
Abstract
Background
Congenital ectopia lentis (CEL) is a rare but serious disease. We use next-generation sequencing to detect genes associated with lens abnormalities in 24 patients with bilateral CEL and search for pathogenic genes and mutation sites.
Materials and methods
A total of 24 patients diagnosed with CEL from January 2019 to November 2019 were enrolled in this study, and their clinical data were collected and genome-wide deoxyribonucleic acid was extracted from peripheral venous blood. Targeted gene capture technology was used to obtain 188 exons of lens abnormality-related genes, which were sequenced using a high-throughput method. The mutation sites were determined through data analysis and verified by the Sanger method. According to the data from previous studies, the association between the genotype and clinical phenotype was analysed.
Result
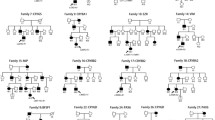
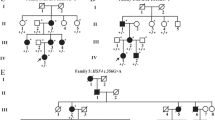
Of the 24 patients, 23 had mutations in the fibrillin-1 (FBN1) gene, and 20 were diagnosed with Marfan syndrome. The 23 cases of FBN1 mutations were all heterozygous mutations, including 17 missense mutations, 3 splicing variants, 2 exon deletion mutations, 1 codon mutation, and 9 new mutations. A total of 17 mutations were located in the calcium-binding epidermal growth factor domain, including 16 mutations that contained missense mutations of cysteine. In addition, a heterozygous mutation of the gap junction protein alpha 8 (GJA8) gene was detected in one patient.
Conclusion
In this study, we identified 23 FBN1 gene mutations and 1 GJA8 gene mutation in 24 patients with CEL. Of these, 9 new FBN1 mutations and 14 known mutations were found. The results expanded the mutation spectrum of the FBN1 gene, suggesting that FBN1 mutation may be the main cause of CEL in Chinese patients.

Similar content being viewed by others

References
Greene VB, Stoetzel C, Pelletier V, Perdomo-Trujillo Y, Liebermann L, Marion V, De Korvin H, Boileau C, Dufier JL, Dollfus H (2010) Confirmation of ADAMTSL4 mutations for autosomal recessive isolated bilateral ectopia lentis. Ophthalmic Genet 31(1):47–51. https://doi.org/10.3109/13816810903567604
Christensen AE, Fiskerstrand T, Knappskog PM, Boman H, Rødahl E (2010) A novel ADAMTSL4 mutation in autosomal recessive ectopia lentis et pupillae. Invest Ophthalmol Vis Sci 51(12):6369–6373. https://doi.org/10.1167/iovs.10-5597
Azar G, Dureau P, Barjol A, Edelson C, Bergès O, Caputo G (2013) Ectopia lentis associated with primary congenital glaucoma. Eur J Ophthalmol 23(4):597–600. https://doi.org/10.5301/ejo.5000264
Chandra A, Ekwalla V, Child A, Charteris D (2014) Prevalence of ectopia lentis and retinal detachment in Marfan syndrome. Acta Ophthalmol 92(1):e82–e83. https://doi.org/10.1111/aos.12175
Sadiq MA, Vanderveen D (2013) Genetics of ectopia lentis. Semin Ophthalmol 28(5–6):313–320. https://doi.org/10.3109/08820538.2013.825276
Aldahmesh MA, Alshammari MJ, Khan AO, Mohamed JY, Alhabib FA, Alkuraya FS (2013) The syndrome of microcornea, myopic chorioretinal atrophy, and telecanthus (MMCAT) is caused by mutations in ADAMTS18. Hum Mutat 34(9):1195–1199. https://doi.org/10.1002/humu.22374
Loeys BL, Dietz HC, Braverman AC, Callewaert BL, De Backer J, Devereux RB, Hilhorst-Hofstee Y, Jondeau G, Faivre L, Milewicz DM, Pyeritz RE, Sponseller PD, Wordsworth P, De Paepe AM (2010) The revised Ghent nosology for the Marfan syndrome. J Med Genet 47(7):476–485. https://doi.org/10.1136/jmg.2009.072785
Mizuguchi T, Collod-Beroud G, Akiyama T, Abifadel M, Harada N, Morisaki T, Allard D, Varret M, Claustres M, Morisaki H, Ihara M, Kinoshita A, Yoshiura K, Junien C, Kajii T, Jondeau G, Ohta T, Kishino T, Furukawa Y, Nakamura Y, Niikawa N, Boileau C, Matsumoto N (2004) Heterozygous TGFBR2 mutations in Marfan syndrome. Nat Genet 36(8):855–860. https://doi.org/10.1038/ng1392
Haji-Seyed-Javadi R, Jelodari-Mamaghani S, Paylakhi SH, Yazdani S, Nilforushan N, Fan JB, Klotzle B, Mahmoudi MJ, Ebrahimian MJ, Chelich N, Taghiabadi E, Kamyab K, Boileau C, Paisan-Ruiz C, Ronaghi M, Elahi E (2012) LTBP2 mutations cause Weill-Marchesani and Weill-Marchesani-like syndrome and affect disruptions in the extracellular matrix. Hum Mutat 33(8):1182–1187. https://doi.org/10.1002/humu.22105
Edwards AO (2008) Clinical features of the congenital vitreoretinopathies. Eye (Lond) 22(10):1233–1242. https://doi.org/10.1038/eye.2008.38
Khan AO, Aldahmesh MA, Noor J, Salem A, Alkuraya FS (2015) Lens subluxation and retinal dysfunction in a girl with homozygous VSX2 mutation. Ophthalmic Genet 36(1):8–13. https://doi.org/10.3109/13816810.2013.827217
Robinson PN, Booms P, Katzke S, Ladewig M, Neumann L, Palz M, Pregla R, Tiecke F, Rosenberg T (2002) Mutations of FBN1 and genotype-phenotype correlations in Marfan syndrome and related fibrillinopathies. Hum Mutat 20(3):153–161. https://doi.org/10.1002/humu.10113
Comeglio P, Evans AL, Brice G, Cooling RJ, Child AH (2002) Identification of FBN1 gene mutations in patients with ectopia lentis and marfanoid habitus. Br J Ophthalmol 86(12):1359–1362. https://doi.org/10.1136/bjo.86.12.1359
Comeglio P, Johnson P, Arno G, Brice G, Evans A, Aragon-Martin J, da Silva FP, Kiotsekoglou A, Child A (2007) The importance of mutation detection in Marfan syndrome and Marfan-related disorders: report of 193 FBN1 mutations. Hum Mutat 28(9):928. https://doi.org/10.1002/humu.9505
Booms P, Cisler J, Mathews KR, Godfrey M, Tiecke F, Kaufmann UC, Vetter U, Hagemeier C, Robinson PN (1999) Novel exon skipping mutation in the fibrillin-1 gene: two “hot spots” for the neonatal Marfan syndrome. Clin Genet 55(2):110–117. https://doi.org/10.1034/j.1399-0004.1999.550207.x
Tiecke F, Katzke S, Booms P, Robinson PN, Neumann L, Godfrey M, Mathews KR, Scheuner M, Hinkel GK, Brenner RE, Hövels-Gürich HH, Hagemeier C, Fuchs J, Skovby F, Rosenberg T (2001) Classic, atypically severe and neonatal Marfan syndrome: twelve mutations and genotype-phenotype correlations in FBN1 exons 24–40. Eur J Hum Genet 9(1):13–21. https://doi.org/10.1038/sj.ejhg.5200582
Palz M, Tiecke F, Booms P, Göldner B, Rosenberg T, Fuchs J, Skovby F, Schumacher H, Kaufmann UC, von Kodolitsch Y, Nienaber CA, Leitner C, Katzke S, Vetter B, Hagemeier C, Robinson PN (2000) Clustering of mutations associated with mild Marfan-like phenotypes in the 3’ region of FBN1 suggests a potential genotype-phenotype correlation. Am J Med Genet 91(3):212–221. https://doi.org/10.1002/(sici)1096-8628(20000320)91:3%3c212::aid-ajmg12%3e3.0.co;2-3
Ponnam SP, Ramesha K, Tejwani S, Ramamurthy B, Kannabiran C (2007) Mutation of the gap junction protein alpha 8 (GJA8) gene causes autosomal recessive cataract. J Med Genet 44(7):e85. https://doi.org/10.1136/jmg.2007.050138
Funding
No funding was received for this research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors certify that they have no affiliations with or involvement in any organisation or entity with any financial interest (such as honoraria; educational grants; participation in speakers’ bureaus; membership, employment, consultancies, stock ownership, or other equity interest; and expert testimony or patent-licensing arrangements), or non-financial interest (such as personal or professional relationships, affiliations, knowledge or beliefs) in the subject matter or materials discussed in this manuscript.
Ethical standards
All procedures performed in studies involving human participants were in accordance with the ethical standards of the Beijing Tongren Hospital, Capital Medical University and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Written informed consent was obtained from all participants.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Qi, M., Wang, C., Liu, Y. et al. Next-generation sequencing panel analysis in 24 Chinese patients with congenital ectopia lentis. Int Ophthalmol 42, 2245–2253 (2022). https://doi.org/10.1007/s10792-022-02224-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10792-022-02224-6