
Abstract
Extracellular vesicles are cell-derived membrane particles ranging from 30 to 5,000 nm in size, including exosomes, microvesicles, and apoptotic bodies. They are released under physiological conditions, but also upon cellular activation, senescence, and apoptosis. They play an important role in intercellular communication. Their release may also maintain cellular integrity by ridding the cell of damaging substances. This review describes the biogenesis, uptake, and detection of extracellular vesicles in addition to the impact that they have on recipient cells, focusing on mechanisms important in the pathophysiology of kidney diseases, such as thrombosis, angiogenesis, tissue regeneration, immune modulation, and inflammation. In kidney diseases, extracellular vesicles may be utilized as biomarkers, as they are detected in both blood and urine. Furthermore, they may contribute to the pathophysiology of renal disease while also having beneficial effects associated with tissue repair. Because of their role in the promotion of thrombosis, inflammation, and immune-mediated disease, they could be the target of drug therapy, whereas their favorable effects could be utilized therapeutically in acute and chronic kidney injury.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Intercellular communication is essential for multicellular organisms and cells communicate by a variety of mechanisms such as direct cell–cell contact, transfer of secreted molecules or intercellular transfer of extracellular vesicles (EVs). EVs are membrane-bound vesicles released by cells under physiological and pathological conditions. As EVs circulate in the blood, they may act as shuttle vectors or signal transducers both locally and at a distance from their site of origin [1]. Another function of EVs is the removal of unwanted molecular material or cellular waste [2], conceivably as a means of maintaining cellular integrity.
Extracellular vesicles are subdivided into exosomes, microvesicles, and apoptotic bodies (Table 1). Exosomes are the smallest vesicles (30–100 nm) released by the fusion of multivesicular bodies containing intraluminal vesicles with the plasma membrane. Microvesicles are vesicular structures (0.1–1.0 μm) shed by outward blebbing of the plasma membrane. The largest EVs (1–5 μm) are apoptotic bodies that are formed during the late stages of apoptosis [5, 10]. These subtypes of extracellular vesicles differ in their mechanism of biogenesis, as described below. This review focuses mainly on exosomes and microvesicles. Certain studies have not specifically analyzed the subtype of vesicle, in which case we refer to the general term EVs.
Although microvesicles and exosomes are structurally similar, they differ in size, lipid composition, content, and cellular origin (Table 1). EVs may be shed, under physiological or pathological conditions, into the extracellular environment either constitutively or upon activation, hypoxia, oxidative stress, senescence or apoptosis [4]. The release of vesicles may be induced by the stimulation of purinergic receptors [11], by shear stress or apoptosis [12, 13] and by proinflammatory mediators [14] or thrombin [15]. In addition, bacterial virulence factors, such as Shiga toxin and lipopolysaccharides [16] and uremic toxins [17] induce the release of EVs.
Microvesicles carry membrane-derived receptors, proteins, including cytokines, chemokines, proteins involved in cellular signaling and/or migration, lipids, carbohydrates, and genetic material including mRNA and microRNAs (miRNAs) [4]. Their contents depend on the parent cell, the microenvironment and on the triggers preceding their release [5, 18,19,20,21]. The transfer of these substances to recipient cells may affect the phenotype of the target cell. EVs transport combinations of multiple mediators and are therefore considered a more powerful means of intercellular communication than the transfer of single molecules. Circulating microvesicles are mainly of platelet, erythrocyte, leukocyte, and endothelial origin [22,23,24,25]. Urinary microvesicles originate mainly from podocytes, tubular cells, and epithelial cells lining the urogenital tract [2].
Extracellular vesicle biogenesis and release
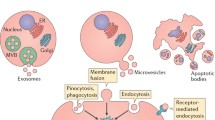
Exosomes are the product of the fusion of a subset of late endosomes, called multivesicular bodies, with the plasma membrane releasing their contents including intraluminal vesicles (ILVs). Once extracellular, these vesicles are termed exosomes (Fig. 1) [3]. ILV formation is regulated via the endosomal sorting complex required for transport (ESCRT, four protein complexes that guide intracellular cargo) [26], and/or by non-ESCRT-related mechanisms, including tetraspanins [27] and membrane lipids [28].

Schematic presentation of the release and uptake of extracellular vesicles. a Exosomes are released from late endosomes termed multivesicular bodies bearing intraluminal vesicles (ILVs) intracellularly. When the multivesicular bodies fuse with the plasma membrane and empty their contents, ILVs are released and are termed exosomes once they are extracellular. Exosomes are the smallest extracellular vesicles (Table 1). b Microvesicles are shed directly from the plasma membrane, thereby carrying membrane markers of the parent cell. Microvesicle formation is calcium-dependent and associated with loss of membrane asymmetry and disruption of the cellular cytoskeleton. c Extracellular vesicle uptake by target cells may occur via fusion of the vesicle membrane with the cell membrane or by endocytosis. The vesicle may also transduce an intracellular signal by ligand binding to a receptor on the recipient cell
Microvesicles are released from cells under physiological conditions, especially during cell growth [29]. Microvesicle shedding is increased when the cells are activated owing to cell injury, proinflammatory stimulants, hypoxia, oxidative stress or shear stress [30, 31]. Microvesicles are formed by outward protrusion or budding of the plasma membrane. This process is initiated by an increase in intracellular cytosolic calcium that activates calpain, a calcium-sensitive protease that detaches membrane proteins from the intracellular cytoskeleton [32], and gelsolin bound to actin filaments [33]. This leads to remodeling of the cytoskeleton, by cleaving the actin protein network, enabling blebbing to occur. Microvesicles are shed from plasma membrane micro-domains known as lipid rafts or caveolae domains [34]. The plasma membrane is composed of a lipid bilayer in which phosphatidylserine is located in the inner leaflet of the resting cell. The enzymes flippase, floppase, and scramblase control phospholipid asymmetry [35]. When the cell is activated, increased cytosolic calcium activates floppase (allowing lipid movement to the outer membrane) and scramblase (enabling bi-directional lipid movement), whereas flippase (allowing lipid movement to the inner membrane) is inactivated, resulting in flopping of negatively charged phosphatidylserine to the outer leaflet of the phospholipid bilayer [20]. This process does not always occur, as some microvesicles do not expose phosphatidylserine on their outer leaflet (Fig. 1) [36]. The presence of phosphatidylserine on the outer leaflet is readily detected, as it binds annexin V.
Microvesicles may express a slightly different repertoire of surface receptors or cytoplasmic components compared with the parent cell owing to a selective process during shedding [34]. Similarly, microvesicles released from activated cells do not express the same surface receptors as microvesicles shed during apoptosis [19] or from resting cells. This was demonstrated in vasculitis patients, microvesicles in patient samples taken during the active phase exhibited more CD62E and CD62P than those taken during remission and control samples [37].
Clearance and uptake of extracellular vesicles
The quantity of EVs in the circulation reflects a balance between their generation and clearance. Microvesicles released into the circulation have a half-life of a couple of minutes to a few hours [38], during which they may be taken up by neighboring or distant cells. In humans undergoing cardiopulmonary bypass, transfusion or dobutamine-induced cardiac stress, elevated levels of circulating microvesicles were detected, but these returned to baseline levels within 15 min to ~6 h [39, 40]. Interestingly, clearance of microvesicles may depend on the cellular origin of the microvesicles as platelet-derived microvesicles were cleared sooner than microvesicles released from red blood cells [40].
There are various mechanisms for the cellular uptake of vesicles depending on the cargo of the vesicle, intercellular communication (e.g. receptor-ligand interactions) and the microenvironment of the cell. The most common mechanism is endocytosis, whereby the extracellular vesicle is engulfed by the recipient cell [38]. There are several mechanisms of endocytosis, such as clathrin-dependent or -independent, caveolin-mediated, macropinocytosis, phagocytosis and lipid raft-mediated [41]. Uptake of EVs seems to depend on the type of recipient cell, its physiological state, and recognition of ligands or receptors on the recipient cell and EVs [41]. For example, vesicles shed from platelets interact with monocytes [42] and endothelial cells [22], but not with neutrophils [42]. Likewise, exosomes exposing the tetraspanin–integrin complex were selectively taken up by endothelial and pancreatic cells [43].
Another mechanism for microvesicle uptake is fusion, whereby the microvesicles fuse with the membranes of the recipient cell and the content of the vesicle is released into the cell. Platelets expressing P-selectin fuse with tissue-factor-rich monocyte-derived microvesicles, increasing the procoagulability of platelets [34]. Fusion efficiency is enhanced in an acidic microenvironment (Fig. 1) [44].
Detection
Extracellular vesicles are mostly detected in blood samples, but also in cerebrospinal fluid [45], urine [46], synovial fluid [47], bronchoalveolar lavage fluid [48], breast milk [49], bile [50], saliva [51], and uterine fluid [52], and the findings may reflect a process occurring on their cells of origin. Techniques for extracellular vesicle detection are listed in Table 2 and briefly described below. Given the heterogeneity of EVs the detection methods vary depending on which vesicle population is studied. The small size of exosomes demands a high sensitivity analysis method, including nanoparticle tracking analysis and electron microscopy [54, 57]. For the detection of microvesicles flow cytometry is the most common technique.
Flow cytometry
The flow cytometer detects microvesicles as small as 150 nm in diameter (depending on the sensitivity of the instrument). The principle of detection is based on vesicles passing through a laser beam. Modern flow cytometers may have many lasers and fluorescence detectors, which allow for labeling with multiple conjugated antibodies in the same sample [64]. Microvesicles may have phosphatidylserine on their outer membrane enabling the use of conjugated annexin V for their detection [65].
Although flow cytometry is widely used to detect microvesicles, it has some limitations. Flow cytometry does not detect the smallest microvesicles as individual events. Multiple microvesicles may be detected collectively as a single event, a phenomenon termed swarm detection (Table 2) [66]. In addition, small microvesicles may have a limited number of antibody binding sites, sterically restricting staining with multiple antibodies [65]. Thus, both the number of small microvesicles and their surface expression may be underestimated.
Transmission electron microscopy
The transmission electron microscope (TEM) visualizes small structures (limited to approximately 1 nm) because of the high resolution of the technique. Immune electron microscopy entails adding a conjugated antibody to detect a specific antigen in the sample [67]. Negative staining is performed when the surrounding medium is stained, leaving the vesicles unstained and the contrast clearly visualizes the vesicles.
Nanoparticle tracking analysis
Nanoparticle tracking analysis (NTA) examines EVs in the liquid phase using a laser beam that determines the size and concentration by filming the light scattering when the particles move under Brownian motion [54]. The technique detects vesicles with a size of 0.05–1 μm (modern instruments may lower the detection limit even further). NTA can be used in fluorescent mode, thus detecting labeled vesicles [54]. NTA with fluorescent mode provides both quantitative and qualitative information on the vesicles in suspension.
Extracellular vesicles in physiological and pathological processes
During physiological and pathological processes, EVs are released and partake in cellular communication affecting processes such as coagulation and thrombosis, angiogenesis, immune modulation and inflammation, which are discussed in the following sections.
Intercellular communication
Extracellular vesicles use various mechanisms to transfer information to recipient cells. They may bind to receptors on target cells, thereby transducing a signal, or transfer functional receptors, proteins, lipids, mRNA or miRNA from parent cells to recipient cells in which they may induce phenotypic changes.
Extracellular vesicles in cell signaling
Extracellular vesicles expose numerous signaling proteins and lipids on their surface and may thus bind to and stimulate target cells directly. For example, microvesicles from platelets exposing P-selectin were shown to bind to P-selectin glycoprotein ligand-1 (PSGL-1) on the surface of leukocytes, leading to leukocyte accumulation and aggregation [68]. During morphogenesis of multicellular organisms, shed microvesicles exposing the morphogen protein “wingless” bind to a family of G protein-coupled receptors called frizzled, thereby forming a gradient necessary for adequate tissue development [69]. Similarly, lymphocyte-derived microvesicles carrying the morphogen “hedgehog” may bind to its receptor on early hematopoietic stem cells and thereby induce differentiation into megakaryocytes [70].
Transfer of receptors
Extracellular vesicles can transfer functional receptors to target cells, allowing cell signaling in cells that originally lacked the receptor or enhancing the number of receptors. For example, microvesicles exposing the kinin B1 receptor transferred a functional receptor to endothelial cells and to human embryonic kidney cells [71]. The transfer of adhesion molecules and receptors from platelets to hematopoietic or malignant cells via platelet-derived microvesicles modulated their adhesion capacity and engraftment [72, 73]. Furthermore, microvesicles released from aggressive glioma cells transferred the oncogenic epidermal growth factor receptor (EGFR) to tumor cells causing a propagation of oncogenic activity [74]. The C-C chemokine receptor type 5 (CCR5) and C-X-C chemokine receptor type 4 (CXCR4) are important for HIV-1 uptake by cells. Microvesicle-mediated transfer of CCR5 and CXCR4 enabled HIV-1 to be internalized in cells previously not susceptible to the virus [75, 76], suggesting that this might be a means of disseminating HIV infection.
Transfer of proteins and lipids
Extracellular vesicles transport proteins such as cytokines, chemokines, and growth factors to neighboring or distant cells, resulting in modulation of the target cell. In addition, EVs may transfer functional channels. Exosomes originating in murine kidney-collecting duct cells (mCCDC11) transfer functional aquaporin 2 (AQP2), increasing water transport in recipient cells [77] and can thus potentially be involved in intra-renal signaling downstream in the nephron. Upon release, EVs may shelter proteins that would otherwise be phagocytosed or neutralized in free form in plasma, thus protecting their content from the host response [22]. This mode of transport can also be utilized by bacterial and viral components to evade the host response [22, 78]. Bioactive lipids, such as sphingosine 1-phosphate and arachidonic acid, are also transported within microvesicles [79]. Lipids in platelet microvesicles can increase adhesion between endothelial cells and monocytes [80]; hence, microvesicles not only affect recipient cells, but also other cells in their microenvironment.
Transfer of mRNA and microRNA
Extracellular vesicles are enriched in mRNA and miRNA, which can be transferred horizontally to and translated in recipient cells, thereby changing the phenotype of the cell. For example, microvesicles shed by endothelial progenitor cells induced activation of quiescent endothelial cells and stimulated angiogenesis by transfer of mRNA [81]. Mesenchymal stem cell (MSC)-derived EVs transfer mRNAs, inducing transcription and proliferation of tubular epithelial cells after in vivo injury [82]. Exosomes may regulate mRNA levels in recipient cells by delivering functional miRNA, thus blocking translation [83,84,85]. Transfer of miRNA by urinary exosomes to tubular cells modulated their function, as exemplified by diminished ROMK1 potassium channel levels in human collecting duct cells [86]. Exosomal transfer of certain miRNAs between immune cells conferred both a proinflammatory and an anti-inflammatory effect in vitro and in mice following endotoxin administration [87]. Likewise, vesicles derived from endothelial progenitor cells contain mRNAs coding for inhibitors of the complement system and anti-apoptotic molecules, thereby inhibiting complement-induced apoptosis and complement deposition on mesangial cells [88]. Interestingly, horizontal transfer of genetic material and the changes seen in the target cells were even demonstrated between cells of different species [89].
Protection against stress and cell death
To what extent EVs contribute to homeostasis and cell survival by ridding cells of unwanted substances is unknown, but may explain why cells release vesicles into their surroundings. The presence of complement C5b-9 on shed microvesicles may preserve the integrity of the parent cells by elimination of complement and the risk of cytolysis [90]. EVs from healthy individuals contain active caspase-3 that was not found in the parental cells, suggesting that caspase-3 might have been removed from the cells to ensure survival [91]. Inhibition of microvesicle release from viable endothelial cells containing active caspase-3 triggered both apoptosis and detachment of the cells [92].
Intriguingly, dying cells release microvesicles bearing the adaptor protein Crkl during the early stages of apoptosis induced by the caspase 3 cascade. These microvesicles were isolated from glomeruli after injury and were shown to induce compensatory proliferation signaling in recipient cells [93, 94]. Taken together, release of microvesicles may rid the cell of toxic substances, but may also induce repair in neighboring cells.
Coagulation and thrombosis
Extracellular vesicles play an important role in coagulation, platelet aggregation, and thrombosis. Pro-thrombotic properties of microvesicles are primarily associated with exposure of negatively charged phosphatidylserine and tissue factor [95]. Phosphatidylserine on circulating platelet- and monocyte-derived microvesicles provides binding sites for the assembly of coagulation factors such as factor IXa, Va, Xa, and VIII followed by thrombin generation [96]. Phosphatidylserine is also present on exosomal membranes [9]. It not only facilitates formation of coagulation complexes, but also promotes tissue factor activity [97]. Tissue factor is normally encrypted, but may be exposed on microvesicles released from platelets, monocytes or endothelial cells [16, 34, 98] and form a complex with factor VII/VIIa, thereby activating the extrinsic pathway of coagulation.
Platelet-derived microvesicles have a significantly higher pro-coagulant activity compared with activated platelets most probably because of their higher surface density of phosphatidylserine, factor Xa, P-selectin, and αIIbβ3 (glycoprotein IIb/IIIa) [99]. At the site of vascular injury, platelet-derived microvesicles support thrombus formation by facilitating the adhesion of platelets to endothelial cell matrix components [100]. The interaction between PSGL-1 on monocyte-derived microvesicles and selectins on platelets, endothelial cells or their shed microvesicles provides a basis for thrombus formation [34].
Angiogenesis
Microvesicles derived from blood and endothelial and tumor cells [101] may possess angiogenic properties, as previously reviewed [102]. The angiogenic effect may be associated with exposure of surface molecules or growth factors within the vesicles. Lymphoid microvesicles induced production of endothelial nitric oxide formation, expression of adhesion molecules, in addition to in vitro angiogenesis and in vivo neovascularization in endothelial cells [103]. Endothelial cell-derived microvesicles induced invasion of endothelial cells into basement membranes followed by capillary-like structure formation in vitro [104]. These properties may be of importance during tissue injury, post-ischemic revascularization and regeneration [105], and thus have importance during acute kidney injury (AKI).
Immune modulation
Extracellular vesicles play an important role in promoting immune responses, affecting both innate and adaptive immunity. Dendritic cell-derived exosomes enhanced the cytotoxic activity of natural killer cells [106]. Moreover, dendritic cell microvesicles stimulated epithelial cells to release pro-inflammatory cytokines [107], leukocyte-derived microvesicles activated the endothelium, upregulating adhesion molecules and releasing cytokines, leading to leukocyte recruitment [108] and platelet microvesicles affected the adhesion of monocytes to the endothelium [80].
Extracellular vesicles may have antigen-presenting properties, exposing major histocompatibility complexes (MHCs). Dendritic cells stimulated with lipopolysaccharide shed vesicles exposing MHC II, CD83, and the co-stimulatory molecule CD40 on their surface initiating a pro-inflammatory response in epithelial cells and T-cell activation [107, 109]. Interestingly, dendritic microvesicles containing tumor necrosis factor-α could initiate an innate immune response in epithelial cells, leading to cytokine release without transfer of antigen-presenting properties [107]. Microvesicles may also affect adaptive immunity, as platelet-derived microvesicles can increase immunoglobulin production by B-cells [110].
Activation of the complement system is usually directed against foreign antigens such as bacteria or damaged host cells. Complement activation and deposition of the membrane attack complex on blood cells is followed by the release of complement-coated microvesicles [111, 112]. Microvesicles bearing C1q reflect activation of the classical pathway of complement on the parent cell [113], whereas the presence of C3 reflects amplification of all three pathways of the complement via the alternative pathway [111]. Direct activation on vesicles, after shedding, may potentially also occur. Blood cell-derived EVs expose complement regulators on their surface such as complement receptor type 1 (C1R), membrane cofactor protein (CD46), decay accelerating factor (DAF/CD55) or CD59 [111, 114], thereby inhibiting assembly of the membrane attack complex (C5b-9) and preventing excessive complement activation. In addition, EVs opsonized by C3b are rapidly cleared from the circulation by phagocytes [115].
Malignancies
Tumor cells release significant numbers of EVs [116] that may influence proliferation, migration, invasion, and immune escape of cancer cells as well as angiogenesis [117] and the tumor environment [118]. EVs may also prime distant organs to a pre-metastatic niche facilitating survival and growth of metastasis [119]. An important step in tumor development is inhibition of immune surveillance. Tumor-derived exosomes can suppress T-cell immunity [120], thereby contributing to tumor progression by modulating and preventing anti-tumor immune reactions. The topic of EVs in malignancies has been reviewed elsewhere [116, 121].
Inflammation
Extracellular vesicles are capable of inducing both inflammatory and anti-inflammatory responses. This may be associated with the transfer of pro- and anti-inflammatory mediators and by inducing the release of cytokines from target cells [108, 122,123,124]. Both leukocyte- and platelet-derived microvesicles induced cytokine release from endothelial cells [122, 125], suggesting that microvesicles might participate in vascular damage and inflammatory disorders. Moreover, EVs may induce chemotaxis. Platelet-derived microvesicles stimulated recruitment of hematopoietic cells [73] and promoted leukocyte migration [126]. Glomerular endothelial cell-derived microvesicles exposing the kinin B1 receptor and interleukin 8 (IL-8) on their surface attracted neutrophils [127, 128]. Proximal tubular cells cultured in the presence of fenoldopam (a dopamine receptor agonist) released exosomes that reduced the production of reactive oxygen species in distal tubule and collecting duct cells [129], indicating the transfer of an anti-inflammatory response.
Anti-microbial effects
Neutrophil-derived microvesicles have been demonstrated to possess antimicrobial properties with a bacteriostatic effect on the uropathogen Escherichia coli [130]. Urinary exosomes also possess antimicrobial peptides, inhibiting the growth of E. coli and inducing bacteriolysis [131]. It has also been postulated that tissue factor-bearing microvesicles may prevent bacteria in the urinary tract from spreading beyond the uroepithelial barrier [132].
Extracellular vesicles as biomarkers and promoters of kidney disease
The prothrombotic, proinflammatory, and immunomodulatory properties associated with EVs, described above, may all contribute to and maintain tissue damage in the kidney and urinary tract during the development of AKI, glomerular and tubular diseases, infections, and chronic renal failure in addition to numerous other conditions affecting the kidney. These aspects have been comprehensively reviewed recently by our group and others [2, 128, 133]. Studies on the role of EVs in AKI have mostly been carried out in patients with sepsis, burns or other forms of acute tubular injury [134, 135]. Our group has focused on the role of microvesicles in hemolytic uremic syndrome and vasculitis, which will be elaborated on below. In Table 3, we summarize various renal conditions in which EVs have been described as biomarkers of disease, in blood or urine, and describe which characteristics contribute to the induction and propagation of tissue injury.
Microvesicles in hemolytic uremic syndrome
Circulating microvesicles are elevated in thrombotic microangiopathies. Microvesicles derived from platelets, neutrophils, monocytes, and red blood cells were detected in blood samples from patients with Shiga toxin-producing E. coli (STEC)-associated hemolytic uremic syndrome (HUS) [16, 22, 23, 111, 219]. Patients with thrombotic thrombocytopenic purpura (TTP) exhibit elevated levels of both platelet and endothelial-derived microvesicles, the latter coated with complement deposits [162, 163, 220].
Our studies have shown that circulating microvesicles in STEC-HUS are pro-thrombotic/procoagulant as they are both tissue factor- and phosphatidylserine-positive. These aspects could be reproduced in vitro when whole blood was stimulated with Shiga toxin and E. coli O157 lipopolysaccharide and shed pro-thrombotic microvesicles were mainly derived from platelets [16]. Similarly, platelet- and monocyte-derived microvesicles in patient samples and in in vitro toxin-stimulated samples were coated with deposits of C3 and C9, suggesting ongoing complement activation.
Patients with STEC-HUS also exhibited elevated C3 and C9 on microvesicles derived from red blood cells, and, interestingly, Shiga toxin could induce complement activation on red blood cells followed by hemolysis, thereby releasing microvesicles from red blood cells with deposits of the membrane attack complex C5b-9 [23].
Shiga toxin is transported in vivo bound to blood cells and after uptake in these cells released within microvesicles (reviewed in Karpman et al. [221]). Blood cell-derived microvesicles transport Shiga toxin to the kidney, where the toxin, within microvesicles, is taken up in glomerular endothelial cells and peritubular capillary endothelial cells. Within the endothelial cells, the microvesicles either empty their cargo or are transcytosed through the cells, and their corresponding basement membranes, into podocytes or tubular cells, respectively. Eventually the microvesicles empty their cargo, although the signal leading to this release of contents is unknown. Intracellular toxin undergoes retrograde transport, binds to ribosomes and induces cell death thus causing renal failure [22]. HUS is characterized by platelet activation and the formation of microthrombi, hemolysis, and acute renal failure. These studies show that microvesicles are not only biomarkers, but actively contribute to disease-specific processes during STEC-HUS by creating a pro-thrombotic environment, partaking in hemolysis, and transporting Shiga toxin into the kidney to induce renal cell death.
In similarity to the pro-thrombotic microvesicles demonstrated in patients with STEC-HUS, serum from patients with aHUS, with mutations in the complement regulator factor H, induced the release of tissue factor- and phosphatidylserine-positive platelet-derived microvesicles from normal washed platelets, effects that could be inhibited by the addition of normal factor H [161].
Microvesicles in vasculitis and inflammatory disorders
Microvesicles shed from endothelial cells, platelets, and leukocytes were increased during the acute phase of vasculitis, returning to normal levels during remission [25, 37, 152]. Endothelial microvesicle levels in pediatric vasculitis correlated with the Birmingham Vasculitis Activity Score (BVAS), C-reactive protein, and erythrocyte sedimentation rate [165]. Likewise, endothelial microvesicles in adults with anti-neutrophil cytoplasmic antibodies (ANCA)-associated vasculitis (AAV) correlated with the BVAS [37] and could thus be used as a biomarker for disease activity [37, 165].
The ANCAs circulating in patients with AAV activated neutrophils, causing them to release microvesicles [167]. In patients with vasculitis, neutrophil microvesicles activated endothelial cells, leading to the release of cytokines [108, 122, 167]. Neutrophil microvesicles may expose proteinase 3 (PR3) and myeloperoxidase (MPO) on their surfaces enabling ANCA to bind. Microvesicles were pro-thrombotic as they promoted the generation of thrombin [167], and could thus contribute to the thromboembolic complications seen in vasculitis.
Our studies have demonstrated systemic activation of the kinin system in children and adults with vasculitis underlying the profound vascular inflammation [222, 223]. We have shown that neutrophil-derived microvesicles bearing the kinin B1 receptor, expressed on cells during chronic inflammation, can transfer the receptor to cells lacking the receptor (demonstrated using transfected and wild-type HEK cells) and to glomerular endothelial cells, thereby promoting the inflammatory response. The phenomenon was confirmed in kidney biopsies showing that B1-receptor-positive neutrophil-derived microvesicles dock on glomerular endothelial cells in vivo during vasculitis [71]. Furthermore, during extensive vascular injury, endothelial microvesicles are released, also bearing the B1 receptor [127]. The B1-receptor-positive endothelial microvesicles recruited neutrophils, thus enhancing the inflammation. Interestingly, C1 inhibitor, the main inhibitor of the kinin system, inhibited the release of the chemotactic glomerular endothelial microvesicles.
In systemic lupus erythematosus (SLE), platelet-derived microvesicles are significantly increased and correlate with thrombin generation, suggesting a role in the thromboembolic state [206]. Other aspects, such as the contribution to immune complex deposition, are presented in Table 3.
Antiphospholipid syndrome is an autoimmune disease associated with antiphospholipid antibodies and thrombotic complications. Patients with antiphospholipid syndrome have elevated endothelial and platelet-derived microvesicles compared with controls and the endothelial vesicles may be pro-thrombotic [208].
The effect of renal replacement therapy and drugs on extracellular vesicles
Treatments given during acute and chronic renal failure may affect levels of EVs. Dialysis treatment (hemodialysis and peritoneal dialysis) not only does not remove EVs, it may increase levels in comparison with healthy controls and after treatment sessions [17, 153]. The same is true for miRNA levels, which do not decrease after hemodialysis [136]. Treatment with recombinant erythropoietin may enhance levels of platelet-derived microvesicles, whereas the presence of an arteriovenous fistula has no effect on microvesicle levels [154].
To our knowledge, the effect of plasma exchange on levels of EVs has not been specifically addressed, but plasma exchange should presumably remove EVs. This has been suggested in the treatment of patients with SLE and antiphospholipid syndrome [224].
Various drugs used in the treatment of renal disease, including anti-hypertensive medications such as calcium channel blockers, amiloride, and beta blockers, or statins, may affect the release of EVs, as reviewed [128, 225]. Amiloride affects both the release and uptake of vesicles [226, 227].
The renal regenerative capacity of extracellular vesicles
Mesenchymal stem cells and endothelial progenitor cells secrete EVs that have been demonstrated to induce nephron regeneration and repair by inhibiting apoptosis and promoting tubular proliferation. These effects have been documented in vitro [228] and in vivo [229] and are attributed to the transfer of both growth factors and RNAs (mRNAs and miRNAs) [230]. As described above, EVs can stimulate angiogenesis, and transfer growth factors such as vascular endothelial growth factor, hepatocyte growth factor [231], insulin-like growth factor-1 (IGF-1), adrenomedullin, and stromal cell-derived factor-1 (SDF1) [4]. Horizontal transfer of the IGF-1 receptor mRNA transcript via MSC EVs to damaged tubular cells induced proliferation [232]. EVs derived from MSCs localize to the kidney [233] and have been extensively investigated, in preclinical studies, for their therapeutic potential to protect tubuli and repair ischemia/reperfusion-induced injury [234].
Extracellular vesicles as vehicles for drug delivery
The capacity of EVs to deliver proteins, lipids, and nucleic acids to recipient cells has therapeutic potential. EVs can be designed to target specific recipient cells. Cells can be genetically altered to express ligands on their membrane that are also present on EVs released from the cells. These ligands can bind to receptors on the target cell [235]. Thus, EVs can be loaded with therapeutic substances for delivery to target cells. These exciting developments in EV-based therapeutics may be used in future clinical trials and have been recently reviewed [236, 237].
Conclusions
Extracellular vesicles play an important role in normal intercellular communication. They can be detected as biomarkers of disease owing to their excessive numbers and their properties and may also contribute to the development of diseases, including kidney disease, by inducing inflammation, vascular injury, and thrombosis in addition to modulating the immune response. Their contribution to the induction and progression of renal diseases may lead to the development of treatments geared toward temporary reduction of EVs systemically in the circulation, or locally in the kidney and urinary tract. Treatments that reduce the release or uptake of EVs need to take into account the notion that EVs may also be cytoprotective, as their release and the removal of unwanted or damaging substances from their parent cells may maintain cellular integrity. EVs may have potentially beneficial properties associated with tubular regeneration and the induction of angiogenesis. The therapeutic potential and nephroprotective effects of EVs, owing to their capacity to shuttle proteins, lipids, and genetic cargo to recipient cells, are being explored in preclinical studies, which may lead to clinical trials in the future.
References
Mittelbrunn M, Sanchez-Madrid F (2012) Intercellular communication: diverse structures for exchange of genetic information. Nat Rev Mol Cell Biol 13:328–335
Pomatto MAC, Gai C, Bussolati B, Camussi G (2017) Extracellular vesicles in renal pathophysiology. Front Mol Biosci 4:37
Colombo M, Raposo G, Thery C (2014) Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol 30:255–289
Camussi G, Deregibus MC, Bruno S, Cantaluppi V, Biancone L (2010) Exosomes/microvesicles as a mechanism of cell-to-cell communication. Kidney Int 78:838–848
Thery C, Ostrowski M, Segura E (2009) Membrane vesicles as conveyors of immune responses. Nat Rev Immunol 9:581–593
Simpson RJ, Lim JW, Moritz RL, Mathivanan S (2009) Exosomes: proteomic insights and diagnostic potential. Expert Rev Proteomics 6:267–283
Zhang HG, Grizzle WE (2014) Exosomes: a novel pathway of local and distant intercellular communication that facilitates the growth and metastasis of neoplastic lesions. Am J Pathol 184:28–41
Cocucci E, Meldolesi J (2015) Ectosomes and exosomes: shedding the confusion between extracellular vesicles. Trends Cell Biol 25:364–372
Skotland T, Sandvig K, Llorente A (2017) Lipids in exosomes: current knowledge and the way forward. Prog Lipid Res 66:30–41
Gyorgy B, Szabo TG, Pasztoi M, Pal Z, Misjak P, Aradi B, Laszlo V, Pallinger E, Pap E, Kittel A, Nagy G, Falus A, Buzas EI (2011) Membrane vesicles, current state-of-the-art: emerging role of extracellular vesicles. Cell Mol Life Sci 68:2667–2688
Wilson HL, Francis SE, Dower SK, Crossman DC (2004) Secretion of intracellular IL-1 receptor antagonist (type 1) is dependent on P2X7 receptor activation. J Immunol 173:1202–1208
Holme PA, Orvim U, Hamers MJ, Solum NO, Brosstad FR, Barstad RM, Sakariassen KS (1997) Shear-induced platelet activation and platelet microparticle formation at blood flow conditions as in arteries with a severe stenosis. Arterioscler Thromb Vasc Biol 17:646–653
Lynch SF, Ludlam CA (2007) Plasma microparticles and vascular disorders. Br J Haematol 137:36–48
Nomura S, Nakamura T, Cone J, Tandon NN, Kambayashi J (2000) Cytometric analysis of high shear-induced platelet microparticles and effect of cytokines on microparticle generation. Cytometry 40:173–181
Heijnen HF, Schiel AE, Fijnheer R, Geuze HJ, Sixma JJ (1999) Activated platelets release two types of membrane vesicles: microvesicles by surface shedding and exosomes derived from exocytosis of multivesicular bodies and alpha-granules. Blood 94:3791–3799
Ståhl AL, Sartz L, Nelsson A, Békássy ZD, Karpman D (2009) Shiga toxin and lipopolysaccharide induce platelet-leukocyte aggregates and tissue factor release, a thrombotic mechanism in hemolytic uremic syndrome. PLoS One 4:e6990
Faure V, Dou L, Sabatier F, Cerini C, Sampol J, Berland Y, Brunet P, Dignat-George F (2006) Elevation of circulating endothelial microparticles in patients with chronic renal failure. J Thromb Haemost 4:566–573
Lai RC, Tan SS, Yeo RW, Choo AB, Reiner AT, Su Y, Shen Y, Fu Z, Alexander L, Sze SK, Lim SK (2016) MSC secretes at least 3 EV types each with a unique permutation of membrane lipid, protein and RNA. J Extracell Vesicles 5:29828
Jimenez JJ, Jy W, Mauro LM, Soderland C, Horstman LL, Ahn YS (2003) Endothelial cells release phenotypically and quantitatively distinct microparticles in activation and apoptosis. Thromb Res 109:175–180
Turturici G, Tinnirello R, Sconzo G, Geraci F (2014) Extracellular membrane vesicles as a mechanism of cell-to-cell communication: advantages and disadvantages. Am J Phys Cell Physiol 306:C621–C633
Mathivanan S, Ji H, Simpson RJ (2010) Exosomes: extracellular organelles important in intercellular communication. J Proteome 73:1907–1920
Ståhl AL, Arvidsson I, Johansson KE, Chromek M, Rebetz J, Loos S, Kristoffersson AC, Békássy ZD, Mörgelin M, Karpman D (2015) A novel mechanism of bacterial toxin transfer within host blood cell-derived microvesicles. PLoS Pathog 11:e1004619
Arvidsson I, Ståhl AL, Hedström MM, Kristoffersson AC, Rylander C, Westman JS, Storry JR, Olsson ML, Karpman D (2015) Shiga toxin-induced complement-mediated hemolysis and release of complement-coated red blood cell-derived microvesicles in hemolytic uremic syndrome. J Immunol 194:2309–2318
Dignat-George F, Boulanger CM (2011) The many faces of endothelial microparticles. Arterioscler Thromb Vasc Biol 31:27–33
Brogan PA, Dillon MJ (2004) Endothelial microparticles and the diagnosis of the vasculitides. Intern Med 43:1115–1119
Colombo M, Moita C, van Niel G, Kowal J, Vigneron J, Benaroch P, Manel N, Moita LF, Thery C, Raposo G (2013) Analysis of ESCRT functions in exosome biogenesis, composition and secretion highlights the heterogeneity of extracellular vesicles. J Cell Sci 126:5553–5565
Van Niel G, Charrin S, Simoes S, Romao M, Rochin L, Saftig P, Marks MS, Rubinstein E, Raposo G (2011) The tetraspanin CD63 regulates ESCRT-independent and -dependent endosomal sorting during melanogenesis. Dev Cell 21:708–721
Trajkovic K, Hsu C, Chiantia S, Rajendran L, Wenzel D, Wieland F, Schwille P, Brugger B, Simons M (2008) Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 319:1244–1247
Ratajczak J, Wysoczynski M, Hayek F, Janowska-Wieczorek A, Ratajczak MZ (2006) Membrane-derived microvesicles: important and underappreciated mediators of cell-to-cell communication. Leukemia 20:1487–1495
Hugel B, Martinez MC, Kunzelmann C, Freyssinet JM (2005) Membrane microparticles: two sides of the coin. Physiology (Bethesda) 20:22–27
VanWijk MJ, VanBavel E, Sturk A, Nieuwland R (2003) Microparticles in cardiovascular diseases. Cardiovasc Res 59:277–287
Pasquet JM, Dachary-Prigent J, Nurden AT (1996) Calcium influx is a determining factor of calpain activation and microparticle formation in platelets. Eur J Biochem 239:647–654
Hartwig JH, Chambers KA, Stossel TP (1989) Association of gelsolin with actin filaments and cell membranes of macrophages and platelets. J Cell Biol 108:467–479
Del Conde I, Shrimpton CN, Thiagarajan P, Lopez JA (2005) Tissue-factor-bearing microvesicles arise from lipid rafts and fuse with activated platelets to initiate coagulation. Blood 106:1604–1611
Daleke DL (2003) Regulation of transbilayer plasma membrane phospholipid asymmetry. J Lipid Res 44:233–242
Connor DE, Exner T, Ma DD, Joseph JE (2010) The majority of circulating platelet-derived microparticles fail to bind annexin V, lack phospholipid-dependent procoagulant activity and demonstrate greater expression of glycoprotein Ib. Thromb Haemost 103:1044–1052
Erdbruegger U, Grossheim M, Hertel B, Wyss K, Kirsch T, Woywodt A, Haller H, Haubitz M (2008) Diagnostic role of endothelial microparticles in vasculitis. Rheumatology (Oxford) 47:1820–1825
Yanez-Mo M, Siljander PR, Andreu Z, Zavec AB, Borras FE, Buzas EI, Casal E, Cappello F, Carvalho J, Colás E, Cordeiro-da Silva A, Fais S, Falcon-Perez JM, Ghobrial IM, Giebel B, Gimona M, Graner M, Gursel I, Gursel M, Heegaard NH, Hendrix A, Kierulf P, Kokubun K, Kosanovic M, Kralj-Iglic V, Krämer-Albers EM, Laitinen S, Lässer C, Lener T, Ligeti E, Linē A, Lipps G, Llorente A, Lötvall J, Manček-Keber M, Marcilla A, Mittelbrunn M, Nazarenko I, Nolte-’t Hoen EN, Nyman TA, O’Driscoll L, Olivan M, Oliveira C, Pállinger É, Del Portillo HA, Reventós J, Rigau M, Rohde E, Sammar M, Sánchez-Madrid F, Santarém N, Schallmoser K, Ostenfeld MS, Stoorvogel W, Stukelj R, Van der Grein SG, Vasconcelos MH, Wauben MH, De Wever O (2015) Biological properties of extracellular vesicles and their physiological functions. J Extracell Vesicles 4:27066
Van den Goor JM, Nieuwland R, Rutten PM, Tijssen JG, Hau C, Sturk A, Eijsman L, de Mol BA (2007) Retransfusion of pericardial blood does not trigger systemic coagulation during cardiopulmonary bypass. Eur J Cardiothorac Surg 31:1029–1036
Augustine D, Ayers LV, Lima E, Newton L, Lewandowski AJ, Davis EF, Ferry B, Leeson P (2014) Dynamic release and clearance of circulating microparticles during cardiac stress. Circ Res 114:109–113
Mulcahy LA, Pink RC, Carter DR (2014) Routes and mechanisms of extracellular vesicle uptake. J Extracell Vesicles. https://doi.org/10.3402/jev.v3.24641
Losche W, Scholz T, Temmler U, Oberle V, Claus RA (2004) Platelet-derived microvesicles transfer tissue factor to monocytes but not to neutrophils. Platelets 15:109–115
Rana S, Yue S, Stadel D, Zoller M (2012) Toward tailored exosomes: the exosomal tetraspanin web contributes to target cell selection. Int J Biochem Cell Biol 44:1574–1584
Parolini I, Federici C, Raggi C, Lugini L, Palleschi S, De Milito A, Coscia C, Iessi E, Logozzi M, Molinari A, Colone M, Tatti M, Sargiacomo M, Fais S (2009) Microenvironmental pH is a key factor for exosome traffic in tumor cells. J Biol Chem 284:34211–34222
Marzesco AM, Janich P, Wilsch-Brauninger M, Dubreuil V, Langenfeld K, Corbeil D, Huttner WB (2005) Release of extracellular membrane particles carrying the stem cell marker prominin-1 (CD133) from neural progenitors and other epithelial cells. J Cell Sci 118:2849–2858
Burger D, Thibodeau JF, Holterman CE, Burns KD, Touyz RM, Kennedy CR (2014) Urinary podocyte microparticles identify prealbuminuric diabetic glomerular injury. J Am Soc Nephrol 25:1401–1407
Gyorgy B, Szabo TG, Turiak L, Wright M, Herczeg P, Ledeczi Z, Kittel A, Polgar A, Toth K, Derfalvi B, Zelenak G, Borocz I, Carr B, Nagy G, Vekey K, Gay S, Falus A, Buzas EI (2012) Improved flow cytometric assessment reveals distinct microvesicle (cell-derived microparticle) signatures in joint diseases. PLoS One 7:e49726
Novelli F, Neri T, Tavanti L, Armani C, Noce C, Falaschi F, Bartoli ML, Martino F, Palla A, Celi A, Paggiaro P (2014) Procoagulant, tissue factor-bearing microparticles in bronchoalveolar lavage of interstitial lung disease patients: an observational study. PLoS One 9:e95013
Zonneveld MI, Brisson AR, van Herwijnen MJ, Tan S, van de Lest CH, Redegeld FA, Garssen J, Wauben MH, Nolte-’t Hoen EN (2014) Recovery of extracellular vesicles from human breast milk is influenced by sample collection and vesicle isolation procedures. J Extracell Vesicles. https://doi.org/10.3402/jev.v3.24215
Witek RP, Yang L, Liu R, Jung Y, Omenetti A, Syn WK, Choi SS, Cheong Y, Fearing CM, Agboola KM, Chen W, Diehl AM (2009) Liver cell-derived microparticles activate hedgehog signaling and alter gene expression in hepatic endothelial cells. Gastroenterology 136:320–330; e322
Xiao H, Wong DT (2012) Proteomic analysis of microvesicles in human saliva by gel electrophoresis with liquid chromatography-mass spectrometry. Anal Chim Acta 723:61–67
Ng YH, Rome S, Jalabert A, Forterre A, Singh H, Hincks CL, Salamonsen LA (2013) Endometrial exosomes/microvesicles in the uterine microenvironment: a new paradigm for embryo-endometrial cross talk at implantation. PLoS One 8:e58502
Robert S, Lacroix R, Poncelet P, Harhouri K, Bouriche T, Judicone C, Wischhusen J, Arnaud L, Dignat-George F (2012) High-sensitivity flow cytometry provides access to standardized measurement of small-size microparticles—brief report. Arterioscler Thromb Vasc Biol 32:1054–1058
Dragovic RA, Gardiner C, Brooks AS, Tannetta DS, Ferguson DJ, Hole P, Carr B, Redman CW, Harris AL, Dobson PJ, Harrison P, Sargent IL (2011) Sizing and phenotyping of cellular vesicles using nanoparticle tracking analysis. Nanomedicine 7:780–788
Lawrie AS, Albanyan A, Cardigan RA, Mackie IJ, Harrison P (2009) Microparticle sizing by dynamic light scattering in fresh-frozen plasma. Vox Sang 96:206–212
Momen-Heravi F, Balaj L, Alian S, Tigges J, Toxavidis V, Ericsson M, Distel RJ, Ivanov AR, Skog J, Kuo WP (2012) Alternative methods for characterization of extracellular vesicles. Front Physiol 3:354. https://doi.org/10.3389/fphys.2012.00354; eCollection 02012
Van der Pol E, Coumans FA, Grootemaat AE, Gardiner C, Sargent IL, Harrison P, Sturk A, van Leeuwen TG, Nieuwland R (2014) Particle size distribution of exosomes and microvesicles determined by transmission electron microscopy, flow cytometry, nanoparticle tracking analysis, and resistive pulse sensing. J Thromb Haemost 12:1182–1192
Liu J, Butte MJ (2012) Single molecule labeling of an atomic force microscope cantilever tip. Appl Phys Lett 101:163705
Sharma S, Rasool HI, Palanisamy V, Mathisen C, Schmidt M, Wong DT, Gimzewski JK (2010) Structural-mechanical characterization of nanoparticle exosomes in human saliva, using correlative AFM, FESEM, and force spectroscopy. ACS Nano 4:1921–1926
Witwer KW, Buzas EI, Bemis LT, Bora A, Lasser C, Lotvall J, Nolte-’t Hoen EN, Piper MG, Sivaraman S, Skog J, Thery C, Wauben MH, Hochberg F (2013) Standardization of sample collection, isolation and analysis methods in extracellular vesicle research. J Extracell Vesicles. https://doi.org/10.3402/jev.v3402i3400.20360
Van der Pol E, Coumans F, Varga Z, Krumrey M, Nieuwland R (2013) Innovation in detection of microparticles and exosomes. J Thromb Haemost 11:36–45
Choi DS, Kim DK, Kim YK, Gho YS (2015) Proteomics of extracellular vesicles: exosomes and ectosomes. Mass Spectrom Rev 34:474–490
Thery C, Amigorena S, Raposo G, Clayton A (2006) Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol 3:22. https://doi.org/10.1002/0471143030.cb0471140322s0471143030
Perfetto SP, Chattopadhyay PK, Roederer M (2004) Seventeen-colour flow cytometry: unravelling the immune system. Nat Rev Immunol 4:648–655
Erdbrugger U, Lannigan J (2016) Analytical challenges of extracellular vesicle detection: a comparison of different techniques. Cytometry A 89:123–134
Van der Pol E, van Gemert MJ, Sturk A, Nieuwland R, van Leeuwen TG (2012) Single vs. swarm detection of microparticles and exosomes by flow cytometry. J Thromb Haemost 10:919–930
Baschong W, Lucocq JM, Roth J (1985) "Thiocyanate gold": small (2-3 nm) colloidal gold for affinity cytochemical labeling in electron microscopy. Histochemistry 83:409–411
Forlow SB, McEver RP, Nollert MU (2000) Leukocyte-leukocyte interactions mediated by platelet microparticles under flow. Blood 95:1317–1323
Greco V, Hannus M, Eaton S (2001) Argosomes: a potential vehicle for the spread of morphogens through epithelia. Cell 106:633–645
Martinez MC, Larbret F, Zobairi F, Coulombe J, Debili N, Vainchenker W, Ruat M, Freyssinet JM (2006) Transfer of differentiation signal by membrane microvesicles harboring hedgehog morphogens. Blood 108:3012–3020
Kahn R, Mossberg M, Ståhl AL, Johansson K, Lopatko Lindman I, Heijl C, Segelmark M, Mörgelin M, Leeb-Lundberg LM, Karpman D (2017) Microvesicle transfer of kinin B1-receptors is a novel inflammatory mechanism in vasculitis. Kidney Int 91:96–105
Janowska-Wieczorek A, Majka M, Kijowski J, Baj-Krzyworzeka M, Reca R, Turner AR, Ratajczak J, Emerson SG, Kowalska MA, Ratajczak MZ (2001) Platelet-derived microparticles bind to hematopoietic stem/progenitor cells and enhance their engraftment. Blood 98:3143–3149
Baj-Krzyworzeka M, Majka M, Pratico D, Ratajczak J, Vilaire G, Kijowski J, Reca R, Janowska-Wieczorek A, Ratajczak MZ (2002) Platelet-derived microparticles stimulate proliferation, survival, adhesion, and chemotaxis of hematopoietic cells. Exp Hematol 30:450–459
Al-Nedawi K, Meehan B, Micallef J, Lhotak V, May L, Guha A, Rak J (2008) Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat Cell Biol 10:619–624
Mack M, Kleinschmidt A, Bruhl H, Klier C, Nelson PJ, Cihak J, Plachy J, Stangassinger M, Erfle V, Schlondorff D (2000) Transfer of the chemokine receptor CCR5 between cells by membrane-derived microparticles: a mechanism for cellular human immunodeficiency virus 1 infection. Nat Med 6:769–775
Rozmyslowicz T, Majka M, Kijowski J, Murphy SL, Conover DO, Poncz M, Ratajczak J, Gaulton GN, Ratajczak MZ (2003) Platelet- and megakaryocyte-derived microparticles transfer CXCR4 receptor to CXCR4-null cells and make them susceptible to infection by X4-HIV. AIDS 17:33–42
Street JM, Birkhoff W, Menzies RI, Webb DJ, Bailey MA, Dear JW (2011) Exosomal transmission of functional aquaporin 2 in kidney cortical collecting duct cells. J Physiol 589:6119–6127
Meckes DG Jr, Raab-Traub N (2011) Microvesicles and viral infection. J Virol 85:12844–12854
Barry OP, Pratico D, Lawson JA, FitzGerald GA (1997) Transcellular activation of platelets and endothelial cells by bioactive lipids in platelet microparticles. J Clin Invest 99:2118–2127
Barry OP, Pratico D, Savani RC, FitzGerald GA (1998) Modulation of monocyte-endothelial cell interactions by platelet microparticles. J Clin Invest 102:136–144
Deregibus MC, Cantaluppi V, Calogero R, Lo Iacono M, Tetta C, Biancone L, Bruno S, Bussolati B, Camussi G (2007) Endothelial progenitor cell derived microvesicles activate an angiogenic program in endothelial cells by a horizontal transfer of mRNA. Blood 110:2440–2448
Bruno S, Grange C, Deregibus MC, Calogero RA, Saviozzi S, Collino F, Morando L, Busca A, Falda M, Bussolati B, Tetta C, Camussi G (2009) Mesenchymal stem cell-derived microvesicles protect against acute tubular injury. J Am Soc Nephrol 20:1053–1067
Montecalvo A, Larregina AT, Shufesky WJ, Stolz DB, Sullivan ML, Karlsson JM, Baty CJ, Gibson GA, Erdos G, Wang Z, Milosevic J, Tkacheva OA, Divito SJ, Jordan R, Lyons-Weiler J, Watkins SC, Morelli AE (2012) Mechanism of transfer of functional microRNAs between mouse dendritic cells via exosomes. Blood 119:756–766
Mittelbrunn M, Gutierrez-Vazquez C, Villarroya-Beltri C, Gonzalez S, Sanchez-Cabo F, Gonzalez MA, Bernad A, Sanchez-Madrid F (2011) Unidirectional transfer of microRNA-loaded exosomes from T cells to antigen-presenting cells. Nat Commun 2:282. https://doi.org/10.1038/ncomms1285
Pegtel DM, Cosmopoulos K, Thorley-Lawson DA, van Eijndhoven MA, Hopmans ES, Lindenberg JL, de Gruijl TD, Wurdinger T, Middeldorp JM (2010) Functional delivery of viral miRNAs via exosomes. Proc Natl Acad Sci U S A 107:6328–6333
Gracia T, Wang X, Su Y, Norgett EE, Williams TL, Moreno P, Micklem G, Karet Frankl FE (2017) Urinary exosomes contain microRNAs capable of paracrine modulation of tubular transporters in kidney. Sci Rep 7:40601
Alexander M, Hu R, Runtsch MC, Kagele DA, Mosbruger TL, Tolmachova T, Seabra MC, Round JL, Ward DM, O’Connell RM (2015) Exosome-delivered microRNAs modulate the inflammatory response to endotoxin. Nat Commun 6:7321
Cantaluppi V, Medica D, Mannari C, Stiaccini G, Figliolini F, Dellepiane S, Quercia AD, Migliori M, Panichi V, Giovannini L, Bruno S, Tetta C, Biancone L, Camussi G (2015) Endothelial progenitor cell-derived extracellular vesicles protect from complement-mediated mesangial injury in experimental anti-Thy1.1 glomerulonephritis. Nephrol Dial Transplant 30:410–422
Lotvall J, Valadi H (2007) Cell to cell signalling via exosomes through esRNA. Cell Adhes Migr 1:156–158
Iida K, Whitlow MB, Nussenzweig V (1991) Membrane vesiculation protects erythrocytes from destruction by complement. J Immunol 147:2638–2642
Abid Hussein MN, Nieuwland R, Hau CM, Evers LM, Meesters EW, Sturk A (2005) Cell-derived microparticles contain caspase 3 in vitro and in vivo. J Thromb Haemost 3:888–896
Abid Hussein MN, Boing AN, Sturk A, Hau CM, Nieuwland R (2007) Inhibition of microparticle release triggers endothelial cell apoptosis and detachment. Thromb Haemost 98:1096–1107
Gupta KH, Goldufsky JW, Wood SJ, Tardi NJ, Moorthy GS, Gilbert DZ, Zayas JP, Hahm E, Altintas MM, Reiser J, Shafikhani SH (2017) Apoptosis and compensatory proliferation signaling are coupled by CrkI-containing microvesicles. Dev Cell 41:674–684; e675
Bussolati B, Camussi G (2017) Renal injury: early apoptotic extracellular vesicles in injury and repair. Nat Rev Nephrol 13:523–524
Satta N, Toti F, Feugeas O, Bohbot A, Dachary-Prigent J, Eschwege V, Hedman H, Freyssinet JM (1994) Monocyte vesiculation is a possible mechanism for dissemination of membrane-associated procoagulant activities and adhesion molecules after stimulation by lipopolysaccharide. J Immunol 153:3245–3255
Owens AP 3rd, Mackman N (2011) Microparticles in hemostasis and thrombosis. Circ Res 108:1284–1297
Wolberg AS, Monroe DM, Roberts HR, Hoffman MR (1999) Tissue factor de-encryption: ionophore treatment induces changes in tissue factor activity by phosphatidylserine-dependent and -independent mechanisms. Blood Coagul Fibrinolysis 10:201–210
Combes V, Simon AC, Grau GE, Arnoux D, Camoin L, Sabatier F, Mutin M, Sanmarco M, Sampol J, Dignat-George F (1999) In vitro generation of endothelial microparticles and possible prothrombotic activity in patients with lupus anticoagulant. J Clin Invest 104:93–102
Sinauridze EI, Kireev DA, Popenko NY, Pichugin AV, Panteleev MA, Krymskaya OV, Ataullakhanov FI (2007) Platelet microparticle membranes have 50- to 100-fold higher specific procoagulant activity than activated platelets. Thromb Haemost 97:425–434
Merten M, Pakala R, Thiagarajan P, Benedict CR (1999) Platelet microparticles promote platelet interaction with subendothelial matrix in a glycoprotein IIb/IIIa-dependent mechanism. Circulation 99:2577–2582
Hood JL, Pan H, Lanza GM, Wickline SA, Consortium for Translational Research in Advanced I, Nanomedicine (2009) Paracrine induction of endothelium by tumor exosomes. Lab Investig 89:1317–1328
Martinez MC, Andriantsitohaina R (2011) Microparticles in angiogenesis: therapeutic potential. Circ Res 109:110–119
Soleti R, Benameur T, Porro C, Panaro MA, Andriantsitohaina R, Martinez MC (2009) Microparticles harboring Sonic Hedgehog promote angiogenesis through the upregulation of adhesion proteins and proangiogenic factors. Carcinogenesis 30:580–588
Taraboletti G, D’Ascenzo S, Borsotti P, Giavazzi R, Pavan A, Dolo V (2002) Shedding of the matrix metalloproteinases MMP-2, MMP-9, and MT1-MMP as membrane vesicle-associated components by endothelial cells. Am J Pathol 160:673–680
Brill A, Dashevsky O, Rivo J, Gozal Y, Varon D (2005) Platelet-derived microparticles induce angiogenesis and stimulate post-ischemic revascularization. Cardiovasc Res 67:30–38
Munich S, Sobo-Vujanovic A, Buchser WJ, Beer-Stolz D, Vujanovic NL (2012) Dendritic cell exosomes directly kill tumor cells and activate natural killer cells via TNF superfamily ligands. Oncoimmunology 1:1074–1083
Obregon C, Rothen-Rutishauser B, Gerber P, Gehr P, Nicod LP (2009) Active uptake of dendritic cell-derived exovesicles by epithelial cells induces the release of inflammatory mediators through a TNF-alpha-mediated pathway. Am J Pathol 175:696–705
Mesri M, Altieri DC (1998) Endothelial cell activation by leukocyte microparticles. J Immunol 161:4382–4387
Segura E, Amigorena S, Thery C (2005) Mature dendritic cells secrete exosomes with strong ability to induce antigen-specific effector immune responses. Blood Cells Mol Dis 35:89–93
Sprague DL, Elzey BD, Crist SA, Waldschmidt TJ, Jensen RJ, Ratliff TL (2008) Platelet-mediated modulation of adaptive immunity: unique delivery of CD154 signal by platelet-derived membrane vesicles. Blood 111:5028–5036
Ståhl AL, Sartz L, Karpman D (2011) Complement activation on platelet-leukocyte complexes and microparticles in enterohemorrhagic Escherichia coli-induced hemolytic uremic syndrome. Blood 117:5503–5513
Sims PJ, Faioni EM, Wiedmer T, Shattil SJ (1988) Complement proteins C5b-9 cause release of membrane vesicles from the platelet surface that are enriched in the membrane receptor for coagulation factor Va and express prothrombinase activity. J Biol Chem 263:18205–18212
Nauta AJ, Trouw LA, Daha MR, Tijsma O, Nieuwland R, Schwaeble WJ, Gingras AR, Mantovani A, Hack EC, Roos A (2002) Direct binding of C1q to apoptotic cells and cell blebs induces complement activation. Eur J Immunol 32:1726–1736
Clayton A, Harris CL, Court J, Mason MD, Morgan BP (2003) Antigen-presenting cell exosomes are protected from complement-mediated lysis by expression of CD55 and CD59. Eur J Immunol 33:522–531
Flaumenhaft R (2006) Formation and fate of platelet microparticles. Blood Cells Mol Dis 36:182–187
Muralidharan-Chari V, Clancy JW, Sedgwick A, D’Souza-Schorey C (2010) Microvesicles: mediators of extracellular communication during cancer progression. J Cell Sci 123:1603–1611
Lopatina T, Gai C, Deregibus MC, Kholia S, Camussi G (2016) Cross talk between cancer and mesenchymal stem cells through extracellular vesicles carrying nucleic acids. Front Oncol 6:125
Kahlert C, Kalluri R (2013) Exosomes in tumor microenvironment influence cancer progression and metastasis. J Mol Med (Berl) 91:431–437
Hoshino A, Costa-Silva B, Shen TL, Rodrigues G, Hashimoto A, Tesic Mark M, Molina H, Kohsaka S, Di Giannatale A, Ceder S, Singh S, Williams C, Soplop N, Uryu K, Pharmer L, King T, Bojmar L, Davies AE, Ararso Y, Zhang T, Zhang H, Hernandez J, Weiss JM, Dumont-Cole VD, Kramer K, Wexler LH, Narendran A, Schwartz GK, Healey JH, Sandstrom P, Labori KJ, Kure EH, Grandgenett PM, Hollingsworth MA, de Sousa M, Kaur S, Jain M, Mallya K, Batra SK, Jarnagin WR, Brady MS, Fodstad O, Muller V, Pantel K, Minn AJ, Bissell MJ, Garcia BA, Kang Y, Rajasekhar VK, Ghajar CM, Matei I, Peinado H, Bromberg J, Lyden D (2015) Tumour exosome integrins determine organotropic metastasis. Nature 527:329–335
Kim JW, Wieckowski E, Taylor DD, Reichert TE, Watkins S, Whiteside TL (2005) Fas ligand-positive membranous vesicles isolated from sera of patients with oral cancer induce apoptosis of activated T lymphocytes. Clin Cancer Res 11:1010–1020
Pap E (2011) The role of microvesicles in malignancies. Adv Exp Med Biol 714:183–199
Mesri M, Altieri DC (1999) Leukocyte microparticles stimulate endothelial cell cytokine release and tissue factor induction in a JNK1 signaling pathway. J Biol Chem 274:23111–23118
Gasser O, Schifferli JA (2004) Activated polymorphonuclear neutrophils disseminate anti-inflammatory microparticles by ectocytosis. Blood 104:2543–2548
Distler JH, Huber LC, Gay S, Distler O, Pisetsky DS (2006) Microparticles as mediators of cellular cross-talk in inflammatory disease. Autoimmunity 39:683–690
Nomura S, Tandon NN, Nakamura T, Cone J, Fukuhara S, Kambayashi J (2001) High-shear-stress-induced activation of platelets and microparticles enhances expression of cell adhesion molecules in THP-1 and endothelial cells. Atherosclerosis 158:277–287
Mause SF, von Hundelshausen P, Zernecke A, Koenen RR, Weber C (2005) Platelet microparticles: a transcellular delivery system for RANTES promoting monocyte recruitment on endothelium. Arterioscler Thromb Vasc Biol 25:1512–1518
Mossberg M, Ståhl AL, Kahn R, Kristoffersson AC, Tati R, Heijl C, Segelmark M, Leeb-Lundberg LMF, Karpman D (2017) C1-inhibitor decreases the release of vasculitis-like chemotactic endothelial microvesicles. J Am Soc Nephrol 28:2472–2481
Karpman D, Ståhl AL, Arvidsson I (2017) Extracellular vesicles in renal disease. Nat Rev Nephrol 13:545–562
Gildea JJ, Seaton JE, Victor KG, Reyes CM, Bigler Wang D, Pettigrew AC, Courtner CE, Shah N, Tran HT, Van Sciver RE, Carlson JM, Felder RA (2014) Exosomal transfer from human renal proximal tubule cells to distal tubule and collecting duct cells. Clin Biochem 47:89–94
Timar CI, Lorincz AM, Csepanyi-Komi R, Valyi-Nagy A, Nagy G, Buzas EI, Ivanyi Z, Kittel A, Powell DW, McLeish KR, Ligeti E (2013) Antibacterial effect of microvesicles released from human neutrophilic granulocytes. Blood 121:510–518
Hiemstra TF, Charles PD, Gracia T, Hester SS, Gatto L, Al-Lamki R, Floto RA, Su Y, Skepper JN, Lilley KS, Karet Frankl FE (2014) Human urinary exosomes as innate immune effectors. J Am Soc Nephrol 25:2017–2027
Kleinjan A, Boing AN, Sturk A, Nieuwland R (2012) Microparticles in vascular disorders: how tissue factor-exposing vesicles contribute to pathology and physiology. Thromb Res 130 [Suppl 1]:S71–S73
Erdbrugger U, Le TH (2016) Extracellular vesicles in renal diseases: more than novel biomarkers? J Am Soc Nephrol 27:12–26
Camussi G, Cantaluppi V, Deregibus MC, Gatti E, Tetta C (2011) Role of microvesicles in acute kidney injury. Contrib Nephrol 174:191–199
Mariano F, Cantaluppi V, Stella M, Romanazzi GM, Assenzio B, Cairo M, Biancone L, Triolo G, Ranieri VM, Camussi G (2008) Circulating plasma factors induce tubular and glomerular alterations in septic burns patients. Crit Care 12:R42
Martino F, Lorenzen J, Schmidt J, Schmidt M, Broll M, Gorzig Y, Kielstein JT, Thum T (2012) Circulating microRNAs are not eliminated by hemodialysis. PLoS One 7:e38269
Tokes-Fuzesi M, Woth G, Ernyey B, Vermes I, Muhl D, Bogar L, Kovacs GL (2013) Microparticles and acute renal dysfunction in septic patients. J Crit Care 28:141–147
Mostefai HA, Meziani F, Mastronardi ML, Agouni A, Heymes C, Sargentini C, Asfar P, Martinez MC, Andriantsitohaina R (2008) Circulating microparticles from patients with septic shock exert protective role in vascular function. Am J Respir Crit Care Med 178:1148–1155
Delabranche X, Boisrame-Helms J, Asfar P, Berger A, Mootien Y, Lavigne T, Grunebaum L, Lanza F, Gachet C, Freyssinet JM, Toti F, Meziani F (2013) Microparticles are new biomarkers of septic shock-induced disseminated intravascular coagulopathy. Intensive Care Med 39:1695–1703
Soriano AO, Jy W, Chirinos JA, Valdivia MA, Velasquez HS, Jimenez JJ, Horstman LL, Kett DH, Schein RM, Ahn YS (2005) Levels of endothelial and platelet microparticles and their interactions with leukocytes negatively correlate with organ dysfunction and predict mortality in severe sepsis. Crit Care Med 33:2540–2546
Trepesch C, Nitzsche R, Glass A, Kreikemeyer B, Schubert JK, Oehmcke-Hecht S (2016) High intravascular tissue factor-but not extracellular microvesicles-in septic patients is associated with a high SAPS II score. J Intensive Care 4:34
Joop K, Berckmans RJ, Nieuwland R, Berkhout J, Romijn FP, Hack CE, Sturk A (2001) Microparticles from patients with multiple organ dysfunction syndrome and sepsis support coagulation through multiple mechanisms. Thromb Haemost 85:810–820
Cantaluppi V, Weber V, Lauritano C, Figliolini F, Beltramo S, Biancone L, De Cal M, Cruz D, Ronco C, Segoloni GP, Tetta C, Camussi G (2010) Protective effect of resin adsorption on septic plasma-induced tubular injury. Crit Care 14:R4
Oehmcke S, Westman J, Malmström J, Mörgelin M, Olin AI, Kreikemeyer B, Herwald H (2013) A novel role for pro-coagulant microvesicles in the early host defense against streptococcus pyogenes. PLoS Pathog 9:e1003529
Collino F, Bruno S, Incarnato D, Dettori D, Neri F, Provero P, Pomatto M, Oliviero S, Tetta C, Quesenberry PJ, Camussi G (2015) AKI recovery induced by mesenchymal stromal cell-derived extracellular vesicles carrying microRNAs. J Am Soc Nephrol 26:2349–2360
Bruno S, Tapparo M, Collino F, Chiabotto G, Deregibus MC, Soares Lindoso R, Neri F, Kholia S, Giunti S, Wen S, Quesenberry P, Camussi G (2017) Renal regenerative potential of different extracellular vesicle populations derived from bone marrow mesenchymal stromal cells. Tissue Eng Part A. https://doi.org/10.1089/ten.TEA.2017.0069
Choi HY, Moon SJ, Ratliff BB, Ahn SH, Jung A, Lee M, Lee S, Lim BJ, Kim BS, Plotkin MD, Ha SK, Park HC (2014) Microparticles from kidney-derived mesenchymal stem cells act as carriers of proangiogenic signals and contribute to recovery from acute kidney injury. PLoS One 9:e87853
du Cheyron D, Daubin C, Poggioli J, Ramakers M, Houillier P, Charbonneau P, Paillard M (2003) Urinary measurement of Na+/H+ exchanger isoform 3 (NHE3) protein as new marker of tubule injury in critically ill patients with ARF. Am J Kidney Dis 42:497–506
Zhou H, Pisitkun T, Aponte A, Yuen PS, Hoffert JD, Yasuda H, Hu X, Chawla L, Shen RF, Knepper MA, Star RA (2006) Exosomal fetuin-A identified by proteomics: a novel urinary biomarker for detecting acute kidney injury. Kidney Int 70:1847–1857
Chen HH, Lai PF, Lan YF, Cheng CF, Zhong WB, Lin YF, Chen TW, Lin H (2014) Exosomal ATF3 RNA attenuates pro-inflammatory gene MCP-1 transcription in renal ischemia-reperfusion. J Cell Physiol 229:1202–1211
Zhou H, Cheruvanky A, Hu X, Matsumoto T, Hiramatsu N, Cho ME, Berger A, Leelahavanichkul A, Doi K, Chawla LS, Illei GG, Kopp JB, Balow JE, Austin HA 3rd, Yuen PS, Star RA (2008) Urinary exosomal transcription factors, a new class of biomarkers for renal disease. Kidney Int 74:613–621
Daniel L, Fakhouri F, Joly D, Mouthon L, Nusbaum P, Grunfeld JP, Schifferli J, Guillevin L, Lesavre P, Halbwachs-Mecarelli L (2006) Increase of circulating neutrophil and platelet microparticles during acute vasculitis and hemodialysis. Kidney Int 69:1416–1423
Burton JO, Hamali HA, Singh R, Abbasian N, Parsons R, Patel AK, Goodall AH, Brunskill NJ (2013) Elevated levels of procoagulant plasma microvesicles in dialysis patients. PLoS One 8:e72663
Ando M, Iwata A, Ozeki Y, Tsuchiya K, Akiba T, Nihei H (2002) Circulating platelet-derived microparticles with procoagulant activity may be a potential cause of thrombosis in uremic patients. Kidney Int 62:1757–1763
Amabile N, Guerin AP, Leroyer A, Mallat Z, Nguyen C, Boddaert J, London GM, Tedgui A, Boulanger CM (2005) Circulating endothelial microparticles are associated with vascular dysfunction in patients with end-stage renal failure. J Am Soc Nephrol 16:3381–3388
Trappenburg MC, van Schilfgaarde M, Frerichs FC, Spronk HM, ten Cate H, de Fijter CW, Terpstra WE, Leyte A (2012) Chronic renal failure is accompanied by endothelial activation and a large increase in microparticle numbers with reduced procoagulant capacity. Nephrol Dial Transplant 27:1446–1453
Boulanger CM, Amabile N, Guerin AP, Pannier B, Leroyer AS, Mallat CN, Tedgui A, London GM (2007) In vivo shear stress determines circulating levels of endothelial microparticles in end-stage renal disease. Hypertension 49:902–908
Amabile N, Guerin AP, Tedgui A, Boulanger CM, London GM (2012) Predictive value of circulating endothelial microparticles for cardiovascular mortality in end-stage renal failure: a pilot study. Nephrol Dial Transplant 27:1873–1880
Lv LL, Cao YH, Ni HF, Xu M, Liu D, Liu H, Chen PS, Liu BC (2013) MicroRNA-29c in urinary exosome/microvesicle as a biomarker of renal fibrosis. Am J Physiol Ren Physiol 305:F1220–F1227
Lv LL, Cao YH, Pan MM, Liu H, Tang RN, Ma KL, Chen PS, Liu BC (2014) CD2AP mRNA in urinary exosome as biomarker of kidney disease. Clin Chim Acta 428:26–31
Ståhl AL, Vaziri-Sani F, Heinen S, Kristoffersson AC, Gydell KH, Raafat R, Gutierrez A, Beringer O, Zipfel PF, Karpman D (2008) Factor H dysfunction in patients with atypical hemolytic uremic syndrome contributes to complement deposition on platelets and their activation. Blood 111:5307–5315
Kelton JG, Warkentin TE, Hayward CP, Murphy WG, Moore JC (1992) Calpain activity in patients with thrombotic thrombocytopenic purpura is associated with platelet microparticles. Blood 80:2246–2251
Tati R, Kristoffersson AC, Ståhl AL, Rebetz J, Wang L, Licht C, Motto D, Karpman D (2013) Complement activation associated with ADAMTS13 deficiency in human and murine thrombotic microangiopathy. J Immunol 191:2184–2193
Jimenez JJ, Jy W, Mauro LM, Horstman LL, Soderland C, Ahn YS (2003) Endothelial microparticles released in thrombotic thrombocytopenic purpura express von Willebrand factor and markers of endothelial activation. Br J Haematol 123:896–902
Brogan PA, Shah V, Brachet C, Harnden A, Mant D, Klein N, Dillon MJ (2004) Endothelial and platelet microparticles in vasculitis of the young. Arthritis Rheum 50:927–936
Clarke LA, Hong Y, Eleftheriou D, Shah V, Arrigoni F, Klein NJ, Brogan PA (2010) Endothelial injury and repair in systemic vasculitis of the young. Arthritis Rheum 62:1770–1780
Hong Y, Eleftheriou D, Hussain AA, Price-Kuehne FE, Savage CO, Jayne D, Little MA, Salama AD, Klein NJ, Brogan PA (2012) Anti-neutrophil cytoplasmic antibodies stimulate release of neutrophil microparticles. J Am Soc Nephrol 23:49–62
Gasser O, Hess C, Miot S, Deon C, Sanchez JC, Schifferli JA (2003) Characterisation and properties of ectosomes released by human polymorphonuclear neutrophils. Exp Cell Res 285:243–257
Huang YM, Wang H, Wang C, Chen M, Zhao MH (2015) Promotion of hypercoagulability in antineutrophil cytoplasmic antibody-associated vasculitis by C5a-induced tissue factor-expressing microparticles and neutrophil extracellular traps. Arthritis Rheumatol 67:2780–2790
Eleftheriou D, Hong Y, Klein NJ, Brogan PA (2011) Thromboembolic disease in systemic vasculitis is associated with enhanced microparticle-mediated thrombin generation. J Thromb Haemost 9:1864–1867
Salih M, Zietse R, Hoorn EJ (2014) Urinary extracellular vesicles and the kidney: biomarkers and beyond. Am J Physiol Ren Physiol 306:F1251–F1259
Duan ZY, Cai GY, Bu R, Lu Y, Hou K, Chen XM (2016) Selection of urinary sediment miRNAs as specific biomarkers of IgA nephropathy. Sci Rep 6:23498
Wang G, Kwan BC, Lai FM, Chow KM, Li PK, Szeto CC (2011) Elevated levels of miR-146a and miR-155 in kidney biopsy and urine from patients with IgA nephropathy. Dis Markers 30:171–179
Moon PG, Lee JE, You S, Kim TK, Cho JH, Kim IS, Kwon TH, Kim CD, Park SH, Hwang D, Kim YL, Baek MC (2011) Proteomic analysis of urinary exosomes from patients of early IgA nephropathy and thin basement membrane nephropathy. Proteomics 11:2459–2475
Gao C, Xie R, Yu C, Wang Q, Shi F, Yao C, Xie R, Zhou J, Gilbert GE, Shi J (2012) Procoagulant activity of erythrocytes and platelets through phosphatidylserine exposure and microparticles release in patients with nephrotic syndrome. Thromb Haemost 107:681–689
Hogan MC, Johnson KL, Zenka RM, Charlesworth MC, Madden BJ, Mahoney DW, Oberg AL, Huang BQ, Leontovich AA, Nesbitt LL, Bakeberg JL, McCormick DJ, Bergen HR, Ward CJ (2014) Subfractionation, characterization, and in-depth proteomic analysis of glomerular membrane vesicles in human urine. Kidney Int 85:1225–1237
Zhou H, Kajiyama H, Tsuji T, Hu X, Leelahavanichkul A, Vento S, Frank R, Kopp JB, Trachtman H, Star RA, Yuen PS (2013) Urinary exosomal Wilms’ tumor-1 as a potential biomarker for podocyte injury. Am J Physiol Ren Physiol 305:F553–F559
Lee H, Han KH, Lee SE, Kim SH, Kang HG, Cheong HI (2012) Urinary exosomal WT1 in childhood nephrotic syndrome. Pediatr Nephrol 27:317–320
Rood IM, Merchant ML, Wilkey DW, Zhang T, Zabrouskov V, van der Vlag J, Dijkman HB, Willemsen BK, Wetzels JF, Klein JB, Deegens JK (2015) Increased expression of lysosome membrane protein 2 in glomeruli of patients with idiopathic membranous nephropathy. Proteomics 15:3722–3730
Hara M, Yanagihara T, Kihara I, Higashi K, Fujimoto K, Kajita T (2005) Apical cell membranes are shed into urine from injured podocytes: a novel phenomenon of podocyte injury. J Am Soc Nephrol 16:408–416
Woei AJFJ, van der Starre WE, Tesselaar ME, Garcia Rodriguez P, van Nieuwkoop C, Bertina RM, van Dissel JT, Osanto S (2014) Procoagulant tissue factor activity on microparticles is associated with disease severity and bacteremia in febrile urinary tract infections. Thromb Res 133:799–803
Pisitkun T, Shen RF, Knepper MA (2004) Identification and proteomic profiling of exosomes in human urine. Proc Natl Acad Sci U S A 101:13368–13373
Gonzales PA, Pisitkun T, Hoffert JD, Tchapyjnikov D, Star RA, Kleta R, Wang NS, Knepper MA (2009) Large-scale proteomics and phosphoproteomics of urinary exosomes. J Am Soc Nephrol 20:363–379
Joo KW, Lee JW, Jang HR, Heo NJ, Jeon US, Oh YK, Lim CS, Na KY, Kim J, Cheong HI, Han JS (2007) Reduced urinary excretion of thiazide-sensitive Na-Cl cotransporter in Gitelman syndrome: preliminary data. Am J Kidney Dis 50:765–773
Kanno K, Sasaki S, Hirata Y, Ishikawa S, Fushimi K, Nakanishi S, Bichet DG, Marumo F (1995) Urinary excretion of aquaporin-2 in patients with diabetes insipidus. N Engl J Med 332:1540–1545
Ben-Dov IZ, Tan YC, Morozov P, Wilson PD, Rennert H, Blumenfeld JD, Tuschl T (2014) Urine microRNA as potential biomarkers of autosomal dominant polycystic kidney disease progression: description of miRNA profiles at baseline. PLoS One 9:e86856
Hogan MC, Bakeberg JL, Gainullin VG, Irazabal MV, Harmon AJ, Lieske JC, Charlesworth MC, Johnson KL, Madden BJ, Zenka RM, McCormick DJ, Sundsbak JL, Heyer CM, Torres VE, Harris PC, Ward CJ (2015) Identification of biomarkers for PKD1 using urinary exosomes. J Am Soc Nephrol 26:1661–1670
Hogan MC, Manganelli L, Woollard JR, Masyuk AI, Masyuk TV, Tammachote R, Huang BQ, Leontovich AA, Beito TG, Madden BJ, Charlesworth MC, Torres VE, LaRusso NF, Harris PC, Ward CJ (2009) Characterization of PKD protein-positive exosome-like vesicles. J Am Soc Nephrol 20:278–288
Preston RA, Jy W, Jimenez JJ, Mauro LM, Horstman LL, Valle M, Aime G, Ahn YS (2003) Effects of severe hypertension on endothelial and platelet microparticles. Hypertension 41:211–217
Wang JM, Su C, Wang Y, Huang YJ, Yang Z, Chen L, Wu F, Xu SY, Tao J (2009) Elevated circulating endothelial microparticles and brachial-ankle pulse wave velocity in well-controlled hypertensive patients. J Hum Hypertens 23:307–315
Huang PH, Huang SS, Chen YH, Lin CP, Chiang KH, Chen JS, Tsai HY, Lin FY, Chen JW, Lin SJ (2010) Increased circulating CD31+/annexin V+ apoptotic microparticles and decreased circulating endothelial progenitor cell levels in hypertensive patients with microalbuminuria. J Hypertens 28:1655–1665
Kwon SH, Woollard JR, Saad A, Garovic VD, Zand L, Jordan KL, Textor SC, Lerman LO (2016) Elevated urinary podocyte-derived extracellular microvesicles in renovascular hypertensive patients. Nephrol Dial Transplant 32:800–807
Al-Massarani G, Vacher-Coponat H, Paul P, Arnaud L, Loundou A, Robert S, Moal V, Berland Y, Dignat-George F, Camoin-Jau L (2009) Kidney transplantation decreases the level and procoagulant activity of circulating microparticles. Am J Transplant 9:550–557
Montecalvo A, Shufesky WJ, Stolz DB, Sullivan MG, Wang Z, Divito SJ, Papworth GD, Watkins SC, Robbins PD, Larregina AT, Morelli AE (2008) Exosomes as a short-range mechanism to spread alloantigen between dendritic cells during T cell allorecognition. J Immunol 180:3081–3090
Walker JD, Maier CL, Pober JS (2009) Cytomegalovirus-infected human endothelial cells can stimulate allogeneic CD4+ memory T cells by releasing antigenic exosomes. J Immunol 182:1548–1559
Cumpelik A, Gerossier E, Jin J, Tsakiris D, Dickenmann M, Sadallah S, Schifferli JA, Zecher D (2015) Mechanism of platelet activation and hypercoagulability by antithymocyte globulins (ATG). Am J Transplant 15:2588–2601
Renner B, Klawitter J, Goldberg R, McCullough JW, Ferreira VP, Cooper JE, Christians U, Thurman JM (2013) Cyclosporine induces endothelial cell release of complement-activating microparticles. J Am Soc Nephrol 24:1849–1862
Dimuccio V, Ranghino A, Pratico Barbato L, Fop F, Biancone L, Camussi G, Bussolati B (2014) Urinary CD133+ extracellular vesicles are decreased in kidney transplanted patients with slow graft function and vascular damage. PLoS One 9:e104490
Pisitkun T, Gandolfo MT, Das S, Knepper MA, Bagnasco SM (2012) Application of systems biology principles to protein biomarker discovery: urinary exosomal proteome in renal transplantation. Proteomics Clin Appl 6:268–278
Alvarez S, Suazo C, Boltansky A, Ursu M, Carvajal D, Innocenti G, Vukusich A, Hurtado M, Villanueva S, Carreno JE, Rogelio A, Irarrazabal CE (2013) Urinary exosomes as a source of kidney dysfunction biomarker in renal transplantation. Transplant Proc 45:3719–3723
Peake PW, Pianta TJ, Succar L, Fernando M, Pugh DJ, McNamara K, Endre ZH (2014) A comparison of the ability of levels of urinary biomarker proteins and exosomal mRNA to predict outcomes after renal transplantation. PLoS One 9:e98644
Sonoda H, Yokota-Ikeda N, Oshikawa S, Kanno Y, Yoshinaga K, Uchida K, Ueda Y, Kimiya K, Uezono S, Ueda A, Ito K, Ikeda M (2009) Decreased abundance of urinary exosomal aquaporin-1 in renal ischemia-reperfusion injury. Am J Physiol Ren Physiol 297:F1006–F1016
Meehan SM, Limsrichamrern S, Manaligod JR, Junsanto T, Josephson MA, Thistlethwaite JR, Haas M (2003) Platelets and capillary injury in acute humoral rejection of renal allografts. Hum Pathol 34:533–540
Nielsen CT, Ostergaard O, Johnsen C, Jacobsen S, Heegaard NH (2011) Distinct features of circulating microparticles and their relationship to clinical manifestations in systemic lupus erythematosus. Arthritis Rheum 63:3067–3077
Nielsen CT, Ostergaard O, Stener L, Iversen LV, Truedsson L, Gullstrand B, Jacobsen S, Heegaard NH (2012) Increased IgG on cell-derived plasma microparticles in systemic lupus erythematosus is associated with autoantibodies and complement activation. Arthritis Rheum 64:1227–1236
Pereira J, Alfaro G, Goycoolea M, Quiroga T, Ocqueteau M, Massardo L, Perez C, Saez C, Panes O, Matus V, Mezzano D (2006) Circulating platelet-derived microparticles in systemic lupus erythematosus. Association with increased thrombin generation and procoagulant state. Thromb Haemost 95:94–99
Sole C, Cortes-Hernandez J, Felip ML, Vidal M, Ordi-Ros J (2015) miR-29c in urinary exosomes as predictor of early renal fibrosis in lupus nephritis. Nephrol Dial Transplant 30:1488–1496
Dignat-George F, Camoin-Jau L, Sabatier F, Arnoux D, Anfosso F, Bardin N, Veit V, Combes V, Gentile S, Moal V, Sanmarco M, Sampol J (2004) Endothelial microparticles: a potential contribution to the thrombotic complications of the antiphospholipid syndrome. Thromb Haemost 91:667–673
Pericleous C, Giles I, Rahman A (2009) Are endothelial microparticles potential markers of vascular dysfunction in the antiphospholipid syndrome? Lupus 18:671–675
Ridger VC, Boulanger CM, Angelillo-Scherrer A, Badimon L, Blanc-Brude O, Bochaton-Piallat ML et al (2017) Microvesicles in vascular homeostasis and diseases. Position Paper of the European Society of Cardiology (ESC) Working Group on Atherosclerosis and Vascular Biology. Thromb Haemost 117:1296–1316
Lakhter AJ, Sims EK (2015) Minireview: emerging roles for extracellular vesicles in diabetes and related metabolic disorders. Mol Endocrinol 29:1535–1548
Sabatier F, Darmon P, Hugel B, Combes V, Sanmarco M, Velut JG, Arnoux D, Charpiot P, Freyssinet JM, Oliver C, Sampol J, Dignat-George F (2002) Type 1 and type 2 diabetic patients display different patterns of cellular microparticles. Diabetes 51:2840–2845
Omoto S, Nomura S, Shouzu A, Nishikawa M, Fukuhara S, Iwasaka T (2002) Detection of monocyte-derived microparticles in patients with type II diabetes mellitus. Diabetologia 45:550–555
Diamant M, Nieuwland R, Pablo RF, Sturk A, Smit JW, Radder JK (2002) Elevated numbers of tissue-factor exposing microparticles correlate with components of the metabolic syndrome in uncomplicated type 2 diabetes mellitus. Circulation 106:2442–2447
Chen Y, Feng B, Li X, Ni Y, Luo Y (2012) Plasma endothelial microparticles and their correlation with the presence of hypertension and arterial stiffness in patients with type 2 diabetes. J Clin Hypertens (Greenwich) 14:455–460
Zubiri I, Posada-Ayala M, Benito-Martin A, Maroto AS, Martin-Lorenzo M, Cannata-Ortiz P, de la Cuesta F, Gonzalez-Calero L, Barderas MG, Fernandez-Fernandez B, Ortiz A, Vivanco F, Alvarez-Llamas G (2015) Kidney tissue proteomics reveals regucalcin downregulation in response to diabetic nephropathy with reflection in urinary exosomes. Transl Res 166:474–484; e474
Barutta F, Tricarico M, Corbelli A, Annaratone L, Pinach S, Grimaldi S, Bruno G, Cimino D, Taverna D, Deregibus MC, Rastaldi MP, Perin PC, Gruden G (2013) Urinary exosomal microRNAs in incipient diabetic nephropathy. PLoS One 8:e73798
Kalani A, Mohan A, Godbole MM, Bhatia E, Gupta A, Sharma RK, Tiwari S (2013) Wilm’s tumor-1 protein levels in urinary exosomes from diabetic patients with or without proteinuria. PLoS One 8:e60177
Ge S, Hertel B, Emden SH, Beneke J, Menne J, Haller H, von Vietinghoff S (2012) Microparticle generation and leucocyte death in Shiga toxin-mediated HUS. Nephrol Dial Transplant 27:2768–2775
Jimenez JJ, Jy W, Mauro LM, Horstman LL, Ahn YS (2001) Elevated endothelial microparticles in thrombotic thrombocytopenic purpura: findings from brain and renal microvascular cell culture and patients with active disease. Br J Haematol 112:81–90
Karpman D, Loos S, Tati R, Arvidsson I (2017) Haemolytic uraemic syndrome. J Intern Med 281:123–148
Kahn R, Herwald H, Muller-Esterl W, Schmitt R, Sjögren AC, Truedsson L, Karpman D (2002) Contact-system activation in children with vasculitis. Lancet 360:535–541
Kahn R, Hellmark T, Leeb-Lundberg LM, Akbari N, Todiras M, Olofsson T, Wieslander J, Christensson A, Westman K, Bader M, Muller-Esterl W, Karpman D (2009) Neutrophil-derived proteinase 3 induces kallikrein-independent release of a novel vasoactive kinin. J Immunol 182:7906–7915
Kronbichler A, Brezina B, Quintana LF, Jayne DR (2016) Efficacy of plasma exchange and immunoadsorption in systemic lupus erythematosus and antiphospholipid syndrome: a systematic review. Autoimmun Rev 15:38–49
Morel O, Toti F, Hugel B, Bakouboula B, Camoin-Jau L, Dignat-George F, Freyssinet JM (2006) Procoagulant microparticles: disrupting the vascular homeostasis equation? Arterioscler Thromb Vasc Biol 26:2594–2604
Chalmin F, Ladoire S, Mignot G, Vincent J, Bruchard M, Remy-Martin JP, Boireau W, Rouleau A, Simon B, Lanneau D, De Thonel A, Multhoff G, Hamman A, Martin F, Chauffert B, Solary E, Zitvogel L, Garrido C, Ryffel B, Borg C, Apetoh L, Rebe C, Ghiringhelli F (2010) Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J Clin Invest 120:457–471
Faille D, El-Assaad F, Mitchell AJ, Alessi MC, Chimini G, Fusai T, Grau GE, Combes V (2012) Endocytosis and intracellular processing of platelet microparticles by brain endothelial cells. J Cell Mol Med 16:1731–1738
Biancone L, Bruno S, Deregibus MC, Tetta C, Camussi G (2012) Therapeutic potential of mesenchymal stem cell-derived microvesicles. Nephrol Dial Transplant 27:3037–3042
Gregorini M, Corradetti V, Pattonieri EF, Rocca C, Milanesi S, Peloso A, Canevari S, De Cecco L, Dugo M, Avanzini MA, Mantelli M, Maestri M, Esposito P, Bruno S, Libetta C, Dal Canton A, Rampino T (2017) Perfusion of isolated rat kidney with mesenchymal stromal cells/extracellular vesicles prevents ischaemic injury. J Cell Mol Med. https://doi.org/10.1111/jcmm.13249
Cantaluppi V, Gatti S, Medica D, Figliolini F, Bruno S, Deregibus MC, Sordi A, Biancone L, Tetta C, Camussi G (2012) Microvesicles derived from endothelial progenitor cells protect the kidney from ischemia-reperfusion injury by microRNA-dependent reprogramming of resident renal cells. Kidney Int 82:412–427
Ju GQ, Cheng J, Zhong L, Wu S, Zou XY, Zhang GY, Gu D, Miao S, Zhu YJ, Sun J, Du T (2015) Microvesicles derived from human umbilical cord mesenchymal stem cells facilitate tubular epithelial cell dedifferentiation and growth via hepatocyte growth factor induction. PLoS One 10:e0121534
Tomasoni S, Longaretti L, Rota C, Morigi M, Conti S, Gotti E, Capelli C, Introna M, Remuzzi G, Benigni A (2013) Transfer of growth factor receptor mRNA via exosomes unravels the regenerative effect of mesenchymal stem cells. Stem Cells Dev 22:772–780
Grange C, Tapparo M, Bruno S, Chatterjee D, Quesenberry PJ, Tetta C, Camussi G (2014) Biodistribution of mesenchymal stem cell-derived extracellular vesicles in a model of acute kidney injury monitored by optical imaging. Int J Mol Med 33:1055–1063
Akyurekli C, Le Y, Richardson RB, Fergusson D, Tay J, Allan DS (2015) A systematic review of preclinical studies on the therapeutic potential of mesenchymal stromal cell-derived microvesicles. Stem Cell Rev 11:150–160
Ohno SI, Takanashi M, Sudo K, Ueda S, Ishikawa A, Matsuyama N, Fujita K, Mizutani T, Ohgi T, Ochiya T, Gotoh N, Kuroda M (2013) Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol Ther 21:185–191
El Andaloussi S, Mager I, Breakefield XO, Wood MJ (2013) Extracellular vesicles: biology and emerging therapeutic opportunities. Nat Rev Drug Discov 12:347–357
Lener T, Gimona M, Aigner L, Borger V, Buzas E, Camussi G, Chaput N, Chatterjee D, Court FA, Del Portillo HA, O’Driscoll L, Fais S, Falcon-Perez JM, Felderhoff-Mueser U, Fraile L, Gho YS, Görgens A, Gupta RC, Hendrix A, Hermann DM, Hill AF, Hochberg F, Horn PA, de Kleijn D, Kordelas L, Kramer BW, Krämer-Albers EM, Laner-Plamberger S, Laitinen S, Leonardi T, Lorenowicz MJ, Lim SK, Lötvall J, Maguire CA, Marcilla A, Nazarenko I, Ochiya T, Patel T, Pedersen S, Pocsfalvi G, Pluchino S, Quesenberry P, Reischl IG, Rivera FJ, Sanzenbacher R, Schallmoser K, Slaper-Cortenbach I, Strunk D, Tonn T, Vader P, van Balkom BW, Wauben M, Andaloussi SE, Théry C, Rohde E, Giebel B (2015) Applying extracellular vesicles based therapeutics in clinical trials—an ISEV position paper. J Extracell Vesicles. https://doi.org/10.3402/jev.v4.30087
Acknowledgements
A portion of the text and figure appeared in the Ph.D. thesis (2017) by Dr Maria Mossberg.
Funding
DK is supported by The Swedish Research Council (K2013-64X-14,008-13-5 and K2015-99X-22,877-01-6), The Knut and Alice Wallenberg Foundation (Wallenberg Clinical Scholar 2015.0320), The Torsten Söderberg Foundation, Skåne Centre of Excellence in Health, IngaBritt and Arne Lundberg Research Foundation, Crown Princess Lovisa’s Society for Child Care, Region Skåne, and The Konung Gustaf V:s 80-års minnesfond. RK is supported by the Swedish Rheumatism Association, The Medical Faculty at Lund University, Alfred Österlund’s Foundation, The Anna-Greta Crafoord Foundation, Greta and Johan Kock Foundation, Samariten Foundation, Sven Jerring Foundation, The Crafoord Foundation, and the Thelma Zoegas Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no conflicts of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Ståhl, Al., Johansson, K., Mossberg, M. et al. Exosomes and microvesicles in normal physiology, pathophysiology, and renal diseases. Pediatr Nephrol 34, 11–30 (2019). https://doi.org/10.1007/s00467-017-3816-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-017-3816-z