
Abstract
Establishing symbiosis between bacteria and invertebrates can significantly enhance energy transfer efficiency between them, which may aid in shaping the flourishing community in deep-sea chemosynthetic ecosystems, including cold seeps, hydrothermal vents, and organic falls. The symbionts utilize the chemical energy from reductive materials to fix carbon, and the hosts absorb the nutrients for growth through farming, milking, or both. Moreover, symbiosis can enhance the sustainability of both participants to survive in harsh conditions. However, the exact process and the regulatory network of symbiosis are still unknown. The cold seeps in the South China Sea offer natural laboratories to study the composition, ecological functions, and regulatory mechanisms of deep-sea symbioses. In this chapter, we focused on two dominant species, a deep-sea mussel Gigantidas platifrons and a squat lobster Shinkaia crosnieri, which represent endosymbiosis and episymbiosis, respectively, at Site F to summarize our understanding of deep-sea chemosymbiosis. We also discussed some promising avenues for future studies, such as deep-sea in situ experiments to show the exact responses of deep-sea organisms, culture-dependent experiments with genetic operations to validate the functions of critical genes, and microscale omics to elucidate the possible interactions at subcellular levels.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


6.1 Introduction
Gravitational and tectonic forces can create leakage pathways, termed cold seeps, that allow subsurface methane to rise from the sea floor (Fig. 6.1; Boetius and Wenzhöfer 2013). Cold seeps are often located on geologically activated and passive continental margins. Leakage fluids are rich in reduced materials, such as methane, short-chain alkanes, and hydrogen sulfide, from deep to shallow sediments or even seawater (Ceramicola et al. 2018). Compared with traditional photosynthetic ecosystems, the particularity of cold seeps is their dependence on chemosynthetic bacteria for primary productivity. The discoveries of such lightless ecosystems have revolutionized our understanding of the origin and energy sources of life on Earth. Since their discovery in the late 1970s and early 1980s (Lonsdale 1979; Paull et al. 1984), an increasing number of cold seeps have been explored with accumulating knowledge of their specialized functions in extreme ecosystems. The most amazing discovery in these ecosystems is the organisms surviving independently of sunlight and form the marvelous landscape of a dense invertebrate community (Fig. 6.2). The macrobenthos fauna is apparently nourished by reduced chemicals seeping out of the sea floor (Kennicutt et al. 1985). To facilitate energy and matter transfer between invertebrates and microbes, they have established close relationships by symbiosis (Husnik et al. 2021). The giant gutless tubeworm Riftia pachyptila is the first discovered deep-sea invertebrate with endosymbionts. They used reduced sulfur compounds to fix carbon dioxide and transported the organic carbon to the hosts (Dubilier et al. 2008). The discovery of R. pachyptila symbiosis has triggered a series of searches for symbiosis in vents and cold seeps in the deep sea.

Schematic view of the cold-seep ecosystem with methane fluxes at continental slopes. Reprinted from Nature Geoscience, 6, Boetius and Wenzhöfer (2013) Seafloor oxygen consumption fuelled by methane from cold seeps, 725–734, Copyright (2022), with permission from Springer Nature

Photos showing the characterized habitat at Site F of the South China Sea. a the assemblage of the squat lobster; b, c mussel beds harboring nails, limpets and Alvin shrimps; d shell debris with tiny sponges; e authigenic carbonate covered with sponges and corals; f bacterial mats. Reprinted from Journal of Marine Systems, 218, Cao et al. (2021) In situ detection of the fine scale heterogeneity of active cold seep environment of the Formosa Ridge, the South China Sea, 103,530, Copyright (2022), with permission from Elsevier
Symbiotic relationships with chemosynthetic bacteria have been widely discovered in diverse groups of macrofauna, including intracellular symbiosis (endosymbiosis) represented by bathymodiolin mussels, thyasirids, and vesicomyids in the phylum Mollusca; tubeworms in the phylum Annelida (Dubilier et al. 2008); and extracellular symbiosis (episymbiosis) represented by galatheid crabs (Watsuji et al. 2010) and alvinocaridid shrimp (Polz and Cavanaugh 1995) in the phylum Arthropoda. The host animal must be able to interact with the symbiont at the tissue, cell, and molecular levels to maintain this high-efficiency chemosymbiosis (Hinzke et al. 2019). The host is able to provide the symbiont with necessary gases, such as sulfide, methane, oxygen, and carbon dioxide, in large quantities to support the fast growth of the symbiont (Hinzke et al. 2021). Moreover, the host also needs to control the symbiont population by selectively digesting a portion of the symbiont and then extracting and transporting the nutrient for its own growth (Gomez-Mendikute et al. 2005; Hirokawa and Noda 2008; Renoz et al. 2015). To date, this unique symbiosis (between invertebrates and chemosynthetic bacteria) has become a model for understanding animal-microorganism interactions and has been intensely studied. Here, we provide a brief review of studies on the composition, function, and regulatory mechanism of symbiosis in cold seeps in the South China Sea (SCS), focusing on two dominant species, G. platifrons and S. crosnieri, at Site F in the SCS.
6.2 Adaptive Characteristics of a Symbiotic Lifestyle
Concerning the location where the symbionts dwell, the symbiotic microbiome can form associations with invertebrates, mainly in two forms, endosymbiosis and episymbiosis. Episymbionts occur on the surface of the host, whereas endosymbionts occur internally, either within a body cavity but outside the cells (extracellular) or within cells (intracellular). Bathymodiolin mussels are the research model of endosymbiosis. These mussels host SOX and/or MOX in specialized gill epithelial cells, namely, bacteriocytes (Halary et al. 2008). To obtain reduced chemicals in deep-sea cold seeps, they have evolved some special characteristics, such as enlarged gills and hypertrophic bacteriocytes (Duperron 2010). The exchange surfaces of the bathymodiolin mussels are approximately 20-fold higher than those found in similar-sized coastal mussels (Duperron et al. 2016). With long-term symbiotic adaptation, the digestive systems of deep-sea mussels have degraded, such as intestinal and lip degradation or disappearance (Fisher and Childress 1986).
These mussels have developed refined structures to sustain the bacteriocytes. The gills of G. platifrons have a typical filibranch bivalve structure involving cilia ventilation that provides symbiotic bacteria with necessary gases, such as methane and oxygen, from the seep fluid (Fig. 6.3). Each gill filament’s lateral face is covered with bacteriocytes, with the exception of the heavily ciliated filament tip. Each bacteriocyte hosts a dense population of a single species of methane-oxidizing symbiont. Symbiont-free intercalary cells also appear between the bacteriocytes. Scanning electron microscopy (SEM) images showed that each bacteriocyte’s apex protrudes slightly above the surrounding intercalary cells (Fig. 6.4). Although the surface of the bacteriocytes is smooth, the intercalary cells have microvilli and cilia on their surfaces, which may help slow down water flow and improve gas exchange (Fig. 6.4b′). A transmission electron microscope (TEM) image showed that the bacteriocytes have a compartmentalized internal ultrastructure (Fig. 6.4c). The methane-oxidizing symbionts (Fig. 6.4d and d′), which have the characteristic intracytoplasmic membrane structure of type I methanotroph bacteria, are engulfed in intracellular vacuoles and aggregated at the apex of the bacteriocytes (Fig. 6.3c and d). Primary and secondary lysosomes are also located in the lower part of the bacteriocytes (Fig. 6.4c).

The symbiosis of the deep-sea mussel G. platifrons. a Sampling of the deep-sea mussel G. platifrons at Site F. b A healthy G. platifrons specimen; the inset shows the dissected gill filaments. c Double-FISH with FITC-labeled eubacteria probe EU338, Cy3-labeled G. platifrons symbiont probe. d Magnified merged image showing the bacteriocytes that host symbionts, nonsymbiotic intercalary cells (white arrowhead), and amoebocytes (white asterisk). Scale bar of Panel (c): 50 μm. Reprinted from Deep-Sea Research Part I, 151, Wang et al. (2019) Comparative transcriptomic analysis 624 illuminates the host-symbiont interactions in the deep-sea mussel Bathymodiolus platifrons, 103,082, Copyright (2022), with permission from Elsevier

Electron microscopy analysis of the gill of G. platifrons a An SEM image of the heavily ciliated tip of the G. platifrons gill tip. Ci: ciliary, CD: ciliary disk. b An SEM image of the lateral face of the G. platifrons gill, showing the bacteriocytes and intercalary cells. b′ A magnified region in b showing the detailed surface structures of the bacteriocyte and intercalary cells. The surfaces of bacteriocytes protrude slightly higher than the surrounding intercalary cells. There are cilia and microvillae on the surface of intercalary cells. MC: mucus cell, IC: intercalary cell. c A TEM image of the longitudinal section of the bacteriocytes and adjacent intercalary cells. d TEM image of methane-oxidizing symbiotic bacteria covered in vacuoles. d′ A magnified image showing the detailed structure of the symbiont. White arrowheads: intracytoplasmic membrane structure, which is a characteristic feature of type I methanotroph bacteria. Black arrowheads: DNA of the symbiotic bacteria. Reprinted from iScience, 24, Wang et al. (2021) Molecular analyses of the gill symbiosis of the bathymodiolin mussel Gigantidas platifrons,101,894, Copyright (2022), with permission from Elsevier
Shinkaia crosnieri is another dominant species at site F and serves as the model for episymbiont studies. S. crosnieri raises complex microbial communities, rather than certain symbiotic species, that mainly attach to the setae of appendages (Fig. 6.5; Xu et al. 2022). Rod-shaped Methylococcales attach to the setae surfaces or via the formation of filamentous aggregations with diameters between 0.8 and 1.5 μm. Both Thiotrichales and Campylobacteriales form filament-like aggregations but differ in diameter from 2 to 3 μm and 0.4 to 0.6 μm, respectively. Flavobacteriales were observed as small, rod-shaped cells attached to other filamentous bacteria, indicating that they may utilize the organic carbon generated by other chemosynthetic bacteria. These associations are consistent with observations in biofilms growing on the surfaces of a black smoker chimney in the Loki’s Castle vent field (Stokke et al. 2015). Episymbiont biofilms tend to develop overlapping colonies with other bacteria that occur in close proximity, thereby facilitating metabolite sharing among symbionts. S. crosnieri can comb out its ventral setae using the third maxilliped to consume the episymbionts. Special ethological features that enhance primary productivity were also recorded for this species. The lobster waves its arm in the reducing fluid to increase the productivity of its epibionts by removing boundary layers that may otherwise limit carbon fixation (Thurber et al. 2011). In contrast, no apparent setae can be observed in another sympatric deep-sea squat lobster, Munidopsis verrilli (Dong and Li 2015).

S. crosnieri setae symbiont sampling information. a The geographic location of the cold seep site studied here (i.e., Site F) and image of the S. crosnieri community adjacent to a methane seepage taken by the ROV Faxian. b Ventral and dorsal views of S. crosnieri, with the setae on the appendages highlighted. c SEM image showing the distribution of key episymbiont taxa on the setae based on FISH analyses. Coloring of taxa is as follows: blue: Methylococcales; red: Thiotrichales; yellow: Campylobacteriales; green: Methylococcales or Thiotrichales. Reprinted from mSystems, 7, Xu et al. (2022) Metabolism Interactions Promote the Overall Functioning of the Episymbiotic Chemosynthetic Community of Shinkaia crosnieri of Cold Seeps, e00320-22, Copyright (2022), with permission from ASM journals
6.3 Taxonomic Composition and Carbon Fixation Pathway of the Symbionts
Although 6 proteobacteria could be recovered from the gills of the G. platifrons, only the methanotrophic bacteria Methyloprofundas spp. were found inside the bacteriocytes with high abundance (up to 95%). Based on the metatranscriptomic dataset, the methane-oxidizing process, which generates formaldehyde, is the most prominent metabolic process in the symbiont (Fig. 6.6; Wang et al. 2021). The three particulate methane monooxygenases that conduct the first step of the reaction by converting methane to methanol were the most highly expressed protein-coding genes. Furthermore, two downstream pathways, the tetrahydromethanopterin (H4MPT) and ribulose monophosphate (RuMP) pathways, which detoxify formaldehyde and generate energy and biomass, were highly active in the symbiont.

An overview of the symbiont methane oxidation and downstream H4MPT and RuMP pathways. The histogram at the bottom shows the relative gene expression levels (log10TPM) of enzymes of these three pathways. Transcripts encoding enzymes in these three metabolic processes dominate the transcriptome of the G. platifrons symbiont. Reprinted from iScience, 24, Wang et al. (2021) Molecular analyses of the gill symbiosis of the bathymodiolin mussel Gigantidas platifrons,101, 894, Copyright (2022), with permission from Elsevier
Complex episymbionts have been observed in the setae of S. crosnieri. Methylococcales (42.3%) was the most abundant order, followed by Thiotrichales (14.8%), Campylobacteriales (4.9%), Flavobacteriales (4.4%), Chitinophagales (3.8%), and Nitrosococcales (2.2%). Metagenomic binning was carried out to further elucidate the taxonomic composition and functions of the symbionts. The MAGs clustered into five monophyletic clades in the orders Thiotrichales, Nitrosococcales, Methylococcales, Chitinophagales, and Flavobacteriales (Fig. 6.7a), which represented all of the dominant taxa recovered in the phyloFlash 16S ribosomal RNA (rRNA) gene-based analysis. Genome Taxonomy Database Toolkit (GTDB-Tk)-based taxonomic assessments indicated that many MAGs were divergent from known reference genomes, suggesting that they may represent unknown genera and even unknown families. The complex episymbiotic community can supply hosts with fixed organic carbons. One-carbon metabolic pathways, including methane oxidation, methanol oxidation, and the RuMP pathway, were the most notable functional categories encoded by the Methylococcales and Nitrosococcales MAGs. Methane oxidation was particularly prominent in the Methylococcales MAGs. Thiotrichales and Campylobacteriales can fix carbon dioxide via the reverse tricarboxylic acid (rTCA) and Calvin-Benson-Bassham (CBB) pathways, respectively, using electrons obtained by the oxidation of reduced sulfur.

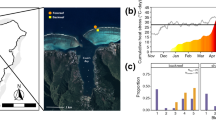
An overview of the symbiont’s composition and metabolic interactions with S. crosnieri. a Phylogenomic assignment, relative abundance, and metabolic potential of the dominant MAGs of the setae along with reference genomes. Phylogeny of 28 high-quality MAGs recovered from the S. crosnieri setae. The colors of the tree represent order-level taxonomic groups for MAGs. Heatmap colors represent the completeness of KEGG modules; b Schematic of the predicted overall community strategy. During the hypoxic phase, the symbionts oxidize methane and sulfide to methanol and sulfur globules by coupling to nitrate respiration. The stable sulfur and methane intermediates can then be stored as inclusions or released to the environment and taken up by other symbionts. During the oxic phase, episymbionts can use oxygen to oxidize sulfur and methanol to conserve additional energy to support cellular growth; c Temporal measurements of DO and methane concentrations inside the S. crosnieri community; d Confocal Raman microscopy imaging showing elemental sulfur in the episymbiont communities. Reprinted from mSystems, 7, Xu et al. (2022) Metabolism Interactions Promote the Overall Functioning of the Episymbiotic Chemosynthetic Community of Shinkaia crosnieri of Cold Seeps, e00320-22, Copyright (2022), with permission from ASM journals
6.4 Nutrient Transfer Between Host and Symbionts
Formations of chemosymbiotic associations, especially endosymbiosis, in the deep sea are mainly nutrient driven. Deep-sea chemosymbiotic invertebrates are suitable for studying metabolic interaction mechanisms due to their exclusive and robust association with chemosynthetic bacteria (Dubilier et al. 2008). With the application of new approaches such as single-cell spatial omics and subcellular imaging, the metabolic interaction mechanisms of these symbiotic systems in cold seeps and hydrothermal vents were revealed (Ponnudurai et al. 2017; Geier et al. 2020). Recently, a comprehensive picture of metabolic cooperation in G. platifrons symbiosis was reported with state-of-the-art single-cell transcriptome and spatial transcriptome sequencing (Chen et al. 2021). It has been demonstrated that deep-sea mussels have reshaped bacteriocyte metabolism remarkably to maximize symbiotic profits for both partners. For example, bacteriocytes encode dozens of genes involved in the biosynthesis and transport of carbohydrates, lipids, amino acids, and vitamins to improve the acquisition of nutrition from symbionts. Nevertheless, the exact metabolites transferred between the host and endosymbionts are less known. It is now widely recognized that methanotrophic endosymbionts can provide sterol intermediates to the mussel host, while endosymbiotic sulfur oxidizers may compensate for the host’s putative deficiency in amino acid and cofactor biosynthesis (Ponnudurai et al. 2017; Takishita et al. 2017). In turn, the host replenishes essential biosynthetic TCA cycle intermediates for their endosymbiont. Recently, it was demonstrated that bacteriocytes could retrieve fructose-6-phosphate (F6P) directly from methanotrophic symbionts by sugar phosphate exchanger genes and convert it into glucose-6-phosphate (G6P), fructose-1,6-bisphosphate (F-1,6-BP) and glyceraldehyde-3-phosphate (G6P) before redistributing them back to both the host and symbionts as supplements of gluconeogenesis and the tricarboxylic acid cycle (TCA cycle). A direct supply of ammonia from the host to symbionts via ammonium transporters was also reported (Chen et al. 2022). The bioconversion and exchange of sugar phosphate and ammonia between the host and symbionts provided an efficient and direct way to coordinate the metabolism of both partners, which greatly promoted the efficiency and profits of symbiosis.
In addition, metatranscriptome sequencing and gene coexpression network analysis of G. platifrons under laboratory maintenance with a gradual loss of symbionts also provided evidence of possible nutrient interactions between the two parts (Sun et al. 2022a). One-day short-term maintenance triggered global transcriptional perturbation in symbionts but little gene expression changes in mussel hosts; the genes that changed were mainly involved in responses to environmental changes (Fig. 6.8a). Long-term maintenance with depleted symbionts induced a metabolic shift in the mussel host. The most notable changes were the suppression of sterol biosynthesis and the complementary activation of terpenoid backbone synthesis in response to the reduction in bacteria-derived terpenoid sources. In addition, gene expression of the host proteasomes responsible for amino acid deprivation caused by symbiont depletion was significantly upregulated. Additionally, a significant correlation between host microtubule motor activity and symbiont abundance was revealed, suggesting the possible function of microtubule-based intracellular trafficking in the nutritional interaction of symbiosis (Fig. 6.8b). Overall, the dynamic transcriptomic changes of G. platifrons during the loss of symbionts highlighted the nutritional dependence of the host on terpenoid compounds and essential amino acids from their endosymbionts and proposed the possible important function of microtubule-based intracellular trafficking in symbiosis.

Schematic diagram summarizing the overall metabolic responses of G. platifrons holobionts to laboratory maintenance. a Transcriptional responses to one-day short-term maintenance. The biosynthesis of essential amino acids and sterol intermediates was downregulated in the endosymbionts after short-term maintenance. The DNA damage repair-related genes and immune-related genes of the mussel host were upregulated. CAD: Cis-aconitate decarboxylase, AP-1: Transcription factor AP-1, IRF2: Interferon regulatory Factor 2, ATM: Serine-protein kinase ATM, GAPR-1: Golgi-associated plant pathogenesis-related protein; b Transcriptional responses to long-term maintenance for 25 and 35 days. The biosynthesis of essential amino acids and sterol intermediates was continuously downregulated in symbionts, and nutrients were deficient due to symbiont depletion. Sterol biosynthesis in the mussel host was suppressed, and the biosynthesis of the terpenoid backbone and proteasomes was complementarily upregulated in response to the reduction in bacteria-derived nutrition sources. The microtubule-based movement and regulatory pathways for DNA replication and cell differentiation or proliferation were also downregulated. In contrast, genes related to protein synthesis and processing, mitochondrial oxidative phosphorylation, and cell redox homeostasis were upregulated. DYH: Dynein heavy chain, KIF: Kinesin-like protein, OST48: Dolichyl-diphosphooligosaccharide-protein glycosyltransferase 48 kDa subunit, HMGCR: 3-hydroxy-3-methylglutaryl-coenzyme a reductase, GST: Glutathione S-transferase, PLCG1: 1-phosphatidylinositol 4,5-bisphosphate phosphodiesterase gamma-1, PLD1: Phospholipase D1, VAV: Proto-oncogene vav. Green arrows indicate the downregulated genes or pathways. Red arrows indicate the upregulated genes or pathways. Reprinted from Molecular Ecology, Sun et al. (2022a, b) Insights into symbiotic interactions from metatranscriptome analysis of deep-sea mussel Gigantidas platifrons under long term laboratory maintenance, https://doi.org/10.1111/mec.16765, Copyright (2022), with permission from John Wiley and Sons
In S. crosnieri, the metabolic potentials of the dominant taxa have been reconstructed based on KEGG module completeness visualizations in Fig. 6.7a (Xu et al. 2022). The episymbionts exhibited the potential to provide most nutrients to their hosts, including hydrocarbons, vitamins, and essential amino acids. In return, invertebrate hosts can supply vitamin B6 to episymbionts, which cannot synthesize it on their own (Lan et al. 2021). The nutrient contributions to the host are also likely complementary. For example, the essential amino acid histidine could only be provided by some Campylobacteriales and Thiotrichales, and the steroid precursor lanosterol could only be synthesized by Methylococcales. The complex functionally complemented symbiotic community may be adaptive and selected by the host. The setae and symbionts can be observed in the intestinal tracts of S. crosnieri, and the symbionts can be digested by intestinal digestive enzymes (Watsuji et al. 2015). The results indicate that S. crosnieri ingests the epibionts using maxillipeds and assimilates them via its digestive organs as a nutrient source.
6.5 The Molecular Basis for Establishing and Maintaining Symbiosis
The establishment of endosymbiosis is undoubtedly the most important step in deep-sea adaptation (Stewart et al. 2005). Bathymodiolin species rely on symbiotic chemoautotrophic bacteria in their specialized gill epithelial cells, which are called bacteriocytes, for all or part of their nutrition as well as environmental adaptations. Bathymodiolin mussel symbiosis is a good model to study host-symbiont interactions, especially how symbionts are actively selected and maintained.
In deep-sea mussels, 1314 positively selected genes were mainly involved in cellular homeostasis, immune response, and transporters. The genes upregulated in the deep-sea mussels were mainly involved in the maintenance of cellular homeostasis, transport, and immune reactions. Based on these results, a conceptual scheme of the interactions of hosts and symbiosis is given (Fig. 6.9). One of the most extraordinary traits of bathymodiolin mussels is their endosymbiosis. Pattern recognition receptors (PRRs) appear to be different in both organisms, and their expression levels also differ between deep-sea and shallow-water mussels. This polymorphism might somehow generate different immune functions and pathogen resistance to support individual adaptation. In summary, chemosynthetic deep-sea mussels might have evolved with reduced PRR patterns and some specific PRRs (e.g., Toll-like receptors, TLR and Complement C1q domain containing, C1qDC proteins) to allow symbiont entry and to successfully maintain them in bacteriocytes. Lineage-specific and positively selected TLRs and highly expressed C1qDC proteins were identified in the gills of the bathymodiolins, suggesting their possible functions in symbiont recognition. However, the PRRs of the bathymodiolins were globally reduced, facilitating the invasion and maintenance of the symbionts obtained by either endocytosis or phagocytosis. Additionally, various transporters were positively selected or more highly expressed in the deep-sea mussels, indicating a means by which necessary materials could be provided for the symbionts. Key genes supporting lysosomal activity were also positively selected or more highly expressed in the deep-sea mussels, suggesting that nutrients fixed by the symbionts can be absorbed in a “farming” way when the symbionts are digested by lysosomes (Zheng et al. 2017). Regulation of key physiological processes, including lysosome activity, apoptosis, and immune reactions, is needed to maintain a stable host-symbiont relationship, but the mechanisms are still unclear.

A conceptual scheme of the host–symbiont interactions in the bacteriocyte. The symbionts are located inside vacuoles (green) surrounded by the bacteriocyte cytosol (blue). An overview of the major processes discussed in this article is shown. The underlined bold indicates the orthologs that are positively selected in deep-sea mussels; the red up arrow indicates the orthologs that are expressed consistently more highly in deep-sea mussels; the blue down arrow indicates the orthologs that are expressed consistently more highly in shallow-sea mussels; the dots inside lysosomes indicate lysosomal acid hydrolase, some of which are positively selected and possess higher expression levels in deep-sea mussels (e.g., cathepsins). Key genes for the regulation of lysosome activities (mTOR and TFEB) are neither positively selected nor highly expressed in deep-sea mussels and are colored gray. TLRs: Toll-like receptors; SvRs: scavenger receptors; SLCs: solute carrier family; ABCs: ATP-binding cassette transporters; SOD: manganese superoxide dismutase; GST: glutathione S-transferase; CYPs: cytochrome P450s; ATGs: autophagy-related proteins; M6P: mannose-6-phosphate; M6PR: cation-independent mannose-6-phosphate receptor-like; ATPeV: vacuolar H + -ATPase; VPSs: vacuolar protein sorting proteins; VAMPs: vesicle-associated membrane proteins; SNAPs: synaptosome-associated proteins; HGSNAT: heparan-α glucosaminide N-acetyltransferase; mTOR: mammalian target of rapamycin; TFEB: transcription factor enriched bacteriocytes. Reprinted from Molecular Ecology, 26, Zheng et al. (2017) Insights into deep-sea adaptations and host symbiont interactions: A comparative transcriptome study on Bathymodiolus mussels and their coastal relatives, 5133–5148, Copyright (2022), with permission from John Willey and Sons
Transcriptomic dynamic analysis during desymbiosis and symbiosis reestablishment was adopted to further depict the possible processes by which the host selects and maintains the specified symbionts. Healthy G. platifrons with dense endogenous symbionts were collected from a cold seep in the SCS and then “desymbionted” by maintaining specimens in an atmospheric pressure lab cultivation system. Furthermore, these conditioned mussels were challenged by either symbionts or environmental bacteria to determine the different host-bacteria recognition mechanisms induced by environmental bacteria or symbionts (Wang et al. 2019). As shown in Fig. 6.10a, the analysis of DEGs between untreated and symbiont-loss samples revealed that metabolic and cellular organizational changes in the gill were associated with increased ribosomal activities, ubiquitin‒proteasome systems, and autophagy. Additionally, the differentially expressed immune genes suggest that the host recognizes and interacts with endosymbionts through PRRs, especially Toll-like receptors. In addition, the DEGs between symbiont-treated and environmental bacteria-treated samples shed light on the mechanism of symbiont recognition (Fig. 6.10b). Symbiont treatment not only partially reversed the transcriptomic changes caused by symbiont loss but also suppressed the host immune system, which might facilitate symbiont entrance into and survival within host bacteriocytes. Taken together, these results suggest that the host-symbiont system in G. platifrons is tightly regulated but also has plasticity to fit environmental fluctuations.

A conceptual scheme of the host-symbiont interaction. a comparison of a, regular bacteriocytes (0Day samples); and in b the symbiont-loss bacteriocytes (integration of 7Day and 12Day_Blank samples). c A proposed model for the different host-bacteria recognition mechanisms induced by environmental bacteria or symbionts. In the environmental bacteria-treated samples, the PRRs were upregulated. The environmental bacteria were first recognized by PRRs and then engulfed by phagocytosis. The internalized bacteria were then eliminated by antibacterial proteins such as C-type lectins, lysozymes, and big defensin. In the symbiont-treated samples, the expression of phagocytosis-related PRRs, such as low density lipoprotein receptors (LDLPR), c-type lectin-like containing (CTLDC), C1qDCs, and mannose receptors, was suppressed. Bacterial permeabilization inducing protein (BPI) and TLRs bind to microbe-associated molecular patterns (MAMP) on symbionts and then induce endocytosis. The internalized symbionts were recognized by TLR13. Reprinted from Deep-Sea Research Part I, 151, Wang et al. (2019) Comparative transcriptomic analysis 624 illuminates the host-symbiont interactions in the deep-sea mussel Bathymodiolus platifrons, 103,082, Copyright (2022), with permission from Elsevier
In the environmental bacteria-treated samples, the PRRs were upregulated. The environmental bacteria were first recognized by PRRs and then engulfed by phagocytosis. The internalized bacteria were then eliminated by antibacterial proteins such as C-type lectins. In the symbiont-treated samples, the expression of phagocytosis-related PRRs, such as low-density lipoprotein receptor (LDLPR) and mannose receptors, was suppressed. Bacterial permeabilization inducing (BPI) protein and TLRs bind to microbe-associated molecular patterns (MAMPs) on symbionts and then induce endocytosis (Fig. 6.10c).
PRRs in the host play irreplaceable roles in the immune recognition of symbionts. To further decode the immune recognition of symbiosis, several PRR proteins that may interact with methanotrophic symbionts were studied to understand their molecular characteristics and expression patterns (Chen et al. 2019; Li et al. 2020, 2021). Six candidate PRRs, including leucine-rich-repeat-domain-containing protein, TLR13, TLR2, integrin, vacuolar sorting protein and matrix metalloproteinase 1, were selected by methane-oxidizing bacteria or lipopolysaccharide pull-down assays, suggesting their key role in symbiont recognition. The key roles of the PRRs were validated by FISH and immunoblotting. Combined with their expression patterns in bacteriocytes, six PRR proteins were shown to be involved in the recognition of symbionts during the establishment of symbiosis between G. platifrons mussels and methane-oxidizing bacteria.
Bacteriocytes are the core units for symbiosis in G. platifrons. However, the homeostasis of the system depends on the cooperation of various cells of the symbiotic tissue, the gill. The gill of the bathymodiolin mussel is a complicated tissue that executes other basic physiological functions, such as respiration, transportation, growth, and immune defense (Neumann and Kappes 2003; Nguyen et al. 2019). The bathymodiolin gill includes a variety of cell types. Beyond its bacteriocytes, the bathymodiolin gill filament has several nonsymbiotic components: the tip and edge of each gill filament are covered with cilia cells and mucous cells, and on the lateral face of the gill filament, numerous intercalary cells fill the intercellular spaces among the bacteriocytes (Fiala-Médioni et al. 1986; Fujiwara et al. 2000; Barry et al. 2002). These accumulating data demonstrate that spatial expression information for genes is crucial for understanding symbiosis. However, because transcriptomic studies use whole-gill tissue homogenate as research material, valuable spatial information is lost. To overcome this drawback, G. platifrons bacteriocytes were enriched for RNA-seq studies (Wang et al. 2021). In comparative analyses of genes enriched in the bacteriocytes and whole gill and in metatranscriptomic analyses of the symbiont, an integrated view of the molecular mechanisms that directly govern symbiosis in G. platifrons was proposed. This breakdown of the complex symbiotic tissue allowed us to characterize the host-symbiont interactions further. GO analyses of the DEGs suggest the presence of transport activities between the bacteriocytes and nonsymbiotic cells. Contrary to expectations, 6 of the top 10 enhanced GO terms in the enriched bacteriocyte samples were involved in microtubule activities. Many of the DEGs in these GO terms encode a variety of tubulins. These results suggest that microtubule activities are significantly enhanced in bacteriocytes. The whole gill samples with enriched GO terms align well with the nonsymbiotic part of the cellular activities of the gill. The genes enriched in “Small molecule transport” and “Transporter activity” indicate that the gill’s nonsymbiotic components actively shuffle or exchange nutrients and metabolites with the bacteriocytes. In addition, several GO terms that were related to DNA replication initiation, including genes involved in controlling cell proliferation, such as DNA replication licensing factors, proliferating cell nuclear antigen, and eukaryotic translation initiation factor, were enriched in whole gill samples. Finally, two terms related to the extracellular matrix, “Extracellular space” and “Extracellular region,” were significantly enriched in whole gill samples, which correlate with the gill’s strong secretion activities. All of the above data showed that the gill’s nonsymbiotic parts play crucial roles in maintaining and protecting the symbiosis; the bacteriocytes supply the symbiont with metabolites, control the symbiont population, and shelter the symbiont from phage infection, and the symbiont contributes products from methane oxidation and energy production.
6.6 Adaptation of Deep-Sea Symbiotic Invertebrates to the Harsh Environment
Due to underground fluid seepage, chemosynthetic ecosystems, including cold seeps and hydrothermal vents, are sometimes hostile to megafauna, as the concentrations of hydrogen sulfide or heavy metals are usually high (Zhou et al. 2020, 2021). Among the deep-sea symbiotic invertebrates, deep-sea bathymodiolin mussels are ubiquitous in most cold seeps and hydrothermal fields, where they can accumulate toxic materials, including metals, and thus could serve as an ideal model to investigate the toxicological responses of deep-sea organisms to metal exposure.
Similar to coastal mussels, deep-sea mussels have adopted similar antioxidant defense mechanisms to detoxify metals. A field investigation of metal concentrations and antioxidant enzymes in deep-sea bathymodiolin mussels from four different deep-sea geochemical settings showed that deep-sea mussel gills generally exhibited higher metal enrichment than the mantle. Mussels from hydrothermal vents usually had higher metal concentrations (Fe, Cr, Cd, and Pb) than those from cold seeps, which could be related to their higher contents in fluids or sediments. However, despite quite different metal loads among the geochemical environment settings, Mn, Zn, and Cu concentrations varied over a smaller range across the sampling sites, implying biological regulation of these elements by deep-sea mussels. Although the vent ecosystem is harsher than the cold seep ecosystem, antioxidant enzyme concentrations in deep-sea mussels were not so different, suggesting that some adaptive or compensatory mechanisms may occur in chronically polluted deep-sea mussels. Principal component analysis allowed for distinguishing different deep-sea settings, indicating that bathymodiolin mussels are robust indicators of their living environments (Fig. 6.11a). To further understand the molecular mechanisms of deep-sea mussels in response to metal exposure, indoor metal exposure experimental studies with deep-sea mussels were conducted. The results showed that metal was able to induce general cellular injury, oxidative stress, and disturbances in the metabolism of amino acids, carbohydrates, and lipids. In addition, different metals activated different molecular responses in deep-sea mussels (Fig. 6.11b). Specifically, the monosaccharide D-allose, which is involved in suppressing mitochondrial reactive oxygen species production, was uniquely downregulated in deep-sea mussels under Cd exposure. Alterations in dopamine, as well as dopamine-related and serotonin-related metabolites, in deep-sea mussels under Cu exposure were found, pointing to perturbation of neurotransmission (Fig. 6.11c).

a Principal component analysis (PCA) ordination plots of metal accumulation and metal biomarker levels in B. mussels from four different deep-sea geothermal sites sampled (CS: one cold seep from the South China Sea; HV1: hydrothermal vent 1 from the Okinawa Trough; HV2: hydrothermal vent 1 from the Okinawa Trough; MN: one hydrothermal vent from Manus). Reprinted from Science of The Total Environment, 707, Zhou et al. (2020) Metal adaptation strategies of deep-sea Bathymodiolus mussels from a cold seep and three hydrothermal vents in the West Pacific,136,046, Copyright (2022), with permission from Elsevier; b Venn diagrams of differentially accumulated metabolites, revealing those commonly or uniquely regulated metabolites occurring in G. platifrons under 7 days of exposure to Cu, Cd, or their mixture; c The top ten metabolic pathways that were significantly affected by exposure of G. platifrons to Cu, Cd. Figure b and c was reprinted from Aquatic Toxicology, 236, Zhou et al. (2021) Biochemical and metabolic responses of the deep sea mussel Bathymodiolus platifrons to cadmium and copper exposure, 105,845, Copyright (2022), with permission from Elsevier
Hydrogen sulfide (H2S) is another highly toxic molecule for animals living in deep-sea environments because it can bind tightly to cytochrome c oxidase (COX) and block mitochondrial oxidative phosphorylation (Kelley et al. 2016). In G. platifrons, the sulfide detoxification and adaptative strategy were detected in both the symbiotic bacterial communities and the host mussels through the comparison of G. platifrons inhabiting hydrothermal vent and methane seep environments with distinct sulfide concentrations. First, symbiotic bacteria, including epibionts and endosymbionts, play a role in sulfide detoxification. As indicated in the analysis of 16S rRNA locus sequencing and metatranscriptome sequencing of G. platifrons collected from a Site F cold methane seep in the SCS and a hydrothermal vent at Iheya North Knoll in the Mid-Okinawa Trough, the composition of bacterial communities in the gills of the vent and seep mussels was different (Sun et al. 2022b). Methane-oxidizing bacteria, endosymbionts belonging to Methyloprofundus (Hirayama et al. 2022), were the most abundant species across all samples. The second most abundant taxon belonged to Campylobacteria, a common sulfur-oxidizing episymbiont bacteria in bathymodiolin mussels (Assié et al. 2016, 2020). Moreover, the relative abundance of Campylobacterota episymbionts was higher in hydrothermal vent mussels (with an average of 39.2%) than in methane seep mussels (with an average of 18.5%). Metatranscriptome sequencing was conducted for each of the three replicates of hydrothermal vent and methane seep mussels, and functional enrichment analysis of the differentially expressed genes (DEGs) in bacterial symbionts revealed that the most significantly enriched pathway was the sulfur metabolism pathway (ko00920, adjusted P value = 1.99E − 10). Notably, a key sulfide-oxidizing gene, sulfide:quinone oxidoreductase (sqr), derived from the methanotrophic endosymbiont, was significantly upregulated in vent mussels, indicating the oxidization of intracellular sulfide by the endosymbiont. In the mussel host, genes participating in mitochondrial oxidative phosphorylation and energy generation, as well as sulfur metabolism, were upregulated in the vent mussels. In summary, the possible adaptations of G. platifrons holobionts for sulfide tolerance and protection were hypothesized (Fig. 6.12): (1) exclusion of environmental sulfide by the presence of abundant sulfur-oxidizing epibiotic bacteria colonizing the surfaces of gill filaments with direct contact with the external reduced environment and forming the front line against the potentially toxic environment; (2) oxidization of the intracellular sulfide in methanotrophic endosymbionts aggregated at the apex of the gill bacteriocytes using the sqr gene, processing additional electron and energy sources to their mussel host, and improved fitness to environmental adaptation; and (3) rapid detoxification through the mitochondrial sulfide oxidation pathway of the mussel host, which oxidizes sulfide to less toxic sulfur compounds and simultaneously provides electrons to mitochondrial oxidative phosphorylation and drives ATP synthesis.

Schematic adaptative strategies of sulfide detoxification hypothesized in the G. platifrons holobiont. The epibionts, endosymbionts, and mussel hosts collaborate on sulfide detoxification from the extracellular to the intracellular space. The extracellular epibiotic bacteria oxidize the environmental sulfide through Sqr and Fcc while simultaneously oxidizing other reduced sulfur compounds (S0 and S2O32−) by the Dsr-Apr-Sat and Sox enzyme system. Methanotrophic endosymbionts are densely aggregated at the apical top of gill bacteriocytes and protect cells by oxidization of intracellular sulfide through the sqr gene. Intracellular sulfide is also oxidized by the mitochondrial sulfide oxidation pathway in the mussel host. Symbols represent bacterial genes involved in the oxidization of reduced sulfide compounds: Sqr: sulfide:quinone reductase; Fcc: sulfide dehydrogenase (flavocytochrome c); Dsr: dissimilatory sulfite reductase; Apr: adenosine phosphosulfate reductase; Sat: sulfate adenylyltransferase; Sox: sox multiple-enzyme system soxABCXYZ. Symbols represent genes in the mitochondrial sulfide oxidation pathway: SQR: sulfide quinone oxidoreductase; PDO: persulfide dioxygenase; TST: thiosulfate sulfurtransferase; SO: sulfite oxidase. Note: In the diagram, the sulfide oxidization process in the epibiont represents genes and pathways found in the sulfur-oxidizing Campylobacterota and Gammaproteobacteria communities instead of a single species of bacteria. Reprinted from Science of the Total Environment, 804, Sun et al. (2022a, b) Adaption to hydrogen sulfide-rich environments: Strategies for active detoxification in deep-sea symbiotic mussels, Gigantidas platifrons, 150,054, Copyright (2022), with permission from Elsevier
The mobile squat lobster S. crosnieri may adopt different strategies to cope with abiotic environmental stress. Environmental conditions are unstable in cold seeps due to temporal variation in reduced compounds that are released from bottom currents (Fig. 6.7c) that could further shape regional chemical spatial distributions in the subseafloor (Girard et al. 2020). Methane concentrations were negatively correlated with oxygen concentrations (Cao et al. 2021). However, lobster symbionts require oxygen and reduced substrates such as methane to efficiently fix carbon, which may be simultaneously unavailable in their natural habitats. Thus, different strategies could be used by chemosynthetic symbionts to solve this paradox and achieve more efficient energy conservation. For example, episymbionts can conserve energy through the oxidation of diverse electron donors (sulfide, thiosulfate, sulfur, methane, ammonia, and hydrogen) coupled with oxygen or nitrate respiration, thereby allowing the cells to utilize adaptations to both oxic and hypoxic environments (Nakagawa and Takai 2008). Sulfur globules were observed in our samples (Fig. 6.7d). Although methanol was not directly quantified in these samples, it is reasonable to speculate that methanol accumulation occurred in the MOBs since pmoABC was expressed at 20-fold higher levels than lanthanide-containing methanol dehydrogenases (xoxF) in hypoxic conditions. In addition, globin-like proteins containing heme domains that may sense O2 were encoded by many of the symbiont MAGs and could facilitate their dynamic responses to fluctuating oxygen environments. Here, a conceptual interactive model was proposed (Fig. 6.7b). During the hypoxic phase, the symbionts oxidize methane and sulfide to methanol and sulfur globules by coupling to nitrate respiration. The stable sulfur and methane intermediates could then be stored as inclusions (Zbinden et al. 2008) or released to the environment and taken up by other symbionts (van Grinsven et al. 2021). During the oxic phase, episymbionts can use oxygen to oxidize sulfur and methanol to conserve additional energy to support cellular growth. Hence, the stable intermediates likely play essential roles in metabolic interactions and adaptations to fluctuating environments that commonly occur at vents and seeps. The cooperation of episymbiotic communities could lead to greater adaptability to chemosynthetic ecosystems, thereby providing greater levels of energy supplies to invertebrate hosts.
6.7 Summary and Perspectives
Deep-sea chemosynthetic symbiotic animals represent a unique model for understanding the interaction between multicellular metazoan and unicellular microorganisms (Dubilier et al. 2008; Moya et al. 2008; Sogin et al. 2021). Several model deep-sea chemosynthetic symbiotic animals, such as squat lobsters, vestimentiferan tubeworms and bathymodiolin mussels, have been intensively studied in the past few decades. Through detailed morphological characterization, ultrastructural imagery analyses, and molecular cloning and characterization, in recent years, with the rapid development of large-scale “OMICs” platforms and computational power, including next-generation direct RNA-seq transcriptomic analysis, large-scale proteomics profiling, and whole-genome sequencing and assembly techniques, the detailed molecular mechanisms governing host-symbiont interactions have been gradually revealed. We now understand that host animals have evolved a sophisticated molecular cascade to communicate and interact with the symbiont. The host also utilizes cellular mechanisms, such as its immune pathways and lysosomes, to recognize, digest and harvest symbionts to harvest nutrients for its growth.
Nevertheless, several fundamental questions in deep-sea symbiosis still need to be answered. For example, it is still largely unknown how host animals recognize symbionts and establish intracellular symbiosis at early developmental stages. How adult host animals produce new bacteriocytes and recruit symbionts is also unclear. Furthermore, at the subcellular scale, how the host senses and regulates the symbiont population, what molecules are used for host-symbiont communication, and what types of metabolites are shared between the symbiont and host still need further investigation. Hopefully, with emerging new technologies, such as deep-sea sampling, single-cell transcriptomics and proteomics sequencing, and high-resolution mass spectrometry, these intriguing scientific questions will be answered in the future.
Symbiosis with chemoautotrophic bacteria was discovered over 40 years ago, and it is now recognized as an important milestone event that shaped the diversity and evolution of deep-sea invertebrates such as sponges, nematodes, crustaceans, annelids, and mollusks. The cold seeps in the SCS provide us with a natural laboratory to understand the sophisticated processes between invertebrates and symbionts. Future studies should focus on discovery-driven expeditions to reveal the full biodiversity of chemosymbiotic associations and their ecological, as well as mechanistic, research to uncover the biochemical, physiological, and molecular mechanisms that govern interactions between symbiotic partners. Revolutionary changes are needed to understand deep-sea organisms. Deep-sea in situ research is undoubtedly the future direction. Moreover, the emerging microscale omics methodology offers us new research opportunities. More works based on experimental verification, such as in vitro culture of symbionts, gene function verification and manipulation of gene expression, should be attempted to better reveal these metabolic interaction mechanisms and confirm similarities and differences between various symbiotic systems.
References
Assié A, Borowski C, van der Heijden K et al (2016) A specific and widespread association between deep-sea Bathymodiolus mussels and a novel family of Epsilonproteobacteria. Environ Microbiol Rep 8(5):805–813
Assié A, Leisch N, Meier DV et al (2020) Horizontal acquisition of a patchwork Calvin cycle by symbiotic and free-living Campylobacterota (formerly Epsilonproteobacteria). ISME J 14(1):104–122
Barry JP, Buck KR, Kochevar RK et al (2002) Methane-based symbiosis in a mussel, Bathymodiolus platifrons, from cold seeps in Sagami Bay. Japan. Invertebr Biol 121(1):47–54
Boetius A, Wenzhöfer F (2013) Seafloor oxygen consumption fuelled by methane from cold seeps. Nat Geosci 6(9):725–734
Cao L, Lian C, Zhang X et al (2021) In situ detection of the fine scale heterogeneity of active cold seep environment of the Formosa Ridge, the South China Sea. J Mar Syst 218:103530
Ceramicola S, Dupré S, Somoza L et al (2018) Cold seep systems. In: Micallef A, Krastel S, Savini A (eds) Submarine Geomorphology. Springer International Publishing, Chem, pp 367–387
Chen H, Wang M, Zhang H et al (2019) An LRR-domain containing protein identified in Bathymodiolus platifrons serves as intracellular recognition receptor for the endosymbiotic methane-oxidation bacteria. Fish Shellfish Immunol 93:354–360
Chen H, Wang M, Zhang H et al (2021) microRNAs facilitate comprehensive responses of Bathymodiolinae mussel against symbiotic and nonsymbiotic bacteria stimulation. Fish Shellfish Immunol 119:420–431
Chen H, Li M, Wang M et al (2022) Single-cell perspectives on the function and development of deep-sea mussel bacteriocytes. bioRxiv https://doi.org/10.1101/2022.05.28.493830
Dong D, Li X (2015) Galatheid and chirostylid crustaceans (Decapoda: Anomura) from a cold seep environment in the northeastern South China Sea. Zootaxa 4057(1):91–105
Dubilier N, Bergin C, Lott C (2008) Symbiotic diversity in marine animals: the art of harnessing chemosynthesis. Nat Rev Microbiol 6(10):725–740
Duperron S (2010) The diversity of deep-sea mussels and their bacterial symbioses. In: Kiel S (ed) The vent and seep biota: aspects from microbes to ecosystems. Springer, Netherlands, Dordrecht, pp 137–167
Duperron S, Quiles A, Szafranski KM et al (2016) Estimating Symbiont Abundances and Gill Surface Areas in Specimens of the Hydrothermal Vent Mussel Bathymodiolus puteoserpentis Maintained in Pressure Vessels. Front Mar Sci 3:1–12
Fiala-Médioni A, Métivier C, Herry A et al (1986) Ultrastructure of the gill of the hydrothermal-vent mytilid Bathymodiolus sp. Mar Biol 92:65–72
Fisher CR, Childress JJ (1986) Translocation of fixed carbon from symbiotic bacteria to host tissues in the gutless bivalve Solemya reidi. Mar Biol 93:59–68
Fujiwara Y, Takai K, Uematsu K et al (2000) Phylogenetic characterization of endosymbionts in three hydrothermal vent mussels: influence on host distributions. Mar Ecol-Prog Ser 208:147–155
Geier B, Sogin EM, Michellod D et al (2020) Spatial metabolomics of in situ host-microbe interactions at the micrometre scale. Nat Microbiol 5(3):498–510
Girard F, Sarrazin J, Olu K (2020) Impacts of an eruption on cold-seep microbial and Faunal Dynamics at a Mud Volcano. Front Mar Sci 7:241. https://doi.org/10.3389/fmars.2020.00241
Gomez-Mendikute A, Elizondo M, Venier P et al (2005) Characterization of mussel gill cells in vivo and in vitro. Cell Tissue Res 321(1):131–140
Halary S, Riou V, Gaill F et al (2008) 3D FISH for the quantification of methane- and sulphur-oxidizing endosymbionts in bacteriocytes of the hydrothermal vent mussel Bathymodiolus azoricus. ISME J 2(3):284–292
Hinzke T, Kleiner M, Meister M et al (2021) Bacterial symbiont subpopulations have different roles in a deep-sea symbiosis. Elife 10:e58371
Hinzke T, Kleiner M, Breusing C et al (2019). Host-microbe interactions in the Chemosynthetic Riftia pachyptila Symbiosis. mBio 10(6):e02243–19
Hirayama H, Takaki Y, Abe M et al (2022) Multispecies populations of methanotrophic methyloprofundus and cultivation of a likely dominant species from the Iheya north deep-sea hydrothermal field. Appl Environ Microbiol 88(2):e0075821
Hirokawa N, Noda Y (2008) Intracellular transport and kinesin superfamily proteins, KIFs: structure, function, and dynamics. Physiol Rev 88(3):1089–1118
Husnik F, Tashyreva D, Boscaro V et al (2021) Bacterial and archaeal symbioses with protists. Curr Biol 31(13):R862–R877
Kelley JL, Arias-Rodriguez L, Martin DP et al (2016) Mechanisms underlying adaptation to life in hydrogen sulfide-rich environments. Mol Biol Evol 33(6):1419–1434
Kennicutt MC, Brooks JM, Bidigare RR et al (1985) Vent-type taxa in a hydrocarbon seep region on the Louisiana slope. Nature 317:351–353
Lan Y, Sun J, Chen C et al (2021) Hologenome analysis reveals dual symbiosis in the deep-sea hydrothermal vent snail Gigantopelta aegis. Nat Commun 12(1):1165
Li M, Chen H, Wang M et al (2020) Identification and characterization of endosymbiosis-related immune genes in deep-sea mussels Gigantidas platifrons. J Oceanol Limnol 38(4):1292–1303
Li M, Chen H, Wang M et al (2021) A Toll-like receptor identified in Gigantidas platifrons and its potential role in the immune recognition of endosymbiotic methane oxidation bacteria. PeerJ 9:e11282
Lonsdale P (1979) A deep-sea hydrothermal site on a strike-slip fault. Nature 281:531–534
Moya A, Peretó J, Gil R et al (2008) Learning how to live together: genomic insights into prokaryote–animal symbioses. Nat Rev Genet 9(3):218–229
Nakagawa S, Takai K (2008) Deep-sea vent chemoautotrophs: diversity, biochemistry and ecological significance. FEMS Microbiol Ecol 65(1):1–14
Neumann D, Kappes H (2003) On the growth of bivalve gills initiated from a Lobule-Producing Budding Zone. Biol Bull 205(1):73–82
Nguyen TTK, Tran HV, Vu TT et al (2019) Peptide-modified electrolyte-gated organic field effect transistor: application to Cu2+ detection. Biosens Bioelectron 127:118–125
Paull CK, Hecker B, Commeau R et al (1984) Biological communities at the florida Escarpment Resemble Hydrothermal Vent Taxa. Science 226(4677):965–967
Polz MF, Cavanaugh CM (1995) Dominance of one bacterial phylotype at a Mid-Atlantic Ridge hydrothermal vent site. Proc Natl Acad Sci U S A 92(16):7232–7236
Ponnudurai R, Kleiner M, Sayavedra L et al (2017) Metabolic and physiological interdependencies in the Bathymodiolus azoricus symbiosis. ISME J 11(2):463–477
Renoz F, Noel C, Errachid A et al (2015) Infection dynamic of symbiotic bacteria in the pea aphid Acyrthosiphon pisum gut and host immune response at the early steps in the infection process. PLoS ONE 10(3):e0122099
Sogin EM, Kleiner M, Borowski C et al (2021) Life in the dark: phylogenetic and physiological diversity of chemosynthetic symbioses. Annu Rev Microbiol 75:695–718
Stewart FJ, Newton ILG, Cavanaugh CM (2005) Chemosynthetic endosymbioses: adaptations to oxic–anoxic interfaces. Trends Microbiol 13(9):439–448
Stokke R, Dahle H, Roalkvam I et al (2015) Functional interactions among filamentous Epsilonproteobacteria and Bacteroidetes in a deep-sea hydrothermal vent biofilm. Environ Microbiol 17(10):4063–4077
Sun Y, Wang M, Chen H et al (2022a) Insights into symbiotic interactions from metatranscriptome analysis of deep-sea mussel Gigantidas platifrons under long-term laboratory maintenance. Mol Ecol. https://doi.org/10.1111/mec.16765
Sun Y, Wang M, Zhong Z et al (2022b) Adaption to hydrogen sulfide-rich environments: Strategies for active detoxification in deep-sea symbiotic mussels, Gigantidas platifrons. Sci Total Environ 804:150054
Takishita K, Takaki Y, Chikaraishi Y et al (2017) Genomic Evidence that Methanotrophic Endosymbionts Likely Provide Deep-Sea Bathymodiolus Mussels with a Sterol Intermediate in Cholesterol Biosynthesis. Genome Biol Evol 9(5):1148–1160
Thurber AR, Jones WJ, Schnabel K (2011) Dancing for food in the deep sea: bacterial farming by a new species of yeti crab. PLoS ONE 6(11):e26243
van Grinsven S, Damste JSS, Harrison J et al (2021) Nitrate promotes the transfer of methane-derived carbon from the methanotroph Methylobacter sp. to the methylotroph Methylotenera sp. in eutrophic lake water. Limnol Oceanogr 66(3):878–891
Wang H, Zhang H, Wang M et al (2019) Comparative transcriptomic analysis illuminates the host-symbiont interactions in the deep-sea mussel Bathymodiolus platifrons. Deep-Sea Res Part I-Oceanogr Res Pap 151:103082
Wang H, Zhang H, Zhong Z et al (2021) Molecular analyses of the gill symbiosis of the bathymodiolin mussel Gigantidas platifrons. iScience 24(1):101894
Watsuji TO, Nakagawa S, Tsuchida S et al (2010) Diversity and function of epibiotic microbial communities on the galatheid crab Shinkaia Crosnieri. Microbes Environ 25(4):288–294
Watsuji T-o, Yamamoto A, Motoki K et al (2015) Molecular evidence of digestion and absorption of epibiotic bacterial community by deep-sea crab Shinkaia crosnieri. ISME J 9(4):821–831
Xu Z, Wang M, Zhang H et al (2022) Metabolism Interactions Promote the Overall Functioning of the Episymbiotic Chemosynthetic Community of Shinkaia crosnieri of Cold Seeps. mSystems 7(4): e00320–22
Zbinden M, Shillito B, Le Bris N et al (2008) New insigths on the metabolic diversity among the epibiotic microbial communitiy of the hydrothermal shrimp Rimicaris exoculata. J Exp Mar Biol Ecol 359(2):131–140
Zheng P, Wang M, Li C et al (2017) Insights into deep-sea adaptations and host-symbiont interactions: A comparative transcriptome study on Bathymodiolus mussels and their coastal relatives. Mol Ecol 26(19):5133–5148
Zhou L, Cao L, Wang X et al (2020) Metal adaptation strategies of deep-sea Bathymodiolus mussels from a cold seep and three hydrothermal vents in the West Pacific. Sci Total Environ 707:136046
Zhou L, Li M, Zhong Z et al (2021) Biochemical and metabolic responses of the deep-sea mussel Bathymodiolus platifrons to cadmium and copper exposure. Aquat Toxicol 236:105845
Acknowledgements
Funding was provided by the NSFC (Grants: 42,030,407 and 42,076,091). We are grateful to the captain and crew of the research vessel (R/V) Kexue, as well as the ROV Faxian for assistance with sample collection. Li Mengna and Ye Ziyun are thanked for her assistance with paper reviewing, which has greatly improved the quality of the chapter.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2023 The Author(s)
About this chapter

Cite this chapter
Li, C. et al. (2023). Symbioses from Cold Seeps. In: Chen, D., Feng, D. (eds) South China Sea Seeps. Springer, Singapore. https://doi.org/10.1007/978-981-99-1494-4_6
Download citation
DOI: https://doi.org/10.1007/978-981-99-1494-4_6
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-99-1493-7
Online ISBN: 978-981-99-1494-4
eBook Packages: Earth and Environmental ScienceEarth and Environmental Science (R0)