
Abstract
CRISPR-Cas9 is an RNA guided endonuclease derived from the bacterium Streptococcus pyogenes. Due to its simplicity, versatility, and high efficiency, it has been widely used for genome editing in a variety of organisms including the protozoan parasite Leishmania, the causative agent of human leishmaniasis. Compared to the traditional homologous recombination gene targeting method, CRISPR-Cas9 has been shown to be a more efficient method to delete or disrupt Leishmania genes, generate point mutations, and add tags to endogenous genes. Notably, the stable CRISPR expression systems were shown to delete multicopy family Leishmania genes and genes present in multiploid chromosomes, identify essential Leishmania genes, and create specific chromosome translocations. In this chapter, we describe detailed procedures on using the stable CRISPR expression system for genome editing in Leishmania. These procedures include CRISPR targeting site selection, gRNA design, cloning single and double gRNA coding sequences into the Leishmania CRISPR vector pLdCN, oligonucleotide donor and drug resistance selection donor design, Leishmania cell transfection, screening, and isolation of CRISPR-edited mutants. As the principles of gene editing are generally similar, many of these procedures could also apply to the transient Leishmania CRISPR systems described by other labs.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others


Key words
1 Introduction
The commonly used Streptococcus pyogenes CRISPR (clustered regularly interspaced short palindromic repeats)-Cas9 is an RNA-guided endonuclease that has been shown to facilitate site-specific DNA cleavage in various organisms [1,2,3,4,5,6]. For gene editing purposes, the CRISPR-Cas9 system requires at least two components; the Cas9 nuclease and a guide RNA (gRNA) (Fig. 1). The gRNA consists of a 20 nucleotide (nt) guide sequence (protospacer) followed by an 82 nt chimeric sequence derived from the bacterial CRISPR RNA and transactivating RNA [1, 2]. The gRNA and Cas9 nuclease form an RNA–protein complex. The gRNA 20-nt guide sequence, which is complementary to the target DNA sequence, directs the Cas9 nuclease to the specific DNA target site. Once the gRNA binds to the complementary target DNA sequence, the Cas9 nuclease becomes active and cleaves the target DNA three base pair (bp) upstream of the triple nucleotides NGG, known as the protospacer-adjacent motif (PAM), to generate a double-stranded break (DSB) (Fig. 1) [7]. DSBs are lethal to the cell and must be repaired by the cellular machinery either through mutation prone DSB repair pathways such as microhomology-mediated end-joining (MMEJ), single strand annealing (SSA) or through homology directed repair (HDR) [8, 9]. Unlike mammalian cells, Leishmania does not have the nonhomologous end-joining (NHEJ) pathway. Instead, Leishmania mainly uses MMEJ or SSA to repair DSBs. Repair through the MMEJ or SSA pathway in Leishmania often leads to deletion of the targeted gene [10,11,12,13]. HDR can be used to introduce a specific DNA sequence with desired mutations or tags [10, 11, 14, 15]. The Cas9 nuclease can be programed to target new sites simply by changing the 20 nt guide sequence of the gRNA. Given its simplicity, efficiency, and versatility, CRISPR-Cas9 has now been widely used for genome engineering in a variety of organisms including human protozoan parasites.

Scheme of CRISPR-Cas9-mediated genome editing in Leishmania. The CRISPR-Cas9 genome editing system consists of at least two components: the gRNA and the Cas9 nuclease. The gRNA forms a ribonucleoprotein complex with Cas9 and guides Cas9 to the specific genomic target site, once the gRNA binds to the complementary target sequence, Cas9 becomes active and generates a double-stranded break (DSB) in the target DNA 3 bp upstream of the NGG PAM site. The DSB is typically repaired in Leishmania by microhomology-mediated end joining (MMEJ) or single strand annealing (SSA) with 5–500 bp homology sequences, which can lead to deletions ranging from 10 bp to more than 7000 bp. However, in the presence of donor DNA (repair template), single- or double-stranded DNA with only 25 nt (bp) homology arms flanking the cleavage site, desired mutations such as stop codons, frame shift, point mutations, tags and selection markers can be precisely inserted by homology directed repair (HDR), SSA or MMEJ into the Leishmania genome
Leishmania protozoans are the causative agents of human leishmaniasis, the second most deadly parasitic disease after malaria. Despite decades of research efforts, there is still no effective vaccine available and the existing antileishmanial drugs are less than ideal with side effects [16]. Due to the lack of an RNA interference pathway in most of the Leishmania species [17], gene targeting in Leishmania traditionally relied on gene replacement by homologous recombination which requires a minimum of 180 bp flanking sequences and two antibiotic selection markers as Leishmania is diploid [18,19,20,21]. For genes present in multiploid chromosomes or present in multiple copies, several rounds of targeting are required which is time consuming and often not possible. Although whole genome sequences have been completed for various Leishmania species, the biological functions of most genes are unknown [22,23,24,25,26]. CRISPR technology has recently been adapted for use in Leishmania and has greatly improved gene editing efficiency [10,11,12,13, 27,28,29,30,31,32,33]. Compared to the traditional gene targeting method, CRISPR is simpler and more efficient; it has been used to generate gene deletion and disruption mutants, single point mutations, and to tag endogenous genes with or without using a selection marker. Importantly, it has been successfully used to delete multicopy family genes, which was often impossible with the traditional method, and to reveal essentiality of several Leishmania genes [10,11,12,13, 30].
Thus far, at least five CRISPR systems have been reported for use in Leishmania which include two stable expression systems and three transient systems [10,11,12, 27,27,29,30,31, 34]. In the two stable CRISPR expression systems, the Cas9 nuclease and gRNA are constitutively expressed in the parasites using expression vectors and gRNA transcribed from either the U6 promoter or the ribosomal RNA promoter [10, 11, 27, 30]. Because of the continuous expression of Cas9 and gRNA in the cell, this stable expression system using the ribosomal RNA promoter has been used to delete multicopy family Leishmania genes, determine gene essentiality and perform basic gene editing [10, 11]. In the transient expression system relying on the T7 RNA polymerase, the Cas9 and T7 RNA polymerase genes are integrated into the parasite genome and constitutively expressed [28, 34]. Gene editing is then achieved by transiently cotransfecting the Cas9 and T7 RNA polymerase expressing promastigotes with gRNA templates and donor DNA containing antibiotic selection markers or fluorescent protein tags [28, 34]. The second transient system recently reported involves directly transfecting the Cas9 expressing Leishmania cells with in vitro synthesized gRNA as well as an antibiotic selection marker donor [31]. The third transient system used a purified recombinant Cas9 protein from Staphylococcus aureus (SaCas9) complexed with in vitro-synthesized gRNA to form the ribonucleoprotein complex (RNP) [29]. Gene editing was achieved by transfecting the parasites with the RNP [29]. Compared with the Cas9 and gRNA constitutive expression systems [10, 11, 27, 29], the major advantages of these transient systems [28, 29, 31, 34] are that, once the Cas9 and T7 RNA polymerase expressing Leishmania cell line has been established or the SaCas9 recombinant protein purified from bacteria [29], the transient systems are more convenient and faster, as they bypass the DNA cloning procedures, and several gRNA species could be delivered simultaneously. The transient systems may also have lower probability to cause off-target mutations. However, the disadvantages of these transient systems are that it is necessary to develop Leishmania lines constitutively expressing T7 polymerase and Cas9 [29, 34] or the need to obtain purified recombinant Cas9 [29]. In the case of SaCas9, the longer PAM (NNGRRT) sequence requirement could also limit the sites available for SaCas9 targeting [12, 29, 35]. Further, how these transient systems will perform on multicopy and essential Leishmania genes remains to be determined.
In this chapter, we describe the detailed procedure for carrying out gene editing using the stable plasmid based CRISPR Cas9 expression system [10,12,12]. This is a versatile system and can be used to delete or disrupt single and multicopy genes, carry out selection with or without antibiotic resistance genes, edit gene codons and establish gene essentiality [10,12,12]. Depending on the experimental requirements and the investigator preference, one of the other transient or stable CRISPR expression systems can also be considered [27,28,29,30,31, 34]. As the principles of gene editing are similar, many of the procedures described within this chapter also apply to the transient systems.
2 Materials
2.1 Molecular Cloning
-
1.
The pLdCN, pLdCN2, pSPBle, and pSPneogRNA241510+MT plasmids have been deposited in Addgene (https://www.addgene.org/) with ID no. 84290, 125186, 63561, and 84292, respectively. The pLdCN plasmid is used to coexpress Cas9 and gRNA in Leishmania. The pLdCN2 plasmid is used to coexpress Cas9 and two gRNAs in Leishmania. The pSPBle plasmid is used as a template to make donor DNA containing the bleomycin-resistant gene for selection of edited clones. The pSPneogRNA241510+MT plasmid can also be used to make dual gRNAs expressing pLdCN plasmids as detailed below.
-
2.
Guide sequences and other primer oligonucleotides need to be ordered from a local supplier; including primers for PCR, donors, and other locus-specific primers for homologous template construction and gene disruption identification.
-
3.
Common molecular biology reagents: such as Bbs I, and other restriction enzymes, T4 DNA ligase, T7 DNA ligase, and T4 Polynucleotide Kinase.
- 4.
-
5.
PCR reagents: high-fidelity DNA polymerases such as the Platinum SuperFi DNA Polymerase, Q5 DNA Polymerase, Taq DNA Polymerase, dNTPs, and other common PCR reagents.
-
6.
Competent bacterial cells such as Escherichia coli DH5α.
-
7.
Plasmid purification kit such as the BIO BASIC Plasmid miniprep kit or GeneJET Plasmid Miniprep.
-
8.
Reagents for agarose gel electrophoresis.
-
9.
PCR purification kit and Gel/DNA extraction kit such as GeneJET PCR Purification Kit, Gel Extraction Kit.
2.2 Leishmania Strains and Culture Medium
The common Leishmania lab strains such as L. donovani 1S/Cl2D, L. major Friedlin V9, and L. mexicana (MNYC/BZ/62/M379). The culture medium is M199 medium (pH 7.4) supplemented with 10% heat-inactivated fetal bovine serum, 40 mM HEPES (pH 7.4), 0.1 mM adenine, 5 mg/L hemin, 1 mg/L biotin, 1 mg/L biopterin, 50 U/ml penicillin, and 50 μg/ml streptomycin. Leishmania promastigotes are routinely cultured at 27 °C and passaged to fresh medium at a 20- to 40-fold dilution once a week.
2.3 Parasite Transfection
-
1.
Depending on the species and strain, Leishmania promastigotes from the middle log phase to early stationary phase are usually used for transfection.
-
2.
Tb-BSF buffer [36]: 90 mM Na2HPO4, 5 mM KCl, 0.15 mM CaCl2, 50 mM HEPES, pH 7.3.
-
3.
Drug(s) for selection such as G418, phleomycin, hygromycin, and puromycin.
-
4.
Tissue culture supplies such as 25 cm2 cell culture flasks (T25), 96-well plates, 24-well plates.
-
5.
Electroporation system such as the Nucleofector™ 2b Device from LONZA (see Note 1).
-
6.
2 mm gap electroporation cuvettes (Thermo Fisher Scientific).
-
7.
Autoclaved glass Pasteur pipette and transfer bulb.
3 Methods
As outlined in Fig. 2a and b (see Drug R donor), the most rapid and direct way to disrupt a gene is to use a single gRNA expressing pLdCN plasmid (follow Subheadings 3.1–3.3) combined with the introduction of a donor DNA fragment containing an antibiotic resistance gene (follow Subheading 3.9). To completely remove a gene from the Leishmania genome as shown in Fig. 3, it is necessary to coexpress two gRNAs targeting the 5′ and 3′ end of the same gene (Subheading 3.4). Generally, it is not necessary to completely remove a gene to disrupt its function; this can more easily be achieved by disrupting the gene with a donor DNA fragment containing a selectable marker as outlined in Subheadings 3.1–3.3 and 3.9.

Gene editing by expressing a single gRNA in Leishmania. (a) Scheme of the Leishmania CRISPR expression vector pLdCN and its guide sequence insertion site. A single gRNA and the Streptococcus pyogenes Cas9 nuclease can be stably expressed in pLdCN under the control of L. donovani ribosome RNA promoter. rRNAP, L. donovani ribosomal RNA promoter; H, Hepatitis delta virus (HDV) ribozyme. The small filled black box in front of Cas9 represents the 92 bp pyrimidine track. A2-IGS A2 intergenic sequence, NEO neomycin resistance gene, AmpR ampicillin resistance gene. The SP6 promoter, which can be used as the sequencing primer to verify the guide sequence insert, is also indicated. The vector size is 9217 bp and the drawing is not to scale. The first nucleotide U of gRNA is highlighted (black) as L. donovani rRNAP initiates transcription at the T residue site. (b) Gene mutants generated by a single gRNA targeting to its coding sequence. The simplest way to inactivate a Leishmania gene is to design a gRNA targeting its coding sequence (ideally in the front half of the gene). MMEJ or SSA often results in large deletions. Single-strand oligonucleotides with stop codons or donors with drug resistant marker genes with 25 nt (bp) homology flanking arms can be used to disrupt the gene following Cas9 double-stranded break to facilitate the isolation of the gene-inactivated mutants. The presence of mutants in a culture can be identified by PCR with a specific primer to the oligo donor, restriction enzyme digestion or drug selection. The oligo donor can also be used to introduce frame shift, point mutations, epitope tags and other modifications. The 92 bp pyrimidine track (the filled black box) is required for trans-splicing of the drug resistance gene. (c) Tagging Leishmania genes. With CRISPR technology, a gene can be tagged in almost any position of its coding sequence. As an example, this illustration shows how a Leishmania gene can be tagged at its 3′ terminus with an oligo donor containing an epitope tag, or GFP together with the drug resistance gene donor. Fluorescence-activated cell sorting may be used to isolate the GFP tagged cells without using the drug resistance gene donor. The primers which can be used to detect these gene editing events are also indicated

Gene editing by coexpressing two gRNAs at the same time in Leishmania (a) Scheme of the Leishmania CRISPR vector pLdCN simultaneously expressing two gRNAs and Cas9. HH Hammerhead ribozyme. See Fig. 2 legend for other abbreviations. (b) Complete gene deletion by two gRNAs targeting respectively the 5′ and 3′ flanking sequences of the gene. Compared with single gRNA, the use of double gRNAs significantly increases the gene targeting efficiency (two to tenfold increase) and can be used to delete the whole coding sequence with or without the drug resistance gene donor. The primers which can be used to detect these gene editing events are also indicated. (c) Double gRNAs can be used to delete multiple copy family genes. (d) Double gRNAs can be used to target two different genes in a single Leishmania chromosome. (e) Double gRNAs can be used to target genes present in two different chromosomes such as the LdBPK_241510.1 gene in chromosome 24 and the miltefosine transporter (MT) gene (LdBPK_131590.1) in chromosome 13. Cotargeting MT and selection for miltefosine resistance cells have been shown to significantly facilitate the isolation of the CRISPR cotargeted gene mutants (see Note 13). Two different chromosomes targeted simultaneously by two gRNAs can also be used to generate the specific chromosome translocation mutants
3.1 Selection of CRISPR Targeting Site(s)
To start a CRISPR experiment, the first thing to consider is where the gRNA will target, and the number of gRNAs to be used. For example, a single gRNA targeted to the 5′ half of a gene or sequence encoding functional protein domains is more likely to cause deletion or disruption mutants with more impact on the gene function. Two gRNAs targeted to the 5′ and 3′ flanking sequences of the gene will usually be required to delete the full coding sequence of a gene. To generate a point mutation, the gRNA/Cas9 cleavage site needs to be as close as possible to the desired mutation site. A gRNA designed to target a conserved sequence can be used in different Leishmania species. Once the targeting region has been identified, its sequence can then be loaded into design tools to search gRNAs with relatively high activity score as described below.
3.2 gRNA Design
Because most current gRNA design tools were developed from the data of higher eukaryotic cells, they may not accurately predict gRNA activity in Leishmania [10, 12]. Nevertheless, to design a gRNA with no off-target site(s) that is likely to be active in Leishmania, we recommend using the Eukaryotic Pathogen CRISPR guide RNA Design Tool (EuPaGDT) (http://grna.ctegd.uga.edu/) which ranks a gRNA based on its activity score, off-target sites in the genome and microhomology sequences flanking the double-strand break (DSB) site [37]. CRISPRater (https://crispr.cos.uni-heidelberg.de/) developed from human and mouse data could also be helpful as it can predict gRNA activity with only the guide sequence information [38]. For convenience, one may also directly use the gRNA sequences designed for targeting the immediate 5′ and 3′ flanking sequences for each of Leishmania genes from (http://leishgedit.net/) [28, 34].
3.3 Clone a gRNA Coding Sequence into the Leishmania CRISPR Vector pLdCN
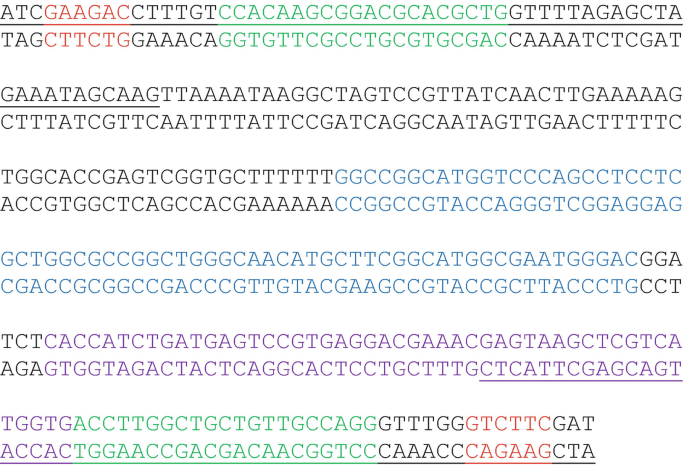
For most gene editing procedures, expression of a single gRNA together with Cas9 is sufficient to enable the introduction of a donor DNA fragment into the target cut site to edit or disrupt a gene or gene family. As illustrated in Fig. 2a, single gRNA guide coding sequences (20 nt) can be ordered as complementary oligos with 5′-TTGT and 5′-AAAC overhangs. For example, for a gRNA targeting site in the genome  (PAM sequence NGG shown in bold), order the following two oligos:
(PAM sequence NGG shown in bold), order the following two oligos:  , and
, and  . Note that the NGG PAM sequence (AGG) is part of the genome sequence but not included in the gRNA sequence (green). Use one of the following two protocols to clone the guide sequences into the Bbs I sites of the pLdCN vector:
. Note that the NGG PAM sequence (AGG) is part of the genome sequence but not included in the gRNA sequence (green). Use one of the following two protocols to clone the guide sequences into the Bbs I sites of the pLdCN vector:
3.3.1 The Traditional Two-Step Digestion and Ligation Cloning Protocol
-
1.
Phosphorylate and anneal each pair of oligos:
3.75 μl
Oligo 1 (100 μM)
3.75 μl
Oligo 2 (100 μM)
1 μl
10× T4 DNA ligase buffer (NEB) (see Note 2)
1 μl
ddH2O
0.5 μl
T4 Polynucleotide Kinase (NEB)
10 μl
Total
Anneal the oligos in a thermocycler using the following parameters:
37 °C
30 min
Then add 90 μl TE to total volume 100 μl (3.75 μM)
95 °C
5 min and then ramp down to 25 °C at 5 °C/min
-
2.
Set up the pLdCN plasmid digestion reaction:
X μl
pLdCN (2 μg) (Addgene #84290)
3 μl
10× FastDigest Buffer
3 μl
FastDigest Bbs I
Y μl
ddH2O
30 μl
Total
Incubate the digestion reaction at 37 °C for 1 h
-
3.
Purify the Bbs I-digested pLdCN vector directly using a PCR product purification kit following manufacturer’s instructions (Thermo Fisher Scientific) and elute the digested pLdCN vector with 30 μl elution buffer.
-
4.
Set up the following ligation reaction:
5.9 μl
Bbs I-digested pLdCN (0.39 μg)
3 μl
Phosphorylated and annealed
Oligo duplex from step 1 (3.75 μM)
1 μl
10× T4 DNA ligase buffer
0.1 μl
T4 DNA ligase (Thermo Fisher Scientific)
10 μl
Total
Incubate the ligation reaction at room temperature for 1–2 h
-
5.
Transform competent E. coli cells with 5 μl of the ligation reaction, plate and grow overnight at 37 °C.
-
6.
Pick and grow four colonies, prepare plasmid DNA using a minipreparation kit and sequence the plasmids using the SP6 promoter primer and conditions for DNA with secondary structure to verify the clones. Purified pLdCN plasmids can be used to transfect Leishmania as detailed below in Subheading 3.5.
3.3.2 The Single-Step Digestion–Ligation Cloning Protocol
The single-step digestion–ligation cloning protocol adapted from Feng Zhang’s lab (http://www.genome-engineering.org/crispr/wp-content/uploads/2014/05/CRISPR Reagent-Description-Rev20140509.pdf) is simpler to set up and may increase cloning efficiency. Due to the simultaneous digestion–ligation step, the guide oligos cannot contain any Bbs I enzyme recognition sites (i.e., the nucleotide sequence “GAAGAC” or ‘GTCTTC’).
-
1.
Phosphorylate and anneal each pair of oligos:
1 μl
Oligo 1 (100 μM)
1 μl
Oligo 2 (100 μM)
1 μl
10× T4 DNA Ligase buffer (NEB)
6.5 μl
ddH2O
0.5 μl
T4 Polynucleotide Kinase (NEB)
10 μl
Total
Anneal in a thermocycler using the following parameters:
37 °C
30 min
95 °C
5 min and then ramp down to 25 °C at 5 °C/min
Dilute the annealed oligo 1:250 (250-fold) in TE buffer.
-
2.
Set up the following digestion–ligation reaction: This reaction combines Bbs I digestion of the pLdCN plasmid and ligation of the oligonucleotide into the Bbs I cut site.
X μl
pLdCN (100 ng) (Addgene #84290)
2 μl
Phosphorylated and annealed
Guide duplex from step 1 (1:250 dilution)
2 μl
10× FastDigest buffer
1 μl
10 mM DTT
1 μl
10 mM ATP
1 μl
FastDigest Bbs I (Thermo Fisher Scientific)
0.5 μl
T7 DNA ligase
Y μl
ddH2O
20 μl
Total
Incubate the ligation reaction in a thermocycler:
37 °C
5 min
23 °C
5 min
Cycle the previous two steps for six cycles (total run time 1 h)
4 °C
Hold until ready to proceed
-
3.
Transform competent E. coli cells with 5 μl of the ligation reaction from step 2, plate and grow overnight at 37 °C.
-
4.
Pick and grow four colonies, prepare plasmid DNA using a minipreparation kit and sequence the plasmids using the SP6 promoter primer and conditions for DNA with secondary structure to verify the clones. Purified pLdCN plasmids can be used to transfect Leishmania as detailed below in Subheading 3.5.
3.4 Clone Two gRNA Guide Coding Sequences into Leishmania CRISPR Vector pLdCN
To delete a complete gene sequence, delete multicopy family genes, cotarget two different genes or generate specific chromosomal translocations, and to improve the gene targeting efficiency, two (or more) gRNAs can be simultaneously expressed in pLdCN vector (Fig. 3a) (see Note 3). One of the following two protocols (3.4.1 or 3.4.2) can be used to express two gRNA sequences from a single vector. The pLdCN2 (Addgene 125186) vector was recently engineered to simplify the cloning of two guide RNA sequences in a single vector (protocol 3.4.2).
3.4.1 The Cloning Protocol Using PCR Generated DNA Cassette
To express two gRNAs in the pLdCN vector, first use PCR to generate a DNA cassette that contains the two gRNA sequences separated by self-splicing ribozyme sequences as shown in Fig. 3a. To insert two gRNA coding sequences into the pLdCN vector with PCR and a single cloning step, an example for targeting the A2 Leishmania gene [11] is shown below:
-
1.
The two A2 gene gRNA targeting sites are as follows: a,
 ; b,
; b,  .
. -
2.
Design and order the following two primers which contain respectively the A2
 guide coding sequence (in Green), a Bbs I site (in Red) and the pSPneogRNA241510+MT plasmid sequence (in Black):
guide coding sequence (in Green), a Bbs I site (in Red) and the pSPneogRNA241510+MT plasmid sequence (in Black):
-
3.
Use the pSPneogRNA241510+MT plasmid (Addgene #84292) as template, carry out a PCR reaction to obtain the following 276 bp PCR product which contains the coding sequences of gRNAa, HDV (in Blue) and Hammerhead (in Purple) ribozymes, and gRNAb guide (see also Fig. 3a). This PCR product is then cloned into the pLdCN vector after Bbs I digestion as cloning the oligo duplex described above in Subheading 3.3.
 ; b,
; b,  .
. guide coding sequence (in Green), a Bbs I site (in Red) and the pSPneogRNA241510+MT plasmid sequence (in Black):
guide coding sequence (in Green), a Bbs I site (in Red) and the pSPneogRNA241510+MT plasmid sequence (in Black):

3.4.2 The Single-Step Digestion–Ligation Cloning Protocol for Two gRNA
To further simplify the cloning of two guide RNA sequences into a single vector, we generated the pLdCN2 plasmid containing Bbs I sites generating four unique ends that ensures the proper insertion and orientation of two pairs of oligonucleotides with the vector and the interspaced ribozyme sequences (required for gRNA processing) as shown in Fig. 4. For versatility and economy, the primer pair used for insertion at site 1 is cross-compatible with the single gRNA expression vector pLdCN. The primer pair used for insertion at site 2 is unique to pLdCN2 and requires the inclusion of a GT:CA pair at the 3′ end of the guide sequence (highlighted in green bold in Fig. 4.) as the Bbs I site in site two cuts into the gRNA scaffold on the vector. Make sure the guide oligos do not contain any Bbs I enzyme recognition sites (i.e., the nucleotide sequence ‘GAAGAC’ or ‘GTCTTC’).

Representation of the pLdCN2 vector used to express two gRNAs using primer pairs as inserts in a single-tube digestion–ligation reaction into a Cas9-expressing plasmid. rRNAP, L. donovani ribosomal RNA promoter; gRNA1, scaffold of the first gRNA; H Hepatitis delta virus (HDV) ribozyme, HH Hammerhead ribozyme, gRNA2 scaffold of the second gRNA. Refer to Fig. 2 legend for other abbreviations. The guide sequence insertion sites 1 and 2 are enlarged to show the Bbs I recognition sites (in red) and cleavage sites (red arrows) with resulting overhangs. Guide sequences (N20) can be inserted into pLdCN2 by using partially complementary primers resulting in overhangs matching insert site 1 or 2 (TTGT/CAAA; GGTG/AAAT, respectively). Overhangs used for insert site 1 are cross compatible with the single insert site pLdCN plasmid. Overhangs used for site 2 are unique to pLdCN2. Insert site 2 further requires a GT:CA 2 bp sequence be appended to the N20 guide sequence for proper insertion (in bold). These 2 base pairs are not part of the recognition sequence and will not affect the targeting of gRNA2
-
1.
Phosphorylate and anneal each pair of oligos in the same reaction:
1 μl
Oligo 1a (100 μM)
1 μl
Oligo 1b (100 μM)
1 μl
Oligo 2a (100 μM)
1 μl
Oligo 2b (100 μM)
1 μl
10× T4 DNA Ligase buffer (NEB)
4.5 μl
ddH2O
0.5 μl
T4 Polynucleotide Kinase (NEB)
10 μl
Total
Anneal in a thermocycler using the following parameters:
37 °C
30 min
95 °C
5 min and then ramp down to 25 °C at 5 °C/min
Dilute the annealed oligos 1:250 (250-fold) in TE buffer.
-
2.
Set up the following digestion–ligation combination reaction: This reaction combines both plasmid digestion with Bbs I and ligation of the two oligonucleotides into site 1 and site 2 of the BbsI-digested plasmid.
X μl
pLdCN2 (100 ng) (Addgene #125186)
2 μl
Phosphorylated and annealed
Guide duplexes from step 1 (1:250 dilution)
2 μl
10× FastDigest buffer
1 μl
10 mM DTT
1 μl
10 mM ATP
1 μl
FastDigest Bbs I
0.5 μl
T7 DNA ligase
Y μl
ddH2O
20 μl
Total
Incubate the digestion–ligation reaction in a thermocycler:
37 °C
5 min
23 °C
5 min
Cycle the previous two steps for six cycles (total run time 1 h)
4 °C
Hold until ready to proceed
-
3.
Transform competent E. coli cells with 5 μl of the ligation reaction from step 2, plate and grow overnight at 37 °C.
-
4.
Pick and grow four colonies, prepare plasmid DNA using a minipreparation kit and sequence the plasmids using the SP6 promoter primer and conditions for DNA with secondary structure to verify the clones. The purified pLdCN2 plasmids can be used to transfect Leishmania as detailed below in Subheading 3.5.
As with the pLdCN vector expressing a single gRNA (Subheading 3.3), an oligonucleotide donor or an antibiotic selection gene containing donor can be designed for integration into the double gRNA targeting sites to improve editing efficiency.
3.5 Transfection of Leishmania with pLdCN Plasmid or Donor DNA
-
1.
2 × 107 Leishmania promastigotes (middle log phase to early stationary phase) are harvested by centrifugation at 4000 rpm (1500 × g) in a benchtop centrifuge for 5 min.
-
2.
Wash once with 200 μl Tb-BSF buffer.
-
3.
Resuspend the parasites in 100 μl Tb-BSF buffer.
-
4.
Add 2–5 μg of the pLdCN or pLdCN2 plasmid from Subheading 3.3 or 3.4 in less than 20 μl volume into the above Leishmania promastigotes suspension and mix well.
-
5.
Transfer the above parasites and DNA mix solution into a 2-mm gap electroporation cuvette.
-
6.
Place the cuvette into the Nucleofector™ 2b Device and perform the transfection using program U-33.
-
7.
Transfer the electroporated parasites into a 25 cm2 flask containing 5 ml culture medium using an autoclaved glass Pasteur pipette or a sterile transfer bulb.
-
8.
Add G418 to the culture at 50 μg/ml the following day, to select for the pLdCN(2) plasmid-transfected cells.
-
9.
Monitor drug selection of the transfection culture daily. If needed, change the culture with fresh medium once every 5 days, and increase the G418 concentration to 100 μg/ml.
-
10.
Once the pLdCN(2) transfected cell culture is established, these cells can be subsequently transfected with the donor DNA (see Subheading 3.8 below for use of donor DNA); 4–8 μl of 100 μM oligonucleotide donor DNA or 2–4 μg of purified PCR product donor DNA is used for each transfection (see Note 4).
-
11.
Cells can be repeatedly transfected with an oligonucleotide donor DNA at 2–3 day intervals, up to a total of four transfections to improve gene editing efficiency [11].
-
12.
The drug selection for integration of the donor DNA (if the donor contains an antibiotic resistance gene) should start 1–2 days after the transfection with a drug resistance gene containing donor to allow time for the donor DNA integration into the genome and expression of the resistance gene. If the bleomycin resistance gene donor is used, add 50 μg/ml phleomycin initially and then increase to 100 μg/ml once the cultures start to look healthy.
-
13.
The transfected Leishmania cultures can sometimes be incubated at 33 °C or 37 °C for 2–3 days to improve gene editing efficiency [10].
3.6 Isolate the CRISPR Edited Mutants
To isolate the CRISPR edited mutants, transfected Leishmania promastigotes can be cloned by limiting dilution in 96-well plates containing G418 (see Note 5). The clones are then expanded in a 24-well plate for genomic DNA extraction [39], PCR, and other analyses. Depending on the editing efficiency, several cloning methods can be used to isolate the edited mutants.
-
1.
For experiments with high editing efficiency (editing frequency > 10%) or if the edited mutants can be selected with an antibiotic selection marker using a donor DNA, the transfected cells or the drug resistant cells can be cloned either by directly inoculating 1–3 promastigotes in 100 μl per well in a 96-well plate or by serial twofold dilution until the single cell per well is reached (limiting dilution) (see Note 6).
-
2.
For experiments with low editing efficiency (editing frequency < 10%) and when the edited mutants cannot be selected with a drug, for example for gene editing with an oligonucleotide donor, the transfectants can be cloned in following way. Inoculate 5–20 promastigotes in 150 μl per well in two to three 96-well plates. Once the cells in these plates have grown up, pool a portion of the cells from 8–12 wells (take 100 μl cells from each of these wells), extract the genomic DNA, and perform PCR analysis to identify the presence of the mutant. If the desired gene editing can be detected in these pooled wells, continue cloning cells from the individual wells until the desired edited mutant is detected by PCR and has been single cell cloned.
-
3.
It is possible that after a first round of cloning, promastigotes with only a single gene allele mutated (deleted or disrupted) are isolated. In this case, the single allele edited mutants should be further cultured and recloned until mutants with all alleles edited have been isolated.
-
4.
Since alteration of the gene sequence (deletion , disruption or point mutation) could result in phenotype changes, pay attention to clones with different phenotypes such as reduced growth rate, changes in size or motility, drug resistance and others. Because the mutant promastigotes often grow slower than the wild-type promastigotes, it is important to clone these CRISPR transfectants early to avoid overgrowth by the wild-type promastigotes.
3.7 Generate Gene Deletion Mutants Without Using a Donor
Generally, it is always better to use a donor DNA to repair the Cas9 cut site to improve the gene targeting efficiency and specifity, since the double strand breaks in Leishmania are mainly repaired by MMEJ or SSA which often cause large deletions [10,11,12,13]. However, it is possible to directly isolate the CRISPR deletion mutants without using a donor, for example in targeting of the miltefosine transporter gene [10, 12], the A2 gene family [11] and the arginine transporter (AAP3) gene [13]. If a donor is not used, it is important to obtain pure clones and to use several PCR primer pairs with various distances flanking the Cas9 cleavage site to identify the deletion junctions (see Fig. 2b) (see Note 7). If a gene targeted is flanked by direct repeated sequences, the gene will most likely be deleted by the SSA mechanism, therefor primers flanking these direct repeat sequences can be conveniently used to detect the gene deletion mutants resulting from SSA [12].
Due to the low efficacy of double-strand DNA break repair in Leishmania, the double allelic mutation frequency using CRISPR without using a donor ranges from <1% to 10% depending on the gRNA used and the length of time post transfection [10]. However, the use of multiple gRNAs, extended culture (months posttransfection) and development of new CRISPR methods could improve the targeting efficiency to ease isolation of the donor-free gene deletion mutants [10, 11].
3.8 Generate Gene Edited Mutants by Using an Oligonucleotide Donor
As discussed above, double strand DNA break repair with MMEJ or SSA can lead to large unpredictable deletions which are often difficult to detect and verify [10, 41]. A single strand sense oligonucleotide with only 25 nt homology arms flanking the Cas9 cleavage site can be used as a source of DNA to repair the double strand break to introduce stop codons, frame shift, single point mutations, restriction sites, small epitope tags and others (Figs. 2b, c and 3) (see Note 8). An example using an oligonucleotide donor with stop codons to disrupt the L. donovani miltefosine transporter gene [10] is shown below.

Sequential transfection of pLdCN plasmid containing promastigotes with an oligonucleotide donor at 2- to 3-day intervals (as soon as enough G418 resistant cells have recovered) up to a total of four transfections can significantly improve the gene editing efficiency [11]. After the last oligonucleotide donor transfection, the Leishmania promastiogtes should be immediately cloned in 96 well plates to avoid overgrowth by the unedited cells. A PCR primer specific to the edited site and/or the inclusion of a restriction site within the oligonucleotide donor can greatly help to identify the gene edited mutants. To avoid the edited site being recut by the gRNA/Cas9 complex, some nucleotides in the seed region of the protospacer and the PAM site should also be mutated or deleted [11]. The oligonucleotide donor can also be used to generate a precise deletion between two gRNA targeting sites (Fig. 3b) or targeted chromosomal translocation between two chromosome targeting sites [10, 11].
3.9 Generate Gene Disrupted Mutants by Using an Antibiotic Resistance Selection Marker Donor
To facilitate the isolation of gene disrupted mutants, donor DNA containing a selection marker such as an antibiotic resistance gene flanked by 25 bp homology arms can be introduced into the Cas9 cleavage site(s) of the open reading frame (ORF) sequence (Figs. 2 and 3) (see Note 9). An example using the bleomycin resistance gene as a donor generated by PCR for disrupting the L. donovani LdBPK_241510.1 gene [10] is shown below.

To generate a donor DNA with a bleomycin resistance gene, it is necessary to start with the pSPBle plasmid that contains the bleomycin resistance gene (Addgene #63561). Design the following two specific donor primers which consist of 20 nt of pSPBle plasmid sequence (in Red) and 25 nt of sequence flanking the Cas9 cleavage site (in Black and Green) (example shown for the LdBPK_241510.1 gene below). The pSPBle plasmid is used as the PCR template to make the 584 bp bleomycin resistance gene donor PCR product (The 92 bp pyrimidine track upstream of the bleomycin resistance gene needed for trans-splicing is underlined).

Following PCR, run the product on an agarose gel to verify the size (584 bp) and purify the PCR product from the agarose gel. Transfect the PCR product containing the bleomycin resistance gene donor into the pLdCN plasmid containing promastigotes as described in Subheading 3.5 (see Note 10).
Likewise, a fluorescent protein coding sequence alone or together with a selection marker can be used to tag the gene of interest (Fig. 2) [10, 28]. As reported by Beneke et al., the use of two selection marker donors can further enhance the gene targeting efficiency [28, 34]. Small epitope tags together with a selection marker can also facilitate isolation of the gene-tagged mutants (Fig. 2).
3.10 Delete Multicopy Family Genes
Compared with the traditional gene targeting methods, one of the major advantages of CRISPR technology is that the gRNA and Cas9 complex stably expressed will continuously scan the genome and generate targeted double-strand breaks until all target sites have been deleted or mutated. The stable CRISPR expression system has been successfully used to delete the multicopy A2 family genes which have more than 11 copies distributed in four clusters on chromosome 22 of L. donovani 1SCL2D parasites [11]. To delete multicopy family genes, it is possible to use either a single gRNA targeted to the gene internally (Fig. 2) or two gRNAs, one targeted to the 5′ flanking sequence of the gene cluster and the other to the 3′ flanking sequence (Fig. 3c). A prolonged culture time and the use of an oligonucleotide donor or drug selection marker donor will increase the efficiency of targeting multicopy genes.
3.11 Target Essential Leishmania Genes
Because some Leishmania chromosomes are multiploid ranging from 3 to 5 chromosomes and Leishmania is well known to be capable of randomly amplifying its genes by using the direct or inverted repeat sequences to form circular or linear amplicons [12, 23, 24, 40, 41], it is often difficult and time consuming (more than two rounds of gene targeting are required) to determine whether a gene is essential for the viability of Leishmania with the traditional homologous recombination gene targeting method. CRISPR technology has greatly reduced the effort and time required to reveal the essentiality of Leishmania genes [10,11,12]. To determine whether a gene is essential for Leishmania, a single or double gRNAs together with an antibiotic selection marker donor should be used for maximal gene targeting efficiency. Once one allele of the essential gene has been disrupted or deleted, as evidenced by PCR analysis, there will be a reduced rate of proliferation compared with targeting nonessential genes. It will be possible to directly observe some of these CRISPR targeted cells dying in culture, especially after cloning into the 96-well plate since the gRNA and Cas9 will continuously scan the genome to search for the last essential gene target site to disrupt resulting in cell death. Depending on the targeting efficiency, 10–50% of these cells could be seen dying after cloning in the 96-well plate. These essential gene knockout promastigotes usually can still grow and divide slowly for about 1–2 weeks forming clumps in the wells before final cell death (lysis) [10,11,12]. This could be due to the time required to dilute and degrade the remaining mRNA and protein after all copies of the essential gene have been knocked out. PCR analysis will show the wild-type gene band persists in surviving single knockout Leishmania cells (see Note 11).
To facilitate the direct observation of dying cells following CRISPR gene targeting of essential genes, add cells into 96-well plates.
-
1.
Inoculate 1–3 promastigotes in 100 μl per well in a 96-well plate.
-
2.
Carefully examine each of the wells under a microscope in the following days and mark the wells with live parasites.
-
3.
Monitor the promastigote growth daily, mark the wells with poorly growing promastigotes, focus on these wells thereafter until all the promastigotes in these wells are dead.
-
4.
Expand the cultures of surviving promastigote wells in a 24-well plate, extract genomic DNA and perform PCR to confirm that the wild-type gene band persists in the surviving clones.
-
5.
If required, additional selection markers or an oligonucleotide donor with silent mutations can be used to further confirm the essentiality of the gene.
3.12 Gene Tagging
As described above, oligonucleotide and PCR products can be used to introduce an epitope tag or a fluorescent protein into the Cas9 cleavage site to tag the gene of interest (Fig. 2c). The ability to tag a gene in its genomic locus will help to accurately measure the native gene expression at the protein level and should be especially useful for genes which are difficult to clone due to their large size or presence of repeated sequences such as the A2 family genes. With the help of cell sorting, cells with GFP or other fluorescent proteins tagged into an endogenous gene could be selected without using an antibiotic selection marker.
3.13 Gene Complementation
Like in mammalian cells, with carefully designed gene-specific gRNAs, CRISPR-Cas9 induced off-target mutations should be low in Leishmania. Nevertheless, to exclude the possibility of an observed phenotype change resulting from off-site targeting and other unexpected genetic changes such as deletion of neighboring genes during the CRISPR process [12], as occurs with traditional gene targeting, it is necessary to add back the wild-type gene to complement the knockout mutant to determine if it is possible to restore the wild-type phenotype. Gene complementation can be achieved by episomal expression of the wild-type gene [12, 25, 41, 42], or by reusing CRISPR technology to insert the wild-type gene back into the locus (see Note 12). Since the selection marker donor (if used) has been inserted into the genome, and the pLdCN CRISPR vector can remain in the parasites for weeks, even in the absence of G418 after gene editing, it is important to consider whether the addback wild-type gene can be similarly targeted by the remaining CRISPR vector, and whether an episomal expression vector with a new selection marker is needed to express the addback gene [10].
3.14 Generate Targeted Chromosome Translocations
One may consider using CRISPR to generate specific chromosome translocations to study polycistronic transcription control, gene amplification, chromosome replication, stability, and others. Two gRNAs each targeting specific sites from two different chromosomes are cloned into the pLdCN2 vector and expressed simultaneously in the cell (Fig. 3e), an antibiotic selection marker donor should help to isolate the cells with targeted chromosome translocation [11].
4 Notes
-
1.
The electroporation method using Bio-Rad Gene Pulser Electroporation System with Cytomix buffer can also be used for labs not yet equipped with LONZA Nucleofector™ System [10].
-
2.
T4 Polynucleotide Kinase is used for 5′ phosphorylation of the oligos for subsequent ligation into the pLdCN(2) vector and requires in the reaction the presence of ATP which is provided by the DNA ligase buffer (1 mM ATP at 1× final concentration). However, the T4 Polynucleotide Kinase buffer itself usually does not contain ATP as T4 Polynucleotide Kinase is mostly used for end radiolabeling of DNA or RNA probes where the [γ-32P]ATP is additionally added for the 5′ End-Labeling reaction.
-
3.
If required, with the PCR cassette approach, more than two gRNAs in tandem array can be cloned into the pLdCN vector. Alternatively, other CRISPR vectors such as pLdCH (Addgene #84291) can be used to clone additional gRNAs.
-
4.
The amount of oligonucleotide donor used has not been systematically tested; one may try different quantities to find the most efficient dose.
-
5.
G418 drug pressure should be maintained in the culture medium to retain the pLdCN plasmid in these Leishmania cells until the desired gene editing mutant has been obtained.
-
6.
Since it is difficult to evenly distribute 1 promastigote per well in a 96-well plate, adding 2–3 promastigotes per well will reduce the number of empty wells (with no promastigotes).
-
7.
Sometimes, due to the presence of wild-type dead promastigotes, the faint wild-type band could be present in PCR products in some of the knockout clones; this can be verified by recloning these promastigotes.
-
8.
Likely because the antisense oligonucleotide could hybridize to the corresponding mRNA to reduce its availability as the repair template, we have successfully generated the single amino acid replacement mutant of the RAD51 protein with the sense oligonucleotide donor but not with the antisense oligo donor [11].
-
9.
For a first-time user of CRISPR gene editing and the experimental goal being simply to knockout (inactivate) a single gene, we recommend to first express a single gRNA in pLdCN (Subheadings 3.1–3.3) and selection with an antibiotic resistance gene donor (Subheading 3.9) because this will be the simplest and most direct way to obtain the disrupted gene mutant. For researchers who already have the first-hand CRISPR experience and are familiar with basic molecular biology and parasitology techniques, they can take more challenging and advanced approaches such as gene targeting without using a donor or using an oligonucleotide donor to tag a gene or edit a gene (Subheading 3.8) or express two-gRNAs from pLdCN2 (Subheading 3.4).
-
10.
Besides the bleomycin resistance gene, other drug resistance genes such as the puromycin and hygromycin resistance genes could also be used as the selection marker donor. As reported by Beneke et al. [28], the raw PCR product donor without purification can be directly used for transfection. As the single-strand DNA donor could be more efficient than the double-strand DNA donor, the PCR product donor can be heat-denatured and sterilized immediately before transfection.
-
11.
Since DSB repair is not efficient in Leishmania and nonrepaired DSB is lethal to the cell, some of these promastigote deaths could also be triggered by DSBs which are not promptly repaired [12]. Cell deaths observed in targeting nonessential genes are most likely caused by nonrepaired DSB. Contrary to cell deaths resulting from knockout of essential genes, cells with nonrepaired DSB usually cannot replicate before death.
-
12.
To re-CRISPR the cell, the pLdCN vector can be removed from the cells by culturing in G418-free medium for about 2 months. Alternatively, other CRISPR vectors such as pLdCH (Addgene#84291) with the hygromycin resistance marker can be used for subsequent CRISPR targeting. In addition, we recently developed a novel Staphylococcus aureus Cas9 (SaCas9) expression vector pLdSaCN (Addgene#123261) which uses NNGRRT PAM sequences, has similar or improved gene editing efficiency compared with the Streptococcus pyogenes Cas9 expression vector pLdCN. The same guide sequence cloning strategy is used for pLdSaCN vector [12].
-
13.
Cotargeting the miltefosine transporter (MT) gene can significantly facilitate isolation of the gene edited mutants. However, mutation of the MT gene could also reduce the parasite infectivity and virulence.
References
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–821
Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339:819–823
Mali P, Esvelt KM, Church GM (2013) Cas9 as a versatile tool for engineering biology. Nat Methods 10(10):957–963
Doudna JA, Charpentier E (2014) Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 346(6213):1258096
Hsu PD, Lander ES, Zhang F (2014) Development and applications of CRISPR-Cas9 for genome engineering. Cell 157(6):1262–1278
Knott GJ, Doudna JA (2018) CRISPR-Cas guides the future of genetic engineering. Science 361(6405):866–869
Nishimasu H, Ran FA, Hsu PD, Konermann S, Shehata SI, Dohmae N, Ishitani R, Zhang F, Nureki O (2014) Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 156(5):935–949
Her J, Bunting SF (2018) How cells ensure correct repair of DNA double-strand breaks. J Biol Chem 293(27):10502–10511
Sallmyr A, Tomkinson AE (2018) Repair of DNA double-strand breaks by mammalian alternative end-joining pathways. J Biol Chem 293(27):10536–10546
Zhang WW, Matlashewski G (2015) CRISPR-Cas9-mediated genome editing in Leishmania donovani. MBio 6(4):e00861
Zhang WW, Lypaczewski P, Matlashewski G (2017) Optimized CRISPR-Cas9 genome editing for Leishmania and its use to target a multigene family, induce chromosomal translocation and study DNA break repair mechanisms. mSphere 2(1):e00340–e00316. https://doi.org/10.1128/mSphere.00340-16
Zhang WW, Matlashewski G (2019) Single-Strand Annealing Plays a Major Role in Double-Strand DNA Break Repair following CRISPR-Cas9 Cleavage in Leishmania. mSphere 4:e00408–19. https://doi.org/10.1128/mSphere.00408-19
Goldman-Pinkovich A, Kannan S, Nitzan-Koren R, Puri M, Bar-Avraham Y, McDonald JA, Sur A, Zhang WW, Matlashewski G, Madhubala R, Michaeli S, Myler PJ, Zilberstein Dan (2019) The hunger games: sensing host arginine is essential for Leishmania parasite virulence. bioRxiv 751610. https://doi.org/10.1101/751610
Böttcher R, Hollmann M, Merk K, Nitschko V, Obermaier C, Philippou-Massier J, Wieland I, Gaul U, Förstemann K (2014) Efficient chromosomal gene modification with CRISPR/cas9 and PCR-based homologous recombination donors in cultured Drosophila cells. Nucleic Acids Res 42(11):e89. https://doi.org/10.1093/nar/gku289
Paix A, Wang Y, Smith HE, Lee CY, Calidas D, Lu T, Smith J, Schmidt H, Krause MW, Seydoux G (2014) Scalable and versatile genome editing using linear DNAs with microhomology to Cas9 sites in Caenorhabditis elegans. Genetics 198(4):1347–1356
Burza S, Croft SL, Boelaert M (2018) Leishmaniasis. Lancet 392(10151):951–970. https://doi.org/10.1016/S0140-6736(18)31204-2
Lye LF, Owens K, Shi H, Murta SM, Vieira AC, Turco SJ, Tschudi C, Ullu E, Beverley SM (2010) Retention and loss of RNA interference pathways in trypanosomatid protozoans. PLoS Pathog 6(10):e1001161
Cruz A, Beverley SM (1990) Gene replacement in parasitic protozoa. Nature 348(6297):171–173
Tobin JF, Laban A, Wirth DF (1991) Homologous recombination in Leishmania enriettii. Proc Natl Acad Sci U S A 88(3):864–868
Cruz A, Coburn CM, Beverley SM (1991) Double targeted gene replacement for creating null mutants. Proc Natl Acad Sci U S A 88(16):7170–7174
Papadopoulou B, Dumas C (1997) Parameters controlling the rate of gene targeting frequency in the protozoan parasite Leishmania. Nucleic Acids Res 25(21):4278–4286
Peacock CS, Seeger K, Harris D, Murphy L, Ruiz JC, Quail MA et al (2007) Comparative genomic analysis of three Leishmania species that cause diverse human disease. Nat Genet 39(7):839–847
Downing T, Imamura H, Decuypere S, Clark TG, Coombs GH, Cotton JA et al (2011) Whole genome sequencing of multiple Leishmania donovani clinical isolates provides insight into population structure and mechanisms of resistance. Genome Res 21(12):2143–2156
Rogers MB, Hilley JD, Dickens NJ, Wilkes J, Bates PA, Depledge DP et al (2011) Chromosome and gene copy number variations allow major structural change between species and strains of Leishmania. Genome Res 21(12):2129–2142
Zhang WW, Ramasamy G, McCall LI, Haydock A, Ranasinghe S, Abeygunasekara P, Sirimanna G, Wickremasinghe R, Myler P, Matlashewski G (2014) Genetic analysis of Leishmania donovani tropism using a naturally attenuated cutaneous strain. PLoS Pathog 10(7):e1004244
Lypaczewski P, Hoshizaki J, Zhang WW, McCall LI, Torcivia-Rodriguez J, Simonyan V, Kaur A, Dewar K, Matlashewski G (2018) A complete Leishmania donovani reference genome identifies novel genetic variations associated with virulence. Sci Rep 8(1):16549
Sollelis L, Ghorbal M, MacPherson CR, Martins RM, Kuk N, Crobu L, Bastien P, Scherf A, Lopez-Rubio JJ, Sterkers Y (2015) First efficient CRISPR-Cas9-mediated genome editing in Leishmania parasites. Cell Microbiol 17(10):1405–1412
Beneke T, Madden R, Makin L, Valli J, Sunter J, Gluenz E (2017) A CRISPR Cas9 high-throughput genome editing toolkit for kinetoplastids. R Soc Open Sci 4:170095. https://doi.org/10.1098/rsos.170095
Soares Medeiros LC, South L, Peng D, Bustamante JM, Wang W, Bunkofske M, Perumal N, Sanchez-Valdez F, Tarleton RL (2017) Rapid, selection-free, high-efficiency genome editing in protozoan parasites using CRISPR-Cas9 ribonucleoproteins. MBio 8:e01788–e01717
Ishemgulova A, Hlaváčová J, Majerová K, Butenko A, Lukeš J, Votýpka J, Volf P, Yurchenko V (2018) CRISPR/Cas9 in Leishmania mexicana: a case study of LmxBTN1. PLoS One 13(2):e0192723
Fernandez-Prada C, Sharma M, Plourde M, Bresson E, Roy G, Leprohon P, Ouellette M (2018) High-throughput Cos-Seq screen with intracellular Leishmania infantum for the discovery of novel drug-resistance mechanisms. Int J Parasitol Drugs Drug Resist 8(2):165–173. https://doi.org/10.1016/j.ijpddr.2018.03.004
Beneke T, Demay F, Hookway E, Ashman N, Jeffery H, Smith J, Valli J, Becvar T, Myskova J, Lestinova T, Shafiq S, Sadlova J, Volf P, Wheeler RJ, Gluenz E, Hill KL (2019) Genetic dissection of a Leishmania flagellar proteome demonstrates requirement for directional motility in sand fly infections. PLOS Pathogens 15(6):e1007828
Giri, S, Shaha, C (2019) Leishmania donovani parasite requires Atg8 protein for infectivity and survival under stress. Cell Death Dis 10:808. https://doi.org/10.1038/s41419-019-2038-7
Beneke T, Gluenz E (2019) LeishGEdit: A Method for Rapid Gene Knockout and Tagging Using CRISPR-Cas9. Methods Mol Biol 1971:189–210. https://doi.org/10.1007/978-1-4939-9210-2_9
Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ, Zetsche B, Shalem O, Wu X, Makarova KS, Koonin EV, Sharp PA, Zhang F (2015) In vivo genome editing using Staphylococcus aureus Cas9. Nature 520(7546):186–191
Schumann Burkard G, Jutzi P, Roditi I (2011) Genome-wide RNAi screens in bloodstream form trypanosomes identifydrug transporters. Mol Biochem Parasitol 175(1):91–94
Peng D, Tarleton R (2015) EuPaGDT: a web tool tailored to design CRISPR guide RNAs for eukaryotic pathogens. Microb Genom 1(4):e000033
Labuhn M, Adams FF, Ng M, Knoess S, Schambach A, Charpentier EM, Schwarzer A, Mateo JL, Klusmann JH, Heckl D (2018) Refined sgRNA efficacy prediction improves large- and small-scale CRISPR–Cas9 applications. Nucleic Acids Res 46(3):1375–1385
Medina-Acosta E, Cross GA (1993) Rapid isolation of DNA from trypanosomatid protozoa using a simple ‘mini-prep’ procedure. Mol Biochem Parasitol 59(2):327–329
Grondin K, Roy G, Ouellette M (1996) Formation of extrachromosomal circular amplicons with direct or inverted duplications in drug-resistant Leishmania tarentolae. Mol Cell Biol 16(7):3587–3595
Ubeda JM, Raymond F, Mukherjee A, Plourde M, Gingras H, Roy G, Lapointe A, Leprohon P, Papadopoulou B, Corbeil J, Ouellette M (2014) Genome-wide stochastic adaptive DNA amplification at direct and inverted DNA repeats in the parasite Leishmania. PLoS Biol 12(5):e1001868
Zhang WW, Matlashewski G (2015) Screening Leishmania donovani complex-specific genes required for visceral disease. Methods Mol Biol 1201:339–361
Acknowledgments
This work was supported by the Canadian Institute of Health Research grant MOP125996 to G.M. and a doctoral training award from the Fonds de recherche du Québec—Santé (FRQS) to P.L.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2020 The Author(s)
About this protocol

Cite this protocol
Zhang, WW., Lypaczewski, P., Matlashewski, G. (2020). Application of CRISPR/Cas9-Mediated Genome Editing in Leishmania. In: Michels, P., Ginger, M., Zilberstein, D. (eds) Trypanosomatids. Methods in Molecular Biology, vol 2116. Humana, New York, NY. https://doi.org/10.1007/978-1-0716-0294-2_14
Download citation
DOI: https://doi.org/10.1007/978-1-0716-0294-2_14
Published:
Publisher Name: Humana, New York, NY
Print ISBN: 978-1-0716-0293-5
Online ISBN: 978-1-0716-0294-2
eBook Packages: Springer Protocols