
Abstract
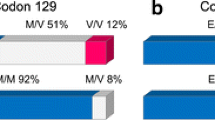
Creutzfeldt-Jakob disease (CJD) is a transmissible neurodegenerative disorder characterized by the accumulation of the amyloid protein PrP in the CNS. Two coding polymorphisms of the PrP gene (PRNP) are a methionine (Met) to valine (Val) change at codon 129, and a deletion in the octapeptide coding region. In the United Kingdom, homozygosity at codon 129 appears to be associated with a predisposition to develop CJD. However, in Japan, where allelic frequencies and genotype distribution are significantly different, such an association has not been demonstrated. To determine whether such deletion(s) or codon 129 polymorphisms of PRNP predispose to the development of CJD in Italian patients, 31 sporadic CJD patients with no known PRNP mutations, and 186 unrelated control subjects were studied. Genotypic frequencies at codon 129 in these Italian CJD patients revealed a significant excess of methionine alleles, and a different genotype distribution in comparison with the normal Italian population. Deletions of a 24-bp segment located in the PrP octapeptide coding region were found in two control subjects, but in none of the sporadic CJD patients. These data suggest that Met homozygosity at codon 129 may contribute, with other enviromental or endogenous factors, to CJD development.
Similar content being viewed by others
References
Baker HF, Poulter M, Crow TJ, Frith CD, Lofthouse R, Ridley RM (1991) Aminoacid polymorphism in human prion protein and age at death in inherited prion disease. Lancet 337:1289
Bosque PJ, Vnencak-Jones CL, Johnson MD, Whitlock JA, McLean MJ (1992) A PrP gene codon 178 base substitution and a 24-bp interstitial deletion in familial Creutzfeldt-Jakob disease. Neurology 42:864–1870
Brown P, Cathala F, Castaigne P, Gajdusek DC (1986) Creutzfeldt-Jakob disease: clinical analysis of a consecutive series of 230 neuropathologically verified cases. Ann Neurol 20:597–602
Brown P, Goldfarb LG, Gibbs CJ, Gajdusek DC (1991) The phenotypic expression of different mutations in transmissible familial Creutzfeldt-Jakob disease. Eur J Epidemiol 7:469–476
Brown P, Cervenakova L, Goldfarb LG, McCombie WR, Rubenstein R, Will RG, Pocchiari M, Martinez-Lage JF, Scalici C, Masullo C, Graupera G, Ligan J, Gajdusek DC (1994) Iatrogenic Creutzfeldt-Jakob disease: an example of the interplay between ancient genes and modern medicine. Neurology 44:Brunori G, Silvestrini C, Pocchiari M (1988) The scrapie agent and the prion hypothesis. Trends Biochem Sci 13:309–313
Bueler H, Aguzzi A, Salier A, Greiner RA, Autenried P, Aguet M. Weissmann C (1993) Mice devoid of PrP are resistant to scrapie. Cell 73:1339–1347
Caughey B, Race R, Ernst D, Buchmeier M, Chesebro B (1989) Prion protein biosynthesis in scrapie-infected and uninfected neuroblastoma cells. J Virol 63:175–181
Collinge J. Palmer M (1991) CJD discrepancy. Nature 353:802
Collinge J, Palmer MS, Dryden AJ (1991) Genetic predisposition to iatrogenic Creutzfeldt-Jakob disease. Lancet 337:1441–1442
Deslys J-P, Marcé D, Dormont D (1994) Similar genetic susceptibility in iatrogenic and sporadic Creutzfeldt-Jakob disease. J Gen Virol 75:23–25
Dietrich JF, Knopman DS, List JF, Olson K, Frey WH II, Emory CR, Sung JH, Haase AT (1992) Deletion in the prion protein gene in a demented patient. Hum Mol Gen 1:443–444
Diringer H (1992) Hidden amiloydoses. Exp Clin Immunogenet 9:212–229
Dlouhy SR, Hsiao K, Farlow MR, Foroud T, Conneally PM, Johnson P, Prusiner SB, Hodes ME, Ghetti B (1992) Linkage of the Indiana kindred of Gerstmann-Straussler-Sheinker disease to the prion protein gene. Nature Genet 1:64–67
Doh-ura K, Tateishi J, Sasaki H, Kitamoto T, Sakaki Y (1989) Pro → leu change at position 102 of prion protein is the most common but not the sole mutation related to Gerstmann-Straussler syndrome. Biochem Biophys Res Commun 163:974–979
Doh-Ura K, Kitamoto T, Sakaki Y, Tateishi J (1991) CJD discrepancy. Nature 353:801–802
Gabizon R, Rosenmann H, Meiner Z, Kahana I, Kahana E, Shugart Y, Ott J, Prusiner SB (1993) Mutation and polymorphism of the prion protein gene in Libyan Jews with Creutzfeldt-Jakob disease (CJD). Am J Hum Genet 53:828–835
Goldfarb LG, Haltia M, Brown P, Nieto A, Kovanen J. McCombie WR, Trapp S, Gajdusek DC (1991a) New mutation in scrapie amyloid precursor gene (at codon 178) in Finnish Creutzfeldt Jakob kindred. Lancet 337:425
Goldfarb LG, Brown P, McCombie WR, Goldgaber D, Swergold GD, Wills PR, Cervenacova L, Baron H, Gibbs CJ Jr. Gajdusek DC (1991b) Transmissible familial Creutzfeldt-Jakob disease associated with five, seven, and eight extra octapeptide coding repeats in the PRNP gene. Proc Natl Acad Sci USA 88:10926–10930
Goldfarb LG, Brown P, Vrbovska A, Baron H, McCombie WR, Cathala F, Gibbs CJ Jr. Gajdusek DC (1992) An insert mutation in the chromosome 20 amyloid precursor gene in a Gerstmann-Straussler-Scheinker family. J Neurol Sci 111:189–194
Goldgaber D, Goldfarb LG, Brown P, Asher DM, Brown WT, Lin S, Teener JW. Feinstone SM, Rubenstein R, Kacsak RJ, Boellaard JW, Gajdusek CD (1989) Mutations in familial Creutzfeldt-Jakob disease and Gerstmann-Straussler-Scheinker's syndrome. Exp Neurol 106:204–206
Hsiao K, Baker HF, Crow TY, Poulter M, Owen F, Terwilliger JD, Westaway W. Ott J, Prusiner SB (1989) Linkage of a prion protein missense variant to Gerstmann-Straussler syndrome. Nature 338:342–345
Hsiao K, Dlouhy SR, Farlow MR, Cass C, Costa M Da, Conneally PM, Hodes ME, Ghetti B, Prusiner SB (1992) Mutant prion proteins in Gerstmann-Straussler-Sheinker disease with neurofibrillary tangles. Nature Genet 1:68–71
Kitamoto T, Doh-Ura K, Muramoto T, Miyazono M, Tateishi J (1992) The primary structure of the prion protein influences the distribution of abnormal Prion protein in the central nervous system. Am J Pathol 141:1–7
Laplanche JL, Chatelain J, Launay JM, Gazengel C, Vidaud M (1990) Deletion in prion protein gene in a Moroccan family. Nucleic Acids Res 18:6745
Laplanche JL, Chatelein J, Thomas S, Brown P, Chatala F (1991) Analyse du gene PrP dans une famille d'origine tunisienne atteinte de Maladie de Creutzfeldt-Jakob. Rev Neurol 147:825–827
Masullo C, Salvatore M, Genuardi M, Pocchiari M, Macchi M (1994) Progressive dementia in a young patient with homozygosity del PrP gene. Ann NY Acad Sci 724:358–360
Medori R, Tritschler H-J (1993) Prion protein gene analysis in three kindreds with fatal familial insomnia (FFI): codon 178 mutation and codon 129 polymorphism. Am J Hum Genet 53:22–827
Miyazono M, Kitamoto T, Doh-Ura K, Iwaki T, Tateishi J (1992) Creutzfeldt-Jakob disease with codon 129 polymorphism (Valine): a comparative study of patients with codon 102 point mutation or without mutations Acta Neuropathol 84:349–354
Oesch B, Westaway D, Walchli M, McKinley MP, Kent SB, Aebersold R, Barry RA, Tempst P, Teplow DB, Hodd LE, Prusiner SB, Weissman C (1985) A cellular gene encodes scrapie PrP27–30 protein. Cell 40:735–746
Owen F, Poulter M, Lofthouse R, Collinge J, Crow TJ, Risby D, Baker HF, Ridley RM, Hsiao K, Prusiner SB (1989) Insertion in prion protein gene in familial Creutzfeldt Jakob. Lancet 1:51–52
Owen F, Poulter M, Collinge J, Crow TJ (1990) Codon 129 changes in the prion protein gene in Caucasians. Am J Hum Genet 46:1215–1216
Palmer MS, Dryden AJ, Hughes JT, Collinge J (1991) Homozygous prion protein predisposes to sporadic Creutzfeldt-Jakob disease. Nature 352:340–342
Pocchiari M, Salvatore M, Cutruzzolá F, Genuardi M, Travaglini-Allocatelli C, Masullo C, Macchi G, Alemá G, Galgani S, Xi YG, Petraroli R, Silvestrini MC, Brunori M (1993) A new point mutation of the prion protein gene in Creutzfeldt-Jakob disease. Ann Neurol 34:802–807
Poulter M, Baker KF, Frith CD, Leach M, Lofthouse R, Ridley RM, Shah T, Owen F, Collinge J, Brown J, Hardy J, Mullan MJ, Harding AE, Bennet C, Doshi R, Crow TJ (1992) Inherited Prion Disease with 144 base pair gene insertion. 1. Genealogical and molecular studies. Brain 115:675–685
Preece M (1993) Human pituitary growth hormone and Creutzfeldt-Jakob disease. Horm Res 39:95–98
Prusiner SB (1993) Genetic and infectious prion diseases. Arch Neurol 50:1129–1153
Puckett C, Concannon P, Casey C, Hood L (1991) Genomic structure of the human prion protein gene. Am J Hum Gen 49:320–329
Ripoll L, Laplance JL, Salzmann M, Jouvet A, Planques B, Dussaucy M, Chatelain J, Beaudry P, Launay JM (1993) A new point mutation in the prion protein gene at codon 210 in Creutzfeldt-Jakob disease. Neurology 43:1934–1938
Sambrook J, Fritsch EF, Maniatis T (1985) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY
Trabattoni G, Lechi A, Bettoni L, Macchi G, Brown P (1990) Consideration on a group of 13 patients with Creutzfeldt-Jakob disease in the region of Parma (Italy). Eur J Epidemiol 6:238–243
Tsuji S, Kuroiwa Y (1983) Creutzfeldt-Jakob disease in Japan. Neurology 33:1503–1506
Vnencak-Jones C, Phillips JA III (1992) Identification of heterogeneous PrP gene deletions in controls by detection of allelespecific heteroduplexes (DASH). Am J Hum Gen 50:871–872
Westaway D, Goodman PA, Mirenda CA, McKinley MP, Carlson GA, Prusiner SB (1987) Distinct prion proteins in short and long scrapie incubation period mice. Cell 51:651–662
Xi YG, Ingrosso L, Ladogana A, Masullo C, Pocchiari M (1992) Amphotericin B treatment dissociates in vivo replication of the scrapie agent from PrP accumulation. Nature 356:598–601
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Salvatore, M., Genuardi, M., Petraroli, R. et al. Polymorphisms of the prion protein gene in Italian patients with Creutzfeldt-Jakob disease. Hum Genet 94, 375–379 (1994). https://doi.org/10.1007/BF00201596
Received:
Revised:
Issue Date:
DOI: https://doi.org/10.1007/BF00201596