using a validated LC–MS/MS assay with a lower limit of quantification of 0.05 ng/mL and imprecision and inaccuracy ≤4.9 and 5.3 %, respectively [14].
2.4 Pharmacokinetic and Pharmacodynamic Analyses
Pharmacokinetic and pharmacodynamic variables were determined by non-compartmental analysis using WinNonlin Professional Version 5.2.1 (Pharsight Corporation, Mountain View, CA, USA). The Cmax or maximum effect (Emax) and corresponding time point (tmax or \( t_{{E_{\hbox{max} } }} \)) were directly obtained from the plasma concentration- or effect-time profiles. The area under the plasma concentration–time curve (AUC) from time 0 to time t of the last measurable concentration (AUC0–t ) was calculated using the linear trapezoidal rule. The AUC from time 0 to infinity (AUC0–∞) was calculated by AUC0–t + C t /λ z , where C t is the last measurable concentration and λ z the terminal elimination rate constant determined by log-linear regression analysis of the measured plasma concentrations in the terminal elimination phase. The elimination half-life (t ½) of S- and R-warfarin was calculated as follows: t ½ = 0.693/λ z . For both INR and factor VII, the AUC was calculated for the period 0–144 h and absolute values are reported.
2.5 Statistical Analysis
The null hypothesis was that one of the 90 % confidence limits (two-sided based on t-distribution) of treatment A versus treatment B for at least one of the five primary pharmacokinetic and pharmacodynamic endpoints (C max and AUC 0–∞ for S- and R-warfarin and AUCINR) was outside the interval 0.8–1.25. The type-I error was set to 0.05 and the power to 90 %. A sample size of 12 provided more than 95 % power to reject the null hypothesis assuming a standard deviation of the difference (in log scale) equal to 0.13 [19].
Treatment differences are displayed using the ratio of the geometric means (treatment A/treatment B) with their corresponding 90 % confidence limits for C max, AUC0–∞, and AUCINR derived from a mixed model analysis of variance with treatment and subject considered fixed effects. The 90 % two-sided confidence limits of the geometric mean ratio were derived using the antilog of the 90 % confidence limits of the difference of the mean between treatment A and treatment B (on the natural logarithmic scale) and were evaluated using the t-distribution. As the null hypothesis of all five primary pharmacokinetic and pharmacodynamic endpoints should have been rejected in order to demonstrate bioequivalence between the two treatments, no correction for multiple testing was needed.
3 Results
3.1 Study Subjects
In this study, 14 healthy male subjects were randomized, and their mean (range) values for age and body mass index were 29.0 (21–44) years and 24.9 (22.9–28.1) kg/m2. Except for one Black subject, all were White/Caucasian. Thirteen subjects completed the study and were included in the per-protocol analysis set. One subject prematurely withdrew from the study in period 1 due to nausea after having received the first dose of almorexant.
3.2 Pharmacokinetics
The mean plasma concentration–time profiles of S- and R-warfarin alone and during concomitant administration of almorexant are superimposable (Fig. 1). The pharmacokinetics of S- and R-warfarin were similar in the absence and presence of almorexant and characterized by a median t max of 2.0 h, C max values of about 1,200 ng/mL and values for t ½ of about 39 h (S-warfarin) and 50 h (R-warfarin) (Table 1).

Arithmetic mean (and standard deviation) plasma concentration–time profile of S-warfarin (top panel) and R-warfarin (bottom panel) after administration of a single dose of 25 mg warfarin alone (treatment B) and in the presence of almorexant 200 mg once daily for 10 days with a single dose of 25 mg warfarin on day 5 (treatment A) to healthy male subjects (n = 13)
Results of the statistical analysis confirmed the absence of a pharmacokinetic interaction between warfarin and almorexant (Table 2). The geometric mean ratios and corresponding 90 % confidence intervals were entirely within the bioequivalence limits of 0.80–1.25 for the variables C max and AUC0–∞ of S- and R-warfarin. No period or sequence effects were observed.
Mean trough plasma concentrations of almorexant showed that steady-state concentrations had been attained by day 4 (mean ± SD, 5.0 ± 2.2 ng/mL) and that the concomitant warfarin dose on day 5 had no effect on the trough plasma concentration of almorexant.
3.3 Pharmacodynamics
A dose of 25 mg warfarin caused an increase in INR that was similar in the absence and presence of almorexant. The maximum increase in INR was observed 24 h after administration, and INR had returned to baseline 144 h after administration (Fig. 2). Derived pharmacodynamic variables of INR did not differ between treatments (Table 3), and the statistical analysis showed that the geometric mean ratio and its 90 % confidence limits for AUCINR were within the limits of 0.80–1.25. No bleeding adverse events were reported during the study (data not shown).

Arithmetic mean (and standard deviation) international normalized ratio (INR) versus time profile after administration of a single dose of 25 mg warfarin alone (treatment B) and in the presence of almorexant 200 mg once daily for 10 days with a single dose of 25 mg warfarin on day 5 (treatment A) to healthy male subjects (n = 13)
Following administration of warfarin, both in the absence and presence of almorexant, factor VII concentrations decreased (Fig. 3). The maximum decrease occurred 24–36 h after administration, and factor VII slowly returned to baseline thereafter. The pharmacodynamic analysis appeared to show a difference in the time to E max between treatments, i.e., 36 h for treatment A and 24 h for treatment B, whereas other variables were similar (Table 3).

Arithmetic mean (and standard deviation) plasma concentration–time profile of factor VII after administration of a single dose of 25 mg warfarin alone (treatment B) and in the presence of almorexant 200 mg once daily for 10 days with a single dose of 25 mg warfarin on day 5 (treatment A) to healthy male subjects (n = 13)
4 Discussion
Almorexant is a dual orexin receptor antagonist and has been shown in vitro to inhibit CYP2C9, CYP2D6, and CYP3A4 (Actelion Pharmaceuticals, data on file). The present study investigated the effects of almorexant on warfarin pharmacokinetics and pharmacodynamics in a randomized, two-way crossover study. Such a design reduces variability as each subject serves as his own control, thereby reducing the number of subjects to be included and is in accordance with current guidelines for in vivo interaction studies [20]. Warfarin was administered when almorexant concentrations were in steady state and any possible inhibition of CYP isoenzymes was maintained during the elimination phase of warfarin by continued administration of almorexant.
The pharmacokinetics of warfarin in the absence of almorexant were in good agreement with previously reported results [19, 21]. Almorexant was not expected to influence the pharmacokinetics of R-warfarin as this compound is mainly metabolized by CYP1A2 [16], an enzyme that is not inhibited by almorexant. However, almorexant also did not exert any effect on S-warfarin pharmacokinetics. Previously, almorexant had been shown to increase exposure to simvastatin, a CYP3A4 substrate, in healthy subjects [14], whereas in vitro it is a more potent inhibitor of CYP2C9, the major metabolizing enzyme of S-warfarin. The inhibition constants of almorexant for CYP2C9 and CYP3A4 (marker: testosterone 6β-hydroxylation) inhibition were 1.6 and 2.9 μM, respectively (Actelion Pharmaceuticals Ltd, data on file). The explanation for these findings lies in the fact that CYP2C9, in contrast to CYP3A4, is not expressed in the gastrointestinal system. Our previous experiments [14, 22] made it plausible that the CYP3A4 inhibitory properties of almorexant are mainly expressed at the gastrointestinal rather than the hepatic level, also related to higher local concentrations. This was delineated by time-separated administration of almorexant and simvastatin [22]. The lack of an effect of almorexant on the pharmacokinetics of S-warfarin is in accordance with insufficient concentrations of almorexant to inhibit CYP2C9. With a dose of 200 mg, a C max value of 93.2 ng/mL or 0.17 μM was observed after 4 days of dosing [11], i.e., well below the inhibitory constant for CYP2C9, particularly when considering free drug concentrations of almorexant. It should be mentioned, however, that plasma concentrations do not necessarily reflect local concentrations in the liver.
In agreement with the lack of an effect on warfarin pharmacokinetics, concomitant administration of almorexant had no effect on the warfarin-induced increase in INR and decrease in factor VII plasma concentrations. Whenever possible, pharmacodynamic variables should be included in drug–drug interaction studies even when no pharmacokinetic interaction is expected as sometimes there may be a disconnect between pharmacokinetics and pharmacodynamics. For example, the intake of cranberry juice enhanced the effect of warfarin on INR in healthy subjects without affecting warfarin pharmacokinetics [18]. The authors explained this observation by an increase in sensitivity to warfarin induced by cranberry, especially in subjects carrying variant genotypes of the vitamin K epoxide reductase subunit 1 gene (VKORC1). No such increase in sensitivity to warfarin was observed in the present study.
The blood sampling scheme applied in the present study was optimized to investigate the pharmacokinetics of warfarin and only few blood samples were taken around the E max of pharmacodynamic variables. This may very well explain the observed increase in \( t_{{E_{\hbox{max} } }} \) of factor VII in the presence of almorexant when compared with warfarin alone. For both treatments, the range of individual \( t_{{E_{\hbox{max} } }} \) values of factor VII was the same (24–36 h). However, during treatment A, seven subjects had a \( t_{{E_{\hbox{max} } }} \) of 36 h and six subjects a \( t_{{E_{\hbox{max} } }} \) of 24 h, whereas numbers were six and seven subjects, respectively, for treatment B, resulting in the observed difference in median \( t_{{E_{\hbox{max} } }} \) .
5 Conclusion
Almorexant has no influence on the pharmacokinetics and pharmacodynamics of warfarin. No dose adjustment of warfarin is necessary when concomitantly administered with almorexant.
References
de Lecea L, Kilduff TS, Peyron C, Gao X, Foye PE, Danielson PE, et al. The hypocretins: hypothalamus-specific peptides with neuroexcitatory activity. Proc Natl Acad Sci USA. 1998;95:322–7.
Sakurai T, Amemiya A, Ishii M, Matsuzaki I, Chemelli RM, Tanaka H, et al. Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell. 1998;92:573–85.
Cao M, Guilleminault C. Hypocretin and its emerging role as a target for treatment of sleep disorders. Curr Neurol Neurosci Rep. 2011;11:227–34.
Nattie E, Li A. Central chemoreception in wakefulness and sleep: evidence for a distributed network and a role for orexin. J Appl Physiol. 2010;108:1417–24.
Tsujino N, Sakurai T. Orexin/hypocretin: a neuropeptide at the interface of sleep, energy homeostasis, and reward system. Pharmacol Rev. 2009;61:162–76.
Scammell TE, Winrow CJ. Orexin receptors: pharmacology and therapeutic opportunities. Annu Rev Pharmacol Toxicol. 2011;51:243–66.
Coleman PJ, Renger JJ. Orexin receptor antagonists: a review of promising compounds patented since 2006. Expert Opin Ther Pat. 2010;20:307–24.
Brisbare-Roch C, Dingemanse J, Koberstein R, Hoever P, Aissaoui H, Flores S, et al. Promotion of sleep by targeting the orexin system in rats, dogs and humans. Nat Med. 2007;13:150–5.
Hoever P, de Haas S, Winkler J, Schoemaker RC, Chiossi E, van Gerven J, Dingemanse J. Orexin receptor antagonism, a new sleep-promoting paradigm: an ascending single-dose study with almorexant. Clin Pharmacol Ther. 2010;87:593–600.
Hoever P, Dorffner G, Beneš H, Penzel T, Danker-Hopfe H, Barbanoj MJ, et al. Orexin receptor antagonism, a new sleep-enabling paradigm: A proof-of-concept clinical trial. Clin Pharmacol Ther. 2012;91:975–85.
Hoever P, de Haas SL, Dorffner G, Chiossi E, van Gerven JM, Dingemanse J. Orexin receptor antagonism: an ascending multiple-dose study with almorexant. J Psychopharmacol. 2012;26:1071–80.
Hoch M, Hoever P, Zisowsky J, Priestley A, Fleet D, Dingemanse J. Absolute oral bioavailability of almorexant, a dual orexin receptor antagonist, in healthy human subjects. Pharmacology. 2012;89:53–7.
Bjornsson TD, Callaghan JT, Einolf HJ, Fischer V, Gan L, Grimm S, Pharmaceutical Research and Manufacturers of America (PhRMA) Drug Metabolism/Clinical Pharmacology Technical Working Group; FDA Center for Drug Evaluation and Research (CDER), et al. The conduct of in vitro and in vivo drug–drug interaction studies: a Pharmaceutical Research and Manufacturers of America (PhRMA) perspective. Drug Metab Dispos. 2003;31:815–32.
Hoch M, Hoever P, Alessi F, Theodor R, Dingemanse J. Pharmacokinetic interactions of almorexant with midazolam and simvastatin, two CYP3A4 model substrates, in healthy male subjects. Eur J Clin Pharmacol. 2013;69:523–32.
Holford NH. Clinical pharmacokinetics and pharmacodynamics of warfarin. Understanding the dose-effect relationship. Clin Pharmacokinet. 1986;11:483–504.
Kaminsky LS, Zhang ZY. Human P450 metabolism of warfarin. Pharmacol Ther. 1997;73:67–74.
Sullivan DM, Ford MA, Boyden TW. Grapefruit juice and the response to warfarin. Am J Health Syst Pharm. 1998;55:1581–3.
Mohammed Abdul MI, Jiang X, Williams KM, Day RO, Roufogalis BD, Liauw WS, et al. Pharmacodynamic interaction of warfarin with cranberry but not with garlic in healthy subjects. Br J Pharmacol. 2008;154:1691–700.
Ouellet D, Bramson C, Carvajal-Gonzalez S, Roman D, Randinitis E, Remmers A, et al. Effects of lasofoxifene on the pharmacokinetics and pharmacodynamics of single-dose warfarin. Br J Clin Pharmacol. 2006;61:741–5.
Draft Guidance for Industry. Drug interaction studies—study design, data analysis, implications for dosing and labeling recommendations. U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER); 2012.
Malhotra B, Alvey C, Gong J, Li X, Duczynski G, Gandelman K. Effects of fesoterodine on the pharmacokinetics and pharmacodynamics of warfarin in healthy volunteers. Br J Clin Pharmacol. 2011;72:257–62.
Hoch M, Hoever P, Theodor R, Dingemanse J. Almorexant effects on CYP3A4 activity studied by its simultaneous and time-separated administration with simvastatin and atorvastatin. Eur J Clin Pharmacol. 2013;69:1235–45.
Acknowledgments
This study was fully funded by Actelion Pharmaceuticals Ltd. Both authors are full-time employees of Actelion Pharmaceuticals Ltd. Jasper Dingemanse and Petra Hoever designed the study and revised the manuscript. Petra Hoever analyzed the data.
The clinical part of the study was conducted at the Privatklinik Leech, Graz, Austria with Fritz Pinl as principal investigator. The stereo-selective bioanalysis of warfarin was performed by Andreas Möller, Bioproof, München, Germany. Almorexant plasma concentrations were determined by Jürgen Karg, Inovalab, Reinach, Switzerland. Editorial assistance for the preparation of the manuscript was provided by Paul van Giersbergen (Van Giersbergen Consulting, Wuenheim, France).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Dingemanse, J., Hoever, P. Absence of Pharmacokinetic and Pharmacodynamic Interactions between Almorexant and Warfarin in Healthy Subjects. Drugs R D 13, 145–151 (2013). https://doi.org/10.1007/s40268-013-0017-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40268-013-0017-5