
Summary
Protein microarrays containing nearly the entire yeast proteome have been constructed. They are typically prepared by overexpression and high-throughput purification and printing onto microscope slides. The arrays can be used to screen nearly the entire proteome in an unbiased fashion and have enormous utility for a variety of applications. These include protein–protein interactions, identification of novel lipid- and nucleic acid-binding proteins, and finding targets of small molecules, protein kinases, and other modification enzymes. Protein microarrays are thus powerful tools for individual studies as well as systematic characterization of proteins and their biochemical activities and regulation.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others



Key words
12.1 Introduction
Protein microarrays contain a large number of proteins that have been spotted in an addressable format and at high density onto microscope slides, thereby allowing the individual analysis of large numbers of proteins simultaneously ( 1, 2 ). There are two types of protein microarrays: antibody microarrays that are used for protein profiling, and functional protein microarrays that contain sets of proteins or, for the case of yeast, nearly an entire yeast proteome. The functional arrays are used for a wide variety of applications and are presented here.
Protein arrays are typically prepared from large overexpression libraries in which proteins are overexpressed, purified in a 96-well format, and then spotted at high density onto microscope slides. The arrays are then probed using molecules containing fluorescent or other probes. To date, protein microarrays have been prepared for yeast (6,000), Arabidopsis (5,000), humans (8,000), and coronaviruses, as well as a number of bacteria ( 3–7 ).
Thus far, protein microarrays have been used for a wide variety of applications. These include enzymatic assays and interactions with proteins, lipids, small molecules, and nucleic acids ( 3, 8 ). One advantage of protein microarrays is that an entire proteome can be screened in an unbiased fashion, thereby leading to the discovery of novel activities that were not anticipated. For example, screening of a yeast protein chip using biotinylated liposomes revealed many new lipid-binding proteins including enzymes involved in glycolysis ( 3 ), and screening of a yeast proteome chip with yeast DNA revealed a novel metabolic enzyme, Arg5,6, that is associated with DNA and appears to regulate mitochondrial gene expression ( 9 ).
Protein microarrays have been used for many other applications as well. Recently, arrays that contain most yeast transcription factors have been prepared and probed using motifs that are conserved across yeast species but whose binding protein was not known ( 10 ). This study assigned candidate binding proteins for specific sequences, one of which was confirmed. Protein microarrays have also been used to assess antibody specificity ( 11 ) and for identification of autoreactive antibody in patients with diseases such as autoimmune diseases and cancer and thereby find candidate biomarkers and insights into the disease state ( 5 ). Lastly, protein microarrays can be used to identify substrates of modification enzymes such as kinases and ubiquitation enzymes ( 12, 13 ).
12.2 Materials
12.2.1 Generation of C-Terminally Tagged Yeast Strains
-
1.
pDONR221 vector containing the open reading frame (ORF) of interest.
-
2.
pBG1805 vector: c-terminal His6X-3C-Protein A “zz” domain or pYES-DEST52 for His6X-V5 fusion proteins.
-
3.
Flanking sequences for integration into Gateway pDONR221 plasmid
-
(a)
att B4: 5′ GGGGCAACTTTGTATAGAAAAGTTG 3′ (added to the 5′ PCR primer).
-
(b)
att B1: 5′ GGGGCTGCTTTTTTGTACAAACTTG 3′ (added to the 3′ PCR primer).
-
(a)
-
4.
Sequencing primers for pBG1805 plasmid
-
(a)
F5: 5′ CATTTTCGGTTTGTATTACTTCTTATTC 3′.
-
(b)
R3: 5′ GGACCTTGAAAAAGAACTTC 3′.
-
(a)
-
5.
S. cerevisiae genomic DNA (strain BY4700, MATa ura3Δ0).
-
6.
S. cerevisiae Y258: MATa, pep4, his4-58, ura3-52, leu 2-3, 112.
-
7.
Pfx polymerase (Invitrogen) or Pfu Ultra polymerase (Stratagene).
-
8.
BsrG1 endonuclease for screening positive clones.
-
9.
Culture tubes.
-
10.
Agarose.
-
11.
LB plus Kanamycin, both liquid, and 2% bacto-agar.
-
(a)
10 g tryptone.
-
(b)
5 g yeast extract.
-
(c)
5 g NaCl.
-
(d)
20 g agar (for plates).
-
(e)
Make up to 1 L with distilled water (dH2O).
-
(f)
Autoclave and allow to cool prior to addition of 50 μg/mL of ampicillin final concentration.
-
(a)
-
12.
DNA ladder
-
13.
E. coli: DH5α
-
14.
TAE
-
(a)
242 g Tris base.
-
(b)
57.1 mL 100% glacial acetic acid.
-
(c)
100 mL 0.5 M ethylenediaminetetraacetic acid (EDTA) (pH 8.0).
-
(d)
Make up with dH2O up to 1 L.
-
(a)
-
15.
LR clonase (Invitrogen).
-
16.
BP clonase (Invitrogen).
12.2.2 Transformation Reagents
-
1.
YPAD (Yeast extract-peptone-adenine-dextrose)
-
(a)
10 g bacto yeast extract.
-
(b)
20 g bacto peptone.
-
(c)
50 mg adenine.
-
(d)
20 g dextrose.
-
(e)
20 g agar (for plates).
-
(f)
Make up with dH2O up to 1 L.
-
(a)
-
2.
1 M lithium acetate (LiAc).
-
3.
50% polyethylene glycol (PEG).
-
4.
10 mg/mL ssDNA.
-
5.
DNA.
-
6.
dH2O.
-
7.
Water bath or heat block.
-
8.
96-well box.
-
9.
96-well polymerase chain reaction (PCR) plate.
-
10.
Synthetic complete minus uracil media (Sc-ura)
-
(a)
1.5 g yeast nitrogen base.
-
(b)
5 g (NH4)2SO4.
-
(c)
2 g Sc-ura drop out mix (commercially available).
-
(d)
20 g raffinose (or dextrose for starter culture media and SD-ura plates).
-
(e)
20 g agar (for plates).
-
(a)
12.2.3 High-Throughput Immunoblot Analysis
-
1.
3× YEP–GAL (yeast extract–peptone–galactose)
-
(a)
30 g yeast extract.
-
(b)
60 g peptone.
-
(c)
Make up to 700 mL with dH2O.
-
(d)
Add 300 mL of sterile filtered 20% galactose to media after autoclaving.
-
(a)
-
2.
0.5 mm zirconia beads or acid-washed glass beads.
-
3.
Lysis buffer 150
-
(a)
50 mM Tris–HCL at pH 7.5.
-
(b)
150 mM NaCl.
-
(c)
1 mM ethyleneglycoltetraacetic acid (EGTA).
-
(d)
10% glycerol.
-
(e)
0.1% Triton X-100.
-
(f)
0.5 mM dithiothreitol (DTT).
-
(g)
1 mM phenylmethylsulfonyl fluoride (PMSF).
-
(h)
1× complete protease inhibitor tablet (Roche).
-
(a)
-
4.
Paint shaker (5G-HD, Harbil) or similar mechanism to break cells.
-
5.
SDS-polyacrylamide gel electrophoresis (SDS-PAGE) gels.
-
6.
Polyvinylidene fluoride (PVDF) or nitrocellulose membranes.
-
7.
Multichannel pipette.
-
8.
Semi-dry transfer apparatus.
-
9.
HRP-IgG.
-
10.
HA antibody (16B12, Covance).
-
11.
Imaging Film (Kodak BioMax MR Film).
12.2.4 Preparation of Proteins for Protein Microarray
-
1.
Wash buffer (150)
-
(a)
50 mM Tris-HCl (pH 7.5).
-
(b)
150 mM NaCl.
-
(c)
10% glycerol.
-
(d)
0.1% and Triton X-100.
-
(a)
-
2.
Elution buffer
-
(a)
50 mM Tris (pH 7.5).
-
(b)
150 mM NaCl.
-
(c)
25% glycerol.
-
(d)
0.1% Triton X-100.
-
(a)
-
3.
96-well box.
-
4.
PVDF 96-well filter plate (1.2-μm pore size).
-
5.
IgG-Fast Flow 6 Sepharose.
-
6.
3C protease.
-
7.
384-well microplate (to array proteins).
-
8.
Adhesive foil lids for sealing microplates.
12.2.5 Printing Arrays
-
1.
Arrays suitable for assay being performed.
-
2.
Bio-Rad ChipWriter Pro or comparable printer.
12.2.6 Purifying V5-Fusion Protein Probe
-
1.
Phosphate buffered saline (PBS) lysis solution
-
(a)
PBS (0.01 M phosphate buffer, 0.0027 M potassium chloride, 0.137 M sodium chloride; pH 7.4).
-
(b)
0.1% Triton X-100.
-
(c)
0.5 mM DTT.
-
(d)
2 mM MgCl2..
-
(e)
500 mM NaCl.
-
(f)
50 mM imidazole (pH 7.4).
-
(g)
1 mM PMSF.
-
(h)
Complete protease inhibitor cocktail – EDTA-free (Roche).
-
(i)
Phosphatase inhibitor Cocktail 1 (Sigma).
-
(a)
-
2.
Ni2+, or Co2+ affinity resin.
-
3.
G25 columns.
-
4.
Microcentrifuge.
-
5.
Wash buffer
-
(a)
PBS (0.01 M phosphate buffer, 0.0027 M potassium chloride, 0.137 M sodium chloride; pH 7.4).
-
(b)
0.1% Triton X-100.
-
(c)
500 mM NaCl.
-
(d)
0.5 mM DTT.
-
(e)
50 mM imidazole (pH 7.4).
-
(f)
2 mM MgCl2.
-
6.
Elution buffer
-
(a)
PBS (0.01 M phosphate buffer, 0.0027 M potassium chloride, 0.137 M sodium chloride; pH = 7.4).
-
(b)
0.1% Triton X-100.
-
(c)
500 mM NaCl.
-
(d)
0.5 mM DTT.
-
(e)
500 mM imidazole (pH 7.4).
-
(f)
2 mM MgCl2..
-
(g)
20% glycerol (if freezing at −80°).
-
(a)
-
(a)
12.2.7 Probing Arrays
-
1.
Blocking buffer
-
(a)
PBS (0.01 M phosphate buffer, 0.0027 M potassium chloride, 0.137 M sodium chloride; pH 7.4).
-
(b)
1% bovine serum albumin (BSA).
-
(c)
0.1% Tween-20.
-
(a)
-
2.
Probe buffer
-
(a)
PBS (0.01 M phosphate buffer, 0.0027 M potassium chloride, 0.137 M sodium chloride; pH 7.4).
-
(b)
2 mM MgCl2..
-
(c)
0.5 mM DTT.
-
(d)
0.05% Triton X-100.
-
(e)
50 mM NaCl.
-
(f)
500 µM ATP (for kinases).
-
(g)
1% BSA.
-
(a)
12.2.8 Identifying Kinase–Substrate Interactions
-
1.
Lysis Buffer
-
(a)
100 mM Tris–HCl pH 7.4.
-
(b)
100 mM NaCl, 1 mM EGTA.
-
(c)
0.1% 2-mercaptoethanol.
-
(d)
0.1% Triton X-100, protease cocktail (Roche).
-
(e)
1 mM EDTA.
-
(f)
50 mM NaF.
-
(g)
10 mM sodium glycerophosphate.
-
(h)
1 mM Na3VO4.
-
(a)
-
2.
Kinase Buffer
-
(a)
100 mM Tris–HCl pH 8.0.
-
(b)
100 mM NaCl.
-
(c)
10 mM MgCl2..
-
(d)
20 mM glutathione.
-
(e)
20% glycerol.
-
(a)
-
3.
Superblock (Pierce)
-
4.
[33P]ATP
12.3 Methods
12.3.1 High-Throughput Preparation of Clones
Protein microarrays are typically constructed using overexpression libraries. To date, two comprehensive expression libraries have been prepared that are suitable for production of yeast protein microarrays. Initially, a library was prepared in which 5,800 yeast ORFs were fused at their amino terminal coding sequences to glutathione-S-transferase and HisX6 coding sequences ( 3 ) . Subsequently, a movable ORF, or MORF, library was prepared in which 5,700 yeast ORFs were fused at their carboxy terminal coding sequences to His6 and IgG binding domain of protein A ( 14 ). The MORF library contains GATEWAY-compatible sequences flanking the inserts, so the yeast ORFs can be shuttled into any vector. In each library the fusion protein is expressed from the GAL1 galactose promoter. A protocol for library construction is as follows:
12.3.1.1 Constructing a Large Number of Clones for Protein Arrays
-
1.
Design forward primers containing the att B4 sequence and reverse primers with the att B1 sequence (see subheading 2.1 and Note 4 ) for the gene of interest.
-
2.
Amplify the gene of interest by PCR using the previously mentioned genomic DNA and polymerase (see subheading 2.1 ).
-
3.
Perform BP clonase reaction (5 µL) using 30–300 ng of PCR product added to 150 ng of pDONR221 plasmid and following manufacturer’s instructions.
-
4.
Incubate the reaction overnight at 25°C.
-
5.
Combine half of the of the BP reaction (3 µL) with 150 ng of pBG1805 DNA or pYES-DEST52 (for V5 fusion) backbone, 0.6 µL of LR clonase, and 0.6 µL of LR clonase 10× buffer.
-
6.
Incubate the reaction overnight at 25°C and use half of the reaction to transform DH5α cells.
-
7.
Plate the cells on LB/ampicillin (see Subheading 2.1 , item 11) plates overnight at 37°C.
-
8.
Miniprep overnight cultures of several colonies of each transformant grown in LB/ampicillin liquid media.
-
9.
Digest with 5 μL of DNA with BsrG1 overnight at 37°C to confirm the presence of insert and sequence clones that contain the insert using sequencing primers (see Subheading 2.1 , item 4).
12.3.1.2 Transforming Yeast with C-Terminal Tagged ORF
-
1.
Inoculate 20 mL YPAD (see subheading 2.2 , item 1) with a starter culture of a fresh Y258 colony from YPAD agarose plate.
-
2.
Inoculate 50 mL of YPAD/10 transformations with enough starter culture to attain an OD600 of 0.1.
-
3.
Grow for several doubling times to an OD600 of ∼0.6–0.8.
-
4.
Transfer the cells to a 50-mL conical tube and spin in a tabletop centrifuge at 3,000 rpm/5 min/4°C.
-
5.
Bring up to a volume of 50 mL with water to wash, and repeat centrifugation.
-
6.
Decant water and add 0.5 mL 100 mM LiAc and transfer to a 1.5-mL snap-cap tube.
-
7.
Spin at 13,000 rpm in a microfuge for 30 s and remove the solution with a pipette.
-
8.
Add enough 100 mM LiAc to bring the volume up to 0.5 mL, and vortex to suspend the cells.
-
9.
Aliquot 50 μL of the suspension into separate snap-cap tubes and centrifuge.
-
10.
Remove liquid as before and prepare pellet for transformation.
-
11.
Add 240 µL of 50% PEG solution, 36 μL of 1 M LiAC, 10 μL of 10 mg/mL ssDNA, 2 μL of miniprepped DNA fusion construct, and 72 μL of dH20.
-
12.
Vortex the mixture and incubate at 30°C for 30 min.
-
13.
Next, move the tubes to a water bath set to 42°C for 30 min.
-
14.
Finally, spin the tubes at 10,000 ´ g for 15 s, and aspirate the solution with a pipette.
-
15.
Add 500 mL of dH2O to each tube and vortex briefly.
-
16.
Pipette 50–100 μL of suspension onto Sc-ura/2% dextrose agar Petri dishes (see subheading 2.2 , item 10).
-
17.
Allow 48 h to recover and screen the transformants by immunoblot.
12.3.1.3 High-Throughput Assaying and Preparation of Protein Methods for Assay Protein Expression and High-Throughput Preparation of Proteins
12.3.1.3.1 High-Throughput Immunoblot Analysis
-
1.
Inoculate 96-well starter cultures in 0.8 mL of SD-ura/2% dextrose.
-
2.
On day 2, wash the cells with Sc-ura/2% raffinose (see subheading 2.2 , item 10), and inoculate 5 μL of starter culture into a new 96-well box (2 mL/well) containing 0.8 mL of Sc-ura/2% raffinose with a 3.5-mm glass ball (PGC scientific) in each well for mixing and aeration.
-
3.
Mix cells for 15 h at 30°C on a platform shaker and then induce by addition of 0.4 mL of 3× YEP-GAL (see subheading 2.3 , item 1) for 6 h, and follow by centrifugation and washing with ice cold water and storage at −80°C.
-
4.
Obtain the lysates by adding 200 μL of lysis buffer 150 (see subheading 2.3 , item 2) and shaking for 6 min in a paint shaker (5G-HD, Harbil) at 4°C with 250 μL of acid-washed glass beads (0.5 mm, Sigma), followed by centrifugation at 1,000 ´ g for 5 min.
-
5.
Combine the crude lysates with 5× SDS loading buffer in a 96-well PCR plate, heat for 5 min at 95°C, and centrifuge again at 2,500 rpm 5 min.
-
6.
Remove 12 μL from each well with a multichannel pipette and load onto SDS-PAGE gels.
-
7.
Western transfer to either PVDF or nitrocellulose membranes using standard wet or dry transfer techniques.
-
8.
Block the membrane with TBS/0.1% Tween–1% milk powder for 1 h.
-
9.
Probe with anti-HA antibodies (16B12, 1:1,000, Covance) overnight in TBS/0.1% Tween–1% milk powder.
-
10.
On day 2, wash the membrane three times with TBS/0.1% Tween, 10 min each wash.
-
11.
Probe for 1 h at room temperature with HRP-conjugated sheep anti-mouse IgG antibody (Amersham).
-
12.
Wash the membrane three times with TBS/0.1% Tween, 10 min each wash, and develop with Supersignal west chemiluminescent substrate (Pierce).
12.3.1.3.2 Purifying Proteins for Arrays
-
1.
Inoculate 96-well starter cultures in 0.8 mL of Sc-ura medium.
-
2.
On day 2, wash the cells with Sc-ura/2% raffinose, and inoculate 5 μL of starter culture into a new 96-well box (2 mL/well) containing 0.8 mL of Sc-ura/2% raffinose with a 3.5-mm glass ball (PGC scientific) in each well for mixing and aeration.
-
3.
Mix cells for 15 h at 30°C on a platform shaker and then induce by addition of 0.4 mL of 3× YEP-GAL (see Subheading 2.3 , item 1) for 6 h followed by centrifugation, washing with ice cold water, and storage at −80°C.
-
4.
Obtain the lysates by adding 200 μL of lysis buffer 150 (see Subheading 2.3 , item 2) and shaking for 6 min in a paint shaker (5G-HD, Harbil) at 4°C with 250 μL of acid-washed glass beads (0.5 mm, Sigma), followed by centrifugation at 2,500 rpm for 5 min.
-
5.
Transfer the cleared lysate to a new 96-well box along with IgG Sepharose resin (6 Fast Flow; GE Biosciences) and incubate at 4°C for several hours with agitation.
-
6.
Afterwards, wash the beads four times with wash buffer 150 (see Subheading 2.4 , item 1)
-
7.
Transfer the beads to a PVDF filter plate (1.2 µm pore size) and spin-through the excess wash. Suspend the resin is in 40 µL of elution buffer containing1 µL of 3C protease for 18 h at 4°C with agitation.
-
8.
The next day, add preconditioned glutathione beads to the reaction to remove the 3C protease and spin the eluted protein through the PVDF membrane into a clean 96-well freezer plate.
-
9.
Array the proteins into 5 μL aliquots on multiple 384-well printing plates and cover with an adhesive aluminum seal for storage at −80°C until printing.
-
10.
Include a control plate with assay-specific controls (see Note 2 ).
12.3.1.4 Printing Protein Microarrays
Once proteins are prepared they must be printed onto surfaces, typically microscope slides. A variety of different surfaces exist for preparing protein microarrays. They include nitrocellulose and aldehyde surface chemistries for chemical attachment through lysines and affinity attachment methods such as nickel-chelated slides for attaching His tagged proteins and glutathione for affinity attachment ( 15, 3, 16 ). The different surfaces have advantages and disadvantages; nitrocellulose allows the attachment of large amounts of protein, and it along with chemical attachment methods results in random orientation of proteins away from the surface. It is likely that the activity of the attached molecule is lost for many molecules attached by these methods. Moreover, binding of probes to regions near the surface is likely to be sterically inhibited. Affinity attachment methods are more likely to retain activity and orient proteins away from the surface, although random presentation of protein surfaces from the slide is not attained. Different surfaces yield different backgrounds and it is prudent to test several surfaces whenever a new assay is employed.
-
1.
Configure the contact arrayer and align the microscope slides (FAST, Path, or other types of slides see Note 1 ) to allow the maximum number of slides to be printed per run. This will vary depending on the arrayer used, but for the Bio-Rad ChipWriter Pro it is approximately 90 arrays/run.
-
2.
Maintain a constant humidity of ∼40% in the printing chamber, and a constant temperature of 4°C (this is achieved by placing the printer in a temperature-controlled cold room).
-
3.
Remove two plates at a time from the −80°C storage, and allow them to thaw on ice for 5–10 min followed by centrifugation at 1,000 × g at 4°C for 2 min.
-
4.
Remove the foil seal from the top of the plate and place the plate into the chamber in the proper orientation for printing (see manufacturer’s instructions).
-
5.
After the print run, cover the plate with a new aluminum seal and return to the −80°C freezer.
-
6.
Repeat for each plate.
-
7.
After printing, store the arrays in a −20°C non-deicing freezer, which can remain stable for up to 1 year.
12.3.1.5 Probing for Protein–Protein Interactions with Protein Microarrays
Protein microarrays can be screened for a wide variety of activities. A common activity is interaction with other proteins. One of the best ways to perform this is to produce the protein of interest with an epitope tag (we prefer to use the V5 epitope for yeast) and probe a yeast proteome chip as follows:
12.3.1.6 Purifying the V5-Fusion Protein Probe
-
1.
Grow 5–20 mL of yeast culture containing V5-fusion protein (pYES-DEST52 vector, Invitrogen, see Note 4 ) probe overnight in Sc-ura/2% dextrose.
-
2.
Inoculate 40–400 mL of the Sc-ura/2% raffinose culture with sufficient starter culture to a final OD600 of 0.1.
-
3.
Grow a large culture for a period of three doubling times and induce with 3×-YEP supplemented with 2% galactose, by adding enough to dilute the induction media by a factor of 3.
-
4.
Induce cells at 30°C for 5 h.
-
5.
Harvest cells using in a JA-10 (or comparable) rotor by spinning ≤400 mL volumes of cell suspension at 4,000 rpm/4°C/5 min.
-
6.
Wash the cells once with 50 mL of cold dH2O and transfer to a 50-mL conical tube. Wash again in cold buffer (without detergents or other additives) used for lysis (e.g., PBS, Tris, HEPES) and transfer to 2-mL snap-cap tubes for lysis.
-
7.
Spin the cells at 20,000 × g/4°C/1 min to a pellet and pipette away the buffer.
-
8.
Place tubes on ice and proceed with lysis step.
-
9.
Lyse the cells with 0.5-mm zirconia beads in a 1:1:1 volumes of cell pellet, beads, and PBS lysis buffer (see Subheading 2.6 , item 1). Vortex the mixture using a paint shaker several times at 2-min intervals at 4°C.
-
10.
Centrifuge the lysate at 20,000 × g in a tabletop microfuge for 10 min at 4°C, followed by ulracentrufugation for 30 min at 150,000g at 4°C.
-
11.
Apply the clarified lysate to the previously mentioned affinity resin.
-
12.
Wash the resin several times with wash buffer (see Subheading 2.6 , item 5), followed by elution with elution buffer (see Subheading 2.6 , item 6)
12.3.1.6.1 Probing the Arrays
-
1.
Dilute the protein probe over a concentration range of 5–500 µg/mL, which must be optimized for each protein–protein interaction assay (see Fig. 1 ). The purified V5-fusion protein is diluted into the probe buffer (see Subheading 2.7 , item 2).
Fig. 1. 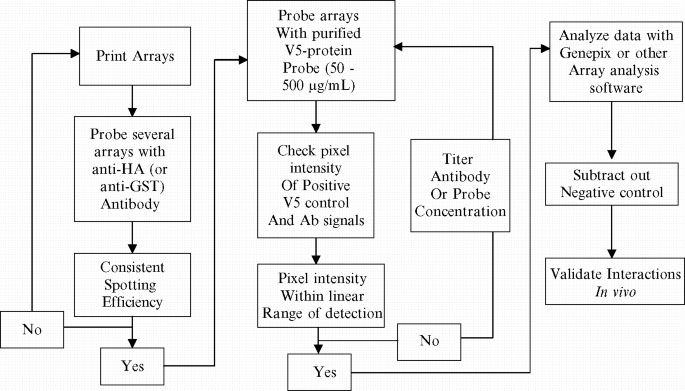
Flowchart for protein–protein interaction assay optimization.
-
2.
Remove the arrays from the freezer (−20°C) and bring to 4°C in the refrigerator, just prior to use, for about 15–20 min. Block the arrays in blocking buffer (see Subheading 2.7 , item 1) for 1 h by shaking at 50 rpm on a stage at 4°C.
-
3.
After blocking, transfer the arrays to a humidity chamber, and add 90 µL of diluted probe directly to the array surface. Overlay the arrays with a raised lifter slip and incubate static (no shaking) in the humidor for 1.5 h.
-
4.
Wash the arrays 3 times for 1 min each in probe buffer in three 50-mL conical tubes (see Note 4 ).
-
5.
To detect interactions, dilute the V5-AlexaFluor 647 antibody to 260 ng/mL in probe buffer and mix thoroughly by shaking.
-
6.
After washing the arrays several times as indicated, add antibody solution directly to array and overlay with a raised lifter slip as before. Incubate the arrays for 30 min/static/4°C.
-
7.
Finally, wash the arrays for 1 min (3×) in probe buffer, and spin in a 50-mL conical tube at 800g in a tabletop centrifuge for 5 min at room temperature. Air-dry the arrays in a slide holder in the dark for 30 min prior to scanning the array at 647 nm.

12.3.1.7 Identifying Protein Kinase Substrate with Protein Arrays
Another useful application of protein microarrays is to identify targets of protein modification enzymes. Recently, a large study has been performed to identify the targets of 87 distinct protein kinases of yeast ( 12 ). A protocol for screening for targets of protein kinases is as follows:
-
1.
Grow cells in 50–500-mL cultures, harvest, and lyse, with glass beads in the lysis buffer (see Subheading 2.7 , item 1) as in “Purifying Proteins for Arrays.”
-
2.
Kinases-GST fusions are bound to glutathione beads and eluted into the kinase buffer (see Subheading 2.7 , item 2).
-
3.
Block the proteome arrays in a Superblock (Pierce) with 0.1% Triton X-100 for 1 h at 4°C and probe in duplicate for every kinase.
-
4.
Optimize conditions: Dilute the kinase into kinase buffer plus 0.5 mg/mL BSA, 0.1% Triton X-100, and 2 L of [33P]ATP (33.3 nM final concentration).
-
5.
Overlay each kinase in buffer on two arrays, cover with a coverslip, and place in a humidified chamber at 30°C for 1 h.
-
6.
Wash the slides twice with 10 mM Tris–HCl pH 7.4, 0.5% SDS and once with double-distilled water before being spun dry and exposed to X-ray film (Kodak).
-
7.
For each experiment, incubate two additional arrays with kinase buffer in the absence of kinase, which will serve as autophosphorylation reference slides.
12.4 Notes
-
1.
Care needs to be taken when printing samples with high glycerol content on surfaces other than nitrocellulose (nickel, ultrgap, etc.) in high humidity >40% because the spot diameter will differ and this could result in samples bleeding together.
-
2.
Probe-specific control spots must be added to each array. These include known concentrations of 3C protease (if used) as well as Alexa Fluor V5 antibody and V5-fusion constructs. These will be used to ascertain optimal probing conditions and antibody titer for protein probing experiments.
-
3.
Primers for sequencing of V5-fusions in pYES-DEST52 can be obtained from Invitrogen’s Website. ORFs can be shuttles from any of the destination (DEST) vectors using the pDONR221 as an intermediary.
-
4.
When probing for protein–protein interactions, it is advisable to wash the arrays using three separate 50-mL conical tubes per slide. Allow the slides to remain submerged in the first chamber until the lifter slip falls off (do not pull it off with force). Afterward, proceed with 1 min washes, gently removing slides with forceps from each bath by gripping the nonmembraneous portion of the slide.
References
Zhu H, et al (2003) Proteomics. Annu. Rev. Biochem. 72:783–812.
Zhu H, Snyder M. (2003) Protein chip technology. Curr. Opin. Chem. Biol. 7(1):55–63.
Zhu H, et al. (2001) Global analysis of protein activities using proteome chips. Science 293(5537):2101–5.
Popescu SC, et al. (2007) Differential binding of calmodulin-related proteins to their targets revealed through high-density Arabidopsis protein microarrays. Proc. Natl. Acad. Sci. U S A 104(11):4730–5.
Hudson ME, et al. (2007) Identification of differentially expressed proteins in ovarian cancer using high-density protein microarrays. Proc. Natl. Acad. Sci. U S A 104(44):17494–9.
Zhu H, et al. (2006) Severe acute respiratory syndrome diagnostics using a coronavirus protein microarray. Proc. Natl. Acad. Sci. U S A 103(11):4011–6.
Rolfs A, et al. (2008) Production and sequence validation of a complete full length ORF collection for the pathogenic bacterium Vibrio cholerae. Proc. Natl. Acad. Sci. U S A 105(11):4364–9.
Huang J, et al. (2004) Finding new components of the target of rapamycin (TOR) signaling network through chemical genetics and proteome chips. Proc. Natl. Acad. Sci. U S A 101(47):16594–9.
Hall DA, et al. (2004) Regulation of gene expression by a metabolic enzyme. Science 306(5695):482–4.
Ho SW, et al. (2006) Linking DNA-binding proteins to their recognition sequences by using protein microarrays. Proc. Natl. Acad. Sci. U S A 103(26):9940–5.
Michaud GA, et al. (2003) Analyzing antibody specificity with whole proteome microarrays. Nat. Biotechnol. 21(12):1509–12.
Ptacek J, et al. (2005) Global analysis of protein phosphorylation in yeast. Nature 438(7068):679–84.
Gupta R, et al. (2007) Ubiquitination screen using protein microarrays for comprehensive identification of Rsp5 substrates in yeast. Mol. Syst. Biol. 3:116.
Gelperin DM, et al. (2005) Biochemical and genetic analysis of the yeast proteome with a movable ORF collection. Genes Dev. 19(23):2816–26.
MacBeath G, Schreiber SL. (2000) Printing proteins as microarrays for high-throughput function determination. Science 289(5485):1760–3.
Ramachandran N, et al. (2004) Self-assembling protein microarrays. Science 305(5680):86–90.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2009 Humana Press, a part of Springer Science+Business Media, LLC
About this protocol
Cite this protocol
Fasolo, J., Snyder, M. (2009). Protein Microarrays. In: Stagljar, I. (eds) Yeast Functional Genomics and Proteomics. Methods in Molecular Biology, vol 548. Humana Press. https://doi.org/10.1007/978-1-59745-540-4_12
Download citation
DOI: https://doi.org/10.1007/978-1-59745-540-4_12
Published:
Publisher Name: Humana Press
Print ISBN: 978-1-934115-71-8
Online ISBN: 978-1-59745-540-4
eBook Packages: Springer Protocols