
Abstract
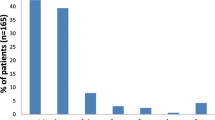
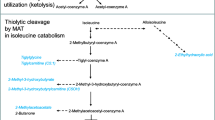
3-Hydroxy-3-methylglutaryl-coenzyme A lyase deficiency (HMG-CoA lyase) is an inborn error of leucine catabolism which often leads to life-threatening illness in the neonatal period. The cardinal clinical features include severe infantile hypoglycemia, metabolic acidosis, hepatomegaly, lethargy or coma and apnea. Hyperammonemia is variable. There is a characteristic absence of ketosis. Considerable heterogeneity has been observed in clinical and biochemical presentation. Acute episodes of illness have been mistaken for Reye syndrome. The pattern of organic acids in the urine includes large amounts of 3-hydroxy-3-methylglutaric, 3-methylglutaconic, 3-methylglutaric and 3-hydroxyisovaleric acids. Smaller, but appreciable levels of glutaric, adipic and other dicarboxylic acids may also be excreted in the urine. Lactic acid may be present in sizable amounts at times of acute illness. The primary defect is a deficiency of 3-hydroxy-3-methylglutaryl-coenzyme A lyase, a key enzyme in the cycle of ketogenesis.
Similar content being viewed by others

Abbreviations
- HMG-CoA lyase:
-
3-hydroxy-3-methylglutaryl-coenzyme A lyase
References
Applegarth DA, MacLeod PM, Toone JR, Kirby LT, MacLean JR, Mamer OA, Montgomery JA (1979) Organic acids and Reye's syndrome. Lancet I:1147
Barritt GL, Zander GL, Utter MF (1976) The regulation of pyruvate carboxylase activity in gluconeogenic tissues. In: Hanson EW, Mehlman MA (eds) Gluconeogenisis: its regulation in mamamalian species. Wiley, New York, p 31
Batshaw ML, Brusilow SW (1980) Treatment of hyperammonemic coma caused by inborn errors of urea synthesis. J Pediatr 97:893–900
Berry HK, Suchy F, Hunt M, Norman E (1981) Treatment of 3-hydroxy-3-methylglutaric aciduria in first cousins. In: Walser M, Williamson JR (eds) Metabolism and clinical implications of branched chain amino and ketoacids. Elsevier/North Holland, New York, pp 405–410
Bier DM, Leake RD, Haymond MW, Arnold KJ, Gruenke LD, Sperling MA, Kipnis DM (1977) Measurement of “true” glucose production rates in infancy and childhood with 6,6-dideuteroglucose. Diabetes 11:1016–1023
Buchanan DN, Thoene JG (1986) Photodiode array detection for liquid chromatographic profiling of carboxylic acids in physiological fluids: 3-hydroxy-3-methylglutaric aciduria. Clin Chem 32: 169–171
Chalmers RA, Lawson AM, Watts RWE (1974) Studies on the urinary acidic metabolites excreted by patients with Beta-methylcrotonylglycinuria, propionic acidaemia and methylmalonic acidaemia, using gas-liquid chromatography and mass spectrometry. Clin Chim Acta 52:43–51
Chalmers RA, Stacey TE, Tracey BM, Sousa C de, Roe CR, Millington DS, Hoppel CL (1984) l-carnitine in disorders of organic acid metabolism: response to l-carnitine by patients with methylmalonic aciduria and 3-hydroxy-3-methylglutaric aciduria. J Inherited Metab Dis [Suppl 2] 7:109–110
Clinkenbeard KD, Reed WD, Mooney RA, Lane MD (1975) Intracellular localization of the 3-hydroxy-3-methylglutaryl Coenzyme A cycle enzymes in liver. J Biol Chem 250:3108–3116
Dasouki M, Buchanan D, Mercer N, Gibson KM, Thoene J (1987) 3-Hydroxy-3-methylglutaric aciduria: response to carnitine therapy and fat and leucine restriction. J Inherited Metab Dis 10: 142–146
Divry P, Rolland MO, Teyssier J, Cotte J, Formosinho Fernandes MC, Tavares de Almeida I, Silveira C de (1981) 3-Hydroxy-3-methylglutaric aciduria combined with 3-methylglutaconic aciduria: a new case: J Inherited Metab Dis 4:173–174
Duran M, Ketting D, Wadman SK, Jakobs C, Schutgens RBH, Veder HA (1978) Organic acid excretion in a patient with 3-hydroxy-3-methylglutaryl-CoA lyase deficiency: facts and artefacts. Clin Chim Acta 90:187–193
Duran M, Schutgens RBH, Ketel A, Heymans H, Berntssen MWJ, Ketting D, Wadman SK (1979) 3-Hydroxy-3-methylglutaryl coenzyme A lyase deficiency: postnatal management following prenatal diagnosis by analysis of maternal urine. J Pediatr 95:1004–1007
Duran M, Beemer FA, Tibosch AS, Bruinvis L, Ketting D, Wadman SK (1982) Inherited 3-methylglutaconic aciduria in two brothers — another defect of leucine metbolism. J Pediatr 101: 551–554
Ellis GH, Mirkin LD, Mills MC (1979) Pancreatitis and Reye's syndrome. Am J Dis Child 133:1014–1016
Faull K, Bolton P, Halpern B, Hammond J, Danks DM, Haehnel R, Wilkinson SP, Wysocki SJ, Masters PL (1976) Patient with defect in leucine metabolism. N Engl J Med 294:1013
Faull KF, Bolton PD, Halpern B, Hammond J, Danks DM (1976) The urinary organic acid profile associated with 3-hydroxy-3-methylglutaric aciduria. Clin Chim Acta 73:553–559
Felig P, Sherwin R, Palaiologos G (1978) Ketone utilization and ketone-amino acid interactions in starvation and diabetes. In: Soeling HD, Seufert CD (eds) Biochemical and clinical aspects of ketone body metabolism. Thieme, Stuttgart, pp 166–173
Francois B, Bachmann C, Schutgens RBH (1981) Glucose metabolism in a child with 3-hydroxy-3-methylglutaryl-coenzyme A lyase deficiency. J Inherited Metab Dis 4:163–164
Gibson KM (1988) Assay of 3-hydroxy-3-methylglutaryl-coenzyme A lyase. In: Colowick SP, Kaplan NO (eds), Harris RA, Sokatch JR (volume eds). Methods in enzymology, branchedchain amino acids. Academic Press, New York (in press)
Gibson KM, Sweetman L, Nyhan WL, Page TM, Greene C, Cann HM (1982) 3-Hydroxy-3-methylglutaric aciduria: a new assay of 3-hydroxy-3-methylglutary-CoA lyase using high performance liquid chromatography. Clin Chim Acta 126:171–181
Gibson KM, Sweetman L, Nyhan WL, Narisawa K, Both K, Lehnert W, Robinson B, Duran M, Wadman SK (1985) 3-Methylglutaconyl-CoA hydratase deficiency: two different clinical and enzymatic phenotypes in 3-methylglutaconic aciduria. Pediatr Res 19:248a
Greene CL, Cann HM, Robinson BH, Gibson KM, Sweetman L, Holm J, Nyhan WL (1984) 3-Hydroxy-3-methylglutaric aciduria. J Neurogenet 1:165–173
Greene C, Blitzer M, Bronfin D, Shapira E (1986) 3-Hydroxy-3-methylglutaric aciduria presenting as Reye syndrome in an infant. Clin Res 34:241a
Greter J, Hagberg B, Steen G, Soderhjelm U (1978) 3-Methylglutaconic aciduria: report on a sibship with infantile progressive encephalopathy. Eur J Pediatr 129:231–238
Haehnel R, Wysocki SJ (1981) Potential prenatal diagnosis of 3-hydroxy-3-methylglutaryl-Coenzyme A lyase deficiency. Clin Chim Acta 111:287–288
Hagberg B, Hjalmarson O, Lindstedt S, Ransnas L, Steen G (1983) 3-Methylglutaconic aciduria in two infants. Clin Chim Acta 134:59–67
Hammond J, Wilcken B (1984) 3-Hydroxy-3-methylglutaric, 3-methyl-glutaconic and 3-methylglutaric acids can be nonspecific indicators of metabolic disease. J Inherited Metab Dis [Suppl 2] 7:117–118
Jakobs C, Bojasch M, Duran M, Ketting D, Wadman SK, Leupold D (1980) 3-Methyl-3-butenoic acid: an artefact in the urinary metabolic pattern of patients with 3-hydroxy-3-methylglutaryl-CoA lyase deficiency. Clin Chim Acta 106:85–89
Ketel A, Ket JL, Schutgens RBH, Duran M, Wadman SK (1980) Clinical and biochemical observations on a child with a deficiency of 3-hydroxy-3-methylglutaryl coenzyme A lyase. J Inherited Metab Dis 3:89–90
Lehnert W, Scharf J, Wendel U (1985) 3-Methylglutaconic and 3-methylglutaric aciduria in a patients with suspected 3-methylglutaconyl-CoA hydratase deficiency. Eur J Pediatr 143:301–303
Leonard JV, Seakins JWT, Griffin NK (1979) β-hydroxy-β-methylglutaricaciduria presenting as Reye's syndrome. Lancet I: 680
Leupold D, Bojasch M, Jakobs C (1982) 3-Hydroxy-3-methylglutaryl-CoA lyase deficiency in an infant with macrocephaly and mild metabolic acidosis. Eur J Pediatr 138:73–76
Lisson G, Leupold D, Bechinger D, Wallesch C (1981) CT findings in a case of deficiency of 3-hydroxy-3-methylglutaryl-CoA-lyase. Neuroradiology 22:99–101
Morens DM, Hammar SL, Heicher DA (1974) Idiopathic acute pancreatitis in children. Association with a clinical picture resembling Reye syndrome. Am J Dis Child 128:401–404
Narisawa K, Gibson KM, Sweetman L, Nyhan WL, Duran M, Wadman SK (1986) Deficiency of 3-methylglutaconyl-coenzyme A hydratase in two siblings with 3-methylglutaconic aciduria. J Clin Invest 77:1148–1152
Norman EJ, Denton MD, Berry HK (1982) Gas-chromatographic/mass spectrometric detection of 3-hydroxy-3-methylglutaryl-CoA lyase deficiency in double first cousins. Clin Chem 28:137–140
Robinson BH, Sherwood WG, Lampty M, Lowden JA (1976) β-Methyl glutaconic aciduria: a new disorder of leucine metabolism. Rediatr Res 10:371
Robinson BH, Oei J, Sherwood WG, Slyper AH, Heininger J, Mamer OA (1980) Hydroxymethylglutaryl CoA lyase deficiency: features resembling Reye syndrome. Neurology 30:714–718
Roe CR, Millington DS, Maltby DA (1986) Identification of 3-methylglutarylcarnitine. A new diagnostic metabolite of 3-hydroxy-3-methylglutaryl-coenzyme A lyase deficiency. J Clin Invest 77:1391–1394
Schutgens RBH, Heymans H, Ketel A, Veder HA, Duran M, Ketting D, Wadman SK (1979) Lethal hypoglycemia in a child with a deficiency of 3-hydroxy-3-methylglutaryl-coenzyme A lyase. J Pediatr 94:89–91
Shilkin R, Wilson G, Owles E (1981) 3-hydroxy-3-methylglutaryl coenzyme A lyase deficiency. Follow-up of first described case. Acta Paediatr Scand 70:265–268
Sovik O, Sweetman L, Gibson KM, Nyhan WL (1984) Genetic complementation analysis of 3-hydroxy-3-methylglutaryl-coenzyme A lyase deficiency in cultured fibroblasts. Am J Hum Genet 36:791–801
Stacey TE, Sousa C de, Tracey BM, Whitelaw A, Mistry J, Timbrell P, Chalmers RA (1985) Dizygotic twins with 3-hydroxy-3-methylglutaric aciduria; unusual presentation, family studies and dietary management. Eur J Pediatr 144:177–181
Sweetman L (1984) Prenatal diagnosis of the organic acidurias. J Inherited Metab Dis [Suppl 1] 7:18–22
Tracey BM, Stacey TE, Chalmers RA (1983) Urinary and plasma organic acids in dizygotic twin siblings with 3-hydroxy-3-methylglutaric aciduria, studied by gas chromatography and mass spectrometry using fused silica capillary columns. J Inherited Metab Dis [Suppl 2] 6:125–126
Truscott RJW, Halpern B, Wysocki SJ, Haehnel R, Wilcken B (1979) Studies on a child suspected of having a deficiency in 3-hydroxy-3-methylglutaryl-CoA lyase. Clin Chim Acta 95:11–16
Walter JH, Clayton PT, Leonard JV (1986) 3-Hydroxy-3-methylglutaryl-CoA lyase deficiency. J Inherited Metab Dis 9:287–288
Wilson WG, Cass MB, Sovik O, Gibson KM, Sweetman L (1984) A child with acute pancreatitis and recurrent hypoglycemia due to 3-hydroxy-3-methylglutaryl-CoA lyase deficiency. Eur J Pediatr 142:289–291
Wolff JA, Thuy LP, Haas R, Carroll JE, Prodanos C, Nyhan WL (1986) Carnitine reduces fasting ketogenesis in patients with disorders of propionate metabolism. Lancet I:289–291
Wysocki SJ, Haehnel R (1976) 3-Hydroxy-3-methylglutaric aciduria: deficiency of 3-hydroxy-3-methylglutaryl coenzyme A lyase. Clin Chim Acta 71:349–351
Wysocki SJ, Haehnel R (1976) 3-Hydroxy-3-methylglutaric aciduria: 3-hydroxy-3-methylglutaryl-coenzyme A lyase levels in leucocytes. Clin Chim Acta 73:373–375
Wysocki SJ, Haehnel R (1978) 3-Methylcrotonylglycine excretion in 3-hydroxy-3-methylglutaric aciduria. Clin Chim Acta 86:101–108
Wysocki SJ, Haehnel R, Truscott RJW, Halpern B, Wilcken B (1979) Hyperammonaemia and urinary organic acids. Lancet II: 371–372
Wysocki SJ, Wilkinson SP, Haehnel R, Wong CYB, Panegyres PK (1976) 3-Hydroxy-3-methylglutaric aciduria, combined with 3-methylglutaconic aciduria. Clin Chim Acta 70:399–406
Yoshida I, Sovik O, Sweetman L, Nyhan WL (1985) Metabolism of leucine in fibroblasts from patients with deficiencies in each of the major catabolic enzymes: branched-chain ketoacid dehydrogenase, isovaleryl-CoA dehydrogenase, 3-methylcrotonyl-CoA carboxylase, 3-methylglutanyl-CoA hydratase, and 3-hydroxy-3-methylglutaryl-CoA lyase. J Neurogenet 2:413–424
Zoghbi HY, Spence JE, Beaudet AL, O'Brien WE, Goodman CJ, Gibson KM (1986) Atypical presentation and neuropathological studies in 3-hydroxy-3-methylglutaryl-CoA lyase deficiency. Ann Neurol 20:367–369
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Gibson, K.M., Breuer, J. & Nyhan, W.L. 3-hydroxy-3-methylglutaryl-coenzyme A lyase deficiency: review of 18 reported patients. Eur J Pediatr 148, 180–186 (1988). https://doi.org/10.1007/BF00441397
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/BF00441397