
Abstract
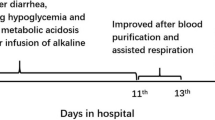
Glutaric aciduria type II, also known as Multiple acyl-CoA Dehydrogenase Deficiency, results from a defect in the mitochondrial electron transport chain resulting in an inability to break down fatty-acids and amino acids. There are three phenotypes- type 1 and 2 are of neonatal onset and severe form, with and without congenital anomalies, respectively, and presents with acidosis, severe hypotonia, cardiomyopathy, hepatomegaly, and non-ketotic hypoglycemia. Type 3 or late-onset Multiple acyl-CoA Dehydrogenase Deficiency usually presents in the adolescent or adult age group with phenotype ranging from mild forms of myopathy and exercise intolerance to severe forms of acute metabolic decompensation on its chronic course. Type 3 Multiple acyl-CoA Dehydrogenase Deficiency rarely presents in infancy and in liver failure. We present a five-month-old developmentally normal female child with acute encephalopathy, hypotonia, non-ketotic hypoglycemia, metabolic acidosis, and liver failure, with a history of sibling death of suspected inborn error of metabolism. The blood acyl-carnitine levels in Tandem Mass Spectrometry and urinary organic acid analysis through Gas Chromatography–Mass Spectrometry were unremarkable. The patient initially responded to riboflavin, CoQ, and supportive management but ultimately succumbed to sepsis with shock and multi-organ dysfunction. The clinical exome sequencing reported a homozygous missense variation in exon 11 of the ETFDH gene (chr4:g.158706270C > T) that resulted in the amino acid substitution of Leucine for Proline at codon 456 (p.Pro456Leu) suggestive of Glutaric aciduria type IIc (OMIM#231,680).

Similar content being viewed by others


Abbreviations
- ETFA :
-
Electron Transfer Flavoprotein Alpha subunit
- ETFB :
-
Electron Transfer Flavoprotein Beta subunit
- ETFDH :
-
Electron Transfer Flavoprotein Coenzyme Q Oxidoreductase
- GCMS:
-
Gas Chromatography-Mass Spectrometry
- FAD:
-
Flavin Adenine Dinucleotide
- MADD:
-
Multiple acyl-CoA Dehydrogenase Deficiency
- TMS:
-
Tandem Mass Spectrometry
References
OMIM [Internet]. [cited 2021 May 6]. Available from: https://www.omim.org/entry/231680
glutaricaciduria-ii [Internet]. [cited 2021 May 6]. Available from: https://rarediseases.org/rare-diseases/glutaricaciduria-ii/
Disease_Search.php [Internet]. [cited 2021 May 6]. Available from: https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=EN&data_id=8766&Disease_Disease_Search_diseaseGroup=231680&Disease_Disease_Search_diseaseType=MIM&Disease%28s%29/group%20of%20diseases=Glutaric-aciduria-type-2&title=Glutaric-aciduria-type-2&searc
Grünert SC. Clinical and genetical heterogeneity of late-onset multiple acyl-coenzyme A dehydrogenase deficiency. Orphanet J Rare Dis. 2014;9:117.
van Rijt WJ, Jager EA, Allersma DP, Aktuğlu Zeybek AÇ, Bhattacharya K, Debray F-G, et al. Efficacy and safety of D, L-3-hydroxybutyrate (D, L-3-HB) treatment in multiple acyl-CoA dehydrogenase deficiency. Genet Med. 2020;22:908–16.
Saral N, Aksungar F, Serteser M. Simplified approach to glutaric acidurias: a Mini-Review. J Rare Dis Res Treat. 2019;4:66–70.
Ding M, Liu R, Qiubo L, Zhang Y, Kong Q. Neonatal-onset multiple acyl-CoA dehydrogenase deficiency (MADD) in the ETFDH gene: a case report and a literature review. Medicine Baltimore. 2020;99:e21944.
Hu G, Zeng J, Wang C, Zhou W, Jia Z, Yang J, et al. A synonymous variant c.579A>G in the ETFDH gene caused exon skipping in a patient with late-onset multiple Acyl-CoA dehydrogenase deficiency: a case report. Front Pediatr. 2020;8:118.
Vieira P, Myllynen P, Perhomaa M, Tuominen H, Keski-Filppula R, Rytky S, et al. Riboflavin-responsive multiple acyl-CoA dehydrogenase deficiency associated with hepatoencephalomyopathy and white matter signal abnormalities on brain MRI. Neuropediatrics. 2017;48:194–8.
Electron transport chain [Internet]. Nilesh. [cited 2021 Sep 17]. Available from: https://microbiochem.weebly.com/electron-transport-chain.html
Missaglia S, Tavian D, Moro L, Angelini C. Characterization of two ETFDH mutations in a novel case of riboflavin-responsive multiple acyl-CoA dehydrogenase deficiency. Lipids Health Dis. 2018;17:254.
VCV000095072.6 - ClinVar - NCBI [Internet]. [cited 2021 Sep 17]. Available from: https://www.ncbi.nlm.nih.gov/clinvar/variation/95072/
Goodman SI, Binard RJ, Woontner MR, Frerman FE. Glutaric acidemia type II: gene structure and mutations of the electron transfer flavoprotein:ubiquinone oxidoreductase (ETF:QO) gene. Mol Genet Metab. 2002;77:86–90.
Lucas TG, Henriques BJ, Gomes CM. Conformational analysis of the riboflavin-responsive ETF:QO-p Pro456Leu variant associated with mild multiple acyl-CoA dehydrogenase deficiency Biochimica et Biophysica Acta (BBA). Proteins and Proteomics. 2020;1868:140393.
Olsen RKJ, Olpin SE, Andresen BS, Miedzybrodzka ZH, Pourfarzam M, Merinero B, et al. ETFDH mutations as a major cause of riboflavin-responsive multiple acyl-CoA dehydrogenation deficiency. Brain. 2007;130:2045–54.
Funding
Nil.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no conflict of interest,
Consent for Publication
Informed written consent was taken from parents for publication.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Keshri, S., Goel, A.K., Johns, J. et al. “Liver Failure in an Infant of Late-Onset Glutaric Aciduria Type II”: Case Report. Ind J Clin Biochem 38, 545–549 (2023). https://doi.org/10.1007/s12291-021-01007-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12291-021-01007-7