
Abstract
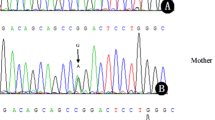
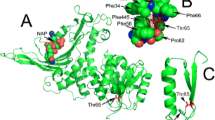
Ornithine transcarbamylase deficiency (OTC), the most common inborn error of the urea cycle, shows an X-linked inheritance with frequent new mutations. Southern blots reveal only a small percent of the mutation, but amplification of cDNA or genomic DNA using the polymerase chain reaction (PCR) followed by DNA sequencing, has contributed greatly to overcoming this difficulty. Problems remaining are the limited availability of fresh liver samples for preparation of intact mRNA in the former case, and there are primer sequences for PCR for only some exons in the latter case. Here, we report the structures of intron sequences which are long enough to analyze all exons and adjacent introns of the OTC gene using PCR and PCR single-strand conformation polymorphisms (PCR-SSCP). We carried out a DNA analysis of findings in five Japanese male patients with neonatal or late onset form. Five patients had mutations in the protein coding region. C to G (S192R), A to T (D196V), A to G (T264A), T to C (M268T), and C to T (R277W) substitutions. The first four of these were novel missense mutations and the presence of the mutation was confirmed in the corresponding families.
Similar content being viewed by others

References
Briand P, Francois B, Rabier D, Cathelineau L (1982) Ornithine transcarbamylase deficiencies in human males: kinetic and immunochemical classification. Biochim Biophys Acta 704:100–106
Brusilow SW, Horwich AL (1989) Urea cycle enzymes. In: Scriber CR, Beaudet AL, Sly WS, Valle D (eds) The metabolic basis of inherited disease, 6th edn. McGraw-Hill, New York, pp 629–663
Carstens RP, Fenton WA, Rosenberg LE (1991) Identification of RNA splicing errors resulting in human ornithine transcarbamylase deficiency. Am J Hum Genet 48:1105–1114
Finkelstein JE, Francomano CA, Brusilow SW, Traystman MD (1990) Use of denaturing gradient gel electrophoresis for detection of mutation and prospective diagnosis in late onset ornithine transcarbamylase deficiency. Genomics 7:167–172
Fox JE, Hack AM, Fenton WA, Rosenberg LE (1986) Identification and application of additional restriction fragment length polymorphisms at the human ornithine transcarbamylase locus. Am J Hum Genet 38:841–847
Grompe M, Muzny DM, Caskey CT (1989) Scanning detection of mutations in human ornithine transcarbamylase by chemical mismatch cleavage. Proc Natl Acad Sci USA 86:5888–5892
Grompe M, Caskey CT, Fenwick RG (1991) Improved molecular diagnostics for ornithine transcarbamylase deficiency. Am J Hum Genet 48:212–222
Haldane JBS (1935) Rate of spontaneous mutation of the human gene. J Genet 31:317–326
Hata A, Tsuzuki T, Shimada K, Takiguchi M, Mori M, Matsuda I (1986) Isolation and characterization of the human ornithine transcarbamylase gene: structure of the 5′-end region. J Biochem 100:717–725
Hata A, Tsuzuki T, Shimada K, Takiguchi M, Mori M, Matsuda I (1988a) Structure of the human ornithine transcarbamylase gene. J Biochem 103:302–308
Hata A, Tsuzuki T, Shimada K, Takiguchi M, Mori M, Matsuda I (1988b) Structural organization of the human ornithine transcarbamylase gene. J Inherited Metab Dis 11:337–340
Hata A, Setoyama C, Shimada K, Takeda E, Kuroda Y, Akaboshi I, Matsuda I (1989) Ornithine transcarbamylase deficiency resulting from a C-to-T substitution in exon 5 of the ornithine transcarbamylase gene. Am J Hum Genet 45:123–127
Hata A, Matsuura T, Setoyama C, Shimada K, Yokoi T, Akaboshi I, Matsuda I (1991) A novel missense mutation in exon 8 of the ornithine transcarbamylase gene in two unrelated male patients with mild ornithine transcarbamylase deficiency. Hum Genet 87:28–32
Hauser ER, Finkelstein JE, Valle DV, Brusilow SW (1990) Allopurinol-induced orotidinuria. A test for mutations at the ornithine carbamoyltransferase locus in women. N Engl J Med 322:1641–1645
Hayashi K (1991) PCR-SSCP: A simple and sensitive method for detection of mutations in the genomic DNA. PCR Methods and Applications 1:34–38
Horwich AL, Fenton WA, Williams K, Kalousek F, Kraus JP, Doolittle RF, Konigsberg W, Rosenberg LE (1984) Structure and expression of a complementary DNA for the nuclear coded precursor of human mitochondrial ornithine transcarbamylase. Science 224:1068–1074
Huygen R, Crabeel M, Glansdorff N (1987) Nucleotide sequence of the ARGS gene of the yeast Saccharomyces cerevisiae encoding ornithine carbamoyltransferase. Comparison with other carbamoyl-transferases. Eur J Biochem 166:371–377
Lyon MF (1972) X-chromosome inactivation and developmental patterns in mammals. Biol Rev 47:1–35
Maddalena A, Sosnoski DM, Berry GT, Nussbaum RL (1988a) Mosaicism for an intragenic deletion in a boy with mild ornithine transcarbamylase deficiency. N Engl J Med 319:999–1003
Maddalena A, Spence JE, O'Brien WE, Nussbaum RL (1988b) Characterization of point mutations in the same arginine codon in three unrelated patients with ornithine transcarbamylase deficiency. J Clin Invest 82:1353–1358
Matsuda I, Hata A, Matsuura T, Tsuzuki T, Shimada K (1989) Inborn enzyme deficiencies; structure of the ornithine transcarbamylase (OTC) gene and DNA diagnosis of OTC deficiency. Clin Chim Acta 185:283–290
Mastuda I, Nagata N, Matsuura T, Oyanagi K, Tada K, Narisawa K, Kitagawa T, Sakiyama T, Yamashita F, Yoshino M (1991) Retrospective survey of urea cycle disorders: Part 1. Clinical and laboratory observations of thirty-two Japanese male patients with ornithine transcarbamylase deficiency. Am J Med Genet 38:85–89
Nagata N, Oyanagi K, Matsuda I (1991a) Estimated frequency of urea cycle enzymopathies in Japan. Am J Med Genet 39:228–229
Nagata N, Matsuda I, Matsuura T, Oyanagi K, Tada K, Narisawa K, Kitagawa T, Sakiyama T, Yamashita F, Yoshino M (1991b) Retrospective survey of urea cycle disorders: Part 2. Neurological outcome in forty-nine Japanese patients with urea cycle enzymopathies. Am J Med Genet 40:477–481
Ng WG, Oizumi J, Koch R, Shaw KNF, McLaren J, Donnel GN, Carter M (1981) Carrier detection of urea cycle disorders. Pediatrics 68:448–452
Old JM, Briand PL, Puvis-Smith S, Howard NJ, Wilcken B, Hammond J, Pearson P, Cathelineau L, Williamson R, Davies KE (1985) Prenatal exclusion of ornithine transcarbamylase deficiency by direct gene analysis. Lancet I:73–75
Orita M, Iwahana H, Kanazawa H, Hayashi K, Sekiya T (1989) Detection of polymorphisms of human DNA by gel elecrophoresis as single-strand conformation polymorphisms. Proc Natl Acad Sci USA 86:2766–2770
Rozen R, Fox J, Fenton WA, Horwich AL, Rosenberg LE (1985) Gene deletion and restriction fragment length polymorphisms at the human ornithine transcarbamylase locus. Nature 313:815–817
Rozen R, Fox JE, Hack AM, Fenton WA, Horwich AL, Rosenberg LE (1986) DNA analysis for ornithine transcarbamylase deficiency. J Inherited Metab Dis 9 [Suppl 1]:49–57
Saiki RK, Scharf S, Faloona, Mullis KB, Horn GT, Erlich HA, Arnheim N (1985) Enzymatic amplification of β-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science 230:1350–1354
Saiki RK, Gelfand DH, Stoffel S, Scharf SJ, Higuchi R, Horn GT, Mullis KB, Erlich HA (1988) Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science 239:487–491
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: A laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY
Veres G, Gibbs RA, Sherer SE, Caskey CT (1987) The molecular basis of the sparse fur mouse mutation. Science 237:415–417
Walser M (1983) Urea cycle disorders and other hereditary hyperammonemic syndromes. In: Stanbury JB, Wyngaarden JB, Fredrickson DS, Goldstein JL, Brown MS (eds) The metabolic basis of inherited disease. McGraw-Hill, New York, pp 402–438
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Matsuura, T., Hoshide, R., Setoyama, C. et al. For novel gene mutations in five Japanese male patients with neonatal or late onset OTC deficiency: application of PCR-single-strand conformation polymorphisms for all exons and adjacent introns. Hum Genet 92, 49–56 (1993). https://doi.org/10.1007/BF00216144
Received:
Issue Date:
DOI: https://doi.org/10.1007/BF00216144