
Abstract
Adsorption of CCl3F, CCl2F2, CClF3, F2, and O2 has been investigated systematically on the polythiophene (PT) moieties using density functional theory (DFT) calculations at the B3LYP/6-31G (d) level. Here, geometry optimizations have been performed on some polythiophene (5PT and 7PT) complexes. Likewise, adsorption energies, dipole moments, HOMO–LUMO orbital analysis, density of states (DOSs), global indices, and UV–vis spectra are calculated using the DFT method. Energies of interaction and HOMO–LUMO gap show that polythiophene has the highest sensitivity toward O2 for B3LYP functional. According to our findings, CFCs, F2, and O2 molecules can be physically adsorbed on the moieties of polythiophene.
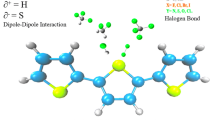
Graphical abstract








Similar content being viewed by others


Data Availability
All data generated or analyzed and related materials (with supplementary materials) are included in this manuscript.
Code availability
The calculations have been carried out using Gaussian 09 and GaussView version 6.0 provided by Gaussian, Inc.
References
Dubbe A (2003) Fundamentals of solid state ionic micro gas sensors. Sensors Actuators B Chem 88:138–148
Zakrzewska K (2001) Mixed oxides as gas sensors. Thin Solid Films 391:229–238
Timmer B, Olthuis W, Van Den Berg A (2005) Ammonia sensors and their applications—a review. Sensors Actuators B Chem 107:666–677
Nicolas-Debarnot D, Poncin-Epaillard F (2003) Polyaniline as a new sensitive layer for gas sensors. Anal Chim Acta 475:1–15
Roncali J (1992) Conjugated poly (thiophenes): synthesis, functionalization, and applications. Chem Rev 92:711–738
Ameer Q, Adeloju SB (2005) Polypyrrole-based electronic noses for environmental and industrial analysis. Sensors Actuators B Chem 106:541–552
Rad AS, Nasimi N, Jafari M et al (2015) Ab-initio study of interaction of some atmospheric gases (SO2, NH3, H2O, CO, CH4 and CO2) with polypyrrole (3PPy) gas sensor: DFT calculations. Sensors Actuators B Chem 220:641–651
Ates M, Sarac TK, AS, (2012) Conducting polymers and their applications. Curr Phys Chem 2:224–240
Halls JJM, Walsh CA, Greenham NC et al (1995) Efficient photodiodes from interpenetrating polymer networks. Nature 376:498–500
Kraft A, Grimsdale AC, Holmes AB (1998) Electroluminescent conjugated polymers—seeing polymers in a new light. Angew Chemie Int Ed 37:402–428
Hepburn AR, Marshall JM, Maud JM (1991) Novel electrochromic films via anodic oxidation of carbazolyl substituted polysiloxanes. Synth Met 43:2935–2938
Dubois JC, Sagnes O, Henry F (1989) Polyheterocyclic conducting polymers and composites derivates. Synth Met 28:871–878
Roncali J, Garreau R, Delabouglise D, et al (1989) Modification of the structure and electrochemical properties of poly (thiophene) by ether groups. J Chem Soc Chem Commun 679–681
Bradley DDC (1991) Molecular electronics. Aspects of the physics. Chem Br 27:
Gas Sensors Fluorine - Gas-Sensing.com. (n.d.). Retrieved September 3, 2021 from (2000) Ultracapacitors: why, how, and where is the technology. In: Journal of Power Sources. pp 37–50.https://www.gassensing.com/information/fluorin
Ullah H, Ayub K, Ullah Z et al (2013) Theoretical insight of polypyrrole ammonia gas sensor. Synth Met 172:14–20
Skotheim TA, Reynolds JR (2007) Handbook of conducting polymers: conjugated polymers processing and applications
Virji S, Huang J, Kaner RB, Weiller BH (2004) Polyaniline nanofiber gas sensors: examination of response mechanisms. Nano Lett 4:491–496
Friend RH, Gymer RW, Holmes AB et al (1999) Electroluminescence in conjugated polymers. Nature 397:121–128
Ma X, Li G, Xu H et al (2006) Preparation of polythiophene composite film by in situ polymerization at room temperature and its gas response studies. Thin Solid Films 515:2700–2704
Molina MJ, Rowland FS (1974) Stratospheric sink for chlorofluoromethanes-chlorine atom catalyzed destruction of ozone. In: International Conference on the Environmental Impact of Aerospace Operations in the High Atmosphere, 2 nd, San Diego, Calif. pp 99–104
Kim K-H, Shon Z-H, Nguyen HT, Jeon E-C (2011) A review of major chlorofluorocarbons and their halocarbon alternatives in the air. Atmos Environ 45:1369–1382
UNEP (2006) The Montreal protocol on substances that deplete the ozone layer
McCulloch A, Ashford P, Midgley PM (2001) Historic emissions of fluorotrichloromethane (CFC-11) based on a market survey. Atmos Environ 35:4387–4397
Metz, Bert M, Kuijpers L, Solomon S, et al (2005) Safeguarding the ozone layer and the global climate system: issues related to hydrofluorocarbons and perfluorocarbons
(2002) Toxicological profile for carbon tetrachloride. In: ATSDR’s Toxicological Profiles
Midgley T Jr (1937) From the periodic table to production. Ind Eng Chem 29:241–244
McCulloch A, Midgley PM, Ashford P (2003) Releases of refrigerant gases (CFC-12, HCFC-22 and HFC-134a) to the atmosphere. Atmos Environ 37:889–902
Siegemund G, Schwertfeger W, Feiring A, et al (2000) Fluorine compounds, organic. Ullmann’s Encycl Ind Chem
Oğuz I-C, Mineva T, Guesmi H (2018) The effect of Pd ensemble structure on the O2 dissociation and CO oxidation mechanisms on Au—Pd (100) surface alloys. J Chem Phys 148:24701
Murray JS, Seybold PG, Politzer P (2021) The many faces of fluorine: some noncovalent interactions of fluorine compounds. J Chem Thermodyn 156:106382. https://doi.org/10.1016/j.jct.2020.106382
Gas sensors fluorine - gas-sensing.com. (n.d.) Retrieved September 3, 2021, https://www.gassensing.com/information/fluorine.
Cabrera N, Mott NF (1949) Theory of the oxidation of metals. Reports Prog Phys 12:163–184
Sun S, Xu P, Ren Y et al (2018) First-principles study of dissociation processes of O2 molecular on the Al (111) surface. Curr Appl Phys 18:1528–1533
Rakib Hossain M, Mehade Hasan M, Ud Daula Shamim S et al (2021) First-principles study of the adsorption of chlormethine anticancer drug on C24, B12N12 and B12C6N6 nanocages. Comput Theor Chem 1197:113156. https://doi.org/10.1016/j.comptc.2021.113156
Shokuhi Rad A, Zardoost MR, Abedini E (2015) First-principles study of terpyrrole as a potential hydrogen cyanide sensor: DFT calculations. J Mol Model 21:273. https://doi.org/10.1007/s00894-015-2814-y
Rad AS, Zardoost MR, Abedini E (2015) First-principles study of terpyrrole as a potential hydrogen cyanide sensor: DFT calculations. J Mol Model 21:1–7
Tenderholt AL, Langner KM, O’Boyle NM (2008) A library for package-independent computational chemistry algorithms. J Comp Chem 839–845
MM Hasan AC Das MR Hossain et al 2021 The computational quantum mechanical investigation of the functionalized boron nitride nanocage as the smart carriers for favipiravir drug delivery: a DFT and QTAIM analysis J BiomolStruct Dyn 1 17. https://doi.org/10.1080/07391102.2021.1982776
MM Hasan MH Kabir MA Badsha MR Hossain 2021 A first-principles DFT study on the adsorption behaviour of CO, CO2, and O3 on pristine B24N24 and silicon-decorated B24N24 nanosheet Phosphorus Sulfur Silicon Relat Elem 1–8. https://doi.org/10.1080/10426507.2021.1988599
Rad AS (2015) Al-doped graphene as modified nanostructure sensor for some ether molecules: Ab-initio study. Synth Met 209:419–425
Kamel M, Raissi H, Morsali A, Shahabi M (2018) Assessment of the adsorption mechanism of Flutamide anticancer drug on the functionalized single-walled carbon nanotube surface as a drug delivery vehicle: an alternative theoretical approach based on DFT and MD. Appl Surf Sci 434:492–503
Saha S, Roy RK, Pal S (2010) CDASE—a reliable scheme to explain the reactivity sequence between Diels-Alder pairs. Phys Chem Chem Phys 12:9328–9338
Sarmah A, Roy RK (2013) Understanding the interaction of nucleobases with chiral semiconducting single-walled carbon nanotubes: an alternative theoretical approach based on density functional reactivity theory. J Phys Chem C 117:21539–21550
MR Hossain MM HasanAshrafi N-E- et al 2021 Adsorption behaviour of metronidazole drug molecule on the surface of hydrogenated graphene, boron nitride and boron carbide nanosheets in gaseous and aqueous medium: a comparative DFT and QTAIM insight Phys E Low-dimensional Syst Nanostructures 126 114483. https://doi.org/10.1016/j.physe.2020.114483
Li J, Lu Y, Ye Q et al (2003) Carbon nanotube sensors for gas and organic vapor detection. Nano Lett 3:929–933
Ahmadi Peyghan A, Hadipour NL, Bagheri Z (2013) Effects of Al doping and double-antisite defect on the adsorption of HCN on a BC2N nanotube: density functional theory studies. J Phys Chem C 117:2427–2432
Ahmadi A, Hadipour NL, Kamfiroozi M, Bagheri Z (2012) Theoretical study of aluminum nitride nanotubes for chemical sensing of formaldehyde. Sensors Actuators B Chem 161:1025–1029. https://doi.org/10.1016/j.snb.2011.12.001
Rad AS, Shabestari SS, Jafari SA et al (2016) N-doped graphene as a nanostructure adsorbent for carbon monoxide: DFT calculations. Mol Phys 114:1756–1762
Maria JP, Nagarajan V, Chandiramouli R (2020) Boron trifluoride interaction studies on graphdiyne nanotubes–a first-principles insight. Chem Phys Lett 738:136841
Diaz AF, Crowley J, Bargon J et al (1981) Electrooxidation of aromatic oligomers and conducting polymers. J Electroanal Chem Interfacial Electrochem 121:355–361
Rad AS, Abedini E (2016) Chemisorption of NO on Pt-decorated graphene as modified nanostructure media: a first principles study. Appl Surf Sci 360:1041–1046
Bulat FA, Murray JS, Politzer P (2021) Identifying the most energetic electrons in a molecule: the highest occupied molecular orbital and the average local ionization energy. Comput Theor Chem 1199:113192. https://doi.org/10.1016/j.comptc.2021.113192
Murray JS, Politzer P (2017) Molecular electrostatic potentials and noncovalent interactions. Wiley Interdiscip Rev Comput Mol Sci 7:e1326
Murray JS, Politzer P (2009) Molecular surfaces, van der Waals radii and electrostatic potentials in relation to noncovalent interactions. Croat Chem Acta 82:267–275
Shamim SUD, Miah MH, Hossain MR et al (2022) Theoretical investigation of emodin conjugated doped B12N12 nanocage by means of DFT, QTAIM and PCM analysis. Phys E Low-dimensional Syst Nanostructures 136:115027. https://doi.org/10.1016/j.physe.2021.115027
Politzer P, Murray JS (2021) Are HOMO–LUMO gaps reliable indicators of explosive impact sensitivity? J Mol Model 27:327. https://doi.org/10.1007/s00894-021-04956-1
Rad AS (2016) Adsorption of C2H2 and C2H4 on Pt-decorated graphene nanostructure: ab-initio study. Synth Met 211:115–120
Rad AS (2015) Application of polythiophene to methanol vapor detection: an ab initio study. J Mol Model 21:1–6
Job G, Herrmann F (2006) Chemical potential—a quantity in search of recognition. Eur J Phys 27:353
Cook G, Dickerson RH (1995) Understanding the chemical potential. Am J Phys 63:737–742
Rad AS (2016) Al-doped graphene as a new nanostructure adsorbent for some halomethane compounds: DFT calculations. Surf Sci 645:6–12
Rad AS (2016) Terthiophene as a model sensor for some atmospheric gases: theoretical study. Mol Phys 114:584–591. https://doi.org/10.1080/00268976.2015.1102348
Rad AS, Valipour P (2015) Interaction of methanol with some aniline and pyrrole derivatives: DFT calculations. Synth Met 209:502–511
Kamel M, Raissi H, Morsali A (2017) Theoretical study of solvent and co-solvent effects on the interaction of Flutamide anticancer drug with carbon nanotube as a drug delivery
H Sajid S Khan K Ayub T Mahmood 2020 High selectivity of cyclic tetrapyrrole over tetrafuran and tetrathiophene toward toxic chemicals; a first-principles study MicroporousMesoporous Mater 299. https://doi.org/10.1016/j.micromeso.2020.110126
Acknowledgements
We highly acknowledge the software support from the Department of Physics, Computational Condensed Matter Physics Laboratory, Jahangirnagar University, Dhaka, Bangladesh. We thank the Jashore University of Science and Technology (JUST) for letting us run the optimization jobs at the Faculty of Science, Department of Physics.
Author information
Authors and Affiliations
Contributions
Md. Mehade Hasan and Shahida Aktar Bithe wrote the first draft of the paper. Besides, the first and corresponding author Md. Mehade Hasan has done the formal analysis, data generation, and investigation of the results and discussion part. All authors revised and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Hasan, M., Bithe, S.A., Neher, B. et al. Polythiophene as a sensor model for chlorofluorocarbon, fluorine, and oxygen gas using DFT calculations. J Mol Model 28, 59 (2022). https://doi.org/10.1007/s00894-022-05048-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-022-05048-4