
Abstract
Drosophila melanogaster, a small dipteran of African origin, represents one of the best-studied model organisms. Early work in this system has uniquely shed light on the basic principles of genetics and resulted in a versatile collection of genetic tools that allow to uncover mechanistic links between genotype and phenotype. Moreover, given its worldwide distribution in diverse habitats and its moderate genome-size, Drosophila has proven very powerful for population genetics inference and was one of the first eukaryotes whose genome was fully sequenced. In this book chapter, we provide a brief historical overview of research in Drosophila and then focus on recent advances during the genomic era. After describing different types and sources of genomic data, we discuss mechanisms of neutral evolution including the demographic history of Drosophila and the effects of recombination and biased gene conversion. Then, we review recent advances in detecting genome-wide signals of selection, such as soft and hard selective sweeps. We further provide a brief introduction to background selection, selection of noncoding DNA and codon usage and focus on the role of structural variants, such as transposable elements and chromosomal inversions, during the adaptive process. Finally, we discuss how genomic data helps to dissect neutral and adaptive evolutionary mechanisms that shape genetic and phenotypic variation in natural populations along environmental gradients. In summary, this book chapter serves as a starting point to Drosophila population genomics and provides an introduction to the system and an overview to data sources, important population genetic concepts and recent advances in the field.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others

Key words
- Drosophila melanogaster
- Population genetics
- Demography
- Recombination
- Selection
- Background selection
- Selective sweeps
- Inversions
- Transposable elements
- Clines
1 Introduction
The fruit fly Drosophila melanogaster is a small Dipteran that originates from sub-Saharan Africa [1] and has since then colonized all continents except for Antarctica as a human commensal [2, 3]. Within the last 15–20,000 years it expanded its range to Europe and Asia and was only recently introduced to Australia and the Americas (~200 years ago according to [1, 4]). Because of its short life cycle and its simple maintenance, it was first adopted as a laboratory model organism by William Castle and later by Thomas Hunt Morgan at the beginning of the twentieth century [3, 5]. At a time when the basic principles of heredity were still under heavy debate, Morgan used the Drosophila system to experimentally prove and extend the fundamental predictions of Mendelian genetics, which led to the discovery of genes and their location on chromosomes. This early work was rewarded with Nobel prizes to Morgan and several of his former students and research assistants and forms the basis of our present day understanding of genetic mechanisms [6]. Subsequently, the Drosophila system was further exploited, and resulted in the development of numerous genetic tools such as balancer chromosomes, gene-specific knockout mutants and other transgenic constructs, including the Gal4/UAS system to study gene expression or more recently, the CRISPR/Cas9 system for site-specific genome engineering. Moreover, with its condensed genome of ~180 Mb, D. melanogaster was among the first eukaryotic organisms whose genome was fully sequenced, assembled and annotated [7].
Beside major advances in functional genetics Drosophila has also proven powerful for population genetic inference. Accordingly, numerous major population genetics discoveries have first been made in flies. Theodosius Dobzhansky, together with coworkers and students, was one of the first to systematically investigate genetic variation in Drosophila—particularly by focusing on chromosomal inversions. His groundbreaking work gave a first insight into the evolutionary processes that shape genetic variation and subsequently paved the ground for the modern synthesis of evolutionary biology (see Note 1) [8, 9]. By sequencing the Adh gene in 11 lines collected in 5 natural populations, Hudson generated the first fruit fly DNA sequence polymorphism data, identifying only one nonsynonymous polymorphism out of 43 SNPs [10]. As early as the 1980s, methods based on restriction enzymes were applied to D. melanogaster to quantify natural genetic variation across multiple loci [11, 12], followed by the first analyses of Sanger sequenced DNA fragments from dozens of genes [13]. These studies provided the first insights into genome-wide patterns of variation in DNA sequences, revealing abundant silent nucleotide site diversity, less abundant nonsynonymous diversity and rarer small insertions and deletions and transposable element insertions [14]. Based on the null hypothesis of neutral evolution, Hudson et al. proposed a first statistical test of selection based on comparing polymorphism and divergence: the Hudson–Kreitman–Aguadé (HKA) test [15], which postulates that genes should all exhibit the same ratio of within-species variability (polymorphism) to between-species divergence at neutral sites. As an extension of the HKA test, McDonald and Kreitman developed a novel test to specifically detect positive selection on protein sequences, first used to detect positive selection at the Adh locus in Drosophila, and which has since become a ubiquitous test of neutrality [16]. The ratio of nonsynonymous to synonymous divergence is expected to be equal to the ratio of nonsynonymous to synonymous polymorphism if nonsynonymous sites are neutral or deleterious, but higher if they are adaptive. Some of the strongest evidence for adaptive molecular evolution documented in all organisms has come from application of the McDonald–Kreitman test and methods based on it (reviewed in [17, 18]). Finally, a major discovery made in D. melanogaster was that the level of nucleotide variability is positively correlated with the local recombination rate [19], suggesting that selection may constitute a major constraint on levels of genomic diversity.
In summary, the fruit fly D. melanogaster is an ideal model for studying neutral and adaptive genome evolution in outbred, sexual organisms since it is characterized by a long history as a genetic model organism [5], exhibits well-documented, rapid, and widespread adaptations over short (<20 generations) timescales in natural populations [20, 21], has powerful genetic tools [5, 22] a well-annotated genome [23], and genome-wide polymorphisms data for several populations (see Subheading 2.2 for details). Moreover, the genomes of over 25 of its congeners have been recently sequenced [24]. In particular, comparative genomics analyses on 12 species provided fundamental new insights into genome evolution [25] and led to the ModENCODE Project [26], which aims at identifying functional elements in the D. melanogaster and Caenorhabditis elegans genomes. In this chapter, we will focus on population genomics studies (see Note 2), mostly based on next generation sequencing data, and review different aspects of both neutral and selective evolution based on the Drosophila system.
2 Data Sources
2.1 Data Acquisition Techniques
One particular strength of the Drosophila system is its simple maintenance under laboratory conditions. Drosophila is commonly propagated as isofemale lines which originate from a single wild-caught and inseminated female. This allows researchers to conduct molecular and phenotypic measurements across several years using the same genetic material and to preserve natural genetic variation under laboratory conditions. In this paragraph we briefly review the nature of the genetic material that has been sequenced in large genome sequencing projects and how these different approaches potentially affect patterns of variation and missing data.
2.1.1 Isofemale Inbred Lines
Isofemale inbred lines are started from single gravid females whose progeny are allowed to interbreed. These lines can be maintained for several years as long as flies are regularly transferred to new vials with fresh fly-food (a well-known task for any student in a Drosophila lab having worked in a fly-room). A high degree of inbreeding due to small population sizes leads to a rapid reduction of genetic variation and heterozygosity at every generation within each isofemale line. Inbred lines are often referred to as F (Filial generations) followed by the number of the generations of full-sib mating (F3, F10, F20, …). Due to their near-complete homozygosity, every line should be considered as contributing a single genome to the total sample (and not two, as it could be assumed for an outbred sample). Since isofemale lines are propagated separately and are not allowed to interbreed, they are a versatile tool to preserve genetic variation under laboratory conditions, given that sufficient isofemale lines per population are maintained [27]. One significant issue with this approach is that lines derived from equatorial populations have shown to be particularly resistant to inbreeding, a problem that has been linked to the presence of inversion polymorphisms hosting recessive lethal mutations. In these lines, large regions (>500 kb) of residual heterozygosity can be observed [28] which complicates the determination of patterns of polymorphism and divergence in this population [29, 30]. Moreover, given the small population sizes at which isofemale lines are usually propagated, novel mutations that appeared after the capture of the wild-caught ancestors are likely to accumulate in each line over time. Isofemale lines that are maintained in the laboratory for long periods of time will thus slightly deviate from their ancestors and be poorer indicators of natural variation compared to recently established lines.
2.1.2 Haploid Embryo Sequencing
To circumvent problems caused by residual heterozygosity, Langley et al. proposed to sequence the amplified genome of a single haploid embryo [29]. Most eggs fertilized by recessive male sterile mutants ms(3)K81 fail to develop [31]. The few that do, however, only contain one haploid maternal genome. Such a single haploid embryo derived from a cross between a female from any line of interest and an ms(3)K81 male provides enough genomic DNA for whole-genome amplification and sequencing [29]. Although whole-genome amplification increases variance in coverage and the frequency of chimeric reads, this technique provides a powerful approach to uniquely generate high-quality sequencing data using standard paired-end sequencing protocols. Similar to isofemale inbred lines, this technique provides a single genome per sequenced individual (female) but allows for obtaining phased DNA sequences even in the presence of inbreeding-resistant polymorphic inversions.
2.1.3 Genomic Sequencing and Phasing of Hemiclones
Whole-genome sequencing of hybrid F1 crosses—the so-called hemiclones—which share one common parent [32], represents an alternative approach to generate phased haplotype sequencing data. Wild-type Drosophila strains are therefore crossed with the same highly inbred or fully isogenic lab-strain that acts as a reference. The resulting F1 hemiclones are then sequenced as single individuals alongside their lab-strain parent to bioinformatically distinguish between the reference and the unknown wild-type allele. This method has been recently employed in D. melanogaster and allowed to combine cytological screens with whole-genome sequencing to generate and analyze fully phased genomes with known inversion polymorphisms [33]. Additionally, this approach was used to sequence and characterize a panel of more than 200 wild-type chromosomes from a North American D. melanogaster population [34].
2.1.4 Pooled Sequencing (Pool-Seq)
Pool-Seq is a sequencing technique, where tissues or whole bodies of multiple individuals are pooled prior to DNA extraction, library preparation, and whole-genome sequencing. In contrast to single individual sequencing, Pool-Seq is very cost-efficient and has proven powerful to accurately estimate population-wide allele frequencies [35,36,37]. However, Pool-Seq also comes at the cost of losing information about individual genotypes and haplotype structure. Moreover, it remains very difficult to distinguish low-frequency variants from sequencing errors, which further complicates population genetics inference [38, 39] and precludes calculating classic population genetic estimators without statistical adjustments (see for example [40,41,42,43]).
It is important to note that these approaches neither allow to measure genotype variation in natural populations, which is the proportion of heterozygote individuals within a population nor the proportion of heterozygote sites within a single diploid individual.
2.2 Consortia and Available Datasets
The first finished genome draft of D. melanogaster was published more than 17 years ago, and was among the very first fully sequenced eukaryotic genomes [7]. Since then, the quality of the reference sequence has further improved, and the number of functional annotations, such as gene models or regulatory elements, keeps increasing continuously. Both sequence and annotation data are publicly available at www.flybase.org, a bioinformatics database that is the main repository of genetic and molecular information for D. melanogaster (and other species from the Drosophilidae family). D. melanogaster was also one of the first species for which full-genome intraspecific variation data was collected. The first whole-genome population genetics study in D. melanogaster surveyed natural variation in three African (Malawi) and six North American (North Carolina) strains using low-coverage sequencing [44].
2.2.1 Drosophila Genetic Reference Panel (DGRP) and Drosophila Population Genomics Project (DPGP)
The first two projects to systematically investigate the genomic variability in natural D. melanogaster populations were the DGRP [45] and DPGP [46] initiatives. Both consortia independently sequenced more than 160 isofemale inbred lines (F20), all sampled in Raleigh, North Carolina, USA; a sample that was later extended to 205 lines [47]. The major aim was to generate whole-genome sequencing data that can be used for genome-wide association studies. The genetic and phenotypic data are available from http://dgrp2.gnets.ncsu.edu. While the DGRP data are well suited for quantitative genetics analyses (using stable, well-described, and homogeneous genetic material), they only provide information about the genetic variation at a single location (North-Eastern USA) although a large portion of the genetic diversity of the species is known to reside in its ancestral range in sub-Saharan Africa [48, 49]. The DGRP data is thus neither suitable for investigating the demographic history of worldwide populations nor the patterns and processes leading to local adaptations that likely facilitated the range expansion and ultimately led to a cosmopolitan distribution of D. melanogaster.
2.2.2 Drosophila Population Genomics Projects
The Drosophila population genomic project (DPGP, http://www.dpgp.org) is an ongoing major population genomic sequencing effort: beside the Raleigh population, the DPGP sequenced a population of Malawi (Africa) that exhibited >40% more polymorphism genome-wide compared to the North-American one [46]. Then, the DPGP2 sequenced 139 wild-derived strains representing 22 populations from sub-Saharan Africa [50]. The analyses of the DPGP2 data confirmed that the most genetically diverse populations are located in Southern Africa (e.g., Zambia). Afterward, the DPGP3 increased the sample size for a Zambian population (Siavonga) up to 197 lines [51]. Most DPGP2 and all DPGP3 lines were sequenced from haploid embryos as described above.
2.2.3 The Drosophila Genome Nexus
The Drosophila Genome Nexus is a population genomic resource that integrates single-individual D. melanogaster genomes from multiple published sources [51, 52], including DPGP and DGRP among others [30, 53,54,55]. The aim was to generate a comprehensive dataset using the same bioinformatics methods to facilitate comparisons among them. The latest iteration (DGN v.1.1 [52]), contains a total of 1121 genomes, from 83 populations in Africa, Europe, North America, and Australia. It especially highlighted differences in levels of heterozygosity among the different datasets. The genome browser PopFly allows for the visualization and retrieval of numerous population genomics statistics, such as estimates of nucleotide diversity, linkage disequilibrium, recombination rates [56].
2.2.4 Dros-RTEC and DrosEU
Complementary to previous efforts, which aim at sequencing single individual genomes in large numbers from a single population (DGRP, DPGP, DPGP3) or in small numbers from multiple locations (DPGP2 [30]), two consortia in North America (Dros-RTEC [57]) and in Europe (DrosEU [43]) recently started to generate Pool-Seq data from wild-caught flies from numerous sampling sites to quantitatively assess genetic variation and differentiation through time and space in natural populations. To date, DrosEU has sequenced and analyzed 48 samples from more than 30 localities all across Europe, which revealed strong and previously unknown population structure—mostly along the longitudinal axis—in Europe. Moreover, population genetic analyses of these data allowed for a description of novel candidates for selective sweeps, to detect previously unknown clines of mitochondrial haplotypes, inversions and transposable elements (TE) and to isolate novel viral species in the microbiome. The Dros-RTEC consortium similarly sequenced 72 samples of D. melanogaster collected from 23 localities mostly in North America [57]. Due to their focus on rapid seasonal adaptation, many localities were sampled at different time points over the course of 1–6 years, which allows for a quantitative investigation of genome-wide seasonal fluctuations in SNPs and inversion polymorphisms. These analyses revealed that previous candidates for seasonality exhibit highly predictable annual allele frequency fluctuations and those signatures of seasonal adaptation parallel spatial differentiation along latitudinal gradients.
2.2.5 Other Data
In addition to these concerted sampling and sequencing efforts, there is a rapidly growing number that similarly sequenced pools of flies from natural populations. For example, Pool-Seq data of populations from the temperature gradients along the North American and Australia were generated [58,59,60]. Large pools of flies collected from Vienna/Austria and Bolzano/Italy were sequenced by [61]. More recently, Kofler and colleagues [62] generated and analyzed Pool-Seq data from more than 550 South African flies. In combination with the aforementioned Pool-Seq data from large consortia, these data represent highly valuable resources to tackle fundamental questions about the adaptive process on complex spatial and temporal scales.
3 Neutral Evolution
3.1 Demographic Analyses
D. melanogaster is one of eight species described in the melanogaster subgroup of the subgenus Sophophora. Within this group, two species are cosmopolitan (D. melanogaster and D. simulans), while the remaining six are endemic to the Afrotropical region (D. sechellia, D. mauritania, D. erecta, D. orena, D. teissieri, D. yakuba). This has led early studies to suggest an Afrotropical origin of D. melanogaster and D. simulans and is now widely accepted [4]. As expected under this hypothesis, the genome-wide average diversity measured in Afrotropical populations of D. melanogaster is higher than in non-African populations [44, 48, 63, 64]. In addition and similarly to Homo sapiens, the genetic variation outside sub-Saharan Africa represents a subset of the diversity found within sub-Saharan African populations, which further suggest that South-Eastern tropical Africa represents the ancestral range of the species [65].
In an influential review summarizing the results of early population variation surveys in D. melanogaster [4], David and Capy categorized worldwide natural populations into three groups: ancestral, ancient, and new populations (Fig. 1). Ancestral populations are located in sub-Saharan Africa, where they probably have split from the sister species D. simulans approximately 2.3 million years ago [71]. Ancient populations are located in Eurasia and migrated out of their ancestral range presumably at the end of the last ice age. The third group, new populations, is located in America and Australia, and represents a blend of ancestral and ancient populations that recently colonized these two continents along European shipping routes during the last centuries. Although these early insights were based on a small number of loci, they have proven to be surprisingly robust and 30 years later, the categorization of David and Capy is still widely accepted. Several studies, however, took advantage of the increasing amount of genetic data and the rapidly developing field of model-based inference in population genetics, to investigate the demographic history of the species within a probabilistic framework. These studies evaluated the likelihood of competing demographic scenarios and provided estimates for demographic parameters such as the age of the split between African and non-African populations, and population sizes at different times of the colonization process. In the next paragraph we review how genome-wide data and statistical modeling updated the insights formulated by David and Capy [4].

Map illustrating worldwide distribution, migration routes and clinal differentiation of the cosmopolitan species D. melanogaster. Populations are separated in ancestral (red), ancient (orange) and newly introduced (blue) populations, according to the categorization in David and Capy [4]. The expected ancestral range (Zambia) is highlighted in dark red. Primary colonization routes across populations are shown by colored arrows: the European colonization started approximately ~10–19,000 years ago [66, 67], followed by a spread to Asia ~5000 years ago [66] and a more recent range expansion to Australia and North America within the last 200 years [2]. Patterns of recent admixture (dotted grey arrows) were documented from European alleles in Africa [50], and from African alleles to North America and Australia [68, 69]. Clinal genetic and phenotypic differentiation (dash-dotted black arrows and color gradient) are documented along latitudinal gradients in North America [58, 68] and Australia [60, 68], and along longitudinal gradients in Africa [70] and in Europe [43]. At the time of the review, no information about demography of South-American populations was available. The dark grey areas depict expected habitable geographic regions and were modeled from 4951 unique worldwide sampling-points in the TaxoDros database (http://taxodros.uzh.ch) and climatic data from the WorldClim database (http://worldclim.org) using the R-package dismo (http://rspatial.org/sdm/). Note that distribution models can be confounded by unequal sampling and may thus explain the missing predicted distribution in South-Western Africa, Central Asia, and Russia
Early population genetics surveys identified East and South African populations to be closer to mutation-drift equilibrium compared to West African populations, which were characterized by higher linkage disequilibrium and lower diversity levels [63,64,65]. These findings suggest that East and South Africa include the ancestral range of the species. Demographic inference using samples from sub-Saharan Africa indicated that the ancestral population has experienced a population size expansion approximately 60,000 years ago (ranging from 26,000 to 95,000 [72]). This ancestral expansion is found in all published models incorporating the African population and is necessary to fit the excess of rare variants measured in samples from the ancestral range (e.g., Zambia, Zimbabwe). These models, however, assume that all sampled mutations are neutral, which is unlikely because of putatively unknown regulatory elements and the presence of background selection [73]. A simulation showed that ignoring background selection in demographic inference leads to an overestimation of growth models [74]. Estimations of the coalescence rate through time using smc++ indicated that the rate of coalescence in a sample from the Zambian population (Siavonga, DPGP3) has been constantly decreasing in the last 100,000 years [75], which is in line with the population expansion scenario suggested by previous studies. Furthermore, Terhorst et al. [75] measured a strong reduction in the coalescent rate for times older than 100,000 years, suggesting either a very large ancestral population size or substantial population structure in the ancestral population [76]. Neither of these two processes is accounted for in current demographic models for D. melanogaster and more work is needed to evaluate whether the decreased ancestral rate measured by [75] is reflecting true ancestral processes or rather aspects of the genomic data that are not accounted by the method. More specifically, it remains to be clarified whether this approach can correctly recover neutral demographic processes when applied to small compact genomes with a high proportion of nonneutral regions. More recently, Kapopoulou et al. [77] estimated the age of the split between ancestral (Zambia) and West African populations to be approximately 72,000 years, which suggests that the population expansion reported by earlier studies could well reflect a genuine early range expansion of the species on the African continent.
3.1.1 Out of Africa
Analysis of European samples revealed that the time of split between African and European populations occurred around 13,000 years [66, 72]. These early studies, however, did not include gene flow between populations in their models and therefore predicted that their estimates were probably younger than the true age of divergence between African and European lineages. Indeed, Kapopoulou et al. [78] recently confirmed this prediction using genome-wide polymorphism data and by explicitly accounting for the effect of gene flow in their inference procedure. Their demographic results identified gene flow as an important factor in the recent history of European and African populations and reported divergence time estimates of approximately 48,000 years. Independently, Pool et al. [50] reported pervasive influence of European admixture in many African populations with greater admixture proportion in urban locations. The “ancient” status of Southeast Asian populations has also been confirmed by [66, 79]. Similarly to the European case described above, divergence time estimates between Asian and European populations strongly depend on whether or not gene flow is taken into account in the inference method (22,000 vs. 5000 years, respectively).
North-American populations are considered as newly introduced because the colonization process has been observed directly by entomologists in the second half of the nineteenth century [2]. Strikingly, D. melanogaster was identified as the most common species across the USA only 25 years after its introduction, suggesting a dramatic population expansion after colonization [2]. A genome-wide analysis of 39 flies sampled as part of the DGRP project [45], using an approximate Bayesian computation method (ABC) revealed the admixed nature of this population with European and African admixture proportions of 85% and 15%, respectively [67]. These estimations confirmed similar conclusions reached earlier using microsatellite data [80]. This very recent secondary contact between African and European lineages is likely responsible for the North-South clinal genetic variation observed in Northern America and Australia (Fig. 1), but local adaptation could contribute to the maintenance of this clinal variation by opposing itself to the homogenizing effect of gene flow [54, 68]. Arguello et al. [79] recently confirmed the importance of Afro-European admixture in the ancestry of North American and Australian flies using a larger dataset and a more precise inference procedure. The mosaic ancestry of American and Australian fly populations therefore represents an exciting opportunity to study how migration and selection interact along a clinal heterogeneous environment. Methods based on hidden Markov models were developed to estimate patterns of local ancestry in samples of North-American populations (where the term local refers to an arbitrary subgenomic unit) [69, 81]. In samples with a predominant European genetic background, their results identify significant differences in the proportion of African ancestry between functional classes of genomic loci.
3.2 Recombination
In most sexually reproducing eukaryotes, recombination ensures both the proper segregation of homologous chromosomes during meiosis and the creation of new combinations of alleles at each generation. During meiosis, a substantial number of double-strand breaks result in meiotic recombination between homologs. These double-strand breaks are repaired either as crossover (CO) or noncrossover (NCO) gene conversions: COs imply reciprocal exchange between flanking regions, whereas NCOs do not. Both forms of recombination are key factors in genome evolution as their rates determine the probability to which extent genomic sites are linked or evolve independently and hence affect the evolutionary fate of the alleles. A fundamental understanding of recombination rates is thus crucial in population genomic studies. In Drosophila, meiotic recombination only occurs in females, but not in males [82], a dimorphism known as “achiasmy” (an extreme case of heterochiasmy observed in many species [83]).
In the 1990s, several studies revolutionized population genetics by showing that the level of genetic diversity in populations of Drosophila species was lower in regions of low recombination [19, 84, 85]. Recombination itself seems to be the major factor determining patterns of nucleotide diversity along the genome. Indeed, mutation associated with recombination can be excluded as the cause of this correlation, at least in Drosophila, given the lack of correlation between recombination and divergence [19, 45]. The frequent occurrence of these patterns [86] has motivated further exploration and estimation of genome-wide patterns of recombination and diversity.
Classically, the estimation of recombination rates generally relies on the “Marey approach” that compares a genetic map, which quantifies distances as CO frequency (in cM) to a physical map (distances in base pairs). A user-friendly web service called MareyMap Online [87] allows to get recombination rate estimates based on such an approach. In their landmark study, Begun and Aquadro [19] found a strong positive correlation between nucleotide diversity estimated at 20 genes and local rates of COs in natural populations of D. melanogaster. They used the coefficient of exchange as a measure of recombination rate, based on the physical distance among cytological markers in combination with DNA content estimates from densities of polytene chromosomes [88]. The fully sequenced Drosophila genome, which became available in 2000 [7], represents a highly accurate physical map that was necessary to generate detailed recombination maps. Marais et al. [89] fitted a third-order polynomial, which provided a first overview of the distribution of COs along each chromosomal arm. They showed that CO rates decline in proximity to telomeres and centromeres. Accounting for specific recombination patterns of the telomeric and centromeric regions, Fiston-Lavier and colleagues provided corrected estimates of local recombination rates in D. melanogaster [90].
Besides classical recombination maps based on crosses, alternative approaches take population genetic variation into account to estimate CO rates. Patterns of linkage disequilibrium (LD) in a population result from historical recombination events. Recombination (CO) rates across the genome can thus be inferred from linkage disequilibrium, through the population-scaled recombination parameter ρ = 4Ner where Ne is the effective population size, and r the CO rate between base pairs per generation [91]. Mc Vean et al. [92] developed a coalescent-based method implemented in the software LDHat for the estimation of local recombination rates (4Ner per kilobase) using a composite likelihood approximation [93], based on the segregation of a high density of physically mapped SNPs. Originally developed for human populations, this method has been applied to many species including Drosophila [46]. Besides providing a recombination map with a higher resolution, Langley et al. showed that r and ρ were strongly positively correlated at a large scale [46], indicating these independent estimates are both capturing heterogeneity in recombination. However, compared to humans, D. melanogaster harbors much higher SNP densities, population recombination parameters are an order of magnitude higher and footprints of positive selection are more widespread. Since the LDhat method assumes neutral evolution, it can infer spurious recombination hotspots under certain conditions of selection. Chan et al. [94] proposed a corrected method (LDhelmet), which is more robust to the effects of selection and computed an improved fine-scale, genome-wide recombination map in D. melanogaster, including a handful of hotspots of at least ten times the background recombination rate.
Combining both crosses and population variation approaches, Comeron et al. [95] proposed a method to distinguish between the two possible outcomes of the repairing of double strand breaks associated with meiotic recombination: CO and NCO gene conversions. While COs involve DNA exchange between chromatid arms of homologous chromosomes on a large-scale, NCOs are nonreciprocal recombination events with a swap of small DNA fragment. First described in Drosophila [96,97,98], CO interference prevents the formation of two COs in close proximity and thus, reduces the probability of double CO events (~1 CO per chromosome per meiosis [99]). Based on the size of genetic regions affected by gene conversion, Comeron et al. [95] estimated separately rates of CO and NCO from crosses, making use of the very high density of SNPs in D. melanogaster (139 million), which allowed them to design a 2 kb-resolution map of recombination. Unlike COs, NCOs appear to be uniformly distributed throughout the genome [95], insensitive to the centromere effect and without interference [100], and more frequent (rates of NCO: CO could reach values over 100 [95]).
While extrapolated and direct recombination estimates are consistent on a large scale, the latter ones show greater variability at the center of the chromosomal arms [101]. Altogether, these recombination maps provide baseline estimates for population genomic studies, especially to model the expected variation under selection at linked sites (see Subheading 4.2 and [102]).
3.3 Biased Gene Conversion
Both CO and NCO recombination involve gene conversion. In particular, the presence of heterozygous sites within heteroduplex DNA results in the formation of mismatches, which lead to the conversion of one allele by the other during the repair. There is evidence, from diverse eukaryotic lineages, that GC:AT mismatches tend to be more often repaired in GC than in AT alleles, a process called GC-biased gene conversion (gBGC [103, 104]). gBGC has been inferred as the main driver of GC-content evolution in vertebrates [103, 105,106,107] and several other taxa [108,109,110,111]. gBGC is a nonadaptive mechanism that mimics natural selection, because it confers a higher transmission probability of GC over AT alleles in heterozygotes. Therefore, gBGC needs to be accounted for in molecular evolution studies to correctly model neutral evolution of the genome [112, 113]. The impact of gBGC in D. melanogaster is, however, less clear: GC content is positively correlated with CO rate [89, 114, 115], but not with NCO rate [95]. Globally, whole-genome polymorphism and divergence data did not support a gBGC model in D. melanogaster [116], except for the X chromosome [117, 118] where it may partly explain the stronger signal of selection on codon usage compared to autosomes [119].
3.4 Population Genetics of Chromosomal Inversions
Chromosomal inversions were first discovered in D. melanogaster almost exactly 100 years ago [120]. They represent structural mutations that result in the reversal of genetic order in the affected genomic region relative to the noninverted (“standard”) arrangement [121, 122]. Inversions can have strong effects on genome evolution in various different ways: breakpoints may disrupt genes (e.g., [118]) or result in gene duplications due to staggered breaks [123, 124]. Moreover, inversions can trigger positional effects, where expression patterns of genes are altered due to changes in their relative chromosomal position ([125,126,127] but see [128]). However, their most fundamental effect is the strong suppression of recombination in heterozygotes, since crossing-over within the inverted region results in abnormal chromatids [129,130,131]. As shown in humans where inversions can cause numerous diseases, many of these effects have deleterious consequences [132]; however there are some rare adaptive cases (reviewed in Subheading 4.5). Upon their discovery, inversions have been predominantly studied in species of the genus Drosophila. Particularly the pioneering work of Theodosius Dobzhansky and colleagues in D. pseudoobscura and D. persimilis [8, 9, 133, 134] gave a first insight into the evolutionary processes that shape genetic variation and differentiation in natural populations [135,136,137]. However, only due to recent advances in whole-genome sequencing technology, it became possible to quantitatively test for different evolutionary models and characterize the genetic effects of inversions on a genome-wide scale. Consistent with the action of spatially varying selection, many inversions in Drosophila are commonly found to exhibit steep clines along environmental gradients [138,139,140,141]. Several of these, such as the latitudinal gradient of the well-studied In(3R)Payne inversion in D. melanogaster, are replicated on multiple continents and persist over time ([33, 142] but see [143]). Recent large-scale genomic datasets of D. melanogaster, for the first time, allow a quantitative assessment of the genetic and evolutionary pattern associated with inversions. Analyses of genome-wide data from African flies allowed for (1) determination of the age and geographic origin of various cosmopolitan and endemic inversions. These analyses revealed that most common cosmopolitan inversions are of African origin and predate the out-of-Africa migration [144]. Furthermore, these data provide (2) a first insight into the amount and distribution of genetic variation and differentiation associated with inversions. Data analyses of the DGRP, for example, found that inversions contribute strongest to genetic differentiation and substructure within a population from Raleigh/North Carolina [47]. Moreover, only with the help of dense genome-wide sequencing, it became possible to show that genetic differentiation is not homogeneously elevated within inversions, but decays toward the inversion center [33, 141, 144,145,146]. Consistent with theoretical predictions [147,148,149], these data suggest that there is a limited amount of genetic exchange among karyotypes rather than a complete inhibition of recombination. In addition, local peaks of strong differentiation close to the inversion center suggest that several inversions, for example In(3R)Payne, contain various adaptive loci which are in strong linkage with the inversion breakpoints [33, 141, 144, 145]. Analyses of genomic data in combination with long-range PCR further helped (3) to reconstruct the exact genetic composition of inversion breakpoints [150] and (4) facilitated the development of inversion-specific marker SNPs, which now make it possible to reliably estimate inversion abundance and frequency in single-individual and Pool-Seq data, respectively [33, 141, 151]. Together, these analyses highlight that whole-genome data for the first time allows to quantitatively elucidate the mechanisms underlying the evolution of chromosomal inversions.
3.5 Population Genomics of Transposable Elements
Transposable elements (TEs) are mobile, self-replicating, repeated DNA sequences found in every eukaryotic genome at varying proportions among taxa [152, 153], among closely related species [24] and among individuals of the same species ([154] for maize; [155] for Arabidopsis; [156] for Drosophila). Because of their mutagenic potential (either by inserting into functional regions or by promoting chromosomal rearrangements via ectopic recombination—Note 3), TEs are thought to play a significant role in populations’ evolution and adaptation [157]. According to the nearly neutral theory, TE insertions are expected to be generally neutral or deleterious to the host genome [158]. However, rare cases of adaptive insertions have also been documented (see Subheading 4.6 for examples in Drosophila). The general model of TE dynamics is the transposition-selection balance model [159]. It assumes that the maintenance of TEs in the population is explained by an equilibrium between (1) the increase in copy number through a constant transposition rate and (2) their removal driven by natural selection, through the combined effect of excision and purifying selection acting against the deleterious effects of inserted TEs [159, 160]. This model predicts that most TEs should be segregating at low TE frequency in D. melanogaster populations (see [161] for detailed review). The burst-transposition model [162] relaxes the assumption of constant transposition rate over time in proposing periods of intense TE transposition (bursts) to explain TE dynamics. According to this model, recent insertions have not yet reached an equilibrium between their transposition rate and negative selection. TEs may thus be at low frequency even under a strictly neutral model. Here, a positive correlation between insertions age and their frequency is expected (recently active TEs should be at low population frequency while long-time inactive TEs could reach fixation).
D. melanogaster has been used as a model species for the study of TE population dynamics for more than 25 years [163] and recent whole-genome population data fuelled this area of research allowing for testing of previous hypotheses. A bulk of new programs was recently developed to estimate TE insertion frequency in a population using NGS datasets (see [164] for review). On top of the 5434 annotated TEs described in the reference genome, 10,208 and 17,639 insertions were discovered in European [165] and North American DGRP [166] populations, respectively. However, these numbers needs to be considered cautiously as the performance of methods detecting polymorphic TE insertions based on short read data depends on many variables, such as the sequencing coverage, the element family, the age of insertion, the size of the copy, the genomic location (see a benchmark in [167]). The large predominance of low frequency insertions along with the scarcity of insertions in exonic regions observed in both datasets supports the transposition-selection balance model. In contrast, Kofler, Betancourt and Schlötterer provided evidence that half of the TE families have had transposition rates that vary with time [165], giving support to the burst-transposition model. However, they also found an excess of rare variants in young TE insertions compared to neutral expectations which suggests the action of purifying selection [166]. Overall, population genomics analyses of TEs provide empirical support for both hypotheses and indicate that they are not mutually exclusive. This is in agreement with previous in situ analyses suggesting that models of evolution could vary among elements and populations [168]. Although dynamics of some TE families can be explained by a neutral model with transposition rates varying over time, purifying selection is necessary to fully explain the patterns of population distribution of TEs [161, 169].
4 Selection
D. melanogaster has been a model species for many studies aiming at describing the genetic basis of adaptation. Comparisons between theoretical models of positive and negative selection with empirical data have started in the early 1980s, when PCR coupled with Sanger sequencing allowed to directly measure natural variation. The positive correlation between local rates of recombination and genetic diversity [19] was among the most important observations made by these early studies and has been interpreted as evidence for the widespread effect of selection along the genome. This postulate challenged the paradigm of the Nearly Neutral extension of the Neutral Theory [170, 171], which assumes that the large majority of polymorphic and divergent sites are neutral or slightly deleterious. Since then, the search for genes underlying adaptation as well as the quantification of the genome-wide impact of selection has stimulated the development of statistical methods aiming at detecting past adaptive processes from DNA polymorphism data. In 1991, McDonald and Kreitman developed their reference test of selection, and detected adaptation on the Adh locus in Drosophila [16]. Based on the McDonald and Kreitman test ratios, the fraction α of nonsynonymous substitutions driven to fixation by position selection can be estimated by 1 − (DsPn)/(DnPs), with Ds and Dn the number of synonymous and nonsynonymous substitutions, respectively and Ps and Pn the number of synonymous and nonsynonymous polymorphisms, respectively ([172] and see Chapter 6 for more details). Numerous studies have provided evidence for pervasive molecular adaptation in D. melanogaster, suggesting that approximately 50% of the amino acid changing substitutions (α = 0.5), and similarly large proportions of noncoding substitutions, were adaptive [173,174,175,176,177,178].
4.1 Hitchhiking Effects
The first mathematical formulation of the effect of a positively selected allele on intra-specific genetic diversity was proposed by Maynard Smith and Haigh in 1974 and coined the “hitchhiking model” [179]. Selection reduces diversity not only at selected sites, but also at linked neutral sites, and the number of variants linked together around a single selected target is inversely proportional to the recombination rate. The hitchhiking model summarizes the relation between the strength of selection on a single adaptive mutant allele, the local recombination rate, and the distribution of surrounding neutral alleles across sites and samples. Under such a linkage model, when a beneficial allele establishes itself in the population, the high rate at which this establishment occurs creates an irregularity in the distribution of neutral alleles around the selected allele. This characteristic signature resulting from positive selection has been coined “selective sweep” (hard sweep), a terminology used to describe both the adaptive process and the resulting signal in genetic data. This model served as basis for the development of statistical tools designed to capture the signal of a selective sweep in the presence of different confounding factors ([180, 181] and see Chapter 5 for more discussion on sweep detection). D. melanogaster has been among the first organisms for which this approach has been used to map selective sweeps [72, 182, 183], eventually yielding to the identification of several candidate genes/regions for adaptations (Table 1) that allowed D. melanogaster to extend its geographic range to very heterogeneous environments and to recent anthropogenic changes [184, 200, 205]. However, the particular demographic history of the species, and especially the severe founding events followed by population expansion should be considered as a confounding factor, strongly increasing the rate of false positives and thus reducing the performance of sweep detection methods in this specific biological system [206,207,208,209]. These insights into the confounding effects of adaptive and neutral processes motivated two lines of research: (a) characterizing neutral models accounting for the major demographic events having affected the genome-wide distribution of neutral alleles (see paragraph above) and (b) more general formulation of the adaptive process initially described by [179].
Soft sweep theory extended the Maynard Smith’s and Haigh’s hitchhiking model, by including the possibility of (1) recurrent mutations leading to beneficial alleles and (2) segregating neutral alleles becoming positively selected (i.e., selection from standing variation; reviewed in [210]). Both cases predict an association of the beneficial alleles with several background haplotypes (versus a single one in the hard sweep model). Garud et al. [204] scanned the DGRP dataset to capture signature of hard and soft sweeps, and found a significantly higher number of candidate genomic regions than expected under the neutral admixture model previously calibrated for this population [67]. Furthermore, they found that among their top 50 candidates most cases were better explained by soft than hard sweeps, suggesting that standing genetic variation and recurrence of beneficial alleles play an important role in real-life adaptive processes in D. melanogaster. However, the statistical significance of their results is highly dependent on an appropriate calibration of neutral demographic models, suggesting that the performance of soft-sweep detection methods still needs to be tested under a large range of demographic models. In the meantime, the results of genome-wide soft-sweep detection studies should be evaluated carefully when used to support claims about adaptive processes [211].
4.2 Recurrent Hitchhiking and Background Selection
Beyond the study of single instances of selective sweeps, D. melanogaster and D. simulans have also been used to investigate the genome-wide effect of recurring sweeps on genetic variation. The relevant model is the recurrent hitchhiking model [212], which describes genome-wide patterns of variation as a function of the occurrence rate of selective sweeps and the distribution of fitness effects of advantageous mutations. Several studies have developed model-based inference approaches to estimate these two parameters using polymorphism and divergence data, reviewed in [213, 214]. All consistent with a strong impact of selection on the pattern of diversity in this species, a wide range of the strength of selection on beneficial mutations (Nes, where Ne is the effective population size and s the selection coefficient) was estimated, ranging from 1–10 [215, 216], ~12 [217], ~40 [218], 350–3500 [72, 172, 219] to ~10,000 [220]. These studies showed that the rate and strength of positive selection was large enough such that a significant amount of neutral alleles in the genome cannot be seen as evolving independently from adaptive sweeping alleles (the dependencies being caused by genetic linkage between beneficial and neutral alleles). Essentially, the disparate estimates reflect variation in the calibration of the different models, in particular according to (1) the type of selection assumed, (2) the modeled relationship between diversity estimates and selection (strength and frequency) through the action of recombination. These results also revealed the difficulty of telling apart whether genome-wide selection is characterized by a small number of large effect or a large number of small effect adaptive alleles.
The relative importance of positive selection in Drosophila has been challenged, however, by studies describing the effect of strictly deleterious alleles on linked neutral variants [73, 221, 222]. This hitchhiking effect caused by selection against recurrent deleterious mutations called background selection has been shown to be a valid alternative explanation for low variability in genome regions with low recombination rates [73, 223]. Importantly, Comeron generated a map describing the strength of background selection along the genome as a function of the local recombination and deleterious mutation rate [224]. This study showed that a large proportion (70%) of the observed variation in the level of diversity across autosomes can be explained by background selection alone and therefore called for the inclusion of background selection in further population genomics analyses. Elyashiv et al. recently proposed a method to jointly estimate the parameters of distinct modes of linked selection, accounting for both positive (selective sweeps) and negative background selection [225]. Applied on D. melanogaster, they showed that negative selection at linked sites has had an even more drastic effect on diversity patterns in D. melanogaster than previously appreciated based on classical selective sweeps models (1.6–2.5-fold). Their results further suggest that 4% of substitutions between D. melanogaster and D. simulans have experienced strong positive selection (s ≈ 10–3.5) and that 35% to 45% of substitutions have been weakly selected (s between 10–5.5 and 10−6).
4.3 Selection on Noncoding DNA
Since the 2000s, whole-genome comparative analyses accumulated evidence that only a small portion of conserved sequences across species (i.e., potentially functional) was composed of protein-coding genes [226, 227]. In the meantime, genomic surveys identified noncoding genomic sequences showing exceptionally high levels of similarity across species, which were termed conserved noncoding elements or CNEs (reviewed in [228]). In Drosophila, CNEs are estimated to cover ~30–40% of the genome [226, 229]. The high levels of evolutionary conservation observed in these regions are postulated to be the result of functional constraints since many CNEs partially overlap with cis-regulatory elements [230] and functional noncoding RNAs [231, 232]. In Drosophila, several classes of noncoding DNA evolve considerably slower than synonymous sites yet show an excess of between-species divergence relative to polymorphism when compared with synonymous sites [175]. While the former observation indicates selective constraints, the latter is a signature of adaptive evolution, which resembles patterns of protein evolution in Drosophila [173, 174]. To quantify the intensity and the relative importance of selection in shaping the evolution of noncoding DNA, several studies applied extensions of the McDonald–Kreitman approach, combining polymorphism and divergence analyses. When analysing noncoding DNA in a population from Zimbabwe, Andolfatto estimated that ~20% of nucleotide divergence in introns and intergenic DNA and ~60% in UTRs were driven to fixation by positive selection [175]. Using a hierarchical Bayesian framework, he estimated that significant positive selection acted on noncoding sequences, especially in UTRs [175]. This was recently supported by a whole-genome survey of 50 European populations that showed that UTRs and noncoding RNAs are the noncoding genomic regions most subjected to adaptive selection, with >40% of divergence being driven by positive selection [229]. Specifically focusing on CNEs of the X chromosome, Casillas et al. [233] observed a large excess of low-frequency derived SNP alleles within CNE relative to non-CNE regions in an African and two European populations. While low levels of purifying and positive selection also act outside of CNEs, Casillas et al. [233] estimated that 85% of the CNEs were functional and evolved under moderately strong purifying selection (Nes ~10–100). Altogether, these studies strongly suggest that CNEs are not solely neutral genomic regions with extremely low mutation rates known as mutation “cold spots” [234] but shaped by both purifying selection and adaptive evolution in Drosophila. Moreover, these findings support the important role of noncoding regulatory changes in evolution.
4.4 Selection on Synonymous Codon Usage
The McDonald and Kreitman test and its extensions are built around the hypothesis that synonymous or fourfold degenerate sites (see Note 4) mostly evolve neutrally, while nonsynonymous sites are under strong purifying or positive selection. However, both synonymous and fourfold degenerate sites might be subject to selection on synonymous codon usage (see original reference for Drosophila by [235], and more recent review by [236]). Comparison of polymorphism and divergence patterns suggested that both strong (4Nes ≫ 1) and weak (4Nes ∼ 1) selection applies to synonymous sites in D. melanogaster [237, 238]. In this species, the level of codon bias is positively correlated to the levels of expression [239], but negatively correlated to the levels of divergence [240, 241]. Both findings suggest selection on codon usage bias. As in most Drosophila species, all preferred codons are GC-ending [239, 242]; selection on codon bias is therefore expected to increase GC content at synonymous sites. Several attempts to detect selection on codon usage bias in D. melanogaster have come to conflicting conclusions. Some studies detected evidence for selection favoring GC-ending codons [119, 243], although the intensity of selection may be weaker in D. melanogaster compared to other Drosophila species [244]. Other studies did not find support for such on-going selection [245, 246], but rather revealed an excess of substitutions toward AT-ending codons. This may either reflect a reduction in selection efficacy (4Nes) or a shift in the mutational bias in D. melanogaster lineage [247]. The population genetics of codon usage bias can however be affected by confounding, nonadaptive processes such as GC-biased gene conversion ([113] but see Subheading 3.3). In a recent study, Jackson et al. [248] modeled base composition evolution, and found evidence for selection on fourfold degenerate sites along both D. melanogaster and D. simulans lineages over a substantial period. They showed that while selection intensity on codon usage was rather stable in D. simulans in the recent past, it was declining in D. melanogaster. In conclusion, the observed AT-biased substitution pattern could not only result from a mutational bias, but likely partially reflects an ancestral reduction in selection intensity.
4.5 Adaptive Chromosomal Inversions
There is ample evidence that inversions play a pivotal role during adaptive processes and various hypotheses have been developed to explain their evolutionary impact [135,136,137, 249]: (1) According to the “coadaptation” model, inversions have higher fitness and spread because they suppress maladaptive crossing-over which would unlink coadapted alleles at epistatically interacting loci with high marginal fitness [9, 250]. Genomic analyses in D. pseudoobscura support this model and provide evidence that loci in tight linkage with an inversion show epistatic interactions [251]. (2) Under the “local adaptation model,” an inversion bears higher fitness because it captures and protects locally adapted loci from recombination with maladaptive migrant haplotypes as initially proposed by [252] and recently revised by [253]. A remarkable conclusion of this model is that the selective advantage of an inversion is determined only by the migration rate of maladapted haplotypes and the amount of linkage among the locally adapted loci. (3) The frequent occurrence of fixed inversions in different species of the genus Drosophila [254,255,256,257] and in other species groups [258, 259] suggests that many divergent inversions evolved by underdominance and are important components of the speciation process by suppressing gene flow among young sym- or parapatric species [260]. Similarly, inversions play a key role in the evolution of sex chromosomes by keeping together alleles in sex determining factors and sexually antagonistic genes [261]. (4) Conversely, inversions can also be maintained due to overdominance or other types of balancing selection. In line with this model, many inversions, particularly in Drosophila, are commonly found to segregate at intermediate frequencies in natural and experimental populations [262].
4.6 Adaptive Insertions of Transposons
Like other type of mutations, TE insertions are expected to be mostly deleterious or evolutionary neutral. However, some transposable elements could be beneficial and positively selected. There are several possible mechanisms by which a TE can be advantageous; either by directly affecting the gene function of individual genes, or by modifying regulatory elements [263, 264]. Due to recent technical advances in sequencing technology (NGS) and due to the rapidly growing number of whole-genome data, the ability to detect selected TE insertions has considerably increased in the past few years. Different methods have been developed to infer selection acting on TE insertions. Villanueva-Cañas et al. [265] provide a detailed overview over the main approaches and their specificities: (1) DNA sequence conservation analyses can be used to detect past events of domestication of TEs as regulatory elements, where TE insertions are conserved among closely related species due to purifying selection (see for example [229]). (2) Methods developed to detect selection on linked polymorphisms from SNPs (see Subheading 4.1 and Chapter 5 for more discussion) can also be applied to identify positively selected TEs. Based on either a bias in frequency spectra or haplotype structure, over 35 putatively adaptive TEs were identified in genome-wide studies in D. melanogaster to date [161, 165, 266, 267]. (3) A third method is built around environmental association analyses that include genome scans for selection performed in parallel in populations from different environments to detect specific adaptation driven by environmental conditions. Using this approach, González et al. [268] discovered several recent TE insertions in D. melanogaster that are putatively involved in local adaptation. These TEs exhibit low population frequencies in ancestral population (Africa) but are common in derived populations (North America and Australia). (4) Using a coalescent framework approach, Blumenstiel et al. [169] identified seven additional putative adaptive insertions exhibiting higher population frequency than expected according to their estimated allele age. (5) Finally, selection on TE insertions should be validated at the phenotypic level using functional assays to identify the molecular and fitness effects. One well-documented example is the insertion Bari-Jheh that was found to affect the level of expression of its nearby genes under oxidative stress conditions and to increase resistance to this stress [269, 270].
Beside the impact of single TE insertions, there is growing evidence for a more global effect of TEs on molecular functions. Especially, in Drosophila, TEs seem to play a role in a diversity of cellular processes [161], such as the establishment of dosage compensation [271], heterochromatin assembly [272] and brain genomic heterogeneity [273].
4.7 Faster-X Evolution
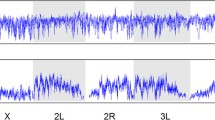
According to a theory proposed by Charlesworth et al. [274], the rates of evolution of X-linked loci are expected to be faster than autosomal ones if mutations are partially recessive (0 < h < 1/2, with h the coefficient of dominance) and expressed in both sexes or males only. In heterogametic males (XY), X-linked mutations are hemizygous and therefore directly exposed to selection, whereas new recessive autosomal mutations are masked from expression in heterozygotes individuals. Moreover, the effective recombination rate is ~1.8-fold greater on the X compared to autosomes [213], which reduces Hill–Robertson interference and increases the efficiency of selection. The increased selection in hemizygous males together with the higher efficiency of selection due to the increased recombination may act synergistically to account for the “faster-X evolution,” which is generally supported by genomic data collected in Drosophila populations (reviewed in [275]). Levels of polymorphism are similar on X-linked loci to autosomal ones in African populations, but lower in derived populations [30, 50], which might be a consequence of selective sweeps in response to the adaptation of new environments [276]. However, recombination seems to play a secondary role in determining pattern of diversity along the X-chromosome. Contrary to autosomes, the X-chromosome exhibits global nucleotide diversity only weakly correlated with recombination rate (Fig. 2), and a nonsynonymous diversity completely independent [277].

Correlation between nucleotide diversity and recombination. Nucleotide diversity (π) calculated in 10 kb nonoverlapping windows was estimated for 48 European populations (DrosEU data, [43] and compared to recombination rate (r) obtained from [95] for the four autosomal arms (2L, 2R, 3L, and 3R) and the X chromosome. We therefore averaged π in equally sized bins according to discrete log-transformed values of r observed in the corresponding genomic regions. The shaded polygons surrounding average values (central lines) for each of the 48 populations show the 95% confidence intervals
In contrast with polymorphism, divergence among Drosophila relatives is greater for the X than for autosomes (reviewed in [275]). Higher efficiency of selection on X is supported by the estimated higher percentage of sites undergoing both strongly deleterious and adaptive evolution than autosomes, and a lower level of weak negative selection in D. melanogaster [45, 46, 277]. Codon usage bias in Drosophila is also higher for X-linked genes than for autosomal ones, possibly due to the higher effective recombination rate and their resulting reduced susceptibility to Hill–Robertson effects [119]. In the end, faster-X evolution also implies that genes for reproductive isolation have a higher probability of being X-linked, what is generally true [161].
5 Perspectives: Temporal and Geographical Clines
Organisms with broad geographic distributions, such as various species of the genus Drosophila, are commonly found along environmental gradients. Such transects have long been in the focus of evolutionary geneticists, as they provide natural test beds to investigate the evolutionary underpinnings of local adaptation [278]. Studying spatially or temporally changing genotypes and phenotypes, which are commonly referred to as “clines” [279], has a long history in D. melanogaster [280,281,282]. While there is growing evidence for longitudinal clines in Africa [70] and in Europe [43], most data have been collected from latitudinal gradients along the North American and Australian east coasts. A large body of literature documents steep and persistent clines in many fitness-related phenotypes, which are often recapitulated on multiple continents. These include, for example, clines in body-, wing- and organ-size [283,284,285,286,287], lifetime fecundity and lifespan [288] as well as heat and cold resistance [289,290,291]. Similarly, various genetic polymorphisms such as microsatellites [292], SNPs [293], TEs [266] and inversions [141,142,143] have been found to vary clinally. Besides these well-defined spatial clines there is growing evidence for rapid adaptation on seasonal timescales leading to temporal clines. These are characterized by predictable annual fluctuations in allele frequencies [20] and variation in life history traits [21] and innate immunity [294].
Ongoing advances in next-generation sequencing technology prompted the development of analytical methods to identify putative targets of local and clinal adaptation (reviewed in [295]) and only recently allowed to extend the hunt for clinal genetic variation from single loci to genome-wide scales. A rapidly increasing number of studies in D. melanogaster have started to comprehensively investigate clinal genomic patterns—mostly by comparing the endpoints of latitudinal gradients from the Australian and North American east coasts [58,59,60, 293]. Many of these pioneering studies identified common patterns in the distribution of genome-wide clinal variation which provide insights, but also raise new questions about the evolutionary mechanisms involved in adaptation: (1) Loci with extensive clinal differentiation are not homogeneously distributed along the genome, but strongly clustered within large inversions [58, 60, 141], which suggests that inversions play an important role during local adaptation—potentially by keeping together coadapted loci associated with polygenic trait variation [252, 253]. However, the identity of these loci and the affected traits remain largely unknown so far. (2) Many clinal polymorphisms, such as the chromosomal inversion In(3R)Payne and variants of the alcohol dehydrogenase (Adh) locus, are paralleled on multiple continents [33, 58, 59, 293] and change frequencies in a predictable fashion. While parallel adaptive evolution due to spatially varying selection along analogous environmental gradients on different continents may shape many clinal patterns, other nonadaptive evolutionary forces could have similar effects. For example, a handful of studies found independent evidence for varying levels of admixture with African genetic variation both in North America [54, 67, 80] and Australia [68]. These findings highlight that clines, which are often considered to be the prime outcome of spatially or temporally varying selection, are potentially confounded with neutral evolutionary processes such as spatially restricted gene flow or admixture [296]. At last, (3) all aforementioned studies failed to identify large numbers of clinal loci with large or even fixed allele differences at the opposite endpoints of the latitudinal gradients. For example, no more than 0.1% of all SNPs exhibited allele frequency differences >0.5 between Florida and Maine, while not a single SNP exceed an allele frequency difference of 0.92 in the analyses of [58]. These findings are consistent with observations from other Drosophila species, which also found moderate and gradual clinal allele frequency changes [140, 297, 298], but in stark contrast to common model expectations for clinal evolution [299]. Together, these first analyses of clinal genomic data clearly show that it still remains challenging to disentangle the evolutionary contribution of selection and demography to clinal variation in natural populations.
Efforts of two large population genomic consortia are currently underway to densely sample natural populations through time and space both in North America [57] and Europe [43]. These comprehensive datasets will markedly extend earlier efforts that focused mostly on the comparison of clinal endpoints. Particularly the analyses of previously largely ignored European D. melanogaster populations will allow to make clear predictions about the adaptive process in derived populations from North America and Australia.
6 Notes
-
1.
The mathematical framework that integrated Darwin’s theory of evolution and the mechanisms of heredity discovered by Mendel.
-
2.
Defined as genome scale analyses of polymorphism including polymorphism/divergence comparisons, but not analyses strictly based on divergence.
-
3.
Ectopic recombination: recombination between two similar nonhomologous sequences, that is, two TE copies of the same family inserted at different genomic locations. Such DNA exchange between nonorthologous regions leads to chromosomal rearrangement.
-
4.
Fourfold degenerate sites consist in sites for which all four possible nucleotides at this position would encode for the same amino acid, representing a subset of all synonymous sites.
References
Lachaise D, Cariou M-L, David JR et al (1988) Historical biogeography of the Drosophila melanogaster species subgroup. In: Evolutionary biology. Springer, Boston, MA, pp 159–225
Keller A (2007) Drosophila melanogaster’s history as a human commensal. Curr Biol 17:R77–R81. https://doi.org/10.1016/j.cub.2006.12.031
Markow TA (2015) The secret lives of Drosophila flies. elife 4:e06793. https://doi.org/10.7554/eLife.06793
David JR, Capy P (1988) Genetic variation of Drosophila melanogaster natural populations. Trends Genet 4:106–111
Hales KG, Korey CA, Larracuente AM, Roberts DM (2015) Genetics on the Fly: a primer on the Drosophila model system. Genetics 201:815–842. https://doi.org/10.1534/genetics.115.183392
Kohler RE (1994) Lords of the fly. University of Chicago Press, Chicago
Adams MD, Celniker SE, Holt RA et al (2000) The genome sequence of Drosophila melanogaster. Science 287:2185–2195
Wright S (1982) Dobzhansky’s genetics of natural populations, I-XLIII. Evolution (N Y) 36:1102. https://doi.org/10.2307/2408088
Dobzhansky T (1970) Genetics of the evolutionary process. Columbia University Press, New York
Hudson RR (1983) Properties of a neutral allele model with intragenic recombination. Theor Popul Biol 23:183–201
Langley CH, Montgomery E, Quattlebaum WF (1982) Restriction map variation in the Adh region of Drosophila. Proc Natl Acad Sci U S A 79:5631–5635. https://doi.org/10.1073/PNAS.79.18.5631
Aquadro CF, Desse SF, Bland MM et al (1986) Molecular population genetics of the alcohol dehydrogenase gene region of Drosophila melanogaster. Genetics 114:1165–1190
Powell JR (1997) Progress and prospects in evolutionary biology: the Drosophila model. Oxford University Press, New York
Charlesworth B, Charlesworth D (2017) Population genetics from 1966 to 2016. Hered 118:2–9. https://doi.org/10.1038/hdy.2016.55
Hudson RR, Kreitman M, Aguade M (1987) A test of neutral molecular evolution based on nucleotide data. Genetics 116:153–159
McDonald J, Kreitman M (1991) Adaptative protein evolution at the Adh locus in Drosophila. Nature 351:652–654
Booker TR, Jackson BC, Keightley PD (2017) Detecting positive selection in the genome. BMC Biol 15:98. https://doi.org/10.1186/s12915-017-0434-y
Kern AD, Hahn MW (2018) The neutral theory in light of natural selection. Mol Biol Evol 35:1366–1371. https://doi.org/10.1093/molbev/msy092
Begun DJ, Aquadro CF (1992) Levels of naturally occurring DNA polymorphism correlate with recombination rates in D. melanogaster. Nature 356:519–520
Bergland AO, Behrman EL, O’Brien KR et al (2014) Genomic evidence of rapid and stable adaptive oscillations over seasonal time scales in Drosophila. PLoS Genet 10:e1004775. https://doi.org/10.1371/journal.pgen.1004775
Behrman EL, Watson SS, O’Brien KR et al (2015) Seasonal variation in life history traits in two Drosophila species. J Evol Biol 28:1691–1704. https://doi.org/10.1111/jeb.12690
St Johnston D (2002) The art and design of genetic screens: Drosophila melanogaster. Nat Rev Genet 3:176–188. https://doi.org/10.1038/nrg751
Hoskins RA, Carlson JW, Wan KH et al (2015) The release 6 reference sequence of the Drosophila melanogaster genome. Genome Res 25:445–458. https://doi.org/10.1101/gr.185579.114
Sessegolo C, Burlet N, Haudry A (2016) Strong phylogenetic inertia on genome size and transposable element content among 26 species of flies. Biol Lett 12:20160407. https://doi.org/10.1098/rsbl.2016.0407
Clark AG, Eisen MB, Smith DR et al (2007) Evolution of genes and genomes on the Drosophila phylogeny. Nature 450:203–218. https://doi.org/10.1038/nature06341
Celniker SE, Dillon LAL, Gerstein MB et al (2009) Unlocking the secrets of the genome. Nature 459:927–930. https://doi.org/10.1038/459927a
David JR, Gibert P, Legout H et al (2005) Isofemale lines in Drosophila: an empirical approach to quantitative trait analysis in natural populations. Heredity (Edinb) 94:3–12. https://doi.org/10.1038/sj.hdy.6800562
Falconer DS (1989) Introduction to quantitative genetics, 3rd edn. Longman, Scientific & Technical, Burnt Mill, Harlow, Essex/New York
Langley CH, Crepeau M, Cardeno C et al (2011) Circumventing heterozygosity: sequencing the amplified genome of a single haploid Drosophila melanogaster embryo. Genetics 188:239–246. https://doi.org/10.1534/genetics.111.127530
Grenier JK, Arguello JR, Moreira MC et al (2015) Global diversity lines - a five-continent reference panel of sequenced Drosophila melanogaster strains. G3 5:593–603. https://doi.org/10.1534/g3.114.015883
Fuyama Y (1984) Gynogenesis in Drosophila melanogaster1. Jpn J Genet 59:91–96
Rice WR, Linder JE, Friberg U et al (2005) Inter-locus antagonistic coevolution as an engine of speciation: assessment with hemiclonal analysis. Proc Natl Acad Sci U S A 102:6527–6534. https://doi.org/10.1073/pnas.0501889102
Kapun M, van Schalkwyk H, McAllister B et al (2014) Inference of chromosomal inversion dynamics from Pool-Seq data in natural and laboratory populations of Drosophila melanogaster. Mol Ecol 23:1813–1827. https://doi.org/10.1111/mec.12594
Gilks WP, Pennell TM, Flis I et al (2016) Whole genome resequencing of a laboratory-adapted Drosophila melanogaster population sample. F1000Res 5:2644. https://doi.org/10.12688/f1000research.9912.1
Zhu Y, Bergland AO, González J, Petrov DA (2012) Empirical validation of pooled whole genome population re-sequencing in Drosophila melanogaster. PLoS One 7:e41901. https://doi.org/10.1371/journal.pone.0041901
Fracassetti M, Griffin PC, Willi Y (2015) Validation of pooled whole-genome re-sequencing in Arabidopsis lyrata. PLoS One 10:e0140462. https://doi.org/10.1371/journal.pone.0140462
Schlötterer C, Tobler R, Kofler R, Nolte V (2014) Sequencing pools of individuals - mining genome-wide polymorphism data without big funding. Nat Rev Genet 15:749–763. https://doi.org/10.1038/nrg3803
Cutler DJ, Jensen JD (2010) To pool, or not to pool? Genetics 186:41–43. https://doi.org/10.1534/genetics.110.121012
Lynch M, Bost D, Wilson S et al (2014) Population-genetic inference from pooled-sequencing data. Genome Biol Evol 6:1210–1218. https://doi.org/10.1093/gbe/evu085
Futschik A (2010) The next generation of molecular markers from massively parallel sequencing of pooled DNA samples. Genetics 186:207–218. https://doi.org/10.1534/genetics.110.114397
Kofler R, Orozco-terWengel P, De Maio N et al (2011) PoPoolation: a toolbox for population genetic analysis of next generation sequencing data from pooled individuals. PLoS One 6:e15925. https://doi.org/10.1371/journal.pone.0015925
Kofler R, Pandey RV, Schlötterer C (2011) PoPoolation2: identifying differentiation between populations using sequencing of pooled DNA samples (Pool-Seq). Bioinformatics 27:3435–3436. https://doi.org/10.1093/bioinformatics/btr589
Kapun M, Barron M, Staubach F, et al (2018) Genomic analysis of European Drosophila melanogaster populations on a dense spatial scale reveals longitudinal population structure and continent-wide selection. bioRxiv. https://doi.org/10.1101/313759
Sackton TB, Kulathinal RJ, Bergman CM et al (2009) Population genomic inferences from sparse high-throughput sequencing of two populations of Drosophila melanogaster. Genome Biol Evol 1:449–465. https://doi.org/10.1093/gbe/evp048
Mackay TF, Richards S, Stone EA et al (2012) The Drosophila melanogaster genetic reference panel. Nature 482:173–178. https://doi.org/10.1038/nature10811
Langley CH, Stevens K, Cardeno C et al (2012) Genomic variation in natural populations of Drosophila melanogaster. Genetics 192:533–598. https://doi.org/10.1534/genetics.112.142018
Huang W, Massouras A, Inoue Y et al (2014) Natural variation in genome architecture among 205 Drosophila melanogaster genetic reference panel lines. Genome Res 24:1193–1208. https://doi.org/10.1101/gr.171546.113
Andolfatto P (2001) Contrasting patterns of X-linked and autosomal nucleotide variation in Drosophila melanogaster and Drosophila simulans. Mol Biol Evol 18:279–290. https://doi.org/10.1093/oxfordjournals.molbev.a003804
Hutter S, Li H, Beisswanger S et al (2007) Distinctly different sex ratios in African and European populations of Drosophila melanogaster inferred from chromosomewide single nucleotide polymorphism data. Genetics 177:469–480. https://doi.org/10.1534/genetics.107.074922
Pool JE, Corbett-Detig RB, Sugino RP et al (2012) Population genomics of sub-Saharan Drosophila melanogaster: African diversity and non-African admixture. PLoS Genet 8:e1003080. https://doi.org/10.1371/journal.pgen.1003080
Lack JB, Cardeno CM, Crepeau MW et al (2015) The Drosophila genome nexus: a population genomic resource of 623 Drosophila melanogaster genomes, including 197 from a single ancestral range population. Genetics 199:1229–1241. https://doi.org/10.1534/genetics.115.174664
Lack JB, Lange JD, Tang AD et al (2016) A thousand fly genomes: an expanded Drosophila genome nexus. Mol Biol Evol 33:3308–3313. https://doi.org/10.1093/molbev/msw195
Campo D, Lehmann K, Fjeldsted C et al (2013) Whole-genome sequencing of two North American Drosophila melanogaster populations reveals genetic differentiation and positive selection. Mol Ecol 22:5084–5097. https://doi.org/10.1111/mec.12468
Kao JY, Zubair A, Salomon MP et al (2015) Population genomic analysis uncovers African and European admixture in Drosophila melanogaster populations from the South-Eastern United States and Caribbean Islands. Mol Ecol 24:1499–1509. https://doi.org/10.1111/mec.13137
Bergman CM, Haddrill PR (2015) Strain-specific and pooled genome sequences for populations of Drosophila melanogaster from three continents. F1000Res 4:31. https://doi.org/10.12688/f1000research.6090.1
Hervas S, Sanz E, Casillas S et al (2017) PopFly: the Drosophila population genomics browser. Bioinformatics 33:2779–2780. https://doi.org/10.1093/bioinformatics/btx301
Machado H, Bergland AO, Taylor R, et al (2018) Broad geographic sampling reveals predictable and pervasive seasonal adaptation in Drosophila. https://doi.org/10.1101/337543
Fabian DK, Kapun M, Nolte V et al (2012) Genome-wide patterns of latitudinal differentiation among populations of Drosophila melanogaster from North America. Mol Ecol 21:4748–4769. https://doi.org/10.1111/j.1365-294X.2012.05731.x
Reinhardt JA, Kolaczkowski B, Jones CD et al (2014) Parallel geographic variation in Drosophila melanogaster. Genetics 197:361–373. https://doi.org/10.1534/genetics.114.161463
Kolaczkowski B, Kern AD, Holloway AK, Begun DJ (2011) Genomic differentiation between temperate and tropical Australian populations of Drosophila melanogaster. Genetics 187:245–260. https://doi.org/10.1534/genetics.110.123059
Bastide H, Betancourt A, Nolte V et al (2013) A genome-wide, fine-scale map of natural pigmentation variation in Drosophila melanogaster. PLoS Genet 9:e1003534. https://doi.org/10.1371/journal.pgen.1003534
Kofler R, Nolte V, Schlötterer C (2015) Tempo and mode of transposable element activity in Drosophila. PLoS Genet 11:e1005406. https://doi.org/10.1371/journal.pgen.1005406
Veuille M, Baudry E, Cobb M et al (2004) Historicity and the population genetics of Drosophila melanogaster and D. simulans. Genetica 120:61–70
Haddrill PR, Thornton KR, Charlesworth B, Andolfatto P (2005) Multilocus patterns of nucleotide variability and the demographic and selection history of Drosophila melanogaster populations. Genome Res 15:790–799
Pool JE, Aquadro CF (2006) History and structure of sub-Saharan populations of Drosophila melanogaster. Genetics 174:915–929. https://doi.org/10.1534/genetics.106.058693
Laurent SJY, Werzner A, Excoffier L, Stephan W (2011) Approximate Bayesian analysis of Drosophila melanogaster polymorphism data reveals a recent colonization of Southeast Asia. Mol Biol Evol 28:2041–2051. https://doi.org/10.1093/molbev/msr031
Duchen P, Zivkovic D, Hutter S et al (2013) Demographic inference reveals African and European admixture in the North American Drosophila melanogaster population. Genetics 193:291–301. https://doi.org/10.1534/genetics.112.145912
Bergland AO, Tobler R, González J et al (2016) Secondary contact and local adaptation contribute to genome-wide patterns of clinal variation in Drosophila melanogaster. Mol Ecol 25:1157–1174. https://doi.org/10.1111/mec.13455
Pool JE (2015) The mosaic ancestry of the Drosophila genetic reference panel and the D. melanogaster reference genome reveals a network of Epistatic fitness interactions. Mol Biol Evol 32:3236–3251. https://doi.org/10.1093/molbev/msv194
Fabian DK, Lack JB, Mathur V et al (2015) Spatially varying selection shapes life history clines among populations of Drosophila melanogaster from sub-Saharan Africa. J Evol Biol 28:826–840. https://doi.org/10.1111/jeb.12607
Li YJ, Satta Y, Takahata N (1999) Paleo-demography of the Drosophila melanogaster subgroup: application of the maximum likelihood method. Genes Genet Syst 74:117–127
Li H, Stephan W (2006) Inferring the demographic history and rate of adaptive substitution in Drosophila. PLoS Genet 2:e166. https://doi.org/10.1371/journal.pgen.0020166
Charlesworth B, Morgan MT, Charlesworth D (1993) The effect of deleterious mutations on neutral molecular variation. Genetics 134:1289–1303
Ewing GB, Jensen JD (2016) The consequences of not accounting for background selection in demographic inference. Mol Ecol 25:135–141. https://doi.org/10.1111/mec.13390
Terhorst J, Kamm JA, Song YS (2017) Robust and scalable inference of population history from hundreds of unphased whole genomes. Nat Genet 49:303–309. https://doi.org/10.1038/ng.3748
Mazet O, Rodríguez W, Grusea S et al (2016) On the importance of being structured: instantaneous coalescence rates and human evolution—lessons for ancestral population size inference? Heredity (Edinb) 116:362–371. https://doi.org/10.1038/hdy.2015.104
Kapopoulou A, Pfeifer SP, Jensen JD, Laurent S (2018) The demographic history of African Drosophila melanogaster. bioRxiv 340406. https://doi.org/10.1101/340406
Kapopoulou A, Kapun M, Pavlidis P, et al (2018) Early split between African and European populations of Drosophila melanogaster. bioRxiv 340422. https://doi.org/10.1101/340422
Arguello JR, Laurent S, Clark AG (2019) Demographic history of the human commensal Drosophila melanogaster. Genome Biol Evol 11:844–854. https://doi.org/10.1093/gbe/evz022
Caracristi G, Schlötterer C (2003) Genetic differentiation between American and European Drosophila melanogaster populations could be attributed to admixture of African alleles. Mol Biol Evol 20:792–799. https://doi.org/10.1093/molbev/msg091
Corbett-Detig R, Nielsen R (2017) A hidden Markov model approach for simultaneously estimating local ancestry and admixture time using next generation sequence data in samples of arbitrary ploidy. PLoS Genet 13:e1006529. https://doi.org/10.1371/journal.pgen.1006529
Morgan TH (1910) Sex limited inheritance in Drosophila. Science 32:120–122. https://doi.org/10.1126/science.32.812.120
Lenormand T, Dutheil J (2005) Recombination difference between sexes: a role for haploid selection. PLoS Biol 3:e63. https://doi.org/10.1371/journal.pbio.0030063
Stephan W, Langley CH (1989) Molecular genetic variation in the centromeric region of the X chromosome in three Drosophila ananassae populations. I. Contrasts between the vermilion and forked loci. Genetics 121:89–99
Aguade M, Miyashita N, Langley CH (1989) Reduced variation in the yellow-achaete-scute region in natural populations of Drosophila melanogaster. Genetics 122:607–615
Cutter AD, Payseur BA (2013) Genomic signatures of selection at linked sites: unifying the disparity among species. Nat Rev Genet 14:262–274. https://doi.org/10.1038/nrg3425
Siberchicot A, Bessy A, Guéguen L, Marais GA (2017) MareyMap online: a user-friendly web application and database service for estimating recombination rates using physical and genetic maps. Genome Biol Evol 9:2506–2509. https://doi.org/10.1093/gbe/evx178
Lindsley DL, Sandler L (1977) The genetic analysis of meiosis in female Drosophila melanogaster. Philos Trans R Soc Lond Ser B Biol Sci 277:295–312. https://doi.org/10.1098/RSTB.1977.0019
Marais G, Mouchiroud D, Duret L (2001) Does recombination improve selection on codon usage? Lessons from nematode and fly complete genomes. Proc Natl Acad Sci U S A 98:5688–5692
Fiston-Lavier AS, Singh ND, Lipatov M, Petrov DA (2010) Drosophila melanogaster recombination rate calculator. Gene 463:18–20. https://doi.org/10.1016/j.gene.2010.04.015
Hudson RR (1987) Estimating the recombination parameter of a finite population model without selection. Genet Res 50:245–250
McVean GAT, Myers SR, Hunt S et al (2004) The fine-scale structure of recombination rate variation in the human genome. Science 304:581–584. https://doi.org/10.1126/science.1092500
Hudson RR (2001) Two-locus sampling distributions and their application. Genetics 159:1805–1817
Chan AH, Jenkins PA, Song YS (2012) Genome-wide fine-scale recombination rate variation in Drosophila melanogaster. PLoS Genet 8:e1003090. https://doi.org/10.1371/journal.pgen.1003090
Comeron JM, Ratnappan R, Bailin S (2012) The many landscapes of recombination in Drosophila melanogaster. PLoS Genet 8:e1002905. https://doi.org/10.1371/journal.pgen.1002905
Sturtevant AH (1913) A third group of linked genes in Drosophila ampelophila. Science 37:990–992. https://doi.org/10.1126/science.37.965.990
Sturtevant AH (1915) The behavior of the chromosomes as studied through linkage. Z Indukt Abstamm Vererbungsl 13:234–287. https://doi.org/10.1007/BF01792906
Muller HJ (1916) The mechanism of crossing-over. Am Nat 50:193–221
Carpenter AT (1975) Electron microscopy of meiosis in Drosophila melanogaster females: II. The recombination nodule--a recombination-associated structure at pachytene? Proc Natl Acad Sci U S A 72:3186–3189. https://doi.org/10.1073/PNAS.72.8.3186
Miller DE, Smith CB, Kazemi NY et al (2016) Whole-genome analysis of individual meiotic events in Drosophila melanogaster reveals that noncrossover gene conversions are insensitive to interference and the centromere effect. Genetics 203:159–171. https://doi.org/10.1534/genetics.115.186486
Fiston-Lavier A-S, Petrov DA. Drosophila melanogaster recombination rate calculator. http://petrov.stanford.edu/cgi-bin/recombination-rates_updateR5.pl
Campos JL, Zhao L, Charlesworth B (2017) Estimating the parameters of background selection and selective sweeps in Drosophila in the presence of gene conversion. Proc Natl Acad Sci 114:E4762–E4771. https://doi.org/10.1073/pnas.1619434114
Duret L, Galtier N (2009) Biased gene conversion and the evolution of mammalian genomic landscapes. Annu Rev Genomics Hum Genet 10:285–311. https://doi.org/10.1146/annurev-genom-082908-150001
Mancera E, Bourgon R, Brozzi A et al (2008) High-resolution mapping of meiotic crossovers and non-crossovers in yeast. Nature 454:479–485. https://doi.org/10.1038/nature07135
Figuet E, Ballenghien M, Romiguier J, Galtier N (2015) Biased gene conversion and GC-content evolution in the coding sequences of reptiles and vertebrates. Genome Biol Evol 7:240–250. https://doi.org/10.1093/gbe/evu277
Bolívar P, Mugal CF, Nater A, Ellegren H (2016) Recombination rate variation modulates gene sequence evolution mainly via GC-biased gene conversion, not Hill–Robertson interference, in an Avian system. Mol Biol Evol 33:216–227. https://doi.org/10.1093/molbev/msv214
Glémin S, Arndt PF, Messer PW et al (2015) Quantification of GC-biased gene conversion in the human genome. Genome Res 25:1215–1228. https://doi.org/10.1101/gr.185488.114
Pessia E, Popa A, Mousset S et al (2012) Evidence for widespread GC-biased gene conversion in eukaryotes. Genome Biol Evol 4:675–682. https://doi.org/10.1093/gbe/evs052
Glémin S, Clément Y, David J, Ressayre A (2014) GC content evolution in coding regions of angiosperm genomes: a unifying hypothesis. Trends Genet 30:263–270. https://doi.org/10.1016/j.tig.2014.05.002
Wallberg A, Glémin S, Webster MT (2015) Extreme recombination frequencies shape genome variation and evolution in the honeybee, Apis mellifera. PLoS Genet 11:e1005189. https://doi.org/10.1371/journal.pgen.1005189
Haudry A, Cenci A, Guilhaumon C et al (2008) Mating system and recombination affect molecular evolution in four Triticeae species. Genet Res (Camb) 90:97–109. https://doi.org/10.1017/S0016672307009032
Romiguier J, Roux C (2017) Analytical biases associated with GC-content in molecular evolution. Front Genet 8:16. https://doi.org/10.3389/fgene.2017.00016
Galtier N, Roux C, Rousselle M et al (2018) Codon usage bias in animals: disentangling the effects of natural selection, effective population size and GC-biased gene conversion. Mol Biol Evol 35(5):1092–1103. https://doi.org/10.1093/molbev/msy015
Singh ND (2005) Genomic heterogeneity of background Substitutional patterns in Drosophila melanogaster. Genetics 169:709–722. https://doi.org/10.1534/genetics.104.032250
Liu G, Li H (2008) The correlation between recombination rate and dinucleotide bias in Drosophila melanogaster. J Mol Evol 67:358–367. https://doi.org/10.1007/s00239-008-9150-0
Robinson MC, Stone EA, Singh ND (2014) Population genomic analysis reveals no evidence for GC-biased gene conversion in Drosophila melanogaster. Mol Biol Evol 31:425–433. https://doi.org/10.1093/molbev/mst220
Galtier N, Bazin E, Bierne N (2006) GC-biased segregation of noncoding polymorphisms in Drosophila. Genetics 172:221–228
Haddrill PR, Waldron FM, Charlesworth B (2008) Elevated levels of expression associated with regions of the Drosophila genome that lack crossing over. Biol Lett 4:758–761. https://doi.org/10.1098/rsbl.2008.0376
Campos JL, Zeng K, Parker DJ et al (2013) Codon usage bias and effective population sizes on the X chromosome versus the autosomes in Drosophila melanogaster. Mol Biol Evol 30:811–823. https://doi.org/10.1093/molbev/mss222
Sturtevant AH (1917) Genetic factors affecting the strength of linkage in Drosophila. Proc Natl Acad Sci U S A 3:555–558. https://doi.org/10.1073/pnas.3.9.555
Sturtevant AH (1919) Contributions to the genetics of Drosophila melanogaster. III. Inherited linkage variations in the second chromosome. Contributions to the genetics of Drosophila melanogaster
Sturtevant AH (1921) A case of rearrangement of genes in Drosophila. Proc Natl Acad Sci U S A 7:235–237. https://doi.org/10.1073/pnas.7.8.235
Mattei JF, Mattei MG, Ardissone JP et al (1980) Pericentric inversion, inv(9) (p22 q32), in the father of a child with a duplication-deletion of chromosome 9 and gene dosage effect for adenylate kinase-1. Clin Genet 17:129–136. https://doi.org/10.1111/j.1399-0004.1980.tb00121.x
Puerma E, Orengo D-J, Aguadé M (2016) Multiple and diverse structural changes affect the breakpoint regions of polymorphic inversions across the Drosophila genus. Sci Rep 6:36248. https://doi.org/10.1038/srep36248
Wargent JM, Hartmann-Goldstein IJ (1974) Phenotypic observations on modification of position-effect variegation in Drosophila melanogaster. Heredity (Edinb) 33:317–326. https://doi.org/10.1038/hdy.1974.98
Salm MPA, Horswell SD, Hutchison CE et al (2012) The origin, global distribution, and functional impact of the human 8p23 inversion polymorphism. Genome Res 22:1144–1153. https://doi.org/10.1101/gr.126037.111
Said I, Byrne A, Serrano V et al (2018) Linked genetic variation and not genome structure causes widespread differential expression associated with chromosomal inversions. Proc Natl Acad Sci U S A 115:5492–5497. https://doi.org/10.1073/pnas.1721275115
Lavington E, Kern AD (2017) The effect of common inversion polymorphisms In(2L)t and In(3R)Mo on patterns of transcriptional variation in Drosophila melanogaster. G3 (Bethesda) 7:3659–3668. https://doi.org/10.1534/g3.117.1133
Dobzhansky T, Sturtevant AH (1938) Inversions in the chromosomes of Drosophila Pseudoobscura. Genetics 23:28–64
Dobzhansky T, Epling C (1948) The suppression of crossing over in inversion heterozygotes of Drosophila Pseudoobscura. Proc Natl Acad Sci U S A 34:137–141. https://doi.org/10.1073/pnas.34.4.137
Garcia C, Valente VLS (2018) Drosophila chromosomal polymorphism: from population aspects to origin mechanisms of inversions. Intech
Puig M, Casillas S, Villatoro S, Cáceres M (2015) Human inversions and their functional consequences. Brief Funct Genomics 14:369–379. https://doi.org/10.1093/bfgp/elv020
Anderson WW, Arnold J, Baldwin DG et al (1991) Four decades of inversion polymorphism in Drosophila pseudoobscura. Proc Natl Acad Sci U S A 88:10367–10371. https://doi.org/10.1073/pnas.88.22.10367
Krimbas CB, Powell JR (1992) The inversion polymorphism of Drosophila subobscura. In: Drosophila inversion polymorphism. CRC Press, Boca Raton, FL, pp 127–220
Hoffmann AA, Sgrò CM, Weeks AR (2004) Chromosomal inversion polymorphisms and adaptation. Trends Ecol Evol 19:482–488. https://doi.org/10.1016/j.tree.2004.06.013
Hoffmann AA, Rieseberg LH (2008) Revisiting the impact of inversions in evolution: from population genetic markers to drivers of adaptive shifts and speciation? Annu Rev Ecol Evol Syst 39:21–42. https://doi.org/10.1146/annurev.ecolsys.39.110707.173532
Kirkpatrick M (2010) How and why chromosome inversions evolve. PLoS Biol 8:e1000501. https://doi.org/10.1371/journal.pbio.1000501
Schaeffer SW (2008) Selection in heterogeneous environments maintains the gene arrangement polymorphism of Drosophila pseudoobscura. Evolution (N Y) 62:3082–3099. https://doi.org/10.1111/j.1558-5646.2008.00504.x
Rezende EL, Balanyà J (2010) Climate change and chromosomal inversions in Drosophila subobscura. Clim Res 43:103–114. https://doi.org/10.3354/cr00869
Rego C, Balanyà J, Fragata I et al (2010) Clinal patterns of chromosomal inversion polymorphisms in Drosophila subobscura are partly associated with thermal preferences and heat stress resistance. Evolution (N Y) 64:385–397. https://doi.org/10.1111/j.1558-5646.2009.00835.x
Kapun M, Fabian DK, Goudet J, Flatt T (2016) Genomic evidence for adaptive inversion clines in Drosophila melanogaster. Mol Biol Evol 33:1317–1336. https://doi.org/10.1093/molbev/msw016
Knibb WR (1982) Chromosome inversion polymorphisms in Drosophila melanogaster II. Geographic clines and climatic associations in Australasia, North America and Asia. Genetica 58:213–221. https://doi.org/10.1007/BF00128015
Umina PA, Weeks AR, Kearney MR et al (2005) A rapid shift in a classic clinal pattern in Drosophila reflecting climate change. Science 308:691–693. https://doi.org/10.1126/science.1109523
Corbett-Detig RB, Hartl DL (2012) Population genomics of inversion polymorphisms in Drosophila melanogaster. PLoS Genet 8:e1003056. https://doi.org/10.1371/journal.pgen.1003056
Rane RV, Rako L, Kapun M, LEE SF (2015) Genomic evidence for role of inversion 3RP of Drosophila melanogaster in facilitating climate change adaptation. Mol Ecol 24:2423–2432. https://doi.org/10.1111/mec.13161/pdf
Fuller ZL, Haynes GD, Richards S, Schaeffer SW (2017) Genomics of natural populations: evolutionary forces that establish and maintain gene arrangements in Drosophila pseudoobscura. Mol Ecol 26:6539–6562. https://doi.org/10.1111/mec.14381
Navarro A, Betrán E, Barbadilla A, Ruiz A (1997) Recombination and gene flux caused by gene conversion and crossing over in inversion heterokaryotypes. Genetics 146:695–709
Andolfatto P, Depaulis F, Navarro A (2001) Inversion polymorphisms and nucleotide variability in Drosophila. Genet Res (Camb) 77:1–8. https://doi.org/10.1017/S0016672301004955
Guerrero RF, Rousset F, Kirkpatrick M (2012) Coalescent patterns for chromosomal inversions in divergent populations. Philos Trans R Soc Lond B Biol Sci 367:430–438. https://doi.org/10.1098/rstb.2011.0246
Corbett-Detig RB, Cardeno C, Langley CH (2012) Sequence-based detection and breakpoint assembly of polymorphic inversions. Genetics 192:131–137. https://doi.org/10.1534/genetics.112.141622
Navarro A, Faria R (2014) Pool and conquer: new tricks for (c)old problems. Mol Ecol 23:1653–1655. https://doi.org/10.1111/mec.12685
Sotero-Caio CG, Platt RN, Suh A, Ray DA (2017) Evolution and diversity of transposable elements in vertebrate genomes. Genome Biol Evol 9:161–177. https://doi.org/10.1093/gbe/evw264
Tenaillon MI, Hollister JD, Gaut BS (2010) A triptych of the evolution of plant transposable elements. Trends Plant Sci 15:471–478. https://doi.org/10.1016/j.tplants.2010.05.003
Tenaillon MI, Hufford MB, Gaut BS, Ross-ibarra J (2011) Genome size and transposable element content as. Mol Biol 3:219–229. https://doi.org/10.1093/gbe/evr008
Stuart T, Eichten SR, Cahn J et al (2016) Population scale mapping of transposable element diversity reveals links to gene regulation and epigenomic variation. Elife 5. https://doi.org/10.7554/eLife.20777
Vieira C, Fablet M, Lerat E et al (2012) A comparative analysis of the amounts and dynamics of transposable elements in natural populations of Drosophila melanogaster and Drosophila simulans. J Environ Radioact 113:83–86. https://doi.org/10.1016/j.jenvrad.2012.04.001
Kidwell MG, Lisch DR (2001) Perspective: transposable elements, parasitic DNA, and genome evolution. Evolution 55:1–24
Charlesworth B, Langley CH, Sniegowski PD (1997) Transposable element distributions in Drosophila. Genetics 147:1993–1995
Charlesworth B, Charlesworth D (1983) The population dynamics of transposable elements. Genet Res 42:1. https://doi.org/10.1017/S0016672300021455
Charlesworth B, Jarne P, Assimacopoulos S (1994) The distribution of transposable elements within and between chromosomes in a population of Drosophila melanogaster. III. Element abundances in heterochromatin. Genet Res 64:183. https://doi.org/10.1017/S0016672300032845
Barrón MG, Fiston-Lavier A-S, Petrov DA, González J (2014) Population genomics of transposable elements in Drosophila. Annu Rev Genet 48:561–581. https://doi.org/10.1146/annurev-genet-120213-092359
Bergman CM, Bensasson D (2007) Recent LTR retrotransposon insertion contrasts with waves of non-LTR insertion since speciation in Drosophila melanogaster. Proc Natl Acad Sci U S A 104:11340–11345. https://doi.org/10.1073/pnas.0702552104
Charlesworth B, Langley CH (1989) The population genetics of Drosophila transposable elements. Annu Rev Genet 23:251–287. https://doi.org/10.1146/annurev.ge.23.120189.001343
Ewing AD (2015) Transposable element detection from whole genome sequence data. Mob DNA 6:24. https://doi.org/10.1186/s13100-015-0055-3
Kofler R, Betancourt AJ, Schlötterer C (2012) Sequencing of pooled DNA samples (Pool-Seq) uncovers complex dynamics of transposable element insertions in Drosophila melanogaster. PLoS Genet 8:e1002487. https://doi.org/10.1371/journal.pgen.1002487
Cridland JM, Macdonald SJ, Long AD, Thornton KR (2013) Abundance and distribution of transposable elements in two Drosophila QTL mapping resources. Mol Biol Evol 30:2311–2327. https://doi.org/10.1093/molbev/mst129
Rishishwar L, Mariño-Ramírez L, Jordan IK (2017) Benchmarking computational tools for polymorphic transposable element detection. Brief Bioinform 18:908–918. https://doi.org/10.1093/bib/bbw072
Vieira C, Lepetit D, Dumont S, Biémont C (1999) Wake up of transposable elements following Drosophila simulans worldwide colonization. Mol Biol Evol 16:1251–1255
Blumenstiel JP, Chen X, He M, Bergman CM (2014) An age-of-allele test of neutrality for transposable element insertions. Genetics 196:523–538. https://doi.org/10.1534/genetics.113.158147
Ohta T (1973) Slightly deleterious mutant substitutions in evolution. Nature 246:96–98. https://doi.org/10.1038/246096a0
Kimura M (1983) The neutral theory of molecular evolution. Cambridge University Press, Cambridge
Eyre-Walker A (2006) The genomic rate of adaptive evolution. Trends Ecol Evol 21:569–575
Fay JC, Wyckoff GJ, Wu C-I (2002) Testing the neutral theory of molecular evolution with genomic data from Drosophila. Nature 415:1024–1026. https://doi.org/10.1038/4151024a
Smith NGC, Eyre-Walker A (2002) Adaptive protein evolution in Drosophila. Nature 415:1022–1024
Andolfatto P (2005) Adaptive evolution of non-coding DNA in Drosophila. Nature 437:1149–1152
Shapiro JA, Huang W, Zhang C et al (2007) Adaptive genic evolution in the Drosophila genomes. Proc Natl Acad Sci 104:2271–2276. https://doi.org/10.1073/pnas.0610385104
Eyre-Walker A, Keightley PD (2009) Estimating the rate of adaptive molecular evolution in the presence of slightly deleterious mutations and population size change. Mol Biol Evol 26:2097–2108. https://doi.org/10.1093/molbev/msp119
Castellano D, Coronado-Zamora M, Campos JL et al (2016) Adaptive evolution is substantially impeded by Hill–Robertson interference in Drosophila. Mol Biol Evol 33:442–455. https://doi.org/10.1093/molbev/msv236
Maynard Smith J, Haigh J (1974) The hitch-hiking effect of a favourable gene. Genet Res 23:23–55
Kim Y, Stephan W (2002) Detecting a local signature of genetic hitchhiking along a recombining chromosome. Genetics 160:765–777
Nielsen R, Williamson S, Kim Y et al (2005) Genomic scans for selective sweeps using SNP data. Genome Res 15:1566–1575. https://doi.org/10.1101/gr.4252305
Glinka S, Ometto L, Mousset S et al (2003) Demography and natural selection have shaped genetic variation in Drosophila melanogaster: a multi-locus approach. Genetics 165:1269–1278
Ometto L, Glinka S, De Lorenzo D, Stephan W (2005) Inferring the effects of demography and selection on Drosophila melanogaster populations from a chromosome-wide scan of DNA variation. Mol Biol Evol 22:2119–2130
Karasov T, Messer PW, Petrov DA (2010) Evidence that adaptation in Drosophila is not limited by mutation at single sites. PLoS Genet 6:e1000924. https://doi.org/10.1371/journal.pgen.1000924
Messer PW, Petrov DA (2013) Population genomics of rapid adaptation by soft selective sweeps. Trends Ecol Evol 28:659–669. https://doi.org/10.1016/j.tree.2013.08.003
Obbard DJ, Jiggins FM, Bradshaw NJ, Little TJ (2011) Recent and recurrent selective sweeps of the antiviral RNAi gene Argonaute-2 in three species of Drosophila. Mol Biol Evol 28:1043–1056. https://doi.org/10.1093/molbev/msq280
Glinka S, De Lorenzo D, Stephan W (2006) Evidence of gene conversion associated with a selective sweep in Drosophila melanogaster. Mol Biol Evol 23:1869–1878. https://doi.org/10.1093/molbev/msl069
Wilches R, Voigt S, Duchen P et al (2014) Fine-mapping and selective sweep analysis of QTL for cold tolerance in Drosophila melanogaster. G3 (Bethesda) 4:1635–1645. https://doi.org/10.1534/g3.114.012757
DuMont VB, Aquadro CF (2005) Multiple signatures of positive selection downstream of notch on the X chromosome in Drosophila melanogaster. Genetics 171:639–653. https://doi.org/10.1534/genetics.104.038851
Magwire MM, Bayer F, Webster CL et al (2011) Successive increases in the resistance of Drosophila to viral infection through a transposon insertion followed by a duplication. PLoS Genet 7:e1002337. https://doi.org/10.1371/journal.pgen.1002337
Schmidt JM, Good RT, Appleton B et al (2010) Copy number variation and transposable elements feature in recent, ongoing adaptation at the Cyp6g1 locus. PLoS Genet 6:e1000998. https://doi.org/10.1371/journal.pgen.1000998
Battlay P, Green L, Leblanc P, et al (2018) Structural variants and selective sweep foci contribute to insecticide resistance in the Drosophila melanogaster genetic reference panel. bioRxiv 301937. https://doi.org/10.1101/301937
Battlay P, Schmidt JM, Fournier-Level A, Robin C (2016) Genomic and Transcriptomic associations identify a new insecticide resistance phenotype for the selective sweep at the Cyp6g1 locus of Drosophila melanogaster. G3 (Bethesda) 6:2573–2581. https://doi.org/10.1534/g3.116.031054
Jensen JD, Bauer DuMont VL, Ashmore AB et al (2007) Patterns of sequence variability and divergence at the diminutive gene region of Drosophila melanogaster: complex patterns suggest an ancestral selective sweep. Genetics 177:1071–1085. https://doi.org/10.1534/genetics.106.069468
Saminadin-Peter SS, Kemkemer C, Pavlidis P, Parsch J (2012) Selective sweep of a cis-regulatory sequence in a non-African population of Drosophila melanogaster. Mol Biol Evol 29:1167–1174. https://doi.org/10.1093/molbev/msr284
Glaser-Schmitt A, Parsch J (2018) Functional characterization of adaptive variation within a cis-regulatory element influencing Drosophila melanogaster growth. PLoS Biol 16:e2004538. https://doi.org/10.1371/journal.pbio.2004538
Svetec N, Pavlidis P, Stephan W (2009) Recent strong positive selection on Drosophila melanogaster HDAC6, a gene encoding a stress surveillance factor, as revealed by population genomic analysis. Mol Biol Evol 26:1549–1556. https://doi.org/10.1093/molbev/msp065
Orengo DJ, Aguade M (2007) Genome scans of variation and adaptive change: extended analysis of a candidate locus close to the phantom gene region in Drosophila melanogaster. Mol Biol Evol 24:1122–1129. https://doi.org/10.1093/molbev/msm032
Beisswanger S, Stephan W (2008) Evidence that strong positive selection drives neofunctionalization in the tandemly duplicated polyhomeotic genes in Drosophila. Proc Natl Acad Sci 105:5447–5452. https://doi.org/10.1073/pnas.0710892105
Voigt S, Laurent S, Litovchenko M, Stephan W (2015) Positive selection at the Polyhomeotic locus led to decreased thermosensitivity of gene expression in temperate Drosophila melanogaster. Genetics 200:591–599. https://doi.org/10.1534/genetics.115.177030
Pool JE, Bauer DuMont V, Mueller JL, Aquadro CF (2006) A scan of molecular variation leads to the narrow localization of a selective sweep affecting both Afrotropical and cosmopolitan populations of Drosophila melanogaster. Genetics 172:1093–1105. https://doi.org/10.1534/genetics.105.049973
Depaulis F, Brazier L, Veuille M (1999) Selective sweep at the Drosophila melanogaster suppressor of hairless locus and its association with the In(2L)t inversion polymorphism. Genetics 152:1017–1024. https://doi.org/10.1017/S0016672398003462
Beisswanger S, Stephan W, De Lorenzo D (2006) Evidence for a selective sweep in the wapl region of Drosophila melanogaster. Genetics 172:265–274. https://doi.org/10.1534/genetics.105.049346
Garud NR, Messer PW, Buzbas EO, Petrov DA (2015) Recent selective sweeps in North American Drosophila melanogaster show signatures of soft sweeps. PLoS Genet 11:e1005004. https://doi.org/10.1371/journal.pgen.1005004
Schlenke TA, Begun DJ (2004) Strong selective sweep associated with a transposon insertion in Drosophila simulans. Proc Natl Acad Sci 101:1626–1631. https://doi.org/10.1073/pnas.0303793101
Teshima KM, Coop G, Przeworski M (2006) How reliable are empirical genomic scans for selective sweeps? Genome Res 16:702–712
Thornton KR, Jensen JD (2007) Controlling the false-positive rate in multilocus genome scans for selection. Genetics 175:737–750
Pavlidis P, Jensen JD, Stephan W (2010) Searching for footprints of positive selection in whole-genome SNP data from nonequilibrium populations. Genetics 185:907–922. https://doi.org/10.1534/genetics.110.116459
Crisci JL, Poh YP, Mahajan S, Jensen JD (2013) The impact of equilibrium assumptions on tests of selection. Front Genet 4:235. https://doi.org/10.3389/fgene.2013.00235
Hermisson J, Pennings PS (2017) Soft sweeps and beyond: understanding the patterns and probabilities of selection footprints under rapid adaptation. Methods Ecol Evol 8:700–716. https://doi.org/10.1111/2041-210X.12808
Jensen JD (2014) On the unfounded enthusiasm for soft selective sweeps. Nat Commun 5:5281. https://doi.org/10.1038/ncomms6281
Kaplan NL, Hudson RR, Langley CH (1989) The “Hitchhiking Effect” revisited. Genetics 123:887–899
Casillas S, Barbadilla A (2017) Molecular population genetics. Genetics 205:1003–1035. https://doi.org/10.1534/genetics.116.196493
Sella G, Petrov DA, Przeworski M, Andolfatto P (2009) Pervasive natural selection in the Drosophila genome? PLoS Genet 5:e1000495. https://doi.org/10.1371/journal.pgen.1000495
Sawyer S, Kulathinal RJ, Bustamante CD, Hartl DL (2003) Bayesian analysis suggests that most amino acid replacements in Drosophila are driven by positive selection. J Mol Evol 57:S154–S164
Schneider A, Charlesworth B, Eyre-Walker A, Keightley PD (2011) A method for inferring the rate of occurrence and fitness effects of advantageous mutations. Genetics 189:1427–1437. https://doi.org/10.1534/genetics.111.131730
Keightley PD, Campos JL, Booker TR, Charlesworth B (2016) Inferring the frequency spectrum of derived variants to quantify adaptive molecular evolution in protein-coding genes of Drosophila melanogaster. Genetics 203:975–984. https://doi.org/10.1534/genetics.116.188102
Andolfatto P (2007) Hitchhiking effects of recurrent beneficial amino acid substitutions in the Drosophila melanogaster genome. Genome Res 17:1755–1762. https://doi.org/10.1101/gr.6691007
Jensen JD, Thornton KR, Andolfatto P (2008) An approximate Bayesian estimator suggests strong, recurrent selective sweeps in Drosophila. PLoS Genet 4:e1000198. https://doi.org/10.1371/journal.pgen.1000198
Macpherson JM, Sella G, Davis JC, Petrov DA (2007) Genomewide spatial correspondence between nonsynonymous divergence and neutral polymorphism reveals extensive adaptation in Drosophila. Genetics 177:2083–2099. https://doi.org/10.1534/genetics.107.080226
Stephan W (2010) Genetic hitchhiking versus background selection: the controversy and its implications. Philos Trans R Soc L B Biol Sci 365:1245–1253. https://doi.org/10.1098/rstb.2009.0278
Charlesworth B (2012) The role of background selection in shaping patterns of molecular evolution and variation: evidence from variability on the Drosophila X chromosome. Genetics 191:233–246
Hudson RR, Kaplan NL (1995) Deleterious background selection with recombination. Genetics 141:1605–1617
Comeron JM (2014) Background selection as baseline for nucleotide variation across the Drosophila genome. PLoS Genet 10:e1004434. https://doi.org/10.1371/journal.pgen.1004434
Elyashiv E, Sattath S, Hu TT et al (2016) A genomic map of the effects of linked selection in Drosophila. PLoS Genet 12:e1006130. https://doi.org/10.1371/journal.pgen.1006130
Siepel A (2005) Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 15:1034–1050. https://doi.org/10.1101/gr.3715005
Bergman CM, Kreitman M (2001) Analysis of conserved noncoding DNA in Drosophila reveals similar constraints in intergenic and intronic sequences. Genome Res 11:1335–1345
Harmston N, Baresic A, Lenhard B (2013) The mystery of extreme non-coding conservation. Philos Trans R Soc Lond Ser B Biol Sci 368:20130021. https://doi.org/10.1098/rstb.2013.0021
Berr T, Peticca A, Haudry A (2019) Evidence for purifying selection on conserved noncoding elements in the genome of Drosophila melanogaster. bioRxiv 623744. https://doi.org/10.1101/623744
Brody T, Yavatkar AS, Kuzin A et al (2012) Use of a Drosophila genome-wide conserved sequence database to identify functionally related cis-regulatory enhancers. Dev Dyn 241:169–189. https://doi.org/10.1002/dvdy.22728
Enright AJ, John B, Gaul U et al (2003) MicroRNA targets in Drosophila. Genome Biol 5:R1. https://doi.org/10.1186/gb-2003-5-1-r1
Lai CK, Miller MC, Collins K (2003) Roles for RNA in telomerase nucleotide and repeat addition processivity. Mol Cell 11:1673–1683
Casillas S, Barbadilla A, Bergman CM (2007) Purifying selection maintains highly conserved noncoding sequences in Drosophila. Mol Biol Evol 24:2222–2234. https://doi.org/10.1093/molbev/msm150
Clark AG (2001) The search for meaning in noncoding DNA. Genome Res 11:1319–1320. https://doi.org/10.1101/gr.201601
Shields DC, Sharp PM, Higgins DG, Wright F (1988) “Silent” sites in Drosophila genes are not neutral: evidence of selection among synonymous codons. Mol Biol Evol 5:704–716. https://doi.org/10.1093/oxfordjournals.molbev.a040525
Hershberg R, Petrov DA (2008) Selection on codon bias. https://doi.org/10.1146/annurev.genet.42.110807.091442
Lawrie DS, Messer PW, Hershberg R, Petrov DA (2013) Strong purifying selection at synonymous sites in D. melanogaster. PLoS Genet 9:e1003527. https://doi.org/10.1371/journal.pgen.1003527
Machado HE, Lawrie DS, Petrov DA (2017) Strong selection at the level of codon usage bias: evidence against the Li-Bulmer model. bioRxiv. https://doi.org/10.1101/106476
Duret L, Mouchiroud D (1999) Expression pattern and, surprisingly, gene length shape codon usage in Caenorhabditis, Drosophila, and Arabidopsis. Evolution (N Y) 96:4482–4487
Powell JR, Moriyama EN (1997) Evolution of codon usage bias in Drosophila. Proc Natl Acad Sci U S A 94:7784–7790
Bierne N, Eyre-Walker A (2006) Variation in synonymous codon use and DNA polymorphism within the Drosophila genome. J Evol Biol 19:1–11
Vicario S, Moriyama EN, Powell JR (2007) Codon usage in twelve species of Drosophila. BMC Evol Biol 7:226. https://doi.org/10.1186/1471-2148-7-226
Zeng K, Charlesworth B (2009) Estimating selection intensity on synonymous codon usage in a nonequilibrium population. Genetics 183:651–662, 1SI-23SI. https://doi.org/10.1534/genetics.109.101782
Andolfatto P, Wong KM, Bachtrog D (2011) Effective population size and the efficacy of selection on the X chromosomes of two closely related Drosophila species. Genome Biol Evol 3:114–128. https://doi.org/10.1093/gbe/evq086
Clemente F, Vogl C (2012) Evidence for complex selection on four-fold degenerate sites in Drosophila melanogaster. J Evol Biol 25:2582–2595. https://doi.org/10.1111/jeb.12003
Poh Y-P, Ting C-T, Fu H-W et al (2012) Population genomic analysis of base composition evolution in Drosophila melanogaster. Genome Biol Evol 4:1245–1255. https://doi.org/10.1093/gbe/evs097
Nielsen R, Bauer DuMont VL, Hubisz MJ, Aquadro CF (2006) Maximum likelihood estimation of ancestral codon usage bias parameters in Drosophila. Mol Biol Evol 24:228–235. https://doi.org/10.1093/molbev/msl146
Jackson BC, Campos JL, Haddrill PR et al (2017) Variation in the intensity of selection on codon bias over time causes contrasting patterns of base composition evolution in Drosophila. Genome Biol Evol 9:102–123. https://doi.org/10.1093/gbe/evw291
Kirkpatrick M, Kern A (2012) Where’s the money? Inversions, genes, and the hunt for genomic targets of selection. Genetics 190:1153–1155. https://doi.org/10.1534/genetics.112.139899
Charlesworth B, Charlesworth D (1973) Selection of new inversions in multi-locus genetic systems. Genet Res (Camb) 21:167. https://doi.org/10.1017/S0016672300013343
Schaeffer SW, Miller JM, Anderson WW (2003) Evolutionary genomics of inversions in Drosophila pseudoobscura: evidence for epistasis. Proc Natl Acad Sci U S A 100:8319–8324. https://doi.org/10.1073/pnas.1432900100
Kirkpatrick M, Barton N (2006) Chromosome inversions, local adaptation and speciation. Genetics 173:419–434. https://doi.org/10.1534/genetics.105.047985
Charlesworth B, Barton NH (2018) The spread of an inversion with migration and selection. Genetics 208:377–382. https://doi.org/10.1534/genetics.117.300426
Bhutkar A, Schaeffer SW, Russo SM et al (2008) Chromosomal rearrangement inferred from comparisons of 12 Drosophila genomes. Genetics 179:1657–1680. https://doi.org/10.1534/genetics.107.086108
Stevison LS, Hoehn KB, Noor MAF (2011) Effects of inversions on within- and between-species recombination and divergence. Genome Biol Evol 3:830–841. https://doi.org/10.1093/gbe/evr081
McGaugh SE, Noor MAF (2012) Genomic impacts of chromosomal inversions in parapatric Drosophila species. Philos Trans R Soc Lond B Biol Sci 367:422–429. https://doi.org/10.1098/rstb.2011.0250
Lohse K, Clarke M, Ritchie MG, Etges WJ (2015) Genome-wide tests for introgression between cactophilic Drosophila implicate a role of inversions during speciation. Evolution (N Y) 69:1178–1190. https://doi.org/10.1111/evo.12650
Lee Y, Collier TC, Sanford MR et al (2013) Chromosome inversions, genomic differentiation and speciation in the African malaria mosquito Anopheles gambiae. PLoS One 8:e57887. https://doi.org/10.1371/journal.pone.0057887
Ayala D, Guerrero RF, Kirkpatrick M (2013) Reproductive isolation and local adaptation quantified for a chromosome inversion in a malaria mosquito. Evolution (N Y) 67:946–958. https://doi.org/10.1111/j.1558-5646.2012.01836.x
Rieseberg LH (2001) Chromosomal rearrangements and speciation. Trends Ecol Evol 16:351–358. https://doi.org/10.1016/S0169-5347(01)02187-5
Abbott JK, Nordén AK, Hansson B (2017) Sex chromosome evolution: historical insights and future perspectives. Proc R Soc B Biol Sci 284:20162806. https://doi.org/10.1098/rspb.2016.2806
Lemeunier F, Aulard S (1992) Inversion polymorphism in Drosophila melanogaster. In: Krimbas CB, Powell JR (eds) Drosophila inversion polymorphism. CRC Press, p 576
Rebollo R, Romanish MT, Mager DL (2012) Transposable elements: an abundant and natural source of regulatory sequences for host genes. Annu Rev Genet 46:21–42. https://doi.org/10.1146/annurev-genet-110711-155621
Casacuberta E, González J (2013) The impact of transposable elements in environmental adaptation. Mol Ecol 22:1503–1517. https://doi.org/10.1111/mec.12170
Villanueva-Cañas JL, Rech GE, de Cara MAR, González J (2017) Beyond SNPs: how to detect selection on transposable element insertions. Methods Ecol Evol 8:728–737. https://doi.org/10.1111/2041-210X.12781
González J, Karasov TL, Messer PW, Petrov DA (2010) Genome-wide patterns of adaptation to temperate environments associated with transposable elements in Drosophila. PLoS Genet 6:e1000905. https://doi.org/10.1371/journal.pgen.1000905
Merenciano M, Ullastres A, de Cara MAR et al (2016) Multiple independent Retroelement insertions in the promoter of a stress response gene have variable molecular and functional effects in Drosophila. PLoS Genet 12:e1006249. https://doi.org/10.1371/journal.pgen.1006249
González J, Lenkov K, Lipatov M et al (2008) High rate of recent transposable element–induced adaptation in Drosophila melanogaster. PLoS Biol 6:e251. https://doi.org/10.1371/journal.pbio.0060251
González J, Macpherson JM, Petrov DA (2009) A recent adaptive transposable element insertion near highly conserved developmental loci in Drosophila melanogaster. Mol Biol Evol 26:1949–1961. https://doi.org/10.1093/molbev/msp107
Guio L, Barrón MG, González J (2014) The transposable element Bari-Jheh mediates oxidative stress response in Drosophila. Mol Ecol 23:2020–2030. https://doi.org/10.1111/mec.12711
Ellison CE, Bachtrog D (2013) Dosage compensation via transposable element mediated rewiring of a regulatory network. Science 342:846–850. https://doi.org/10.1126/science.1239552
Sentmanat MF, Elgin SCR (2012) Ectopic assembly of heterochromatin in Drosophila melanogaster triggered by transposable elements. Proc Natl Acad Sci U S A 109:14104–14109. https://doi.org/10.1073/pnas.1207036109
Perrat PN, DasGupta S, Wang J et al (2013) Transposition-driven genomic heterogeneity in the Drosophila brain. Science 340:91–95. https://doi.org/10.1126/science.1231965
Charlesworth B, Coyne JA, Barton NH (1987) The relative rates of evolution of sex chromosomes and autosomes. Am Nat 130:113–146. https://doi.org/10.1086/284701
Charlesworth B, Campos JL, Jackson BC (2018) Faster-X evolution: theory and evidence from Drosophila. Mol Ecol 27:3753–3771. https://doi.org/10.1111/mec.14534
Betancourt AJ, Kim Y, Orr HA (2004) A pseudohitchhiking model of X vs. autosomal diversity. Genetics 168:2261–2269. https://doi.org/10.1534/genetics.104.030999
Campos JL, Halligan DL, Haddrill PR, Charlesworth B (2014) The relation between recombination rate and patterns of molecular evolution and variation in Drosophila melanogaster. Mol Biol Evol 31:1010–1028. https://doi.org/10.1093/molbev/msu056
Endler JA (1977) Geographic variation, speciation, and clines. Princeton University Press, Princeton, NJ
Huxley JS (1938) Clines: an auxiliary taxonomic principle. Nature 142:219–220. https://doi.org/10.1038/142219a0
de Jong G, Bochdanovits Z (2003) Latitudinal clines in Drosophila melanogaster: body size, allozyme frequencies, inversion frequencies, and the insulin-signalling pathway. J Genet 82:207–223. https://doi.org/10.1007/BF02715819
Adrion JR, Hahn MW, Cooper BS (2015) Revisiting classic clines in Drosophila melanogaster in the age of genomics. Trends Genet 31:434–444. https://doi.org/10.1016/j.tig.2015.05.006
Flatt T (2016) Genomics of clinal variation in Drosophila: disentangling the interactions of selection and demography. Mol Ecol 25:1023–1026. https://doi.org/10.1111/mec.13534
Rosenzweig ML (1968) The strategy of body size in mammalian carnivores. Am Midl Nat 80:299
Blanckenhorn WU, Demont M (2004) Bergmann and converse bergmann latitudinal clines in arthropods: two ends of a continuum? Integr Comp Biol 44:413–424. https://doi.org/10.1093/icb/44.6.413
Stillwell RC (2010) Are latitudinal clines in body size adaptive? Oikos 119:1387–1390. https://doi.org/10.1111/j.1600-0706.2010.18670.x
Kivelä SM, Välimäki P, Carrasco D et al (2011) Latitudinal insect body size clines revisited: a critical evaluation of the saw-tooth model. J Anim Ecol 80:1184–1195. https://doi.org/10.2307/41332025?ref=no-x-route:0c31195f82739061ed649686a0a9f292
Colombo PC, Remis MI (2015) Morphometric variation in chromosomally polymorphic grasshoppers (Orthoptera: Acrididae) from South America: Bergmann and Converse Bergmann patterns. Florida Entomol 98:570–574. https://doi.org/10.1653/024.098.0228
Schmidt PS, Paaby AB (2008) Reproductive diapause and life-history clines in North American populations of Drosophila melanogaster. Evolution (N Y) 62:1204–1215. https://doi.org/10.1111/j.1558-5646.2008.00351.x
Addo-Bediako A, Chown SL, Gaston KJ (2000) Thermal tolerance, climatic variability and latitude. Proc R Soc B Biol Sci 267:739–745. https://doi.org/10.1098/rspb.2000.1065
Hoffmann AA, Anderson A, Hallas R (2002) Opposing clines for high and low temperature resistance in Drosophila melanogaster. Ecol Lett 5:614–618. https://doi.org/10.1046/j.1461-0248.2002.00367.x
Castañeda LE, Lardies MA, Bozinovic F (2005) Interpopulational variation in recovery time from chill coma along a geographic gradient: a study in the common woodlouse, Porcellio laevis. J Insect Physiol 51:1346–1351
Gockel J, Kennington WJ, Hoffmann AA et al (2001) Nonclinality of molecular variation implicates selection in maintaining a morphological cline of Drosophila melanogaster. Genetics 158:319–323
Turner TL, Levine MT, Eckert ML, Begun DJ (2008) Genomic analysis of adaptive differentiation in Drosophila melanogaster. Genetics 179:455–473. https://doi.org/10.1534/genetics.107.083659
Behrman EL, Howick VM, Kapun M et al (2018) Rapid seasonal evolution in innate immunity of wild Drosophila melanogaster. Proc R Soc B Biol Sci 285:20172599. https://doi.org/10.1098/rspb.2017.2599
De Mita S, Thuillet A-C, Gay L et al (2013) Detecting selection along environmental gradients: analysis of eight methods and their effectiveness for outbreeding and selfing populations. Mol Ecol 22:1383–1399. https://doi.org/10.1111/mec.12182
Vasemägi A (2006) The adaptive hypothesis of clinal variation revisited: single-locus clines as a result of spatially restricted gene flow. Genetics 173:2411–2414. https://doi.org/10.1534/genetics.106.059881
Prevosti A, Serra L, Segarra C et al (1990) Clines of chromosomal arrangements of Drosophila subobscura in South America evolve closer to Old World patterns. Evolution (N Y) 44:218. https://doi.org/10.2307/2409539
Machado HE, Bergland AO, O’Brien KR et al (2016) Comparative population genomics of latitudinal variation in Drosophila simulans and Drosophila melanogaster. Mol Ecol 25:723–740. https://doi.org/10.1111/mec.13446
Barton NH (1999) Clines in polygenic traits. Genet Res 74:223–236
Acknowledgments
We are very grateful to Laurent Duret, Jeffrey Jensen, Gabriel Marais, Alan Moses, Cristina Vieira, and Stephen Wright for their helpful comments on the manuscript. We are very thankful to Julien Dutheil for the invitation to write this chapter.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2020 The Author(s)
About this protocol

Cite this protocol
Haudry, A., Laurent, S., Kapun, M. (2020). Population Genomics on the Fly: Recent Advances in Drosophila. In: Dutheil, J.Y. (eds) Statistical Population Genomics. Methods in Molecular Biology, vol 2090. Humana, New York, NY. https://doi.org/10.1007/978-1-0716-0199-0_15
Download citation
DOI: https://doi.org/10.1007/978-1-0716-0199-0_15
Published:
Publisher Name: Humana, New York, NY
Print ISBN: 978-1-0716-0198-3
Online ISBN: 978-1-0716-0199-0
eBook Packages: Springer Protocols