
Abstract
A carbided molybdena on sulfated zirconia support has been prepared by in situ exposure to a methane/hydrogen mixture at 650 °C and the characteristics established using N2 adsorption and X-ray diffraction. The activity of the catalyst in the hydroconversion of C6 to C9n-alkanes was investigated in the temperature range 350–450 °C at 1 atm pressure. Activity and selectivity were found to show strong dependence on the choice of n-alkane with reaction rates found to be higher for the shorter alkanes. n-Hexane and n-heptane were hydroisomerised to the corresponding C6 and C7 isomers with high selectivity while the higher alkanes, especially nonane, produced mainly hydrocracking products. The catalyst offers a potential system for the upgrading of light naphtha range paraffins.
Similar content being viewed by others
Introduction
The phasing out of tetraethyl lead, along with other gasoline additives such as methylcylopentadienyl manganese tricarbonyl and aromatics, following legislation prompted by environmental concerns has led to the search for alternative method of increasing the octane number of gasoline (Mokaya et al. 1997). This can be achieved by converting the linear alkanes in the gasoline pool into their corresponding isomers, as the latter have higher octane number. For example, the respective octane numbers of n-octane and n-heptane are −15 and 0 compared to 100 and 89.0 for 2,2,4-trimethylpentane and 2,3-dimethylpentane, respectively. Hydroisomerisation of n-alkanes is carried out at high hydrogen pressure (>20 bar) over a bifunctional catalyst containing a metal function and appropriate support material. The metal catalyses the dehydrogenation of the n-alkane to the corresponding n-alkene and the hydrogenation of the iso-alkene into iso-alkane in the final stage of the reaction. In addition to the good surface area to maximize the dispersion of the metal particles, the support requires Brönsted acid sites to convert the n-alkene into n- and iso-carbenium ions at the intermediate stage. To minimize the extent of hydrogenolysis reaction on the metal sites, hydrocracking on the Brönsted acid sites as well as other side reactions such as oligomerisation, dehydrocyclisation and aromatisation, the catalyst and the reaction conditions must be designed such that the metal function and the Brönsted acid sites are present in an appropriate quantity. Several industrial catalysts had been tested and abandoned due to associated challenge(s). Freidel Crafts and chlorinated alumina catalysts were the most widely accepted materials by most petroleum refineries during the earlier stages due to their low temperature activity; however, these were abandoned due to corrosion and disposal problems (Iglesia et al. 1993; Ono 2003). Noble metal supported heteropoly acid such as H3PMo12O40, H4SiW12O40, H3PW12O40 and H4SiMo12O40 have been explored. For example, Kozhevnikov (1998) correlated the Brönsted acidity of these compounds with activity for hydroisomerisation at temperatures up to 465 °C, and compared the behavior with other solid acids such as HX and HY zeolites, SiO2–Al2O3 and H3PO4/SiO2. Among these catalysts, those based on heteropoly acids showed the highest selectivity and yields toward branched isomers. The main problems associated with these compounds are poor thermal stability at high temperatures and loss of acidity with time (Kozhevnikov 1998; Travers et al. 2001). A slight deactivation leading to decreased acidity was observed for operational temperatures greater than 350 °C, although catalysts regeneration was possible. Catalysts based on sulfated zirconia show good activity for the isomerization of lighter alkanes, especially n-butane. These materials, however produce large carbonaceous deposits with higher alkanes, they have poor resistance to catalyst poisons such as water and are associated with significant loss of acidity at higher temperatures due to loss of sulfate species (Vera et al. 1999; Comelli et al. 1995; Ahmad et al. 2003; Ono 2003). Pt and Pd supported zeolites have been the most widely studied catalysts for hydroisomerisation in the recent years. They can provide optimal yields of mono- and multiple branched-alkanes and their properties could be modified by treatment with both solid and gaseous modifiers (Lei et al. 1999; De Lucas et al. 2004, 2005; Woltz et al. 2006). Like many other catalysts, the key challenges associated with zeolites in hydroisomerisation include the prevention of cracked hydrocarbon formation and loss of acidity due to coke formation.
The numerous challenges associated with the various catalysts necessitate the search for more appropriate materials. Catalysts based on molybdenum carbides have been tested in a number of hydrotreating reactions (especially hydrodenitrogenation and hydrodesulfuration) and give excellent selectivities, thermal stability and resistance to catalyst poisons (Bussell et al. 2003; Da Costa et al. 2004). They are also considerably cheaper than the bifunctional zeolites. These advantages suggest that if molybdenum carbides could be appropriately designed that they might form good hydroisomerisation catalysts. This study reports on the hydroisomerisation of C6 to C9 alkanes over β-Mo2C supported on sulfated ZrO2 prepared by in situ carburisation.
Supported molybdenum oxide catalysts are often prepared by impregnating the support with a solution of soluble molybdenum compound, including ammonium heptamolybdate due to its appreciable solubility (0.43 g mL−1) in water at room temperature. The ammonium ions are readily removed during calcination. An alternative to this is the “slurry method”, which involves direct impregnation of MoO3 oxide slurry with the support. Although the solubility of MoO3 in water (about 0.001 g mL−1) is very low, sufficient deposition over the support can be achieved. As the dissolved species are deposited, more solid MoO3 is dissolved, and at the end of the impregnation all the MoO3 species may be deposited. This method requires no calcination (Zdrazil 2001). In this study the former widely performed method was employed. The surface molybdenum based species present in the air-exposed and hydrated catalyst is a function of the pH in the hydrated layer during impregnation. Highly dispersed species can be formed over sulfated zirconia due to high surface density of hydroxyl groups present on this material (Cheng et al. 1999). Carburisation of the molybdena formed after calcination should be carried out in situ to avoid contact of the generated β-Mo2C with air. In the presence of air, even in trace quantities, transition metal carbides oxidize rapidly to yield an oxy-carbide phase (York et al. 1997). Various methods for preparing molybdenum carbide (Mo2C) catalysts have been reported. These cover carburization of pure or supported molybdenum metal, molybdenum oxides or molybdenum nitride with an appropriate carbon source (Saito and Anderson 1980; Miyao et al. 1997; Tack Jung et al. 2004; Wang et al.2006). In many reports hydrocarbon/H2 mixtures are employed but these reagents may give rise to low specific area materials at low flow rates. To obtain a high surface area material, high flow rates are required coupled with temperatures closer to 1,000 °C. This high temperature condition can be significantly reduced by selecting an appropriate H2/CH4 flow ratio (Miyao et al. 1997; Wang et al.2006). In this study, a 4:1 (H2:CH4) ratio was selected and the carburization process was conducted at 650 °C.
Experimental
Catalyst preparation
Preparation of MoO3/SO42−–ZrO2
The sulfated zirconia was prepared by calcination of a sample of sulfated zirconium hydroxide (MEL chemicals, Analar grade) by heating at 500 °C for 3 h in a flow (50 ml min−1) of air. An amount (4.60 g) of ammonium heptamolybdate tetrahydrate ([(NH4)6Mo7O24·4H2O], Fischer Scientific, Laboratory grade reagent) to give 37.5 wt% of MoO3/SO42−–ZrO2 (25 wt% based on Mo content) was completely dissolved in 100 mL of distilled water with the aid of stirring, after which an appropriate mass of sulfated zirconia was added. Excess water was removed by means of rotary evaporation at 80 °C and the resulting solid dried in an oven at 120 °C overnight. The resulted material was then calcined in air flow (50 mL min−1) at 550 °C for 3 h and then stored in a sample vial.
Preparation of β-Mo2C/(S)ZrO2
The active phase of the catalyst was generated by in situ carburization of MoO3/SO42−–ZrO2. Generally, 0.2 g of the precursor diluted with 0.5 g of SiC as an inert diluent was loaded into a quartz reactor between two quartz wool plugs. The carburization treatment was carried out at 650 °C using a 1:4 volumetric flow of CH4:H2 for 1 h 15 min before cooling to the desired reaction temperature in 20 ml min−1 H2.
Catalyst characterization
BET surface area
Nitrogen adsorption experiments to determine the surface area were carried out using a Beckman Coulter SA 3100 instrument with the sample held at liquid nitrogen temperature. Samples were previously degassed overnight at 200 °C under vacuum to remove physically adsorbed water molecules and any other adsorbed gases.
X-ray diffraction studies
Structural characterization of the samples was carried out using powder X-ray diffraction (XRD). About 0.5 g of a sample was loaded into a Bruker-D2 phaser XRD difractometer employing Cu Kα monochromatic radiation. Measurements were performed at 22 °C with steps of 0.02° using a 2θ range of 2–80° and 0.15°/step. The phases were identified with the aid of a program developed in Bruker-D8 software stored in a PC connected to the diffractometer.
Catalytic measurements
Hydroconversion of the C6 to C9 alkanes was carried out with a quartz reactor (1.0 cm i.d., 25-cm long) at atmospheric pressure. The catalyst bed consisted of 200 mg catalyst diluted with 0.5 g SiC in order to stabilize flow conditions and was fixed between two quartz wool plugs. Hydrogen was employed as a carrier gas normally at a flow of 20 ml min−1. The n-alkane flow was generally fixed at 15.0 μL min−1. The corresponding GHSVs were 2.95, 3.07, 3.16 and 3.23 h−1 for n-hexane, n-heptane, n-octane and n-nonane, respectively. Reaction temperature was varied from 350 to 450 °C. The products were analysed with a gas chromatograph (Perkin-Elmer 8410 fitted with a 5.6-m long 15% squalane on chromosorb WHP column and equipped with flame ionization detector). Sampling was carried out with the aid of a ten-port multi-loop VICI Valco sampling valve. Reaction rates and rate constants (kT) for the reaction at various temperatures were computed as follows. Reaction rates were calculated from conversions, molar flow rates and mass of catalysts employed. The rate constants at various temperatures were derived from the reaction rates and partial pressures of both hydrogen and alkanes. Values obtained were employed using an Arrhenius plot to determine the apparent activation energies.
Results
Physicochemical properties
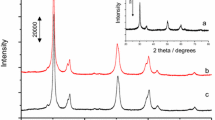
Physicochemical properties derived from the adsorption of nitrogen on calcined support, MoO3/SO42−–ZrO2 and β-Mo2C/SO42−–ZrO2 are reported in Table 1. Data indicate that the surface area and pore distributions were not significantly altered by the addition of molybdena (followed by calcination) to the calcined sulfated zirconia support. On the other hand, carburisation treatment led to a loss in the BET surface area from 124 to 98 m2 g−1. However, the microporosity, mesoporosity, macroporosity, pore volume all increased quite significantly as a result of the treatment in methane and hydrogen at 650 °C. The loss in surface area had the consequence of increasing the density of accessible surface Mo atoms per unit area. X-ray diffractograms (Fig. 1) show the precursor MoO3/SO42−–ZrO2 materials to contain weak broad features suggesting poorly crystalline molybdena phase (pattern A). The carburised material was more crystalline with sharper peaks (pattern B). The intense peaks in the diffractogram are features of the SiC diluent. The key features are the loss of reflections due to molybdena which were present in the diffractogram of the precursor materials and the appearance of peaks at 2θ values corresponding to 34.5, 37.9, 39.5, 61.5, and 69.6° which can be attributed to reflections of the β form of Mo2C (Zhu et al. 2007).

X-ray powder diffraction patterns of MoO3/SO42−–ZrO2 (pattern A) and SiC diluted β-Mo2C/SO42−–ZrO2 catalyst (pattern B). W = MoO3, X = SiC, Y = Mo2C and Z = ZrO2
n-Alkane hydroconversion
Catalysts were studied in the n-alkane conversion over a period of 6 h. During this period, the catalyst showed good stability independent of the n-alkane employed as feed over the 6 h period with no signs of significant deactivation or changes in selectivity patterns. An example of this behavior is illustrated by the use of n-heptane conversion at 450 °C. Under constant conditions, both conversion (Fig. 2) and products selectivities (Fig. 3) remained almost invariant. The steady state activities for C6 to C9n-alkane hydroconversion over the β-Mo2C/SO42−–ZrO2 catalyst in the temperature range of 350–450 °C are presented in Fig. 4. A maximum rate of 314.0 μmol min−1g−1 was obtained with n-hexane at 450 °C (Fig. 4) while the lowest rate of 5.88 μmol min−1g−1 was observed with n-nonane at 350 °C. At a particular temperature, the rate decreased with increasing n-alkane chain length. Table 2 shows the selectivity the reaction of the different alkanes toward hydrocracking and hydroisomerisation products. Note that under the reaction conditions employed, no cyclised or dehydrocyclised products were detected. This contrast with the behavior of Pt-based catalysts under equivalent conditions (Anderson et al. 1989). With n-octane and n-nonane, the selectivity to hydrocracking products reached 100% at all the temperatures. Butanes formed the dominant species with n-octane while propane and methane are the major reaction products for n-nonane. On the other hand n-hexane and n-heptane produced mainly hydroisomerisation products with selectivity ≥85.0%. The breakdown of the products from hexane and heptanes reactions is shown in Table 3. Increasing the temperature increased conversion but led to slight loss of hydroisomerisation selectivity (Table 2) as a result of secondary reactions. The results for the apparent activation energies of the various reactions are presented in Table 4. For the n-alkanes, the Ea values increased with chain length. The activation energy for n-hexane was 98.0 kJ mol−1 and this increased to 122.1 kJ mol−1 for n-nonane. Values are in reasonable agreement those reported in the literature for other, mainly zeolitic type catalysts.

n-Heptane conversion over 0.2 g catalyst at 450 °C, 1 atm pressure

Products selectivities during n-heptane conversion over 0.2 g catalyst at 450 °C, 1 atm pressure where the square block represents isomers and circular block represents selectivity to hydrocracking

Relationship between n-alkane chain length and reaction rates over 0.2 g catalyst at temperatures of (square block ) 350 (circular block) 400 and (triangular block) 450 °C and 1 atm pressure
2-Methylheptane reaction
An exception to the trend of increasing apparent activation energy with chain length was observed when 2-methyl heptanes, as an example of a branched alkane, was tested. The value determined for this 8-carbon molecule lay between the values for n-hexane and n-heptane, thus showing the relative ease with which the hydrocarbon may be activated over this catalyst when it contains a carbon atom attached to more than two carbons. The results of the reactions of 2-methylheptane over β-Mo2C/SO42−–ZrO2 are presented in Figs. 5, 6, 7, 8. Figures 5 and 6 show the effect of temperature on conversion and products selectivity, respectively. 2-methylheptane conversion and selectivity to hydrocracking increased with temperature while the selectivity to hydroisomerisation decreased as temperature was increased, consistent with behavior of the n-alkanes. Consistent with expectation, increasing the flow of H2 decreased the conversion of 2-methylheptane and hydrocracking selectivity but increased the production of hydroisomerisation products (Figs. 7, 8).

2-Methylheptane conversion at temperatures of 250–450 °C, 1 atm pressure and 0.2 g catalyst loading

2-Methylheptane products selectivities to isomers (square block) and (circular block) total hydrocracking products for reactions over 0.2 g of catalyst at 250–450 °C and 1 atm pressure

Influence of H2 flow rate on 2-methylheptane conversion using 0.2 g catalyst at 400 °C, 1 atm pressure

Influence of H2 flow on 2-methylheptane products selectivities over 0.2 g catalyst at 400 °C and 1 atm pressure where (square block) represents total isomers and (circular block) represents selectivity to hydrocracking
Discussion
Catalyst synthesis and characterisation
The selection of the most appropriate carburization conditions is necessary to avoid excessive coke deposition and sintering of the active surface area that could lead to loss of catalyst activity. Although higher temperatures such as 750 °C with n-octane as carburizing agent achieved conversion to the molybdenum carbide, significant levels of carbonaceous deposition also occurred. Carburisation was optimized using a 4:1 (H2/CH4) reagent ratio at 650 °C. An additional, potential advantage of this moderate temperature condition is in avoiding excessive sulfate loss through reduction and subsequent H2S evolution which is known to occur for sulfated zirconias at temperatures above 700 °C (Bensitel et al. 1988; Xu and Sachtler 1997; Rosenberg et al., 2003). TPR of the sulfated zirconia employed here showed a maximum at 660 °C. In addition to partial sulfate loss, the BET surface area of the sample decreased after the carburisation treatment suggesting perhaps pore mouth blockage or pore filling by carbonaceous deposition. However, data indicates that the carbided phase had a greater internal surface area than its precursor phase mainly arising from increased mesoporosity (0.1213 ml g−1), along with increased concentration of micropores (0.0144 ml g−1) and macropores (0.0370 ml g−1). Such enhancement in mesoporous volume may be beneficial as hydroconversion of n-alkanes over mesoporous catalysts has been reported to yield high concentration of both mono- and multiple branched alkanes because of their ability to diffuse faster through the pores. Recently, Moushey and Smirniotis (2009) reported mesoporous ZSM-12 and Beta zeolites were able to yield ~90% n-heptane hydroisomerisation selectivity.
The appearance of X-ray diffraction patterns of MoO3 are influenced by both loading and crystallite sizes. Using a trial loading of ca. 15 wt% MoO3, the supported phase could not be detected by XRD suggesting the presence of a highly dispersed species. For a support of ca 100 m2g−1, this loading is above the value of 9 wt% which is required for theoretical monolayer (Meunier et al. 1997). The diffraction pattern of the 37.5 wt% of MoO3/SO42−-ZrO2 (Fig. 1) shows weak signals below 30o, 2θ (25.7, 27.3 and 29.4o), indicating that the loading employed results mainly in the formation of structures which are poorly crystalline and/or consist of many crystallites close to or below 4 nm (Chary et al. 2004). Use of Debye–Scherer equation indicates that the starting material consists of molybdena structures with crystallite sizes close to 9 nm. The reflections due to ZrO2 in the same diffractogram are also weak and of low intensity and correspond with 8 nm crystallite size. Similar diffractograms for materials of similar high loadings have been interpreted to infer strong Mo/Zr interactions (Christodoulakis and Boghison 2008). ZrO2 material may exist as one or more of the three well-studied type of phases; tetragonal, cubic and monoclinic (Chary et al. 2004). The diffraction pattern shows diffraction angles of 2θ at 30.2, 35.4, 50.2 and 60.1o, among which the strongest peak is 30.2o (Fig. 1) confirming the presence of tetragonal zirconia. This indicates that the crystallographic form of the SO42−–ZrO4 material is stable under the carburization conditions employed. Although the monoclinic sulfated zirconia has been demonstrated to show some activity for alkane isomerisation, the tetragonal phase is reported to be a more active form, especially at lower temperatures (Stichert et al. 2001). However, when support in the absence of molybdena was carburized in methane and hydrogen at 650 °C (i.e., same conditions employed for supported catalyst), only ca. 1% n-hexane conversion was obtained at 450 °C. The reaction produced cracking products with 30 and 70% selectivity to methane and propane, respectively. Therefore the 55% conversion and high hydroisomerisation selectivity (~90%) achieved over the supported catalyst under similar conditions can be deduced to require the presence of the β-Mo2C phase. The low contribution from the support alone was at least in part due to the deactivation of the sulfated zirconia due to sulfate loss rather than being related to a requirement to have bifunctional active components. When the sulfated zirconia was pretreated at 450 °C in octane/hydrogen rather than 650 °C in methane/hydrogen, a conversion of 30% was attained for similar reaction conditions.
Molybdenum carbide exhibits six different phase, with varying stabilities depending on conditions. These phases are α-Mo2C, β-Mo2C, γ-MoC and the other three different phases represented as δ-MoC1−X, γI-MoC1−X and η-MoC1−X with similarity in carbon content and thermal transition properties. Among the various phases, only β-Mo2C and γ-MoC are stable at room temperature. α-Mo2C is stable only at temperatures above 1,000 °C while δ-MoC1−X and η-MoC1−X are stable only at temperatures closer to 1,700 °C, which greatly hinders their exploitation for commercial applications. The weak signals at 34.5, 37.9, 39.5, 61.5 and 69.60 in diffractograms of samples after carburisation (Fig. 1) are characteristic of β-Mo2C (Zhu et al. 2007). In hydrogen alone at 823 K, reduction may be limited to MoO (Faraldos et al.1996). The formation of Mo2C may proceed via the reduction of MoO3 to MoO2 and then to Mo by both H2 and CH4 (Xiao et al. 2000; Hanif et al. 2002). The disappearance of features due to MoO3 in the XRD patterns (Fig. 1) is consistent with either of these transformations. The final state is β-Mo2C (Fig. 1) according to powder diffraction patterns. Although transition to oxycarbide is possible on exposure to air, the low temperature oxidation process can be inhibited by catalyst exposure to hydrocarbon reactants and/or passivation with O2/He atmosphere prior to air exposure (Lu et al. 2000; Hanif et al. 2002; Solymosi and Barthos 2005).
Catalytic reactions
Effect of chain length on reaction rates
The rate of the hydroconversion was found to vary significantly, depending upon the length of the n-alkane (Fig. 2). Slow hydroconversion rates of 5.88, 7.56 and 11.3 μmol min−1g−1 were measured for n-nonane at 350, 400 and 450 °C, respectively. However, as the carbon number decreased, the hydroconversion rate increased and using n-hexane for example, the rates were found to be 43.7, 37.7 and 27.8 times faster than the corresponding rates measured using n-nonane under similar conditions. For porous materials such as zeolites, the pore distribution of the active catalyst may be important as samples of higher mesoporosity allow faster reactions due to reduced diffusion limitations (Lungstein et al. 1999; Komatsu 2010). The rates were found to increase with temperature for all the n-alkanes, but with larger proportional increases found for the longer alkanes, consistent with their higher activation energies (Table 4). While the reaction rates were influenced by chain length, the stability of the catalyst was similar with all the n-alkanes and showed remarkable stability and resistance to deactivation. At 450 °C the conversion of n-heptane varied from 55.00 to 55.05% (Fig. 2) and the selectivities ranged from 84.89 to 85.04% and 14.98 to 15.03% (Fig. 3) for the hydroisomerisation and hydrocracking products, respectively. The trend was found to be similar for the other alkanes, indicating very good stability over a 6-h period.
Effect of chain length and reactant structure on products distribution
Results for the distribution of the categories of reaction products for the various alkanes (Table 2) indicate that, n-nonane and n-octane produced 100% hydrocracking products at all temperatures studied while n-heptane and n-hexane produced hydroisomerisation products with a high selectivity (Table 2 and 3). A broad range of C1 to C6 products was found for n-nonane while n-octane produced C1 to C5 species. The presence of isobutanes, isopentanes and isohexanes among the reaction products suggests that the iso-nonanes and iso-octanes may have been produced initially but that these primary products underwent facile hydrocracking to give lighter alkanes. The test results using 2-methyl heptane (Fig. 5) suggests that branched hydrocarbons are more reactive than n-alkanes. This is consistent with the findings that the activation energy required for n-octane was 110.38 compared to 100.11 kJ mol−1 for 2-methylheptane (Table 3). This indicates that isoalkanes are, at least for the longer alkanes, more active than the n-alkanes themselves and undergo rapid conversion, thereby severely limiting the selectivity to isomers that can be obtained at high conversions. Groten and Wojciechowski (1993) and De Lucas et al. (2005) have shown that n-nonane and n-octane are prone to hydrocracking (≥90%) over metal supported zeolites at temperatures greater than 350 °C. At GHSV of 639 and 852 h−1. Here, n-octane produced traces (~3%) of 2,2,4-trimethylpentane, the hydrocracking of which normally produces propane, butanes and pentanes. The higher space velocity reduced the extent of hydrocracking and shifted the reaction toward hydroisomerisation indicating again that hydrocracking resulted from conversion of primary isomerisation products. Reduced contact times and higher hydrogen partial pressure diminish the extent to which carbonaceous deposits are generated from oligomerisation and/or dehydrocyclisation reactions (Lungstein et al. 1999; Komatsu 2010). Hydroisomerisation of n-hexane and n-heptane produced 2,2-dimethylbutane and 2,2-dimethylpentane as the major respective reaction products with traces of monobranched isomers (Table 3). Selectivity to isohexanes was greater than selectivity to iso-heptanes at all the temperatures. n-Heptane produced methane, propane and butanes while n-hexane produced methane, propane and pentanes as the only hydrocracking species. Work by Hunter and East (2002) would suggest that the bond strengths of the central C–C for the C6 to C10n-alkanes do not differ significantly and thus cannot be used to explain difference in behavior of the series. On the other hand, Brönsted acid mediated fission reactions involving the central C–C bonds become more favorable as chain length increases. This does not account, however, for the relative ease of activation of the shorter, compared with longer alkanes or the relative selectivities observed. It is clear that in order to account for both, isomerisation has to be favored, irrespective of chain length for all n-alkanes but with the shorter alkanes being more readily activated (Table 3), whereas the isomers formed are more stable for those derived from short chain alkanes and consequently undergo secondary reactions at a lesser rate. Work is ongoing to establish the contributions to the overall mechanism played by the residual sulfate on the zirconia and whether this carbided MoO3/SO42−–ZrO2 system does operate as a truly bifunctional catalyst.
Conclusions
β-Molybdenum carbide supported tetragonal sulfated zirconia was successfully synthesized from in situ carburization of 37.51 wt% MoO3/SO42−–ZrO2 at lower temperatures (650 °C) than those routinely employed (1,000 °C). The catalyst activity was highly dependent on the length of the n-alkane with shorter alkanes undergoing more rapid reaction. Hydroisomerisation products dominate for the shorter alkanes while the higher alkanes produce products resulting from hydrocracking. Although n-nonane and noctane were converted mainly to hydrocracking products, the high selectivities to hydroisomerisation obtained with n-heptane and n-hexane (≥90%) at all the temperatures employed suggests the catalyst to have a good potential as hydroisomerisation material and possibly a replacement to the currently used catalysts that are either very expensive or rapidly deactivate due to poor thermal stability, coke deposition or interaction with catalyst poison(s) even in trace quantities.
References
Ahmad R, Melsheimer J, Johntoff FC, Schlogl R (2003) Isomerisation of n-butane and n-pentane in the presence of sulphated zirconia: formation of surface deposits investigated by In situ UV–vis diffuse reflectance spectroscopy. J Catal 218(2):365–374
Allain JF, Magnoux R, Schulz MPh, Guisnet M (1997) Hydroisomerization of n-hexane over platinum mazzite and platinum mordenite catalysts kinetics and mechanism. Appl Catal A: Gen 152:221–235
Anderson JA, Mordente MGV, Rochester CH (1989) Effects of oxidation/reduction on Pt/Al2O3 on catalytic activity and selectivity for hexane reforming. J Chem Soc Faraday Trans 1(85):2991–2998
Bensitel M, Saur O, Lavelley JC (1988) An infrared study of sulfated zirconia. Mater Chem Phys I 9(1988):141–156
Bussell ME, Sawhill SJ, Layman KA, Burns AW (2003) New catalysts for. hydrodesulfurization. Fuel Chem Div Prepr 48(1):177–179
Chary KVR, Reddy KR, Kishan G, Niemantsverdriet JW, Mestl G (2004) Structure and catalytic properties of molybdenum oxide catalysts supported on zirconia. J Catal 226:283–291
Cheng MF, Kumata T, Saito T, Komatsu T, Yashima (1999) Preparation and characterization of Mo catalysts over AlMCM-41/γ-Al2O3 extruded supports. Appl Catal A: Gen 183(1):199–208
Christodoulakis A, Boghison S (2008) Molecular structure and activity of molybdena catalysts supported on zirconia for ethane oxidative dehydrogenation studied by Operando Raman Spectroscopy. J Catal 260:178–187
Comelli RA, Vera CR, Parera JM (1995) Influence of ZrO2 crystalline structure and sulfate ion concentration on the catalytic activity of SO42−- ZrO2. J Catal 151:96–101
Da Costa P, Potvin C, Manoli J, Genin B, Djega-Mariadassuo G (2004) Deep hydrodesulphurization and hydrogenation of diesel fuels on alumina-supported and bulk molybdenum carbide catalysts. Fuel 83(13):1717–1726
De Gauw, F.J.M.M (2002) Kinetic studies of alkane hydroisomerization over solid acid catalysts. PhD Thesis, Eindhoven University of Technology, Netherlands. Available from: http://alexandria.tue.nl/extra2/200210842.pdf
De Lucas A, Valverde JL, Sánchez P, Dorado F, Ramos MJ (2004) Influence of the binder on the n-octane hydroisomerization over palladium-containing zeolite catalyststs. Ind Eng Chem Res 43:8217
De Lucas A, Valverde JL, Sánchez P, Dorado F, Ramos MJ (2005) Hydroisomerization of n-octane over platinum catalysts with or without binder. Appl Catal 282:15–24
De Lucas A, Sánchez P, Dorado F, Ramos MJ, Valverde JL (2006) Kinetic model of the n-octane hydroisomerization on ptbeta agglomerated catalyst: influence of the reaction conditions. Ind Eng Chem Res 45(3):978–985
Faraldos M, Bañares MA, Anderson JA, Hu H, Wachs IE, Fierro JLG (1996) Comparison of silica supported MoO3 and V2O5 catalysts in the selective oxidation of methane. J Catal 160:214–221
Groten WA, Wojciechowski BW (1993) The kinetics of hydrocarbon cracking : the cracking of n-nonane. J Catal 140:262–280
Hanif A, Xiao T, York APE, Sloan J, Gren MLH (2002) Study on the structure and formation mechanism of molybdenum carbides. Chem Mater 14:1009–1015
Hunter KC, East ALL (2002) Properties of C–C bonds in n-Alkanes: Relevance to cracking mechanisms. J Phys Chem A 106(7):1346–1356
Iglesia E, Soled SL, Kramer GM (1993) Isomerization of alkanes on sulfated zirconia: promotion by Pt and by adamantyl hydride transfer species. J Catal 144:238–253
Jung KT, Kim WB, Rhee CH, Lee JS (2004) Effects of transition metal addition on the solid state transformation of molybdenum trioxide to molybdenum carbides. Chem Mater 16:307–314
Kinger G, Vinek H (2001) n-Nonane hydroconversion on Ni and Pt containing HMFI, HMOR and HBEA. Appl Catal A: Gen 218(2001):139–149
Kinger G, Majdal D, Vinek H (2002) n-Heptane hydroisomerization over Pt-containing mixtures of zeolites with inert materials. Appl Catal A: Gen 225(2002):301–312
Komatsu T (2010) Catalytic cracking of paraffins on zeolite catalysts for the production of light olefins. In: 20th Annual Saudi-Japan Symposium; Catalysts in Petroleum Refining and Petrochemicals, Dharan, Saudi-Arabia, December, 2010
Kozhevnikov IV (1998) Catalysis by heteropoly acids and multicomponent polyoxometalates in liquid-phase reactions. Chem Rev 98:171–198
Lei T, Xu JS, Gao Z (1999) Acidity enhancement of H-mordenite by sulfation. Mater Chem Phys 60(2):177–181
Lu L, Hugosson H, Eriksson O, Nordstrom L, Jansson U (2000) Chemical vapour deposition of molybdenum carbides: aspects of phase stability. Thin Solid Films 370(17):203–212 (1–2)
Lungstein A, Jentys A, Vinek H (1999) Hydroisomerization and cracking of n-octane and C8 isomers on Ni-containing zeolites. Appl Catal A: Gen 176:119–128
Meunier FC, Yasmeen A, Ross JRH (1997) Oxidative dehydrogenation of propane over molybdena-containing catalysts. Catal Today 37:33–43
Miyao T, Shishikura I, Matuoka M, Magai M, Oyama ST (1997) Preparation and characterization of alumina-supported molybdenum carbide. Appl Catal A: Gen 165:419–428
Mokaya R, Jones W, Moreno S, Poncelet G (1997) n-Heptane hydroconversion over aluminosilicate mesoporous molecular sieves. Catal Lett 49:87–94
Moushey DL, Smirniotis PG (2009) n-Heptane hydroisomerisation over mesoporous zeolites made by utilizing carbon particles as the template for mesoporosity. Catal Lett 129:20–25
Ono Y (2003) A survey of the mechanism in catalytic isomerization of alkanes. Catal Today 81:3–16
Rosenberg DJ, Bachiller-Baeza B, Dines TJ, Anderson JA (2003) Nature of surface sulfate species and the generation of active sites on silica-zirconia mixed-oxide catalysts. J Phys Chem B 107:6526
Saito M, Anderson RB (1980) The activity of several molybdenum compounds for the methanation of CO. J Catal 63:438–446
Solymosi F, Barthos R (2005) Aromatization of n-hexane on Mo2C catalysts. Catal Letters. 101(3–4):235–239
Stichert W, Schüth F, Kuba S, Knözinger H (2001) Monoclinic and tetragonal high surface area sulfated zirconias in butane isomerization: CO adsorption and catalytic results. J Catal 198:277–285
Travers C, Essayemb N, Delaga M, Quelen S (2001) Heteropolyanions based catalysts for paraffins isomerization. Catal Today 65:355–361
Vera CR, Pieck CL, Shimizu K, Querini CA, Parera JM (1999) Coking of SO42−–ZrO2 catalysts during isomerization of n-butane and its relation to the reaction mechanism. J Catal 187:39–49
Wang X, Hao H, Zhang M, Li W, Tao K (2006) Synthesis and characterization of molybdenum carbides using propane as carbon source. J Solid State Chem 179:538–543
Woltz C, Jentys A, Lercher JA (2006) Improving bifunctional zeolite catalysts for alkane hydroisomerization via gas phase sulfation. J Catal 237:337–348
Xiao T, York APE, Williams VC, Almegren H, Hani A, Zhou X, Green MLH (2000) Preparation of molybdenum carbides using butane and their catalytic performance. Chem Mater 12:3896–3905
Xu B, Sachtler WMH (1997) Reduction of SO =4 ions in sulfated zirconia catalysts. J Catal 167(1):224–233
York APE, Pham-Huu C, Gallo PE, Ledoux MJ (1997) Molybdenum oxycarbide hydrocarbon isomerization catalysts: cleaner fuels for the future. Catal Today 35:51–57
Zdrazil M (2001) Supported MoO3 catalysts: preparation by the new “slurry impregnation” method and activity in hydrodesulphurization. Catal Today 65:301–306
Zhu Q, Chen Q, Yang X, Ke D (2007) A new method for the synthesis of molybdenum carbide. Mater Letters 61:5173–5174
Acknowledgments
The authors are grateful to Petroleum Technology Development Fund (PTDF), Nigeria for the award of a scholarship (to A. G.).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Galadima, A., Wells, R.P.K. & Anderson, J.A. n-Alkane hydroconversion over carbided molybdena supported on sulfated zirconia. Appl Petrochem Res 1, 35–43 (2012). https://doi.org/10.1007/s13203-011-0004-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13203-011-0004-0