
Abstract
Venous thromboembolism (VTE) is a disease of blood coagulation that occurs in the veins, most often in the calf veins first, from where it may extend and cause deep vein thrombosis (DVT) or pulmonary embolism (PE). The first described case of venous thrombosis that we know of dates back to the thirteenth century, when deep vein thrombosis was reported in the right leg of a 20-year-old man [1].
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


1 Introduction
Venous thromboembolism (VTE) is a disease of blood coagulation that occurs in the veins, most often in the calf veins first, from where it may extend and cause deep vein thrombosis (DVT) or pulmonary embolism (PE). The first described case of venous thrombosis that we know of dates back to the thirteenth century, when deep vein thrombosis was reported in the right leg of a 20-year-old man [1].
The risk of thrombosis is influenced by both genetic and environmental factors. The risk factors for venous thrombosis are immobility, major surgery, underlying medical conditions like malignancies, medication use such as hormonal therapies, obesity, and genetic predisposition. In contrast to that, the major risk factor for arterial thrombosis is atherosclerosis [2].
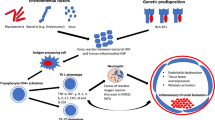
In 1859, a German scientist, Rudolf Virchow, elucidated the mechanism of pulmonary embolism and hence deduced the major pathogenic determinants for DVT and PE, named as Virchow’s triad that comprised (1) blood stasis, (2) changes in the vessel wall, and (3) hypercoagulability. This triad still applies, with essentially all prothrombotic factors, whether systemic or molecular, influencing one of these three mechanisms [3] (Fig. 12.1).

Virchow’s triad and some inflammatory changes and their association with venous and arterial thrombosis
DVT and PE are extremely common medical problems, and they are among the major cause of morbidity and death worldwide [3].
About 1–2 per 1000 individuals are affected by VTE per year with PE being the lethal complication and is associated with a high mortality rate that exceeds 15% in the first 3 months after diagnosis [4].
Although disturbance of the coagulation and anticoagulation mechanisms is a very important risk factor for VTE, several studies suggest the role of innate immunity in the development of VTE [4].
Venous thrombosis results from multiple interactions between acquired and inherited risk factors [4].
In this chapter, we discuss the association of thrombosis with autoimmune rheumatologic disorders. The pathophysiology of thrombosis, effects of inflammation, endothelial dysfunction, some novel factors on promoting thrombosis in different rheumatic disorders, diagnostic strategies for thromboembolic disease, and its treatment, management, as well as preventive measures will be addressed in detail. This chapter is useful for students, residents, fellows, and physicians interested in learning about rheumatic disease and thrombosis. The main objective of this chapter is to make the readers able to achieve the following goals:
-
1.
Explain and discuss the pathophysiology of thrombosis in rheumatic diseases.
-
2.
Recognize the multifactorial role of inflammation in inducing the hypercoagulable state which promote thrombosis in autoimmune rheumatic disorders.
-
3.
Classify thrombosis in patients with rheumatologic diseases (arterial or venous) according to the presence of different risk factors and type of the disorders.
-
4.
Identify the role of different autoantibodies in specific rheumatic diseases such as systemic lupus erythematosus and antiphospholipid syndrome that contribute to the high risk of thrombosis in these conditions.
-
5.
Describe the effects of different therapies commonly used in patients with rheumatic disorders and their role in thrombosis promotion or prevention.
-
6.
Recognize the clinical features of thromboembolic diseases, as well as formulate a comprehensive history of thrombosis from patients with rheumatic diseases.
-
7.
Judge when to select specific assays for thrombophilia screening, clotting factors, autoantibodies, and other tests for thrombotic episodes in rheumatic diseases and choose the appropriate investigations necessary for the diagnosis of thrombosis in these patients.
-
8.
Construct an approach to the diagnosis of thrombosis in rheumatic diseases based on patient’s clinical presentation, pretest probability scores, and investigations.
-
9.
Describe therapeutic regimens for thrombosis in rheumatic diseases.
-
10.
Discuss the role of adjunctive and preventive therapy in thrombosis in rheumatic disease.
-
11.
Identify conditions that mandate prophylaxis with antithrombotic medications in patients with rheumatic diseases.
To achieve these purposes, this chapter is written in three sections.
In Sect. 2 of this chapter, we will discuss the mechanism and pathophysiology of thrombosis in individual rheumatic disorders, i.e., systemic lupus erythematosus (SLE), antiphospholipid syndrome (APS), rheumatoid arthritis (RA), vasculitis, and Behçet’s disease; this includes the effects and role of different medications used specifically in rheumatic disorders in promoting or preventing thrombosis.
In Sect. 3, we discussed in general the approach and the strategies for diagnosing VTE (DVT and PE) and arterial thrombi in different autoimmune rheumatic disorders.
In Sect. 4, we will discuss updates about the management of thrombosis in rheumatic diseases and recommendations for prophylaxis and secondary thrombosis prevention in rheumatic disorders.
2 Pathophysiology of Thrombosis in Rheumatic Disorders
Arterial and venous thrombosis and systemic inflammatory diseases are highly linked, and the systemic inflammation promotes an extensive cross-link to exist between inflammation and hemostasis [5]. Systemic inflammation disturbs the natural tight balance between the procoagulants and the anticoagulants in the blood by release of certain inflammatory markers and cytokines like tumor necrosis factor alpha (TNF-α) and interleukin-1 (IL-1) and interleukin-6 (IL-6) that finally promote a prothrombotic state [5] (Fig. 12.2).

Close relationship between mechanisms of inflammation and thrombosis in Rheumatic diseases
Inflammation is a common feature of many rheumatic and immune-mediated disorders; systemic inflammation modulates thrombotic responses by suppressing fibrinolysis, upregulating procoagulants, and downregulating anticoagulants. Several studies indicate the role of innate immunity in promoting thrombosis as it was shown that coagulation and innate immunity have a common evolutionary origin; this leads to the concept that the immune system and coagulations system are linked [4]. These findings conclude that autoimmune disorders such as SLE, APS, Behçet’s disease, RA, and vasculitis syndromes like Wegener’s granulomatosis have been linked to an increased risk of venous thrombosis [4].
RA, as well as other types of arthritides and connective tissue diseases, are associated with accelerated atherosclerosis and increased cardiovascular morbidity and mortality [6].
Chronic systemic inflammation predisposes to accelerated atherosclerosis, a risk that is well-known in systemic lupus erythematosus (SLE) and in rheumatoid arthritis (RA) patients [7].
The mechanisms for an enhanced and premature atherosclerosis in autoimmune rheumatic disorders such as RA, SLE, and systemic sclerosis (SS) include chronic inflammatory process, immune dysregulation, and the classical risk factors; this explains the very high risk of cardiovascular disease in patients with SLE and RA and some other autoimmune diseases [8].
The coagulation factor XII (FXII, Hageman factor) activity correlates with fibrinolysis [9]. Some studies had found the association between pulmonary embolism and decreased levels of FXII; one study was done on large cohort of patients in Japan with different rheumatic disorder reports that FXII reduction coexisted with the presence of antiphospholipid antibodies (a PL) in most thrombotic patients with rheumatic disorders; they conclude the presence of anti-FXII autoantibodies as a cause of FXII deficiency in the presence of aPL antibodies [9].
2.1 SLE and Thrombosis
Thrombosis in SLE is multifactorial, and hence SLE patients are at significantly increased risk of thrombosis and atherosclerosis. Arterial and/or venous thrombosis is a well-known clinical entity in SLE, with a prevalence >10%. This prevalence may even exceed 50% in high-risk patients [10]. The incidence of thrombosis in SLE patients according to two studies was 26.8 and up to 51.9 per 1000 patient-years, according to the disease duration [10]. Other study reported that the incidence of thrombosis was 36.3 per 1000 patient years [10]. In a 10-year prospective cohort study of patients with SLE, the most frequent causes of death were active SLE (26.5%), thrombosis (26.5%), and infection (25%), with thrombosis dominating the second 5-year period of follow-up [11]. Patients with SLE have thrombosis at an early age than the age of thrombosis occurrence in the general population, with the incidence being increased in the first year, which may be explained by high disease activity, circulating immune complexes, cytotoxic antibodies, or a higher inflammatory state at first year of SLE diagnosis [10].
Thrombosis is the most frequent cause of death in SLE. With its frequent manifestation in patients with SLE, thrombosis contributes significantly to high morbidity and mortality [11].
Several studies showed that atherosclerotic cardiovascular and cerebrovascular diseases are more common causes of late deaths than active SLE itself. Some studies revealed that subclinical coronary heart disease and carotid plaque were present in a significantly higher proportion of SLE patients than in control subjects of similar age and sex with similar risk factors. Compared with individuals without SLE, the risk of myocardial infarction in SLE patients is 2–50 times higher, and the risk of stroke is 2–10 times higher. The prevalence of symptomatic coronary heart disease in SLE patients has been reported to be 6–20%, depending on the characteristics of the cohort, disease duration, study design, prevalence of antiphospholipid antibodies (aPL), and ethnic composition. 3–15% of SLE patients have a nonfatal stroke [12].
2.1.1 Risk Factor and Etiology of Thrombosis in SLE
2.1.1.1 Inflammation and Disease Activity
Inflammation promotes thrombosis through its several effects on blood coagulation [10, 13].
Inflammation induces the expression of tissue factor (TF), which is an important factor in coagulation initiation [10, 13]. The production of plasminogen activator inhibitor (PAI) is upregulated in inflammation which leads to decreased fibrinolysis activity and increases the risk of thrombosis [10]; high levels of C-reactive protein (CRP) released in inflammatory conditions facilitate the interaction between the monocyte and the endothelial and promote plasminogen activator inhibitor-1 (PAI-1) and TF [13]. In inflammation, fibrinogen, an acute phase reactant, is secreted in higher concentrations which further increase the risk of thrombosis in patients with SLE [13]. Inflammation impairs protein C pathway and decreases protein S level, thus worsening the risk of thrombosis in SLE patients who might have thrombotic events early in the disease as compared to patients without SLE [10, 13]. It was found that SLE patients with lupus nephritis have a high disease activity and inflammation, and this is associated with increase risk of DVT and renal vein thrombosis; they also frequently have systemic hypertension and hyperlipidemia which further worsen thrombotic risk [10].
2.1.1.2 Antiphospholipid (aPL) Antibodies
aPL antibodies are type of autoantibodies that directed towards phospholipid binding proteins, anionic phospholipids, or a combination of the two [9]; they include anticardiolipin antibodies (ACA), lupus anticoagulant (LA), and anti ß2-glycoprotein I (anti-ß2-GPI) [14]. aPL antibodies induce platelet activation, interfere with coagulation inhibitors such as protein C, inhibit antithrombin and fibrinolysis, and then initiate the formation of a thrombus [10]; they are associated with an increased risk of arterial and venous thrombosis in addition to recurrent pregnancy loss in which they comprise an antiphospholipid syndrome (APS) which could occur as a primary disease (primary APS) or associated with several autoimmune disorders, most frequently in SLE patients, where it is named as secondary APS [15]. Lupus anticoagulant is considered as significant risk factor for stroke and myocardial infarction [10] as well as a strongest predictor of thrombosis [15].
There is a significant occurrence of aPL antibodies among SLE patients [16]; about one-third of patients with SLE show aPL positivity, but not all of them have the clinical presentation of thrombosis or APS [14].
In one retrospective study of 42 SLE patients, 60% were positive for one or two aPL antibodies, but only 27% of them (10 patients) had a history of APS. The most common clinical presentation was DVT/PE in eight patients. Less common was arterial thrombosis and pregnancy loss. One patient with a history of PE developed autoimmune hemolytic anemia. Another patient without history of DVT/PE presented with thrombocytopenia [16].
The risk of thrombosis in LA and ACA positive patients has been addressed by many researchers. In patients with SLE, 42% of LA-positive and 40% of ACA-positive individuals had a history of thrombosis; in contrast, the prevalence of thrombosis in LA-negative or ACA-negative SLE patients was only10–18% [10].
APS is the main cause of thrombosis and a major predictor of irreversible organ damage and death in SLE patients [15].
ACA might be transiently positive, or persistently positive, and considered significant when it tests positive on at least two occasions, 12 weeks apart. The risk of thrombosis was significantly higher in persistently positive ACA antibodies (33% risk) versus 3% risk in those with transiently positive ACA as shown in the prospective, observational cohort study by A Martinez-Berriotxoa et al. (2007) [15].
-
Persistently positive ACA: patients are positive for IgG and/or IgM ACA at medium-high levels (titers ≥20 GPL and/or MPL) in whom more than two-thirds of the ACA determinations were positive; ACA were measured four or more times in all patients [15].
-
Transiently positive ACA: patients are positive for IgG and/or IgM aCL in which less than two-thirds of the ACA determinations were positive; ACA were measured four or more times in all patients [15].
In all patients with SLE, even if there are no clinical manifestations, aPL antibodies should be done as they are considered part of American College of Rheumatology (ACR) classification criteria for SLE, and they have been associated with increased risk of thrombosis [10], as the first presentation can be fatal presenting with a CVA. Diagnosis allows prophylactic measures to be instituted in high-risk situations, e.g., prolonged immobility and postoperative states; increased awareness of APS should lead to earlier recognition of associated episodes and laboratory screening for all SLE patients to allow for prophylactic anticoagulation in high-risk situations [16].
2.1.1.3 Protein C and S and Antithrombin Deficiencies
They are rare but carry a higher risk for venous thrombosis [10].
2.1.1.4 Factor V Leiden
Activation of factor V leads to the formation of a cross-linked fibrin clot. Factor V Leiden (FVL) is the most common inherited risk factor for venous thrombosis in the general population and is an important factor for thrombosis in patients with SLE. FVL polymorphism is considered to be risk variant for thrombosis and confers resistance to activated protein C, thus shifting the balance towards thrombosis in the clotting cascade. FVL variant is found in 20–60% of patients with idiopathic DVT but without SLE. Patients with SLE and/or aPL positivity who have the FVL polymorphism have at least two times the odds of thrombosis compared to patients without this polymorphism. This observation places FVL to be an independent risk factor for thrombosis in SLE [11].
2.1.1.5 Hyperhomocysteinemia
Hyperhomocysteinemia is a strong and independent factor for increased risk of atherosclerosis, mainly of the carotid and coronary arteries, as well as venous thrombosis to some extent [10, 17]. 27.3% of SLE patients with thrombosis have hyperhomocysteinemia, which is significantly higher than those without thrombosis in whom it is detected at 16.9% [10]. Patients with shortened APTT have a hypercoagulable state and were found to have high levels of homocysteine that place them at a higher risk of thrombotic events, as shown in a study done by T. M. K. Refai et al. (2002) [17]. In this study, the researchers found that 21% of SLE patients had elevated levels of homocysteine; interestingly, the level was higher in male patients more than in female ones and also those on prednisolone; they observed that lupus patients with hyperhomocysteinemia had a threefold increase in odds ratio of thrombotic episodes. This is partly because of the direct toxic effect of homocysteine on endothelium and partly indirect effects, such as induction of a vascular-endothelial-cell activator, promotion of vascular smooth muscle proliferation, and an inhibitory effect on endothelial cell growth; these findings support the hypothesis that hyperhomocysteinemia is an independent risk factor for thrombosis in young patients with SLE [17].
2.1.1.6 Traditional Risk Factors
Smoking is associated with worse outcome and mainly venous thrombosis by inducing endothelial damage; patients with SLE may have hypertension (HTN), diabetes mellitus (DM), and dyslipidemia which predispose them to thrombotic events. Older patients have more endothelial damage and vascular morbidity, and hence age is considered to be a risk factor for thrombosis in SLE [10].
2.1.2 Medication and Thrombosis in SLE
-
Glucocorticoids have been used frequently in SLE; they mediate endothelial damage and hence lead to accelerated atherosclerosis; high doses of glucocorticoids are associated with abnormalities of the coagulation system [10]. Chronic glucocorticoid consumption has been reported to increase plasma von Willebrand factor (VWF) levels, endothelial dysfunction, increased oxidative stress, and insulin resistance. Glucocorticoids use also increases (PAI-1); it was found that secretion of t-PA levels is limited in patients receiving glucocorticoids, which further worsen the coagulation system and cause hypercoagulable state which further enhances thrombosis risk in SLE patients [18].
-
Hydroxychloroquine (HCQ) is an antimalarial agent used in patients with SLE; it has antithrombotic effect by inhibiting platelet aggregation and adhesion and arachidonic acid release from stimulated platelets; it also decreases the thrombus size and the time of thrombus development which is dose dependent. HCQ inhibits GPIIb/IIIa receptor expression that is induced by aPL antibodies. Its role is more extended in the protection from thrombosis by lowering cholesterol level and lowering LDL; it also reduces interleukin-6 levels and decreases SLE flare episodes [10, 19].
-
Aspirin (ASA) inhibits platelet aggregation through inhibition of cyclooxygenase enzyme and hence the synthesis of thromboxane A2 [10].
Table 12.1 summarizes the risk factors of thrombosis and accelerated atherosclerosis in SLE patients.
2.2 RA and Thrombosis
RA is a common chronic systemic inflammatory disease with worldwide distribution. There is increased incidence of premature cardiovascular disease (CVD) and venous thrombosis in patients with rheumatoid arthritis and hence increased premature mortality and death on average 2.5 years earlier in community-based studies and approximately 18 years earlier in hospital-based studies than non-RA patients [20, 21]. The risk of VTE in patients with RA is increased to more than threefold than non-RA, as shown by Bacani A et al. (2012); they found that RA patients had a higher VTE cumulative incidence at 10 years than non-RA patients (6.7 in RA versus 2.8 in non-RA), and the risk of VTE was significantly higher within 90 days following hospitalization [20]. RA patients showed a higher age- and sex-adjusted increased risk of mortality (60%) and thromboembolic events (30%–50%) during a 5-year follow-up compared to non-RA patients. Similarly significant elevated risks (70% for death and 30%–40% for thromboembolic events) were seen when compared to OA patients. Several studies have shown that RA patients are 30%–60% more likely to suffer a cardiovascular event [19].
2.2.1 Risk Factor and Etiology of Thrombosis in RA
2.2.1.1 Lifestyle in RA
Patients with RA are physically less active due to their disease, and they may suffer from obesity, diabetes mellitus (DM), and hypertension that may result from medication use like steroids; some of RA patients are smokers as well; all these factors contribute to the accelerated atherosclerosis in RA subjects. Obesity is a time-dependent risk factor for development of VTE in RA patients, as shown by Bacani et al. in their study [20].
2.2.1.2 Inflammation
RA is characterized by a chronic inflammation that results in impaired immune system as well as persistent endothelial dysfunction, which predisposes to vascular wall damage and accelerated atherosclerosis. Such damage can be detectable by ultrasound measurement of carotid intima-media thickness (IMT) in a preclinical stage of the disease. Carotid IMT in RA patients is associated with markers of systemic inflammation and disease duration [22]. CD4+ are subsets of T cells that lack surface CD28 molecule (CD4 + CD28-) and expand when stimulated by endothelial autoantigens, in a subgroup of RA patients. Moreover, they infiltrate the atherosclerotic plaques and pose high pro-inflammatory and tissue-damaging properties which promote vascular injury. The role of these cells in contributing to early development of atherosclerosis in RA has been confirmed by recent studies which showed that RA patients with CD4 + CD28- cell expansion have a higher degree of endothelial dysfunction and a higher carotid IMT than patients without expansion of these cells [22].
2.2.1.3 High Disease Activity and High Levels of Inflammatory Markers
RA patients with high ESR and/or high CRP were found to have increased carotid artery IMT as well as increased probability of vessel plaque, which supports hypotheses of the relationship between systemic inflammation and atherosclerosis. ESR primarily reflects increased fibrinogen levels in response to systemic inflammation. The association between fibrinogen, as measured by the ESR, and increased carotid IMT suggests that inflammation-coagulation interactions may also have a role in atherogenesis. CRP is produced by the liver in response to interleukin-6, an earlier inflammation mediator, and can be found in the atheromatous lesions, suggesting its pathogenic role in atherothrombosis [23]; CRP is an independent risk factor for atherothrombotic disease [19].
2.2.1.4 Hospitalization
The relative risk factor for VTE is increased within 90 days post-hospitalization in RA patients. Orthopedic surgeries are reported to be a time-dependent cofactor risks for VTE development in RA patients that may develop within 90 days following lower extremity arthroplasty [20].
2.2.1.5 aPL Antibodies
RA patients may develop APL antibodies in 5%–75%, which increases the risk of VTE in these patients [20].
2.2.1.6 TNF-α
It causes endothelium damage and promotes blood coagulation through monocyte activation by increasing the TF levels [19].
2.2.1.7 Fibrinogen, VWF, Tissue Plasminogen Activator (t-PA) Antigen, and D-Dimer
Levels of these thrombotic variables are significantly higher in patients with RA [21].
2.2.1.8 Leukocytosis, Thrombocytosis, Increasing Platelet Activity, and Low Serum Albumin
These inflammatory markers are associated with increased cardiovascular risk and accelerated atherosclerosis in RA patients [19, 21].
2.2.1.9 High Systolic Blood Pressure (SBP) and Low Levels of High Density Lipoprotein (HDL)
High SBP in RA patients is mostly a result of a widespread use of NSAIDs and rarely can be caused by renal vasculitis and amyloidosis; HDL has a cardioprotective role against ischemia; low levels of HDL are found in RA. So, it is considered that both high SBP and low HDL are cardiovascular risk factors in patients with RA [21].
2.2.1.10 Rheumatoid Factor (RF)
RF positivity is associated with vascular injury and vasculitis, which increases plasma levels of VWF and t-PA that further enhance the thrombotic risks in RA patients [21].
2.2.1.11 Prothrombotic Condition in RA
Homocysteine (Hcy), in patients with RA the degree of inflammation is found to be correlated with Hcy levels. A positive relationship was found between the Hcy concentration and some parameters of inflammation, such as adhesion molecules and CPR [19].
Microparticles (MP) are membrane-bound vesicles that circulate in the blood and mediate inflammation and thrombosis. The most abundant MP in the blood come from platelets, and high levels of platelets MP were found in RA patients and correlated to high disease activity as measured by disease activity score (DAS 28). MP derived from granulocytes and monocytes have been found in the synovial fluid of RA patients as well; they stimulate TF and factor VII-dependent thrombin generation and lead to intra-articular inflammation and formation of fibrin clots, known as rice bodies [19].
2.2.2 Medications and Thrombosis in RA
-
Nonsteroidal anti-inflammatory drugs (NSAIDs) are widely used for pain management in RA, associated with enhance cardiovascular events risk through rising the blood pressure, especially indomethacin and piroxicam, which rise the mean arterial BP by approximately 5 mm Hg [21].
-
Glucocorticoids, through their effects on blood pressure, insulin resistance, lipid profile, body weight, coagulation, and endothelial dysfunction, might significantly increase the risk of CVD in RA patients [19].
-
Disease-modifying anti-rheumatic drugs (DMARDs), methotrexate (MTX), the most common DMARDs used in RA, inhibit the homocysteine-methionine pathway which leads to hyperhomocysteinemia, but the concomitant use of folic acid reduces homocysteine level, thus decreasing the risk of CVD in AR patients; a long-term follow-up of RA patients has shown that the use of MTX is related to reduced cardiovascular mortality, probably related to a reduction of disease activity. Leflunomide and cyclosporine can cause hypertension which increases the risk of cardiovascular disease in RA patients; the suboptimal control of inflammation by both these drugs also increases the risk of thrombosis. Antimalarials, such as HCQ, have a beneficial effect in decreasing the serum cholesterol and low-density proteins [19].
-
Biologic Therapy with TNF-α Blockers.
A recent study suggested that the risk of developing any CV event in RA is lower in patients who receive TNF-α blockers. One study reported that TNF-α blockade using infliximab improves endothelial function after 12 weeks of therapy. This improvement depends on the clinical improvement of the joint manifestations and on a decrease in the CRP and ESR levels. Other studies have shown the potential effect of short-term adalimumab therapy on endothelial function in RA patients with long-standing disease [19].
2.3 Vasculitis and Thrombosis
Vasculitides are a heterogeneous group of diseases characterized by the presence of vascular inflammation, which can lead to either a vessel wall destruction (leading to aneurysm or rupture) or a vessel stenosis (leads to tissue ischemia and necrosis) [24].
2.3.1 Large Vessel Vasculitis
They include Takayasu’s (TAK) and giant cell arteritis (GCA). Chronic vascular inflammation leads to endothelial dysfunction that results in a premature atherosclerosis. The risk of arterial thrombosis is increased; strokes and transient ischemic attacks (TIA) have a similar rate of occurrence in this form of vasculitis, but there has been no clear increased risk of venous thrombosis [5].
2.3.2 Medium Vessel Vasculitis
Polyarteritis nodosa (PAN) is associated with increased risk of both arterial thrombi and VTE. This risk is high during active disease (3.27 events/person/year versus 0.58 in patients with inactive disease) and independent of hepatitis B status. The coronary arteries are affected mainly by arterial thrombosis, but there is no clear association between ischemic strokes and PAN [5].
2.3.3 Small Vessel Vasculitis
They include granulomatosis with polyangiitis (GPA), microscopic polyangiitis (MPA), and Churg-Strauss syndrome. This type of vasculitis is associated with high risk of arterial as well as venous thrombosis. The risk of a first-time symptomatic VTE has been considered by some researcher to be as seven times first symptomatic VTE risk in SLE [5]. The prevalence of cardiovascular disease is high and biphasic; the highest risk of cardiac ischemia appears either within 4 years of diagnosis or after 10 years of diagnosis. Prospective data from four European Vasculitis Study Group trials found that 14% of patients with GPA and MPA will have a cardiovascular event within 5 years of diagnosis [5]. The age-standardized annual cardiovascular mortality rate was found to be 3.7 times higher than expected in the general population. The presence of proteinase 3 (PR3) antineutrophil cytoplasmic antibodies (ANCA) was found to be protective, whereas a positive myeloperoxidase ANCA test was associated with an increased risk of cardiovascular events. There is no evidence of increased risk of ischemic stroke in small vessel vasculitis [5].
2.3.4 Risk Factors for Thrombosis in Vasculitis
2.3.4.1 Changes in Endothelial Function and Hypercoagulability
The endothelium loses its anti-thrombogenic activity which results from its damage and activation during inflammation. During inflammation, several cytokines are released; these cytokines along with vessel ischemia cause endothelial damage. Circulating ANCAs also cause endothelial damage, and circulating endothelial cells as a marker for endothelial damage have been detected in ANCA-associated vasculitis (AAV) patients, especially when AAV is active [25].
2.3.4.2 Hypercoagulability
Hypercoagulable state is present in patients with active AAV, and it is triggered by pro-inflammatory cytokines, such as TNF-α and IL-1. It is manifested by the presence of high levels of D-dimers and thrombin-antithrombin III complexes which reflects activated clotting system, increased expression of tissue factor, which activates factor VIII factor which in turn increases VTE risk in these patients. Increased platelet aggregation and reduced fibrinolytic capacity during active disease are among the other hypercoagulable causes of thrombosis in AAV patients [25].
2.3.4.3 Hypereosinophilia in Churg-Strauss
Eosinophils contain preformed protein-containing granules which are released when activated. Some of these proteins have prothrombotic effects through releasing tissue factor and several other proteins and enzymes which result in decrease fibrinolysis and block the anticoagulant effects of endothelial bound and exogenous heparin, stimulate the production of platelet factor 4 from platelets, and inhibit protein C activation [5].
2.3.4.4 aPL Autoantibodies
They are well-known cause of VTE, and they are detectable in some patients with vasculitis (especially AAV); aPL antibodies were detected in 19% in patients with GPA (formerly known as Wegener’s granulomatosis) according to one study [25].
2.3.5 Medications and Thrombosis in Vasculitis
-
Cyclophosphamide, the most commonly drug used in the treatment of vasculitis, is associated with an increased risk of VTE in patients with AAV, through induction of vascular endothelial damage, endothelial cells apoptosis, platelet activation, and cytokines release [5].
-
Corticosteroids, particularly in high doses, can be thrombogenic through induction of high levels of factor VIII and lower fibrinolytic activity [25].
-
Low-dose ASA is proven to be beneficial in the prevention of cerebrovascular insults and visual loss in GCA and thus is recommended to use for same purpose in TAK [5].
2.4 Behçet’s Disease (BD)
BD is a chronic inflammatory disorder of unknown cause; its manifestations are considered to be caused by an underlying vasculitis [26], characterized by recurrent oral aphthous ulcers, genital ulcers, and uveitis, followed by involvement of other systems causing thrombophlebitis, arthritis, pulmonary and neurological involvement, erythema nodosum, and gastrointestinal disease [27]. Vascular involvement is common in BD; it affects up to 40% of patients resulting in arterial and venous thrombosis and aneurysms particularly of pulmonary arteries [5, 27]; vessels of all sizes are involved, both in the arterial and venous systems [28]. Venous thrombi are more common with involvement of DVT and superficial thrombophlebitis [27]. Asymptomatic DVT of the extremities in patients with BD with no history of vascular thrombosis is reported to be 6% which is higher than that seen in a healthy population [5]. BD is associated with low rate of pulmonary thrombosis (between 4 and 10%); this is because of tight adhesions of the peripheral thrombosis to the venous walls [5]. Other sites of venous thrombi in BD are vena cava thrombosis, Budd-Chiari syndrome (which coexists with inferior vena cava and portal vein thrombosis), and cerebral venous sinus thrombosis. BD complicated with Budd-Chiari syndrome is associated with poor and mean survival of 10 months compared with 16 months in patients affected with Budd-Chiari syndrome without BD. Cerebral venous sinus thrombosis (CVT) is estimated to occur in 8% of BD patients and in about 13% of BD with neurologic involvement. CVT most commonly manifests as intracranial hypertension. CVT in BD more likely affects male gender, presents at a younger age, and less likely develops venous infarcts [5]. The mortality rate is higher in patients with BD who had venous thrombosis, especially if large vessel is involved (mortality rate reached 12.1%) than in those without VTE [28].
2.4.1 Risk Factors for Thrombosis in BD
2.4.1.1 Endothelial Cell Dysfunction
Inflammation and resultant endothelial dysfunction suppresses nitric oxide (NO) secretion in patients with active BD, thereby impairing its normal function of vasodilation and inhibition of platelet aggregation, which in turn increases the risk of thrombosis in BD [5].
2.4.1.2 Low Protein C
BD is associated with a significant reduction in activated protein C, lower endothelial protein C receptor levels, and increased resistance to activated protein C, leading to significant impairment of anticoagulation as well as anti-inflammatory properties of protein C. Lower levels of activated protein C is found in patients with a history of VTE as compared to those without VTE history, which increase the risk of recurrent thrombi further [5].
2.4.1.3 Activated Platelets and Microparticles (MP)
MP are small membrane particles derived from platelets, monocytes, and leukocytes; they are secreted in higher levels during active inflammation and lead to the expression of TF tissue factor and anionic phospholipids which trigger the coagulation cascade and increase the risk of thrombosis in patients with BD [5].
2.4.1.4 Vascular Endothelial Growth Factor (VEGF)
VEGF levels in BD patients with acute thrombosis were higher than those of BD patients in chronic stage. Also, higher levels of MCP-1 were found in BD patients with acute thrombosis as compared with healthy controls. The positive correlation of the elevated levels of various factors with venous thrombosis can be a useful marker to predict the likelihood of thrombosis in BD [29].
2.4.1.5 HLA-B51 and HLA-B35 Positivity
BD patient positive for HLA-B51 are at increased risk of VTE, while those with HLA-B35 are protective from VTE [55].
2.4.2 Medications and Thrombosis in BD
-
Azathioprine, immunosuppression with azathioprine 2.5 mg/kg per day, decreased the rate of DVT according to one control trial [5].
-
Glucocorticoids, cyclophosphamide, one large study had found that immunosuppressives and glucocorticoids significantly decreased the risk of recurrent DVT in 807 patients with BD [5]. The European League Against Rheumatism (EULAR) in 2008 recommended the use of immunosuppressants (glucocorticoids, azathioprine, cyclophosphamide, and cyclosporine A) in the management of acute DVT in patients with BD disease [5]. Immunosuppressive agents improve prognosis in patients with BD by decreasing the odds of venous thrombosis relapse in BD by fourfold; immunosuppression in Budd-Chiari syndrome is associated with a significant improvement in prognosis as shown in a study done by Desbois et al. [28]. A retrospective study of 37 patients with venous thrombosis in BD compared immunosuppressive agents, anticoagulation treatment, and the combination of immunosuppressive agents and anticoagulation treatment; 3 of the 4 patients in the anticoagulant-treated group (75%) developed new thromboses, compared to 2 of 16 patients in the immunosuppressive agent-treated group (12.5%) and 1 of 17 patients in the combination-treated group (5.9%) [28].
2.5 Antiphospholipid Syndrome (APS) and Thrombosis
APS is characterized by recurrent venous and arterial thrombosis and/or fetal loss in combination with the persistent presence of circulating aPL antibodies, which comprise LA, ACA, and/or anti β2GPI antibodies [30].
DVT is the most frequent clinical manifestation of APS. Larger veins like subclavian, iliofemoral, upper abdomen, portal, and axillary veins may be affected as well. Thrombosis of almost every organ has been described in APS, which result in different clinical conditions and syndromes, such as superficial thrombophlebitis; superior vena cava syndrome; renal vein thrombosis; adrenal infarction; Addison’s syndrome; Budd-Chiari syndrome; pulmonary hypertension, due to recurrent pulmonary embolism; and diffuse pulmonary hemorrhage, due to microthromboses [31].
Arterial thrombosis consists a main clinical feature of APS, but appears less frequently than Venous, the most common site of arterial thrombosis is the cerebral circulation, leading to stroke or transient ischemic attack (TIA), CVT, coronary, renal and mesenteric arteries thrombosis has been observed also. In women under 50 years, LA is considered to be a major risk factor for arterial thrombosis as shown in RATIO study (Risk of Arterial Thrombosis In Relation to Oral Contraceptives). CNS involvement in APS mainly strokes and TIAs is associated with high morbidity and mortality [31].
Other manifestations of hypercoagulopathy and thrombosis in APS include thrombocytopenia, hemolytic anemia, pregnancy loss, eclampsia, livedo reticularis, purpura, Libman-Sacks valvulopathy, amaurosis fugax, retinal vessels thrombosis, and avascular necrosis of the bones [32].
2.5.1 Risk Factors for Thrombosis in APS
2.5.1.1 aPL Autoantibodies
aPL antibodies are a heterogeneous group of different autoantibodies with distinct specificity for cardiolipin or for plasma proteins with affinity for anionic phospholipids such as β2 GPI, prothrombin, or annexin A5 [33]. Oxidized β2 GPI is able to bind to and activate dendritic cells which results in autoantibodies production [32]. aPL antibodies are highly thrombotic. The ACA are directed against cardiolipin and b2GPI, the anti-β2GPI antibody is directed against β2GPI, while the LA measures functional anti-β2GPI antibodies and antiprothrombin antibodies. β2GPI antibodies are responsible for the increased risk of thromboembolic complications; patients positive for all three aPL antibodies have a significant increased risk of recurrence of thromboembolic disease, while patients positive for only one of the three aPL antibodies hardly have a significant increase in recurrence compared to patients with thrombosis but without aPL antibodies as shown by Pengo et al. (2011) [34].
The presence of LA, triple positivity (combination of LA, aCL and β2GPI antibodies), isolated, but, persistently positive aCL at medium–high levels are conditions considered as a high risk serological aPL profile for thrombosis. Patients with triple positivity have aPL levels much higher than others, thus making thrombosis highest risk of thrombosis in this group. Patients with isolated aCL or β2GPI at low-medium titers, particularly if intermittently positive, are considered to have a low-risk profile for thrombosis [30] (Table 12.2).
LA positivity increased the risk of stroke 48-fold and the risk of myocardial infarction 11-fold, while β2GPI antibodies are associated with double risk for stroke as shown by Urbanus RT et al. in the RATIO study [30].
2.5.1.2 The Effects of aPL Antibodies on Endothelial Cells
Anti-β2GPI antibodies result in increased expression of adhesion molecules (ICAM-1, VCAM-1, E-selectin); aPL autoantibodies increase the synthesis and secretion of pro-inflammatory cytokines IL-1, IL-6, and IL-8 and increase TF expression and upregulation of tissue factor messenger RNA (mRNA) as well as enhancement of endothelin-1 levels [32]. aPL antibodies cause defective apoptosis of endothelial cells, which exposes membrane phospholipids to the binding of various plasma proteins [31, 32].
2.5.1.3 Hypercoagulable Effect of aPL Antibodies
Production of antibodies against coagulation factors, including prothrombin, protein C, protein S, and annexins, platelets activation to enhance endothelial adherence, activation of vascular endothelium, which, facilitates the binding of platelets and monocytes that result in a hypercoagulable state. aPL antibodies react with oxidized LDL and predispose to atherosclerosis. Moreover, complement activation by aPL has been recognized as a possible significant cause in APS pathogenesis. Emerging evidence from murine models suggests that APL-mediated complement activation may be a primary event in pregnancy loss [32].
2.5.1.4 Platelet Activation and Aggregation by aPL
β2GPI antibodies activate platelets aggregation and release of platelets factor 4 (PF4) and thromboxane B2; aPL cannot bind to the surface of “intact” platelets, while they have the ability to bind to platelets with exposed negatively charged phospholipids in their membranes [31]. Bleeding time is prolonged in about 40% of patients with APS, without accompanying bleeding tendency, which indicates impaired platelet function in APS as a result of platelet activation by aPL. The expression of platelet membrane glycoproteins, particularly GPIIb-IIIa (fibrinogen receptor, critical in platelet aggregation) and GPIIIa, is also increased that enhance platelets aggregation further [31]. Another mechanism of platelets activation is the production of high plasma levels of active VWF in patients with β2GPI antibodies. In the normal conditions, binding of β2GPI to VWF results in inhibiting its ability to promote adhesion and platelet aggregation, but in the presence of anti-β2GPI antibodies, this anticoagulant effect is blocked [31]. β2GPI antibodies and LA induce the formation of stable thrombi and large aggregates, as shown by Jankowski et al. in animal model [31].
2.5.1.5 β2GPI Binding with Platelet Factor 4 (PF4)
PF4 is recognized recently as the dominant β2GPI-binding protein. PF4 binds in vitro, with high-affinity, recombinant β2GPI; PF4 tetramers can bind two β2GPI molecules simultaneously. Anti-β2GPI antibodies selectively interact with complexes composed of (β2GPI)2-(PF4)4. This reaction is higher against PF4-β2GPI complex than against β2GPI alone. Anti-β2GPI-β2GPI-PF4 complex significantly induced platelet p38 MAPK phosphorylation and thromboxane A2 production [37]. p38 MAPK is mitogen-activated protein (MAP) kinase that controls many cellular responses, such as proliferation, migration, differentiation, and apoptosis. In platelets, p38 MAPK regulates platelet adhesion to collagen and aggregation [37]. β2GPI antibodies form stable complexes with PF4, leading to the stabilization of β2GPI, which facilitates antibody recognition. This interaction is found to be involved in the procoagulant tendency of APS [36].
2.5.1.6 Activation of Monocytes by aPL Antibodies
This will result in increased TF expression and activity as well as increased production of pro-inflammatory cytokines, which increases the risk of thrombosis in APS patients; many researchers had found high levels of soluble TF (sTF) in the peripheral blood of patients with a history of thrombosis and aPL [31, 35].
2.5.1.7 Other Risk Factors for Thrombosis in APS
Hypertension, smoking, hypercholestrolemia or estrogen use: the coexistent presence of these factors is associated with thrombosis. The interaction between aPL, smoking, and oral contraceptive pills (OCP) has been identified and clarified in the case-control study; the risk for suffering a stroke doubled among smoking LA-positive women, as compared with non-smokers; the risk of stroke among OCP users is increased to sevenfold. One study showed that all smoker women who had LA suffered a myocardial infarction [29]. Concomitant SLE in APS (SAPS) increases the risk of thrombosis further in these patients (see SLE and thrombosis in this chapter) [29].
3 Approach and Diagnosis of Thrombosis in Rheumatic Diseases
The clinical features of thrombosis in patients with rheumatic diseases are similar to that in patients with other diseases and the general population; therefore it is very important for the physician who attends such patients to take a careful detailed history, perform a thorough physical examination, and do an appropriate workup (laboratory and imaging).
Unprovoked (idiopathic) venous thrombotic events are defined as venous thrombosis that occurs in the absence of any of the known risk factors; about 50% of patients presenting with a first idiopathic venous thrombosis have an underlying thrombophilia [38]; therefore special attention is needed to consider thrombophilia in the history and the workup of thrombosis.
3.1 History Taking
Symptoms of PE and DVT are not specific, so it is important to ask about the risk factors such as the age, previous history of VTE, recent long-distance travelling for active malignancy, coagulation disorders, hormone replacement therapy (HRT) in postmenopausal women, use of an oral contraceptive pills (OCP) in a female of childbearing age, abdominal/pelvic surgery/knee joint replacement, and diseases or conditions that lead to limited mobility [39,40,41,42], as well as a comprehensive history of rheumatic disorders that associated with increased risk of thrombosis, especially if VTE is recurrent, or unusual presentation in the absence of the known risk factors, or if the patient is a young one with no known predisposing factors for thrombosis, or if the patient presents with multiple thrombi at different sites or had both arterial and venous thrombosis.
Detailed history of medications must be taken in patients with rheumatic disorders, as some of them have thrombotic risks, while others have a protective role (see details of thrombosis and medications in individual disorder in Sect. 1).
Inquire about the constitutional symptoms in rheumatic disorders, this includes fever, sweats, loss of appetite and weight.
History of skin rashes, such as malar rash, photosensitivity, raynaud’s phenomenon if SLE is considered in the differential diagnosis of VTE. Mouth and genital ulcers and visual complaints may provide a clue to the presence of BD or other rheumatic diseases.
A careful obstetrical history is mandatory if APS (either primary or secondary) is suspected to be the cause of thrombosis in a young female in the absence of the traditional risk factors.
Inquiries about the renal complications must be included in the history in patients with SLE who presents with thrombotic episodes.
Patients with PE present to the emergency room (ER) and may complain of sudden dyspnea, pleuritic and non-pleuritic chest pain, cough or hemoptysis, fever, and diaphoresis; they may have cyanosis or syncope especially if massive PE [41, 43, 44]. Patients may have DVT at the same time which can be asymptomatic, since less than 25% of PE patients presented with symptoms and signs of DVT [41].
Patients suffering from DVT may complain of sudden ipsilateral leg swelling, redness, intermittent cramps, and pain in the calf or leg [45,46,47]. Presence of risk factors will increase suspicious of DVT which include prior history of DVT or PE, recent surgery, active cancer, trauma, hospitalization, immobility, co morbidities, family history of VTE, advanced age, current pregnancy, hormonal contraceptives and hormonal replacement therapy, and obesity [42, 45, 47, 48].
DVT and PE are considered combined emergency problem; 70% of patients diagnosed with PE have DVT in their leg and 50% of DVT patients established asymptomatic PE [47]. For that, we must diagnose DVT to prevent PE by history, physical examination, and investigations [48].
3.2 Physical Examination
The physical examination of patients with PE reveals some signs, of which tachypnea is the most common [43, 49]; other signs such as hypotension and cardiogenic shock may present in massive PE. Signs of right ventricular failure (RVF) such as tachycardia, distended neck veins, and tricuspid regurgitation may be there; wheezes, loud pulmonary component of second heart sound (loud P2), and pleural rub may also be heard sometimes [44]. Signs of DVT must be looked for [43]; they include leg redness, edema, warmth, tenderness, superficial dilation of veins, and fever; tachycardia and sign of pulmonary embolism should be looked for as well even in the absence of symptoms [47, 50].
Rheumatic disorders should be considered as the top most differential diagnosis in unusual presentation of different forms of thromboembolic diseases such as young patients with no known risk factors, or thrombosis in unusual sites, or in case of multiple or mixed arterial and venous thrombi; thus, a careful examination should be done.
Many diseases mimic the sign and symptoms of DVT and PE, rendering the physical examination to be not enough for diagnosis, although very essential to perform [47, 50]. Therefore, another methods need to be used before going further to work up these patients.
3.3 Clinical Pretest Probability (CPTP) for VTE
CPTP includes Wells and modified Wells criteria [44, 51]; it is a useful and important method to determine the probability of DVT and PE, respectively, and to classify the risk as low, medium, and high and is helpful in selecting the proper test for workup [41].
Wells criteria is based on history and physical examination and classifies the patient as high risk if score is between 3 and 8 points, moderate risk if 1–2 points, and low risk if 0 to −2 points [44, 45, 48, 50, 52]. For details see Fig. 12.3 and Table 12.3.

Wells Criteria for DVT Probability
In modified Wells criteria, the total point is 12, based on symptoms and signs and risk to get the disease. Patients are considered low risk if less than 2 points, intermediate risk if 2 to 6 points, and high risk if more than 6 points [8, 16]; the probability of PE is also categorized as likely and less likely; if it was more than 4 points, the patients are likely to have PE [44]. Clinical sign and symptoms of DVT (3points), other diagnosis less likely than pulmonary embolism (3 points), heart rate (HR) > 100/min (1.5 points), immobilization >3 days or surgery in previous 4 weeks (1.5 points), previous PE or DVT (1.5 point), hemoptysis (1 point) and malignancy (1 point) [44, 51]. Figure 12.4 and Table 12.4.

Modified Wells Criteria for PE Probability
3.4 Laboratory and Radiology Workup
Duplex ultrasound (DUS) is recommended as an initial imaging test for patients with high and moderate clinical probability of DVT, if DUS is positive then DVT is established, but if negative then D-dimer should be obtained, positive D-dimer and negative DUS after that follow up the patient and repeat the ultrasound, but if both DUS and D dimer are negative, DVT is ruled out, while a low clinical probability of DVT require D-dimer as initial test, if negative, DVT is ruled out, but if positive, DUS should be done [44]. Venography is indicated in patients with high clinical probability and negative DUS or if DUS cannot be done [48] (Fig. 12.5).

Probability of DVT algorithm
D-dimer is recommended for patients with low or intermediate risk for PE, and, if negative, PE is ruled out, but if positive, then CT pulmonary angiography (the gold standard) [39] is required. If patient’s risk for PE is high, CT pulmonary angiography should be done, but in situation where contraindication for contrast is used in CT angiogram such as in patients with renal failure, ventilation/perfusion (V/Q) scan can be done [43, 51, 53] (Fig. 12.6).

Clinical probability of PE
Pulmonary embolism rule out criteria (PERC) is used in patients who are less likely to have PE (4 points or less); factors of PERC are age < 50 years, HR < 100/min, oxygen saturation >94%, no unilateral leg swelling, no hemoptysis, no surgery or trauma within 4 weeks, no prior DVT or PE, and no estrogens or progestin use. If patients met these criteria, they are regarded as negative for PE, and no further investigations are needed, but if PERC is not met, they are considered positive for PE, and D-dimer should be tested; if it is negative, PE is ruled out [44, 54] (Fig. 12.7).

Probability of Pulmonary Embolism algorithm
Other imaging such as chest X-ray may be required to confirm the diagnosis; chest X-ray is usually normal in PE, but it is important to exclude other diseases. Signs of PE on chest radiograph are atelectasis, pleural-based infiltrates, or effusions, there may be a wedge shaped opacity and oligemia - cut off - of arteries, and right descending enlargement of the pulmonary artery) [39, 43, 44].
In PE, ECG may show supraventricular arrhythmia, signs of right ventricular (RV) strains, right axis derivation, T-wave inversion in V1–V4, right bundle branch block, S1Q3T3, and QR in V1 or P pulmonale [39, 43, 44].
Arterial blood gases can show hypoxemia, hypocapnia, and widened (A-a) O2 gradient [44].
In patients with PE, check troponin and brain natriuretic peptide (BNP) levels, since their high levels are associated with RV strain and linked to increased mortality in PE [44].
Laboratory investigations include general and specific workup. Generally, for all patients with thrombosis, full blood count, renal and liver function, and coagulation profile need to be done. Specific workup for thrombophilia and hereditary hypercoagulable disorders, this includes: Factor V Leiden, Prothrombin 20210A, Protein C and S, Antithrombin III [38], as they also can present in autoimmune rheumatic disorders such as SLE. Acquired hypercoagulable states includes anti- aPL antibodies (LA, ACA, β2GP1) should be done in idiopathic thrombosis or if there multiple thrombi, HLA-B51 test for BD if thrombosis is at an usual site (Bud Chairi syndrome) or both arterial and venous thrombi or idiopathic CVT should be considered [55].
Other blood workup includes acute phase reactants that measure disease activity (CRP, ESR), since their high levels correlate with high risk of thrombosis and atherosclerosis in certain diseases such as RA [19].
4 Management of Thrombosis in Rheumatic Diseases, Prophylaxis, and Secondary Prevention of Thrombosis
4.1 Management of Thrombosis in Rheumatic Diseases
There are no specific recommendations for the management of thrombosis in patients with rheumatic disorders, the same plan of treatment as of patients with other diseases and general population. Anticoagulation with intravenous unfractionated heparin (UFH) or subcutaneous low molecular weight heparin (LMWH), followed by warfarin, is the initial treatment strategy for cases with acute thrombosis [57].
Treatment of patients with PE depends on clinical probability of pulmonary embolism and hemodynamic stability of patients, initial treatment with anticoagulant if clinical probability is high or intermediate and cannot get the investigation within 4 h and in case of low probability and investigation deferred for 24 hours, anticoagulant therapy include LMWH, or fondaparinux, both are administered subcutaneously, do not require monitoring of PT and APTT, and not to be used in renal failure, UFH is administered intravenously and it is preferred in massive PE. If there is contraindication to anticoagulant, then inferior vena cava filter should be considered. Thrombolytic therapy is used in patients who have hemodynamic instability [40, 41] (Fig. 12.8).

Treatment of PE
In patients with DVT, LMWH is recommended, as it is superior to UFH especially in pregnant and patient with cancer, but it should not be used in patient with renal failure; they should be treated with unfractionated heparin (Fig. 12.9); warfarin should be started together with LMWH until targeted INR is reached; inferior vena cava filter is indicated in patient with contraindication to anticoagulation therapy [45, 48, 56] (Fig. 12.10).

Characteristics of special population and types of heparin

Management of DVT algorithm.
Compression stocking is used within 1 month of DVT diagnosis to prevent post-thrombotic syndrome and for at least 1 year after diagnosis [45, 56].
Anticoagulation with warfarin had been associated with several disadvantages related to the drug itself such as slow onset of action, variable pharmacologic effects, food-drug interactions, prolonged half-life, and the need for close monitoring of INR [65]. However, several large studies have been done in this field, and researchers had found that the NOACs are now emerging as the alternative anticoagulation therapy to conventional therapy for patients with acute VTE; the advantages of these novel anticoagulant therapy are many and overcome the troubles of warfarin therapy, such as the fixed therapeutic dose, without the need of dose adjustment; they do not require routine laboratory monitoring of PT and INR. They reach their peak efficacy within 1 to 4 h after ingestion; thus a prolonged period of bridging therapy is not required when switching from initial treatment with UFH or LMWH to these novel agents and less risks of major bleeding. Unfortunately, the antidote for bleeding events is not available yet [64,65,66]. No data is available regarding the safety of NOACs in pregnancy, for which it should be avoided in a pregnant patient and also in some other conditions such as patients with mechanical heart valves and in severe renal insufficiency [65].
Two groups of NOACs are available, factor Xa inhibitors (rivaroxaban, apixaban) and direct thrombin inhibitors (dabigatran). The safety and efficacy of these agents for the treatment and prevention of recurrent VTE have been studied by large randomized prospective trials [64].
Apixaban has a rapid onset of action and is approved for use in the prevention of stroke and systemic embolism in adult patients with nonvalvular atrial fibrillation (AF) and in the primary prevention of VTE in adult patients who have undergone elective total hip or total knee arthroplasty [65].
Apixaban is effective for the prevention of recurrent VTE if patients complete 6 to 12 months of anticoagulant therapy for acute VTE, with major bleeding risk similar to those for placebo.
Therapy with apixaban was compared with conventional anticoagulant therapy in patients with acute symptomatic VTE in the AMPLIFY trial (Apixaban for the Initial Management of Pulmonary Embolism and Deep-Vein Thrombosis as First-Line Therapy) [66]. The AMPLIFY study has very impressive results and concluded that a fixed-dose oral apixaban alone was as effective as conventional treatment which consists of enoxaparin followed by warfarin and was associated with a clinically relevant reduction of 69% in major bleeding, and its efficacy in patients with PE was similar to that in the patients with DVT. Moreover, the efficacy and the reduction in major bleeding with apixaban were consistent with that of warfarin, but clinically relevant non-major bleeding were less. Interestingly, The efficacy and safety of apixaban were consistent in all patients participated in the trial including patients older than 75 years, obese patients of more than 100 kg, use of parenteral anticoagulant treatment before randomization, and treatment duration. AMPLIFY trial results are very promising and encouraging to consider apixaban a safe and effective regimen for the initial and long-term VTE treatment [66].
4.1.1 Special Consideration for Thrombosis in Rheumatic Disorders
-
Patients with rheumatic diseases and thrombosis need a long-term management (indefinite) for thrombosis especially those with aPL autoantibodies to prevent recurrent thrombosis, that is, secondary thrombosis prevention; they require the optimal intensity of anticoagulation with warfarin [30, 57].
-
Several studies had proven that high-intensity treatment with warfarin to maintain INR = 3.0 with or without low-dose aspirin was more effective than moderate-intensity warfarin or low-dose aspirin for the prevention of recurrent thrombosis in aPL-positive patients, but, recently, some trials demonstrated that high-intensity anticoagulation (INR 3.1–4.0) was no better than moderate intensity (INR 2.0–3.0). So, moderate-intensity anticoagulation is the current standard of treatment of first venous thrombosis [57,58,59,60] (Fig. 12.9).
-
For the management of acute DVT in BD immunosuppressive agents such as corticosteroids, azathioprine, cyclophosphamide, and cyclosporine A are recommended, but there is no evidence of benefit from, and uncontrolled experience with anticoagulation, use of antiplatelet or antifibrinolytic agents in the management of DVT or for the use of anticoagulation in arterial thrombosis in patients with BD, in patients with CVT (dural sinus thrombosis) treatment with is corticosteroids is recommended (modified EULAR 2008 recommendation) [59]. Anticoagulant therapy must be used cautiously and only after systemic immunosuppressant, and if thrombi are not extensive, antiplatelet treatment with low-dose aspirin is probably sufficient [61].
-
• In patients with a low-risk aPL profile, who had first venous thrombosis in the presence of a known transient risk factor, anticoagulation could be limited to 3–6 months [30].
4.2 Prophylaxis and Secondary Thrombosis Prevention in Rheumatic Disorders
-
Daily ASA in doses of 75–325 mg are suitable for inhibition of platelet aggregation for prophylaxis against cardiovascular events in RA patients [19].
-
Prophylaxis use of LMWH to prevent venous thrombosis during periods of immobilization, as immobilization in RA patients is related to disease activity and inflammation [19].
-
ASA (75–150 mg/day) is recommended for the prevention of cerebrovascular events and vision loss in GCA, and it should be also considered for the primary prevention of cardiovascular events in TAK [5].
4.2.1 Primary Prophylaxis in SLE Patients
-
HCQ reduces thrombotic risk and disease-related morbidity and mortality in SLE and is recommended for all patients unless it is contraindicated [10].
-
HCQ plus low-dose ASA is recommended for SLE patients with positive LA or ACA (medium-high titers) [30].
4.2.2 Primary Prophylaxis in APS Patients
-
In asymptomatic individuals, aPL antibodies positivity is an incidental finding; thus, primary prophylaxis can be considered with ASA 81 mg per day [59, 60].
-
Healthy individuals, with positive aPL antibodies in high titers and with no thrombotic manifestations, should be advised for a primary prophylaxis with ASA 325 mg orally daily [60].
-
HCQ 400 mg orally daily decreases aPL antibody titers and thus protects from further thrombotic episode; that is based on trials in animal models and an indirect evidence from human studies, so more studies are needed to prove this effect of HCQ for standard recommendation in healthy aPL-positive patients [60] (Fig. 12.11).

Recommendation for treatment and prevention of thrombosis in patients with APS
4.2.3 Primary Prophylaxis in High-Risk Situations
All patients with aPL positivity should receive usual doses of LMWH in high-risk situations, such as surgery, prolonged immobilization, and puerperium [30]; the same is applied for all patients with other rheumatic disorders.
4.2.4 Secondary Prophylaxis in Patients with Positive aPL Antibodies
-
Patient who suffered from either arterial or venous thrombosis and aPL who do not fulfill criteria for APS should be managed in the same manner as aPL-negative patients with similar thrombotic events [30].
-
Recurrent venous thrombosis has been reported in patients with APS at 3% to 24%. Secondary prophylaxis with high-intensity warfarin (INR = 3–4), or moderate-intensity warfarin (INR = 2–3) plus ASA is recommended [60].
-
Treatment of APS patients with arterial thrombosis is controversial, and only ASA 325 mg/day can be given or moderate-intensity warfarin (INR = 2–3) alone or combined low-dose ASA or high-intensity warfarin (INR > 3) [30, 60].
-
In pregnant women, with recurrent fetal loss, a combination of ASA and heparin is recommended. ASA 81 mg/day should be started when attempting conception, and when the pregnancy is confirmed, heparin subcutaneously should be started as LMW (enoxaparin 1 mg/kg/day, dalteparin 5000 units/day, or nadroparin 3800 units/day) or as unfractionated (5000–10,000 units 12 hourly) [60].
-
In catastrophic APS, a combination therapy is required with (1) anticoagulation with intravenous (IV) heparin for 7–10 days, (2) steroids in high doses with (IV) methylprednisolone 1 g daily for 3 or more days, (3) IV immune globulin (IVIG) 0.4 mg/kg/body weight/day for 4–5 days, and/or (4) plasmapheresis for 3–5 days at least with fresh frozen plasma replacement [60].
(See Fig. 12.11 for treatment and secondary prophylaxis of thrombosis in APS)
-
In patients with a low-risk aPL profile without SLE, who have first non-cardioembolic cerebral arterial event due to one of the reversible risk factors, antiplatelet agents are considered for the secondary prophylaxis [30].
4.2.5 Refractory and Difficult Situations in aPL-Positive Patients
In patients with difficult management due to recurrent thrombosis, fluctuating INR levels, major bleeding, or a high risk for major alternative therapies with a long-term low LMWH, HCQ or statins are needed for the management of acute thrombosis as well as the secondary prophylaxis [30].
4.2.6 Statin Role for the Prophylaxis against Thrombosis in Rheumatic Diseases
Statins have pleiotropic effect (anti-inflammatory, antioxidant, and potent antithrombotic) in addition to a lipid-lowering effect. Thus, by inhibition of atherosclerosis progression, statin decreases the cardiovascular risk for arterial thrombosis in rheumatic and other diseases [62]. Several studies revealed that the use of statins is associated with decrease levels of inflammatory markers such as IL-6, IL-8, MCP-1, and CRP which cause endothelial dysfunction. In addition to that, statins were found to exert an antioxidant function by increasing nitric oxide synthase level. In the JUPITER trial, use of rosuvastatin in patients with elevated CRP levels results in a significant reduction of DVT [63]. Knowing these advantageous effects of statin, it is advised to consider it for prophylaxis of venous thrombosis in rheumatic diseases, but further trials are needed in this field [63].
References
Lijfering WM, Rosendaal FR, Cannegieter SC. Risk factors for venous thrombosis – current understanding from an epidemiological point of view. Br J Haematol. 2010;149:824–33.
Konkle B. Part 2. Cardinal manifestations and presentation of diseases. Section 10, hematologic alterations. In: Online, Harrison’s principles of internal medicine. 18th ed. Chapter 58, Bleeding and Thrombosis.
López JA, Kearon C, Lee AYY. Deep venous thrombosis. Journal of Hematology. 2004:439–56.
Zöller B, Li X, Sundquist J, Sundquist K. Autoimmune diseases and venous thromboembolism: a review of the literature. Am J Cardiovasc Dis. 2012;2(3):171–83.
Springera J, Villa-Forteb A. Thrombosis in vasculitis. Curr Opin Rheumatol. 2013;25(1):19–25.
Kerekes G, et al. Validated methods for assessment of subclinical atherosclerosis in rheumatology. Nat Rev Rheumatol. 2012;8:224–34.
Santos MJ, et al. Early vascular alterations in SLE and RA patients—a step towards understanding the associated cardiovascular risk. 2012;7(9):1–6.
Cojocaru M, Cojocaru IM, Silosi I, Vrabie CD. Accelerated atherosclerosis in autoimmune rheumatic diseases. J Clin Exp Invest. 2010;1(3):232–4.
Takeishi M, Mimori A, Nakajima K, Mimura T, Suzuki T. 2003. Reduction of factor XII in antiphospholipid antibody-positive patients with thrombotic events in the rheumatology clinic. Clin Rheumatol. 2003;22:40–4.
Al-Homood IA. Thrombosis in Systemic Lupus Erythematosus: A Review Article. ISRN Rheumatology. 2012;428269:6. https://doi.org/10.5402/2012/428269. ISSN 2090-5467
Kaiser R, et al. Factor V Leiden and thrombosis in patients with systemic lupus erythematosus: a meta-analysis. Genes Immun. 2009;10:495–502.
Mok CC, Tang SSK, Chi Hung To, Petri M. Incidence and risk factors of thromboembolism in systemic lupus Erythematosus. Arthritis & Rheumatism. 2005;52(9):2774–82.
Esmon CT. The interactions between inflammation and coagulation. Br J Haematol. 2005;131:417–30.
Hakim A, Clunie G, Haq I. Antiphospholipid (antibody) syndrome and systemic lupus erythematosus. Third ed: Oxford Handbook of Rheumatology; 2011. p. 329.
Martinez-Berriotxoa A, et al. Transiently positive anticardiolipin antibodies and risk of thrombosis in patients with systemic lupus erythematosus. Lupus. 2007;16:810–6.
McMahon MA, Keogan M, O'Connell P, Kearns G. The prevalence of antiphospholipid antibody syndrome among systemic lupus erythematosus patients. Ir Med J Nov-Dec. 2006;99(10):296–8.
Refai TMK, et al. Hyperhomocysteinaemia and risk of thrombosis in systemic lupus Erythematosus patients. Clin Rheumatol. 2002;21:457–61.
van Zaane B, et al. Systematic review on the effect of glucocorticoid use on procoagulant, anti-coagulant and fibrinolytic factors. J Thromb Haemost. 2010;8:2483–93.
Mameli A, Barcellona D, Marongiu F. Review: rheumatoid arthritis and thrombosis. Clin Exp Rheumatol. 2009;27:846–55.
Kirstin Bacani A, et al. Noncardiac vascular disease in rheumatoid arthritis. Increase in venous thromboembolic events? Arthritis & Rheumatism. 2012;64(1):53–61.
McEntegart A, et al. Cardiovascular risk factors including thrombotic variables, in a population with rheumatoid arthritis. Rheumatology. 2001;40:640–4.
Shoenfeld, et al. Accelerated Atherosclerosis in Autoimmune Rheumatic Diseases. Circulation. 2005:3337–47.
Del R’n, et al. Association between carotid atherosclerosis and markers of inflammation in rheumatoid arthritis patients and healthy subjects. Arthritis & Rheumatism. 2003;48(7):1833–40.
Hakim A, Clunie G, Haq I. Primary vasculitides. Third ed: Oxford Handbook of Rheumatology; 2011. p. 406.
Stassen PM, et al. Venous thromboembolism in ANCA-associated vasculitis—incidence and risk factors. Rheumatology. 2008;47:530–4.
John H. Klippel. 2008. Vasculitides, E. Miscellaneous Vasculitis. Primer on the Rheumatic Diseases. 13, chapter 21, p 435.
Ames PRJ, et al. Thrombosis in Behçet’s disease: a retrospective survey from a single UK centre. Rheumatology. 2001;40:652–5.
Desbois, et al. Immunosuppressants reduce venous thrombosis relapse in Behçet’s disease. Arthritis & Rheumatism. 2012;64(8):2753–60.
Shankar S, Gill S. Predicting thrombosis in Behçet’s disease with novel biomarkers. Indian J Rheumatol. 2011;6(4):166–7.
Ruiz-Irastorza G, et al. Evidence-based recommendations for the prevention and long-term management of thrombosis in antiphospholipid antibody-positive patients: report of a task force at the 13th international congress on Antiphospholipid antibodies. Lupus. 2011;20:206–18.
Marina P. Sikara, Eleftheria P. Grika and Panayiotis G. Vlachoyiannopoulos. 2011. Pathogenic Mechanisms of Thrombosis in Antiphospholipid Syndrome (APS). ISBN: 978-953-307-872-4, Thrombophilia book http://www.intechopen.com/books/thrombophilia/pathogenicmechanisms-of-thrombosis-in-antiphospholipid-syndrome-aps.
http://emedicine.medscape.com/article/333221-overview#a0104. Date of citation Feb 05, 2014.
de Groot PG, et al. Pathophysiology of thrombotic APS: where do we stand? Lupus. 2012;21:704–7.
Groot PGDE, Meijers JCM, Urbanus RT. Recent development in understanding of APS. Int J Lab Hematol. 2012;34:223–231.nternational.
Zhou, et al. Characterization of monocyte tissue factor activity induced by IgG antiphospholipid antibodies and inhibition by dilazep. Blood. 2004;104(8):2353–8.
Sikara, et al. β2 glycoprotein I (β2GPI) binds platelet factor 4 (PF4): implications for the pathogenesis of antiphospholipid syndrome. Blood. 2010;115(3):713–23.
N Rukoyatkina et al. 2013. p38 MAPK in platelet apoptosis. Online citation from: Cell Death and Disease (2013) 4, e931. https://doi.org/10.1038/cddis.2013.459. www.nature.com/cddis. Date of citation Feb 22, 2014 G.
Paul Schick. Updated: Feb 19, 2013. Hypercoagulability - Hereditary Thrombophilia and Lupus Anticoagulants Associated With Venous Thrombosis and Emboli. http://emedicine.medscape.com/article/211039-overview. Date of citation March 3, 2014.
Bĕlohlávek J, Dytrych V, Linhart A. Pulmonary embolism, part I: epidemiology, risk factors and risk stratification, pathophysiology, clinical presentation, diagnosis and nonthrombotic pulmonary embolism, experimental and clinical cardiology journal, PubMed. NCBI. 2013;18(2):129–38.
Campbell IA, Fennerty A, Miller AC. Guidelines for the management of suspected acute pulmonary embolism. British Thoracic Society Thorax. 2003;58(6):470–84.
Takach Lapner S, Kearon C. Diagnosis and management of pulmonary embolism. BMJ. 2013;346:f757.
Goldhaber SZ. Risk factors for venous thromboembolism. J Am Coll Cardiol. 2010;56(1):1–7.
Gregory Piazza, Samuel Z, Goldhaber. 2006. Acute Pulmonary Embolism Part I: Epidemiology and Diagnosis, American heart association,114: e28-e32. Date of citation 5June 2013.
Dupras D et al., 2013, Venous Thromboembolism Diagnosis and Treatment. Thirteenth Edition. N. Institute for Clinical Systems Improvement. http://bit.ly/VTE0113
Goodacre S. In the clinic deep venous thrombosis. Annal of Intern Med. 2008;149(5):ITC3–1.
Strijkers RHW, ten Cate-Hoek AJ, Bukkems SFFW, Wittens CHA. Management of deep vein thrombosis and prevention of post-thrombotic syndrome. BMJ. 2011;343:d5916.
Saeed M. Deep vein thrombosis and its diagnosis. Techniques in Orthopaedics, Lippincott Williams & Wilkins, Inc. 2008;23(3):273–85.
Kyrle PA, Eichinger S. Deep vein thrombosis. Lancet. 2005;365:1163–74.
B Taylor Thompson, Charles A Hales. 2013. Overview of acute pulmonary embolism, up to date. Date of citation 5 June 2013.
Brydon JB Grant. 2013. Diagnosis of suspected deep vein thrombosis of the lower extremity, up to date. Date of citation June 9, 2013.
Wells PS, et al. Derivation of a simple clinical model to categorize patients probability of pulmonary embolism: increasing the models utility with the SimpliRED D-dimer. J Thromb Haemost. 2000;83(3):358–519.
Fancher TL, White RH, Kravitz RL. Combined use of rapid D-dimer testing and estimation of clinical probability in the diagnosis of deep vein thrombosis: systematic review. BMJ. 2004;329:821.
BMJ Publishing Group. Pulmonary embolism, best practice. BMJ. 2013;
Hugli O, et al. The pulmonary embolism rule-out criteria (PERC) rule does not safely exclude pulmonary embolism. J Thromb Haemost. 2011;9(2):300–4.
Rocha, et al. Risk-assessment algorithm and recommendations for venous thromboembolism prophylaxis in medical patients. Vascular Health and Risk Management. 2007;3(4):533–53.
Snow V, et al. Management of Venous Thromboembolism: a clinical practice guideline from the American College of Physicians and the American Academy of family physicians. ACP. 2007;146(3):204–10.
Michael H. Weisman et al. 2010. Targeted Treatment of the Rheumatic Diseases. 1st edition. Chapter 11. Management of acute thrombosis and secondary thrombosis prevention in APS. ISBN 978-1-4160-9993-2. Date of online citation March 05, 2014.
Khamashta MA. Management of Thrombosis in the Antiphospholipid-antibody syndrome. N Engl J Med. 1995;332(15):993–7.
Kelley’s text book of Rheumatology, ninth edition. Antiphospholipid Syndrome, chapter 82, pages 1331–1341, and Behçet’s Disease, chapter 93, pages 1525–1532.
Leslie Kahl.2012. Antiphospholipid syndrome. The Washington manual rheumatology subspecialty consult, second edition, chapter 39, pages 333–334.
Emad Y, Abdel-Razek N, Gheita T, El-Wakd M, El Gohary T, Samadoni A. Multislice CT pulmonary findings in Behçet’s disease. Clin Rheumatol. 2007;26:879–84.
Bielinska A, Gluszko P. Statins – are they potentially useful in rheumatology? Pol Arch Med Wewn. 2007;117(9):420–5.
Rodriguez, et al. Statins, inflammation and deep vein thrombosis: a systematic review. J Thromb Thrombolysis. 2012;33(4):371–82.
Gregory YH Lip, Russell D Hull. 2014. Treatment of lower extremity deep vein thrombosis. UpTodate. www.uptodate.com/contents/treatment-of-lower-extremity-deep-vein-thrombosis#H32722067. Date of citation March 13, 2014.
Gonsalves WI, Pruthi RK, Patnaik MM. The new Oral anticoagulants in clinical practice. Mayo Clin Proc. 2013;88(5):495–511.
Agnelli G, et al. Oral Apixaban for the treatment of acute venous thromboembolism. NEJM. 2013;369(9):799–808.
Acknowledgments
The author gratefully acknowledges Dr. Albatool Algailani (medical intern graduated from Umm Al-Qura University, Makkah, KSA) for her help in the preparation of this chapter. The authors also would like to thank Dr. Waleed Hafiz for his assistance in the development of this chapter.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2021 The Author(s)
About this chapter

Cite this chapter
Bashal, F. (2021). Thrombosis in Rheumatological Diseases. In: Almoallim, H., Cheikh, M. (eds) Skills in Rheumatology . Springer, Singapore. https://doi.org/10.1007/978-981-15-8323-0_12
Download citation
DOI: https://doi.org/10.1007/978-981-15-8323-0_12
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-15-8322-3
Online ISBN: 978-981-15-8323-0
eBook Packages: MedicineMedicine (R0)