
Abstract
α-Helical coiled coils constitute one of the most diverse folds yet described. They range in length over two orders of magnitude; they form rods, segmented ropes, barrels, funnels, sheets, spirals, and rings, which encompass anywhere from two to more than 20 helices in parallel or antiparallel orientation; they assume different helix crossing angles, degrees of supercoiling, and packing geometries. This structural diversity supports a wide range of biological functions, allowing them to form mechanically rigid structures, provide levers for molecular motors, project domains across large distances, mediate oligomerization, transduce conformational changes and facilitate the transport of other molecules. Unlike almost any other protein fold known to us, their structure can be computed from parametric equations, making them an ideal model system for rational protein design. Here we outline the principles by which coiled coils are structured, review the determinants of their folding and stability, and present an overview of their diverse architectures.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
4.1 Introduction
Coiled-coil proteins were the earliest biological macromolecules studied at high resolution. Following the discovery of X-rays at the end of the nineteenth century, it was soon recognized that these provided a powerful new tool for probing the structure of biological matter. In an X-ray beam, crystalline materials produce diffraction patterns from which the position of individual atoms can be inferred, an insight that ushered in the era of structural biology at atomic resolution. Proteins remained too large for this method for several decades, requiring many technological and computational developments before the first atomic structure could be determined (myoglobin; Kendrew et al. 1958), but protein fibres were rapidly established as a rewarding target for X-ray diffraction. From the mid 1920s on, William Astbury probed systematically many natural fibres, including wool in the native and denatured state, porcupine quills, horn, nails, muscle, tendons, even DNA (Astbury 1938, 1946). He discovered that the diffraction spectra of protein fibres fell into a small number of spectral forms: an α-form exemplified by wool, a β-form exemplified by silk, and third form corresponding to tendon. He further found that proteins of the α-form could be converted to the β-form by stretching, or to yet another form, cross-β, by heat denaturation (“supercontraction”). All these forms showed simple diffraction patterns, illustrating that the underlying fibres were built by the regular repetition of simple structural motifs, a realization that invited numerous modelling efforts.
The most common diffraction pattern was the α-form, generated by a group of proteins that Astbury came to refer to as ‘k-m-e-f’, for keratin, myosin, epidermin, and fibrinogen. The hallmark of this pattern were prominent meridional arcs at 5.15 Å, indicating the repeating unit of the structure. Numerous attempts were made to derive molecular models from this pattern, including efforts by Astbury himself. These led to a race for the correct structural model between Bragg, Perutz and Kendrew at Cambridge and Pauling and Corey at Caltech (Judson 1979), a race that Linus Pauling won in 1950 with the announcement of the α-helix (Pauling and Corey 1950; Pauling et al. 1951). His structure, however, did not show a periodicity of 5.15 Å, as in the fibre diffraction pattern of the k-m-e-f proteins, but rather of 5.4 Å. Pauling had taken the confidence to ignore the meridional arcs of the α-form because the diffraction pattern of a synthetic fibre, poly-γ-methyl-L-glutamate, which was clearly of the α-type, also showed reflections at 5.4 Å away from the meridian. Pauling and Corey’s α-helix was rapidly confirmed by diffraction data from the Cambridge group, but the meridional arcs of k-m-e-f proteins remained unexplained and continued to preoccupy both groups. Undoubtedly these proteins were α-helical, but their constituent helices had to be distorted in some way relative to the ideal structure. Independently of each other, Linus Pauling at Caltech and Francis Crick at Cambridge found a solution to this problem through packing-induced superhelical distortions of the α-helices in the native proteins (Crick 1952, 1953a, b; Pauling and Corey 1953). Pauling called these supercoiled structures compound helices, Crick called them coiled coils.
In modelling the α-helix, Pauling and Corey (1950) had understood that the structure did not require an integer or a ratio of small integers for the number of residues per turn. Nevertheless, they had allowed for the possibility that, in a crystalline arrangement, the regular packing of the helices could favour deformations into configurations with a rational number of residues per turn, such as 11/3 (11 residues over 3 turns), 15/4, and 18/5. Two years later they returned to this idea, proposing that such superhelical deformation, resulting from the α-helices twisting around each other, could account for the meridional arc in the diffraction spectrum of k-m-e-f proteins (Pauling and Corey 1953). They discussed several sequence periodicities (4/1, 7/2, 15/4) with supercoil twists in the same or in the opposite sense to those of the constituent helices. They also discussed a number of different stoichiometries, including one with six helices coiling around a straight seventh one. This article did not offer quantitative parameters for the model structures and did not consider sidechain packing, proposing instead that supercoiling resulted from the exact repetition of short sequences that caused periodic fluctuations in backbone hydrogen-bond lengths.
Whereas Pauling and Corey only considered backbone configurations in their article, Crick placed sidechain geometry at the centre of his modelling effort (Crick 1952, 1953a, b). His key insight was that α-helices, when twisted around each other at an angle of about 20°, would interlock their sidechains systematically along the core of the structure, repeating the same interactions every seven residues (or two turns of the α-helix). Crick offered a full parameterization for such a structure with sequence periodicity of 7/2, which he called the ‘coiled-coil’, and referred to the systematic interlocking of sidechains as ‘knobs’ into ‘holes’. He conjectured that the energy required to distort the helices could be provided by the packing interactions of the sidechains, if these were hydrophobic, leading him to predict that the sequence of coiled coils would show a periodic recurrence of hydrophobic residues with a periodicity of 7/2 = 3.5 (a periodicity that in time became known as the ‘heptad repeat’).
Although the heptad periodicity predicted by Crick was only confirmed more than 20 years later with the determination of the tropomyosin sequence (Stone et al. 1975) and knobs-into-holes packing with the structure of influenza hemagglutinin (Wilson et al. 1981), the elegance and simplicity of Crick’s parameterization was immediately convincing and became the canonical account of coiled-coil structure. As an intriguing aside, the first coiled-coil protein of known sequence was murein lipoprotein (Braun and Bosch 1972) and the first with a high-resolution structure catabolite gene activator protein (McKay and Steitz 1981), but the coiled coils in these proteins were only recognized years later.
Following Crick’s model of the coiled coil, the number of papers on this topic saw an explosive increase, also relative to other topics in the rapidly expanding field of protein biochemistry, but started losing ground after the early 1960s (Fig. 4.1). Interest was rekindled by the growing availability of protein sequences, which opened coiled coils to computational sequence analysis and modelling (e.g. McLachlan and Stewart 1975, 1976; Parry 1975; Parry et al. 1977), and by the first high-resolution structures, which led Carolyn Cohen and David Parry to propose that an understanding of coiled coils would not only be relevant for long fibrous proteins, but also for globular and membrane proteins (Cohen and Parry 1986, 1990). Their insight was proven visionary just 2 years later with the leucine zipper hypothesis (Landschulz et al. 1988), which fundamentally altered the perception of coiled coils in protein science (see e.g. Cohen and Parry 1994). This led to a second phase of explosive growth for this topic (Fig. 4.1) and coiled coils and leucine zippers are now household terms in protein science. There have been many excellent reviews on their various aspects over the years (Lupas 1996; Cohen 1998; Burkhard et al. 2001; Gruber and Lupas 2003; Mason and Arndt 2004; Lupas and Gruber 2005; Woolfson 2005; Parry et al. 2008), so we will place more emphasis here on developments of the last decade and refer our readers to these reviews for further information.

Number of coiled-coil publications over the years. The number of documents in the Web of Science (Thomson Reuters) obtained with the Topic query [(“coiled coil*” OR “leucine zipper”) AND protein*] over 5-year intervals starting with 1951–1955 are shown with a dashed line (right axis). These numbers, divided by the number of documents obtained with the Topic query [protein*] over the same intervals, are shown with a solid line (left axis). More than 5000 coiled-coil publications have appeared every year for the last two decades, amounting to about 0.5 % of publications in the protein sciences
4.2 Structural Parameters
4.2.1 The Standard Model
Coiled coils are α-helical structures in which helices are wound around each other to form superhelical bundles. They usually consist of two or three helices in parallel or antiparallel orientation, but structures with seven and more helices have been determined. They are usually oligomers either of the same (homo) or of different chains (hetero), but can also be formed by consecutive helices from the same polypeptide chain, which in that case almost always have an antiparallel orientation.
The constituent helices of coiled coils interact via a knobs-into-holes geometry of amino-acid sidechains at their interface (Crick 1952, 1953b). In this geometry, a residue from one helix (knob) packs into a space surrounded by four sidechains of the facing helix (hole). Because the residue thus comes to be located next to the equivalent residue from the facing helix, this geometry is sometimes referred to as in-register. This geometry contrasts with the more irregular packing of helices in globular proteins and non-coiled-coil bundles. In that packing mode, referred to as ridges-into-grooves (Chothia et al. 1977, 1981), a residue packs above or beneath the equivalent residue from the facing helix and is therefore also called out-of-register. The two packing modes impose fundamentally different constraints on the α-helices. Whereas ridges-into-grooves packing can be formed with undistorted helices and a multitude of sidechain positions, the regular meshing of knobs-into-holes packing requires precisely recurrent positions of the side-chains every seven residues along the helix interface (the heptad repeat). Because these seven positions, arranged over two turns of the helix, yield a periodicity of 3.5 residues per turn and an undistorted helix has 3.63 residues per turn, the helices must bend to reduce the periodicity to 3.5 relative to a central axis. This bending is called supercoiling and occurs in the sense opposite to that of the individual helices; this means that, because α-helices are right-handed, the supercoil of heptad coiled coils must be left-handed. The regularity of side-chain positions is reflected at the level of their biophysical properties: coiled coils show seven-residue sequence repeats whose positions are labelled a–g; the core-forming positions (a and d) are usually occupied by hydrophobic residues, whereas the remaining, solvent-exposed positions (b, c, e, f, and g) are dominated by hydrophilic residues.
The consequence of the regular nature of coiled coils is that their structures can be described fully by parametric equations (Crick 1953a; Offer et al. 2002; Lupas and Gruber 2005). The coiled-coil parameters characterize individual helices and their orientation in a superhelical bundle (Fig. 4.2). These parameters can be used to calculate a Cα trace of a coiled-coil bundle, which upon backbone reconstruction and side-chain placement, can serve as a starting point for design and modelling procedures (see Chap. 2 by Woolfson 2017, in this issue).

Schematic representation of a trimeric coiled coil, showing the main parameters. O n marks the centre of one α-helix, A n the Cα position of a constituent residue, and C n the superhelix axis. The distance required for the superhelix to complete a full turn is called the pitch, and the angle of a helix relative to the superhelical axis is α, the pitch angle (sometimes also called superhelix crossing angle or tilt angle). The angle between two neighbouring helices is Ω, the pairwise helix-crossing angle. The vector connecting the centre of a helix to the superhelical axis gives r 0 , the superhelix radius, and that connecting the centre of a helix to the Cα carbons of its constituent residues gives r 1 , the α-helix radius. The angle between the α-helix radius and superhelix radius vectors for the same residue is φ, the positional orientation angle, or Crick angle (which is sometimes confusingly denoted α as well); it gives the location of a given residue relative to the supercoil axis. The angle between the α-helix radius vectors for two consecutive residues is the phase shift of the α-helix (Δφ) and the angle between two consecutive superhelix radius vectors is the phase shift of the supercoil (Δω)
4.2.2 Prediction and Analysis Programs
Thanks to the regularity of the repeating pattern, coiled coils can be predicted directly from their sequence to a level of detail that permits the assignment of individual residues to the positions of the heptad repeat. The most popular tools for such predictions follow the idea of scoring the sequence against a matrix of residue frequencies derived from known coiled coils (Parry 1982). This approach was implemented in the COILS program by substituting residue preferences for frequencies (i.e. dividing residue frequencies by their background frequencies in the sequence database), introducing a scanning window, and scaling scores against reference databases to obtain probabilities (Lupas et al. 1991; Gruber et al. 2006). A variant of this approach, using pairwise residue correlations, was implemented in the program PairCoil (Berger et al. 1995; McDonnell et al. 2006). More recently, another approach based on Hidden Markov Models led to the development of MARCOIL (Delorenzi and Speed 2002), which operates without a scanning window and appears to offer the best combination of sensitivity and speed among the currently available methods (Gruber et al. 2006). Conceptually similar methods have also been implemented to predict the oligomerization state of coiled coils from sequence data, yielding the programs SCORER (Woolfson and Alber 1995; Armstrong et al. 2011) and Multicoil (Wolf et al. 1997; Trigg et al. 2011), and, through use of support vector machine-based classification, PrOCoil (Mahrenholz et al. 2011). These programs all distinguish only between parallel dimers and trimers, and are unable to identify coiled coils that are neither, thus being somewhat limited in their use. A step forward has been taken with LOGICOIL (Vincent et al. 2013), a program that combines Bayesian variable selection with multinomial probit regression for prediction; this program is able to additionally distinguish antiparallel dimers and tetramers, thus considerably extending its range. For all programs discussed here, it is important to keep in mind that they are designed to analyze unbroken heptad repeats and therefore have limited applicability to coiled coils with other periodicities (see Sect. 4.2.4). In addition, the sequences of coiled coils are similar to those of natively unstructured proteins, leading to an overlap between the two types of predictions. The reasons for this similarity and ways to deal with the resulting false negative and false positive predictions will be discussed in Sect. 4.3.2.
The ability to describe coiled coils with parametric equations has also led to the development of a large number of useful programs that analyse and model coiled-coil structures. Socket (Walshaw and Woolfson 2001) detects knobs-into-holes packing and Twister (Strelkov and Burkhard 2002), SamCC (Dunin-Horkawicz and Lupas 2010a), and CCCP fit (Grigoryan and DeGrado 2011) quantify the properties of experimental structures, while BeammotifCC (Offer et al. 2002), CCCP generate (Grigoryan and DeGrado 2011), and CCBuilder (Wood et al. 2014) allow users to generate models for coiled coils with predefined properties. Socket relies on a user-adjustable geometric definition of knobs-into-holes packing and on a simplified structure representation, considering sidechains only by their centre of mass. A scan of the Protein Structure Databank with Socket allowed Woolfson and coworkers to classify numerous coiled-coil structures according to their architecture in a relational database, CC+ (Testa et al. 2009). In contrast to Socket, Twister ignores the sidechains completely and considers only Cα carbons of a coiled-coil structure to determine the position of the central axis for each helix which, in turn, determines the location of the supercoil axis. Once the axes are traced, all structural parameters defined by the Crick parameterization can be calculated at the resolution of individual residues. Twister is very well suited to track local fluctuations in coiled-coil structures, but is limited by the requirement that the helices be in a parallel orientation and symmetric (because only averaged results are emitted). This issue has been addressed in the SamCC program, which can be used to measure antiparallel and asymmetric coiled coils with four or more helices. A different approach is used in the CCCP suite of programs, which does not divide the structure into consecutive layers, but rather globally fits Crick parameters to a given backbone structure. CCCP can not only be used to analyse structures, but also to produce coordinates for the main chains of coiled coils based on specified Crick parameters. Such modelling can be also performed with BeammotifCC, which is uniquely able to model transitions between regions of different periodicity. Finally, modelling of coiled coils can also be performed in CCBuilder, which is available as a web service only and provides the user with a complete pipeline for coiled coil design and model validation.
4.2.3 Coiled Coils with Variant Core Geometry
While most coiled coils follow the canonical knobs-into-holes packing described by Crick, two variant packing modes are sometimes observed in antiparallel structures. The first, complementary x–da, is brought about by global axial rotation of all helices, in the ideal case by about 26°. This rotation shifts the relative position of residues and leads to a variant of knobs-into-holes packing, in which the hydrophobic core is formed by three positions, rather than two. When viewed from the N-terminus of a helix, clockwise rotation moves position d into the centre of the core, position a outward and position g inward to yield an a–d–g core; counter-clockwise rotation has the opposite effect, moving position a to the centre and yielding an a–d–e core (Fig. 4.3a). In both cases, the positions of the extended hydrophobic core assume two distinct geometries: we refer to one geometry as x, where the sidechains point towards the centre of the coiled-coil bundle, and to the other as da, where two sidechains point side-ways, enclosing a central cavity. Importantly, x and da represent structural nomenclature and da should not be confused with positions d and a of a heptad repeat. In the a–d–g core, positions g and a assume da geometry, whereas position d assumes x geometry. Similarly, in the a–d–e core, positions d and e assume da geometry and position a assumes x geometry (Fig. 4.3a). Complementary x–da packing is almost invariably found in antiparallel four-helical bundles, because x and da alternate along the helices and therefore the geometrically most favourable packing is achieved when the two pairs of diagonally opposite helices run in inverse directions and the x positions in one pair are combined with da positions in the other. The only case known to us where complementary x–da packing occurs in a parallel coiled coil is in the HAMP domain, a homodimer of two consecutive helices in which complementarity is achieved by the N-terminal helix having an a–d–e core and the C-terminal one an a–d–g core (Hulko et al. 2006). Intriguingly, HAMP can assume both canonical and complementary x–da packing (Ferris et al. 2011), suggesting that it could relay conformational signals in transmembrane receptors by the concerted axial rotation of its helices (Mondejar et al. 2012; Ferris et al. 2012, 2014).

Variant core geometries. (a) Complementary x–da packing via axial helix rotation in antiparallel four-helical coiled coils, showing helical wheel-diagrams of a–d–e (left) and a–d–g (right) hydrophobic cores. Canonical hydrophobic core positions (a and d) and positions co-opted to the core in x–da packing (e or g) are highlighted. (b) Alacoil interactions in the context of four-helical bundles. The heptad positions of the helices are denoted relative to the overall register of the bundle. The Alacoil interactions can occur along either the d–g (left) or the a–e (right) edge; the participating positions are highlighted
The extent to which helices are axially rotated is captured by the Crick angle of the standard model, which describes the orientation of a residue relative to the bundle axis. Measurements of Crick angles in experimentally determined structures using SamCC showed that coiled coils rarely assume an ideal x–da packing, in which helices are rotated by +26° or −26°. Rather, a continuum of axial rotational states was observed, predominantly in coiled coils with a–d–e cores (Dunin-Horkawicz and Lupas 2010a); in contrast, coiled coils with a–d–g cores are rarely observed and their rotation angles are typically <10° (Szczepaniak et al. 2014). A survey of natural and computed structures found that small hydrophobic residues in position e and hydrophilic residues in position g favour a–d–e cores; the opposite distribution favours a–d–g cores (Dunin-Horkawicz and Lupas 2010b; Szczepaniak et al. 2014). Depending on the sidechain size of the residues co-opted into the core (e in the case of a–d–e cores, g for a–d–g cores) the cross-sections of the resulting bundles may range from square (e.g. GCN4-pV, Table 4.1) to distinctly rectangular (e.g. in Lac21 and GCN4-pAeLV, where the co-opted positions e are occupied by alanines, Table 4.1).
The second variant packing mode, Alacoil, refers to an arrangement of two tightly associated, antiparallel α-helices (Gernert et al. 1995); we will address the unique case of an engineered, parallel Alacoil, which assembles into sheets, in Sect. 4.3.1. The exceptionally small distance of ~8 Å between helical axes is possible owing to the overrepresentation of small residues on one side of the hydrophobic core, typically alanine (hence the name). If the small residues occur in position a (defined with respect to the pairwise interaction), the resulting Alacoil is considered to be of the ferritin type, if in d, of the ROP type (Fig. 4.4; Table 4.1). The residues in the other core position (d in ferritins and a in ROPs) are typically large and interdigitate to form a continuous ridge. The two Alacoil types can also be distinguished by the relative shift between the interacting helices: when viewed from the side, helical turns in ROP-like structures appear to lie directly across each other, whereas in ferritin-like structures they appear offset (Gernert et al. 1995).

Alacoil interactions between antiparallel helices. In ROP-type Alacoils (left column) small residues are localized in position d of the heptad repeat, whereas in Ferritin-type (right column) in position a. Positions occupied by small resides are highlighted. Note the difference in the relative axial shift between the interacting helices
In the original description of the Alacoil (Gernert et al. 1995), all examples were pairwise helical interactions extracted from bundles comprising four α-helices. The heptad register used to describe them referred to the pairwise interaction, ignoring the register of the parent bundle. Since Alacoils can form along either the a–e or d–g edges of a bundle (Fig. 4.3b; Table 4.1), the pairwise register has to be related to the overall register on a case-by-case basis. This has proven a consistent source of confusion, further enhanced in many studies by a failure to distinguish between Alacoil interactions and x–da packing. Although some coiled coils with x–da packing resemble Alacoils in having a seam of small residues that leads to close, pairwise association of their helices, the two packing modes differ fundamentally in the axial rotation state of their helices with respect to the bundle. Whereas helices showing Alacoil interactions are essentially unrotated (Dunin-Horkawicz and Lupas 2010a), helices with x–da interactions are rotated by > ±10° with respect to the bundle axis (Szczepaniak et al. 2014). An example is provided by the four-helical bundle formed by the Lac repressor tetramer, Lac21, which was described as a ferritin-like Alacoil (Solan et al. 2002), but, in fact, shows complementary x–da packing with an a–d–e core, due to the ~16° counter-clockwise rotation of its helices (Table 4.1).
4.2.4 Non-heptad Coiled Coils
Coiled coils typically show a high degree of regularity, but are rarely without any discontinuities. The most common of these result from the insertion of three (stammer) or four (stutter) residues into the heptad repeat. Both are close enough to the periodicity of canonical coiled coils (7/2 = 3.5) to be accommodated without disrupting the constituent helices. By inserting more than 3.5 residues, stutters raise the periodicity locally to 3.67 residues per turn (7 + 4 = 11 -> 11/3), leading to an unwinding of the left-handed supercoil; stammers have the opposite effect and reduce the periodicity to 3.33 (7 + 3 = 10 -> 10/3), causing overwinding. Stutters, in particular, are frequently seen in coiled coils and the 11-residue segments they generate are often referred to as hendecads. These local changes in supercoiling affect core packing in the same way as the global rotation of the helices (Sect. 4.2.3), leading to the local adoption of x–da geometry: stutters are analogous to counter-clockwise rotation and stammers to clockwise rotation, as seen from the N-terminus of the helix. In coiled coils that are parallel and in-register, residues in positions x point towards the supercoil axis and residues in da interact in a ring around a central cavity; both depart from knobs-into-holes geometry to form knobs-to-knobs interactions. In dimeric coiled coils, these knobs-to-knobs interactions are particularly constrained, as residues in x point directly at each other, causing steric clashes if the residues are larger than glycine or alanine. Several strategies to accomodate x layers are observed (Lupas and Gruber 2005): the coiled coils may become locally asymmetric to avoid clashes; they may form higher oligomers, where the centre of the bundle offers increasingly more space; they may move locally out of register towards ridges-into-grooves geometry; or they may form antiparallel bundles, which have more space for residues in x positions because sidechains are angled towards the N-terminus and thus point in opposite directions (also, here x and da geometries can be combined to form complementary x–da layers, as discussed in Sect. 4.2.3).
Insertions may be delocalized over several heptads; for example, a stutter can result in higher periodicities like 18 residues over 5 turns (14 + 4 = 18 -> 18/5 = 3.6), or 25 residues over 7 turns (21 + 4 = 25 -> 25/7 = 3.57). Delocalization over more than one heptad also allows for accommodation of discontinuities other than stutters or stammers, such as insertions of 1 (skip) or 5 residues. In the context of a single heptad, these would lead to periodicities of 2.66 ((7 + 1)/3), 3.0 ((7 + 5)/4), or 4.0 ((7 + 1)/2; (7 + 5)/3), none of which fall into the range accessible to coiled coils. However, if delocalized over at least two heptads they produce periodicities such as 3.67 ((21 + 1)/6), 3.71 ((21 + 5)/7), 3.75 ((14 + 1)/4), or 3.8 ((14 + 5)/5), all of which can result in stable structures. Fundamentally, all periodicities accessible to coiled coils without disruption of the constituent helices can be described as combinations of three- and four-residue segments that start with a core residue (Hicks et al. 2002; Gruber and Lupas 2003). This is evidently true for heptads (7), stutters (4), and stammers (3), but also for skips, which in the context of a heptad are equivalent to two stutters (7 + 1 = 4 + 4), and 5-residue insertions, which, in the same context, are equivalent to three stutters (7 + 5 = 4 + 4 + 4). When elements of 3 and 4 alternate regularly, perfect knobs-into-holes packing is achieved. In places where consecutive elements of the same kind (3–3 or 4–4) occur, the packing locally requires knobs-to-knobs interactions.
There are limits to the periodicities that coiled coils can assume, resulting from the supercoil strain imposed on the constituent helices. The further a periodicity is from the 3.63 residues per turn of an undistorted helix, the stronger the supercoiling needed to form a coiled coil. At some point, the strain is sufficient to break the helix. Indeed, two crystal structures of stammers in heptad coiled coils show that the strain these introduce is accommodated by the formation of local 310-helical segments (Hartmann et al. 2009, 2016). It thus appears that 3.33 residues per turn (or 0.3 less than an undistorted helix) mark the lower boundary of periodicities accessible to coiled coils. By extension, we anticipate the upper boundary to be around 3.95. Indeed, if a skip residue cannot be accommodated by delocalization over at least two heptads, the local periodicity of 4.0 it causes is too high for a coiled coil and the residue is looped out into a π-turn, leaving the remaining coiled coil largely unperturbed (Lupas 1996; Lupas and Gruber 2005).
If the insertion of 3 residues marks a lower limit for the supercoiling accessible to a coiled coil, what happens if 2x3 or 3x3 residues are inserted (corresponding to the deletion of 1 or insertion of 2 residues in a heptad frame, respectively)? Surprisingly, in trimeric coiled coils, these were recently shown to cause the local formation of short β-strands, which move the path of each chain in the trimer by 120° around the supercoil axis (Hartmann et al. 2016), resulting in an α/β coiled coil that represents a substantially new kind of fibre (Hartmann et al. 2016) (Fig. 4.5).

Coiled coil structures with different periodicities. Top row: The left-handed supercoil of GCN4-pII (PDB: 1GCM) with a periodicity of seven residues over two helical turns (7/2), the straight helices of tetrabrachion (PDB: 1FE6) with 11/3 periodicity, and the right-handed supercoil of human VASP (PDB: 1USD) with 15/4 periodicity. Bottom row: Two mildly left-handed supercoils created by a 18/5 periodicity in the influenza hemagglutinin, pH 4 (PDB: 1HTM) and a 25/7 periodicity in the Sendai virus phosphoprotein (PDB: 1EZJ). In TCAR0761 from Thermosinus carboxydivorans (pdb: 5APZ), the 9/3 periodicity leads to the formation of an α/β coiled coil. Grey backgrounds indicate the extent of a single repeat
When the local discontinuities described here are repeated regularly along the helix, they lead to the formation of coiled coils whose periodicity deviates globally from the standard model. For these, the handedness of the supercoil is given by the difference of their periodicity to the 3.63 of an undistorted α-helix: coiled coils with periodicities of 3.4 (17/5) or 3.57 (25/7) are left-handed; with periodicities of 3.6 (18/5) or 3.67 (11/3) essentially straight, and with periodicities of 3.75 (15/4) or 3.8 (19/5) right-handed (Fig. 4.5). Since only heptads lead to continuous knobs-into-holes packing, all these periodicities cause the periodic formation of knobs-to-knobs interactions. Because of the constraints resulting from them, coiled coils that deviate globally from the heptad pattern are essentially never two-helical. Such coiled coils were anticipated by Pauling and Corey (1953), but not considered by Crick, who placed knobs-into-holes interactions at the centre of his model. Nevertheless, the Crick equations fully account for coiled coils with non-heptad periodicities and their implementation into computational tools like CCCP or BeammotifCC, can be used to measure and model such coiled coils. BeammotifCC, though, is the only tool that can model transitions between different types of periodicities such as seen in many natural coiled coils, for example in the rod of the trimeric adhesin YadA, which goes from 15/4 to 19/5 to 7/2 (Koretke et al. 2006).
4.3 Structural Determinants of Folding and Stability
4.3.1 Number and Orientation of Helices
The sidechains at the helical interfaces of coiled coils are the main determinants of their oligomeric state and of the orientation of the helices in the bundle. We will outline a number of factors here that can lead to a specific structural state, but individually these should be seen as context-dependent preferences, which may be overridden by other factors.
In parallel coiled coils, an important role is played by the core residues in positions a and d, which show distinct preferences for specific sidechains in two-, three-, and four-helical coiled coils. This is because in position a of dimeric coiled coils, the Cα-Cβ vector of residues is parallel to that of the peptide bond facing them across the interface, while in position d it is perpendicular. This provides more space at the centre of the interface for position a, favouring the β-branched residues isoleucine and valine, and a more constrained central space for position d, favouring the γ-branched residue leucine. In trimeric coiled coils, the Cα-Cβ vectors of positions a and d have a similar, acute angle with respect to the interface and do not show a preference for specific sidechains. In tetrameric coiled coils, the geometry of the positions at the interface is reversed, with a having a perpendicular Cα-Cβ vector and a preference for leucine, and d having a parallel vector and a preference for isoleucine and valine. For this reason, switching the position of specific sidechains in the core can lead to a switch in oligomer states between dimers, trimers, and tetramers in otherwise identical sequences (Harbury et al. 1993). These preferences may, however, not be sufficient to specify the oligomeric state in some cases (Armstrong et al. 2009; Zaytsev et al. 2010). The size of sidechains at core positions also plays a role in establishing the oligomeric state. Whereas the aliphatic residues are compatible with all forms, coiled coils with phenylalanine or tryptophan at all core positions form pentamers, although the presence of a single methionine in the core reduces the phenylalanine pentamer to a tetramer (Liu et al. 2004, 2006a). The oligomeric state can be further influenced by polar residues in the core. Thus, asparagine in position a is a determinant of dimeric structure in leucine zippers (Harbury et al. 1993; Gonzalez et al. 1996; Akey et al. 2001), but in position d it specifies trimerization in a range of surface proteins (Hartmann et al. 2009). As a general rule, polar and charged residues favour dimers, where they can be more easily solvated, but exceptions abound.
Next to the core residues, the residues in the positions flanking the core (e and g) also influence oligomerization and orientation. Specifically, if they include a preponderance of hydrophobic residues in one of the positions, they favour tetramers over dimers or trimers, frequently in antiparallel orientation. The resulting, broader hydrophobic core can be described as having two seams of core residues, which overlap by one position, i.e. g–d and d–a, or d–a and a–e (Walshaw et al. 2001; Woolfson et al. 2012). Axially symmetric packing of such a larger interface causes the shared position to point towards the central axis (x geometry) and the two other positions to point side-ways, enclosing a central cavity (da geometry). We have already encountered this packing in Sects. 4.2.3 and 4.2.4, in the context of complementary x–da interactions, and of stutters and stammers.
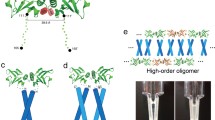
As the two seams move further apart on the face of the helices, they favour increasingly higher oligomers, marking a transition from fibres to tubes (see also Sect. 4.4.2). Thus, adjacent seams (g-d and a-e) lead to the formation of pentamers, hexamers, and heptamers, and seams separated by an intervening residue (g-d and e-b, separated by a; or c-g and a-e, separated by d) to even higher oligomers, the largest of which is the antiparallel barrel of 12 helices in the multidrug efflux protein TolC (Koronakis et al. 2000). Helices with two seams have been called “bifaceted” and the three types have been denoted I, II, and III in the order of increasing separation (Walshaw et al. 2001; Woolfson et al. 2012). Thomson et al. (2014) have recently developed a quantitative description of type II bifaceted helices, which has allowed them to engineer penta- to heptameric barrels by computational design. Egelman et al. (2015) have gone one step further by designing a type III bifaceted helix that assembles into sheets (Table 4.1). This open-ended architecture is brought about by two factors: (a) a heterotypic association between the g–d seam of one helix and the e–b seam of the next, such that the growing sheet always presents a g–d seam at one edge and an e–b seam at the other; and (b) the placement of alanines in position g of the g–d seam and position b of the e–b seam, so that a parallel Alacoil interaction can form. Since, in Alacoils, the two helices approach each other more closely along the ridge of alanines than along the other ridge of residues, the angle between the two seams (theoretically 155°) is widened to approximately 180°, allowing the sheet to remain open.
The interface residues thus not only determine the oligomerization and orientation of coiled-coil helices, they also set the geometry of packing interactions (canonical, Alacoil, partially rotated, fully x–da). In doing so they navigate an energy landscape in which the different structural states are often nearly isoenergetic and separated by low energy barriers. In our discussion of structural diversity in coiled coils (Sect. 4.4.1), we will review in detail how minor changes to the sequence of the GCN4 leucine zipper can lead to a range of different forms; here we would just like to point out a side-effect of this flat energy landscape, namely that coiled-coil fragments crystallized outside their native protein context often assume non-physiological structures. Instances of this abound in the Protein Structure Databank. For example: (a) the S-helix of the beta-1 subunit of a soluble guanylyl cyclase (3HLS) forms an antiparallel homotetramer, while the parent protein is a parallel heterodimer (with a minor homodimeric form); (b) the pilin subunit of Neisseria gonorrhoeae (1AY2) forms an antiparallel dimer, while the parent structure is an extended spiral of offset, parallel subunits; (c) a fragment of the SARS coronavirus spike glycoprotein forms an antiparallel tetramer with x–da packing (1ZV7), while the parent protein is a parallel trimer with canonical packing; (d) the cytosolic coiled-coil segments of ion channels have been variously solved as parallel tetramers (3BJ4, 2OVC), antiparallel tetramers (3E7K), and parallel trimers (3HFE, 2PNV), even though the parent proteins are all parallel tetramers. A fair number of other such cases could be listed here.
4.3.2 Folding and Stability
Due to the regularity of their interactions, coiled coils are often very stable proteins. Indeed, the most stable protein reported to date may well be the surface-layer protein of the archaeon Staphylothermus marinus, whose stalk domain – a homotetrameric coiled coil – withstands heating to 130 °C in 6 M guanidinium hydrochloride and requires 70 % sulphuric acid for denaturation (Peters et al. 1995). Since this treatment leads to hydrolysis of the peptide bonds, tetrabrachion may be the only known protein where the primary structure appears to be less stable than the secondary, tertiary, and quaternary structures. This is certainly exceptional, but many coiled coils have been reported to withstand extreme chemical and thermal conditions, including some comprising only a few heptads, such as variants of the GCN4 leucine zipper. The unusual stability that can be achieved by natural coiled coils has been recently reproduced by computational design in engineered coiled coils (Huang et al. 2014). Factors that promote stability are the propensity of the constituent helices to adopt a helical structure, the tightness of their packing interactions, the hydrophobicity of the resulting core, and a shell of favourable polar and ionic interactions that shield this core from solvent. It follows that, all things being equal, the stability of coiled coils should increase with the number of helices, as this provides them with a broader hydrophobic core (at least until the number of helices turns them from bundles into barrels and causes the formation of a central, solvent-filled pore). Indeed, this increase in stability can be observed between the dimeric, trimeric, and tetrameric variants of the GCN4 leucine zipper (Harbury et al. 1993).
Given their thermodynamic stability, it may come as a surprise that coiled coils are close to the unfolded state; in fact, it is not uncommon for them to be mistaken for natively unstructured polypeptides by disorder prediction programs. We are not aware of any systematic study of this, but we have observed it ourselves in many cases over the years, for example with the myosin heavy chain, whose extended stalk is both predicted confidently as a coiled coil and seen as intrinsically disordered by the respective prediction programs (Fig. 4.6). This failure cuts both ways: highly charged sequences that are largely devoid of hydrophobic residues and lack sequence repeats indicative of coiled-coil structure are often predicted as coiled coils, even though they are most likely unstructured (Gruber et al. 2006). This is because coiled coils are highly repetitive, largely solvent-exposed structures and therefore have reduced sequence complexity and a low proportion of hydrophobic to hydrophilic residues relative to globular proteins - just like many natively unstructured sequences. The five most frequent residues in coiled coils (E, L, K, A, Q) comprise more than half of the total (with the charged residues E and K alone constituting more than a quarter), whereas they represent less than a third of residues in globular proteins. Conversely, the five rarest residues in coiled coils (F, H, C, W, P) barely add up to 2 %, whereas they constitute more than 15 % in globular proteins. The similarity between coiled coils and natively unstructured sequences offers a substantial challenge in structure prediction. We find a number of questions helpful in discriminating between the two: (1) Is the sequence repetitive and, if yes, is the periodicity compatible with coiled-coil structure? (2) Does the repetition entail a preponderance of hydrophobic residues at core positions and hydrophilic residues at all other positions? (3) Is the structural context indicative of, or at least compatible with, a coiled coil (for example, does the sequence belong to a protein family that is known to be oligomeric and fibrous)? (4) Is an elevated coiled-coil propensity conserved in homologues? Answering Yes to these questions progressively increases the confidence that a given sequence forms a coiled coil.

Predictions of coiled-coil propensity and disorder in human myosin heavy chain. The boundary between the globular head domain and the fibrous stalk is marked by a vertical dotted line. The output of FoldIndex is shown on an inverted scale in order to make it directly comparable to the two other programs. The graphs show that the rod is recognized both as a coiled coil (COILS) and as natively unstructured (IUPred, Dosztányi et al. 2005; FoldIndex, Prilusky et al. 2005)
In our opinion, coiled-coil sequences have evolved to resemble unstructured polypeptides because they need to ensure in-register folding of rods that are sometimes many hundreds of residues long. Since packing interactions are structurally the same all along the rod, coiled coils are confronted with many, essentially isoenergetic intermediates that could trap the folding chains out of register if they formed spontaneously. To prevent this, coiled coils have evolved sequences that allow them to be quite stable thermodynamically, once folded, but have kinetic folding barriers that maintain them in an unstructured state until folding has been initiated at a nucleation site and is therefore guaranteed to be in register. Thus, fragments of natural coiled coils, even of considerable size, often do not fold: for example, myosin rod fragments hundreds of residues long remain soluble, but unstructured, if they do not include a nucleation site (Trybus et al. 1997). The concept of nucleation sites as initiators of coiled-coil folding was pioneered by Kammerer, Steinmetz and co-workers, who called them ‘trigger sequences’ (Steinmetz et al. 1998; Kammerer et al. 1998) and characterized them in a number of different coiled coils. Subsequent identifications of further trigger sequences (Frank et al. 2000; Wu et al. 2000; Alfadhli et al. 2002; Araya et al. 2002) showed that these nucleation sites do not adhere to a particular consensus sequence, but rather represent short segments of high α-helical propensity, capable of forming many stabilizing interactions in the correct oligomeric form through optimized electrostatic interactions and hydrophobic packing (Lee et al. 2001).
4.4 Structural Diversity
4.4.1 Fibres and Zippers
For decades after their initial description, the concept of coiled coils was closely associated with long fibres, such as the keratins, myosins, epidermins, and fibrinogens of the k-m-e-f class, which were all two- or three-helical. Their properties had greatly helped the original fibre diffraction studies, but caused major impediments to their analysis by X-ray crystallography. Today, there is still only one structure of a k-m-e-f class protein known to high resolution (fibrinogen at 2.7 Å, 1M1J; Yang et al. 2001), while information on the others is available for fragments only or at considerably lower resolution, such as for tropomyosin at 7.0 Å (Whitby and Phillips 2000).
As the X-ray crystallographic analysis of proteins developed, it emerged gradually that coiled-coil interactions were also observed in much shorter helical bundles, often embedded within globular proteins, such as in the catabolite gene activator protein (McKay and Steitz 1981; Nilges and Brunger 1991). These observations led to the proposal that an understanding of coiled-coil structure would be of substantial significance not only for long fibres, but also for globular and membrane proteins, as well as for structures with larger numbers of helices, such as four-helical bundles (Cohen and Parry 1986). Soon afterwards, the discovery of the leucine zipper and particularly of its prototypical representative in the yeast transcription factor GCN4 (Landschultz et al. 1988) provided a tractable model system of great biological importance for the high-resolution analysis and manipulation of coiled coils (O’Shea et al. 1991; Harbury et al. 1993). Today it is widely recognized that coiled coils not only comprise the long fibrous proteins that form filaments, tethers, stalks, levers, and large, mechanically rigid assemblies (e.g. hair, feathers, horn, blood clots), but also many shorter structures, which – among other activities – mediate oligomerization, transduce signals, and facilitate the transport of small molecules (see also Chap. 3 by Hartmann 2017, in this issue).
As an example for the structural diversity accessible to coiled coils we will discuss here the GCN4 leucine zipper, which can be converted to a broad range of structural forms by minor changes to its sequence, illuminating the versatility of the fold and the closeness of its different variants in the energy landscape (Table 4.2). In the native state, it forms a parallel, two-helical coiled coil of 30 residues (GCN4-p1), whose dimerization depends largely on an asparagine residue in position a of the heptad repeat (N16). This residue represents an instance of ‘negative design’ since it provides structural specificity at the expense of stability; its replacement generally increases stability, but leads to trimers, or mixtures of dimers and trimers (Harbury et al. 1993; Gonzalez et al. 1996; Akey et al. 2001). All these forms are parallel, but in one case an antiparallel trimer was obtained by mutating N16 to alanine. This mutation caused the structure to become trimeric and invert the orientation of one helix, such that the A16 residues form mixed a-d core layers with L12, filling the cavity caused by the small alanine sidechains. The parallel orientation of the helices could be rescued by adding benzene to the solvent, which filled the hydrophobic cavity in the core (Holton and Albert 2004). The oligomeric state of GCN4 could also be altered by other substitutions in the hydrophobic core: as outlined in Sect. 4.3.1, the geometry of packing interactions leads coiled-coil dimers to favour β-branched residues in position a and γ-branched or unbranched residues in position d, and tetramers to favour the reverse; trimers show no preference. This rule was, in fact, derived from GCN4 variants, for which isoleucines in a and leucines in d produced dimers (GCN4-IL) and the reverse distribution tetramers (GCN4-LI; Harbury et al. 1993). A retro-GCN4, consisting of an inversion of the GCN4 sequence, therefore unsurprisingly yielded a tetramer (Mittl et al. 2000). Other combinations of isoleucine, leucine, and valine typically resulted in trimers or, rarely, mixtures of dimers and trimers (Harbury et al. 1994).
Structural diversity was also brought about by mutating positions e and g, flanking the hydrophobic core (Table 4.2). For example, introducing an ionic bond between neighbouring chains via an arginine in position g and a glutamate in position e overrode the the effect of N16 and converted GCN4-p1 to a trimer, but only when this was done at the trigger site of the coiled coil (Ciani et al. 2010). A more comprehensive set of polar mutations in positions e and g, aimed at obtaining negatively and positively charged variants of the helices, resulted in heterodimers between these (Keating et al. 2001). Most other mutations in the flanking positions, however, involved an increase in hydrophobicity and generally produced tetramers, albeit with great heterogeneity in orientation and core packing geometry (Table 4.2). Thus, mutating all polar residues in position e to valine (GCN4-pVe) yielded an odd, parallel four-helical bundle with offset helices that engage in a mixture of canonical and complementary x–da interactions, whereas the equivalent mutation of polar residues in position g (GCN4-pVg) yielded a symmetrical, antiparallel tetramer with near-ideal x–da packing (Liu et al. 2006b; Deng et al. 2006; Dunin-Horkawicz and Lupas 2010a). A similar replacement of polar residues in position g with alanine (GCN4-pAg) also yielded an antiparallel tetramer, but the axial rotation of the helices went so far that a new canonical core was formed between positions d and g, resulting in the only GCN4 variant with Alacoil packing (Deng et al. 2006; Table 4.1). As we discussed in Sect. 4.2.3, placing small, hydrophobic residues in positions e or g biases the helices towards complementary x–da packing with an a–d–e or a–d–g core, respectively. Deng et al. (2008) exploited this in order to obtain heteromeric antiparallel tetramers with mixed a–d–g and a–d–e cores by combining either GCN4-pVe with GCN4-pVg, or the equivalent variants carrying alanine in place of valine. Combining hydrophobic mutations in e and g into the same helix (GCN4-pAA) yielded the most unusual zipper variant yet: a heptameric coiled-coil tube with the helices staggered in phase with the heptad repeat (Liu et al. 2006c).
The diversity of GCN4 variants highlights how close these are in their overall energy. The fact that the N16A mutant can be switched from an antiparallel to a parallel trimer by the addition of benzene is evidence of this, as is the observation of Yadav et al. (2006) that the tetrameric GCN4 variant, GCN4-pLI, when carrying a single point mutation from glutamate to serine in position e, can be switched between a parallel form with canonical packing and an antiparallel form with x–da packing (Table 4.2), simply by varying the buffer. Collectively, GCN4 variants map out how simple changes can lead from a plain dimeric fibre to the higher-order assemblies seen in many proteins.
4.4.2 Tubes, Sheets, Spirals, and Rings
As discussed in Sect. 4.3.1, coiled coils can progressively increase the number of helices by broadening their hydrophobic core, then splitting it into two adjacent cores. Starting with four-helical bundles, the space along their central axis increases to form cavities, frequently containing water or other solvent molecules; from five-helical bundles onward, the cavities merge into a continuous, solvent-filled pore. The resulting structures have been referred to variously as tubes (Lupas and Gruber 2005), α-barrels (Koronakis et al. 2000), or α-cylinders (Walshaw et al. 2001; Walshaw and Woolfson 2003). As the diameter of the pore widens and packing interactions become more irregular, the constituent helices may also start to deviate from the perpendicular, yielding funnel-shaped tubes, for example in the upper collar protein of the Bacillus bacteriophage φ29 (Simpson et al. 2000). The potential of coiled-coil tubes to mediate solute transport when embedded in the membrane has been recognized for at least two decades (Malashkevich et al. 1996) and recently Joh et al. (2014) designed the first synthetic ion antiporter by exploiting the axial cavities of a membrane-embedded four-helical coiled coil. The differential chemical accessibility of the central channel in aqueous environment has also suggested a potential to engineer new enzymes (Burton et al. 2013). So far, tubes with five, six, ten, and twelve helices have been identified in nature, and this range has been complemented by computational design to include engineered five-, six-, and seven-helical structures (Thomson et al. 2014; Huang et al. 2014).
When the interaction seams of bifaceted helices are maximally separated, they may come to specify angles close to 180°, depending on the size distribution of residues at the interfaces. In such cases, the helices assemble into open α-sheets, rather than into circular tubes. Similarly, pairs of helices that neutralize each other’s angular offset, such as in the archaeal protein pT26-6p (2WB7; Table 4.1), also lead to open structures (Walshaw et al. 2001; Walshaw and Woolfson 2003). Most natural examples consist of three or four helices, but recently, computational design has resulted in sheets with a much higher number of helices, as discussed in Sect. 4.3.1; four of these sheets assemble to form a novel type of fibre (Egelman et al. 2015) (Fig. 4.7). Maintaining knobs-into-holes contacts in a sheet requires all but the edge helices to supercoil in opposite directions. The strain resulting from this conformational conflict distorts the packing interactions and gradually moves the helices out of register, restricting the length of these associations to typically less than four heptads.

Diversity of coiled-coil structures. The figure shows a fibre (tropomyosin), a zipper (the Fos b-Zip domain bound to DNA), a tube in side and top view (TolC), a sheet in side and top view (colicin IA), a spiral (phage PF1 coat protein B), a synthetic nanotube assembled from sheets, and a ring (apolipoprotein A-I)
Sheets meet tubes in the formation of spirals. Compared to tubes, the helices of spirals are noticeably offset from each other along the axis of the coiled coil. For example, the major coat proteins of filamentous bacteriophages form staggered and slightly curved sheets, which then assemble into a multi-stranded spiral (Fig. 4.7), an architecture also found in proteins of the bacterial flagella and pili (reviewed in more detail in Lupas and Gruber 2005). Another example is the multidrug resistance protein MexA, which was crystallized as a complex, tail-to-tail assembly of two spirals, one with six and the other with seven subunits. Each subunit contributes a helical hairpin to the spiral and the interaction between adjacent hairpins is set off by one heptad. This structure probably represents another instance of the crystallization artefacts discussed in Sect. 4.3.1, as MexA most likely assembles into a funnel in vivo (Symmons et al. 2009). Some of the GCN4 variations that we have discussed in Sect. 4.4.1 are staggered as well, most notably the heptameric GCN4-pAA. The mechanism, however, differs. Spirals make knobs-into-holes contacts between core residues in different heptads, whereas these coiled coils make homotypic contacts but are axially shifted by the equivalent of one residue per helix (Liu et al. 2006c).
An even more intriguing case is that of the apolipoproteins, which, despite being clearly homologous, form structures differing substantially in packing, supercoiling, and oligomer state. Apolipoproteins A-I and A-II oligomerize into ring-shaped four-helical bundles with an underlying 22/6 periodicity (Fig. 4.7), but show only local knobs-into-holes packing and supercoil angles similar to undistorted helices. In contrast, apolipoprotein E and apolipophorin III form monomeric left-handed coiled coils with regular knobs-into-holes interactions (Boguski et al. 1985; reviewed in detail in Lupas and Gruber 2005).
4.5 Evolution and Phylogenetic Diversity
The large diversity of coiled-coil structures discussed above begs the question of their evolutionary origins. Some clearly form ancient families, which can occasionally be traced as far as the root of a kingdom (witness the leucine zipper transcription factors of eukaryotes), but in most cases sequence similarity searches do not uncover homologues beyond individual phyla and usually not even that far. Only in exceptional cases can a coiled coil be traced back all the way to the time of the Last Universal Common Ancestor (e.g. in seryl-tRNA synthetase). We would like to offer two reasons why this might be the case. One is that coiled coils diverge faster than most proteins, due to their primarily structural function, which has lower constraints than catalytic activity, and their highly repetitive nature, which makes them more resilient against point mutations than other structures. At the same time, they converge faster than most other proteins, due to their repetitive structure, which favours the same patterns of hydrophobic and hydrophilic residues, and the same restricted alphabet of residues with high helical propensity. For these reasons, the point in the past where it becomes impossible to distinguish homology from analogy in statistical sequence analyses is more recent for coiled coils than for other proteins. This does not mean that individual coiled-coil families are not older than this point, it just means that we can’t trace their origins beyond it. Additionally, as discussed in Sect. 4.3.2, coiled coils seem to have been under evolutionary pressure to evolve towards unstructured sequences for reasons of folding specificity, which has further lowered their sequence complexity and increased their rate of divergence. In this context, we note the existence of coiled coils with highly biased residue distributions, for example in a family of proteins we have analyzed in great detail, the trimeric autotransporter adhesins (TAAs; Szczesny and Lupas 2008; Bassler et al. 2015); here, one can find proteins such as Bcep18194_B0441 of Burkholderia lata, which has two extensive coiled-coil segments of ~700 and ~1200 residues, respectively, consisting to two-thirds of serine and threonine, or the putative adhesin VEIDISOL_00919 of Veillonella dispar, in whose stalk of more than 2000 residues a quarter are asparagine. Other examples of this kind can be readily found, also in eukaryotic proteins.
The second reason why coiled-coil domains might be difficult to trace far back in time is their rapid turnover in many families. Like many other repetitive structures, they seem to constantly evolve by amplification and divergence from individual repeats but, unlike most other repetitive structures, their repeat unit is so short that it is easy to recruit from sequences not part of a coiled coil, or even from non-coding sequence. Coiled coils thus seem to readily evolve de novo. As evidence for this we see coiled coils, primarily in prokaryotes, whose repeat units are essentially identical to each other over hundreds of residues, not only in the protein sequence, but also at the level of the DNA; such coiled coils typically occur only in a few closely related species and are therefore probably not more than a few million years old. We see further evidence for de novo evolution in the many protein families that are generally globular, but in which one or a few members have “grown” coiled coils through the extension of helices that are an integral part of the family fold (Lupas and Gruber 2005). Although more difficult to establish, it seems reasonable to assume that the de novo acquisition of coiled coils is paralleled by a corresponding rate of loss.
These considerations, of course, proceed from sequences that we can recognize as coiled coils, either because they are considered to assume this structure by prediction programs, or (much more rarely) because we have structural information. Not considered are an unknown number of sequences that form coiled coils, but remain unpredicted because of an odd residue composition or an odd periodicity. The surprising recent discovery of α/β coiled coils (Hartmann et al. 2016) as a substantially new variant of a fold we thought we understood comprehensively suggests that there might be yet more to this seemingly simple fold than we realize.
References
Akey DL, Malashkevich VN, Kim PS (2001) Buried polar residues in coiled-coil interfaces. Biochemistry 40:6352–6360
Alfadhli A, Steel E, Finlay L, Bächinger HP, Barklis E (2002) Hantavirus nucleocapsid protein coiled-coil domains. J Biol Chem 277:27103–27108
Araya E, Berthier C, Kim E, Yeung T, Wang X, Helfman DM (2002) Regulation of coiled-coil assembly in tropomyosins. J Struct Biol 137:176–183
Armstrong CT, Boyle AL, Bromley EH, Mahmoud ZN, Smith L, Thomson AR, Woolfson DN (2009) Rational design of peptide-based building blocks for nanoscience and synthetic biology. Faraday Discuss 143:305–317
Armstrong CT, Vincent TL, Green PJ, Woolfson DN (2011) SCORER 2.0: an algorithm for distinguishing parallel dimeric and trimeric coiled-coil sequences. Bioinformatics 27:1908–1914
Astbury, W. T. (1938). The fourth Spiers memorial lecture. X-ray adventures among the proteins. Transactions of the Faraday Society 34, 378–388.
Astbury WT (1946) X-ray studies of nucleic acids. Sym Soc Exp Biol (1):66–76
Bassler J, Alvarez BH, Hartmann MD, Lupas AN (2015) A domain dictionary of trimeric autotransporter adhesins. Int J Med Microbiol 305:265–275
Berger B, Wilson DB, Wolf E, Tonchev T, Milla M, Kim PS (1995) Predicting coiled coils by use of pairwise residue correlations. Proc Natl Acad Sci 92:8259–8263
Boguski MS, Elshourbagy N, Taylor JM, Gordon JI (1985) Comparative analysis of repeated sequences in rat apolipoproteins AI, A-IV, and E. Proc Natl Acad Sci 82:992–996
Braun V, Bosch V (1972) Repetitive sequences in the murein-lipoprotein of the cell wall of Escherichia coli. Proc Natl Acad Sci 69:970–974
Burkhard P, Stetefeld J, Strelkov SV (2001) Coiled coils: a highly versatile protein folding motif. Trends Cell Biol 11:82–88
Burton AJ, Thomas F, Agnew C, Hudson KL, Halford SE, Brady RL, Woolfson DN (2013) Accessibility, reactivity, and selectivity of side chains within a channel of de novo peptide assembly. J Am Chem Soc 135:12524–12527
Chothia C, Levitt M, Richardson D (1977) Structure of proteins: packing of α-helices and pleated sheets. Proc Natl Acad Sci 74:4130–4134
Chothia C, Levitt M, Richardson D (1981) Helix to helix packing in proteins. J Mol Biol 145:215–250
Ciani B, Bjelic S, Honnappa S, Jawhari H, Jaussi R, Payapilly A, Jowitt T, Steinmetz MO, Kammerer RA (2010) Molecular basis of coiled-coil oligomerization-state specificity. Proc Natl Acad Sci 107:19850–19855
Cohen C (1998) Why fibrous proteins are romantic. J Struct Biol 122:3–16
Cohen C, Parry DAD (1986) α-helical coiled coils - a widespread motif in proteins. Trends Biochem Sci 11:245–248
Cohen C, Parry DAD (1990) α-helical coiled coils and bundles: how to design an α-helical protein. Proteins 7:1–15
Cohen C, Parry DAD (1994) Alpha-Helical Coiled Coils-More Facts and Better Predictions. Science 263:488–489
Crick FHC (1952) Is α-keratin a coiled coil? Nature 170:882–883
Crick FHC (1953a) The Fourier transform of a coiled-coil. Acta Crystallogr 6:685–689
Crick FHC (1953b) The packing of α-helices: simple coiled-coils. Acta Crystallogr 6:689–697
Delorenzi M, Speed T (2002) An HMM model for coiled-coil domains and a comparison with PSSM-based predictions. Bioinformatics 18:617–625
Deng Y, Liu J, Zheng Q, Eliezer D, Kallenbach NR, Lu M (2006) Antiparallel four-stranded coiled coil specified by a 3-3-1 hydrophobic heptad repeat. Structure 14:247–255
Deng Y, Liu J, Zheng Q, Li Q, Kallenbach NR, Lu M (2008) A Heterospecific Leucine Zipper Tetramer. Chem Biol 15:908–919
Dosztányi Z, Csizmok V, Tompa P, Simon I (2005) IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics 21:3433–3434
Dunin-Horkawicz S, Lupas AN (2010a) Measuring the conformational space of square four-helical bundles with the program samCC. J Struct Biol 170:226–235
Dunin-Horkawicz S, Lupas AN (2010b) Comprehensive analysis of HAMP domains: implications for transmembrane signal transduction. J Mol Biol 397:1156–1174
Egelman EH, Xu C, DiMaio F, Magnotti E, Modlin C, Yu X, Wright E, Baker D, Conticello VP (2015) Structural plasticity of helical nanotubes based on coiled-coil assemblies. Structure 23:280–289
Ferris HU, Dunin-Horkawicz S, Mondéjar LG, Hulko M, Hantke K, Martin J, Schultz JE, Zeth K, Lupas AN, Coles M (2011) The mechanisms of HAMP-mediated signaling in transmembrane receptors. Structure 19:378–385
Ferris HU, Dunin-Horkawicz S, Hornig N, Hulko M, Martin J, Schultz JE, Zeth K, Lupas AN, Coles M (2012) Mechanism of regulation of receptor histidine kinases. Structure 20:56–66
Ferris HU, Zeth K, Hulko M, Dunin-Horkawicz S, Lupas AN (2014) Axial helix rotation as a mechanism for signal regulation inferred from the crystallographic analysis of the E. coli serine chemoreceptor. J Struct Biol 186:349–356
Frank S, Lustig A, Schulthess T, Engel J, Kammerer RA (2000) A distinct seven-residue trigger sequence is indispensable for proper coiled-coil formation of the human macrophage scavenger receptor oligomerization domain. J Biol Chem 275:11672–11677
Gernert KM, Surles MC, Labean TH, Richardson JS, Richardson DC (1995) The Alacoil: a very tight, antiparallel coiled-coil of helices. Protein Sci 4:2252–2260
Gonzalez L Jr, Woolfson DN, Alber T (1996) Buried polar residues and structural specificity in the GCN4 leucine zipper. Nat Struct Mol Biol 3:1011–1018
Grigoryan G, DeGrado WF (2011) Probing designability via a generalized model of helical bundle geometry. J Mol Biol 405:1079–1100
Gruber M, Lupas AN (2003) Historical review: another 50th anniversary - new periodicities in coiled coils. Trends Biochem Sci 28:679–685
Gruber M, Söding J, Lupas AN (2006) Comparative analysis of coiled-coil prediction methods. J Struct Biol 155:140–145
Harbury PB, Zhang T, Kim PS, Alber T (1993) A switch between two-, three-, and four-stranded coiled coils in GCN4 leucine zipper mutants. Science 262:1401–1407
Harbury PB, Kim P, Alber T (1994) Crystal structure of an isoleucine-zipper trimer. Nature 371:80–83
Hartmann MD (2017) Functional and structural roles of coiled coils. Fibrous proteins: structures and mechanisms. Springer, Dordrecht
Hartmann MD, Ridderbusch O, Zeth K, Albrecht R, Testa O, Woolfson DN, Sauer G, Dunin-Horkawicz S, Lupas AN, Alvarez BH (2009) A coiled-coil motif that sequesters ions to the hydrophobic core. Proc Natl Acad Sci 106:16950–16955
Hartmann MD, Mendler CT, Bassler J, Karamichali I, Ridderbusch O, Lupas AN, Hernandez Alvarez B (2016) α/β coiled coils. eLife 5:e11861
Hicks MR, Walshaw J, Woolfson DN (2002) Investigating the tolerance of coiled-coil peptides to nonheptad sequence inserts. J Struct Biol 137:73–81
Holton J, Alber T (2004) Automated protein crystal structure determination using ELVES. Proc Natl Acad Sci 101:1537–1542
Huang PS, Oberdorfer G, Xu C, Pei XY, Nannenga BL, Rogers JM, DiMaio F, Gonen T, Luisi B, Baker D (2014) High thermodynamic stability of parametrically designed helical bundles. Science 346:481–485
Hulko M, Berndt F, Gruber M, Linder JU, Truffault V, Schultz A, Martin J, Schultz JE, Lupas AN, Coles M (2006) The HAMP domain structure implies helix rotation in transmembrane signaling. Cell 126:929–940
Joh NH, Wang T, Bhate MP, Acharya R, Wu Y, Grabe M, Hong M, Grigoryan G, DeGrado WF (2014) De novo design of a transmembrane Zn2+-transporting four-helix bundle. Science 346:1520–1524
Judson HF (1979) The eighth day of creation. Simon & Schuster, New York
Kammerer RA, Schulthess T, Landwehr R, Lustig A, Engel J, Aebi U, Steinmetz MO (1998) An autonomous folding unit mediates the assembly of two-stranded coiled coils. Proc Natl Acad Sci 95:13419–13424
Keating AE, Malashkevich VN, Tidor B, Kim PS (2001) Side-chain repacking calculations for predicting structures and stabilities of heterodimeric coiled coils. Proc Natl Acad Sci 98:14825–14830
Kendrew JC, Bodo G, Dintzis HM, Parrish RG, Wyckoff H, Phillips DC (1958) A three-dimensional model of the myoglobin molecule obtained by x-ray analysis. Nature 181:662–666
Koretke KK, Szczesny P, Gruber M, Lupas AN (2006) Model structure of the prototypical non-fimbrial adhesin YadA of Yersinia enterocolitica. J Struct Biol 155:154–161
Koronakis V, Sharff A, Koronakis E, Luisi B, Hughes C (2000) Crystal structure of the bacterial membrane protein TolC central to multidrug efflux and protein export. Nature 405:914–919
Landschulz WH, Johnson PF, McKnight SL (1988) The leucine zipper: a hypothetical structure common to a new class of DNA binding proteins. Science 240:1759–1764
Lee DL, Lavigne P, Hodges RS (2001) Are trigger sequences essential in the folding of two-stranded α-helical coiled-coils? J Mol Biol 306:539–553
Liu J, Yong W, Deng Y, Kallenbach NR, Lu M (2004) Atomic structure of a tryptophan-zipper pentamer. Proc Natl Acad Sci 101:16156–16161
Liu J, Zheng Q, Deng Y, Kallenbach NR, Lu M (2006a) Conformational transition between four and five-stranded phenylalanine zippers determined by a local packing interaction. J Mol Biol 361:168–179
Liu J, Deng Y, Zheng Q, Cheng CS, Kallenbach NR, Lu M (2006b) A parallel coiled-coil tetramer with offset helices. Biochemistry 45:15224–15231
Liu J, Zheng Q, Deng Y, Cheng CS, Kallenbach NR, Lu M (2006c) A seven-helix coiled coil. Proc Natl Acad Sci 103:15457–15462
Liu J, Zheng Q, Deng Y, Qunnu L, Kallenbach NR, Lu M (2007) Conformational specificity of the lac repressor coiled-coil tetramerization domain. Biochemistry 46:14951–14959
Lupas A (1996) Coiled coils: new structures and new functions. Trends Biochem Sci 21:375–382
Lupas AN, Gruber M (2005) The structure of α-helical coiled coils. Adv Protein Chem 70:37–78
Lupas A, Van Dyke S, Stock J (1991) Predicting coiled coils from protein sequences. Science 252:1162–1164
Mahrenholz CC, Abfalter IG, Bodenhofer U, Volkmer R, Hochreiter S (2011) Complex networks govern coiled-coil oligomerization–predicting and profiling by means of a machine learning approach. Mol Cell Proteomics 10:M110–004994
Malashkevich VN, Kammerer RA, Efimov VP, Schulthess T, Engel J (1996) The crystal structure of a five-stranded coiled coil in COMP: a prototype ion channel? Science 274:761–765
Mason JM, Arndt KM (2004) Coiled coil domains: stability, specificity, and biological implications. Chembiochem 5:170–176
McDonnell AV, Jiang T, Keating AE, Berger B (2006) Paircoil2: improved prediction of coiled coils from sequence. Bioinformatics 22:356–358
McKay DB, Steitz T (1981) Structure of catabolite gene activator protein at 2.9 Å resolution suggests binding to left-handed B-DNA. Nature 290:744–749
McLachlan AD, Stewart M (1975) Tropomyosin coiled-coil interactions: evidence for an unstaggered structure. J Mol Biol 98:293–304
McLachlan AD, Stewart M (1976) The 14-fold periodicity in α-tropomyosin and the interaction with actin. J Mol Biol 103:271–298
Mittl PRE, Deillon C, Sargent D, Liu N, Klauser S, Thomas RM, Gutte B, Grütter MG (2000) The retro-GCN4 leucine zipper sequence forms a stable three-dimensional structure. Proc Natl Acad Sci 97:2562–2566
Mondéjar LG, Lupas A, Schultz A, Schultz JE (2012) HAMP domain-mediated signal transduction probed with a mycobacterial adenylyl cyclases as a reporter. J Biol Chem 287:1022–1031
Nilges M, Brünger AT (1991) Automated modeling of coiled coils: application to the GCN4 dimerization region. Protein Eng 4:649–659
Offer G, Hicks MR, Woolfson DN (2002) Generalized Crick equations for modeling noncanonical coiled coils. J Struct Biol 137:41–53
O’Shea EK, Klemm JD, Kim PS, Alber T (1991) X-ray structure of the GCN4 leucine zipper, a two-stranded, parallel coiled coil. Science 254:539–544
Parry DAD (1975) Analysis of the primary sequence of α-tropomyosin from rabbit skeletal muscle. J Mol Biol 98:519–535
Parry DAD (1982) Coiled-coils in α-helix-containing proteins: analysis of the residue types within the heptad repeat and the use of these data in the prediction of coiled-coils in other proteins. Biosci Rep 2:1017–1024
Parry DAD, Crewther WG, Fraser RD, MacRae TP (1977) Structure of α-keratin: structural implication of the amino acid sequences of the type I and type II chain segments. J Mol Biol 113:449–454
Parry DAD, Fraser RB, Squire JM (2008) Fifty years of coiled-coils and α-helical bundles: A close relationship between sequence and structure. J Struct Biol 163:258–269
Pauling L, Corey RB (1950) Two hydrogen-bonded spiral configurations of the polypeptide chain. J Am Chem Soc 72:534
Pauling L, Corey RB (1953) Compound helical configurations of polypeptide chains: structure of proteins of the α-keratin type. Nature 171:59–61
Pauling L, Corey RB, Branson HR (1951) The structure of proteins: two hydrogen-bonded helical configurations of the polypeptide chain. Proc Natl Acad Sci 37:205–211
Peters J, Nitsch M, Kuhlmorgen B, Golbik R, Lupas A, Kellermann J, Engelhardt H, Pfander JP, Müller S, Goldie K, Engel A, Stetter K-O, Baumeister W (1995) Tetrabrachion: a filamentous archaebacterial surface protein assembly of unusual structure and extreme stability. J Mol Biol 245:385–401
Prilusky J, Felder CE, Zeev-Ben-Mordehai T, Rydberg EH, Man O, Beckmann JS, Silman I, Sussman JL (2005) FoldIndex©: a simple tool to predict whether a given protein sequence is intrinsically unfolded. Bioinformatics 21:3435–3438
Simpson AA, Tao Y, Leiman PG, Badasso MO, He Y, Jardine PJ, Olson NH, Morais MC, Grimes S, Anderson DL, Baker TS (2000) Structure of the bacteriophage ϕ29 DNA packaging motor. Nature 408(6813):745–750
Solan A, Ratia K, Fairman R (2002) Exploring the role of alanine in the structure of the Lac repressor tetramerization domain, a ferritin-like Alacoil. J Mol Biol 317:601–612
Steinmetz MO, Stock A, Schulthess T, Landwehr R, Lustig A, Faix J, Gerisch G, Aebi U, Kammerer RA (1998) A distinct 14 residue site triggers coiled-coil formation in cortexillin I. EMBO J 17:1883–1891
Stone D, Sodek J, Johnson P, Smillie LB (1975) Tropomyosin: correlation of amino acid sequence and structure. FEBS Proc 31:125–136
Strelkov SV, Burkhard P (2002) Analysis of α-helical coiled coils with the program TWISTER reveals a structural mechanism for stutter compensation. J Struct Biol 137:54–64
Symmons MF, Bokma E, Koronakis E, Hughes C, Koronakis V (2009) The assembled structure of a complete tripartite bacterial multidrug efflux pump. Proc Natl Acad Sci 106:7173–7178
Szczepaniak K, Lach G, Bujnicki JM, Dunin-Horkawicz S (2014) Designability landscape reveals sequence features that define axial helix rotation in four-helical homo-oligomeric antiparallel coiled-coil structures. J Struct Biol 188:123–133
Szczesny P, Lupas A (2008) Domain annotation of trimeric autotransporter adhesins—daTAA. Bioinformatics 24:1251–1256
Testa OD, Moutevelis E, Woolfson DN (2009) CC+: a relational database of coiled-coil structures. Nucleic Acids Res 37:D315–D322
Thomson AR, Wood CW, Burton AJ, Bartlett GJ, Sessions RB, Brady RL, Woolfson DN (2014) Computational design of water-soluble α-helical barrels. Science 346:485–488
Trigg J, Gutwin K, Keating AE, Berger B (2011) Multicoil2: predicting coiled coils and their oligomerization states from sequence in the twilight zone. PLoS One 6:e23519
Trybus KM, Freyzon Y, Faust LZ, Sweeney HL (1997) Spare the rod, spoil the regulation: necessity for a myosin rod. Proc Natl Acad Sci 94:48–52
Vincent TL, Green PJ, Woolfson DN (2013) LOGICOIL—multi-state prediction of coiled-coil oligomeric state. Bioinformatics 29:69–76
Walshaw J, Woolfson DN (2001) SOCKET: a program for identifying and analysing coiled-coil motifs within protein structures. J Mol Biol 307:1427–1450
Walshaw J, Woolfson DN (2003) Extended knobs-into-holes packing in classical and complex coiled-coil assemblies. J Struct Biol 144:349–361
Walshaw J, Shipway JM, Woolfson DN (2001) Guidelines for the assembly of novel coiled-coil structures: α-sheets and α-cylinders. Biochem Soc Symp 68:111–123
Whitby FG, Phillips GN (2000) Crystal structure of tropomyosin at 7 Ångstroms resolution. Proteins 38:49–59
Wilson IA, Skehel JJ, Wiley DC (1981) Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 Å resolution. Nature 289:366–373
Wolf E, Kim PS, Berger B (1997) MultiCoil: a program for predicting two- and three-stranded coiled coils. Protein Sci 6:1179–1189
Wood CW, Bruning M, Ibarra AÁ, Bartlett GJ, Thomson AR, Sessions RB, Brady RL, Woolfson DN (2014) CCBuilder: an interactive web-based tool for building, designing and assessing coiled-coil protein assemblies. Bioinformatics 30:3029–3035
Woolfson DN (2005) The design of coiled-coil structures and assemblies. Adv Protein Chem 70:79–112
Woolfson DN (2017) Coiled-coil design: updated and upgraded. Fibrous proteins: structures and mechanisms. Springer, Dordrecht
Woolfson DN, Alber T (1995) Predicting oligomerization states of coiled coils. Protein Sci 4:1596–1607
Woolfson DN, Bartlett GJ, Bruning M, Thomson AR (2012) New currency for old rope: from coiled-coil assemblies to α-helical barrels. Curr Opin Struct Biol 22:432–441
Wu KC, Bryan JT, Morasso MI, Jang S-I, Lee J-H, Yang J-M, Marekov LN, Parry DAD, Steinert PM (2000) Coiled-coil trigger motifs in the 1B and 2B rod domain segments are required for the stability of keratin intermediate filaments. Mol Biol Cell 11:3539–3558
Yadav MK, Leman LJ, Price DJ, Brooks CL 3rd, Stout CD, Ghadiri MR (2006) Coiled coils at the edge of configurational heterogeneity. Structural analyses of parallel and antiparallel homotetrameric coiled coils reveal configurational sensitivity to a single solvent-exposed amino acid substitution. Biochemistry 45:4463–4473
Yang Z, Kollman JM, Pandi L, Doolittle RF (2001) Crystal structure of native chicken fibrinogen at 2.7 Å resolution. Biochemistry 40:12515–12523
Zaytsev DV, Xie F, Mukherjee M, Bludin A, Demeler B, Breece RM, Tierney DL, Ogawa MY (2010) Nanometer to millimeter scale peptide-porphyrin materials. Biomacromolecules 11:2602–2609
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Lupas, A.N., Bassler, J., Dunin-Horkawicz, S. (2017). The Structure and Topology of α-Helical Coiled Coils. In: Parry, D., Squire, J. (eds) Fibrous Proteins: Structures and Mechanisms. Subcellular Biochemistry, vol 82. Springer, Cham. https://doi.org/10.1007/978-3-319-49674-0_4
Download citation
DOI: https://doi.org/10.1007/978-3-319-49674-0_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-49672-6
Online ISBN: 978-3-319-49674-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)