
Abstract
Tuberculosis (TB) is still the number one cause of death due to an infectious disease. Pharmacokinetics and pharmacodynamics of anti-TB drugs are key in the optimization of TB treatment and help to prevent slow response to treatment, acquired drug resistance, and adverse drug effects. The aim of this review was to provide an update on the pharmacokinetics and pharmacodynamics of anti-TB drugs and to show how population pharmacokinetics and Bayesian dose adjustment can be used to optimize treatment. We cover aspects on preclinical, clinical, and population pharmacokinetics of different drugs used for drug-susceptible TB and multidrug-resistant TB. Moreover, we include available data to support therapeutic drug monitoring of these drugs and known pharmacokinetic and pharmacodynamic targets that can be used for optimization of therapy. We have identified a wide range of population pharmacokinetic models for first- and second-line drugs used for TB, which included models built on NONMEM, Pmetrics, ADAPT, MWPharm, Monolix, Phoenix, and NPEM2 software. The first population models were built for isoniazid and rifampicin; however, in recent years, more data have emerged for both new anti-TB drugs, but also for defining targets of older anti-TB drugs. Since the introduction of therapeutic drug monitoring for TB over 3 decades ago, further development of therapeutic drug monitoring in TB next steps will again depend on academic and clinical initiatives. We recommend close collaboration between researchers and the World Health Organization to provide important guideline updates regarding therapeutic drug monitoring and pharmacokinetics/pharmacodynamics.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Information on pharmacokinetics and pharmacodynamics of anti-tuberculosis drugs can help to optimize treatment. |
Population pharmacokinetics and Bayesian dose adjustment can help to optimize dose adjustment. |
There is still a significant knowledge gap for many of the tuberculosis drugs. |
1 Introduction
Tuberculosis (TB) is still the number one cause of death due to an infectious disease. The World Health Organization estimated that about 1.6 million individuals died from TB in 2018. Of the 10 million people that developed TB in 2018, 3.4% of new cases and 18% of previously treated TB cases were estimated to carry a drug-resistant TB strain [1]. Most of the new cases with drug-resistant TB were diagnosed with multi-drug-resistant (MDR)-TB, defined as TB that is resistant to at least isoniazid and rifampicin. Despite all current efforts, the global burden of disease is not falling fast enough to reach the milestones of the ‘WHO End TB Strategy’ [1]. Next to that, the COVID-19 pandemic will further jeopardize TB control [2]. Providing patients with more active and less toxic treatment may help to achieve goals. In addition to the development of new drugs, optimization of current treatment is important to ensure that drugs for drug-susceptible (DS) and drug-resistant TB are given at doses (and achieve exposures) most likely to be effective without causing significant toxicity. The aim of this narrative review is to summarize the knowledge on the pharmacokinetics and pharmacodynamics of the currently available anti-TB drugs and show how population pharmacokinetics coupled with therapeutic drug monitoring (TDM) can be used to optimize treatment for individual patients.
2 Pharmacokinetics and Pharmacodynamics of Anti-TB Drugs
Pharmacokinetics describes the behavior of a drug in the patient’s body, including absorption, distribution, metabolism, and excretion, whereas pharmacodynamics describes the biochemical or pharmacological effect of a drug at the site of action in the patient’s body. Over the past decade, pharmacokinetic (PK) and pharmacodynamic (PD) science has greatly contributed to the understanding of TB treatment response and outcome [3]. In infectious diseases, it is common to link exposure of the drug to the sensitivity of the bacteria, the minimum inhibitory concentration (MIC). In TB, the PK/PD indexes are similar to those for other infectious diseases; maximal plasma concentration (Cmax)/MIC, area under the concentration–time curve of a dosing interval (AUC0–τ)/MIC, and time of the concentration over MIC (T > MIC). Often, the total concentration, i.e., the sum of the unbound and plasma protein bound fraction of the drug, is determined. Only the free (f) drug can diffuse through biological membranes to the site of infection and exert its pharmacological effect [4]. Therefore, especially for highly bound drugs, the above indexes should be converted to fCmax/MIC and fAUC0–τ/MIC. For dose optimization, most studies aim at achieving drug exposure associated with efficacy in 80% of the patients. In clinical practice, MIC determinations and free drug concentrations are not measured because of logistical or financial limitations. In such cases, low exposure of a certain drug is often reported as a risk factor for suboptimal treatment. As there is no formal definition of ‘low concentration’, we have defined low exposure to be < 50% inhibitory concentration, which is the concentration that causes 50% of the maximum kill. This is, in our opinion, more meaningful than expressing it as a percentage of exposure achieved in healthy volunteers because that does not include a measure of response.
For most drugs, specific PK/PD indexes have been linked to their efficacy, reflecting their time- or concentration-dependent activity. However, it is important to note that Cmax and AUC0–τ are closely linked and therefore their PK/PD indexes as well.
Hollow fiber infection models have been very useful to explore PK/PD relationships as it is possible to simulate human concentration–time profiles, and test different dosing strategies [5]. Although hollow fiber infection models have limitations, i.e., a lack of human immune response and a maximum number of drugs that can be tested simultaneously, the results correlate well with treatment response [6], which resulted in endorsement by the European Medicines Agency to make use of these models to guide dose selection for clinical trials [7].
3 Population Pharmacokinetics
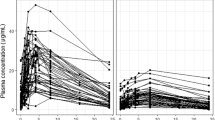
The typical PK profile of a drug and the variability among individuals can be characterized with population PK models. Also known as nonlinear mixed-effect models, this is a type of compartmental model consisting of a structural part describing the typical behavior, and a stochastic part accounting for random variability [8]. The structural part includes the primary PK parameters such as the rate of absorption, volume of distribution, and clearance, as well as a quantitative estimate of the influence of patient characteristics (covariates, e.g., body size, age, genetic polymorphisms) on those parameters. The stochastic part describes the magnitude and distribution of random variability between individuals and within an individual between different dosing occasions. It also contains the unexplained variability, i.e., the residual error. Population PK models are developed using drug concentration measurements gathered after controlled dosing. The development and evaluation of the population PK models are done according to widely accepted standards in the field [9], generally employing software packages such as NONMEM (ICON plc, Gaithersburg, MD, USA) and Monolix (Lixoft, Antony, France). An overview of the set-up and development using population PK models is presented in Fig. 1.

Population pharmacokinetic (PK) modeling. incl. including, MIC minimum inhibitory concentration, PD pharmacodynamic, TDM therapeutic drug monitoring. Created with Biorender.com
4 TDM and Dose Adjustment
Therapeutic drug monitoring is a tool used to integrate PK and PD knowledge to optimize and personalize drug therapy. Therapeutic drug monitoring uses drug plasma concentrations to personalize drug therapy to bring and keep the concentration within the targeted therapeutic range. Below this range, the treatment is less effective, whereas high concentrations may result in toxicity [4]. When selecting a population PK model for TDM purposes, it is important to consider if the population in which the model was developed is representative for the population where TDM is to be conducted.
Therapeutic drug monitoring is mostly performed using blood samples, i.e., serum, plasma, or dried blood spot, while the TB infection is located elsewhere, in the lungs or extra-pulmonary, for instance, in the central nervous system in the case of TB meningitis. Drug penetration at the site of mycobacterial infection may be hampered, if the disease progresses and caseous lesions are formed [10]. Strydom et al. investigated the penetration of nine anti-TB drugs in resected lung tissue and showed that, irrespective of the drug choice, patients with cavitary disease were at risk for subtherapeutic concentrations [10].
Derived or secondary PK parameters commonly used in TDM such as AUC0–τ, Cmax, and trough concentration (Cmin) can be predicted for specific individuals based on a population PK model, relevant dosing, and covariate information plus any observed drug concentrations. Correct handling of inter-occasion variability is important to maximize the precision of the prediction [11].
For most anti-TB drugs, the AUC0–τ (often AUC0–24 h)/MIC is the most predictive PK/PD index. However, a specific AUC0–24 h/MIC target value has not yet been established for all anti-TB drugs. Here, it must be addressed that individual MIC values might not always be reliable and often are not available early in treatment. Moreover, MIC values might be significantly different when using different methods (e.g., solid or liquid media) [5, 12, 13]. This has important implications for the interpretation of AUC0–24 h/MIC target values when taking into account how the MIC was determined in that study.
Obtaining a full PK curve to calculate the AUC0–24 h using non-compartmental analysis techniques is a laborious and expensive procedure and is thus not feasible in clinical practice. A limited sampling strategy (LSS) either based on a population PK model or on multiple linear regression may help to overcome these problems. This method implies that a limited number, usually one to three, of appropriately timed blood samples is needed to adequately predict the AUC0–24 h as a measure for drug exposure [14]. Population PK models are often used in clinical dosing software that aids in estimating PK parameters such as AUC and that uses Bayesian simulations to estimate appropriate drug exposure [15, 16].
Bayesian-informed dosing takes into account the prior information (including patient-specific values of covariates such as body size, age, and genetics) from a population PK model and uses individual measurements, usually drug concentrations, to provide a patient-tailored posterior dosing recommendation. This allows individual patients plasma drug exposures to meet the PK/PD target [17]. The advantages of the Bayesian approach are the flexible timing of samples, as the population PK model can correct for deviations, and that it takes a number of covariates into account leading to a more accurate estimation of AUC0–24 h [18]. The advantage of multiple linear regression-based LSSs is that these do not require modeling software and AUC0–24 h can be easily estimated using only an equation and the measurement of drug concentrations. The disadvantage is that samples should be taken as close as possible to predefined schedules, and the population of interest should be comparable to the population in which the multiple linear regression has been performed because patient characteristics are not included in the equations to estimate drug exposure [18].
5 Anti-TB Drugs
In the section below, we summarize the mechanism of action, PK/PD drivers from preclinical models, human pharmacokinetics (with sources of variability, where known), and clinical PK/PD targets for each drug that is used in the treatment of drug-susceptible or MDR-TB. An overview on relevant PK models is included and recommendations on sampling strategies and dosing strategies are summarized. Table 1 gives an overview of the published population PK models per anti-TB drug. Table 2 indicates PK/PD targets and normal ranges of exposures that have been identified for the different drugs, this is not repeated in the text.
We used a non-systematic literature search in PubMed using a combination of the following keywords: tuberculosis, [drug name], pharmacokinetics, pharmacodynamics, sampling, and concentration. Case reports, abstracts, and posters were excluded. Included articles were limited to the English language. References of included articles were scanned for possible relevant articles. The search was last performed on 4 August, 2020.
5.1 Isoniazid
5.1.1 Preclinical Pharmacokinetics/Pharmacodynamics
Isoniazid is active against Mycobacterium tuberculosis (MTB), inhibiting the mycolic acid synthesis, which affects cell wall synthesis, resulting in a bactericidal effect [19]. Furthermore, isoniazid has an effect on DNA biosynthesis and has it has been shown that early bacterial activity (EBA) on the first 2 days of therapy is related to the acetylator type and 600 mg achieves the highest EBA [19, 20]. Hollow fiber infection models showed that isoniazid has bactericidal activity that is driven by AUC0–24 h/MIC [21, 22]. Studies in the mouse confirm this bactericidal activity, but isoniazid appears to be a weak sterilizing drug [23]. In the mouse, the activity of isoniazid appeared to be more concentration than time dependent, with Cmax/MIC and AUC0–24 h/MIC having a strong association with activity [24].
5.1.2 Clinical Pharmacokinetics/Pharmacodynamics
Isoniazid is fairly hydrophilic, with moderate clinical plasma concentrations 2 h after ingestion (C2) of 3–6 mg/L after a 300-mg dose, and 9–15 mg/L after a 900-mg dose [25, 26]. Isoniazid is unable to penetrate and accumulate in caseous granulomas [10]. In healthy volunteers and patients with TB starting on therapy, the administration of isoniazid with a high-fat meal modestly reduced Cmax and bioavailability [27, 28]. Both orange juice and aluminium–magnesium antacid produced little effect on the bioavailability of isoniazid [27]. Isoniazid metabolism is genetically determined based on N-acetyl transferase 2 activity [25, 27]. It has been proposed that patients be split between having none, one or two rapid N-acetyl transferase 2 alleles and that dosing could be then adapted to 2.5 (slow acetylators), 5.0, and 7.5 mg/kg (fast acetylators), respectively [29]. Pharmacokinetic data for isoniazid in adults and children with TB suggested that low and variable concentrations are common [30, 31]. In 142 drug-susceptible patients with TB, Pasipanodya et al. showed that to achieve a favorable outcome, in order of importance, AUC0–24 h of pyrazinamide, rifampicin, and isoniazid should be over 363, 13, and 52 mg h/L respectively. Low Cmax of rifampicin (< 6.6 mg/L) and isoniazid (< 8.8 mg/L) preceded all (three) cases of acquired drug resistance [31].
5.1.3 Population Pharmacokinetics, TDM, and Dosing
Standardized doses may not be adequate for isoniazid, especially given the differences between fast and slow acetylators. Therapeutic drug monitoring could be used to rapidly find the correct dose for each patient [32].
Twelve population PK models were found from the time period of 1997–2019 (Table 1). Most of the models were built with NONMEM and included the acetylation status/N-acetyl transferase 2 genotype as a covariate on clearance.
Limited sampling strategies were developed for all four first-line TB drugs and moxifloxacin in an ethnically heterogenous population, using intensive PK sampling after the intake of drugs on an empty stomach and after more than 2 weeks of therapy [33]. For isoniazid, LSSs of 1, 2.5, and 6 h post-dose performed best [33]. Recently, several LSSs were proposed for isoniazid alone or in combination with other first-line drugs. Limited sampling strategies were derived from a study with intense sampling of 20 patients conducted in Indonesia, who started on TB medication [34]. Both studies used the best subset selection, multiple linear regression to calculate LSS and the chosen LSS performed best at estimating the AUC0–24 h.
5.2 Rifampicin
5.2.1 Preclinical Pharmacokinetics/Pharmacodynamics
Rifampicin binds to and inhibits the action of the DNA-dependent RNA polymerase of mycobacteria. It has both bactericidal and sterilizing activity. In the past decades, it was assumed that the efficacy of rifampicin is associated with Cmax/MIC, but recent preclinical studies have established that the microbial killing can also be explained by AUC/MIC [35, 36]. Given the short half-life of rifampin of around 2 h at steady state, there is a high correlation between Cmax and AUC. Moreover, in one of these studies, it was well presented that in a mouse model with dose fractionation and ranging, the PK/PD parameter that correlated best with a decrease of bacterial counts was AUC/MIC [36].
5.2.2 Clinical Pharmacokinetics/Pharmacodynamics
Rifampicin is preferably taken on an empty stomach, as intake with food reduces Cmax and causes a modest decrease in AUC0–24 h [28, 37]. However, in the case of gastrointestinal adverse effects, concomitant intake with a light meal is recommended to prevent or alleviate these effects. The distribution of rifampicin is fast and includes all fluids and organs, but only about 5% of rifampicin reaches the cerebrospinal fluid [38].
Repeated daily administration of rifampicin results in a decrease in exposure and half-life of the drug, owing to an increased rate of drug clearance caused by auto-induction [39]. This is separate from induction of the cytochrome P450 system by rifampicin (see below). Increasing the dose of rifampicin results in a more than proportional increase of exposure in plasma (non-linear pharmacokinetics). Rifampicin is excreted mainly through the bile. Urinary excretion is about 10–15% of biliary excretion but increases with the dose.
Rifampicin is a potent inducer of several phase I and II metabolic enzymes and drug transporter proteins, often resulting in a decrease in exposure of concomitantly used drugs [40]. This is especially true for cytochrome P450 3A4, a major drug-metabolizing pathway. Significant induction after starting rifampicin is reached after a few doses and is complete after a week [40]. Baseline enzyme activity after discontinuing rifampicin is attained in about 2 weeks.
The drug is currently used in a dose of 10 mg/kg daily, but this is at the lower end of the dose–response curve. A dose of 35 mg/kg daily, resulting in a ten-fold higher AUC0–24 h value in plasma, is safe and tolerable for 3 months [41, 42]. Moreover, higher dosing has shown a significant relationship between exposure to rifampicin and EBA [43]. Higher rifampicin exposures also reduce the time to culture conversion in pulmonary TB [44]. A meta-analysis on the exposure of rifampicin reported that at least 25-mg/kg dosing is required to achieve PK/PD targets [45].
5.2.3 Population Pharmacokinetics, TDM, and Dosing
Nineteen population PK models for rifampicin in adult humans with fully re-implementable parameter values could be identified in the time period from 1997 up to 2018 (Table 1). These models differed with respect to the number of compartments, the inclusion of components for non-linearity and for auto-induction, which are both typical to rifampicin. Only one model was based on higher doses of rifampicin up to 40 mg/kg [46]. Most of the models were built in NONMEM.
Limited sampling strategies were developed using the best subset selection, multiple linear regression for all four first-line TB drugs and moxifloxacin in an ethnically heterogenous population, using intensive PK sampling after the intake of drugs on an empty stomach and after more than 2 weeks of therapy [33]. Limited sampling at various fixed sampling points up to 6 h enabled an accurate and precise prediction of AUC0–24 h of all drugs separately and simultaneously. The limited sampling formula called for 2-, 4-, and 6-h samples [33]. Another study also applied a linear regression approach, but focused on the prediction of the AUC0–24 h of rifampicin and other TB drugs during the first 3 days of treatment [34]. In the homogenous Indonesian population studied, limited sampling at 2, 4, and 8 h post-dose performed best to predict AUC0–24 h values of all first-line TB drugs.
Sturkenboom et al. were the first to use a model-based (maximum a posteriori Bayesian fitting) approach to derive a LSS for rifampicin [14]. They extended the population used by Magis-Escurra et al. [33], which took rifampicin on an empty stomach, to patients who ingested the drug with a light meal and derived a population PK model in MWPharm (Mediware, Zuidhorn, the Netherlands). This study showed that rifampin AUC0–24 h could be predicted with acceptable bias and precision with optimal sampling at time points 1, 3, and 8 h post-dose. Furthermore, a sampling strategy using blood collection at convenient 2- and 4-h sampling times has been shown in another study to be the most suitable [47]. This NONMEM model was successfully implemented in the InsightRX (San Francisco, CA, USA) precision dosing platform.
5.3 Pyrazinamide
5.3.1 Preclinical Pharmacokinetics/Pharmacodynamics
Pyrazinamide is a prodrug, which is activated to pyrazinoic acid inside the MTB by the bacterial pyrazinamidase [48]. It is mainly active against non-replicating bacilli, especially at low pH [49], which allows for accumulation of pyrazinoic acid inside the MTB [50]. In pre-clinical hollow fiber infection models, the sterilizing effect of pyrazinamide was most closely related to the AUC0–24 h/MIC ratio [51]. The MIC determination is challenging, owing to the difficulty of reproducibly growing MTB at low pH across laboratories.
5.3.2 Clinical Pharmacokinetics/Pharmacodynamics
Pyrazinamide is used both against drug-sensitive [52] and resistant TB [53] and it is often dosed 20–30 mg/kg bodyweight. It should be noted that the original British Medical Research Council studies used fixed dosing of 1500 mg for patients less than 46 kg, and 2000 mg for larger patients, with an average dose of 35 mg/kg. A large number of PK studies have been published on pyrazinamide at the 20–30 mg/kg dose: its pharmacokinetics is described by a one-compartment open model [54], it reaches Cmax approximately 2 h post-dose and with a terminal half-life of approximately 9 h it only moderately accumulates, reaching steady state in 2–3 days. It does not significantly bind to plasma proteins [55] and it is metabolized by liver deaminase [56]. A recent review summarizing exposures in patients (human immunodeficiency virus positive/negative) reported median AUC0–24 h in the range of 250–450 mg h/L and Cmax in the range of 25–55 mg/L [57]. Several reports indicated that the current weight-banded dosing approach achieves lower concentrations in patients with lower weight [58,59,60,61].
In clinical studies in patients with drug-susceptible TB, worse outcomes were observed in patients with Cmax < 35 mg/L, while a positive association between AUC0–24h > 363 mg h/L and long-term TB treatment outcome has been reported [31, 62]. The main adverse reactions to pyrazinamide are arthralgia and hepatotoxicity [63]. Doses higher than the current doses have previously been used, and there is a debate regarding increasing toxicity with increasing doses [64].
5.3.3 Population Pharmacokinetics, TDM, and Dosing
Nine population PK models were found, from which three were made with NONMEM, three with Monolix, one with ADAPT5, and one with NPEM2 (Table 1). Only one model included a covariate sex on clearance [65]. Although a robust target for efficacy has not been established and safety is not of concern at the current levels, a TDM strategy could be useful to identify patients with low exposure using, for example, LSSs of 0, 2, and 6 h or 2, 4, and 8 h post-dose [33, 34].
5.4 Ethambutol
5.4.1 Preclinical Pharmacokinetics/Pharmacodynamics
The precise mode of action of ethambutol is not fully understood, but it appears to inhibit mycobacterial cell wall arabino-transferases, leading to depletion of arabinogalactan and lipoarabinomannan. In an early study by Dickinson et al. in guinea pigs, the effects of ethambutol were concentration dependent (Cmax/MIC) [66]. More recent experiments in the hollow fiber infection model showed that the microbial kill to ethambutol was linked to AUC0–24 h/MIC, although in some experiments Cmax/MIC could also have explained the kill, whereas T > MIC was relevant for suppressing resistance [67].
5.4.2 Clinical Pharmacokinetics/Pharmacodynamics
It often is used in a dose of 15–20 mg/kg daily, with a maximum of 25 mg/kg daily. Higher doses and exposures, especially in patients with decreased renal function, are associated with a higher incidence of retrobulbar (optic) neuritis [68]. The EBA of the 15–20 mg/kg dose is negligible and the drug has very little sterilizing activity. However, ethambutol is added to other first-line TB drugs during the first weeks of treatment to prevent rifampin resistance in cases where there may be unrecognized isoniazid resistance. A clinical trial in Indian patients with TB showed that microbiological response was AUC/MIC driven [69, 70].
Ethambutol can be taken with or without food, as food modestly reduces Cmax but does not affect the AUC0–24 h of the drug [28, 37]. It has rapid good distribution throughout the body (including in large lung lesions), but it only penetrates the cerebrospinal fluid in the presence of inflammation, probably having a minor contribution to the treatment of TB meningitis [71]. A recent evaluation using ultrafiltration showed an average protein binding of 12% [55]. Ethambutol is largely excreted unchanged by the kidneys. Patients with a creatinine clearance less than 30 mL/min are at risk for accumulation of the drug and resulting retrobulbar neuritis. They should receive a longer interval between doses with three times a week administration of the same 15–20 mg/kg dose. In persons with good renal function, ethambutol shows a biphasic decline in plasma concentration with a terminal half-life of about 10–12 h. The drug shows no relevant drug interactions, apart from a reduction in its Cmax caused by aluminum-containing antacids [72].
5.4.3 Population Pharmacokinetics, TDM, and Dosing
Traditionally, limited sampling of ethambutol aimed at ‘catching’ and estimating the Cmax of the drug in an individual by sampling at 2 and 6 h after dosing. Recently developed LSSs aim to predict the AUC0–24 h for ethambutol as well. As described earlier, Magis-Escurra et al. developed an LSS using best subset selection, multiple linear regression for all first-line TB drugs (including ethambutol) separately and simultaneously [33]. Saktiawati et al. also applied a linear regression approach, but focused on prediction of the AUC0–24 h of first-line TB drugs during the first 3 days of treatment, based on data from Indonesian patients with TB [34]. Horita et al. were the only investigators to use a Bayesian approach to estimate AUC0–24 h of ethambutol in children [30].
Ten population PK models for ethambutol could be identified (Table 1) [30, 72,73,74,75,76,77,78,79]. These models differ with respect to the number of compartments, the inclusion of a lag time, or transit compartment absorption. Most of the models were built in NONMEM. It is important to note that a decreased renal function may be the most relevant indication for TDM of ethambutol, but relatively few patients with renal dysfunction were included in the various cohorts. This means that the effect of this covariate on clearance has not been adequately evaluated in population PK models. Similarly, only one model was derived in obese patients taking ethambutol [75].
5.5 Levofloxacin/Moxifloxacin
5.5.1 Preclinical Pharmacokinetics/Pharmacodynamics
Fluoroquinolones are amongst the most potent drugs against MTB. They produce their rapid killing effect by inhibiting the bacterial DNA replication and transcription through affecting the type II topoisomerases [80].
The efficacy of fluoroquinolones is correlated with the ratio of the fAUC0–24 h/MIC [81,82,83]. In addition, an 800-mg daily dose for moxifloxacin is suggested to achieve maximal kill and suppress resistance [84, 85].
5.5.2 Population Pharmacokinetics, TDM, and Dosing
Therapeutic drug monitoring of moxifloxacin and levofloxacin is proposed because of high PK variability and a substantial proportion of patients showing low drug exposure [86,87,88]. Fluoroquinolones are well tolerated and adverse effects do not require TDM (e.g., QT interval prolongation by moxifloxacin). Therapeutic drug monitoring is helpful to ensure adequate exposure after dose adjustments of levofloxacin due to kidney failure or in the case of drug–drug interactions. For instance, moxifloxacin exposure is approximately 25% decreased if concomitantly used with rifampicin owing to an increase of moxifloxacin clearance [18]. Four population PK models were identified for levofloxacin and six for moxifloxacin (Table 1). The models were made with NONMEM, MWPharm, NPEM2, and Monolix and the majority were one-compartment models. To reduce the burden and costs of TDM, LSSs can be applied. The under the concentration–time curve from time 0 to 24 h can be adequately estimated using a population PK model and two optimally timed samples for levofloxacin (0 and 5 h) and for moxifloxacin (0 and 6 h, either with or without rifampicin) [18, 89].
5.6 Bedaquiline
5.6.1 Preclinical Pharmacokinetics/Pharmacodynamics
Bedaquiline is a diarylquinoline that targets mycobacterial ATP synthase, disrupting the bacteria’s energy metabolism [90]. Dose-fractionation experiments in mice identified AUC/MIC as the main driver of bactericidal effect [91].
5.6.2 Clinical Pharmacokinetics/Pharmacodynamics
The standard dosing regimen in the treatment of MDR-TB consists of a 2-week loading phase with 400 mg daily followed by 22 weeks with 200 mg dosed three times per week. An alternative dosing regimen with 200 mg daily during the first 8 weeks, thereafter 100 mg daily, has been utilized in recent clinical trials [92]. Uptake of bedaquiline is increased when given together with food and maximal concentrations are expected 4–6 h after dosing. The binding to plasma proteins is very high, more than 99.9%, and the distribution to tissues is extensive, resulting in an extremely long terminal half-life [93]. One case report showed undetectably low bedaquiline concentrations in cerebrospinal fluid [94].
Bedaquiline is mainly hepatically cleared. Cytochrome P450 3A4 (primarily) transforms bedaquiline into the N-monodesmethyl metabolite M2, which in turn is metabolized in the same way to M3 [93, 95].
A model-based PK/PD analysis of phase II data found a weekly average concentration (analogous to weekly AUC) to be the best driver and estimated that a concentration of 1.42 mg/L would give half of the maximal possible effect [96]. The relationship has also been confirmed in a separate study [97]. With the standard regimen typical (mean ± one standard deviation) maximal, average, and trough concentrations at day 14 are 1.6–3.2 mg/L, 0.96–2.1 mg/L, and 0.44–1.4 mg/L, respectively [98]. At the end of the continuation phase, the corresponding values are 0.9–2.1 mg/L, 0.41–1.2 mg/L, and 0.26–0.91 mg/L [98].
5.6.3 Pharmacokinetics, TDM, and Dosing
There are two published population PK models of bedaquiline in patients with drug-resistant pulmonary TB, partly based on the same data and both developed in NONMEM (Table 1). The first only describes bedaquiline [99], while the second also incorporates the main metabolite M2 [98]. There are no published evaluations of TDM sampling strategies for bedaquiline. The time on treatment needs to be considered, accounting for the loading phase and the accumulation. Assessing the Cmin at the end of the loading phase is a practically feasible option. A retrospective study described potential risk factors of low and high bedaquiline concentrations, where administration of food, drug–drug interactions, gastrointestinal complaints, lower body weight, and age > 70 years were among the named factors [100]. In addition, black race has been connected to approximately 50% clearance [99]. Therapeutic drug monitoring could be of benefit for specific patients using bedaquiline; however, standardized drug susceptibility testing must first be developed [101]. The M2 metabolite is the main driver of side effects such as QT prolongation and phospholipidosis [102].
5.7 Linezolid
5.7.1 Preclinical Pharmacokinetics/Pharmacodynamics
Linezolid exerts its antibacterial effect by binding 70S ribosomal components and disrupting the initiation of protein synthesis. A hollow fiber infection model study has indicated optimal mycobacterial kill to be associated with fAUC0–24 h/MIC [103]. In murine models, its sterilizing activity is dose related and can occur within 2 months of effective combination therapy, leading some investigators to consider short high-dose therapy followed by discontinuation or intermittent dosing to retain efficacy whilst reducing toxicity [104, 105].
5.7.2 Clinical Pharmacokinetics/Pharmacodynamics
The bioavailability after oral administration of linezolid is nearly 100%, and it shows good tissue penetration into tuberculous lung cavities caseating granulomas and cerebrospinal fluid [10, 106]. The standard adult dose (600 mg twice daily) is associated with myelosuppression, lactic acidosis, and neurotoxicity during long-term use. Consequently, a dose of 600 mg, or occasionally 300 mg, once daily is often recommended for patients with TB [104, 107].
The PK/PD target for myelotoxicity is suggested as a Cmin of 2 mg/L [108]. While meta-analyses and simulations from some of these data indicate high attainment of putative efficacy targets at 600 mg once or twice daily, Cmin regularly exceeds the toxicity threshold with twice-daily dosing [109, 110]. One study reported that higher plasma linezolid exposure and greater toxicity risk are associated with advancing age and lower weight [111]. It has been reported that 300 mg twice daily might be effective [104]. Studies looking into 300-mg daily dosing have shown promising results in some patients [112, 113]; however, until larger trials show results, 600 mg has been suggested to keep the balance between efficacy and toxicity [105]. No association between human immunodeficiency virus or concurrent anti-retroviral therapy and linezolid exposure has been described [111]. An approach of short-duration higher dose linezolid to maximize efficacy whilst avoiding toxicities associated with cumulative dose requires further evaluation; however, an interim analysis of the Nix-TB trial showed a significant amount of patients experiencing linezolid toxicity [114].
5.7.3 Population Pharmacokinetics, TDM, and Dosing
The narrow therapeutic window has prompted suggestion that TDM might be beneficial. An LSS consisting of 0 and 2 h has been described to predict the plasma AUC0–24 h with acceptable bias and precision [115]. Three population PK linezolid models were identified (Table 1), which were all one-compartment models and two were developed with MWPharm and one with NONMEM.
5.8 Clofazimine
5.8.1 Preclinical Pharmacokinetics/Pharmacodynamics
Studies in the mouse model show that clofazimine did not have EBA, but subsequently demonstrated potent, dose- and concentration-independent bactericidal activity [116]. The authors suggested that much lower doses could be used effectively for TB and that clofazimine exhibits slow elimination [116]. In another mouse model, the activity of clofazimine was confirmed to be best when administered with other first-line anti-TB drugs [117].
5.8.2 Clinical Pharmacokinetics/Pharmacodynamics
Most clinical studies have used 100-mg daily doses, thus the literature lacks human dose-ranging data for TB. Very limited PK/PD data are available for clofazimine, with most only available as meeting abstracts.
Clofazimine is highly lipophilic, leading to high accumulation in fat tissues and relatively low serum concentrations (0.7–1.0 mg/L) [118]. Clofazimine is unable to penetrate and accumulate in caseous granulomas, but it does accumulate in the highly cellular peripheral zone of the granuloma, consisting of macrophages, epithelioid macrophages, and lymphocytes [118]. In healthy volunteers, the administration of clofazimine with a high-fat meal provided the greatest bioavailability. However, bioavailability was associated with high inter- and intra-subject variability. Both orange juice and an aluminum-magnesium antacid reduced a mean bioavailability of clofazimine [119].
5.8.3 Population Pharmacokinetics, TDM, and Dosing
Overall, three NONMEM population PK models were identified (Table 1). A recent population PK model on NONMEM described clofazimine disposition being strongly affected by body fat, thus clofazimine showed lower plasma concentrations in women [120]. Additionally, it was suggested that patients with extreme body composition might need to have individualized dosing and a high-fat content might require longer loading periods [120]. As mentioned, specific PK/PD targets for clofazimine are scarce. Clofazimine is suggested to be sampled 2–3 h after the dose and 6 h to assess delayed absorption, whereas Cmax is expected at 0.5–2.0 mg/L [26, 121, 122].
5.9 Cycloserine/Terizidone
5.9.1 Preclinical Pharmacokinetics/Pharmacodynamics
Cycloserine is an amino-acid derivative, which suppresses the growth of MTB by inhibiting the enzymes that produce peptidoglycan causing changes in the cell wall [123]. Terizodone contains two cycloserine molecules. Hollow fiber infection models have proposed that T > MIC predicts efficacy best [124,125,126].
5.9.2 Clinical Pharmacokinetics/Pharmacodynamics
Cycloserine has been suggested to be used in patients with rifampicin-resistant TB as a Group B drug. It may also be of value in the therapy of TB meningitis owing to its penetration into cerebrospinal fluid [126].
As cycloserine has a long half-life, different Cmax ranges have been reported. To determine exposure, the first measurement should be done after 3–4 days of therapy and Cmax is expected to be around 20–35 mg/L [26, 127, 128].
5.9.3 Population Pharmacokinetics, TDM, and Dosing
Two population PK models have used the preclinical established PK/PK index T > MIC over 30% model [125, 129]. Five cycloserine/terizidone population PK models were identified and all of these were one-compartment models made with different software, Monolix, NONMEM, and MWPharm (Table 1). Recently, a sampling strategy using blood collection at a convenient 4 h has been proposed in combination with a population PK model to estimate AUC0–24h obtaining a low relative error [125].
5.10 Delamanid
5.10.1 Preclinical
Delamanid is a nitroimidazole antibiotic with mechanisms of action including inhibition of mycolic acid synthesis and generation of nitrous oxide in anaerobic conditions [130]. In animal models, the activity of delamanid activity appears to be time dependent [130].
5.10.2 Clinical Pharmacokinetics/Pharmacodynamics
The licensed dose is 100 mg twice daily, for 24 weeks, but an alternative dosing regimen of 100 mg twice daily for 8 weeks followed by 200 mg once daily for 16 weeks was used in the recent phase III trial [131]. The main route of metabolism of delamanid is purported to be via albumin, with cytochrome P450 3A playing a minor role [132]. It has several metabolites, but DM-6705 circulates at the highest concentrations. Delamanid and DM-6705 have half-lives of 30–38 h and 121–425 h, respectively, and the latter is linked to the key, albeit modest, adverse effect of delamanid—prolongation of the cardiac QT interval [133]. The bioavailability of delamanid is enhanced two- to four-fold by food, exposures are less than dose proportional, the drug is highly protein bound (> 99.5%), and the Tmax is at 4 h [134, 135]. This drug appears to reach high concentrations in the brain; its utility in central nervous system TB is being explored [136]. Delamanid is not an inducer or inhibitor of cytochrome P450 enzymes, and drug–drug interaction liability is low. In human EBA studies, doses of 200–300 mg total daily dose produced higher microbiologic activity than 100 mg per day [134]; in a phase II trial, there was a trend towards better activity of delamanid 200 mg twice daily compared with 100 mg twice daily on solid medium [137].
5.10.3 Population Pharmacokinetics, TDM, and Dosing
Median delamanid AUC0–24 h after 56 days of dosing is 7.9 mg h/L (coefficient of variation 37.5%). Maximum plasma concentration values are 0.4 mg/L (coefficient of variation 40.5%) at 56 days [132, 136]. There are no clear target concentrations owing to a lack of clinical PK/PD data. It has been reported that delamanid acquires resistance quickly, thus optimization of therapy using TDM may help in reducing the acquired resistance [138].
5.11 Pretomanid
5.11.1 Preclinical
Pretomanid was introduced with the BPal regimen (bedaquiline and pretomanid with linezolid). An in vivo model presented activity comparable to isoniazid, where the suggested reasoning was lower bioavailability in necrotic lung granulomas [139]. In another mouse model, the addition of pretomanid showed bactericidal activity increase while preventing emergence of resistance to bedaquiline and duration of therapy [140]. Later, a synergistic three-drug combination, including pretomanid, was suggested in a time kill assay combined with population modeling [141].
5.11.2 Clinical Pharmacokinetics/Pharmacodynamics
Pretomanid has shown to have higher bioavailability when taken with food [142]. In healthy adults it has been shown that AUC and Cmax could be almost doubled [142]. Moreover, drug interactions have been described with rifampicin, lopinavir/ritonavir, and efavirenz—all of these reduce the AUC of pretomanid [143].
5.11.3 Population Pharmacokinetics, TDM, and Dosing
Two population PK models were identified, one in NONMEM and the other developed on GNU MCSim modeling and simulation suite (Table 1).
5.12 Meropenem/Imipenem-Cilastatin/Ertapenem
5.12.1 Preclinical Pharmacokinetics/Pharmacodynamics
Meropenem, a carbapenem, is a member of the β-lactam class of antibiotics. It inhibits cell wall synthesis through inhibition of classical D, D-transpeptidases, and non-classical L, D transpeptidases [144]. Clavulanate (not available except co-formulated with amoxicillin) irreversibly inhibits MTB β-lactamase, potentiating the activity of meropenem. Ertapenem has shown to a have sterilizing effect against M. tuberculosis in a hollow fiber infection model; however, falsely high MIC values might have been reported because of ertapenem degradation in acidic conditions, which could have led to underuse of ertapenem in TB therapy [145, 146].
5.12.2 Clinical Pharmacokinetics/Pharmacodynamics
In an EBA study, meropenem given at a dose of 2 g three times daily intravenously (supplemented with twice-daily amoxicillin/clavulanate) produced measurable activity in patients with pulmonary TB [147]. Recently presented EBA dose-finding trial results suggest that there is a significant drop in microbiologic activity when the meropenem dose is reduced from 6 g per day (given as 2 g three times daily) to 3 g per day (with 1 g three times daily performing modestly better than 3 g once daily). Meropenem, together with amoxicillin-clavulanate, is used off-label for TB. Doses for other bacterial infections range from 1.5 to 6 g per day, given in divided doses to maximize the activity of this time-dependent drug. Most (around 70%) of an intravenous meropenem dose is excreted unchanged in the urine [148]. Protein binding is very low (2%). Ertapenem in a dose of 1 g once daily intramuscularly in combination with amoxicillin-clavulanic acid showed no EBA [149]. The lack of activity of this dose is in line with an earlier published study using the hollow fiber infection model indicating that a dose of at least 2 g once daily was required [146].
5.12.3 Population Pharmacokinetics, TDM, and Dosing
Mean meropenem AUC0–24 h given at 2 g three times daily is 573 mg h/L, with a Cmax of 133 mg/L; at that dose, with an MIC of 1 mg/L, T > MIC is appropriately 75% [134]. In the absence of a PK target that maximizes efficacy against slow-growing MTB, it seems reasonable to strive for these average PK values. Collecting samples for the PK pre-dose and mid-dosing interval is most informative. While carbapenems are generally well tolerated, there is an increased risk of neurotoxicity with Cmin > 16 mg/L (and the dose-limiting toxicity is chiefly diarrhea from the amoxicillin–clavulanate). Therapeutic drug monitoring of meropenem in TB could thus serve to help with individualized dosing of meropenem, achieving exposures similar to those seen with maximal EBA activity and avoiding toxic Cmin.
Ertapenem in a 2000-mg once-daily pharmacokinetic exploratory study showed that f40% of T > MIC was reached for most patients [150]. A PK model based on data from patients with MDR-TB proposed a limited sampling strategy of 1 h and 5 h [16].
5.13 Amikacin/Streptomycin
5.13.1 Preclinical Pharmacokinetics/Pharmacodynamics
In a hollow fiber infection model for TB, the local amikacin Cmax/MIC ratio of 10 was determined to be the effective PK/PD index, closely followed by AUC/MIC [151, 152].
5.13.2 Clinical Pharmacokinetics/Pharmacodynamics
Aminoglycoside use is limited by both the parenteral administration and significant toxicity that is observed after prolonged use [153] and should only be used if drug susceptibility testing results confirm susceptibility and audiometry monitoring can be ensured [53]. A 10% probability of ototoxicity occurred with a threshold cumulative AUC > 87 g h/L [154]. It has been revealed that the probability of ototoxicity increased sharply starting after 6 months of amikacin therapy to near maximum at 9 months. Considering the poor penetration of aminoglycosides in lung tissue, this translates into a serum Cmax/MIC ratio of 70–90 or an AUC0–24 h/MIC ratio of 103 [151].
5.13.3 Population Pharmacokinetics, TDM, and Dosing
Therapeutic drug monitoring of aminoglycosides is indicated to ensure efficacy and should be combined with determination of MIC as the risk of ototoxicity can be reduced if the MIC is low enough to lower the (cumulative) dose and exposure [155]. A one-compartment population PK model has been reported for amikacin. This included a LSS of 1 h and 4 h suggested for predicting AUC0–24 h [156].
5.14 Ethionamide/Prothionamide
5.14.1 Preclinical Pharmacokinetics/Pharmacodynamics
The thiomides ethionamide and its propyl-analog prothionamide are drugs with bactericidal activity against MTB [157]. They are considered interchangeable within anti-tubercular regimens [158]. In hollow fiber systems, the target exposure was identified as AUC0–24 h/MIC [159].
5.14.2 Clinical Pharmacokinetics/Pharmacodynamics
It is currently used for MDR-TB at 15–20 mg/kg daily (maximum 1000 mg) [53] and for drug-susceptible TB in children or TB meningitis. The plasma protein binding of ethionamide is approximately 30% [160] and it is metabolized by hepatic monooxygenases [161].
5.14.3 Population Pharmacokinetics, TDM, and Dosing
Three population PK ethionamide models were identified, from which all were one-compartment models using NONMEM and Pmetrics software (Table 1). Clinically, after administration of 250–500 mg of ethionamide, a Cmax in the range of 1–5 mg/L has been suggested [26]. A TDM strategy could identify patients with low concentrations by evaluating two samples at 2 h and 6 h with a Cmax of 1–5 mg/L [26, 162, 163].
5.15 P-Aminosalicylic Acid
5.15.1 Preclinical Pharmacokinetics/Pharmacodynamics
The mechanism of action of P-amino salicylic acid (PAS) remains unknown but it is assumed that it interferes with bacterial folate synthesis [164].
5.15.2 Clinical Pharmacokinetics/Pharmacodynamics
Although one study described a bactericidal effect of PAS at high (20 g) once-daily dosing [165], it is generally regarded as a bacteriostatic agent. It is associated with considerable gastrointestinal intolerance and was quickly replaced by isoniazid, rifampicin, and pyrazinamide in first-line anti-TB therapy. It has recently been downgraded in the World Health Organization MDR-TB guidelines but retains a role as component of treatment for patients with extensively drug resistant TB (XDR-TB) and limited other options [53]. Current PAS dosing for adults is 150 mg/kg/day in two to four divided oral doses (usually 8–12 g/day), and 200–300 mg/kg/day in two to four divided doses for children. Absorption is improved by consumption with acidic food or yoghurt [166]. It attains its maximum serum concentration within about 2 h, and has a fairly short half-life [165]. Protein binding is between 50 and 60% [167]. Current granular formulations are designed to provide slow release, assuming a fCmin > 1 mg/L will be maintained [167]. Some authors have reported that a high Cmax may be more important for prevention of resistance to partner drugs [168].
5.15.3 Population Pharmacokinetics, TDM, and Dosing
Three NONMEM population PK models were identified, which were all one-compartment models (Table 1). A role for TDM in PAS dosing has not yet been explored.
6 Concluding Remarks
In this review, over 80 different population PK models for 14 anti-TB drugs are presented.
As different models are difficult to compare and because the true model cannot be identified, there is no such thing as the ‘best’ or most recommended model. This was already stated in 1976, in one of the most famous adages in pharmacokinetics: ‘all models are wrong, some are useful’ [169]. Our personal preference would be using a validated model and performing TDM with Bayesian estimation to most accurately estimate AUC for the individual patient and situation. This can be done using different modeling and Bayesian simulation software. A good overview of different available methods has recently been presented [170].
By including TDM as an opportunity for optimizing TB treatment in TB guidelines [53, 171, 172], the first step towards implementation of precision medicine has been made. The guidelines include indications and clinical conditions for which TDM may be useful. To be of practical use, guidance is required on how drug exposure should be evaluated and how dosages could be adjusted [173]. We therefore performed this review evaluating the available literature on pharmacokinetics and PK modeling of anti-TB drugs. For the first-line anti-TB drugs, it is clear that studies focussed mainly on rifampicin and isoniazid [25, 174,175,176]. Pharmacokinetic variability is substantial and can be explained by pharmacogenetic differences and other factors such as co-morbidities [45, 177, 178]. With respect to dose optimization, there is a clear rationale based on in vitro PK/PD and clinical studies. Unfortunately, a well-designed study comparing TDM with standard of care is lacking. Although higher dosages are currently being trailed for rifampicin, TDM will likely still have a role as PK variability is still high, which leads to the conclusion that a high dose does not automatically result in a high exposure in every patient.
For anti-TB drugs used for the treatment of M/XDR-TB, there is a significant knowledge gap for many of the drugs. Only for the fluoroquinolones, linezolid, and aminoglycosides does a substantial body of evidence support TDM. The new drugs such as bedaquiline, delamanid, and pretomanid lack this abundance of data but publications on acquired resistance, drug–drug interactions, and significant variability of bioavailability based on concomitant food intake already provide a rationale that TDM may be of use for the newer drugs as well. However, it needs to be mentioned again that a well-designed prospective study comparing TDM in M/XDR-TB is still lacking. As researchers, we ourselves often struggle with conducting prospective studies to show the value of TDM. Recently, a review by Märtson et al. identified knowledge gaps and provided guidance with an appropriate checklist and data elements that are suggested to take into account when designing and conducting high-quality TDM studies in the field of infectious diseases [179]. Briefly, the focus was on estimating drug exposure and assessing the susceptibility of the pathogen, but also suggestions on how to select study endpoints and clinical trial design were provided [179].
Logistical issues regarding TDM samples seem to be a hurdle that will be overcome in the next couple of years owing to the introduction of dried blood spot monitoring, point of care saliva tests, and urine testing [173, 180]. To support a physician in making informed dosing decisions in individual patients, easy-to-use model-informed precision dosing software has to be made freely accessible via mobile platforms such as smartphones and tablets. On-site patient covariates and drug concentrations can be used to simulate dosing regimens to maximize the likelihood to attain the therapeutic target. Such software is currently under development for drugs such as vancomycin but there is a clear need to expand the panel with anti-TB drugs.
As TB treatment duration is long, there is an urgent need to find biomarkers that predict clinical outcome. A combination of dose optimization based on measured drug concentration and pathogen susceptibility with a sensitive and responsive biomarker would be helpful to optimize treatment.
Since the introduction of TDM for TB over 3 decades ago, further development of TDM in TB next steps will again depend on academic and clinical initiatives supported by funding institutions such as EDCTP or the Bill & Melinda Gates Foundation, as the pharmaceutical industry is not interested in personalized prescribing for the novel compounds and the generic pharmaceutical industry has no role in drug development or treatment optimization. We recommend that researchers work closely together with guideline issuing bodies like the World Health Organization to ensure the evidence generated will be reviewed during guideline updates. With such an approach, TDM will become available within the next 5 years and can help to contribute to END TB.
References
World Health Organization (WHO). Global tuberculosis report 2019. Geneva: WHO; 2019.
Migliori GB, Thong PM, Akkerman O, et al. Worldwide effects of coronavirus disease pandemic on tuberculosis services, January-April 2020. Emerg Infect Dis. 2020;26:2709–12.
Gumbo T, Alffenaar J-WC. Pharmacokinetic/pharmacodynamic background and methods and scientific evidence base for dosing of second-line tuberculosis drugs. Clin Infect Dis. 2018;67:S267–73.
Veringa A, Sturkenboom MG, Dekkers BG, et al. LC-MS/MS for therapeutic drug monitoring of anti-infective drugs. Trends Anal Chem. 2016;84:34–40.
Alffenaar J-WC, Gumbo T, Dooley KE, et al. Integrating pharmacokinetics and pharmacodynamics in operational research to end tuberculosis. Clin Infect Dis. 2020;70:1774–80.
Gumbo T, Pasipanodya JG, Romero K, et al. Forecasting accuracy of the hollow fiber model of tuberculosis for clinical therapeutic outcomes. Clin Infect Dis. 2015;61(Suppl. 1):S25-31.
Cavaleri M, Manolis E. Hollow fiber system model for tuberculosis: the European Medicines Agency experience. Clin Infect Dis. 2015;61(Suppl. 1):S1-4.
Ette EI, Williams PJ. Pharmacometrics: the science of quantitative pharmacology. Hoboken: Wiley; 2007.
Nguyen THT, Mouksassi M-S, Holford N, et al. Model evaluation of continuous data pharmacometric models: metrics and graphics. CPT Pharmacometrics Syst Pharmacol. 2017;6:87–109.
Strydom N, Gupta SV, Fox WS, et al. Tuberculosis drugs’ distribution and emergence of resistance in patient’s lung lesions: a mechanistic model and tool for regimen and dose optimization. PLoS Med. 2019;16:e1002773.
Abrantes JA, Jönsson S, Karlsson MO, et al. Handling interoccasion variability in model-based dose individualization using therapeutic drug monitoring data. Br J Clin Pharmacol. 2019;85:1326–36.
Schön T, Matuschek E, Mohamed S, et al. Standards for MIC testing that apply to the majority of bacterial pathogens should also be enforced for Mycobacterium tuberculosis complex. Clin Microbiol Infect. 2019;25:403–5.
World Health Organization (WHO). Technical report on critical concentrations for drug susceptibility testing of medicines used in the treatment of drug-resistant tuberculosis. Geneva: WHO; 2018.
Sturkenboom MG, Mulder LW, de Jager A, et al. Pharmacokinetic modeling and optimal sampling strategies for therapeutic drug monitoring of rifampin in patients with tuberculosis. Antimicrob Agents Chemother. 2015;59:4907–13.
Savic RM, Ruslami R, Hibma JE, et al. Pediatric tuberculous meningitis: model-based approach to determining optimal doses of the anti-tuberculosis drugs rifampin and levofloxacin for children. Clin Pharmacol Ther. 2015;98:622–9.
van Rijn SP, Zuur MA, van Altena R, et al. Pharmacokinetic modeling and limited sampling strategies based on healthy volunteers for monitoring of ertapenem in patients with multidrug-resistant tuberculosis. Antimicrob Agents Chemother. 2017;61:e01783-e1816.
Roberts JA, Abdul-Aziz MH, Lipman J, et al. Individualised antibiotic dosing for patients who are critically ill: challenges and potential solutions. Lancet Infect Dis. 2014;14:498–509.
van den Elsen SHJ, Sturkenboom MGG, Akkerman OW, et al. Limited sampling strategies using linear regression and the Bayesian approach for therapeutic drug monitoring of moxifloxacin in tuberculosis patients. Antimicrob Agents Chemother. 2019;63:e00384-e419.
Vilchèze C, Jacobs WR Jr. The isoniazid paradigm of killing, resistance, and persistence in Mycobacterium tuberculosis. J Mol Biol. 2019;431:3450–61.
Donald PR, Sirgel FA, Botha FJ, et al. The early bactericidal activity of isoniazid related to its dose size in pulmonary tuberculosis. Am J Respir Crit Care Med. 1997;156:895–900.
Gumbo T, Louie A, Liu W, et al. Isoniazid bactericidal activity and resistance emergence: integrating pharmacodynamics and pharmacogenomics to predict efficacy in different ethnic populations. Antimicrob Agents Chemother. 2007;51:2329–36.
Gumbo T. New susceptibility breakpoints for first-line antituberculosis drugs based on antimicrobial pharmacokinetic/pharmacodynamic science and population pharmacokinetic variability. Antimicrob Agents Chemother. 2010;54:1484–91.
Ahmad Z, Klinkenberg LG, Pinn ML, et al. Biphasic kill curve of isoniazid reveals the presence of drug-tolerant, not drug-resistant, Mycobacterium tuberculosis in the guinea pig. J Infect Dis. 2009;200:1136–43.
Jayaram R, Shandil RK, Gaonkar S, et al. Isoniazid pharmacokinetics-pharmacodynamics in an aerosol infection model of tuberculosis. Antimicrob Agents Chemother. 2004;48:2951–7.
Peloquin CA, Jaresko GS, Yong CL, et al. Population pharmacokinetic modeling of isoniazid, rifampin, and pyrazinamide. Antimicrob Agents Chemother. 1997;41:2670–9.
Alsultan A, Peloquin CA. Therapeutic drug monitoring in the treatment of tuberculosis: an update. Drugs. 2014;74:839–54.
Peloquin CA, Namdar R, Dodge AA, et al. Pharmacokinetics of isoniazid under fasting conditions, with food, and with antacids. Int J Tuberc Lung Dis. 1999;3:703–10.
Saktiawati AM, Sturkenboom MG, Stienstra Y, et al. Impact of food on the pharmacokinetics of first-line anti-TB drugs in treatment-naive TB patients: a randomized cross-over trial. J Antimicrob Chemother. 2016;71:703–10.
Kinzig-Schippers M, Tomalik-Scharte D, Jetter A, et al. Should we use N-acetyltransferase type 2 genotyping to personalize isoniazid doses? Antimicrob Agents Chemother. 2005;49:1733–8.
Horita Y, Alsultan A, Kwara A, et al. Evaluation of the adequacy of WHO revised dosages of the first-line anti-tuberculosis drugs in children with tuberculosis using population pharmacokinetic modeling and simulations. Antimicrob Agents Chemother. 2018;62:e00008-18.
Pasipanodya JG, McIlleron H, Burger A, et al. Serum drug concentrations predictive of pulmonary tuberculosis outcomes. J Infect Dis. 2013;208:1464–73.
Peloquin C. The role of therapeutic drug monitoring in mycobacterial infections. Microbiol Spectr. 2017;5:TNMI7-0029–2016.
Magis-Escurra C, Later-Nijland HMJ, Alffenaar JWC, et al. Population pharmacokinetics and limited sampling strategy for first-line tuberculosis drugs and moxifloxacin. Int J Antimicrob Agents. 2014;44:229–34.
Saktiawati AMI, Harkema M, Setyawan A, et al. Optimal sampling strategies for therapeutic drug monitoring of first-line tuberculosis drugs in patients with tuberculosis. Clin Pharmacokinet. 2019;58:1445–54.
Gumbo T, Louie A, Deziel MR, et al. Concentration-dependent Mycobacterium tuberculosis killing and prevention of resistance by rifampin. Antimicrob Agents Chemother. 2007;51:3781–8.
Jayaram R, Gaonkar S, Kaur P, et al. Pharmacokinetics-pharmacodynamics of rifampin in an aerosol infection model of tuberculosis. Antimicrob Agents Chemother. 2003;47:2118–24.
Lin MY, Lin SJ, Chan LC, et al. Impact of food and antacids on the pharmacokinetics of anti-tuberculosis drugs: systematic review and meta-analysis. Int J Tuberc Lung Dis. 2010;14:806–18.
Svensson EM, Dian S, Te Brake L, et al. Model-based meta-analysis of rifampicin exposure and mortality in Indonesian tuberculosis meningitis trials. Clin Infect Dis. 2020;71:1817–23.
Chirehwa MT, Rustomjee R, Mthiyane T, et al. Model-based evaluation of higher doses of rifampin using a semimechanistic model incorporating autoinduction and saturation of hepatic extraction. Antimicrob Agents Chemother. 2016;60:487–94.
Niemi M, Backman JT, Fromm MF, et al. Pharmacokinetic interactions with rifampicin: clinical relevance. Clin Pharmacokinet. 2003;42:819–50.
Boeree MJ, Diacon AH, Dawson R, et al. A dose ranging trial to optimize the dose of rifampin in the treatment of tuberculosis. Am J Respir Crit Care Med. 2015;191:1058–65.
Boeree MJ, Heinrich N, Aarnoutse R, et al. High-dose rifampicin, moxifloxacin, and SQ109 for treating tuberculosis: a multi-arm, multi-stage randomised controlled trial. Lancet Infect Dis. 2017;17:39–49.
Svensson RJ, Svensson EM, Aarnoutse RE, et al. Greater early bactericidal activity at higher rifampicin doses revealed by modeling and clinical trial simulations. J Infect Dis. 2018;218:991–9.
Svensson EM, Svensson RJ, Te Brake LHM, et al. The potential for treatment shortening with higher rifampicin doses: relating drug exposure to treatment response in patients with pulmonary tuberculosis. Clin Infect Dis. 2018;67:34–41.
Stott KE, Pertinez H, Sturkenboom MGG, et al. Pharmacokinetics of rifampicin in adult TB patients and healthy volunteers: a systematic review and meta-analysis. J Antimicrob Chemother. 2018;73:2305–13.
Svensson RJ, Aarnoutse RE, Diacon AH, et al. A population pharmacokinetic model incorporating saturable pharmacokinetics and autoinduction for high rifampicin doses. Clin Pharmacol Ther. 2018;103:674–83.
van Beek SW, Ter Heine R, Keizer RJ, et al. Personalized tuberculosis treatment through model-informed dosing of rifampicin. Clin Pharmacokinet. 2019;58:815–26.
Scorpio A, Zhang Y. Mutations in pncA, a gene encoding pyrazinamidase/nicotinamidase, cause resistance to the antituberculous drug pyrazinamide in tubercle bacillus. Nat Med. 1996;2:662–7.
Mitchison DA. The action of antituberculosis drugs in short-course chemotherapy. Tubercle. 1985;66:219–25.
Whitfield MG, Soeters HM, Warren RM, et al. A global perspective on pyrazinamide resistance: systematic review and meta-analysis. PLoS ONE. 2015;10:1–16.
Gumbo T, Dona CS, Meek C, et al. Pharmacokinetics-pharmacodynamics of pyrazinamide in a novel in vitro model of tuberculosis for sterilizing effect: a paradigm for faster assessment of new antituberculosis drugs. Antimicrob Agents Chemother. 2009;53:3197–204.
World Health Organization (WHO). Treatment of tuberculosis guidelines. 4th ed. Geneva: WHO; 2010.
World Health Organization (WHO). WHO consolidated guidelines on drug-resistant tuberculosis treatment. Geneva: WHO; 2019.
Wilkins JJ, Langdon G, McIlleron H, et al. Variability in the population pharmacokinetics of pyrazinamide in South African tuberculosis patients. Eur J Clin Pharmacol. 2006;62:727–35.
Alghamdi WA, Al-Shaer MH, Peloquin CA. Protein binding of first-line antituberculosis drugs. Antimicrob Agents Chemother. 2018;62:e00641-e718.
Konno K, Feldmann FM, McDermott W. Pyrazinamide susceptibility and amidase activity of Tubercle bacilli. Am Rev Respir Dis. 1967;95:461–9.
Daskapan A, Idrus LR, Postma MJ, et al. A systematic review on the effect of HIV infection on the pharmacokinetics of first-line tuberculosis drugs. Clin Pharmacokinet. 2019;58:747–66.
Chirehwa MT, McIlleron H, Rustomjee R, et al. Pharmacokinetics of pyrazinamide and optimal dosing regimens for drug-sensitive and -resistant tuberculosis. Antimicrob Agents Chemother. 2017;61:e00490-e517.
Sekaggya-Wiltshire C, Chirehwa M, Musaazi J, et al. Low antituberculosis drug concentrations in HIV-tuberculosis-coinfected adults with low body weight: is it time to update dosing guidelines? Antimicrob Agents Chemother. 2019;63:e02174-e2218.
Muliaditan M, Della PO. How long will treatment guidelines for TB continue to overlook variability in drug exposure? J Antimicrob Chemother. 2019;74:3274–80.
McIlleron H, Rustomjee R, Vahedi M, et al. Reduced antituberculosis drug concentrations in HIV-infected patients who are men or have low weight: implications for international dosing guidelines. Antimicrob Agents Chemother. 2012;56:3232–8.
Chideya S, Winston CA, Peloquin CA, et al. Isoniazid, rifampin, ethambutol, and pyrazinamide pharmacokinetics and treatment outcomes among a predominantly HIV-infected cohort of adults with tuberculosis from Botswana. Clin Infect Dis. 2009;48:1685–94.
Girling DJ. The hepatic toxicity of antituberculosis regimens containing isoniazid, rifampicin and pyrazinamide. Tubercle. 1977;59:13–32.
Pasipanodya JG, Gumbo T. Clinical and toxicodynamic evidence that high-dose pyrazinamide is not more hepatotoxic than the low doses currently used. Antimicrob Agents Chemother. 2010;54:2847–54.
Vinnard C, Ravimohan S, Tamuhla N, et al. Pyrazinamide clearance is impaired among HIV/tuberculosis patients with high levels of systemic immune activation. PLoS ONE. 2017;12:e0187624.
Dickinson JM, Ellard GA, Mitchison DA. Suitability of isoniazid and ethambutol for intermittent administration in the treatment of tuberculosis. Tubercle. 1968;49:351–66.
Srivastava S, Musuka S, Sherman C, et al. Efflux-pump-derived multiple drug resistance to ethambutol monotherapy in Mycobacterium tuberculosis and the pharmacokinetics and pharmacodynamics of ethambutol. J Infect Dis. 2010;201:1225–31.
Radenbach KL. Minimum daily efficient dose of ethambutol: general review. Bull Int Union Tuberc. 1973;48:106–11.
Anonymous. Ethambutol plus isoniazid for the treatment of pulmonary tuberculosis: a controlled trial of our regimens. Tubercle. 1981;62:13–29.
Pasipanodya J, Gumbo T. An oracle: antituberculosis pharmacokinetics-pharmacodynamics, clinical correlation, and clinical trial simulations to predict the future. Antimicrob Agents Chemother. 2011;55:24–34.
Donald PR. Cerebrospinal fluid concentrations of antituberculosis agents in adults and children. Tuberculosis (Edinb). 2010;90:279–92.
Peloquin CA, Bulpitt AM, Jaresko GS, et al. Pharmacokinetics of ethambutol under fasting conditions, with food, and with antacids. Antimicrob Agents Chemother. 1999;43:568–72.
Zhu M, Burman WJ, Starke JR, et al. Pharmacokinetics of ethambutol in children and adults with tuberculosis. Int J Tuberc Lung Dis. 2004;8:1360–7.
Jonsson S, Davidse A, Wilkins J, et al. Population pharmacokinetics of ethambutol in South African tuberculosis patients. Antimicrob Agents Chemother. 2011;55:4230–7.
Hall RG, Swancutt MA, Meek C, et al. Ethambutol pharmacokinetic variability is linked to body mass in overweight, obese, and extremely obese people. Antimicrob Agents Chemother. 2012;56:1502–7.
Denti P, Jeremiah K, Chigutsa E, et al. Pharmacokinetics of isoniazid, pyrazinamide, and ethambutol in newly diagnosed pulmonary TB patients in Tanzania. PLoS ONE. 2015;10:e0141002.
Mehta K, Ravimohan S, Pasipanodya JG, et al. Optimizing ethambutol dosing among HIV/tuberculosis co-infected patients: a population pharmacokinetic modelling and simulation study. J Antimicrob Chemother. 2019;74:2994–3002.
Sundell J, Bienvenu E, Birgersson S, et al. Population pharmacokinetics and pharmacogenetics of ethambutol in adult patients coinfected with tuberculosis and HIV. Antimicrob Agents Chemother. 2020;64:e01583-e1619.
Abdelwahab MT, Leisegang R, Dooley KE, et al. Population pharmacokinetics of isoniazid, pyrazinamide, and ethambutol in pregnant South African women with tuberculosis and HIV. Antimicrob Agents Chemother. 2020;64:e01978-e2019.
Wohlkonig A, Chan PF, Fosberry AP, et al. Structural basis of quinolone inhibition of type IIA topoisomerases and target-mediated resistance. Nat Struct Mol Biol. 2010;17:1152–3.
Shandil RK, Jayaram R, Kaur P, et al. Moxifloxacin, ofloxacin, sparfloxacin, and ciprofloxacin against Mycobacterium tuberculosis: evaluation of in vitro and pharmacodynamic indices that best predict in vivo efficacy. Antimicrob Agents Chemother. 2007;51:576–82.
Gumbo T, Louie A, Deziel MR, et al. Selection of a moxifloxacin dose that suppresses drug resistance in Mycobacterium tuberculosis, by use of an in vitro pharmacodynamic infection model and mathematical modeling. J Infect Dis. 2004;190:1642–51.
Deshpande D, Pasipanodya JG, Mpagama SG, et al. Levofloxacin pharmacokinetics/pharmacodynamics, dosing, susceptibility breakpoints, and artificial intelligence in the treatment of multidrug-resistant tuberculosis. Clin Infect Dis. 2018;67:S293-302.
Heinrichs MT, Drusano GL, Brown DL, et al. Dose optimization of moxifloxacin and linezolid against tuberculosis using mathematical modeling and simulation. Int J Antimicrob Agents. 2019;53:275–83.
Louie A, Duncanson B, Myrick J, et al. Activity of Moxifloxacin against Mycobacterium tuberculosis in acid phase and nonreplicative-persister phenotype phase in a hollow-fiber infection model. Antimicrob Agents Chemother. 2018;62:e01470-e1518.
Pranger AD, van Altena R, Aarnoutse RE, et al. Evaluation of moxifloxacin for the treatment of tuberculosis: 3 years of experience. Eur Respir J. 2011;38:888–94.
Ghimire S, Maharjan B, Jongedijk EM, et al. Levofloxacin pharmacokinetics, pharmacodynamics and outcome in multidrug-resistant tuberculosis patients. Eur Respir J. 2019;53:1802107.
Van’t Boveneind-Vrubleuskaya N, Seuruk T, van Hateren K, et al. Pharmacokinetics of levofloxacin in multidrug- and extensively drug-resistant tuberculosis patients. Antimicrob Agents Chemother. 2017;61:e00343-e417.
van den Elsen SHJ, Sturkenboom MGG, Van’t Boveneind-Vrubleuskaya N, et al. Population pharmacokinetic model and limited sampling strategies for personalized dosing of levofloxacin in tuberculosis patients. Antimicrob Agents Chemother. 2018;62:e01092-e1118.
Koul A, Dendouga N, Vergauwen K, et al. Diarylquinolines target subunit c of mycobacterial ATP synthase. Nat Chem Biol. 2007;3:323–4.
Rouan M-C, Lounis N, Gevers T, et al. Pharmacokinetics and pharmacodynamics of TMC207 and its N-desmethyl metabolite in a murine model of tuberculosis. Antimicrob Agents Chemother. 2012;56:1444–51.
Salinger DH, Nedelman JR, Mendel C, et al. Daily dosing for bedaquiline in patients with tuberculosis. Antimicrob Agents Chemother. 2019;63:e00463-e519.
van Heeswijk RPG, Dannemann B, Hoetelmans RMW. Bedaquiline: a review of human pharmacokinetics and drug–drug interactions. J Antimicrob Chemother. 2014;69:2310–8.
Akkerman OW, Odish OFF, Bolhuis MS, et al. Pharmacokinetics of bedaquiline in cerebrospinal fluid and serum in multidrug-resistant tuberculous meningitis. Clin Infect Dis. 2016;62:523–4.
Svensson EM, Aweeka F, Park J-G, et al. Model-based estimates of the effects of efavirenz on bedaquiline pharmacokinetics and suggested dose adjustments for patients coinfected with HIV and tuberculosis. Antimicrob Agents Chemother. 2013;57:2780–7.
Svensson EM, Karlsson MO. Modelling of mycobacterial load reveals bedaquiline’s exposure-response relationship in patients with drug-resistant TB. J Antimicrob Chemother. 2017;72:3398–405.
Tanneau L, Karlsson MO, Svensson EM. Understanding the drug exposure-response relationship of bedaquiline to predict efficacy for novel dosing regimens in the treatment of multidrug-resistant tuberculosis. Br J Clin Pharmacol. 2020;86:913–22.
Svensson E, Dosne A-G, Karlsson M. Population pharmacokinetics of bedaquiline and metabolite M2 in patients with drug-resistant tuberculosis: the effect of time-varying weight and albumin. CPT Pharmacometrics Syst Pharmacol. 2016;5:682–91.
McLeay SC, Vis P, van Heeswijk RPG, et al. Population pharmacokinetics of bedaquiline (TMC207), a novel antituberculosis drug. Antimicrob Agents Chemother. 2014;58:5315–24.
Alffenaar J-WC, Akkerman OW, Tiberi S, et al. Should we worry about bedaquiline exposure in the treatment of multidrug-resistant and extensively drug-resistant tuberculosis? Eur Respir J. 2020;55:1901908.
Nguyen TVA, Anthony RM, Bañuls A-L, et al. Bedaquiline resistance: its emergence, mechanism, and prevention. Clin Infect Dis. 2018;66:1625–30.
US FDA, Center for Drug Evaluation and Research. Application number 204384Orig1s000.
Srivastava S, Magombedze G, Koeuth T, et al. Linezolid dose that maximizes sterilizing effect while minimizing toxicity and resistance emergence for tuberculosis. Antimicrob Agents Chemother. 2017;61:e00751-e817.
Millard J, Pertinez H, Bonnett L, et al. Linezolid pharmacokinetics in MDR-TB: a systematic review, meta-analysis and Monte Carlo simulation. J Antimicrob Chemother. 2018;73:1755–62.
Bolhuis MS, Akkerman OW, Sturkenboom MGG, et al. Linezolid-based regimens for multidrug-resistant tuberculosis (TB): a systematic review to establish or revise the current recommended dose for TB treatment. Clin Infect Dis. 2018;67:S327–35.
Sun F, Ruan Q, Wang J, et al. Linezolid manifests a rapid and dramatic therapeutic effect for patients with life-threatening tuberculous meningitis. Antimicrob Agents Chemother. 2014;58:6297–301.
Singh B, Cocker D, Ryan H, et al. Linezolid for drug-resistant pulmonary tuberculosis. Cochrane Database Syst Rev. 2019;3:CD012836.
Song T, Lee M, Jeon H-S, et al. Linezolid trough concentrations correlate with mitochondrial toxicity-related adverse events in the treatment of chronic extensively drug-resistant tuberculosis. EBioMedicine. 2015;2:1627–33.
Zhao W, Guo Z, Zheng M, et al. Activity of linezolid-containing regimens against multidrug-resistant tuberculosis in mice. Int J Antimicrob Agents. 2014;43:148–53.
Tasneen R, Betoudji F, Tyagi S, et al. Contribution of oxazolidinones to the efficacy of novel regimens containing bedaquiline and pretomanid in a mouse model of tuberculosis. Antimicrob Agents Chemother. 2016;60:270–7.
Wasserman S, Denti P, Brust JCM, et al. Linezolid pharmacokinetics in South African patients with drug-resistant tuberculosis and a high prevalence of HIV coinfection. Antimicrob Agents Chemother. 2019;63:e02164-e2218.
Bolhuis MS, Tiberi S, Sotgiu G, et al. Is there still room for therapeutic drug monitoring of linezolid in patients with tuberculosis? Eur Respir J. 2016;47:1288–90.
Bolhuis MS, van der Werf TS, Kerstjens HAM, et al. Treatment of multidrug-resistant tuberculosis using therapeutic drug monitoring: first experiences with sub-300 mg linezolid dosages using in-house made capsules. Eur Respir J. 2019;54:1900580.
Conradie F, Diacon AH, Ngubane N, et al. Treatment of highly drug-resistant pulmonary tuberculosis. N Engl J Med. 2020;382:893–902.
Kamp J, Bolhuis MS, Tiberi S, et al. Simple strategy to assess linezolid exposure in patients with multi-drug-resistant and extensively-drug-resistant tuberculosis. Int J Antimicrob Agents. 2017;49:688–94.
Swanson RV, Adamson J, Moodley C, et al. Pharmacokinetics and pharmacodynamics of clofazimine in a mouse model of tuberculosis. Antimicrob Agents Chemother. 2015;59:3042–51.
Ammerman NC, Swanson RV, Bautista EM, et al. Impact of clofazimine dosing on treatment shortening of the first-line regimen in a mouse model of tuberculosis. Antimicrob Agents Chemother. 2018;62:e00636-e718.
Cholo MC, Mothiba MT, Fourie B, et al. Mechanisms of action and therapeutic efficacies of the lipophilic antimycobacterial agents clofazimine and bedaquiline. J Antimicrob Chemother. 2017;72:338–53.
Nix DE, Adam RD, Auclair B, et al. Pharmacokinetics and relative bioavailability of clofazimine in relation to food, orange juice and antacid. Tuberculosis. 2004;84:365–73.
Abdelwahab MT, Wasserman S, Brust JCM, et al. Clofazimine pharmacokinetics in patients with TB: dosing implications. J Antimicrob Chemother. 2020;75:3269–77.
Garrelts JC. Clofazimine: a review of its use in leprosy and Mycobacterium avium complex infection. DICP. 1991;25:525–31.
Holdiness MR. Clinical pharmacokinetics of clofazimine: a review. Clin Pharmacokinet. 1989;16:74–85.
Arbex MA, Varella Mde CL, Siqueira HR, et al. Antituberculosis drugs: drug interactions, adverse effects, and use in special situations. Part 2: second line drugs. J Bras Pneumol. 2010;36:641–56.
Alghamdi WA, Alsultan A, Al-Shaer MH, et al. Cycloserine population pharmacokinetics and pharmacodynamics in patients with tuberculosis. Antimicrob Agents Chemother. 2019;63:e00055-e119.
van der Galiën R, Boveneind-Vrubleuskaya NV, Peloquin C, et al. Pharmacokinetic modeling, simulation, and development of a limited sampling strategy of cycloserine in patients with multidrug-/extensively drug-resistant tuberculosis. Clin Pharmacokinet. 2020;59:899–910.
Deshpande D, Alffenaar J-WC, Köser CU, et al. d-Cycloserine pharmacokinetics/pharmacodynamics, susceptibility, and dosing implications in multidrug-resistant tuberculosis: a Faustian deal. Clin Infect Dis. 2018;67:S308–16.
Hung W-Y, Yu M-C, Chiang Y-C, et al. Serum concentrations of cycloserine and outcome of multidrug-resistant tuberculosis in Northern Taiwan. Int J Tuberc Lung Dis. 2014;18:601–6.
Zhu H, Guo S-C, Liu Z-Q, et al. Therapeutic drug monitoring of cycloserine and linezolid during anti-tuberculosis treatment in Beijing, China. Int J Tuberc Lung Dis. 2018;22:931–6.
Alghamdi WA, Antwi S, Enimil A, et al. Population pharmacokinetics of efavirenz in HIV and TB/HIV coinfected children: the significance of genotype-guided dosing. J Antimicrob Chemother. 2019;74:2698–706.
Matsumoto M, Hashizume H, Tomishige T, et al. OPC-67683, a nitro-dihydro-imidazooxazole derivative with promising action against tuberculosis in vitro and in mice. PLoS Med. 2006;3:e466.
von Groote-Bidlingmaier F, Patientia R, Sanchez E, et al. Efficacy and safety of delamanid in combination with an optimised background regimen for treatment of multidrug-resistant tuberculosis: a multicentre, randomised, double-blind, placebo-controlled, parallel group phase 3 trial. Lancet Respir Med. 2019;7:249–59.
Sasahara K, Shimokawa Y, Hirao Y, et al. Pharmacokinetics and metabolism of delamanid, a novel anti-tuberculosis drug, in animals and humans: importance of albumin metabolism in vivo. Drug Metab Dispos. 2015;43:1267–76.
Ferlazzo G, Mohr E, Laxmeshwar C, et al. Early safety and efficacy of the combination of bedaquiline and delamanid for the treatment of patients with drug-resistant tuberculosis in Armenia, India, and South Africa: a retrospective cohort study. Lancet Infect Dis. 2018;18:536–44.
Diacon AH, Dawson R, Hanekom M, et al. Early bactericidal activity of delamanid (OPC-67683) in smear-positive pulmonary tuberculosis patients. Int J Tuberc Lung Dis. 2011;15:949–54.
Delamanid, Deltyba 50 mg coated tablet, Summary of Product Characteristics. https://www.ema.europa.eu/en/documents/product-information/deltyba-eparproduct-information_en.pdf. Accessed 17 Feb 2021.
Tucker EW, Pieterse L, Zimmerman MD, et al. Delamanid central nervous system pharmacokinetics in tuberculous meningitis in rabbits and humans. Antimicrob Agents Chemother. 2019;63:e00913-e919.
Gler MT, Skripconoka V, Sanchez-Garavito E, et al. Delamanid for multidrug-resistant pulmonary tuberculosis. N Engl J Med. 2012;366:2151–60.
Nguyen TVA, Anthony RM, Cao TTH, et al. Delamanid resistance: update and clinical management. Clin Infect Dis. 2020;71:3252–9.
Dutta NK, Karakousis PC. PA-824 is as effective as isoniazid against latent tuberculosis infection in C3HeB/FeJ mice. Int J Antimicrob Agents. 2014;44:564–6.
Xu J, Li S-Y, Almeida DV, et al. Contribution of pretomanid to novel regimens containing bedaquiline with either linezolid or moxifloxacin and pyrazinamide in murine models of tuberculosis. Antimicrob Agents Chemother. 2019;63:e00021-e119.
Drusano GL, Neely MN, Kim S, et al. Building optimal 3-drug combination chemotherapy regimens. Antimicrob Agents Chemother. 2020;64:e01610-e1620.
Winter H, Ginsberg A, Egizi E, et al. Effect of a high-calorie, high-fat meal on the bioavailability and pharmacokinetics of PA-824 in healthy adult subjects. Antimicrob Agents Chemother. 2013;57:5516–20.
Dooley KE, Luetkemeyer AF, Park J-G, et al. Phase I safety, pharmacokinetics, and pharmacogenetics study of the antituberculosis drug PA-824 with concomitant lopinavir-ritonavir, efavirenz, or rifampin. Antimicrob Agents Chemother. 2014;58:5245–52.
Gupta R, Lavollay M, Mainardi J-L, et al. The Mycobacterium tuberculosis protein LdtMt2 is a nonclassical transpeptidase required for virulence and resistance to amoxicillin. Nat Med. 2010;16:466–9.
Srivastava S, van Rijn SP, Wessels AMA, et al. Susceptibility testing of antibiotics that degrade faster than the doubling time of slow-growing Mycobacteria: ertapenem sterilizing effect versus Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2016;60:3193–5.
van Rijn SP, Srivastava S, Wessels MA, et al. Sterilizing effect of ertapenem-clavulanate in a hollow-fiber model of tuberculosis and implications on clinical dosing. Antimicrob Agents Chemother. 2017;61:e02039-e2116.
Diacon AH, van der Merwe L, Barnard M, et al. β-Lactams against tuberculosis: new trick for an old dog? N Engl J Med. 2016;375:393–4.
Mouton JW, van den Anker JN. Meropenem clinical pharmacokinetics. Clin Pharmacokinet. 1995;28:275–86.
de Jager VR, Vanker N, van der Merwe L, et al. Optimizing β-lactams against tuberculosis. Am J Respir Crit Care Med. 2020;201:1155–7.
Zuur MA, Ghimire S, Bolhuis MS, et al. Pharmacokinetics of 2,000 milligram ertapenem in tuberculosis patients. Antimicrob Agents Chemother. 2018;62:e02250-e2317.
Srivastava S, Modongo C, Siyambalapitiyage Dona CW, et al. Amikacin optimal exposure targets in the hollow-fiber system model of tuberculosis. Antimicrob Agents Chemother. 2016;60:5922–7.
Sturkenboom MGG, Simbar N, Akkerman OW, et al. Amikacin dosing for MDR tuberculosis: a systematic review to establish or revise the current recommended dose for tuberculosis treatment. Clin Infect Dis. 2018;67:S303–7.
Peloquin CA, Berning SE, Nitta AT, et al. Aminoglycoside toxicity: daily versus thrice-weekly dosing for treatment of mycobacterial diseases. Clin Infect Dis. 2004;38:1538–44.
Modongo C, Pasipanodya JG, Zetola NM, et al. Amikacin concentrations predictive of ototoxicity in multidrug-resistant tuberculosis patients. Antimicrob Agents Chemother. 2015;59:6337–43.
van Altena R, Dijkstra JA, van der Meer ME, et al. Reduced chance of hearing loss associated with therapeutic drug monitoring of aminoglycosides in the treatment of multidrug-resistant tuberculosis. Antimicrob Agents Chemother. 2017;61:e01400-e1416.
Dijkstra JA, van Altena R, Akkerman OW, et al. Limited sampling strategies for therapeutic drug monitoring of amikacin and kanamycin in patients with multidrug-resistant tuberculosis. Int J Antimicrob Agents. 2015;46:332–7.
Heifets LB, Lindholm-Levy PJ, Flory M. Comparison of bacteriostatic and bactericidal activity of isoniazid and ethionamide against Mycobacterium avium and Mycobacterium tuberculosis. Am Rev Respir Dis. 1991;143:268–70.
Thee S, Garcia-Prats AJ, Donald PR, et al. A review of the use of ethionamide and prothionamide in childhood tuberculosis. Tuberculosis. 2016;97:126–36.
Deshpande D, Pasipanodya JG, Mpagama SG, et al. Ethionamide pharmacokinetics/pharmacodynamics-derived dose, the role of MICs in clinical outcome, and the resistance arrow of time in multidrug-resistant tuberculosis. Clin Infect Dis. 2018;67:S317–26.
Buchanan N, Van Der Walt LA. The binding of antituberculous drugs to normal and kwashiorkor serum. S Afr Med J. 1977;52:522–5.
Henderson MC, Siddens LK, Morré JT, et al. Metabolism of the anti-tuberculosis drug ethionamide by mouse and human FMO1, FMO2 and FMO3 and mouse and human lung microsomes. Toxicol Appl Pharmacol. 2008;233:420–7.
Zhu M, Namdar R, Stambaugh JJ, et al. Population pharmacokinetics of ethionamide in patients with tuberculosis. Tuberculosis (Edinb). 2002;82:91–6.
Lee SH, Seo K-A, Lee YM, et al. Low serum concentrations of moxifloxacin, prothionamide, and cycloserine on sputum conversion in multi-drug resistant TB. Yonsei Med J. 2015;56:961–7.
Zheng J, Rubin EJ, Bifani P, et al. para-Aminosalicylic acid is a prodrug targeting dihydrofolate reductase in Mycobacterium tuberculosis. J Biol Chem. 2013;288:23447–56.
Jindani A, Aber VR, Edwards EA, et al. The early bactericidal activity of drugs in patients with pulmonary tuberculosis. Am Rev Respir Dis. 1980;121:939–49.
Peloquin CA, Zhu M, Adam RD, et al. Pharmacokinetics of para-aminosalicylic acid granules under four dosing conditions. Ann Pharmacother. 2001;35:1332–8.
De Kock L, Sy SKB, Rosenkranz B, et al. Pharmacokinetics of para-aminosalicylic acid in HIV-uninfected and HIV-coinfected tuberculosis patients receiving antiretroviral therapy, managed for multidrug-resistant and extensively drug-resistant tuberculosis. Antimicrob Agents Chemother. 2014;58:6242–50.
Singh B, Mitchison DA. Bactericidal activity of streptomycin and isoniazid in combination with p-aminosalicylic acid against Mycobacterium tuberculosis. J Gen Microbiol. 1955;12:76–84.
Bonate PL. Pharmacokinetic-pharmacodynamic modeling and simulation. 2nd ed. New York: Springer; 2011.
Kantasiripitak W, Van Daele R, Gijsen M, et al. Software tools for model-informed precision dosing: how well do they satisfy the needs? Front Pharmacol. 2020;11:620.
Nahid P, Mase SR, Migliori GB, et al. Treatment of drug-resistant tuberculosis: an official ATS/CDC/ERS/IDSA clinical practice guideline. Am J Respir Crit Care Med. 2019;200:e93-142.
Nahid P, Dorman SE, Alipanah N, et al. Official American Thoracic Society/Centers for Disease Control and Prevention/Infectious Diseases Society of America clinical practice guidelines: treatment of drug-susceptible tuberculosis. Clin Infect Dis. 2016;63:e147–95.
Alffenaar J-WC, Heysell SK, Mpagama SG. Therapeutic drug monitoring: the need for practical guidance. Clin Infect Dis. 2019;68:1065–6.
Wilkins JJ, Langdon G, McIlleron H, et al. Variability in the population pharmacokinetics of isoniazid in South African tuberculosis patients. Br J Clin Pharmacol. 2011;72:51–62.
Zvada SP, Denti P, Donald PR, et al. Population pharmacokinetics of rifampicin, pyrazinamide and isoniazid in children with tuberculosis: in silico evaluation of currently recommended doses. J Antimicrob Chemother. 2014;69:1339–49.
Wilkins JJ, Savic RM, Karlsson MO, et al. Population pharmacokinetics of rifampin in pulmonary tuberculosis patients, including a semimechanistic model to describe variable absorption. Antimicrob Agents Chemother. 2008;52:2138–48.
Azuma J, Ohno M, Kubota R, et al. NAT2 genotype guided regimen reduces isoniazid-induced liver injury and early treatment failure in the 6-month four-drug standard treatment of tuberculosis: a randomized controlled trial for pharmacogenetics-based therapy. Eur J Clin Pharmacol. 2013;69:1091–101.
Naidoo A, Chirehwa M, Ramsuran V, et al. Effects of genetic variability on rifampicin and isoniazid pharmacokinetics in South African patients with recurrent tuberculosis. Pharmacogenomics. 2019;20:225–40.
Märtson A-G, Sturkenboom MGG, Stojanova J, et al. How to design a study to evaluate therapeutic drug monitoring in infectious diseases? Clin Microbiol Infect. 2020;26:1008–16.
Kim HY, Heysell SK, Mpagama S, et al. Challenging the management of drug-resistant tuberculosis. Lancet. 2020;395:783.
Hiruy H, Rogers Z, Mbowane C, et al. Subtherapeutic concentrations of first-line anti-TB drugs in South African children treated according to current guidelines: the PHATISA study. J Antimicrob Chemother. 2015;70:1115–23.
Seng K-Y, Hee K-H, Soon G-H, et al. Population pharmacokinetic analysis of isoniazid, acetyl-isoniazid and isonicotinic acid in healthy volunteers. Antimicrob Agents Chemother. 2015;59:6791–9.
Lalande L, Bourguignon L, Bihari S, et al. Population modeling and simulation study of the pharmacokinetics and antituberculosis pharmacodynamics of isoniazid in lungs. Antimicrob Agents Chemother. 2015;59:5181–9.
Rockwood N, Meintjes G, Chirehwa M, et al. HIV-1 coinfection does not reduce exposure to rifampin, isoniazid, and pyrazinamide in South African tuberculosis outpatients. Antimicrob Agents Chemother. 2016;60:6050–9.
Vinnard C, Ravimohan S, Tamuhla N, et al. Isoniazid clearance is impaired among human immunodeficiency virus/tuberculosis patients with high levels of immune activation. Br J Clin Pharmacol. 2017;83:801–11.
Chirehwa MT, McIlleron H, Wiesner L, et al. Effect of efavirenz-based antiretroviral therapy and high-dose rifampicin on the pharmacokinetics of isoniazid and acetyl-isoniazid. J Antimicrob Chemother. 2019;74:139–48.
Aruldhas BW, Hoglund RM, Ranjalkar J, et al. Optimization of dosing regimens of isoniazid and rifampicin in children with tuberculosis in India. Br J Clin Pharmacol. 2019;85:644–54.
Goutelle S, Bourguignon L, Maire PH, et al. Population modeling and Monte Carlo simulation study of the pharmacokinetics and antituberculosis pharmacodynamics of rifampin in lungs. Antimicrob Agents Chemother. 2009;53:2974–81.
Smythe W, Khandelwal A, Merle C, et al. A semimechanistic pharmacokinetic-enzyme turnover model for rifampin autoinduction in adult tuberculosis patients. Antimicrob Agents Chemother. 2012;56:2091–8.
Milán Segovia RC, Domínguez Ramírez AM, Jung Cook H, et al. Population pharmacokinetics of rifampicin in Mexican patients with tuberculosis. J Clin Pharm Ther. 2013;38:56–61.
Jeremiah K, Denti P, Chigutsa E, et al. Nutritional supplementation increases rifampin exposure among tuberculosis patients coinfected with HIV. Antimicrob Agents Chemother. 2014;58:3468–74.
Seng K-Y, Hee K-H, Soon G-H, et al. Population pharmacokinetics of rifampicin and 25-deacetyl-rifampicin in healthy Asian adults. J Antimicrob Chemother. 2015;70:3298–306.
Jing Y, Zhu LQ, Yang JW, et al. Population pharmacokinetics of rifampicin in Chinese patients with pulmonary tuberculosis. J Clin Pharmacol. 2016;56:622–7.
Denti P, Martinson N, Cohn S, et al. Population pharmacokinetics of rifampin in pregnant women with tuberculosis and HIV coinfection in Soweto, South Africa. Antimicrob Agents Chemother. 2015;60:1234–41.
Schipani A, Pertinez H, Mlota R, et al. A simultaneous population pharmacokinetic analysis of rifampicin in Malawian adults and children. Br J Clin Pharmacol. 2016;81:679–87.
Zhu M, Starke JR, Burman WJ, et al. Population pharmacokinetic modeling of pyrazinamide in children and adults with tuberculosis. Pharmacotherapy. 2002;22:686–95.
Mugabo P, Mulubwa M. Population pharmacokinetic modelling of pyrazinamide and pyrazinoic acid in patients with multi-drug resistant tuberculosis. Eur J Drug Metab Pharmacokinet. 2019;44:519–30.
Peloquin CA, Hadad DJ, Molino LP, et al. Population pharmacokinetics of levofloxacin, gatifloxacin, and moxifloxacin in adults with pulmonary tuberculosis. Antimicrob Agents Chemother. 2008;52:852–7.
Denti P, Garcia-Prats AJ, Draper HR, et al. Levofloxacin population pharmacokinetics in South African children treated for multidrug-resistant tuberculosis. Antimicrob Agents Chemother. 2018;62:e01521-e1617.
Al-Shaer MH, Alghamdi WA, Alsultan A, et al. Fluoroquinolones in drug-resistant tuberculosis: culture conversion and pharmacokinetic/pharmacodynamic target attainment to guide dose selection. Antimicrob Agents Chemother. 2019;63:e00279-e319.
Pranger AD, Kosterink JG, van Altena R, et al. Limited-sampling strategies for therapeutic drug monitoring of moxifloxacin in patients with tuberculosis. Ther Drug Monit. 2011;33:350–4.
Zvada SP, Denti P, Sirgel FA, et al. Moxifloxacin population pharmacokinetics and model-based comparison of efficacy between moxifloxacin and ofloxacin in African patients. Antimicrob Agents Chemother. 2014;58:503–10.
Chang MJ, Jin B, Chae J-W, et al. Population pharmacokinetics of moxifloxacin, cycloserine, p-aminosalicylic acid and kanamycin for the treatment of multi-drug-resistant tuberculosis. Int J Antimicrob Agents. 2017;49:677–87.
Alffenaar JW, Kosterink JG, van Altena R, et al. Limited sampling strategies for therapeutic drug monitoring of linezolid in patients with multidrug-resistant tuberculosis. Ther Drug Monit. 2010;32:97–101.
Garcia-Prats AJ, Schaaf HS, Draper HR, et al. Pharmacokinetics, optimal dosing, and safety of linezolid in children with multidrug-resistant tuberculosis: combined data from two prospective observational studies. PLoS Med. 2019;16:e1002789.
Alghamdi WA, Al-Shaer MH, An G, et al. Population pharmacokinetics of linezolid in tuberculosis patients: dosing regimen simulation and target attainment analysis. Antimicrob Agents Chemother. 2020;64:e01174-e1220.
Faraj A, Svensson RJ, Diacon AH, et al. Drug effect of clofazimine on persisters explains an unexpected increase in bacterial load in patients. Antimicrob Agents Chemother. 2020;64:e01905-e1919.
Mulubwa M, Mugabo P. Steady-state population pharmacokinetics of terizidone and its metabolite cycloserine in patients with drug-resistant tuberculosis. Br J Clin Pharmacol. 2019;85:1946–56.
Chirehwa MT, Court R, De Kock M, et al. Population pharmacokinetics of cycloserine, and pharmacokinetic/pharmacodynamic target attainment, in MDR-tuberculosis patients dosed with terizidone. Antimicrob Agents Chemother. 2020;64:e01381-e1420.
Lyons MA. Modeling and simulation of pretomanid pharmacokinetics in pulmonary tuberculosis patients. Antimicrob Agents Chemother. 2018;62:e02359-e2417.
Salinger DH, Subramoney V, Everitt D, et al. Population pharmacokinetics of the antituberculosis agent pretomanid. Antimicrob Agents Chemother. 2019;63:e00907-e919.
Bjugård Nyberg H, Draper HR, Garcia-Prats AJ, et al. Population pharmacokinetics and dosing of ethionamide in children with tuberculosis. Antimicrob Agents Chemother. 2019;63:e01984-e2019.
Al-Shaer MH, Märtson A-G, Alghamdi WA, et al. Ethionamide population pharmacokinetic model and target attainment in multidrug-resistant tuberculosis. Antimicrob Agents Chemother. 2020;64:e00713-e720.
Abulfathi AA, Assawasuwannakit P, Donald PR, et al. Probability of mycobactericidal activity of para-aminosalicylic acid with novel dosing regimens. Eur J Clin Pharmacol. 2020;76(11):1557–65.
Zheng X, Bao Z, Forsman LD, et al. Drug exposure and minimum inhibitory concentration predict pulmonary tuberculosis treatment response. Clin Infect Dis. 2020;ciaa1569. https://doi.org/10.1093/cid/ciaa1569.
Diacon AH, Pym A, Grobusch M, et al. The diarylquinoline TMC207 for multidrug-resistant tuberculosis. N Engl J Med. 2009;360:2397–405.
Moellering RC Jr. Linezolid: the first oxazolidinone antimicrobial. Ann Intern Med. 2003;138:135–42.
Linezolid, Zyvox 600 mg film-coated tablets, Summary of Product Characteristics. https://www.medicines.org.uk/emc/product/1688/smpc. Accessed 17 Feb 2021.
Zítková L, Tousek J. Pharmacokinetics of cycloserine and terizidone: a comparative study. Chemotherapy. 1974;20:18–28.
van Rijn SP, van Altena R, Akkerman OW, et al. Pharmacokinetics of ertapenem in patients with multidrug-resistant tuberculosis. Eur Respir J. 2016;47:1229–34.
Modongo C, Pasipanodya JG, Magazi BT, et al. Artificial intelligence and amikacin exposures predictive of outcome in multidrug-resistant tuberculosis patients. Antimicrob Agents Chemother. 2016;60:5928–32.
Park S-I, Oh J, Jang K, et al. Pharmacokinetics of second-line antituberculosis drugs after multiple administrations in healthy volunteers. Antimicrob Agents Chemother. 2015;59:4429–35.
Acknowledgements
Anne-Grete Märtson was funded by Marie Skłodowska-Curie Actions [Grant agreement no. 713660—PRONKJEWAIL—H2020-MSCA-COFUND-2015].
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Marieke G.G. Sturkenboom, Anne-Grete Märtson, Elin M. Svensson, Derek J. Sloan, Kelly E. Dooley, Simone H.J. van den Elsen, Paolo Denti, Charles A. Peloquin, Rob E. Aarnoutse, and Jan-Willem C. Alffenaar have no conflicts of interest that are directly relevant to the content of this article.
Funding
No funding was received for the preparation of this article.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Code availability
Not applicable.
Authors’ contributions
JWCA and MGGS conceived the presented idea. AGM, MGGS, JWCA, EMS, DJS, KED, SHJvdE, PD, CAP, and REA undertook the search and wrote the different parts of the first draft of the manuscript. MGGS and AGM constructed the tables. AGM constructed the figure. Critical revision was undertaken by MGGS, AGM, JWCA, EMS, DJS, KED, SHJvdE, PD, CAP, and REA. All authors read and approved the final version of the manuscript.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Sturkenboom, M.G.G., Märtson, AG., Svensson, E.M. et al. Population Pharmacokinetics and Bayesian Dose Adjustment to Advance TDM of Anti-TB Drugs. Clin Pharmacokinet 60, 685–710 (2021). https://doi.org/10.1007/s40262-021-00997-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-021-00997-0