
Abstract
Guillain-Barré syndrome (GBS) is one of the classic diseases in neurology with an easily recognized clinical pattern of acral paresthesias, ascending generalized weakness, and areflexia. This syndrome has typical presentations and several variants, as well as variable electrodiagnostic features, and treatment modalities including the selection of immunomodulating agents. This chapter reviews the current views of the pathogenesis and clinical presentations of GBS, its electrodiagnostic features, differential diagnosis, treatment and prognosis. A review of GBS variants follows the discussions of classical GBS.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
- Guillain-Barré syndrome
- Landry-Guillain-Barré-Strohl syndrome
- acute inflammatory demyelinating polyneuropathy
- acute motor axonal neuropathy
- acute motor sensory axonal neuropathy
- Miller-Fisher syndrome
- facial diplegia with paresthesias
- pharyngeal-cervical-brachial form
- pure dysautonomia
Introduction
Guillain-Barré syndrome (GBS), also known as Landry-Guillain-Barré-Strohl syndrome, was described in 1916 [1, 2]. GBS is usually a predominantly motor disorder with areflexia and subjective more than objective sensory symptoms. GBS is the most common cause of acute flaccid paralysis worldwide, particularly with the almost complete eradication of poliomyelitis. Its incidence in North America and Europe ranges from 0.8 to 1.9 cases per 100,000 population. GBS affects individuals of all races and ages, but is more common in subjects above 50-year-old [3, 4], and the incidence increases by 20 % for every 10-year increase in age [5]. The annual incidence of GBS increased with age from 1.7/100,000 before 50 years to 3.3/100,000 after 50 years. The annual incidence in children less than 15 years old is between 0.34 and 1.34 per 100,000 population [4]. GBS affects males more than females with a male to female ratio of 1.78.
For many years since its original description, GBS was considered to be a single disorder and interchangeably named acute inflammatory demyelinating polyradiculoneuropathy (AIDP), based on evidence of acute immune attack on myelin and resemblance to experimental allergic neuritis. In addition, criteria for the published diagnosis of GBS have been based on AIDP clinical and electrophysiological features (Table 28.1). More recently, it has become clear that AIDP represent only the prototype of GBS, and other related immune polyneuropathies that cause acute generalized weakness but with different etiologies and pathophysiologies have been grouped together and called GBS. These GBS types comprise most commonly the axonal subtypes of GBS, in which the primary pathology is axonal degeneration and not segmental demyelination, including acute motor axonal neuropathy (AMAN), a pure motor disorder, and acute motor-sensory axonal neuropathy (AMSAN), an acute mixed sensorimotor axonopathy (Table 28.2).
In addition to these three major subtypes of GBS, several GBS variants have been described. These variants deviate significantly from the flaccid weakness and areflexia of typical GBS. Their link to GBS is supported by preceding infectious episodes, diminished reflexes, elevated CSF protein levels, and a presumed immune-mediated origin. Of these, Miller Fisher syndrome is the most widely known, but others include facial diplegia with paresthesias, pure sensory or ataxic forms, a pharyngeal-cervical-brachial regional form, and an acute dysautonomia (see Table 28.2).
Etiology
Although the topography, pathological features, pathophysiology, prognostic features and, more recently, immunopathogenesis of GBS are well understood, the etiology of the disease remains uncertain. There are no genetic factors that are known to predispose individuals to develop GBS, nor is there evidence that GBS is communicable. There have been reports of clusters of cases without an obvious source, but these have been thought to represent random variations [7].
The association of GBS with vaccinations has been suspected and debated for more than four decades. Reports of a definite association of GBS and conventional influenza, hepatitis B, and Gardasil vaccines have been published [8–10]. An “epidemic” of GBS, more accurately, an approximately fivefold increase in the incidence of cases over the estimated natural occurrence, was reported following the swine influenza vaccination program in 1976 [11]. However, lack of clarity in case ascertainment still generates controversy regarding the validity of this association [12–14]. Nonetheless, the risk of developing GBS is one to two additional GBS cases per one million vaccinated persons. The incidence of GBS was higher among conventional influenza vaccinees that were younger than 65 years, but the morbidity was higher among those older than 65 years [10]. Following the recent pandemic swine flu in 2009, another specially designed H1N1-influenza vaccine was introduced. Though epidemiological studies from North America and Europe did not find an increase in incidence of typical GBS among contemporary H1N1-influenza vaccinees [15, 16], atypical GBS cases and cases with GBS variants were recently reported [17].
An upper respiratory tract viral illness, diarrhea, or other infectious illness occurs 1–4 weeks before the onset of GBS in approximately two-thirds of patients (Table 28.3) [2, 18]. In any individual case, the relationship between an antecedent illness and GBS may be less certain. Respiratory or gastrointestinal syndromes are the most common antecedent illnesses. In most patients, the infection resolves by the time the neurological condition develops.
Campylobacter jejuni (C. jejuni) is the most frequently identified bacterial infection preceding GBS. In addition to its peculiarity of being a bacterial rather than a viral infection, it is implicated in up to 30 % of GBS cases studied prospectively with serological studies [19, 20]. Patients with C. jejuni enteritis develop fever, watery diarrhea, and abdominal cramping with GBS typically developing days later. However, a substantial number of patients have only serological evidence of recent C. jejuni infection without enteritis [19]. C. jejuni-related GBS has more severe axonal loss on electrodiagnostic (EDX) studies, elevated anti-GM1 antibodies, and a more protracted recovery compared to cases without the infection (see Sect. “Acute Motor Axonal Neuropathy” below) [19, 21]. The immunologic implications of C. jejuni as a triggering agent for GBS are of great interest because this sequence of events supports postinfectious nerve inflammation as a pathogenetic theory. Certain strains of C. jejuni are the cause of enteritis in a disproportionate number of GBS patients [22]. The lipopolysaccharides of these organisms share ganglioside-like epitopes with peripheral nerves (such as GM1, GQ1b, and GalNAc-GD1a) and are thought to induce a form of molecular mimicry in which the immune system, in its efforts to eradicate C. jejuni, elaborates antibodies against neural antigens and secondarily produces GBS [21, 23].
The most commonly identified viral infection is cytomegalovirus (CMV), with serological evidence of preceding infection in 10–15 % of cases [18, 24]. In some instances, the only indication that CMV is the preceding infectious agent may be an elevation in liver enzymes concomitant with the onset of GBS symptoms. GBS triggered by CMV infection tends to occur in younger individuals and to produce a more severe course with respiratory failure, prominent sensory loss, more frequent cranial nerve involvement, and raised antibodies directed against the ganglioside GM2 [18, 25]. Similarly, judging by serological studies, Epstein-Barr virus infection precedes GBS in approximately 10 % of patients, and the infectious clinical syndrome varies from mononucleosis to pharyngitis or hepatitis [18]. The relationship between GBS and other viruses reported to precede the condition, such as respiratory syncytial virus, parainfluenza virus, echovirus, coxsackie virus, measles, mumps, rubella, herpes zoster and simplex virus, influenza, and hepatitis A and B, is less certain (see Table 28.3). GBS may occur soon after seroconversion with human immunodeficiency virus (HIV) [26]. There are no clinical or EMG features that distinguish these patients from non-HIV-related forms of GBS, except for a prominent lymphocytic pleocytosis in the spinal fluid of patients with HIV, which may therefore complicate the CSF formula in GBS [26]. GBS has also been reported following immune reconstitution from highly active retroviral immunotherapy [27].
Among other bacteria, Mycoplasma pneumoniae (M. pneumoniae) has been reported to precede GBS in approximately 5 % of cases and should be considered when weakness develops after a prodromal illness characterized by fever, headache, and severe dry cough [18]. Infection with M. pneumoniae is supported by the presence of cold agglutinin antibodies in the serum and is confirmed by complement-fixing antibody tests. Lyme disease is a bacterial illness caused by a spirochete, Borrelia burgdorferi in the United States and Borrelia afzelii in Europe. It is transmitted by infected hard ticks belonging to a few species of the genus Ixodes. Lyme disease may cause a chronic axonal sensorimotor polyneuropathy, a painful polyradiculitis (Bannwarth’s syndrome), or acute facial diplegia [28, 29]. This may mimic GBS, but the presence of a true postinfectious polyneuritis with Lyme is still somewhat uncertain. As with HIV, a lymphocytic pleocytosis in the spinal fluid may distinguish these patients from typical cases of GBS [28, 29]. Shigella, salmonella, typhoid, brucella, cyclospora, and yersinia enterocolitica also have preceded GBS during epidemics or in single cases [30].
Several systemic illnesses also have been tenuously linked to acute GBS, but most of these are implicated more often with CIDP. For example, a GBS-like syndrome has been described in patients with Hodgkin’s disease, lung cancer, thyroid disease, systemic lupus erythematosus, paraproteinemia, and sarcoidosis in single case reports or small series [31, 32]. It is difficult to be certain that these are anything more than chance associations.
Many GBS series have included a small proportion of cases that occurred after surgery, and only few cases genuinely appear to have been triggered by an operation. It is now considered that some of these patients who develop weakness after being admitted to the intensive care unit have a “critical illness polyneuropathy” rather than GBS as a consequence of multiorgan failure and sepsis or other factors associated with a prolonged postoperative course in an intensive care unit (see Chap. 76). It is also unlikely that the axonal loss and prognosis in GBS patients worsen after admission to the intensive care unit due to concomitant critical illness polyneuropathy [33]. Trauma has rarely been reported as a precipitant to GBS, and we have seen several such cases, but the association remains uncertain. In one study reported in 2006, 16 patients receiving tumor necrosis factor-alpha antagonist therapy developed GBS and were reported to the US Food and Drug Administration [34]. Other medications, drugs of abuse, bone marrow transplantation, and spinal epidural anesthesia all have been reported to precede GBS, but these connections also remain unproven. GBS may occur at any time during pregnancy, but the risk is maximal during the first 2 weeks after delivery [35].
Pathogenesis
A number of immune mechanisms involving humoral and cellular immunity, complement deposition, proinflammatory cytokines, and other inflammatory mediators are theorized to be involved in the pathogenesis of GBS [36]. Many of these purported mechanisms have been gleaned from studies in an experimental model of the disease—experimental autoimmune neuritis (EAN) that represents a fair version of the human disease. The extent to which each of these immune processes is related to various clinical and electrophysiological patterns, and the implications for treatment and prognosis, is of great interest but has not been fully studied. In EAN, rabbits, guinea pigs, or rats are immunized with autologous peripheral nerve tissue and Freund’s adjuvant (a nonspecific stimulator of immune reactions). After a latency of several days, they develop a rapidly progressive paralytic illness with the pathologic features of endoneurial inflammation and demyelination, identical to the clinical and pathological manifestations of GBS [37]. This inflammatory response is mediated by T cells that are directed against epitopes on peripheral nerve myelin including PO, P2, and PMP 22, and by implication, this immune attack leads to macrophage invasion and demyelination [38].
In classical pathological studies of GBS, demyelination was most prominent adjacent to regions of intense perivenular inflammation [39, 40]. Pathological material from patients with GBS shows a similar accumulation of lymphocytes and macrophages in a perivascular distribution scattered throughout the peripheral nervous system with a predilection for spinal roots [39–41]. Macrophages and T cells express major histocompatibility (MHC) class II antigens which are upregulated on Schwann cells in the region of inflammatory lesions in patients with GBS. One hypothetical sequence that has been offered is that activated T cells, stimulated by a preceding infection and by an interaction with antigen presenting cells that express MHC class II antigens, disrupt the blood nerve barrier, attack endoneurial antigens, and release inflammatory cytokines such as interleukin-2 and tumor necrosis factor (TNF) [36]. In keeping with this hypothesis, several investigators have demonstrated elevated levels of TNF and soluble TNF receptor in the serum of patients with acute GBS [42, 43]. These cytokines attract macrophages that are capable of producing nerve demyelination and damage Schwann cells and axons [36, 42, 43].
There is also evidence that humoral factors are central to the development of GBS. Passive transfer studies have shown that sera from GBS patients injected into the nerves of animals induces local demyelination. Furthermore, the observed clinical recovery following removal or neutralization of autoantibodies (or other pathogenic humoral factors) by plasmapheresis or intravenous immune globulin (IVIG) supports a role for B-cell-mediated processes in the pathogenesis of GBS [44]. Koski et al. demonstrated that elevated serum levels of complement-fixing anti-myelin antibodies correlated with disease activity in patients with GBS [45, 46]. Recent autopsy studies revealed that local complement activation occurs at the site of nerve lesion, such as the axolemma in patients with AMAN and the Schwann cell membrane in patients with AIDP [47, 48]. There is now evidence, using high-resolution immunocytochemistry, of early complement activation and deposition of activated complement components along the outer surface of the Schwann cell. The presence of terminal complement complex is associated with vesiculation of the outermost myelin lamellae. This occurs before and within 1 week of invasion of macrophages [48]. In patients with AMAN associated with axonal loss, the complement activation product binds to the axolemma of motor fibers and, in severe cases, immunoglobulin and activated complement within the periaxonal space of myelinated internodes [47].
Numerous studies have demonstrated the presence of anti-neural antibodies directed against acidic glycoconjugates in the serum of patients with GBS. These peripheral nerve antigens are usually gangliosides (GM1, GD1a, GQ1b, and GT1a) which differ with regard to the position and number of their sialic acid. There is solid indication that anti-ganglioside antibodies play a pathogenic role in the pathophysiology of GBS [49]. In clinical practice, these antibodies are found in only a minority of patients since autoantigens have not been well identified in AIDP. The anatomic distribution of the gangliosides within the peripheral nervous system may explain some of the observed clinical variants of GBS [36, 49]. For example, GQ1b is strongly expressed in the oculomotor, trochlear and abducens nerves, as well as the muscle spindles in the limbs, which explain the distinct Miller Fisher syndrome (ophthalmoplegia, ataxia, and areflexia), frequently associated with anti-GQ1b antibody [50–52]. There is also strong evidence currently that the axonal subtypes of GBS, particularly AMAN and less commonly AMSAN, are associated with antibodies directed against GM1 and GD1a at the axolemma. This eventually attracts macrophages invading the nodes of Ranvier and inserting between the axon and the axolemma, resulting in axonal degeneration. In AMAN, the myelin sheath also remains intact, and there is no lymphocytic inflammation [53]. In AMAN, only the axons of the ventral roots are involved, while in AMSAN, both the dorsal and ventral roots are affected [38, 41].
Increasing evidence supports that molecular mimicry plays an important role in the pathogenesis of GBS. It is likely that genetic polymorphism in various strains of C. jejuni determines the specificity of the antiganglioside antibodies and the associated variant of GBS. Lipooligosaccharide, a major component of the outer membrane of C. jejuni, has ganglioside-like products; sensitization with GM1-like lipooligosaccharide by injecting it, rabbits induced a neuropathy resembling AMAN [54]. C. jejuni bacterial isolates from patients with AMAN have GM1-like and GD1a-like lipopolysaccharides, whereas bacterial isolates from patients with the Miller Fisher syndrome usually express GQ1b-like lipopolysaccharides [55, 56]. It is now evident that C. jejuni is composed of several classes that have diverse lipooligosaccharide biosynthesis genes. C. jejuni is now grouped into several classes based on the organization of these genes. A specific class carrying a sialyltransferase gene (cst-II) is associated with the development of GBS. Patients infected with a specific strain (Thr51), which expressed both GM1-like and GD1a-like lipooligosaccharides, had anti-GM1 or anti-GD1a IgG antibodies and present with typical GBS manifestations including limb weakness. In contrast, patients infected with another strain (Asn51), which expresses GT1a-like or GD1c-like lipooligosaccharides, have anti-GQ1b IgG antibodies and present with the Miller Fisher syndrome including ophthalmoplegia and ataxia [55–58]. On the basis of the above findings, one may conclude that C. jejuni-associated/GM1-related GBS represents at least true instance of a molecular-mimicry-related disease.
Clinical Features
GBS in its typical form is a predominantly motor neuropathy, although acral paresthesias are almost always present at the onset of the illness (Table 28.4). Tingling, prickling, or pins and needles sensations are usually followed within hours or days by symmetrical leg weakness and trouble walking. The presence of acral paresthesias increases the probability of the correct diagnosis of GBS [2]. Difficulty climbing stairs or arising from a chair or commode is typical. Weakness of the upper limbs, ocular, oropharyngeal, and facial muscles develops with variable frequency and severity.
The weakness is often bilateral but some degree of asymmetry is common. Rarely, the weakness begins in one limb hours or a day before involving the contralateral limb. Proximal weakness is more frequent than distal and often more severe. In contrast to diseases that affect the muscle or neuromuscular junction, weakness rarely remains restricted to the shoulder or hip girdle muscles; some degree of hand or distal leg weakness develops after the proximal muscles. The weakness often moves to the upper limbs resulting in an ascending paralysis. Weakness that remains limited to the legs or, alternatively, weakness that begins in the hands or shoulder girdle and involves the legs may occur as the condition advances. A pattern of descending paralysis occurs in 10–15 % of cases, with symptoms beginning in the cranial nerves or arms and spreading to the legs. Approximately one-third of fully developed cases have a degree of weakness that is similar in the arms and legs. Fasciculations or myokymia are observed in a small number of patients.
The second hallmark sign of GBS is reduced or absent deep tendon reflexes, presumably reflecting desynchronization or dispersion of impulses carried by myelinated fibers in the afferent arm of the reflex arc. Approximately 70 % of patients have absent deep tendon reflexes at the time they are first examined. Reflexes occasionally remain elicitable until weakness or large fiber sensory loss advances. Reflexes are almost always unobtainable in limbs that are too weak to resist gravity. Yuki et al. found that the myotatic reflexes were normal or exaggerated during the entire clinical course in approximately 10 % of GBS patients, more commonly in patients with AMAN than AIDP [60]. However, the diagnosis of GBS must remain questionable in this group, and upper motor neuron causes of weakness should be excluded.
More than half of GBS patients experience paresthesias of the distal extremities as the initial symptom. Most patients complain of “pins and needles,” “prickling,” or “tingling” feelings, likened to an “asleep feeling” in an arm or leg following compression of the limb. In contrast to most length-dependent axonal polyneuropathies, patients with GBS often develop paresthesias in the fingertips soon after the feet are affected and sometimes beforehand. The sensory symptoms are symmetric and often precede weakness by a few days, ascending to the ankles and wrists as the illness progresses. Paresthesias of the trunk or face are infrequent. Patients may also describe an acral numb, heavy, or dead sensation as the disease evolves. Some experience sensory loss over the trunk, and a well-demarcated sensory level simulating spinal cord disease has been described, but only to the extent of a noticeable change in sensation, not analgesia below the level. The detection of a genuine thoracic sensory level should prompt further evaluation with an MRI of the spinal cord to exclude a myelopathy including transverse myelitis. Reduced vibration sense and proprioception in the distal limbs are the most common findings. A substantial number have sensory ataxia that is soon obscured by weakness. Pinprick sensation may also be impaired in distal parts in severely affected patients.
Pain is a common but underappreciated symptom in GBS. Pain may precede the onset of weakness by 2 weeks in 1/3 of patients [61], and about 2/3 of patients have modest discomfort early in the illness [62]. Pain, when not severe, may be overlooked by medical staff who are preoccupied with more pressing medical complications, and intubated patients often are unable to convey their discomfort. The discomfort in GBS has been described as (1) aching, usually confined to muscles of the back, hips, or upper legs (the most common type); (2) shooting or stabbing, radicular pain radiating from the back to one or both legs; or (3) chronic and unrelenting, burning, dysesthetic feelings in the distal limbs [61–63]. Rarely, back and radicular pain can precede weakness and paresthesias and be attributed to sciatica or a spinal condition [63]. At 1-year follow-up, pain is reported in 38 % of patients, and the pain intensity was highest in patients with typical GBS and in those with sensory disturbances and with higher level of weakness and disability [61].
Approximately one-half of GBS patients will have cranial nerve involvement at some time in the course. The facial nerve is most commonly affected, and facial weakness typically occurs when there is substantial limb weakness. Conversely, lack of facial weakness in a patient with severe generalized paralysis should at least raise concerns about the accuracy of the diagnosis. As with limb weakness, facial paralysis is often bilateral, but occasionally asymmetrical, and rarely unilateral. Weakness of the ocular muscles arises in 10–20 % of patients, the abducens nerve being most commonly affected. Impaired abduction is usually bilateral and occasionally asymmetrical. Oropharyngeal weakness occurs in up to one-half of patients during the course of the illness and presents great problems in terms of aspiration. In severely affected patients, there may be paralysis of all the cranial muscles, ventilatory failure, and flaccid paralysis of all the limbs, simulating the “locked-in” state [64].
Weakness of the diaphragm that leads to respiratory failure and a requirement for ventilator support occurs in approximately 20–30 % of patients with GBS [65–67]. Most such patients are quadriparetic, although patients with a bibrachial pattern of weakness may also have pronounced oropharyngeal and respiratory muscle involvement (see Sect. “Guillain-Barré Syndrome Variants”). Weakness of the neck muscles, tongue, and palate tends to parallel involvement of the diaphragm and respiratory muscles. Diaphragmatic weakness, which causes reduced vital capacity, inspiratory force, and tidal volume, invariably causes atelectasis. Coughing and clearing of oral secretions are then impaired, generating progressive atelectasis, arteriovenous shunting, and mild hypoxia. These changes further aggravate ventilatory failure by causing tachypnea and an increased work of breathing. As the respiratory rate increases, levels of carbon dioxide actually may be reduced in the early stages of respiratory compromise. However, as the diaphragm, intercostal, and accessory muscles become further exhausted, hypercapnea ensues and patients may rapidly deteriorate with hypercarbia and respiratory arrest. If diaphragmatic and respiratory muscle weakness have not occurred 2 weeks into the course of the illness, assisted ventilation should not be necessary unless other pulmonary or medical complications ensue. The main predictors of mechanical ventilation include shorter days between onset of weakness and admission, higher Medical Research Council (MRC) sum score, and presence of facial and/or bulbar weakness (see Sect. “Prognosis”) [65]. Patients with GBS who require ventilator support have a less favorable prognosis for neurologic recovery, longer hospitalization, and higher mortality.
Dysautonomia is a less common but well-recognized feature in patients with fully developed GBS, occurring up to 65 % of cases [68, 69]. This number is certainly overestimated if one considers only changes of clinical significance. Autonomic nervous system complications tend to occur more frequently in those with severe paralysis and ventilatory difficulties, but rarely may develop in otherwise mild cases. The most common cardiac manifestations include sinus tachycardia, sinus bradycardia, sinus arrest and other supraventricular arrhythmias, paroxysmal hypertension, hypotension (especially postural hypotension), and so-called vagal spells that consist of bronchorrhea, bradycardia, and hypotension. Infection, hypoxia, pulmonary embolus, and other medical complications should be excluded before attributing cardiovascular disturbances to dysautonomia. Because of the potential for complete heart block, sinus arrest or other life-threatening cardiac arrhythmias (e.g., ventricular tachycardia), and the risk of rapidly progressive respiratory failure, most patients with GBS require monitoring in an intensive care setting early in the illness (see Sect. Supportive Care in under the Sect. Treatment and Management). Other features of autonomic instability include ileus, urinary retention (surprisingly common—seen in one-quarter of patients and suggesting a myelopathy), and inappropriate antidiuretic hormone secretion leading to hyponatremia [70]. Many patients have minor aspects of dysautonomia that are clinically insignificant, such as altered sweating, mild orthostatic hypotension, and acral cyanosis from vasomotor instability.
GBS can have sometimes unusual features such hearing loss, meningeal signs, vocal cord paralysis, papilledema, and mental status changes [71]. Recently, cases of GBS has been associated with posterior reversible encephalopathy syndrome (PRES) [72]. One explanation for the PRES is that the cytokines, produced in the context of GBS, may increase the permeability of the blood–brain barrier.
Guillain-Barré Syndrome Subtypes
For many years, the term AIDP was interchangeably used with GBS. It is now well recognized, particularly during the last two decades, that axonal forms of GBS exist and these are distinguished from AIDP using electrophysiological and pathological characteristics. These disorders, AMAN and AMSAN, remain under the same umbrella term of GBS because they share many of the clinical findings, including flaccid weakness and areflexia, preceding infectious episode, and a presumed immune-mediated origin (see Table 28.2). These disorders are also often preceded by infections, but may be occasionally associated with connective tissue disorders [73, 74]. Also, these disorders may be difficult to distinguish on early electrodiagnostic studies since the nerve conduction changes may overlap and sequential studies are often necessary.
Acute Inflammatory Demyelinating Polyradiculoneuropathy (AIDP)
AIDP is the prototype of GBS and characterized by peripheral nerve and spinal root demyelination. This disorder accounts for up to 90 % of GBS cases in North America and Europe [75] but only 22–46 % of cases in China, Japan, India, Southeast Asia, and Mexico [76, 77]. This disorder is characterized by vesicular degeneration of myelin triggered by membrane-attack complex formation on the outer surface of Schwann cells. AIDP has not been strongly associated with antiganglioside antibodies [78].
Acute Motor Axonal Neuropathy (AMAN)
McKhann, Griffin, and colleagues described an acute paralytic syndrome in patients from regions of northern China and coined the term acute motor axonal neuropathy (AMAN) [79–81]. It is now clear that this disorder is as common as AIDP in Mexico, China, Japan, India, and Southeast Asia, accounting for 30–65 % of cases [76, 77]. In contrast, AMAN is rare in North America and Europe, probably accounting for less than 10 % of cases [75].
The disorder primarily afflicts children and young adults [79, 82] and causes symmetrical limb weakness, areflexia, facial diplegia, and oropharyngeal and respiratory muscle weakness that evolves over several weeks. The extraocular muscles are spared. There are no sensory features although mild changes in sensory nerves may occur [83]. The condition occurs as an annual epidemic during summer months. Most cases are preceded by a gastrointestinal illness with abdominal pain, cramps, and diarrhea and elevated antibody titers to Campylobacter jejuni, anti-GM1, and anti-GD1a [84–86]. The spinal fluid protein concentration is usually slightly elevated after several days of the illness, but EDX studies show reduced or absent compound muscle action potential amplitudes with normal conduction velocities, no conduction blocks, normal sensory potentials, and early active denervation, thus implicating a process similar or identical to AMSAN, with the exception of the normal sensory potentials (see below) [79–81].
Autopsy findings have confirmed widespread axonal degeneration with little demyelination or inflammation [48, 81]. Pathologic studies using electron microscopy have demonstrated the presence of macrophages in the periaxonal space of myelinated internodes [48, 53, 87]. The pathogenesis of AMAN has not been fully elucidated, but there is convincing evidence for an antibody- and complement-mediated process directed primarily at motor axons. There is evidence to suggest that IgG anti-GM1 or anti-GD1a antibodies bind to the axolemma at the node of Ranvier leading to membrane-attack complex formation. This results in the loss of voltage-gated sodium channels and leads to conduction failure. These rapidly reversible immune-mediated changes at the nodes of Ranvier may explain the puzzling speedy recovery that occurs in some patients with AMAN, a rate that is comparable to patients with AIDP. Another explanation for the rapid recovery is selective degeneration and subsequent quick regeneration of intramuscular motor nerve terminals in AMAN [86, 88, 89].
Acute Motor-Sensory Axonal Neuropathy (AMSAN)
In 1986, Feasby and coworkers described an axonal form of GBS, challenging the existent notion of GBS being a primarily demyelinating disease. These patients developed rapidly progressive paralysis, areflexia, and distal sensory loss [90, 91]. All of their patients required assisted ventilation and recovery was poor. The spinal fluid protein level was increased, but in contrast to the demyelinating features of typical GBS, EDX evaluation showed numerous inexcitable nerves, widespread active denervation, and no evidence of demyelination. An autopsy in one case showed axonal degeneration without inflammation or primary demyelination in the spinal roots and peripheral nerves. Since then, several studies have suggested that this syndrome, now termed acute motor-sensory axonal neuropathy (AMSAN), represents a variant of GBS that is clinically indistinguishable from typical, albeit very acute, cases but in which axons are the targets of the immune reaction. Virtually all patients become quadriplegic within days and require ventilator support and most have substantial residual weakness after recovery from the acute illness; some remain ventilator dependent for prolonged periods. Nerve conduction studies indicate an acute and widespread axonal sensory and motor neuropathy without demyelinating features.
It has also been argued that complete, distal conduction block and reversible conduction failure can simulate the finding of nerve inexcitability that is at the core of the diagnosis [92]. Subsequent pathological material from a few cases has shown axonal degeneration in the motor nerves with macrophages insinuated in the periaxonal space of internodes; some patients also had axonal loss in the spinal roots [41].
Recent evidence suggests that AMAN and AMSAN share a common immunological profile and represent a continuum within the spectrum of axonal GBS [41]. Anti-GM1, anti-GM1b, and anti-GD1a, immunological markers for AMAN, are seen in high percentage of patients with AMSAN [93]. Also, sensory fiber involvement which distinguishes AMSAN from AMAN has been shown to be often involved subclinically in AMAN patients [83].
Evaluation and Diagnosis
Electrodiagnostic Studies
Electrodiagnostic (EDX) studies are very important in the diagnosis of GBS. Abnormalities on nerve conduction studies (NCS) are seen in up to 95 % of cases, and these findings are diagnostic in large number of GBS patients at some time during the course of the illness [94–96]. Unfortunately, NCS may be normal or show only modest nondiagnostic changes early in the course of GBS, at a time when treatment decisions have to be made. Repeat studies are often necessary, particularly when initial NCS findings are not specific [94]. The nature of the abnormalities detected by NCS depends upon the timing of the study in relation to disease onset and the number of nerves studied. Extensive testing of multiple nerves and multiple nerve segments in multiple limbs including evaluation of F-waves, H-reflexes, and blink reflexes is essential. The aim of NCS is to show evidence of multifocal acquired nerve demyelination, the hallmarks of AIDP, which represents the majority of patients with GBS in the Western World [75].
Abnormal Electrodiagnostic Parameters in GBS
The EDX studies in GBS include a variety of NCS parameters which may become abnormal during the course of illness. These include motor distal latencies, motor conduction velocities, CMAP amplitude and waveform configuration, sensory studies, late responses, and needle EMG. Although these EDX abnormalities are common in GBS, they vary in specificity which renders some of them less useful than others. The following are the most common abnormalities seen in GBS, with varying degree of specificity:
Abnormal H-Reflex
The tibial H-reflex is a sensitive test for detecting abnormalities of the S1 nerve root and early polyneuropathy and correlates fairly well with the Achilles reflex [97]. Absent H-reflexes correlate well with the areflexia in the lower extremities of GBS patients. The H-reflexes are absent bilaterally in almost all patients with GBS, including in 95–100 % of patients during the first 1–2 weeks of illness [98–100]. Hence, the H-reflex is the most sensitive EDX test. However, absent H-reflexes are not specific for GBS since it a common finding in the elderly and occurs in the majority of large fiber sensory and sensorimotor peripheral polyneuropathy such as diabetic and critical illness polyneuropathies, as well as S1 radiculopathies, cauda equina, and conus medullaris lesions.
Abnormal F-Waves
Multifocal acquired nerve demyelination, the hallmark of GBS [6], preferentially affects proximal and distal portions of the peripheral nerves [101]. Therefore, a common finding in early GBS is prolonged F-waves [94–96]. Prolonged or absent F-responses have been reported in as many as 40–80 % of GBS patients early in the illness [100, 102]. It may be the sole electrodiagnostic abnormality in about one-fourth of patients [101, 102]. The yield of F-wave studies improves by assessment of additional multiple F-wave parameters, including chronodispersion, mean latency, and mean amplitude [101]. However, these parameters are difficult to quantitate and are subject to variability.
Multiple and Complex A-Waves
A-waves are reproducible intermediate-to-late responses that are distinguished from F-waves and H-reflexes and usually seen during routine F-wave studies. A-waves may be recorded in normal individuals of the foot muscles while stimulating the tibial nerve. A-waves are commonly seen in multiple nerves and often with complex morphology in about 2/3 of patients with GBS [103, 104]. Their precise mechanism is not known, but they may be due to ephaptic transmission between axons or proximal re-excitation of the axon. Although prevalent in GBS, A-waves are not specific for GBS since they may be seen in other acquired and inherited demyelinating polyneuropathies (such as chronic inflammatory demyelinating polyneuropathy and Charcot-Marie-Tooth disease type I) and, less often, in axonal polyneuropathies, radiculopathies and motor neuron disease [103].
Motor Conduction Blocks
This is defined as a reduction (usually >20–50%) in the amplitude and area of the compound motor action potential (CMAP) following proximal nerve stimulation (Fig. 28.1). To be more specific and to avoid confusing GBS with polyneuropathies associated with compressive mononeuropathies, the conduction blocks in GBS should be located at non-entrapment sites (Fig. 28.2). This is a highly specific finding but is found in only about one-third of patients with GBS, dependent on the number of nerves and nerve segments studied [95, 98, 100]. Conduction block is often a sign of segmental demyelination, but transient conduction and block may be seen as the early manifestations of axonal loss such during the early stages of AMAN [105, 106]. Sequential studies are necessarily before a final diagnosis of axonal loss is made. Also, low distal CMAPs may be due to distal demyelination with distal conduction block and often mimic axonal loss. Rapid recovery of low distal CMAPs and SNAPs on sequential studies is a necessary confirmatory sign of distal demyelination [106].

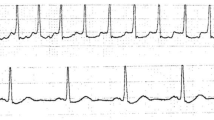
Median motor nerve conduction study in a 35 year old patient with GBS examined at week 2 of illness. Note the forearm conduction block as evidenced by the significant drop in CMAP amplitude (80 %) and area (76 %) when CMAP obtained with distal stimulation at the wrist (upper tracing) is compared to proximal stimulation at the elbow (lower tracing). There is also marked slowing of forearm conduction velocity (18.9 m/s). dLAT/CV distal latency/conduction velocity, AMP amplitude

Peroneal motor nerve conduction study in a 55 year old patient with GBS examined at week 4 of illness. Note the significant drop in CMAP amplitude (84 %) and area (88 %), with CMAP dispersion, when CMAP obtained with distal stimulation at the ankle (upper tracing) is compared to proximal stimulation below fibular neck (middle tracing) and popliteal fossa (lower tracing). The conduction block is distal to the fibular neck, not at a common entrapment site (i.e., fibular tunnel). dLAT/CV distal latency/conduction velocity, AMP amplitude
Nerve Conduction Velocities Slowing
Significant slowing of nerve conduction velocities was first reported by Lambert and Mulder who reported in 60 % of their patients with GBS. During the early stage of disease, however, 80–90 % of patients with AIDP have normal conduction velocities [95]. It is now clear that prominent slowing of nerve conduction velocities is uncommon and is detected in only approximately 25 % of cases [98, 100]. Slowing is particularly uncommon during the first 2 weeks of illness in AIDP, and conduction velocities become paradoxically slower between 3 and 6 weeks from onset and during the recovery phase, presumably due to nerve fiber remyelination [95]. Additionally, controversies remain regarding the exact cutoff of conduction velocity that distinguishes primary demyelination from axonal loss, particularly in the presence of low CMAP amplitude. These values have varied from 60 to 80 % of the lower limits of the normal conduction velocity values [94, 95, 107].
Distal Latency Slowing
Since multifocal acquired nerve demyelination in GBS preferentially affects proximal and distal portions of the peripheral nerves, common findings in early GBS are prolonged distal motor latencies [94, 95]. Although this finding is common, it lacks specificity except when the latencies are significantly delayed and are within the demyelinating ranges. Similar to velocities, these ranges have also been controversial since demyelinating ranges have varied from 120 to 150 % of the upper limit of normal values [107–110].
Compound Muscle Action Potentials (CMAPs) Dispersion
The CMAP duration may be prolonged in GBS, and this is attributed to varying degrees of conduction slowing in demyelinated motor nerve fibers. Dispersion of the CMAP has only been considered as a criterion of demyelination in one study [108, 109]. Adding this fairly specific finding to other EDX parameters improved the sensitivity of EDX studies without worsening its specificity [111]. Distal CMAP durations of median, ulnar, peroneal, or tibial nerves measuring more than 8.5 ms are strong evidence for the presence of demyelination [111, 112].
Abnormal Sensory Nerve Action Potentials (SNAPs)
Earlier EDX criteria of GBS overlooked sensory nerve conduction studies by emphasizing abnormalities of motor NCS, such as conduction block, CMAP dispersion, abnormal F-waves, and slowing of latencies and velocities. It is now clear that SNAPs abnormalities are common and sometimes specific for AIDP. SNAP are abnormal in about 75 % of patients sometimes during the illness [94, 95, 100, 102]. The most common findings are reduced SNAPs amplitudes. This may reflects axon loss but is more likely caused by conduction block and phase cancellation. Slowing of sensory nerve conduction velocities is detected less frequently because the SNAP potential usually drops out before severe slowing is found.
Sensory NCS is now recognized to be important in providing EDX evidence that might distinguish primary demyelinating from axonal polyneuropathy. A sural-sparing pattern, also known as a “normal sural-abnormal median” pattern, is now recognized to be a common and specific finding in GBS and in particular AIDP [94, 100, 113, 114]. In contrast to the majority of length-dependent axonal polyneuropathies, the median and ulnar SNAPs are frequently reduced or absent when the sural nerve is normal (“sural sparing”), presumably as a consequence of random, multifocal demyelination. This pattern is the most specific sensory abnormality in AIDP and is present in about 50 % of patients during the first 2 weeks of illness [98, 100].
There are several limitations of the sural-sparing pattern which affects its sensitivity and specificity in the diagnosis of GBS. First, the sural SNAP may be either low in amplitude or absent in elderly patients and in those with underlying diabetic polyneuropathy [98]. Second, technical considerations in hospitalized or critically ill patients or those on mechanical ventilation render it difficult to study the sural SNAP. Third, the median sensory study may be abnormal in patients with preexisting carpal tunnel syndrome. Hence, sural sparing is better defined by preservation of sural SNAP in the presence of normal or near normal median and ulnar SNAPs in the upper extremity [98, 100].
Comparing several SNAP amplitudes in the upper and lower extremities is a useful exercise, particularly in elderly where the sural may be unobtainable. Among them, a sensory ratio (sural + radial SNAPs/median + ulnar SNAPs) is a good substitute for sural-sparing pattern. Patients with AIDP are 12 times more likely to have an elevated sensory ratio (>1) compared to patients with axonal polyneuropathies such as diabetic or critical illness polyneuropathies [115]. It is not known whether another lower limb SNAP, such as the superficial peroneal, could substitute for the sural.
Needle Electromyography (EMG)
Needle EMG is the least helpful EDX tool in the evaluation of patients with GBS, mostly since the EDX studies are often done early in the disease before signs of axonal degeneration are apparent on needle EMG. The initial finding in patients with GBS is reduced recruitment with the degree of abnormality proportional to the degree of muscle weakness. Early on, the combination of normal or virtually normal motor unit action potentials (MUAPs), reduced MUAP recruitment, and absent fibrillation potentials detected by needle EMG is characteristic. However, similar needle EMG findings are also seen in other acute axonal nerve lesions. The detection of abnormal spontaneous activity (fibrillation potentials) indicates axonal damage and occurs in 20–60 % of GBS patients in the first 4 weeks of the illness [95, 107]. This is seen in the demyelinating and axonal subtypes of GBS and signifies a variable element of axonal loss in all patients. Abnormal spontaneous activity is found more often during follow-up studies, 2–4 months after onset, and may be observed in proximal and distal muscles, consistent with multifocal nerve degeneration [95]. MUAP morphology changes start to occur after the fourth week of illness with an increased percentage of polyphasic MUAPs as the early change. Myokymia may be found in limb or facial muscles in some GBS patients, usually early in the course of the disease [94].
Electrodiagnostic Criteria in GBS
Various sets of EDX criteria for the detection of demyelination have been developed. These were mostly made by consensus or as part of the methods utilized in GBS studies [94, 95, 107, 109, 110]. However, most of these criteria were not subjected to vigorous scrutiny or applied to other neuropathies, and their specificities in GBS diagnosis were not well tested. For example, classic published diagnostic criteria of AIDP in GBS are fulfilled in 20–70 % of the cases based on the specific criteria utilized [99, 116]. This variation depends on how strict these criteria are in excluding patients with equivocal EDX findings.
Although it is intuitive to conclude that EDX studies are sensitive in confirming demyelination, EDX studies often reveal abnormalities that are not specific of primary demyelinating polyneuropathy during the early phases of the disease. Common abnormalities seen during the first few weeks of illness include absent or delayed H-reflexes, F-responses or blink reflexes, “sural sensory sparing,” distal CMAP temporal dispersion, or frequent A-waves [95, 98–100]. During the first 4 days of weakness GBS, about 1/2 of the patients have normal NCSs (except for absent H-reflex in the majority of them), while only about 10 % of them have normal studies by the first week of illness [98]. Another 5–10 % have only nonspecific nerve conduction abnormalities, such as mild slowing, absent and/or prolonged H-reflexes or F-waves (due to spinal root demyelination), or low-amplitude CMAPs (due to intramuscular motor nerve terminals involvement).
We recommend using EDX criteria that include these different variables and have increasing levels of certainty in confirming the diagnosis of demyelination in AIDP [100] (Table 28.5). These criteria are designed to grade the level of confidence of the EDX studies, ranging from normal study to definite findings of acquired multifocal demyelination. Using these criteria, about 2/3 of patients with GBS meet the highly suggestive or definite criteria for AIDP during the first two weeks of illness with a very high specificity of 96–100 %. The remainder likely includes patients with the axonal subtypes of GBS, AMAN, and AMSAN, who do not have evidence of demyelination.
Electrodiagnostic Studies in GBS Subtypes
The EDX studies in GBS and its subtype may be confusing at the early stage of the disease when it is often difficult to determine the subtype classification of the disease (axonal vs. demyelinating) [105]. AIDP, AMAN, and AMSAN may have similar findings during the first weeks of illness. As outlined above, sensory conduction abnormalities are seen in patients with AMAN, making the distinction with AMSAN sometimes difficult [83]. Also, transient conduction slowing and block may be encountered during the early stages of AMAN, leading to incorrect diagnosis of AIDP [106, 117]. Similarly, some patients initially classified as “axonal” by nerve conduction studies have distal conduction block, and follow-up studies demonstrate rapid recovery of motor and/or sensory amplitudes typical of distal demyelination as seen in AIDP [92]. A useful diagnostic clue is the time course of EDX abnormalities. The nadir of conduction slowing in AIDP is 3–6 weeks after onset of symptoms, and this corresponds with the beginning of the clinical improvement [95]. In contrast, when conduction slowing or block is present in AMAN, it is evident early during the first 3 weeks of illness and rapidly resolves in parallel with clinical improvement [106, 117].
Prognosis of GBS Using Electrodiagnostic Studies
It has been long known that this presence of axonal damage in GBS is generally associated with worse outcome. In contrast, there is no association between slowing of nerve conduction velocities or F-wave latencies and clinical recovery. Early studies indicated a relationship between the detection of fibrillation potentials and poor outcome [118], but others have failed to confirm this finding as fibrillation potentials may occur when minimal amount of axonal loss has occurred [75, 107]. Also, fibrillation potentials may take several weeks to appear which renders needle EMG findings less useful.
Reduced CMAP amplitude is the most important predictor of outcome in GBS. Reduced mean CMAP amplitude (<20 % of the lower limit of normal) or absent CMAPs are strongly associated with a poor prognosis [66, 75, 119]. Hadden and colleagues demonstrated that 42 % of patients with an axonal loss pattern, on follow-up studies, were nonambulatory after 48 weeks [75]. On individual basis, one should be careful in making definite prognostic implications based on EDX studies only during the first 2–3 weeks of illness for several reasons: (1) Patients with axonal GBS studied early may show normal CMAP amplitude before the onset and completion of Wallerian degeneration [120]; (2) EDX evidence of conduction block when the site of stimulation is advanced proximally does not always imply segmental demyelination since primary axonal degeneration may manifest with conduction block before the completion of Wallerian degeneration [121]; and (3) although low distal CMAP amplitudes imply almost always axonal loss, a severe distal demyelinative conduction block may mimic axonal degeneration and shows improvement of CMAP over a short period of time with good prognosis [94].
Serial EDX studies are extremely useful in GBS for the accurate diagnosis and prognosis of GBS. The disorder often evolves over several days to weeks, and a single early EDX study and before the illness reaches its nadir may be misleading. This lone study may miss the pathological changes that may have not been completed including Wallerian degeneration and sometimes rapid remyelination. Also, signs of segmental demyelination are most evident during the third and fourth weeks of illness [95]. These serial EDX studies are also mandatory for proper diagnosis and classification of GBS subtypes (AIDP, AMAN, and AMSAN). In many cases, the relative contributions of primary demyelination and axonal degeneration, or a combination of the two, cannot be determined with any certainty except if EDX studies are performed many weeks after the onset of disease and corroborated by the outcome.
Cerebrospinal Fluid Studies
Typically, the CSF protein level is elevated in the majority of patients at some time during the course of the illness. The protein concentration is elevated in only 50 % of patients during the first week of illness and is elevated in 75 % by the third week [2]. Usually, the protein level peaks in the second or third weeks of the illness followed by a slow decline towards normal that may take several months. The cause of increased CSF protein is not known but presumably results from abnormalities in the blood-CSF barrier due to inflammation at the level of the spinal nerve roots. Patients with GBS and exceptionally high protein levels (e.g., 1,500 mg/dL) may develop papilledema and symptoms of pseudotumor cerebri. Patients with AMAN and AMSAN also tend to have elevated CSF protein levels, but the frequency of this finding and the protein concentration is usually lower compared to patients with AIDP; normal values are not unusual. There is also correlation between the presence of demyelination on EDX studies and the CSF protein concentration during the first 2 weeks of illness [100]. There is no apparent correlation between the CSF protein level and clinical findings or outcome.
The increase in CSF protein is not usually associated with a cellular response. This “cyto-albuminologic dissociation” was observed first by Guillain-Barré and Strohl in their first cases and made the disease credible by differentiating it from a number of febrile paralytic disorders, particularly poliomyelitis [1]. However, in most large series, minorities, usually less than 10 %, of patients have a slight lymphocytic CSF pleocytosis greater than 10 cells/mm. GBS that follows Lyme or HIV infection often has a more prominent pleocytosis that reflects a concurrent meningeal reaction [26, 122]. Therefore, the presence of cells in the CSF certainly does not exclude the diagnosis, but other infectious disorders, such as Lyme and HIV [26, 122], or malignant conditions, such as lymphoma [32], should be excluded.
Antiganglioside Antibodies
Gangliosides are a large family of glycosphingolipids, predominantly distributed on the cell-surface membrane. Although antibodies against many of these gangliosides have been detected in sera of GBS patients, including LM1, GM1, GM1b, GM2, GD1a, GalNAc-GD1a, GD1b, GD2, GD3, GT1a, and GQ1b, the pathological significance of some of these antibodies is not known. In addition, most of these antibodies are found in subgroups of GBS patients. Antibodies to GD3, GT1a, and GQ1b are often seen in high percentage of patients with GBS associated with ophthalmoplegia, and antibodies to GQ1b are detected in 95 % of patients with MFS [123]. Antibodies to GM1, GM1b, GD1a, or GalNAc-GD1a are associated with about 50 % of patients with AMAN or axonal variants of GBS [78]. When strict criteria for GBS subtypes are used, IgG autoantibodies against GM1 or GD1a are associated with AMAN, AMSAN, and acute motor-conduction-block neuropathy (see below), but not with AIDP. No specific ganglioside antibody appears to be associated with AIDP, the prototypical and most common form of GBS in North America and Europe. This explains why these ganglioside antibodies are not clinically useful in clinical practice in these countries.
Imaging Studies
Magnetic resonance imaging (MRI) is most useful in excluding central nervous system disorders which may mimic GBS in their presentations. This includes MRI of the brain to rule out brainstem pathology and MRI of the cervical and thoracic spine to exclude cord compression or transverse myelitis. MRI of the lumbar spine, however, is often abnormal and shows nerve root enhancement of the cauda equina with gadolinium. This occurs in up to 80–90 % of patients particularly in children and in patients with severe weakness and severe leg and back pain (Fig. 28.3) [124, 125]. Occasionally, the facial nerves may also enhance with gadolinium in GBS patients with facial palsies [126].

MRI of lumbar spine in a 4 year old with GBS showing enhancement of cauda equina. T1 weighted image (left panel) is normal while T1 weighted image following contrast shows enhancement of nerve roots (white arrow, right panel)
Other Laboratory Studies
Routine laboratory studies are usually normal in patients with GBS. The erythrocyte sedimentation rate or liver function studies are infrequently elevated, although these findings most likely reflect a recent, preceding infectious illness and have little diagnostic utility. Viral antibody titers, particularly EBV and CMV, may be increased and thus help to identify the triggering infectious agent in individual cases. Prior infection with C. jejuni may be confirmed by detecting elevated serum IgM antibodies or culturing the bacteria from the stool [19].
An HIV titer should be obtained in patients with GBS who have CSF pleocytosis, risk factors for HIV or reside in geographic regions where AIDS is prevalent. Urine porphyrin screen, heavy metal testing, and Lyme titer may be indicated in selected cases but are rarely necessary. Stool culture and serum titers for C. botulinum and anti-acetylcholine receptor antibodies are helpful in patients when the ocular-pharyngeal-brachial variant is considered or when electrophysiological studies are consistent with a disorder of neuromuscular transmission. Creatine kinase (CK) may be slightly elevated, particularly in patients with muscle pain and tenderness [63]. A very high serum CK level distinguishes GBS from rhabdomyolysis or acute myopathies. Severe electrolyte imbalance may cause generalized weakness that rarely mimics GBS, and levels of magnesium, phosphorus, and potassium should be checked.
Differential Diagnosis
Acute GBS is easily recognized in typical cases, but unusual presentations expand the differential diagnosis to many other central nervous system and neuromuscular diseases (Table 28.6). With several subtypes and variant syndromes (see below), GBS may mimic a variety of neurological disorders [127]. Careful history and examination, coupled with cautious interpretation of diagnostic testing, is often necessary for accurate diagnosis. The first step is to establish that the clinical features are a consequence of a peripheral nerve condition.
Acute Peripheral Neuropathies and Poliomyelitis
Apart for GBS, the majority of acute peripheral neuropathies are toxic in nature. Acute toxic neuropathies are axonal and evolve in a subacute or chronic fashion. There are numerous environmental, industrial (heavy metals), and occupational toxins that cause a neuropathy, resembling GBS, following an acute exposure, including if a history of ingestion is lacking. Acute arsenic poisoning may have a presentation that is indistinguishable from GBS and AIDP. The EDX studies may show findings of acquired demyelinating polyneuro-pathy, which when repeated, convert to a dying-back neuropathy [128]. Thallium, lead, n-hexane (glue-sniffers neuropathy), and organophosphate poisoning are other examples of acute toxic neuropathies. In most instances, intoxication is heralded by gastrointestinal symptoms, and usually there is involvement of other organ systems, skin lesions, alopecia, and encephalopathy, coma, or other features of central nervous system toxicity. Acute toxic neuropathies are distinguished from GBS by the history of preceding toxic ingestion and by detecting suspected toxins in the serum or urine. Acute demyelinating neuropathies mimicking GBS have been linked to certain medications, such as amiodarone, perhexiline, and gold therapy for rheumatoid arthritis. An acute, rapidly progressive neuropathy, similar to GBS, has been described in alcoholics [129, 130]. These patients invariably have a long-standing history of alcohol abuse prior to onset of the acute neuropathy. Most develop progressive generalized weakness, severe distal sensory loss, and areflexia over days to weeks. The condition is distinguished from GBS by a normal CSF protein concentration and axonal features on EDX studies.
Patients with acute intermittent porphyria (AIP) may develop a neuropathy that resembles GBS [131–133]. A variety of medications or infections may trigger an acute attack. Initial symptoms include vomiting, constipation, and abdominal pain. Seizures occur in 10–20 % of cases and delirium or other psychiatric symptoms occur in most. The weakness is symmetric and begins in proximal muscles of the arms, but widespread weakness develops in most as the syndrome progresses. Hypertension, arrhythmias, or other features of dysautonomia are common. The cranial nerves are typically spared. EMG shows an axonopathy rather than demyelination, thus differentiating this condition from typical GBS. Increased urinary excretion of delta-aminolevulinic acid and porphobilinogen during an acute attack of AIP establishes the diagnosis.
Neuropathy is a common complication of systemic vasculitis and usually evolves in a subacute fashion; rarely, acute mononeuritis multiplex may have a fulminant course that simulates an acute polyneuropathy. Polyarteritis nodosa, and hepatitis B or C-associated vasculitis Churg-Strauss syndrome are the vasculitides most likely to cause a rapidly progressive polyneuropathy [134]. Focal or multifocal onset, severe pain, lack of diaphragmatic weakness or cranial nerve involvement, and electrophysiological features of a multifocal axonopathy distinguish acute vasculitic neuropathy from GBS in most cases. Furthermore, the CSF protein level is normal in the former condition. The diagnosis is established by pathologic evidence of vasculitis on biopsy material. Nonsystemic vasculitic neuropathy is generally an indolent condition that is rarely mistaken for GBS.
Lyme disease, a tick-borne illness, may be a consideration in cases of GBS presenting with facial diplegia or when there is a CSF pleocytosis. However, as already noted, most cases of Lyme neuropathy are characterized by chronic, slowly progressive, distal sensory loss or an asymmetric, painful polyradiculopathy.
Poliomyelitis, once the most common cause of acute paralysis in the world, is now a rarity except in few underdeveloped countries (see Chap. 19). The condition is most commonly contracted by non-vaccinated individuals after exposure to infants who recently were vaccinated against the poliovirus. A number of other viruses, typically the enteroviruses such as the West Nile virus, may produce an identical poliomyelitis syndrome and are a more common cause of acute motor neuronopathy simulating GBS. Affected individuals have a febrile illness, usually a gastroenteritis, followed by a paralytic phase 7–14 days later. The neurologic syndrome begins with fever, headache, and neck stiffness, followed by muscle pain, asymmetric flaccid limb paralysis, and fasciculations, and reaches a nadir within 4 weeks. Mild confusion may occur early in the illness. Diaphragmatic and oropharyngeal weakness are common but the extraocular muscles and sensory functions are spared. The CSF shows a lymphocytic pleocytosis and a normal or mildly elevated protein concentration, in contrast to GBS. Similarly, nerve conduction studies in poliomyelitis show reduced or absent motor amplitudes with normal conduction velocities and no conduction block. Sensory potentials are normal. Fibrillations are prominent in weak muscles early in poliomyelitis. Culture of the poliovirus from the pharynx or stool or detection of elevated antibodies directed against poliovirus with acute and convalescent sera establishes the diagnosis.
During the West Nile encephalitis epidemic in North America in the late 1990s and early 2000s, many patients with weakness were diagnosed mistakenly as GBS. Upon further studies, it was clear that these were cases of West Nile-induced poliomyelitis with evidence of anterior horn cell damage [135, 136]. When motor weakness occurs in West Nile, it may be asymmetric with paralysis of one limb (monoparesis) or fairly symmetric affecting four limbs (quadriparesis), with or without brainstem involvement and respiratory failure. EDX studies also reveal normal SNAPs, low-amplitude CMAPs recorded from weak muscles, signs of diffuse active denervation (including the paraspinal muscles), and generalized or focal loss of motor units [137]. The pathology is consistent with poliomyelitis with identified neuronal loss, perivascular chronic inflammation, and microglial proliferation in the ventral horns of the spinal cord, especially in the cervical and lumbar segments [138].
Diphtheria is also a rare cause of acute polyneuropathy in developed countries because of effective vaccination programs. It is also reported in closed communities in countries where diphtheria remains endemic despite prior vaccination [139]. Infection with Corynebacterium diphtheria produces a febrile syndrome with severe pharyngitis, followed by palatal weakness and descending paralysis that develops weeks later. A membranous exudate over the tonsils and pharynx is present in most cases, and cervical adenopathy may be prominent. Bulbar weakness is present in the majority of patients. Over half the patients develop paralysis of accommodation. Symmetric limb weakness may be mild or severe with a predilection for proximal muscles. There is generalized areflexia, minimal sensory loss, and no dysautonomia. The course is slower than GBS, with maximal paralysis developing as long as 3 months after the onset of palatal weakness. The CSF profile demonstrates a cyto-albuminologic dissociation, similar to GBS, and EDX studies show an acute demyelinating neuropathy. Differentiation from GBS may be impossible if the early facial involvement is not recognized, but the low prevalence of diphtheria in developed countries makes GBS a more likely cause of acute, generalized, areflexic paralysis.
Critical illness polyneuropathy (CIP) was once frequently mistaken for GBS in the intensive care setting (see Chap. 76). It is now recognized as a common cause of limb weakness and failure to wean in ventilated patients in intensive care units (ICU). CIP is encountered most often in patients who have had sepsis and multiorgan failure [140–142]. Flaccid limb weakness, muscle atrophy, and generalized hypo- or areflexia are found in most patients. A small minority have facial weakness or ophthalmoparesis. Accurate sensory examination may be difficult in ventilated ICU patients, but most patients appear to have distal loss of all sensory modalities. Dysautonomia does not occur. The CSF protein concentration is normal and EDX studies demonstrate axonal loss with diffuse denervation without demyelination. Critical illness myopathy and prolonged exposure to neuromuscular blocking agents are other conditions that may simulate GBS in the ICU.
Disorders of the Neuromuscular Junction
Botulism is a rare condition that begins with cranial nerve dysfunction followed by generalized paralysis and can be confused with the oropharyngeal or ophthalmoparetic regional variants of GBS (see Chap. 50). Food-borne botulism usually begins hours to days after the ingestion of the neurotoxin produced by Clostridium botulinum types A, B, or E by way of contaminated food. Nausea and vomiting are followed by constipation and neurological symptoms. Blurred vision is an early complaint. Initial findings include dilated pupils in most patients, with paralysis of accommodation, ptosis, and oropharyngeal weakness. Diaphragmatic weakness with ventilatory failure is common and may be more severe than limb weakness. In contrast to GBS, the deep tendon reflexes are usually preserved. EDX studies show reduced amplitudes of the motor potentials with an incremental increase of the amplitude (usually >100 % above baseline) following high-frequency repetitive nerve stimulation, indicating a presynaptic neuromuscular junction abnormality. The CSF is normal. Detection of botulinum toxin in the serum, contaminated food source, or culture of Clostridium botulinum from the stool confirms the diagnosis.
In myasthenia gravis (MG), the slow onset of weakness, prominent fluctuation, fatigue, and the regional pattern of involvement usually poses little difficulty distinguishing these cases from GBS, but rarely patients with MG have a fulminant course with rapidly progressive limb and respiratory muscle weakness resembling an ocular-pharyngeal-brachial variant of GBS (see below) [143]. Although ptosis occurs in a minority of patients with GBS and may be transiently responsive to edrophonium, though it does not fatigue. Similarly, oropharyngeal weakness, nasal speech, and hypophonia do not fluctuate in GBS patients. Preserved deep tendon reflexes and lack of sensory symptoms or signs, dysautonomia, or an elevated CSF protein level are other features that differentiate MG from variant patterns of GBS. Patients with MG usually have transient improvement in strength following administration of edrophonium. EDX studies easily differentiate the two conditions; in MG, nerve conduction studies are normal and low-frequency repetitive nerve stimulation recording from a clinically affected muscle demonstrates a decrement (>10 %) of the amplitude characteristic of a postsynaptic neuromuscular junction abnormality.
Tick paralysis causes rapidly progressive paralysis that perhaps simulates GBS more closely than any other condition [144]. The illness is rare and affects mostly children in the northwestern United States in the spring and summer. A short prodrome characterized by fatigue, paresthesias, and ataxia is followed by generalized, areflexic weakness that occurs 3–5 days after attachment of the tick. The CSF is normal throughout the illness. The EDX studies show reduced motor amplitudes without demyelinating features; sensory studies are normal. The diagnosis is established by finding the tick, usually located at the hairline, and removal is followed by rapid recovery.
Central Nervous System Disorders
Occlusion of the basilar artery with pontine infarction may produce flaccid quadriparesis with ocular and bulbar findings. The symptoms usually begin suddenly and often there is a history of preceding transient ischemic attacks. Deep tendon reflexes may be reduced at the initial evaluation although hyperreflexia develops after a few days or weeks. Babinski signs are usually present. Vertical eye movements are preserved and other cranial nerve findings may be present but are usually asymmetrical. Most patients with brainstem stroke are somnolent or comatose because of involvement of the ascending reticular activating system.
Acute cervical transverse myelitis may cause a rapidly progressive quadriparesis. The detection of a spinal sensory level on the trunk and upper motor neuron findings (hyperreflexia, extensor plantar responses) differentiates this condition from GBS, but in the spinal shock stage of an acute myelopathy, flaccid limb weakness and areflexia simulate a lower motor neuron condition. In the acute inflammatory myelopathies (multiple sclerosis, neuromyelitis optica, or acute disseminated encephalomyelitis), weakness is often asymmetric, bowel and bladder dysfunction is an early and prominent finding, and the cranial nerves are normal. There is usually an inflammatory response with lymphocytes in the spinal fluid. Increased signal abnormalities on T2-weighted MRI or gadolinium enhancement of the cervical or thoracic cord establish the diagnosis.
Anxiety or a panic attack may be considered early in the illness when paresthesias are the only symptom of emerging GBS and deep tendon reflexes may be preserved. These patients may be labeled as “anxious” with symptoms attributed to hyperventilation and are often discharged from the emergency room, only to return later with generalized and diaphragmatic weakness.
Treatment and Management
Supportive Care
Most patients with GBS require admission to an intensive care unit (ICU) under the care of physicians who are familiar with the medical complications that develop in paralyzed ICU patients (Table 28.7) [146, 147]. The timely and skillful management of medical problems is as important as immune therapy in the outcome of patients with GBS.
Ventilatory failure is a central issue and should be anticipated in any GBS patient with progressive limb or oropharyngeal weakness. About 15–30 % of patients with GBS will need ventilatory support [65, 147]. Atelectasis develops early and leads to mild hypoxemia. Hypercarbia and hypoxemia is a later finding as ventilatory failure advances; therefore, arterial blood gases are not as helpful as respiratory mechanics in monitoring the evolution of diaphragmatic weakness in GBS patients. Vital capacity (VC), tidal volume, and negative inspiratory force are reasonably sensitive reflections of diaphragmatic power, and progressive decline in these values indicates impending mechanical respiratory failure and the need for ventilatory support (Fig. 28.4) [146]. These measures should be obtained early on in the course and repeated as frequently as dictated by the clinical state (up to every 4–6 h). Bulbar dysfunction including swallowing problems with difficulty in clearing secretions increases the odds of needing ventilator support. In general, if the forced vital capacity is less than 15–20 mL/kg, the maximum expiratory pressure is less than 30 cmH2O and the maximum inspiratory pressure is less than 30 cmH2O, impending respiratory arrest is present and intubation should be performed [148]. Also, PCO2 above 48 mmHg or PO2 less than 56 mmHg on room air are definite indications for intubation [2, 149].

Schematic diagram of pathophysiologic events in patients with ventilatory failure and Guillain-Barré syndrome, with corresponding suggested management (Reprinted with permission from Ropper AH [190])
Prediction for respiratory failure in patients with GBS is important in order to improve outcome and reduce mortality. Several studies have recently tried to answer this question. Walgaard et al. found that patients who required ventilatory support had facial and/or bulbar weakness, shorter days between onset of weakness and admission, and more limb weakness (higher Medical Research Council sum score) [65]. Sharshar et al. proposed that patients should be monitored in ICU setting if at least one of the following predictors of respiratory failure is present: (1) time from onset to admission of <7 days, (2) inability to cough, (3) inability to stand, (4) inability to lift the elbows, (5) inability to lift or head, or (6) vital capacity <60 % of predicted. More than 85 % of patients with 4 out of these 6 predictors were intubated [150]. Moreover, Durand et al. found that the EDX is a predictor for mechanical ventilation. They found that the risk of respiratory failure was very low in GBS patients with less than 55.6 % conduction block of the common peroneal nerve [151].
Incentive spirometry is useful in the early stages of the illness to prevent atelectasis. Frequent suctioning and chest physiotherapy minimize the accumulation of secretions and prevent aspiration and bronchopneumonia, but patients with moderate oropharyngeal weakness (e.g., those who cannot safely swallow liquids) will probably require intubation for protection of the airway. Most patients with severe, ventilator-dependent GBS who have no improvement after 2 weeks require tracheostomy to secure long-term airway management, avoid tracheal stenosis, facilitate suctioning, and maximize patient comfort. However, tracheostomy can be deferred for another week if pulmonary function tests show any significant improvement form baseline. Older patients with preexisting pulmonary disease are more likely to require tracheostomy [152].
Autonomic dysfunction is a well-recognized feature of GBS and a significant source of mortality. It is present in 70 % of patients, but serious and potentially fatal dysautonomia occurs in 20 % of patients. Severe autonomic disturbances affect mainly patients with severe weakness and those with respiratory failure. Many of the features of autonomic dysfunction are self-limited and require no intervention. For example, resting tachycardia is common in GBS patients and does not require treatment except in those with active coronary artery disease and acute myocardial ischemia. Hypertension often develops as a consequence of a procedure (e.g., suctioning or catheter placement) but is usually transient and does not require therapy. Sustained high blood pressure (e.g., >180/95) may be managed with angiotensin-converting enzyme inhibitors or beta-blocking agents. Short-acting intravenous medications, such as nitroprusside or esmolol, are preferred for patients who have severe, labile hypertension and require immediate therapy. Conversely, postural hypotension, often precipitated by only minor position changes, can be effectively treated with a bolus of intravenous saline or by placing the patient in the supine position. Norepinephrine and other sympathomimetic agents generally should be avoided because of the risk of rebound hypertension, but vasopressors may be necessary for those with persistent supine hypotension. Invasive procedures or cholinergic medications may trigger excessive vagal discharges (“vagal spells”) and precipitate bradycardia, asystole, or other vagally mediated arrhythmias [153, 154]. These episodes are usually transient, but anti-arrhythmic medications (atropine) or cardiac pacing may be necessary [154]. Micturitional disturbances are common and can be managed with intermittent catheterization or an indwelling catheter [147, 155].
Nosocomial infections are probably the most common medical complication that develops in GBS patients in the ICU with pulmonary infections predominating [156]. Other infections include urinary tract infections and central venous catheters sepsis. Tracheitis and sinusitis are other considerations in intubated patients who have a persistent fever and no apparent source of infection. Every effort should be made to identify the organism to guide appropriate antibiotic therapy. Routine monitoring of the chest X-ray and sputum and urine cultures are useful. However, bacterial colonization occurs frequently, and patients should receive antibiotic therapy only when there is clinical evidence of an infection; inappropriate treatment only increases the risk of infection with resistant pathogens. Although indwelling urinary catheters are often necessary in GBS patients, the risk of colonization and infection increases after several days.
Immobilization that is associated with GBS predisposes to deep venous thrombosis and pulmonary embolism. Subcutaneous heparin or intermittent pneumatic compression boots should be used routinely, and chronic anticoagulation with warfarin may be considered for those who are bedbound or ventilator dependent for long periods. Low molecular weight heparin has become popular, but the value of neither the old nor new type of heparin has been established by a trial; they are presumed from experiences in general medicine to be helpful. Although mucous plugging is probably the most common cause of acute respiratory decompensation in ventilated patients with GBS, any unexplained episode of acute oxygen desaturation requires evaluation for pulmonary embolism.
Patients who are intubated or have dysphagia due to oropharyngeal weakness require nasogastric tube feedings to maintain long-term nutritional support. A gastrostomy tube can be placed at the time of tracheostomy in patients with prolonged recovery. Hyperalimentation may be necessary in patients with an ileus. Neostigmine or erythromycin can be effective to treat ileus [147]. Moreover, daily abdominal auscultation and monitoring of opioid administration should be performed. Judicious intravenous hydration is essential because insensible fluid losses may be substantial, leading to dehydration and exacerbating autonomic instability. Hyponatremia may develop, usually as a consequence of inappropriate antidiuretic hormone secretion. Hyponatremia is an independent indicator of poor prognosis [70].
Pain is a common symptom in patients with GBS and is often underappreciated by the medical staff [61, 63]. Narcotics are almost always needed to manage pain, and the use of nonnarcotic, chronic analgesics often provides additional relief. Carbamazepine and gabapentin are usually successful in alleviating pain during the acute phase of GBS and may be used for long-term management of neuropathic pain [147]. An excellent alternative for severe pain in the back or legs is epidural analgesia [157]. Most patients benefit from early physical and occupational therapy [158]. For example, passive range of motion of paralyzed joints prevents contractures, foot boards minimize the risk of foot drop and shortening of the Achilles tendon, and frequent turning and air mattresses reduce the risk of skin breakdown and development of decubitus ulcers. Symptoms of anxiety and depression are also common, occur at all stages of the illness, and should not be overlooked. Those who are paralyzed, intubated, and unable to communicate with their caregivers are most likely to experience fear and helplessness. Communication boards allow patients to maintain a connection with their families, nurses, and medical staff. Facilitating contact with recovered GBS patients provides additional psychological support.
Immunotherapy
Several large, randomized, controlled trials have demonstrated the benefits of plasma exchange when performed within the first 2 weeks of the illness. The North American study involved 245 patients randomized to receive plasma exchange, 200–250 mL/kg over five sessions within 2 weeks, or supportive care [159]. Treated patients improved more rapidly and regained the ability to walk earlier (an average of 53 days for the plasma exchange group vs. 85 days in untreated patients). Furthermore, those who required ventilator assistance and were treated with plasma exchange weaned sooner than controls (24 vs. 48 days). Similar findings were reported from French and Swedish trials [160, 161]. Severely affected patients with features that are associated with a poor prognosis (see below) also benefited from treatment, but probably less so than others. The efficacy of plasma exchange is reduced if therapy is initiated after 3 weeks from the onset of symptoms [159, 162]. Plasma exchange is most effective when started within the first 2 weeks of symptom onset [159], but benefits are still seen if started within 30 days of symptom onset [163]. Improvement may occur after as few as two exchanges in patients with mild GBS, but four exchanges, performed approximately on alternate days, appear necessary in those with moderate or severe forms of the illness [164]. Adding more exchanges is not indicated, since six exchanges are not superior to four in severe GBS [163, 164]. A recent report of the therapeutics and technology assessment subcommittee of the American Academy of Neurology found that plasmapheresis is effective for the treatment of moderate and severe GBS (class I studies, level A) and probably effective in mild disease (level B) [165].
Patients treated with plasma exchange generally experience few serious adverse effects. Treatment-related complications include pneumothorax at the time of placement of central venous catheters, line infection with bacteremia or sepsis, cardiac arrhythmias, hypotension from rapid fluid shifts, and excessive bleeding. In some patients, generally large men, the high volume of exchange necessary may be accomplished through antecubital veins, thus obviating the risk of a central venous catheter. Plasma exchange is difficult to conduct in young children and patients with severe dysautonomia, especially those with hypotension, or with active cardiac disease, coagulopathy, or hepatic failure. Plasma exchange is safe to perform in children and pregnant women.
Intravenous immune globulin was introduced as an alternative to plasma exchange because of its efficacy in other immune-mediated disorders, its relative safety, and ease of administration. A randomized trial comparing IVIG (400 mg/kg/day for 5 days) to plasma exchange in 150 patients with GBS established that (1) IVIG is an effective therapy for GBS, (2) the efficacy is comparable to plasma exchange, and (3) there was a low frequency of adverse effects [166]. A larger, international, randomized, multicenter, controlled trial of 379 patients with GBS (the “Sandoglobulin trial”) subsequently compared IVIG (400 mg/kg/day for 5 days), plasma exchange (50 mL/kg exchanges over 8–13 days), and combined therapy. It established that IVIG and plasma exchange had equivalent efficacy and that plasma exchange followed by IVIG provided no additional benefit [167]. There was no difference in functional disability scores at 4 and 48 weeks between IVIG, plasma exchange, and combined therapy, nor was there any difference in secondary outcome measures (time required to wean from mechanical ventilation, number of days to recover ambulation, and the proportion of patients unable to walk after 48 weeks). These findings were subsequently confirmed by several studies and meta-analysis, despite the lack of placebo studies in IVIG [163, 168, 169]. A recent report of the therapeutics and technology assessment subcommittee of the American Academy of Neurology concurred also that there is strong evidence (level A) to support the use of IVIG in GBS which should be initiated during the first 2 weeks of disease onset [169]. Also, the analysis concluded that the combination of plasmapheresis and IVIG is probably not better than either treatment alone.
The beneficial immunomodulator effects of IVIG are complex and not completely understood, but probably include neutralization of proinflammatory cytokines (especially tumor necrosis factor-alpha and interleukin-1 beta), downregulation of pathogenic antibodies, modulation of Fc-receptor-mediated phagocytosis, inhibition of complement deposition, and promotion of remyelination [36, 44].
Headache is probably the most common side effect of IVIG occurring in up to 16 % of patients [169]. It may be prevented by premedication with analgesics or corticosteroids and by slowing the rate of infusion. Other minor complications include transient fever, chills, and transient hypertension. Serious adverse effects are currently rare and occur in less than 5 % of patients. These include serum sickness reaction, aseptic meningitis, acute renal tubular necrosis (possibly due to a high osmotic load), and a hypercoagulable state with risk of deep vein thrombosis, especially in nonambulatory patients, and myocardial infarction or stroke, especially in patients with risk factors for cardiovascular disease. IVIG should not be administered to patients with known IgA deficiency, a rare genetic trait, because of a high risk of developing an anaphylactic reaction. Transmission of HIV has not been reported, and infection with hepatitis C virus has been reported during the 1990s but not since. Recent improvement of IVIG manufacturing using nanofiltration or caprylate (a saturated medium-chain fatty acid) has essentially eliminated the risk of viral transmission. IVIG can be administered safely to pregnant patients [170]. Because IVIG is generally well tolerated and easy to administer, it has become the preferred therapy for GBS in the United States [171].
The optimal IVIG dose needed for patients with GBS remains somehow controversial. The dose of IVIG was set arbitrarily at 400 mg/kg/day for 5 days (total dose 2 g/kg) based on its use in hematologic disorders. This dose was used in most subsequent studies of IVIG in patients with GBS. A longer duration or higher dose of IVIG treatment in patients with severe disease may be beneficial. Indeed, Raphael et al. found that, in select patients on mechanical ventilation, a 6-day course of IVIG (total dose 2.4 g/kg) is more beneficial than a 3-day treatment (total dose 1.2 g/kg) with more rapid rate of recovery [172]. More recently, Kuitwaard et al. compared IgG level in serum before and 2 weeks after infusion with the standard dose of IVIG (total dose 2 g/kg) [173]. They identified a large variation of IgG levels at 2 weeks following the infusion of the same standard dose and a dose–response relationship. More significantly, patients with slight or no increase in serum IgG at 2 weeks had more severe clinical deficit at nadir, and fewer were able to walk at 6 months. An international study is currently assessing whether additional IVIG in patients who do not show a meaningful rise in IVIG level is beneficial.
Approximately 10 % of GBS patients treated with plasma exchange or IVIG relapse after initial treatment. This rate is equal with the use of plasma exchange or IVIG [167, 174]. The cause of these relapses is unknown but may be related to persistence of active disease after completing therapy or a rebound in antibody production. Patients with a more protracted course may be at higher risk for a relapse, but no other predictive factors have been identified [175]. Occasionally, a relapse may be triggered by an intercurrent infection or an ongoing CMV or EBV infection. A single study indicated that a repeated course of IVIG administered to patients who do not respond or relapse following the initial treatment may be of some benefit [176], but this regimen is currently being evaluated by a controlled study. A GBS relapse raises the concern of more chronic disorder, namely, CIDP. Ruts and colleagues found recently that GBS patients deteriorate more rapidly (15–27 days) while CIDP worsen much slower (31–63 days) [177]. Also, CIDP is the likely diagnosis if patients diagnosed with GBS exhibit more than two treatment-related fluctuations.
The difficult issue of how to treat patients with severe GBS who have not improved with standard therapies remains not well clarified. The “Sandoglobulin trial” showed that a course of plasma exchange followed by IVIG is not better than plasma exchange of IVG alone [167]. A repeated dose of IVIG has only been shown to be effective in a small study [176]. An international study is being conducted in assessing the use of additional IVIG in unresponsive server GBS patients or those who relapse.
A discussion on GBS treatment cannot be completed without addressing corticosteroids. Anecdotal reports indicated for decades that corticosteroids were beneficial to patients with GBS, but larger, prospective studies showed subsequently that oral and intravenous high-dose corticosteroids made no difference in outcome [178, 179]. There is also limited evidence suggesting that oral corticosteroids may slow recovery from GBS [180]. In combination with intravenous immunoglobulin, intravenous methylprednisolone may have a short-term effect and hasten recovery but does not significantly affect the long-term outcome [181].
The efficacy of other pharmacological agents is unknown. A small randomized controlled trial demonstrated that interferon beta 1a (Rebif®) was not associated with significant clinical improvement [182]. A recent meta-analysis concluded that there are no beneficial effects on GBS from brain-derived neurotrophic factor or cerebrospinal fluid filtration [183].
Prognosis
The majority of patients with GBS have a rapidly progressive course followed by a plateau phase of varying duration. Three-quarters of patients reach a nadir by 1 week and virtually all (98 %) do so by 4 weeks (Fig. 28.5) [77, 184–186]. Approximately 15 % of patients have a mild condition, remain ambulatory, and recover after a few weeks. Conversely, 5–20 % of patients have a fulminant course and develop flaccid quadriplegia, ventilator dependence, and axonal degeneration, often within 2 days from the onset of symptoms [110, 187]. This group is often caused by axonal loss (AMSAN subtype); The recovery is delayed and virtually always incomplete [90]; at 1-year follow-up, most have substantial residual motor deficits [187]. In contrast to AMSAN, patient with AMAN have a prognosis which is comparable to patients AIDP. This may be due to rapidly reversible immune-mediated changes at the nodes of Ranvier (conduction failure) that occur in some patients with AMAN or the selective degeneration and subsequent quick regeneration of intramuscular motor nerve terminals [87, 89, 188].

Time from onset of the illness to onset of improvement (end of the plateau phase) in the Massachusetts General Hospital retrospective series. (Reprinted with permission from Ropper AH et al. [145])
Neurologic disability advances steadily until a plateau is reached. The plateau phase usually lasts several weeks but may be only a few days in mild cases or persist for months in patients with quadriplegia and ventilator dependence. At the time of the maximum deficit, approximately one-third of patients require assisted ventilation, almost half are wheelchair or bedbound, 7 % have trouble walking, and the remainder are ambulatory [189]. Once recovery begins, improvement follows a predictable course; patients who were quadriparetic often recover to walk in a few months, although about half have persistent symptoms at 1 year. An earlier study reported that at 1-year follow-up, 62 % had recovered completely, 14 % could walk but not run, 9 % could not walk without assistance, 4 % remained bedbound or ventilated, and 8 % died [190]. In a more recent study, full recovery or minor deficits were observed in 41 % of patients in the first month, 71 % in the third month, 86 % in the sixth month, and 92 % by the first year [184].
Features that predicted a poor recovery (inability to walk independently) in various studies include older age (>60 years), history of preceding diarrheal illness, recent CMV infection, fulminant and rapidly progressive course, ventilator dependence, hyponatremia, peroneal nerve conduction block, and greatly reduced CMAP mean amplitudes (<20 % of the lower limit of normal) or inexcitable nerves [66, 70, 159, 191].
Several clinical scoring systems to predict prognosis have been used in the past. The Erasmus University scales have been validated. The scales uses the GBS disability score [179] (Table 28.8) and the Medical Research sum (MRC) score [166] (Table 28.9), as well as several simple and easily obtainable clinical data. The Erasmus GBS Outcome Score (EGOS) was developed based on a prognostic model that uses easy-to-obtain patients’ clinical characteristics during the acute phase of illness to accurately predict the chance of being able to walk independently at 6 months. The EGOS is based on age, diarrhea, and GBS disability score at 2 weeks after hospital admission that accurately predicts the chance of being able to walk independently at 6 months [185]. A final score higher than four correlates with poor recovery (Table 28.10). The modified EGOS (mEGOS) model replaced the GBS disability score by the more detailed MRC sum score [186]. It is also applicable at hospital admission as well as 1 week after hospital admission (see Table 28.10). It is used to predict inability to walk independently at 4 weeks, 3 months, and 6 months. The mEGOS is advantageous because it can predict groups with poor prognosis early in the disease when treatment is considered to be most effective. In addition, the Erasmus respiratory insufficiency score was also validated with specific parameters which predict mechanical ventilation at the time of hospital admission. These include the time between onset of weakness and admission, the MRC sum score, and presence or absence of facial and/or bulbar weakness (see Table 28.10) [65].
The mortality from GBS, once reported to be as high as 10–20 % [192, 193], has been reduced to 1–5 % in North America and Europe, most likely because of the advent of critical care units with experience in the management of this disorder [194–196]. The predictors of death are similar to the predictors of poor disability outcome [194].
Approximately 3–6 % of patients with typical GBS develop a chronic relapsing course consistent with CIDP. These cases are referred to as acute-onset CIDP. There are no distinctive features that allow early recognition of these patients; however, CIDP should be considered very likely in patients with GBS whose relapses reach nadir after 8 weeks or have more than two relapses [177]. Some patients with a progressive phase of 4–8 weeks and a monophasic course have been labeled subacute inflammatory demyelinating polyneuropathy (SIDP) [197]. These patients fall between CIDP and a relapsing GBS. These patients often do not relapse and treatment could be completely tapered over several months. A few patients had bouts of recurrent GBS completely remitting and separated by long asymptomatic intervals [198, 199].
Guillain-Barré Syndrome Variants
Miller Fisher Syndrome
The triad of acute ophthalmoplegia, ataxia, and areflexia, initially described by Fisher in 1956, has been recognized as a variant of GBS on the basis of shared clinical features with the typical disease and EDX findings indicating an acute sensory neuropathy [200]. Miller Fisher syndrome accounts for approximately 5 % of GBS cases in North America and Europe but may represent as high as 20–25 % of GBS patients in Eastern Asia [201, 202]. In some patients, the illness progresses to generalized GBS that overtakes the other features [184, 203].
Diplopia is usually the first symptom, followed by limb or gait ataxia that appears within days. Additionally, there may be mild distal paresthesias, dysphagia, or mild proximal limb weakness in up to one-third to one-half of cases. In some patients, frank GBS develops with respiratory failure requiring ventilator support. In patients with the classical syndrome, respiratory failure is, however, rare [201, 202]. Unilateral or bilateral asymmetric abducens weakness is a common initial finding and often evolves to complete external ophthalmoplegia. Ptosis is present in the majority of patients but pupillary function is usually preserved (a small proportion do have internal ophthalmoplegia). Ataxia typically involves all the limbs and gait but may be asymmetric early in the illness. Large amplitude, discoordinated, and poorly metricated limb movements are indistinguishable from a cerebellar efferent ataxia. The deep tendon reflexes are absent in all fully developed cases. The Fisher syndrome can be confused with brainstem lesions such as encephalitis or infarction, but the presence of other central nervous system features (confusion, seizures, alternating hemiparesis, Babinski signs, etc.) and normal electrophysiologic studies in the latter conditions clarify the diagnosis.
The CSF protein level may be elevated in Fisher syndrome but less frequently than in typical cases of GBS [201]. The EDX findings are abnormal in half of the patients with absent or low-amplitude SNAPs or a sural spaing pattern. Motor con-duction abnormalities are rare [204]. In almost all patients (95–98 %) with Fisher syndrome studied, there has been elevated antiganglioside antibodies directed against the epitope GQ1b (Table 28.11) [51, 205]. The observation that paranodal regions of the oculomotor, trochlear, and abducens nerves are enriched with GQ1b ganglioside supports a role of anti-GQ1b antibodies in the pathogenesis of ophthalmoplegia. Indeed, patients with incomplete forms (acute ataxic neuropathy and acute ophthalmoplegia), generalized GBS with ophthalmoplegia, and Bickerstaff’s encephalitis also display the antibody [51, 205, 206]. Sera from patients with anti-GQ1b antibodies and Fisher syndrome induced rapid and reversible failure of release of acetylcholine from presynaptic motor nerve terminals and motor nerve terminal blockade [207, 208]. The ataxia is believed to be secondary to selective involvement of muscle spindle afferents [52, 205, 206].
About 20 % of patients with Miller Fisher syndrome have followed C. jejuni infection and 8 % followed H. Influenzae infection [209, 210]. The majority of patients peak at a median of 1 week and improvement starts at a median of 2 weeks [211]. Most patients are recovered completely by 6 months.
Miller Fisher Syndrome (MFS) and Bickerstaff’s encephalitis with variable CNS and PNS involvement are considered now part of a continuous clinical spectrum with a common pathogenesis related to the presence of anti-GQ1b antibody. Ataxia and ophthalmoplegia are common to both conditions. Impaired consciousness and upper motor neuron signs are more characteristic of Bickerstaff’s encephalitis. The current hypothesis is that this antibody reaches the brainstem via area postrema, attacking the reticular formation and corticospinal tract, among other structures. Hyperreflexia and Babinski’s sign are present in 1/3 of the Bickerstaff’s encephalitis patients. CSF albuminocytological dissociation occurs in around 25 % of patients during the first week of illness, increasing to 50 % in the second week. Pleocytosis is present in 30 % of cases, much more frequently than in the Fisher syndrome (around 5 %). MRI detects CNS lesions (brainstem and cerebellum) in 1/10 of the Bickerstaff’s encephalitis patients [205]. Elevated anti-GQ1b antibody is seen in up to 70 % of patients with Bickerstaff’s encephalitis (see Table 28.11).
Immunotherapy has been found to be effective only in retrospective studies [202, 212, 213]. IVIG slightly hastens the recovery of ophthalmoplegia and ataxia, but does not seem to influence patients’ outcomes, presumably because of the good natural history of Fisher syndrome. However, plasma exchange or IVIG should be administered as early as possible when the disorder overlaps with GBS or Bickerstaff brainstem encephalitis, because these conditions may not have as good a natural course as the Fisher syndrome [202]. Animal models of MFS with transference of GQ1b antibodies have showed efficacy of C5 complement inhibitors as promising treatment. Clinical application in human being is yet to be reported [214, 215].
Ataxic Variant (Acute Ataxic Neuropathy)
In 1962, Richter quoted the term ataxic GBS and described a patient with acute severe “cerebellar type ataxia” without proprioceptive sensory loss or ophthalmoplegia [216]. This group of patients presents with a rapid-onset ataxia, hypo- or areflexia, distal paresthesias, and CSF albuminocytological dissociation [206]. They have negative Romberg test with intact proprioception, and 65 % of them have elevated GQ1b antibodies (see Table 28.11). SNAPs are intact in 60 % and reduced or absent in the rest [206]. Clinically, this variant is similar to the Miller Fisher syndrome, except for the lack of ophthalmoplegia and reduced occurrence of elevated GQ1b antibodies (65 % vs. 98 % in Miller Fisher syndrome). Another group of acute ataxic patients, labeled “acute sensory ataxic neuropathy”, has similar presentation but positive Romberg test and marked proprioceptive sensory loss [217, 218]. About 90 % of these patients have low-amplitude or absent SNAPs [206]. Antibodies against the epitopes GQ1b or GD1b are present in about 50 % of these patients. It is now likely that the ataxic GBS, originally described by Richter, and the acute sensory axonal neuropathy form a continuous spectrum and represent an incomplete form of the Miller Fisher syndrome with similarly good prognosis. Some authors recently proposed the terms “acute ataxic neuropathy” [206] or “acute sensory axonopathy-ganglionopathy” to encompass both of these disorders [219]. The main differential diagnoses of ataxic GBS are subacute paraneoplastic sensory neuronopathy (anti-Hu syndrome), Sjögren syndrome, pyridoxine intoxication, and vitamin B12 and thiamine deficiency. Anecdotal reports have shown improvement with IVIG or plasma exchange [206].
Pharyngeal-Cervical-Brachial Variant
A regional pattern of weakness of the cervical, brachial, and oropharyngeal muscles exclusively is a rare variant of GBS [143, 220]. The clinical picture is characterized by a recent history of viral illness followed by severe weakness limited to pharyngeal and neck muscles at the onset, with spread to the arms and legs only after several weeks. However, upper limb weakness may sometimes precede the dysphagia. Muscle power, reflexes, and sensation are entirely spared in the legs. Facial weakness and respiratory failure also may occur, thus simulating a disorder of neuromuscular transmission. Curiously, ptosis is almost universal, further emulating myasthenia gravis [143, 184]. Ophthalmoparesis is rare. Aspiration is a common complication and most require intubation for airway protection. The CSF protein concentration is usually slightly increased. About half of the patients exhibit elevated anti-GT1a antibodies, perhaps reflecting an immune attack that is restricted to nerves with that particular epitope serving as an antigenic target [221]. Antibodies against GQ1b are also present in around a third of patients [221]. Nerve conduction studies may demonstrate demyelinating and axonal changes limited to the upper limbs. The blink reflexes are very useful and often show demyelinative changes that are commensurate with the severity of facial paralysis. Some patients with the pharyngeal-cervical-brachial variant of GBS show low-amplitude distal CMAPs and SNAPs and partial motor conduction blocks which normalized within 4 weeks [222]. This is consistent with reversible conduction failure in both motor and sensory fibers which can account for a better prognosis and similar to what is observed in AMAN. In the rest of the patients, recovery may take months and many patients need a gastrostomy tube for nutritional support and a tracheostomy for airway management. Case reports have shown improvement with IVIG or plasma exchange [223].
Multiple Cranial Neuropathy Variant
This GBS variant is characterized by acute onset of multiple cranial nerve dysfunction, multiple ocular motor nerve palsies, and facial and bulbar dysfunction [224]. This form accounts for around 5 % of GBS patients in Taiwan [224]. The findings are often due to involvement of individual cranial nerves, often bilaterally and symmetrically. This also helps distinguish this from the pharyngeal-cervical-brachial variant of GBS. Bilateral IX, X, and XI cranial nerve impairment, resulting in dysphagia, laryngopharyngeal discomfort, and slurred speech, is the initial symptom in most cases [203]. The second most common mode of presentation is facial nerve palsy, which is usually bilateral and of the peripheral type [223]. Complete or partial bilateral extraocular nerve palsies may later develop. Although not initially present, within 2–3 weeks most patients will develop sensory symptoms and limb weakness. Areflexia is usually present.
CSF analysis shows the characteristic albuminocytologic dissociation in half of the patients in the first week after disease onset, a somewhat higher rate and earlier occurrence than that seen in typical GBS [223]. Most case series report abnormalities in motor and sensory conduction velocities and abnormal F-wave responses. The characteristic rapid, progressive course with respiratory paralysis makes early recognition and prompt treatment very important. IVIG was effective in most case series and this is considered the preferred therapy [223, 224].
Facial Diplegia with Paresthesias
A syndrome of acute-onset facial diplegia often associated with distal limb paresthesias is another GBS variant [225]. Other cranial nerve involvements, limb weakness, or ataxia are absent. The majority of patients have hyporeflexia, while hyperreflexia has been described in this syndrome [226, 227]. More than 2/3 of them have a preceding infectious illness. One-third of the patients have positive serology for a recent CMV infection. All the patients had elevated CSF protein. Nerve conduction studies shows demyelinating changes in limb nerves in 60 % of patients. Favorable outcome occurred in the majority of patients and in some have residual facial weakness [225].
Paraparetic Variant
Another regional variant of GBS has features of isolated leg weakness and areflexia simulating a cauda equina or spinal cord syndrome [143]. The arms, ocular, facial, and oropharyngeal muscles are spared, and sphincteric function is normal. Radicular leg pain is common and may be severe, but other sensory features are more variable. The spinal fluid protein level is elevated in the majority. EDX studies show typical demyelinating features of GBS restricted to the legs. MRI of the spinal cord and lumbar roots should be obtained in these patients to exclude a lesion of the caudal spinal cord or cauda equina. Imaging may also show gadolinium enhancement of the lumbosacral nerve roots, as also occurs in many typical GBS cases [124].
Acute Pandysautonomia (Acute Autonomic Neuropathy)
An acute autonomic neuropathy is thought to be another rare variant of GBS [184, 228, 229]. Gastrointestinal symptoms are commonly the initial features, namely, abdominal pain, vomiting, constipation, or diarrhea, all following a viral syndrome. Gastroparesis or abdominal distention with an ileus may develop. Patients complain of lightheadedness due to orthostatic changes in blood pressure following positional changes, and in extreme forms, severe orthostatic hypotension with recurrent syncope are seen. Additional manifestations that are acquired in the first week or two include erectile dysfunction, urinary frequency, urgency, and retention; vasomotor instability with acrocyanosis; and reduced salivation, lacrimation, or sweating. Sensation is normal initially but most patients develop various degrees of sensory impairment without motor dysfunction [230]. The sensory symptoms tend to be segmental and asymmetrical, often associated with pain. The majority of patients have reduced or absent deep tendon reflexes after several weeks. Routine nerve conduction studies are typically normal or show reduced SNAPs particularly in patients with sensory ataxia. Specialized testing of autonomic functions (heart rate variability testing, quantitative axon reflex studies, tilt table testing) are abnormal. The CSF protein concentration is usually mildly elevated. Combined sensory and autonomic GBS variants are probably common [219, 231].
In order to diagnose this condition, one has to rule out a large number of disorders that may cause autonomic dysfunction (cancer, multiple system atrophy, Parkinson’s disease, amyloidosis, and others). Autoantibodies specific for nicotinic acetylcholine receptors in the autonomic ganglia are present in about 30 % of the paraneoplastic form and 50 % of the idiopathic autonomic ganglionopathies [232]. The seropositive group has less frequently a preceding viral illness but more frequently a subacute onset, abnormal pupillary responses, sicca complex, and lower gastrointestinal dysautonomia [233].
This GBS variant characteristically progresses and then plateaus after a few weeks, and approximately half the patients recover slowly after several months. There have been case reports of improvement with immunomodulatory therapy, including plasmapheresis or infusion of IVIG [234–236].
Other Possible GBS Variants
A number of other peculiar constellations of neuropathic disorders that develops acutely, often after a minor infectious illness, are possible variants of GBS. These conditions begin acutely, are mostly bilateral in one region, evolve over days or weeks, and are monophasic. Most are associated with an elevated spinal fluid protein concentration without a cellular response and with features of demyelination on EDX studies. These syndromes include acral paresthesias with diminished reflexes in either the arms or legs, abducens nerve palsies with distal paresthesias and hypo- or areflexia, isolated ophthalmoplegia (e.g., asymmetric oculomotor nerve palsies), and bilateral foot drop with upper limb paresthesias [237, 238]. A small fiber sensory neuropathy was also described and considered by some authors to be a GBS variant [239, 240]. The outcome in these patients was favorable.
References
Guillain G, Barré JA, Strohl A. Sur un syndrome de radiculonevrite avec hyper-albuminose du liquide cephalo-rachidien sans reaction cellulaire: remarques sur les caracterres cliniques et graphiques des reflexes tendineux. Bulletins et memoires de la societe des Medecins des Hopitaux de Paris. 1916;40:1462–70.
Yuki N, Hartung HP. Guillain-Barré syndrome. N Engl J Med. 2012;366(24):2294–304. Epub 2012/06/15.
van der Maas NA, Kramer MA, Jacobs BC, van Soest EM, Dieleman JP, Kemmeren JM, et al. Guillain-Barré syndrome: background incidence rates in The Netherlands. J Peripher Nerv Syst. 2011;16(3):243–9. Epub 2011/10/19.
McGrogan A, Madle GC, Seaman HE, de Vries CS. The epidemiology of Guillain-Barré syndrome worldwide. A systematic literature review. Neuroepidemiology. 2009;32(2):150–63. Epub 2008/12/18.
Sejvar JJ, Baughman AL, Wise M, Morgan OW. Population incidence of Guillain-Barré syndrome: a systematic review and meta-analysis. Neuroepidemiology. 2011;36(2):123–33. Epub 2011/03/23.
Asbury AK, Cornblath DR. Assessment of current diagnostic criteria for Guillain-Barré syndrome. Ann Neurol. 1990;27(Suppl):S21–4. Epub 1990/01/01.
Kaplan JE, Poduska PJ, McIntosh GC, Hopkins RS, Ferguson SW, Schonberger LB. Guillain-Barré syndrome in Larimer County, Colorado: a high-incidence area. Neurology. 1985;35(4):581–4. Epub 1985/04/01.
Souayah N, Michas-Martin PA, Nasar A, Krivitskaya N, Yacoub HA, Khan H, et al. Guillain-Barré syndrome after Gardasil vaccination: data from Vaccine Adverse Event Reporting System 2006–2009. Vaccine. 2011;29(5):886–9. Epub 2010/09/28.
Haber P, DeStefano F, Angulo FJ, Iskander J, Shadomy SV, Weintraub E, et al. Guillain-Barré syndrome following influenza vaccination. JAMA. 2004;292(20):2478–81. Epub 2004/11/25.
Souayah N, Nasar A, Suri MF, Qureshi AI. Guillain-Barré syndrome after vaccination in United States a report from the CDC/FDA Vaccine Adverse Event Reporting System. Vaccine. 2007;25(29):5253–5. Epub 2007/06/15.
Lehmann HC, Hartung HP, Kieseier BC, Hughes RA. Guillain-Barré syndrome after exposure to influenza virus. Lancet Infect Dis. 2010;10(9):643–51. Epub 2010/08/28.
Schonberger LB, Bregman DJ, Sullivan-Bolyai JZ, Keenlyside RA, Ziegler DW, Retailliau HF, et al. Guillain-Barré syndrome following vaccination in the National Influenza Immunization Program, United States, 1976–1977. Am J Epidemiol. 1979;110(2):105–23. Epub 1979/08/01.
Sejvar JJ, Pfeifer D, Schonberger LB. Guillain-Barré syndrome following influenza vaccination: causal or coincidental? Curr Infect Dis Rep. 2011;13(4):387–98. Epub 2011/06/18.
Kurland LT, Wiederholt WC, Kirkpatrick JW, Potter HG, Armstrong P. Swine influenza vaccine and Guillain-Barré syndrome. Epidemic or artifact? Arch Neurol. 1985;42(11):1089–90. Epub 1985/11/01.
Dieleman J, Romio S, Johansen K, Weibel D, Bonhoeffer J, Sturkenboom M, et al. Guillain-Barré syndrome and adjuvanted pandemic influenza A (H1N1) 2009 vaccine: multinational case–control study in Europe. BMJ. 2011;343:d3908. Epub 2011/07/14.
Deeks SL, Lim GH, Simpson MA, Rosella L, Mackie CO, Achonu C, et al. Estimating background rates of Guillain-Barré Syndrome in Ontario in order to respond to safety concerns during pandemic H1N1/09 immunization campaign. BMC Public Health. 2011;11:329. Epub 2011/05/19.
Shaikh AG, Termsarasab P, Nwankwo C, Rao-Frisch A, Katirji B. Atypical forms of Guillain-Barré syndrome and H1N1-influenza vaccination. Vaccine. 2012;30(22):3251–4. Epub 2012/03/21.
Jacobs BC, Rothbarth PH, van der Meche FG, Herbrink P, Schmitz PI, de Klerk MA, et al. The spectrum of antecedent infections in Guillain-Barré syndrome: a case–control study. Neurology. 1998;51(4):1110–5. Epub 1998/10/22.
Rees JH, Soudain SE, Gregson NA, Hughes RA. Campylobacter jejuni infection and Guillain-Barré syndrome. N Engl J Med. 1995;333(21):1374–9. Epub 1995/11/23.
Poropatich KO, Walker CL, Black RE. Quantifying the association between Campylobacter infection and Guillain-Barré syndrome: a systematic review. J Health Popul Nutr. 2010;28(6):545–52. Epub 2011/01/26.
Sheikh KA, Ho TW, Nachamkin I, Li CY, Cornblath DR, Asbury AK, et al. Molecular mimicry in Guillain-Barré syndrome. Ann N Y Acad Sci. 1998;845:307–21. Epub 1998/07/21.
Kuroki S, Saida T, Nukina M, Haruta T, Yoshioka M, Kobayashi Y, et al. Campylobacter jejuni strains from patients with Guillain-Barré syndrome belong mostly to Penner serogroup 19 and contain beta-N-acetylglucosamine residues. Ann Neurol. 1993;33(3):243–7. Epub 1993/03/01.
Feasby TE, Hughes RA. Campylobacter jejuni, antiganglioside antibodies, and Guillain-Barré syndrome. Neurology. 1998;51(2):340–2. Epub 1998/08/26.
Hadden RD, Karch H, Hartung HP, Zielasek J, Weissbrich B, Schubert J, et al. Preceding infections, immune factors, and outcome in Guillain-Barré syndrome. Neurology. 2001;56(6):758–65. Epub 2001/03/29.
Visser LH, van der Meche FG, Meulstee J, Rothbarth PP, Jacobs BC, Schmitz PI, et al. Cytomegalovirus infection and Guillain-Barré syndrome: the clinical, electrophysiologic, and prognostic features. Dutch Guillain-Barré Study Group. Neurology. 1996;47(3):668–73. Epub 1996/09/01.
Cornblath DR, McArthur JC, Kennedy PG, Witte AS, Griffin JW. Inflammatory demyelinating peripheral neuropathies associated with human T-cell lymphotropic virus type III infection. Ann Neurol. 1987;21(1):32–40. Epub 1987/01/01.
Piliero PJ, Fish DG, Preston S, Cunningham D, Kinchelow T, Salgo M, et al. Guillain-Barré syndrome associated with immune reconstitution. Clin Infect Dis. 2003;36(9):e111–4. Epub 2003/04/26.
Logigian EL, Steere AC. Clinical and electrophysiologic findings in chronic neuropathy of Lyme disease. Neurology. 1992;42(2):303–11. Epub 1992/02/01.
Thaisetthawatkul P, Logigian EL. Peripheral nervous system manifestations of lyme borreliosis. J Clin Neuromuscul Dis. 2002;3(4):165–71. Epub 2002/06/01.
Richardson Jr RF, Remler BF, Katirji B, Murad MH. Guillain-Barré syndrome after Cyclospora infection. Muscle Nerve. 1998;21(5):669–71. Epub 1998/05/08.
Zhang H, Wu J. Systemic lupus erythematosus associated relapsing polyneuropathy: recurrent Guillain-Barré syndrome or chronic inflammatory demyelinating polyradiculoneuropathy? Muscle Nerve. 2011;44(4):609–10. Epub 2011/09/17.
Lisak RP, Mitchell M, Zweiman B, Orrechio E, Asbury AK. Guillain-Barré syndrome and Hodgkin’s disease: three cases with immunological studies. Ann Neurol. 1977;1(1):72–8. Epub 1977/01/01.
Ho D, Thakur K, Gorson KC, Ropper AH. Influence of critical illness on axonal loss in Guillain-Barré syndrome. Muscle Nerve. 2009;39(1):10–5. Epub 2008/12/17.
Shin IS, Baer AN, Kwon HJ, Papadopoulos EJ, Siegel JN. Guillain-Barré and Miller Fisher syndromes occurring with tumor necrosis factor alpha antagonist therapy. Arthritis Rheum. 2006;54(5):1429–34. Epub 2006/04/29.
Cheng Q, Jiang GX, Fredrikson S, Link H, de Pedro-Cuesta J. Increased incidence of Guillain-Barré syndrome postpartum. Epidemiology. 1998;9(6):601–4. Epub 1998/11/03.
Hartung HP, Pollard JD, Harvey GK, Toyka KV. Immunopathogenesis and treatment of the Guillain-Barré syndrome – part I. Muscle Nerve. 1995;18(2):137–53. Epub 1995/02/01.
Willison HJ. Biomarkers in experimental models of antibody-mediated neuropathies. J Peripher Nerv Syst. 2011;16 Suppl 1Suppl 1:60–2. Epub 2011/07/01.
Hughes RA, Cornblath DR. Guillain-Barré syndrome. Lancet. 2005;366(9497):1653–66. Epub 2005/11/08.
Asbury AK, Arnason BG, Adams RD. The inflammatory lesion in idiopathic polyneuritis. Its role in pathogenesis. Medicine (Baltimore). 1969;48:173–215.
Honavar M, Tharakan JK, Hughes RA, Leibowitz S, Winer JB. A clinicopathological study of the Guillain-Barré syndrome. Nine cases and literature review. Brain. 1991;114(Pt 3):1245–69. Epub 1991/06/01.
Griffin JW, Li CY, Ho TW, Tian M, Gao CY, Xue P, et al. Pathology of the motor-sensory axonal Guillain-Barré syndrome. Ann Neurol. 1996;39(1):17–28. Epub 1996/01/01.
Zhu J, Mix E, Link H. Cytokine production and the pathogenesis of experimental autoimmune neuritis and Guillain-Barré syndrome. J Neuroimmunol. 1998;84(1):40–52. Epub 1998/05/26.
Sharief MK, Ingram DA, Swash M. Circulating tumor necrosis factor-alpha correlates with electrodiagnostic abnormalities in Guillain-Barré syndrome. Ann Neurol. 1997;42(1):68–73. Epub 1997/07/01.
Hartung HP, Pollard JD, Harvey GK, Toyka KV. Immunopathogenesis and treatment of the Guillain-Barré syndrome – part II. Muscle Nerve. 1995;18(2):154–64. Epub 1995/02/01.
Koski CL, Gratz E, Sutherland J, Mayer RF. Clinical correlation with anti-peripheral-nerve myelin antibodies in Guillain-Barré syndrome. Ann Neurol. 1986;19(6):573–7. Epub 1986/06/01.
Koski CL, Chou DK, Jungalwala FB. Anti-peripheral nerve myelin antibodies in Guillain-Barré syndrome bind a neutral glycolipid of peripheral myelin and cross-react with Forssman antigen. J Clin Invest. 1989;84(1):280–7. Epub 1989/07/01.
Hafer-Macko C, Hsieh ST, Li CY, Ho TW, Sheikh K, Cornblath DR, et al. Acute motor axonal neuropathy: an antibody-mediated attack on axolemma. Ann Neurol. 1996;40(4):635–44. Epub 1996/10/01.
Hafer-Macko CE, Sheikh KA, Li CY, Ho TW, Cornblath DR, McKhann GM, et al. Immune attack on the Schwann cell surface in acute inflammatory demyelinating polyneuropathy. Ann Neurol. 1996;39(5):625–35. Epub 1996/05/01.
Kaida K, Kusunoki S. Antibodies to gangliosides and ganglioside complexes in Guillain-Barré syndrome and Fisher syndrome: mini-review. J Neuroimmunol. 2010;223(1–2):5–12. Epub 2010/02/23.
Willison HJ, Veitch J, Paterson G, Kennedy PG. Miller Fisher syndrome is associated with serum antibodies to GQ1b ganglioside. J Neurol Neurosurg Psychiatry. 1993;56(2):204–6. Epub 1993/02/01.
Chiba A, Kusunoki S, Obata H, Machinami R, Kanazawa I. Serum anti-GQ1b IgG antibody is associated with ophthalmoplegia in Miller Fisher syndrome and Guillain-Barré syndrome: clinical and immunohistochemical studies. Neurology. 1993;43(10):1911–7. Epub 1993/10/01.
Liu JX, Willison HJ, Pedrosa-Domellof F. Immunolocalization of GQ1b and related gangliosides in human extraocular neuromuscular junctions and muscle spindles. Invest Ophthalmol Vis Sci. 2009;50(7):3226–32. Epub 2009/03/04.
Griffin JW, Li CY, Macko C, Ho TW, Hsieh ST, Xue P, et al. Early nodal changes in the acute motor axonal neuropathy pattern of the Guillain-Barré syndrome. J Neurocytol. 1996;25(1):33–51. Epub 1996/01/01.
Yuki N, Susuki K, Koga M, Nishimoto Y, Odaka M, Hirata K, et al. Carbohydrate mimicry between human ganglioside GM1 and Campylobacter jejuni lipooligosaccharide causes Guillain-Barré syndrome. Proc Natl Acad Sci U S A. 2004;101(31):11404–9. Epub 2004/07/28.
Koga M, Takahashi M, Masuda M, Hirata K, Yuki N. Campylobacter gene polymorphism as a determinant of clinical features of Guillain-Barré syndrome. Neurology. 2005;65(9):1376–81. Epub 2005/09/16.
Yuki N, Koga M. Bacterial infections in Guillain-Barré and Fisher syndromes. Curr Opin Neurol. 2006;19(5):451–7. Epub 2006/09/14.
Koga M, Gilbert M, Takahashi M, Li J, Koike S, Hirata K, et al. Comprehensive analysis of bacterial risk factors for the development of Guillain-Barré syndrome after Campylobacter jejuni enteritis. J Infect Dis. 2006;193(4):547–55. Epub 2006/01/21.
Jacobs BC, Koga M, van Rijs W, Geleijns K, van Doorn PA, Willison HJ, et al. Subclass IgG to motor gangliosides related to infection and clinical course in Guillain-Barré syndrome. J Neuroimmunol. 2008;194(1–2):181–90. Epub 2008/01/08.
Ropper AH. Weekly clinicopathological exercises. N Eng J Med. 1999;341:1996–2003.
Yuki N, Kokubun N, Kuwabara S, Sekiguchi Y, Ito M, Odaka M, et al. Guillain-Barré syndrome associated with normal or exaggerated tendon reflexes. J Neurol. 2012;259(6):1181–90. Epub 2011/12/07.
Ruts L, Drenthen J, Jongen JL, Hop WC, Visser GH, Jacobs BC, et al. Pain in Guillain-Barré syndrome: a long-term follow-up study. Neurology. 2010;75(16):1439–47. Epub 2010/09/24.
Moulin DE, Hagen N, Feasby TE, Amireh R, Hahn A. Pain in Guillain-Barré syndrome. Neurology. 1997;48(2):328–31. Epub 1997/02/01.
Ropper AH, Shahani BT. Pain in Guillain-Barré syndrome. Arch Neurol. 1984;41(5):511–4. Epub 1984/05/01.
Bakshi N, Maselli RA, Gospe Jr SM, Ellis WG, McDonald C, Mandler RN. Fulminant demyelinating neuropathy mimicking cerebral death. Muscle Nerve. 1997;20(12):1595–7. Epub 1997/12/09.
Walgaard C, Lingsma HF, Ruts L, Drenthen J, van Koningsveld R, Garssen MJ, et al. Prediction of respiratory insufficiency in Guillain-Barré syndrome. Ann Neurol. 2010;67(6):781–7. Epub 2010/06/03.
McKhann GM, Griffin JW, Cornblath DR, Mellits ED, Fisher RS, Quaskey SA. Plasmapheresis and Guillain-Barré syndrome: analysis of prognostic factors and the effect of plasmapheresis. Ann Neurol. 1988;23(4):347–53. Epub 1988/04/01.
Gracey DR, McMichan JC, Divertie MB, Howard Jr FM. Respiratory failure in Guillain-Barré syndrome: a 6-year experience. Mayo Clin Proc. 1982;57(12):742–6. Epub 1982/12/01.
Asahina M, Kuwabara S, Suzuki A, Hattori T. Autonomic function in demyelinating and axonal subtypes of Guillain-Barré syndrome. Acta Neurol Scand. 2002;105(1):44–50. Epub 2002/03/21.
Zochodne DW. Autonomic involvement in Guillain-Barré syndrome: a review. Muscle Nerve. 1994;17(10):1145–55. Epub 1994/10/01.
Saifudheen K, Jose J, Gafoor VA, Musthafa M. Guillain-Barré syndrome and SIADH. Neurology. 2011;76(8):701–4. Epub 2011/02/23.
Bensa S, Hadden RD, Hahn A, Hughes RA, Willison HJ. Randomized controlled trial of brain-derived neurotrophic factor in Guillain-Barré syndrome: a pilot study. Eur J Neurol. 2000;7(4):423–6. Epub 2000/09/06.
Van Diest D, Van Goethem JW, Vercruyssen A, Jadoul C, Cras P. Posterior reversible encephalopathy and Guillain-Barré syndrome in a single patient: coincidence or causative relation? Clin Neurol Neurosurg. 2007;109(1):58–62. Epub 2006/02/21.
Ubogu EE, Zaidat OO, Suarez JI. Acute motor-sensory axonal neuropathy associated with active systemic lupus erythematosus and anticardiolipin antibodies. J Clin Rheumatol. 2001;7(5):326–31. Epub 2006/10/14.
Awad A, Mathew S, Katirji B. Acute motor axonal neuropathy in association with Sjogren syndrome. Muscle Nerve. 2010;42(5):828–30. Epub 2010/10/27.
Hadden RD, Cornblath DR, Hughes RA, Zielasek J, Hartung HP, Toyka KV, et al. Electrophysiological classification of Guillain-Barré syndrome: clinical associations and outcome. Plasma Exchange/Sandoglobulin Guillain-Barré Syndrome Trial Group. Ann Neurol. 1998;44(5):780–8. Epub 1998/11/18.
Ho TW, Mishu B, Li CY, Gao CY, Cornblath DR, Griffin JW, et al. Guillain-Barré syndrome in northern China. Relationship to Campylobacter jejuni infection and anti-glycolipid antibodies. Brain. 1995;118(Pt 3):597–605. Epub 1995/06/01.
Hiraga A, Mori M, Ogawara K, Hattori T, Kuwabara S. Differences in patterns of progression in demyelinating and axonal Guillain-Barré syndromes. Neurology. 2003;61(4):471–4. Epub 2003/08/27.
Hiraga A, Kuwabara S, Ogawara K, Misawa S, Kanesaka T, Koga M, et al. Patterns and serial changes in electrodiagnostic abnormalities of axonal Guillain-Barré syndrome. Neurology. 2005;64(5):856–60. Epub 2005/03/09.
McKhann GM, Cornblath DR, Griffin JW, Ho TW, Li CY, Jiang Z, et al. Acute motor axonal neuropathy: a frequent cause of acute flaccid paralysis in China. Ann Neurol. 1993;33(4):333–42. Epub 1993/04/01.
McKhann GM, Cornblath DR, Ho T, Li CY, Bai AY, Wu HS, et al. Clinical and electrophysiological aspects of acute paralytic disease of children and young adults in northern China. Lancet. 1991;338(8767):593–7. Epub 1991/09/07.
Griffin JW, Li CY, Ho TW, Xue P, Macko C, Gao CY, et al. Guillain-Barré syndrome in northern China. The spectrum of neuropathological changes in clinically defined cases. Brain. 1995;118(Pt 3):577–95. Epub 1995/06/01.
Takahashi M, Koga M, Yokoyama K, Yuki N. Epidemiology of Campylobacter jejuni isolated from patients with Guillain-Barré and Fisher syndromes in Japan. J Clin Microbiol. 2005;43(1):335–9. Epub 2005/01/07.
Capasso M, Notturno F, Manzoli C, Uncini A. Involvement of sensory fibres in axonal subtypes of Guillain-Barré syndrome. J Neurol Neurosurg Psychiatry. 2011;82(6):664–70. Epub 2011/03/30.
Jacobs BC, Schmitz PI, van der Meche FG. Campylobactery jejuni infection and treatment for Guillain-Barré syndrome. N Engl J Med. 1996;335(3):208–9. Epub 1996/07/18.
Rees JH, Gregson NA, Hughes RA. Anti-ganglioside GM1 antibodies in Guillain-Barré syndrome and their relationship to Campylobacter jejuni infection. Ann Neurol. 1995;38(5):809–16. Epub 1995/11/01.
Ho TW, Willison HJ, Nachamkin I, Li CY, Veitch J, Ung H, et al. Anti-GD1a antibody is associated with axonal but not demyelinating forms of Guillain-Barré syndrome. Ann Neurol. 1999;45(2):168–73. Epub 1999/02/16.
Ho TW, Hsieh ST, Nachamkin I, Willison HJ, Sheikh K, Kiehlbauch J, et al. Motor nerve terminal degeneration provides a potential mechanism for rapid recovery in acute motor axonal neuropathy after Campylobacter infection. Neurology. 1997;48(3):717–24. Epub 1997/03/01.
Kuwabara S, Yuki N, Koga M, Hattori T, Matsuura D, Miyake M, et al. IgG anti-GM1 antibody is associated with reversible conduction failure and axonal degeneration in Guillain-Barré syndrome. Ann Neurol. 1998;44(2):202–8. Epub 1998/08/26.
Ho TW, Li CY, Cornblath DR, Gao CY, Asbury AK, Griffin JW, et al. Patterns of recovery in the Guillain-Barré syndromes. Neurology. 1997;48(3):695–700. Epub 1997/03/01.
Feasby TE, Gilbert JJ, Brown WF, Bolton CF, Hahn AF, Koopman WF, et al. An acute axonal form of Guillain-Barré polyneuropathy. Brain. 1986;109(Pt 6):1115–26. Epub 1986/12/01.
Feasby TE, Hahn AF, Brown WF, Bolton CF, Gilbert JJ, Koopman WJ. Severe axonal degeneration in acute Guillain-Barré syndrome: evidence of two different mechanisms? J Neurol Sci. 1993;116(2):185–92. Epub 1993/06/01.
Triggs WJ, Cros D, Gominak SC, Zuniga G, Beric A, Shahani BT, et al. Motor nerve inexcitability in Guillain-Barré syndrome. The spectrum of distal conduction block and axonal degeneration. Brain. 1992;115(Pt 5):1291–302. Epub 1992/10/01.
Yuki N, Kuwabara S, Koga M, Hirata K. Acute motor axonal neuropathy and acute motor-sensory axonal neuropathy share a common immunological profile. J Neurol Sci. 1999;168(2):121–6. Epub 1999/10/20.
Albers JW, Kelly Jr JJ. Acquired inflammatory demyelinating polyneuropathies: clinical and electrodiagnostic features. Muscle Nerve. 1989;12(6):435–51. Epub 1989/06/01.
Albers JW, Donofrio PD, McGonagle TK. Sequential electrodiagnostic abnormalities in acute inflammatory demyelinating polyradiculoneuropathy. Muscle Nerve. 1985;8(6):528–39. Epub 1985/07/01.
Cornblath DR, Mellits ED, Griffin JW, McKhann GM, Albers JW, Miller RG, et al. Motor conduction studies in Guillain-Barré syndrome: description and prognostic value. Ann Neurol. 1988;23(4):354–9. Epub 1988/04/01.
Katirji B, Weissman JD. The ankle jerk and the tibial H-reflex: a clinical and electrophysiological correlation. Electromyogr Clin Neurophysiol. 1994;34(6):331–4. Epub 1994/09/01.
Gordon PH, Wilbourn AJ. Early electrodiagnostic findings in Guillain-Barré syndrome. Arch Neurol. 2001;58(6):913–7. Epub 2001/06/19.
Vucic S, Cairns KD, Black KR, Chong PS, Cros D. Neurophysiologic findings in early acute inflammatory demyelinating polyradiculoneuropathy. Clin Neurophysiol. 2004;115(10):2329–35. Epub 2004/09/08.
Al-Shekhlee A, Hachwi RN, Preston DC, Katirji B. New criteria for early electrodiagnosis of acute inflammatory demyelinating polyneuropathy. Muscle Nerve. 2005;32(1):66–72. Epub 2005/05/10.
Kiers L, Clouston P, Zuniga G, Cros D. Quantitative studies of F responses in Guillain-Barré syndrome and chronic inflammatory demyelinating polyneuropathy. Electroencephalogr Clin Neurophysiol. 1994;93(4):255–64. Epub 1994/08/01.
Ropper AH, Wijdicks EF, Shahani BT. Electrodiagnostic abnormalities in 113 consecutive patients with Guillain-Barré syndrome. Arch Neurol. 1990;47(8):881–7. Epub 1990/08/01.
Rowin J, Meriggioli MN. Electrodiagnostic significance of supramaximally stimulated A-waves. Muscle Nerve. 2000;23(7):1117–20. Epub 2000/07/07.
Kornhuber ME, Bischoff C, Mentrup H, Conrad B. Multiple A waves in Guillain-Barré syndrome. Muscle Nerve. 1999;22(3):394–9. Epub 1999/03/23.
Uncini A, Kuwabara S. Electrodiagnostic criteria for Guillain-Barré syndrome: a critical revision and the need for an update. Clin Neurophysiol. 2012;123:1487–95. Epub 2012/04/07.
Uncini A, Manzoli C, Notturno F, Capasso M. Pitfalls in electrodiagnosis of Guillain-Barré syndrome subtypes. J Neurol Neurosurg Psychiatry. 2010;81(10):1157–63. Epub 2010/09/28.
Cornblath DR. Electrophysiology in Guillain-Barré syndrome. Ann Neurol. 1990;27(Suppl):S17–20. Epub 1990/01/01.
Meulstee J, van der Meche FG. Electrodiagnostic studies in the Dutch multicentre Guillain-Barré study: a review. J Peripher Nerv Syst. 1997;2(2):143–50. Epub 1997/01/01.
Meulstee J, van der Meche FG. Electrodiagnostic criteria for poly-neuropathy and demyelination: application in 135 patients with Guillain-Barré syndrome. Dutch Guillain-Barré Study Group. J Neurol Neurosurg Psychiatry. 1995;59(5):482–6. Epub 1995/11/01.
The Italian Guillain-Barré Study Group. The prognosis and main prognostic indicators of Guillain-Barré syndrome. A multicentre prospective study of 297 patients. Brain. 1996;119(Pt 6):2053–61. Epub 1996/12/01.
Cleland JC, Malik K, Thaisetthawatkul P, Herrmann DN, Logigian EL. Acute inflammatory demyelinating polyneuropathy: contribution of a dispersed distal compound muscle action potential to electrodiagnosis. Muscle Nerve. 2006;33(6):771–7. Epub 2006/03/09.
Clouston PD, Kiers L, Zuniga G, Cros D. Quantitative analysis of the compound muscle action potential in early acute inflammatory demyelinating polyneuropathy. Electroencephalogr Clin Neurophysiol. 1994;93(4):245–54. Epub 1994/08/01.
Wee AS, Abernathy SD. The sural sensory nerve is usually spared in Guillain-Barré syndrome. J Miss State Med Assoc. 2003;44(8):251–5. Epub 2003/10/09.
Bromberg MB, Albers JW. Patterns of sensory nerve conduction abnormalities in demyelinating and axonal peripheral nerve disorders. Muscle Nerve. 1993;16(3):262–6. Epub 1993/03/01.
Al-Shekhlee A, Robinson J, Katirji B. Sensory sparing patterns and the sensory ratio in acute inflammatory demyelinating polyneuropathy. Muscle Nerve. 2007;35(2):246–50. Epub 2006/09/14.
Alam TA, Chaudhry V, Cornblath DR. Electrophysiological studies in the Guillain-Barré syndrome: distinguishing subtypes by published criteria. Muscle Nerve. 1998;21(10):1275–9. Epub 1998/09/15.
Hong YH, Sung JJ, Oh MY, Moon HJ, Park KS, Lee KW. Axonal conduction block at intermediate nerve segments in pure motor Guillain-Barré syndrome. J Peripher Nerv Syst. 2011;16(1):37–46. Epub 2011/04/21.
Eisen A, Humphreys P. The Guillain-Barré syndrome. A clinical and electrodiagnostic study of 25 cases. Arch Neurol. 1974;30(6):438–43. Epub 1974/06/01.
Miller RG, Peterson GW, Daube JR, Albers JW. Prognostic value of electrodiagnosis in Guillain-Barré syndrome. Muscle Nerve. 1988;11(7):769–74. Epub 1988/07/01.
Brown WF, Feasby TE, Hahn AF. Electrophysiological changes in the acute “axonal” form of Guillain-Barré syndrome. Muscle Nerve. 1993;16(2):200–5. Epub 1993/02/01.
van der Meche FG, Meulstee J, Kleyweg RP. Axonal damage in Guillain-Barré syndrome. Muscle Nerve. 1991;14(10):997–1002. Epub 1991/10/01.
Sterman AB, Nelson S, Barclay P. Demyelinating neuropathy accompanying Lyme disease. Neurology. 1982;32(11):1302–5. Epub 1982/11/01.
Kanzaki M, Kaida K, Ueda M, Morita D, Hirakawa M, Motoyoshi K, et al. Ganglioside complexes containing GQ1b as targets in Miller Fisher and Guillain-Barré syndromes. J Neurol Neurosurg Psychiatry. 2008;79(10):1148–52. Epub 2008/03/15.
Gorson KC, Ropper AH, Muriello MA, Blair R. Prospective evaluation of MRI lumbosacral nerve root enhancement in acute Guillain-Barré syndrome. Neurology. 1996;47(3):813–7. Epub 1996/09/01.
Mulkey SB, Glasier CM, El-Nabbout B, et al. Nerve root enhancement on spinal MRI in pediatric Guillain-Barré syndrome. Pediatr Neurol. 2010;43(4):263–9.
Yardimci N, Avci AY, Kayhan E, Benli S. Bilateral facial nerve enhancement demonstrated by magnetic resonance imaging in Guillain-Barré syndrome. Neurol Sci. 2009;30(5):431–3. Epub 2009/08/29.
Levin KH. Variants and mimics of Guillain-Barré syndrome. Neurologist. 2004;10(2):61–74. Epub 2004/03/05.
Donofrio PD, Wilbourn AJ, Albers JW, Rogers L, Salanga V, Greenberg HS. Acute arsenic intoxication presenting as Guillain-Barré-like syndrome. Muscle Nerve. 1987;10(2):114–20. Epub 1987/02/01.
Tabaraud F, Vallat JM, Hugon J, Ramiandrisoa H, Dumas M, Signoret JL. Acute or subacute alcoholic neuropathy mimicking Guillain-Barré syndrome. J Neurol Sci. 1990;97(2–3):195–205. Epub 1990/07/01.
Wohrle JC, Spengos K, Steinke W, Goebel HH, Hennerici M. Alcohol-related acute axonal polyneuropathy: a differential diagnosis of Guillain-Barré syndrome. Arch Neurol. 1998;55(10):1329–34. Epub 1998/10/21.
McEneaney D, Hawkins S, Trimble E, Smye M. Porphyric neuropathy – a rare and often neglected differential diagnosis of Guillain-Barré syndrome. J Neurol Sci. 1993;114(2):231–2. Epub 1993/02/01.
Cuquemelle E, Ehrmann S, Razazi K, Deybach JC, Brun-Buisson C, Thille AW. An atypical case of Guillain-Barré syndrome: acute intermittent porphyria. Intensive Care Med. 2012;38(5):913–4. Epub 2012/01/26.
Suarez JI, Cohen ML, Larkin J, Kernich CA, Hricik DE, Daroff RB. Acute intermittent porphyria: clinicopathologic correlation. Report of a case and review of the literature. Neurology. 1997;48(6):1678–83. Epub 1997/06/01.
Briemberg HR, Levin K, Amato AA. Multifocal conduction block in peripheral nerve vasculitis. J Clin Neuromuscul Dis. 2002;3(4):153–8. Epub 2002/06/01.
Jeha LE, Sila CA, Lederman RJ, Prayson RA, Isada CM, Gordon SM. West Nile virus infection: a new acute paralytic illness. Neurology. 2003;61(1):55–9. Epub 2003/07/09.
Leis AA, Van Gerpen JA, Sejvar JJ. The aetiology of flaccid paralysis in West Nile virus infection. J Neurol Neurosurg Psychiatry. 2004;75(6):940. Epub 2004/05/18.
Al-Shekhlee A, Katirji B. Electrodiagnostic features of acute paralytic poliomyelitis associated with West Nile virus infection. Muscle Nerve. 2004;29(3):376–80. Epub 2004/02/26.
Fratkin JD, Leis AA, Stokic DS, Slavinski SA, Geiss RW. Spinal cord neuropathology in human West Nile virus infection. Arch Pathol Lab Med. 2004;128(5):533–7. Epub 2004/04/17.
Krumina A, Logina I, Donaghy M, Rozentale B, Kravale I, Griskevica A, et al. Diphtheria with polyneuropathy in a closed community despite receiving recent booster vaccination. J Neurol Neurosurg Psychiatry. 2005;76(11):1555–7. Epub 2005/10/18.
Bolton CF, Laverty DA, Brown JD, Witt NJ, Hahn AF, Sibbald WJ. Critically ill polyneuropathy: electrophysiological studies and differentiation from Guillain-Barré syndrome. J Neurol Neurosurg Psychiatry. 1986;49(5):563–73. Epub 1986/05/01.
Bolton CF, Breuer AC. Critical illness polyneuropathy. Muscle Nerve. 1999;22(3):419–24. Epub 1999/03/23.
Bird SJ. Diagnosis and management of critical illness polyneuropathy and critical illness myopathy. Curr Treat Options Neurol. 2007;9(2):85–92. Epub 2007/02/15.
Ropper AH. Unusual clinical variants and signs in Guillain-Barré syndrome. Arch Neurol. 1986;43(11):1150–2. Epub 1986/11/01.
Vedanarayanan VV, Evans OB, Subramony SH. Tick paralysis in children: electrophysiology and possibility of misdiagnosis. Neurology. 2002;59(7):1088–90. Epub 2002/10/09.
Ropper AH, Wijdicks EFM, Truax BT. Guillain-Barré syndrome. Philadelphia: F.A. Davis; 1991.
Ropper AH. Intensive care of acute Guillain-Barré syndrome. Can J Neurol Sci. 1994;21(2):S23–7. Epub 1994/05/01.
Hughes RA, Wijdicks EF, Benson E, Cornblath DR, Hahn AF, Meythaler JM, et al. Supportive care for patients with Guillain-Barré syndrome. Arch Neurol. 2005;62(8):1194–8. Epub 2005/08/10.
Lawn ND, Fletcher DD, Henderson RD, Wolter TD, Wijdicks EF. Anticipating mechanical ventilation in Guillain-Barré syndrome. Arch Neurol. 2001;58(6):893–8. Epub 2001/06/19.
Burakgazi AZ, Hoke A. Respiratory muscle weakness in peripheral neuropathies. J Peripher Nerv Syst. 2010;15(4):307–13. Epub 2011/01/05.
Sharshar T, Chevret S, Bourdain F, Raphael JC. French Cooperative Group on Plasma Exchange in Guillain-Barré S. Early predictors of mechanical ventilation in Guillain-Barré syndrome. Crit Care Med. 2003;31(1):278–83. Epub 2003/01/25.
Durand MC, Porcher R, Orlikowski D, Aboab J, Devaux C, Clair B, et al. Clinical and electrophysiological predictors of respiratory failure in Guillain-Barré syndrome: a prospective study. Lancet Neurol. 2006;5(12):1021–8. Epub 2006/11/18.
Ropper AH, Kehne SM. Guillain-Barré syndrome: management of respiratory failure. Neurology. 1985;35(11):1662–5. Epub 1985/11/01.
Lawn ND, Wijdicks EF. Tracheostomy in Guillain-Barré syndrome. Muscle Nerve. 1999;22(8):1058–62. Epub 1999/07/27.
Minahan Jr RE, Bhardwaj A, Traill TA, Hanley DF. Stimulus-evoked sinus arrest in severe Guillain-Barré syndrome: a case report. Neurology. 1996;47(5):1239–42. Epub 1996/11/01.
Khan F, Ng L, Amatya B, Brand C, Turner-Stokes L. Multidisciplinary care for Guillain-Barré syndrome. Eur J Phys Rehabil Med. 2011;47(4):607–12. Epub 2011/09/14.
Henderson RD, Lawn ND, Fletcher DD, McClelland RL, Wijdicks EF. The morbidity of Guillain-Barré syndrome admitted to the intensive care unit. Neurology. 2003;60(1):17–21. Epub 2003/01/18.
Genis D, Busquets C, Manubens E, Davalos A, Baro J, Oterino A. Epidural morphine analgesia in Guillain Barré syndrome. J Neurol Neurosurg Psychiatry. 1989;52(8):999–1001. Epub 1989/08/01.
Khan F, Ng L, Amatya B, Brand C, Turner-Stokes L. Multidisciplinary care for Guillain-Barré syndrome. Cochrane Database Syst Rev. 2010;10:CD008505. Epub 2010/10/12.
The Guillain-Barré syndrome Study Group. Plasmapheresis and acute Guillain-Barré syndrome. Neurology. 1985;35(8):1096–104. Epub 1985/08/01.
French Cooperative Group on Plasma Exchange in Guillain-Barré syndrome. Efficiency of plasma exchange in Guillain-Barré syndrome: role of replacement fluids. Ann Neurol. 1987;22(6):753–61. Epub 1987/12/01.
Osterman PO, Fagius J, Lundemo G, Pihlstedt P, Pirskanen R, Siden A, et al. Beneficial effects of plasma exchange in acute inflammatory polyradiculoneuropathy. Lancet. 1984;2(8415):1296–9. Epub 1984/12/08.
McKhann GM, Griffin JW. Plasmapheresis and the Guillain-Barré syndrome. Ann Neurol. 1987;22(6):762–3. Epub 1987/12/01.
Hughes RA, Swan AV, Raphael JC, Annane D, van Koningsveld R, van Doorn PA. Immunotherapy for Guillain-Barré syndrome: a systematic review. Brain. 2007;130(Pt 9):2245–57. Epub 2007/03/06.
The French Cooperative Group on Plasma Exchange in Guillain-Barré syndrome. Appropriate number of plasma exchanges in Guillain-Barré syndrome. Ann Neurol. 1997;41(3):298–306. Epub 1997/03/01.
Cortese I, Chaudhry V, So YT, Cantor F, Cornblath DR, Rae-Grant A. Evidence-based guideline update: plasmapheresis in neurologic disorders: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology. 2011;76(3):294–300. Epub 2011/01/19.
van der Meche FG, Schmitz PI. A randomized trial comparing intravenous immune globulin and plasma exchange in Guillain-Barré syndrome. Dutch Guillain-Barré Study Group. N Engl J Med. 1992;326(17):1123–9. Epub 1992/04/23.
Plasma Exchange/Sandoglobulin Guillain-Barré Syndrome Trial Group. Randomised trial of plasma exchange, intravenous immunoglobulin, and combined treatments in Guillain-Barré syndrome. Lancet. 1997;349(9047):225–30. Epub 1997/01/25.
Hughes RA, Raphael JC, Swan AV, van Doorn PA. Intravenous immunoglobulin for Guillain-Barré syndrome. Cochrane Database Syst Rev. 2006;(1):CD002063. Epub 2006/01/27.
Patwa HS, Chaudhry V, Katzberg H, Rae-Grant AD, So YT. Evidence-based guideline: intravenous immunoglobulin in the treatment of neuromuscular disorders: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology. 2012;78(13):1009–15. Epub 2012/03/29.
Clark AL. Clinical uses of intravenous immunoglobulin in pregnancy. Clin Obstet Gynecol. 1999;42(2):368–80. Epub 2012/03/29.
Alshekhlee A, Hussain Z, Sultan B, Katirji B. Immunotherapy for Guillain-Barré syndrome in the US hospitals. J Clin Neuromuscul Dis. 2008;10(1):4–10. Epub 2008/09/06.
Raphael JC, Chevret S, Harboun M, Jars-Guincestre MC. French Guillain-Barré Syndrome Cooperative G. Intravenous immune globulins in patients with Guillain-Barré syndrome and contraindications to plasma exchange: 3 days versus 6 days. J Neurol Neurosurg Psychiatry. 2001;71(2):235–8. Epub 2001/07/19.
Kuitwaard K, de Gelder J, Tio-Gillen AP, Hop WC, van Gelder T, van Toorenenbergen AW, et al. Pharmacokinetics of intravenous immunoglobulin and outcome in Guillain-Barré syndrome. Ann Neurol. 2009;66(5):597–603. Epub 2009/11/26.
French Cooperative Group on Plasma Exchange in Guillain-Barré syndrome Plasma exchange in Guillain-Barré syndrome: One-year follow-up. Ann Neurol. 1992;32(1):94–7. Epub 1992/07/01.
Visser LH, van der Meche FG, Meulstee J, van Doorn PA. Risk factors for treatment related clinical fluctuations in Guillain-Barré syndrome. Dutch Guillain-Barré Study Group. J Neurol Neurosurg Psychiatry. 1998;64(2):242–4. Epub 1998/03/07.
Farcas P, Avnun L, Frisher S, Herishanu YO, Wirguin I. Efficacy of repeated intravenous immunoglobulin in severe unresponsive Guillain-Barré syndrome. Lancet. 1997;350(9093):1747. Epub 1997/12/31.
Ruts L, Drenthen J, Jacobs BC, van Doorn PA, Dutch GBSSG. Distinguishing acute-onset CIDP from fluctuating Guillain-Barré syndrome: a prospective study. Neurology. 2010;74(21):1680–6. Epub 2010/04/30.
Hughes RA, Swan AV, van Koningsveld R, van Doorn PA. Corticosteroids for Guillain-Barré syndrome. Cochrane Database Syst Rev. 2006;(2):CD001446. Epub 2006/04/21.
Hughes RA, Newsom-Davis JM, Perkin GD, Pierce JM. Controlled trial prednisolone in acute polyneuropathy. Lancet. 1978;2(8093):750–3. Epub 1978/10/07.
Hughes RA, Swan AV, van Doorn PA. Corticosteroids for Guillain-Barré syndrome. Cochrane Database Syst Rev. 2010;(2):CD001446. Epub 2010/02/19.
van Koningsveld R, Schmitz PI, Meche FG, Visser LH, Meulstee J, van Doorn PA, et al. Effect of methylprednisolone when added to standard treatment with intravenous immunoglobulin for Guillain-Barré syndrome: randomised trial. Lancet. 2004;363(9404):192–6. Epub 2004/01/24.
Pritchard J, Gray IA, Idrissova ZR, Lecky BR, Sutton IJ, Swan AV, et al. A randomized controlled trial of recombinant interferon-beta 1a in Guillain-Barré syndrome. Neurology. 2003;61(9):1282–4. Epub 2003/11/12.
Hughes RA, Pritchard J, Hadden RD. Pharmacological treatment other than corticosteroids, intravenous immunoglobulin and plasma exchange for Guillain Barré syndrome. Cochrane Database Syst Rev. 2011;(3):CD008630. Epub 2011/03/18.
Soysal A, Aysal F, Caliskan B, Dogan Ak P, Mutluay B, Sakalli N, et al. Clinico-electrophysiological findings and prognosis of Guillain-Barré syndrome – 10 years’ experience. Acta Neurol Scand. 2011;123(3):181–6. Epub 2010/05/26.
van Koningsveld R, Steyerberg EW, Hughes RA, Swan AV, van Doorn PA, Jacobs BC. A clinical prognostic scoring system for Guillain-Barré syndrome. Lancet Neurol. 2007;6(7):589–94. Epub 2007/06/01.
Walgaard C, Lingsma HF, Ruts L, van Doorn PA, Steyerberg EW, Jacobs BC. Early recognition of poor prognosis in Guillain-Barré syndrome. Neurology. 2011;76(11):968–75. Epub 2011/03/16.
Dyck PJ. Is there an axonal variety of GBS? Neurology. 1993;43(7):1277–80. Epub 1993/07/01.
Kuwabara S, Kokubun N, Misawa S, Kanai K, Isose S, Shibuya K, et al. Neuromuscular transmission is not impaired in axonal Guillain-Barré syndrome. J Neurol Neurosurg Psychiatry. 2011;82(10):1174–7. Epub 2010/11/13.
Winer JB, Hughes RA, Greenwood RJ, Perkin GD, Healy MJ. Prognosis in Guillain-Barré syndrome. Lancet. 1985;1(8439):1202–3. Epub 1985/05/25.
Ropper AH. The Guillain-Barré syndrome. N Engl J Med. 1992;326(17):1130–6. Epub 1992/04/23.
Rees JH. Risk factors for treatment related clinical fluctuations in Guillain-Barré syndrome. J Neurol Neurosurg Psychiatry. 1998;64(2):148–9. Epub 1998/03/07.
Haymaker W, Kernohan JW. The Landry Guillain-Barré syndrome; a clinicopathologic study of 50 fatal cases. Trans Am Neurol Assoc. 1948;73(73 Annual Meet):17–20. Epub 1948/01/01.
Ravn H. The Landry-Guillain-Barré syndrome. A survey and a clinical report of 127 cases. Acta Neurol Scand. 1967;43 Suppl 30Suppl 30:1–64. Epub 1967/01/01.
Alshekhlee A, Hussain Z, Sultan B, Katirji B. Guillain-Barré syndrome: incidence and mortality rates in US hospitals. Neurology. 2008;70(18):1608–13. Epub 2008/04/30.
Bogliun G, Beghi E, Italian GBS Registry Study Group. Incidence and clinical features of acute inflammatory polyradiculoneuropathy in Lombardy, Italy, 1996. Acta Neurol Scand. 2004;110(2):100–6. Epub 2004/07/10.
Hughes RA, Hadden RD, Rees JH, Swan AV. The Italian Guillain-Barré Study Group. The prognosis and main prognostic indicators of Guillain-Barré syndrome: a multicentre prospective study of 297 patients. Brain. 1998;121(Pt 4):767–9. Epub 1998/05/13.
Oh SJ, Kurokawa K, de Almeida DF, Ryan Jr HF, Claussen GC. Subacute inflammatory demyelinating polyneuropathy. Neurology. 2003;61(11):1507–12. Epub 2003/12/10.
al-Hakim M, Cohen M, Daroff RB. Postmortem examination of relapsing acute Guillain-Barré syndrome. Muscle Nerve. 1993;16(2):173–6. Epub 1993/02/01.
Wijdicks EF, Ropper AH. Acute relapsing Guillain-Barré syndrome after long asymptomatic intervals. Arch Neurol. 1990;47(1):82–4. Epub 1990/01/01.
Fisher M. An unusual variant of acute idiopathic polyneuritis (syndrome of ophthalmoplegia, ataxia and areflexia). N Engl J Med. 1956;255(2):57–65. Epub 1956/07/12.
Lyu RK, Tang LM, Cheng SY, Hsu WC, Chen ST. Guillain-Barré syndrome in Taiwan: a clinical study of 167 patients. J Neurol Neurosurg Psychiatry. 1997;63(4):494–500. Epub 1997/10/29.
Mori M, Kuwabara S. Fisher syndrome. Curr Treat Options Neurol. 2011;13(1):71–8. Epub 2010/11/26.
Ter Bruggen JP, van der Meche FG, de Jager AE, Polman CH. Ophthalmoplegic and lower cranial nerve variants merge into each other and into classical Guillain-Barré syndrome. Muscle Nerve. 1998;21(2):239–42. Epub 1998/02/18.
Lo YL. Clinical and immunological spectrum of the Miller Fisher syndrome. Muscle Nerve. 2007;36(5):615–27. Epub 2007/07/28.
Ito M, Kuwabara S, Odaka M, Misawa S, Koga M, Hirata K, et al. Bickerstaff’s brainstem encephalitis and Fisher syndrome form a continuous spectrum: clinical analysis of 581 cases. J Neurol. 2008;255(5):674–82. Epub 2008/02/16.
Ito M, Matsuno K, Sakumoto Y, Hirata K, Yuki N. Ataxic Guillain-Barré syndrome and acute sensory ataxic neuropathy form a continuous spectrum. J Neurol Neurosurg Psychiatry. 2011;82(3):294–9. Epub 2011/01/22.
Uncini A, Lugaresi A. Fisher syndrome with tetraparesis and antibody to GQ1b: evidence for motor nerve terminal block. Muscle Nerve. 1999;22(5):640–4. Epub 1999/05/20.
Roberts M, Willison H, Vincent A, Newsom-Davis J. Serum factor in Miller-Fisher variant of Guillain-Barré syndrome and neurotransmitter release. Lancet. 1994;343(8895):454–5. Epub 1994/02/19.
Koga M, Gilbert M, Li J, Koike S, Takahashi M, Furukawa K, et al. Antecedent infections in Fisher syndrome: a common pathogenesis of molecular mimicry. Neurology. 2005;64(9):1605–11. Epub 2005/05/11.
Koga M, Koike S, Hirata K, Yuki N. Ambiguous value of Haemophilus influenzae isolation in Guillain-Barré and Fisher syndromes. J Neurol Neurosurg Psychiatry. 2005;76(12):1736–8. Epub 2005/11/18.
Mori M, Kuwabara S, Fukutake T, Yuki N, Hattori T. Clinical features and prognosis of Miller Fisher syndrome. Neurology. 2001;56(8):1104–6. Epub 2001/04/26.
Mori M, Kuwabara S, Fukutake T, Hattori T. Intravenous immunoglobulin therapy for Miller Fisher syndrome. Neurology. 2007;68(14):1144–6. Epub 2007/04/04.
Mori M, Kuwabara S, Yuki N. Fisher syndrome: clinical features, immunopathogenesis and management. Expert Rev Neurother. 2012;12(1):39–51. Epub 2011/12/14.
Halstead SK, Humphreys PD, Goodfellow JA, Wagner ER, Smith RA, Willison HJ. Complement inhibition abrogates nerve terminal injury in Miller Fisher syndrome. Ann Neurol. 2005;58(2):203–10. Epub 2005/07/29.
Halstead SK, Humphreys PD, Zitman FM, Hamer J, Plomp JJ, Willison HJ. C5 inhibitor rEV576 protects against neural injury in an in vitro mouse model of Miller Fisher syndrome. J Peripher Nerv Syst. 2008;13(3):228–35. Epub 2008/10/11.
Richter RB. The ataxic form of polyradiculoneuritis (Landry-Guillain-Barré syndrome). Clinical and pathologic observations. J Neuropathol Exp Neurol. 1962;21:171–84. Epub 1962/04/01.
Notturno F, Caporale CM, Uncini A. Acute sensory ataxic neuropathy with antibodies to GD1b and GQ1b gangliosides and prompt recovery. Muscle Nerve. 2008;37(2):265–8. Epub 2007/09/08.
Pan CL, Yuki N, Koga M, Chiang MC, Hsieh ST. Acute sensory ataxic neuropathy associated with monospecific anti-GD1b IgG antibody. Neurology. 2001;57(7):1316–8. Epub 2001/10/10.
Uncini A, Yuki N. Sensory Guillain-Barré syndrome and related disorders: an attempt at systematization. Muscle Nerve. 2012;45(4):464–70. Epub 2012/03/21.
Ropper AH. Miller Fisher syndrome and other acute variants of Guillain-Barré syndrome. Baillieres Clin Neurol. 1994;3(1):95–106. Epub 1994/04/01.
Nagashima T, Koga M, Odaka M, Hirata K, Yuki N. Continuous spectrum of pharyngeal-cervical-brachial variant of Guillain-Barré syndrome. Arch Neurol. 2007;64(10):1519–23. Epub 2007/10/10.
Capasso M, Notturno F, Manzoli C, Yuki N, Uncini A. Reversible conduction failure in pharyngeal-cervical-brachial variant of Guillain-Barré syndrome. Muscle Nerve. 2010;42(4):608–12. Epub 2010/09/30.
Unal-Cevik I, Onal MZ, Odabasi Z, Tan E. IVIG- responsive multiple cranial neuropathy: a pharyngo-facial variant of Guillain-Barré syndrome. Acta Neurol Belg. 2009;109(4):317–21. Epub 2010/02/03.
Lyu RK, Chen ST. Acute multiple cranial neuropathy: a variant of Guillain-Barré syndrome? Muscle Nerve. 2004;30(4):433–6. Epub 2004/09/17.
Susuki K, Koga M, Hirata K, Isogai E, Yuki N. A Guillain-Barré syndrome variant with prominent facial diplegia. J Neurol. 2009;256(11):1899–905. Epub 2009/07/28.
Susuki K, Atsumi M, Koga M, Hirata K, Yuki N. Acute facial diplegia and hyperreflexia: a Guillain-Barré syndrome variant. Neurology. 2004;62(5):825–7. Epub 2004/03/10.
Sethi NK, Torgovnick J, Arsura E, Johnston A, Buescher E. Facial diplegia with hyperreflexia – a mild Guillain-Barré syndrome variant, to treat or not to treat? J Brachial Plex Peripher Nerve Inj. 2007;2:9. Epub 2007/04/12.
Young RR, Asbury AK, Corbett JL, Adams RD. Pure pan-dysautonomia with recovery. Description and discussion of diagnostic criteria. Brain. 1975;98(4):613–36. Epub 1975/12/01.
Thomashefsky AJ, Horwitz SJ, Feingold MH. Acute autonomic neuropathy. Neurology. 1972;22(3):251–5. Epub 1972/03/01.
Koike H, Atsuta N, Adachi H, Iijima M, Katsuno M, Yasuda T, et al. Clinicopathological features of acute autonomic and sensory neuropathy. Brain. 2010;133(10):2881–96. Epub 2010/08/26.
Yasuda T, Sobue G, Mokuno K, Hakusui S, Ito T, Hirose Y, et al. Clinico-pathophysiological features of acute autonomic and sensory neuropathy: a long-term follow-up study. J Neurol. 1995;242(10):623–8. Epub 1995/10/01.
Vernino S, Low PA, Fealey RD, Stewart JD, Farrugia G, Lennon VA. Autoantibodies to ganglionic acetylcholine receptors in autoimmune autonomic neuropathies. N Engl J Med. 2000;343(12):847–55. Epub 2000/09/21.
Sandroni P, Vernino S, Klein CM, Lennon VA, Benrud-Larson L, Sletten D, et al. Idiopathic autonomic neuropathy: comparison of cases seropositive and seronegative for ganglionic acetylcholine receptor antibody. Arch Neurol. 2004;61(1):44–8. Epub 2004/01/21.
Klein CM. Evaluation and management of autonomic nervous system disorders. Semin Neurol. 2008;28(2):195–204. Epub 2008/03/21.
Ishitobi M, Haginoya K, Kitamura T, Munakata M, Yokoyama H, Iinuma K. Acute dysautonomia: complete recovery after two courses of IVIg. Brain Dev. 2004;26(8):542–4. Epub 2004/11/10.
Venkataraman S, Alexander M, Gnanamuthu C. Postinfectious pandysautonomia with complete recovery after intravenous immunoglobulin therapy. Neurology. 1998;51(6):1764–5. Epub 1998/12/17.
Tatsumoto M, Odaka M, Hirata K, Yuki N. Isolated abducens nerve palsy as a regional variant of Guillain-Barré syndrome. J Neurol Sci. 2006;243(1–2):35–8. Epub 2006/01/13.
Ropper AH. Further regional variants of acute immune polyneuropathy. Bifacial weakness or sixth nerve paresis with paresthesias, lumbar polyradiculopathy, and ataxia with pharyngeal-cervical-brachial weakness. Arch Neurol. 1994;51(7):671–5. Epub 1994/07/01.
Seneviratne U, Gunasekera S. Acute small fibre sensory neuropathy: another variant of Guillain-Barré syndrome? J Neurol Neurosurg Psychiatry. 2002;72(4):540–2. Epub 2002/03/23.
Dabby R, Gilad R, Sadeh M, Lampl Y, Watemberg N. Acute steroid responsive small-fiber sensory neuropathy: a new entity? J Peripher Nerv Syst. 2006;11(1):47–52. Epub 2006/03/08.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Piccione, E.A., Salame, K., Katirji, B. (2014). Guillain-Barré Syndrome and Related Disorders. In: Katirji, B., Kaminski, H., Ruff, R. (eds) Neuromuscular Disorders in Clinical Practice. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-6567-6_28
Download citation
DOI: https://doi.org/10.1007/978-1-4614-6567-6_28
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-6566-9
Online ISBN: 978-1-4614-6567-6
eBook Packages: MedicineMedicine (R0)