
Abstract
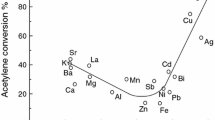
The recent literature concerning the use of supported gold catalysts is reviewed. In particular, two main uses of gold catalysts are considered, namely (a) the hydrochlorination of ethyne using supported chlorides, and (b) the oxidation of carbon monoxide at ambient temperature. For the hydrochlorination of ethyne a correlation of catalytic activity with the metal standard electrode potential predicted that gold would be the most active catalyst for this reaction, and subsequent experimental work confirmed this prediction. Low temperature oxidation of carbon monoxide is a reaction of current environmental interest and gold catalysts are effective at ambient conditions. For both these reactions, gold is found to be the most active catalyst and is therefore the metal of choice.
Similar content being viewed by others

References
G.C. Bond,Gold Bull., 1972,5, 11
J. Schwank,Gold Bull., 1985,18, 1
F. Gasparrini, M. Giovannoli, D. Misiti, G. Natile and G. Palmierri,Tetrahedron, 1983,39, 3181
F. Gasparrini, M. Giovannoli, D. Misiti, G. Natile and G. Palmierri,Tetrahedron, 1984,40, 165
J.W.A. Sachtler, M.A. Van Hove, J.P. Biberian and G.A. Somorjai,Phys. Rev. Lett., 1980,45, 1601
J.R.H. Schaik, R.P Dressing and V. Ponec,J. Catal., 1975,38, 273
D. Thompson,Gold Bull., 1996,39, 94
A.I. Gel’bshtein, M.I. Siling, G.G. Shcheglova and I.B. Vasil’eva,Kinet. Catal., 1964,5, 402
D.M. Smith, P.M. Walsh and T.L. Slager,J. Catal., 1968,11, 113
K. Shinoda,Chem. Lett., 1975, 219
G.J. Hutchings andD. Grady Appl Catal., 1985,16, 441
G.J. Hutchings and D. Grady,AppL Catal., 1985,17, 155
A.I. Gel’bshtein, M.I. Siling, G.A. Sergeeva and G.G. Shcheglova,Kinet Catal., 1963,4, 123
A.I. Gel’bshtein and M.I. Siling,Kinet Catal., 1963,4, 262
A.I. Gel’bshtein, G.G. Shcheglova and A.A. Khomenko,Kinet Catal., 1964,4, 543
G.J. Hutchings,J. Catal., 1985,96, 292
B. Nkosi, N.J. Coville, and G.J. Hutchings,J. Chem. Soc, Chem. Commun., 1988, 71
B. Nkosi, N.J. Coville and G.J. Hutchings,AppL Catal., 1988,43, 33
B. Nkosi, N.J. Coville, G.J. Hutchings, M.D. Adams, J. Friedl and F. Wagner,J. Catal., 1991,128, 366
B. Nkosi, M.D. Adams, N.J. Coville and G.J. Hutchings,J. Catal., 1991,128, 378
S. Veprek, D.L. Cocke, S. Kehl and H.R. Oswald,J. Catal., 1986,100, 250
G.J Hutchings, A.A Mirzaei, R.W Joyner, M.R.H. Siddiqui and S.H. Taylor,Catal. Lett., in press
M. Haruta, T. Kobayashi, H. Sano and N. Yamada,Chem. Lett., 1987, 405
M. Haruta, N. Yamada, T. Kobayashi and S. Ijima,J. Catal., 1989,115, 301
M. Haruta, S. Tsubota, T. Kobayashi, H. Kageyama, M.J. Genet and B. Delmon,J. Catal., 1993,144, 175
M. Haruta, A. Ueda, R.M. Torres Sanchez and K. Tanaka, Prp. Pet. Div., ACS Symp., New Orleans, 1996; T. Hayashi, K. Tanaka and M. Haruta, Prp. Pet. Div., ACS Symp., New Orleans, 1996
S.D. Gardner, G.B. Hoflund, B.T Upchurch, D.R. Schryer, E.J. Kielen and J. Schryer,J. Catal., 1991,129, 114
G. Srinivas, J. Wright, C.-S. Bai and R. Cook,StulSurf. Sci. Catal., 1996,101. 427
R.W. Joyner, F. King, M.A. Thomas and G. Roberts,Catal. Today, 1991,10, 417
A.A. Mirzai, M.R.H. Siddiqui, S.H. Taylor, C.J Kieley, A. Burrows, R.W. Joyner and G.J Hutchings, in preparation
G.J. Hutchings, M.R.H. Siddiqui, A. Burrows C.J. Kiely and R. Whyman,J. Chem. Soc. Faraday Trans., in press
F. Boccuzzi, A. Chiorino, S. Tsubota and M. Haruta,Catal. Lett., 1994,29, 225
H. Sakurai, S. Tsubota and M. Haruta,Appl. Catal., 1993,102, 125
H. Sakurai and M. Haruta,Catal. Today, 1996,29, 361
M. Haruta, A. Ueda, S. Tsubota and R.M. Torres Sanchez,Catal. Today, 1996,29, 443
T. Kobayashi, M. Haruta, S. Tsubota and H. Sano,Sensors. Act., 1987,B1, 222
M. Ando, T. Kobayashi and M. Haruta,5th Im. Meet Chem. Sensors, 1994, 1156
F. Boccuzzi, A. Chiorino, S. Tsubota and M. Haruta,Sensors. Act., 1995,B24–25, 540
A. Ueda, T. Oshima and M. Haruta,Environ. Catal., 1995, 343
Author information
Authors and Affiliations
Additional information
Professor Graham Hutchings is Deputy Director of the Leverhulme Centre for Innovative Catalysis at the University of Liverpool. He has experience in both academic and industrial catalysis research and has a current research interest in oxidation chemistry.
Rights and permissions
About this article
Cite this article
Hutchings, G.J. Catalysis: A golden future. Gold Bull 29, 123–130 (1996). https://doi.org/10.1007/BF03214746
Issue Date:
DOI: https://doi.org/10.1007/BF03214746