
Abstract
The Domain Boundary Prediction is a crucial task for functional classification of proteins, homology-based protein structure prediction and for high-throughput structural genomics. Each amino acid is represented using a set of physico-chemical properties. Random Forest Classifier is explored for accurate prediction of domain regions by training on the curated dataset obtained from CATH database. The software is tested on proteins of CASP-6, CASP-8, CASP-9 and CASP-10 targets in order to evaluate its prediction accuracy using three fold cross validation experiments. Finally, a consensus approach is used to combine results of the classifiers obtained through the cross-validation experiments. The average recall and precision scores achieved by the developed consensus based Random Forest classifiers (PDP-RF) are 0.98 and 0.88 respectively for prediction of CASP targets. The overall accuracy and F-scores of the PDP-RF are observed as 0.87 and 0.91 respectively.
You have full access to this open access chapter, Download conference paper PDF
Similar content being viewed by others


Keywords
- Random Forest
- Random Forest Classifier
- Consensus Approach
- Position Specific Scoring Matrix
- Trained Classifier
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
A domain is a segment of a polypeptide chain that can fold into a three dimensional structure irrespective of the presence of other segments of the chain [1]. Some simple combinations of protein secondary structure elements are referred to as ‘super-secondary structure’, or ‘motifs’. Several motifs pack together to form compact, local, semi-independent units called domains. The overall 3D structure of the polypeptide chain is referred to as the protein’s tertiary structure, whereas the domain is the fundamental building block of tertiary structure. So, a domain is a structural and functional unit of protein. To predict the tertiary structure of a protein, it is useful to segment the protein by identifying domain boundaries in it. A number of methods so far have been developed to identify protein domains starting from their primary sequences which are mainly developed for prediction of multi-domains in protein chains.
Galzitskaya et al. [2] considered conformational entropy for each amino acid and searches for a global minimum on an entropy profile constructed for the whole protein chain from its amino acid sequence. Based on the difference in amino acid compositions between domain and linker regions, a method DOMCUT [3] has been developed to predict linker regions among domains. CHOPnet [4] uses evolutionary information, predicted secondary structure, solvent accessibility, amino acid flexibility and amino acid composition for predicting domains in protein chains. Armadillo [5], the another domain predictor uses any amino acid index named as Domain Linker propensity Index (DLI) to convert a protein sequence to a smoothed numeric profile, from which domains and domain boundaries may be predicted. The Position Specific Scoring Matrix (PSSM)of the target protein obtained through PSI-BLAST, has also been used for domain boundary prediction by PPRODO [6] using Artificial neural network as a classifier. A machine learning predictor DOMpro [7] uses a combination of evolutionary information (in the form of profiles), predicted secondary structures, predicted solvent accessibility of the protein chains.
In the work of Sikder and Zomaya [8], the performance of DomainDiscovery of protein domain boundary assignment is improved significantly by including inter domain linker index value along with PSSM, predicted secondary structures, solvent accessibility information. Support Vector Machine (SVM) is used to predict possible domain boundaries for target sequences. Based on the application of secondary structure element alignment (SSEA) and profile-profile alignment (PPA) in combination with InterPro pattern searches, a protein domain prediction approach, called SSEP-Domain, is proposed by Gewehr and Zimmer [9]. Cheng [10] proposed a hybrid domain prediction web service, called DOMAC, by integrating template-based and ab initio methods. The template-based method is used in DOMAC to predict domains for proteins having homologous template structures in protein Data Bank [11]. If no significant homologous template is found, DOMAC invokes the ab initio domain predictor DOMpro to predict domains. To achieve a more accurate and stable predictive performance than the existing state-of –the-art models, a new machine learning based domain predictor, viz., DomNet [12] is trained using a novel compact domain profile, predicted secondary structure, solvent accessibility information and inter-domain linker index. FIFEDom [13] is other type of multi-domain prediction where prediction is done using fuzzy mean operator. This fuzzy operator assigns a membership value for each residue as belonging to a domain boundary thus finding contiguous boundary regions. Eickholt et al. propose a new method DoBo [14] where machine learning approach with evolutionary signals is used. It first extracts putative domain boundary signals from MSA between sequence and its homologs. Then those sites are classified by SVM where sequence profiles, secondary structures or solvent accessibility are used as features. Another SVM predictor DROP [15] empowered with 25 optimal features distinguish linkers from non-linkers effectively. In the first step, a random forest algorithm was used to evaluate 3000 features. In the next step, a selection protocol was used to select optimal features. Based on a creating hinge region strategy, a new approach DomHR [16] predicts domain boundary by means of constructing profiles of domain Hinge-boundary (DHB) features. Besides these, improvement in contact prediction provides a new source of domain boundary prediction. In the work of Sadowski [17], kernel smoothing based method and methods based on building alpha carbon models onto this contact information. A recent template based method on this field is ThreaDom [18] proposed by Xue et al. in which protein domain boundary information is extracted from multiple threading alignments. The core of the method is use of domain conservation score that combines information from template domain structures and terminal and internal alignment gaps.
It appears from the above discussion that there are still some scopes for improvement in protein domain prediction. The rationale behind the choices of the feature sets and classifiers for prediction of domain boundaries are discussed in the following sections.
2 Materials and Methods
An attempt has been made under the present work to employ Random Forest Classifier as a machine learning algorithm for protein domain boundary prediction on the basis of an effective feature set consisting of hydrophobicity, linker index, polarity, ordered or disordered region of protein sequence and flexibility. Different methods [3, 6, 19] use different sliding window sizes for domain boundary prediction. Studies say that prediction within ± 20 residues from the true boundary position are considered as successful with existing evaluation criteria for boundary prediction methods. These studies motivate us to test the prediction performance of our domain predictor PDP-RF with optimal residue windows, since larger window size is useful to predict multi-domain proteins.
Features Set
Five types of features, viz., predicted ordered or disordered region, normalized flexibility parameters (B-values), polarity, linker index, modified Kyte-Doolittle hydrophobicity scale are used for this work. The last four features for the current experiment are chosen from (exactly 544 in the selected version) AAIndex database [20] release 9.0 (http://www.genome.jp/aaindex/). From experimental findings, it is known that large ordered region when they are divided by shorter parts of disordered regions in a protein chain, are likely to be separate domains [21]. For this reason, ordered or disordered region predicted by disprot tool [22] is taken as a feature. On the other hand, the presence of multiple domains in proteins gives rise to a great deal of flexibility and mobility [23]. The Debye-Waller factor (B-value) (ACC No: VINM940101) which measures average flexibility parameters is used as one of the five features. The distribution of polar and non-polar side chains is one of the most important factors governing the folding of a protein into 3D structure [24]. Latest polarity (ACC No: GRAR740102)feature is taken as a feature in this work. To represent the preference for amino acid residues in linker or regions, a parameter called the linker index is defined by Sumaya and Ohara [3]. From the AAINDEX, linker index(Acc No: BAEK050101) is taken as a feature. The more exposed the linker, the more likely it is to contain hydrophilic residues. Greater hydrophobicity is found in more linker connections between two domains. Modified Kyte-Doolittle hydrophobicity scale (Acc No: JURD980101) is taken as a feature in the current work, which is also from the AAIndex dataset.
Experimentation
In this work, we have taken Random Forest (RF) Classifier and a consensus scheme. Random Forest is a popular ensemble algorithm based on decision trees [25]. It is commonly used in bioinformatics, as it is relatively easy to apply and robust against many kinds of noisy and incomplete data characteristic for experimental biological problems [26]. In this work we trained Random Forest with 100 trees with \( \surd d \) attributes considered for each split (d – number of all attributes). The implementation we used came from scikit-learn library [27].
It is conducted in two stages. In the first stage 354 protein chains of the CATH database (version 2.5.1) are used to perform a three-fold cross validation experiment where in each experimental fold, 67 % of the positive/negative samples are used for training and the rest of the samples for testing. Each domain region residue is considered a positive sample, and non-domain residues are considered negatives. RF based classifiers are trained to generate three trained classifiers from three cross-validated experiments.
In the second stage of the experiment, we consider a consensus approach on the basis of the trained classifiers to generate test results on 19 protein sequences, taken from the CASP-6 dataset [28], 109 protein sequences from the CASP-8 dataset [29], 100 protein sequences from the CASP-9 dataset [30] and 59 protein sequences from CASP-10 dataset [31]. According to the consensus strategy, for each classifier, \( 1 - star \), \( 2 - star \) and \( 3 - star \) consensus classifiers are designed. At next step, a \( n - star \) consensus strategy (here, n = 3, as number of classifiers are 3) is applied [32] to three classifiers. Thus we obtained \( 1 - star \), \( 2 - star \) and \( 3 - star \) classifiers. As a result, 3 consensus classifiers are also designed to achieve improved performance. Here we define a 3-star quality consensus scheme as \( C_{\text{n}}^{\text{N}} \), where N is the number of classifiers of a particular type participating in the specific consensus strategy, and n (\( 1 \le n \le N \)) is the quality of prediction [32]. More specifically, \( 1 - star \) prediction says that any one of possible N classifiers predicts the test sequence to be positive for the domain region under consideration, and \( N - star \) represents that all classifiers agreed to the decision. Along this principle, we define the \( 3 - star \) consensus over 3-variations of training on three fold cross-validation data of a special type classifier. Subsequently, \( C_{\text{n }}^{3} \) is defined as the consensus among three classifiers. Question arises as to how \( n - star \) consensus relates to Random Forest, which is already an ensemble algorithm. In Random Forest decision is made through weighted voting. Our consensus approach is equivalent to standard (equal weights) voting with a variable threshold. This allows choosing a tradeoff between precision and recall of the ensemble.
3 Results and Discussion
The current experiment is conducted in two stages. In the first stage 354 protein chains of the CATH database (version 2.5.1) are used to perform a three-fold cross validation experiment where in each experimental fold, 67 % of the positive/negative samples are used for training and the rest of the samples for testing. RF based classifiers are trained to generate three trained classifiers from three cross-validated experiments. In the second stage of the experiment, we consider a consensus approach on the basis of the trained classifiers to generate test results on 19 protein sequences, taken from the CASP-6 dataset [28], 109 protein sequences from the CASP-8 dataset [29],100 protein sequences from the CASP-9 dataset [30] and 59 protein sequences from CASP-10 dataset [31]. According to the consensus strategy, for three classifiers of each classifier, \( 1 - star \), \( 2 - star \) and \( 3 - star \) consensus classifiers, namely, PDP-RF-1,PDP-RF-2 and PDP-RF-3 are designed. In case of sequence based prediction, the length of sequence fragment whose central amino acid is being predicted as domain or linker region is very crucial. Different methods use different sliding window sizes for domain boundary prediction. Studies say that prediction within ± 20 residues from the true boundary prediction are considered as successful with existing evaluation criteria for domain boundary prediction methods. To determine the length of the sequence fragment or window, prediction results are observed for classifiers only on a single fold among three cross validated datasets. Among 13, 15, 17, 19, 21, 25 and 29 window sizes, performance of classifier at 17 window size is the best. So, this window size is made fixed for this work.
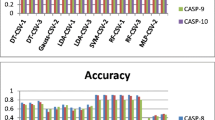
From three cross validated experiments, three classifiers are designed and their performance is observed. Outstanding performance is observed in Random Forest Classifiers in prediction of CASP-6, CASP-8, CASP-9 and CASP-10 targets. For CASP-6, CASP-8 and CASP-9 targets its behavior is found to be consistent whereas prediction results are somewhat less in CASP-10 targets. Table 1 shows the average performance of 3 classifiers.
As three classifiers are taken, so, \( 1 - star \), \( 2 - star \), \( 3 - star \) consensus strategy may be adopted as already defined in the previous section. The performance of consensus classifier must demand the good predictive accuracy in comparison to single classifier. From Table 2, it can be observed that with the introduction of consensus classifier, the performance of each type classifier is increased in a large scale.
As performance of single RF classifier is found to be the best whereas consensus classifier uplifts its accuracy up to its highest limit. Table 2 shows overall performance of consensus classifiers of RF. In case of RF classifiers, performance of \( 1 - star \), \( 2 - star \) and \( 3 - star \) consensus schemes are found to be the same which indicate the prediction decisions among three classifiers at higher confidence. In Table 3, it is seen that consensus classifier improves the accuracy of single classifier a little.
As mentioned earlier, we have taken domain as positive class and linker as negative class. Since the proportion of domain and linker in our dataset is not equal i.e., domain residue represents majority class and non-domain or linker residue represents minority class, the prediction results may turn out to be biased towards majority class. For this reason, we reverse the role of domain and linker residue by taking linker residue as positive and domain residue as negative class. The overall performance of PDP-RF is found to be the somewhat less compared to former performance (Accuracy in prediction of CASP targets using majority class training is 0.88 whereas is 0.85 using minority class training).
A Robust Consensus Classifier
In this work, an attempt has been done to choose random Forest, as effective machine learning classifier, to exploit strong multi facet feature sets and by applying a novel consensus approach. Thus objective is to design a strong robust classifier which enables the system to predict targets very efficiently and effectively. In prediction of CASP targets, in most of the cases, RF classifier offers the best predictive ability. Inclusion of the novel \( 3 - {\text{star}} \) consensus approaches further improves the classifiers’ performances.
We have taken PPRODO [6], DomPro [7], DROP [15], FIFEDom [13], ThreaDom [18] as existing methods for comparison because most of the methods are freely available. PPRODO, DomPro are not recent but they are based on machine learning method. DROP is recent machine learning method as well. On the other hand, Threadom is recent template based method which predicts multi domain proteins of CASP targets very well. Overall, the successful performance of most of the classifiers in CASP competition is found. Performances of PDP-RF classifiers are analyzed with ThreaDom1, ThreaDom2 [18], FIFEDom [13], Pfam [33], DROP [15], DOMPro [7], PPRODO [6], DoBo [14] in prediction of CASP-9 targets and CASP-10 targets. Finding the appropriate robust machine learning classifier, use of significant feature set, selection of optimal window and finally incorporation of consensus approach into three classifiers of each type of classifier is a very challenging task in prediction of domain boundaries along protein sequences. Learning patterns is a very challenging issue for any classifier in case of binary classification where proportion of positive and negative samples is not equal. Moreover, a novel \( 3 - star \) consensus approach is applied to further improve the prediction accuracy. We finally conclude that the designed feature set; alongside with Random Forest based classifier based consensus approach effectively predicts the domain regions in multi-domain protein chains. The cross-validated experimental setup with standard CATH database establishes our claims. Prediction decisions from the three experimental folds are combined to design \( n - star \) quality consensus strategies. Here, \( 3 - star \) quality consensus is designed by combining the decisions of the three classifiers from each of the three sets of cross validation experiments. The consensus strategy is found to be superior in comparison with the performances of the best single classifier.
Prediction is done on residue level i.e. whether a residue belongs to domain or linker region but not on domain boundary based. Domain prediction methods vary in the procedure, i.e. either they are template based (e.g., Threadom or FIFEDOM) or ab initio based (e.g. DomPro, DROP etc.). Some Predictors predicts domain boundary (DOMPRo, Threadom) and some of them predicts linkers. The goal of the current state of the art and our proposed method is more or less same but difference lies in the domain boundary definition (e.g. DomPro considers the residues in the range of 20 residues around the center of domain region the domain boundary residues from the CATH assignment). In this work, we take domain regions from CATH by considering domain number starting/end positions of each domain sequentially. As a result, our dataset contains domain residue serving as majority class. So, it cannot be compared with current state of the art in terms of performance metrics. Here, recall scores of PDP-RFs on CASP-9 and CASP-10 targets are 0.98 and 0.97 whereas precision scores of PDP-RFs on the same are 0.89 and 0.79. Template based method Threadom2, Threadom1, FIFEDOM predicts CASP-9 targets at 0.534, 0.397, 0.233 recall scores and 0.764, 0.636, 0.34 precision scores. PFAM, DROP (linker based), and DomPro, PPRODO (ab initio) predict CASP9-targets at recall of 0.548, 0.26, 0.219, 0.397 and precision of 0.5,0. 679, 0.727 and 0.56.
In prediction of CASP-10 targets, Threadom2 and Threadom1 predict targets well (recall score: 0.625, 0.625 and precision score: 0.796, 0.732). But FIFEDOM predict targets at low recall and precision score (0.188, 0.28). On the other hand, PFAM, DROP (linker based), and DomPro, PPRODO (ab initio) predict CASP10-targets at recall of 0.547, 0.156, 0.109 and 0.406 and precision of 0.466, 0.714, 0.44 and 0.591which is better than that of CASP-9 targets. Recall and precision score of PDP-RF are reported but not compared with these methods as it is not fair to compare a residue based prediction scheme with domain boundary based or linker based prediction method or with template based method where there lies a difference in evaluation criteria.
Methods for building feature importance rankings based on Random Forest can also be used to gain more insights into amino acid properties correlated with domain boundaries. To support validity of our method we also plan to include comparison with other machine learning algorithms in our next work.
References
Mount, D.: Bioinformatics: Sequence and Genome Analysis, p. 416. Cold Spring Harbor Laboratory Press, New York (2004)
Melnik, B.S., Galzitskaya, O.V.: Prediction of protein domain boundaries from sequence alone. Protein Sci. 12, 696–701 (2003)
Suyama, M., Ohara, O.: Domcut: prediction of inter-domain linker regions in amino acid sequences. Bioinformatics 19, 673–674 (2003)
Liu, J., Rost, B.: Sequence-based prediction of protein domains. Nucleic Acids Res. 32, 3522–3530 (2004)
Dumontier, M., Yao, R., Feldman, H.J., Hoque, C.W.: Armadillo: domain boundary prediction by amino acid composition. J. Mol. Biol. 350, 1061–1073 (2005)
Sim, J., Kim, S.Y., Lee, J.: PPRODO: prediction of protein domain boundaries using neural networks. Proteins. 59, 627–632 (2005)
Cheng, J., Sweredoski, M.J., Baldi, P.: DOMpro: Protein domain prediction using profiles, secondary structure, relative solvent accessibility, and recursive neural networks. Data Min. Knowl. Discov. 13, 1–10 (2006)
Sikder, A.R., Zomaya, A.Y.: Improving the performance of domaindiscovery of protein domain boundary assignment using inter-domain linker index. BMC Bioinformatics. 7(Suppl 5), S6 (2006)
Gewehr, J.E., Zimmer, R.: SSEP-Domain: Protein domain prediction by alignment of secondary structure elements and profiles. Bioinformatics 22, 181–187 (2006)
Cheng, J.: DOMAC: An accurate, hybrid protein domain prediction server. Nucleic Acids Res. 35, W354–W356 (2007)
Berman, H.M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T.N., Weissig, H., Shindyalov, I.N., Bourne, P.E.: The Protein Data Bank. Nucleic Acids Res. 28, 235–242 (2000)
Yoo, P.D., Sikder, A.R., Taheri, J., Zhou, B.B., Zomaya, A.Y.: DomNet: protein domain boundary prediction using enhanced general regression network and new profiles. NanoBioSci. IEEE Trans. 7, 172–181 (2008)
Bondugula, R., Lee, M.S., Wallqvist, A.: FIFEDom: a transparent domain boundary recognition system using a fuzzy mean operator. Nucleic Acids Res. 37, 452–462 (2009)
Eickholt, J., Deng, X., Cheng, J.: DoBo: protein domain boundary prediction by integrating evolutionary signals and machine learning. BMC Bioinformatics 12, 43 (2011)
Ebina, T., Toh, H., Kuroda, Y.: DROP: an SVM domain linker predictor trained with optimal features selected by random forest. Bioinformatics 27, 487–494 (2011)
Zhang, X.Y., Lu, L.J., Song, Q., Yang, Q.Q., Li, D.P., Sun, J.M., Li, T.H., Cong, P.S.: DomHR: accurately identifying domain boundaries in proteins using a hinge region strategy. PLoS One 8, e60559 (2013)
Sadowski, M.I.: Prediction of protein domain boundaries from inverse covariances. Proteins 81, 253–260 (2013)
Xue, Z., Xu, D., Wang, Y., Zhang, Y.: ThreaDom : extracting protein domain boundary information from multiple threading alignments. Bioinformatics 29, 247–256 (2013)
Galzitskaya, O.V., Dovidchenko, N.V., Lobanov, M., Garbuzinskii, S.A.: Prediction of protein domain boundaries from statistics of appearance of amino acid residues. Mol. Biol (Mosk). 40, 96–107 (2006)
Kawashima, S., Ogata, H., Kanehisa, M.: AAindex: amino acid index database. Nucleic Acids Res. 27, 368–369 (1999)
Wyrwicz, L.S., Koczyk, G., Rychlewski, L., Plewczynski, D.: ProteinSplit: splitting of multi-domain proteins using prediction of ordered and disordered regions in protein sequences for virtual structural genomics. J. Phys. Condens. Matter 19, 285222 (2007)
Sickmeier, M., Hamilton, J.A., LeGall, T., Vacic, V., Cortese, M.S., Tantos, A., Szabo, B., P, Tompa, Chen, J., Uversky, V.N., Obradovic, Z., Dunker, A.K.: DisProt: The database of disordered proteins. Nucleic Acids Res. 35, D786–93 (2007)
Bu, Z., Callaway, D.J.: Proteins move! protein dynamics and long range allostery in cell signaling. Adv. Protein Chem. Struct. Biol. 83, 163–221 (2011)
Cordes, M.H., Davidson, A.R., Sauer, R.T.: Sequence space, folding and protein design. Curr. Opin. Struct. Biol. 6, 3–10 (1996)
Breiman, L.: Random forests. Mach. Learn. 45, 5–32 (2001)
Yang, P., Yang, Y.H., Zhou, B.B., Zomaya, A.Y.: A review of ensemble methods in bioinformatics. Curr. Bioinform. 5, 296–308 (2010)
Pedregosa, F., Varoquaux, G., Gramfort, A., Michel, V., Thirion, B., Grisel, O., Blondel, M., Prettenhofer, P., Weiss, R., Dubourg, V., Vanderplas, J., Passos, A., Cournapeau, D., Brucher, M., Perrot, M., Duchesnay, E.: Scikit-learn: machine learning in python. J. Mach. Learn. Res. 12, 2825–2830 (2011)
Moult, J., Fidelis, K., Rost, B., Hubbard, T., Tramontano, A.: Critical assessment of methods of protein structure prediction (CASP)–round 6. Proteins. 61(Suppl 7), 3–7 (2005)
Moult, J., Fidelis, K., Kryshtafovych, A., Rost, B., Tramontano, A.: Critical assessment of methods of protein structure prediction (CASP)-round VIII. Proteins 77, 1–4 (2009)
Moult, J., Fidelis, K., Kryshtafovych, A.: Critical assessment of methods of protein structure prediction (CASP)–round IX. Proteins. 79(Suppl 10), 1–5 (2011)
Moult, J., Fidelis, K., Kryshtafovych, A., Schwede, T., Tramontano, A.: Critical assessment of methods of protein structure prediction (CASP)-round X. Proteins. 82(Suppl 1), 1–6 (2014)
Plewczynski, D., Basu, S., Saha, I.: AMS 4.0: consensus prediction of post-translational modifications in protein sequences. Amino Acids 43, 573–582 (2012)
Finn, R.D., Bateman, A., Clements, J., Coggill, P., Eberhardt, R.Y., Eddy, S.R., Heger, A., Hetherington, K., Holm, L., Mistry, J., Sonnhammer, E.L., Tate, J., Punta, M.: Pfam: the protein families database. Nucleic Acids Res. 42, D222–D230 (2014)
Acknowledgments
The paper is co-funded by the European Union from financial resources of the European Social Fund. Project PO KL “Information technologies: Research and their interdisciplinary applications”. This work was partially supported by the Polish National Science Centre (Grant number 2014/15/B/ST6/05082 and UMO-2013/09/B/NZ2/00121), and COST BM1405 EU action. Authors are thankful to the “Center for Microprocessor Application for Training Education and Research” for providing infrastructure facility during progress of the work. It is also co-funded by UPE-II, PURSE project, Govt. Of India at Department of Computer Science & Engineering, Jadavpur University.
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this paper
Cite this paper
Chatterjee, P., Basu, S., Zubek, J., Kundu, M., Nasipuri, M., Plewczynski, D. (2015). PDP-RF: Protein Domain Boundary Prediction Using Random Forest Classifier. In: Kryszkiewicz, M., Bandyopadhyay, S., Rybinski, H., Pal, S. (eds) Pattern Recognition and Machine Intelligence. PReMI 2015. Lecture Notes in Computer Science(), vol 9124. Springer, Cham. https://doi.org/10.1007/978-3-319-19941-2_42
Download citation
DOI: https://doi.org/10.1007/978-3-319-19941-2_42
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-19940-5
Online ISBN: 978-3-319-19941-2
eBook Packages: Computer ScienceComputer Science (R0)
