
Abstract
Hereditary myopathies characterized by the development of autophagic vacuoles can be categorized into three groups: rimmed vacuolar myopathies, acid maltase deficiency (glycogen storage disease type II), and myopathies characterized by the autophagic vacuoles with unique vacuolar membranes. Rimmed vacuolar myopathies are most likely secondary lysosomal myopathies because all of the identified causative genes encode extralysosomal proteins. Deficiency of acid maltase, a lysosomal enzyme, has been well characterized clinically, pathologically, biochemically, and genetically, and may become treatable in the near future. The diseases in the last category are relatively rare, but appear to be genetically heterogeneous and the list of these diseases is expanding. Danon disease, the bestcharacterized disorder in this group, is caused by primary deficiency of a lysosomal membrane protein, LAMP-2. Therefore, diseases in this category are expected to be primary lysosomal disease.
Similar content being viewed by others

References and Recommended Reading
Alberts B: Molecular Biology of the Cell, edn 4. New York: Garland Science; 2001.
Nonaka I, Sunohara N, Ishiura S, et al.: Familial distal myopathy with rimmed vacuole and lamellar (myeloid) body formation. J Neurol Sci 1981, 51:141–155.
Argov Z, Yarom R: ‘Rimmed vacuole myopathy’ sparing the quadriceps: a unique disorder in Iranian Jews. J Neurol Sci 1984, 64:33–43.
Ikeuchi T, Asaka T, Saito M, et al.: Gene locus for autosomal recessive distal myopathy with rimmed vacuoles maps to chromosome 9. Ann Neurol 1997, 41:432–437.
Mitrani-Rosenbaum S, Argov Z, Blumenfeld A, et al.: Hereditary inclusion body myopathy maps to chromosome 9p1-q1. Hum Mol Genet 1996, 5:159–163.
Eisenberg I, Avidan N, Potikha T, et al.: The UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase gene is mutated in recessive hereditary inclusion body myopathy. Nat Genet 2001, 29:83–87. This paper reports that GNE gene is implicated in hereditary inclusion body myopathy.
Nishino I, Noguchi S, Murayama K, et al.: Distal myopathy with rimmed vacuoles is allelic to hereditary inclusion body myopathy. Neurology 2002, in press.
McFerrin J, Engel WK, Askanas V: Impaired innervation of cultured human muscle overexpressing beta APP experimentally and genetically: relevance to inclusion-body myopathies. Neuroreport 1998, 9:3201–3205.
Sugarman MC, Yamasaki TR, Oddo S, et al.: Inclusion body myositis-like phenotype induced by transgenic overexpression of bAPP in skeletal muscle. Proc Natl Acad Sci U S A 2002, 99:6334–6339. This paper suggests that amyloid deposition may be responsible for inclusion body mysositis pathology, although abnormalities of lysosomes are not described.
DiMauro S, Musumeci O: Metabolic myopathies. In Neuromuscular Disorders in Clinical Practice. Edited by Katirji B. Boston: Butterworth-Heinemann; 2002.
Danon MJ, Oh SJ, DiMauro S, et al.: Lysosomal glycogen storage disease with normal acid maltase. Neurology 1981, 31:51–57.
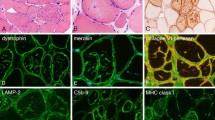
Nishino I, Fu J, Tanji K, et al.: Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature 2000, 406:906–910. The autophagic vacuolar myopathy with unique vacuolar membrane was shown to be due to the deficiency of lysosome-associated membrane protein-2 (LAMP-2). This finding was also confirmed by LAMP-2 knockout mouse in another report in the same issue of the jounal.
Sugie K, Yamamoto A, Murayama K, et al.: Clinicopathological features of genetically confirmed Danon disease. Neurology 2002, 58:1773–1778. Clinicopathologic review of Danon disease.
Murakami N, Goto YI, Itoh M, et al.: Sarcolemmal indentation in cardiomyopathy with mental retardation and vacuolar myopathy. Neuromusc Disord 1995, 5:149–155.
Muntoni F, Catani G, Mateddu A, et al.: Familial cardiomyopathy, mental retardation and myopathy associated with desmin-type intermediate filaments. Neuromusc Disord 1994, 4:233–241.
Kannan K, Divers SG, Lurie AA, et al.: Cell surface expression of lysosome-associated membrane protein-2 (lamp2) and CD63 as markers of in vivo platelet activation in malignancy. Eur J Haematol 1995, 55:145–151.
Reddy A, Caler EV, Andrews NW: Plasma membrane repair is mediated by Ca2+-regulated exocytosis of lysosomes. Cell 2001, 106:157–169.
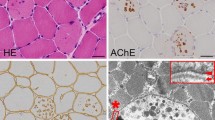
Kalimo H, Savontaus ML, Lang H, et al.: X-linked myopathy with excessive autophagy: a new hereditary muscle disease. Ann Neurol 1988, 23:258–265.
Yamamoto A, Morisawa Y, Verloes A, et al.: Infantile autophagic vacuolar myopathy is distinct from Danon disease. Neurology 2001, 57:903–905.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Nishino, I. Autophagic vacuolar myopathies. Curr Neurol Neurosci Rep 3, 64–69 (2003). https://doi.org/10.1007/s11910-003-0040-y
Issue Date:
DOI: https://doi.org/10.1007/s11910-003-0040-y