
Abstract
Neurodegenerative disorders such as Huntington’ disease and frontotemporal lobar degeneration (FTLD) are often caused by aggregation of causative proteins and co-morbid with mental disorders of unknown origin. Although toxic protein aggregates may entrap proteins involved in psychiatric disorders and elicit deleterious effects, candidate mechanisms for this co-aggregation hypothesis remain unknown. Here we show a novel cytosolic protein aggregate in FTLD brain composed of DISC1, a biological mediator for mental illnesses and TDP-43, the major protein constituent of insoluble inclusions in FTLD. FTLD model mice with DISC1/TDP-43 co-aggregates in frontal cortex showed hyperactivity and social interaction deficits that were rescued by exogenous DISC1 expression. At the cellular level, the amounts of newly synthesized proteins in dendrites are reduced in the neurons that lack DISC1 expression or contain DISC1/TDP-43 co-aggregates. These results support our novel hypothesis that co-aggregation of risk factors for psychiatric diseases may compromise local translation in dendrites and mental conditions.
You have full access to this open access chapter, Download conference paper PDF
Similar content being viewed by others


Keywords
1 Introduction
Neurodegenerative disorders are associated with a wide spectrum of behavioral abnormalities including sensory, motor, memory and mental deficits (Orr and Zoghbi 2007). The neurodegenerative disease, frontotemporal lobar degeneration (FTLD), is characterized by neuronal atrophy in frontal and temporal brain lobes associated with marked deficits in cognitive behavior (Rascovsky et al. 2011; Irish et al. 2012). In FTLD brain, TAR DNA-binding protein 43 (TDP-43) is a major protein component of ubiquitin-positive hyper-phosphorylated inclusions (Neumann et al. 2006), suggesting that its aggregation is implicated in FTLD pathogenesis (Cohen et al. 2011; Ling et al. 2013). TDP-43 is a heterogeneous ribonucleoprotein containing two highly conserved RNA recognition motifs that are believed to selectively regulate RNA processing, such as exon splicing, mRNA transport and microRNA biogenesis (Lee et al. 2012) In neurons, TDP-43 is a constituent of neuronal RNA granules in dendrites (Lee et al. 2012; Wang et al. 2008) where it interacts with a complex of RNA binding proteins including fragile X mental-retardation protein (FMRP) and Staufen1, which collectively regulate mRNA transport and local translation (Wang et al. 2008), the processes that are critical for synaptic function and plasticity (Bramham and Wells 2007; Holt and Schuman 2013; Buffington et al. 2014). Considerable progress has been made to understand the molecular mechanism of neurodegeneration caused by TDP-43 aggregation (Baralle et al. 2013). However, it remains unclear how TDP-43 aggregation mediates complex behavioral phenotypes in FTLD including progressive impairment in mental function (Rascovsky et al. 2011; Irish et al. 2012). In this study, we investigate the causal mechanism for psychiatric symptoms in FTLD by testing the general hypothesis that, at least in some neurodegenerative diseases, co-aggregation of casual disease factors like TDP-43, with genetic risk factors for psychiatric disease may alter cellular and behavioral phenotypes, resulting in overt manifestation of psychiatric symptoms.
2 Results
2.1 TDP-43 Forms Co-Aggregates with DISC1 in Neurons
We examined DISC1 as a possible binding partner for TDP-43 due to its aggregation-prone nature and well-established genetic role in mental illness (Leliveld et al. 2008; Brandon and Sawa 2011; Tanaka et al. 2017). First, we examined the co-immunoprecipitation of DISC1 and TDP-43 in vivo. DISC1 formed a complex with TDP-43 in both mouse and human brain and the binding interaction was higher in cerebral cortex than in cerebellum. DISC1 also bound to fused in sarcoma/translated in liposarcoma (FUS/TLS) and FMRP, both of which interact with TDP-43. Aggregation of a C-terminal fragment of TDP-43 in the cytosol is a hallmark of FTLD neurons (Neumann et al. 2006). Therefore, we examined whether DISC1 forms co-aggregates with the C-terminal fragment of TDP-43. Using a lentivirus system, we cultured cortical neurons that expressed an N-terminal Venus-tagged C-terminal fragment (residues 220–414) of TDP-43, termed TDP-220C that formed round-shaped aggregates in both cytosol and neurites typical of those observed in FTLD neurons (Fig. 18.1a) (Lee et al. 2012). These aggregates were ubiquitinated and highly phosphorylated (Ser409/Ser410) characteristic of aggregated TDP-43 in neurons of FTLD brain (Neumann et al. 2006). Neurons with TDP-220C aggregates were immunostained with an antibody against DISC1, demonstrating that endogenous DISC1 co-localizes with TDP-220C aggregates (Fig. 18.1b). Furthermore, the filter trap assay showed that detergent-resistant insoluble DISC1 was increased in neurons expressing TDP-220C. Collectively, these results are consistent with the aggregation-prone nature of DISC1 (Leliveld et al. 2008; Tanaka et al. 2017), although physiological consequences of DISC1 aggregation in neuropsychiatric diseases remain poorly understood.

DISC1 and TDP-43 form co-aggregates in neurons
Cultured cortical neurons were infected with a lentivirus expression vector encoding Venus-TDP-220C (green) and endogenous DISC1 was immunostained with an anti-DISC1 antibody (red). Nuclei were stained with DAPI (blue) and dendrites were stained with an anti-MAP 2 antibody (blue). A representative image is shown for the whole neuron (Top) and dendrite (Bottom), respectively. Scale bar represents 5 μm
Several RNA-binding proteins such as TDP-43, FUS/TLS and FMRP were co-immunoprecipitated with DISC1. So, we asked whether RNA is critical for this interaction. To examine this possibility, we treated mouse brain homogenates with or without RNase, followed by co-immunoprecipitation with an anti-DISC1 antibody. RNase treatment abolished the binding of DISC1 to RNA-binding proteins, indicating RNA-dependence of the DISC1 association. This result suggests that DISC1 has a functional role in RNA metabolism to regulate neuronal functions. Furthermore, we investigated whether DISC1 regulates the abundance of synaptic proteins. When we examined amounts of postsynaptic proteins in synaptosomal fractions by western blotting, we found that the DISC1 depletion reduced levels of several key postsynaptic proteins in dendrites.
2.2 Role of DISC1 in Local Translation in Dendrites
To investigate whether the reduced levels of synaptic proteins in synaptosomal fractions might be caused by impaired local translation in dendrites, we examined the involvement of DISC1 in mRNA translation. Previous studies indicated that TDP-43 is associated with a translational machinery and polyribosomes (Higashi et al. 2013; Coyne and Siddegowda 2014). DISC1 can interact with TDP-43, therefore we examined whether DISC1 is also associated with a translational machinery by polysome gradient centrifugation analysis. DISC1 was detected in polyribosome fractions, as observed for TDP-43 (Coyne and Siddegowda 2014). After either EDTA or RNase treatment (Stefani et al. 2004), DISC1 in the polyribosome fractions co-migrated with TDP-43 and ribosomal S6 (RS6), indicating that, like TDP-43, DISC1 interacts with polyribosomes. Immunoprecipitation experiments demonstrated that DISC1 forms a complex with TDP-43 in the polyribosome fraction and that this binding was more significant in cerebral cortex than that cerebellum. Together, these data suggest that the DISC1-TDP-43 interaction is crucial for translation in cerebral cortex.
To further investigate the possible roles of DISC1 in local translation in dendrites, we examined whether DISC1 regulates translation of synaptic mRNAs in neurons by Surface Sensing of Translation (SUnSET), a method to monitor new protein synthesis by incorporation of puromycin into newly synthesized polypeptides (Schmidt et al. 2009). First, we assessed whether DISC1 might regulate local translation of synaptic mRNAs which are bound with DISC1. We labeled newly synthesized proteins in cultured cortical neurons with puromycin, and puromycin-incorporated proteins were immunoprecipitated from the synaptosomal fraction, followed by detection of newly synthesized postsynaptic NR2B and PSD95 proteins by western blotting. Remarkably, DISC1 knockdown significantly reduced synthesis of these synaptic proteins, which were restored by co-expression of an RNAi-resistant form of DISC1. These results suggest that DISC1 plays a critical role in dendrite local translation.
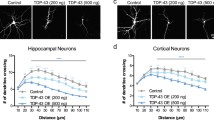
Neuronal stimulation induces local protein synthesis in dendrites (Ramocki and Zoghbi 2008; Ebert and Greenberg 2013). Therefore, we examined the effects of DISC1 depletion on new protein synthesis induced by neuronal stimulation. We evaluated new protein synthesis in a synaptosomal fraction with or without neuronal stimulation with KCl (Wang et al. 2008; Bading et al. 1993). In basal culture conditions without stimulation (5 mM KCl), newly synthesized protein levels were similar between control and DISC1-knockdown neurons. We confirmed that neuronal stimulation with 55 mM KCl enhanced protein synthesis in control neurons. In contrast, such an increase in new protein synthesis was not observed in DISC1-knockdown neurons, but the defect was restored by co-expression of the RNAi-resistant form of DISC1. To confirm the involvement of DISC1 in local translation in dendrites, we visualized puromycin-conjugated nascent polypeptides. We found the DISC1 depletion reduced new protein synthesis in dendrites under stimulation (Fig. 18.2), consistent with the observation by western blotting. Total mRNA levels in the synaptosomal fraction were not altered by the deletion of DISC1, suggesting that the decreased activity-dependent protein synthesis in DISC1-knockdown neurons is not caused by defects of mRNA transport but by impaired translation activity. Together, these results showed a novel function of DISC1 in regulating dendrite local translation that is dependent on neuronal activity.

Local translation in dendrites is regulated by DISC1
Neurons were immunostained with anti-puromycin (gray) and anti-MAP 2 (red) antibodies. The fluorescent intensities in MAP 2-labeled dendrites were measured and normalized by those in 5 mM KCl treated-control neurons (right). (n = 19,18,25,21 for Scramble RNAi +5 mM KCl, DISC1 RNAi +5 mM KCl, Scamble RNAi +55 mM KCl and DISC1 RNAi +55 mM KCl respectively. F(3,79) = 74.17, P < 0.0001, one-way ANOVA; ∗∗∗P < 0.001, Bonferroni’s multiple comparison test post hoc). Scale bar represents 25 μm
2.3 TDP-43-DISC1 Co-Aggregation Inhibits Local Translation in Dendrites
Since endogenous DISC1 is readily sequestered into TDP-220C aggregates, we hypothesized that the co-aggregation between DISC1 and TDP-220C might impair local translation, similar to the DISC1 depletion. Overexpression of TDP-220C in neurons decreased synaptic protein levels in synaptosomal fractions, including surface-localized neuronal receptors. However, co-expression of DISC1 normalized the reduction of synaptic protein levels. Furthermore, the overexpression of TDP-220C in neurons impaired induction of new protein synthesis in synaptosomal fractions by neuronal stimulation, which was also restored by co-expression of DISC1. These rescue effects were not caused simply by reduction in TDP-220C protein levels due to the co-expression of DISC1. These results indicate that the loss of functional DISC1 mediates the impairment of local translation in the neurons containing TDP-220C aggregates. Together, our findings suggest that co-aggregation of DISC1 with TDP-220C selectively perturbs the role of DISC1 in local translation in dendrites.
2.4 DISC1-Dependent Behavioral Impairment and Rescue in TDP-220C Mice
We examined in vivo behavioral consequences of the co-aggregation of DISC1 with TDP-220C. To examine this issue, we stereotaxically injected adeno-associated virus (AAV) encoding N-terminally Venus tagged TDP-220C into the frontal cortex of mice (hereafter termed TDP-220C mice) at 6 weeks old. The AAV infection resulted in formation of highly phosphorylated TDP-220C aggregates in neurons of frontal cortex. We observed endogenous DISC1 and full-length TDP-43 sequestered into the TDP-220C aggregates. Protein levels of synaptic genes were significantly reduced in TDP-220C mice, consistent with the results from cultured neurons.
Next, we examined selected behavioral repertoire of TDP-220C mice after 2 weeks following AAV injection. TDP-220C mice showed significantly increased locomotor activity (hyperactivity) in the open field test while no change was observed in the time spent in a center region of the field between control and TDP-220C mice. In social interaction tests, control mice interacted with an unfamiliar stranger mouse than a familiar mouse for a longer time, but TDP-220C mice did not show significant differences (Fig. 18.3). Importantly, the time spent in each area was comparable for control and TDP-220C mice, indicating that the social interaction defect of TDP-220C mice was not caused simply by their hyperactive phenotypes. We then examined whether the hyperactivity and the impaired sociability in TDP-220C mice could be normalized by DISC1 co-expression. We found that the rescue of the behavioral phenotypes could be achieved by DISC1 co-expression, indicating that restoration of DISC1 function was critical to normative behavior. Furthermore, we performed a Morris water maze test in order to evaluate the potential impact of TDP-220C-DISC1 co-aggregation on acquisition of spatial memory. TDP-220C mice, but not control mice, showed a significant learning impairment. We found that co-expression of DISC1 did not rescue the spatial learning impairment, indicating that the defect in the hippocampal-dependent learning task in TDP-220C mice is independent of DISC1 dysfunction. As an additional control, the grip strength test revealed that neuromuscular function was not affected in TDP-220C mice. Collectively, these results suggest that hyperactivity and disturbed sociability in TDP-220C mice are selectively mediated by the loss of DISC1 function due to the co-aggregation, providing support for the role of TDP-43/DISC1 interactions in mental conditions.

Social deficits in FTLD model TDP-220C mice are mediated by DISC1 dysfunction
In the social interaction test, TDP-220C mice were less interested in novel stranger mice, which was rescued by the co-expression of DISC1. During the first encounter, all mice group spent more time in the area with first stranger mice (S1) compared to empty cage (E). During the second encounter, EGFP control mice spent more time with novel, unfamiliar mice (S2) than with familiar mice (F) whereas TDP-220C mice spend equal time with unfamiliar and familiar mice. This behavior was normalized by co-expression of DISC1. n = 23, 23, 21, 24 for EGFP (white), TDP-220C + EGFP (black), TDP 220C + DISC1 (dark gray), DISC1 + EGFP (light gray) mice, respectively (EGFP: F(5,132) = 9.048, P < 0.0001; TDP-220C + EGFP: F(5,132) = 4.875, P = 0.0004; TDP 220C + DISC1: F(5,120) = 3.666, P = 0.004; DISC1 + EGFP: F(5,138) = 19.95, P < 0.0001, one-way ANOVA, ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001, Bonferroni’s multiple comparison test post hoc)
3 Discussion
In this study, we demonstrate that TDP-43, the causal protein in FTLD, co-aggregates with DISC1, a biological mediator for major mental illnesses, resulting in impaired local dendritic translation and disturbed social behaviors. The TDP-43-DISC1 co-aggregation compromised activity-dependent local dendritic translation and elicited mental deficits in FTLD model mice. Importantly, the psychiatric behaviors in the disease model mice were normalized by exogenous expression of DISC1 in frontal cortex, demonstrating that the loss of soluble, functional DISC1 due to co-aggregation is causally responsible for disturbed mental conditions in FTLD model mice. It has remained unclear why abnormal mental disorders are frequently observed in many neurodegenerative disorders. Given that local translation in dendrites is a critical process in higher brain functions (Buffington et al. 2014), our study has solved the long-standing puzzle of this neurological/psychiatric phenotypic overlap.
DISC1 was previously proposed to regulate synaptic plasticity and mental conditions, but the underlying molecular mechanisms have remained unclear (Tsuboi et al. 2015; Hayashi-Takagi et al. 2010). Our study reveals that DISC plays an unexpected role in determining activity-dependent local translation in dendrites, suggesting that DISC1 regulates the expression and hence function of postsynaptic genes in dendrites. Thus, our finding provides a candidate molecular basis for impaired synaptic plasticity and mental conditions observed in DISC1-knockdown neurons and DISC1 mutant mice (Hayashi-Takagi et al. 2010; Kvajo et al. 2011; Kuroda et al. 2011). Our results show that DISC1 regulates translation of mRNAs of critical synaptic genes, some of which are associated with psychiatric disorders. Our study validated, for TDP-43 and DISC1, the novel hypothesis that the co-aggregation of risk molecules for psychiatric diseases may account for latent psychiatric symptoms in neurodegenerative disorders.
References
Bading H, Ginty DD, Greenberg ME (1993) Regulation of gene expression in hippocampal neurons by distinct calcium signaling pathways. Science 260:181–186
Baralle M, Buratti E, Baralle FE (2013) The role of TDP-43 in the pathogenesis of ALS and FTLD. Biochem Soc Trans 41:1536–1540
Bramham CR, Wells DG (2007) Dendritic mRNA: transport, translation and function. Nat Rev Neurosci 8:776–789
Brandon NJ, Sawa A (2011) Linking neurodevelopmental and synaptic theories of mental illness through DISC1. Nat Rev Neurosci 12:707–722
Buffington SA, Huang W, Costa-Mattioli M (2014) Translation control in synaptic plasticity and cognitive dysfunction. Annu Rev Neurosci 37:17–38
Cohen TJ, Lee VM, Trojanowski JQ (2011) TDP-43 functions and pathogenic mechanisms implicated in TDP-43 proteinopathies. Trends Mol Med 11:659–667
Coyne AN, Siddegowda BB (2014) Futch/MAP 1B mRNA is a translational target of TDP-43 and is neuroprotective in a Drosophila model of amyotrophic lateral sclerosis. J Neurosci 34:15962–15974
Ebert DH, Greenberg ME (2013) Activity-dependent neuronal signalling and autism spectrum disorder. Nature 493:327–337
Hayashi-Takagi A et al (2010) Disrupted-in-schizophrenia 1 (DISC1) regulates spines of the glutamate synapse via Rac1. Nat Neurosci 13:327–332
Higashi S, Kabuta T, Nagai Y, Tsuchiya Y, Akiyama H et al (2013) TDP-43 associates with stalled ribosomes and contribute to cell survival during cellular stress. J Neurochem 126:288–300
Holt CE, Schuman EM (2013) The central dogma decentralized: new perspectives on RNA function and local translation in neurons. Neuron 80:648–657
Irish M, Piguet O, Hodges JR (2012) Self-projection and the default network in frontotemporal dementia. Nat Rev Neurol 8:152–161
Kuroda K et al (2011) Behavioral alterations associated with targeted disruption of exon 2 and 3 of the Disc1 gene in the mouse. Hum Mol Genet 20:4666–4683
Kvajo M et al (2011) Altered axonal targeting and short-term plasticity in the hippocampus of Disc1 mutant mice. Proc Natl Acad Sci U S A 108:1349–1358
Lee EB, Lee VM, Trojanowski JQ (2012) Gains or losses: molecular mechanisms of TDP43-mediated neurodegeneration. Nat Rev Neurosci 13:38–50
Leliveld SR et al (2008) Insolubility of disrupted-in-schizophrenia 1 disrupts oligomer-dependent interactions with nuclear distribution element 1 and is associated with sporadic mental disease. J Neurosci 28:3839–3845
Ling SC, Polymenidou M, Cleveland DW (2013) Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 3:416–438
Neumann M et al (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133
Orr HT, Zoghbi HY (2007) Trinucleotide repeat disorders. Annu Rev Neurosci 30:575–621
Ramocki MB, Zoghbi HY (2008) Failure of neuronal homeostasis results in common neuropsychiatric phenotypes. Nature 455:912–918
Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH et al (2011) Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 134:2456–2477
Schmidt EK, Clavarino G, Ceppi M, Pierre P (2009) SUnSET, a nonradioactive method to monitor protein synthesis. Nat Methods 6:275–277
Stefani G, Fraser CE, Darnell JC, Darnell RB (2004) Fragile X mental retardation protein is associated with translating polyribosomes in neuronal cells. J Neurosci 24:7272–7276
Tanaka M et al (2017) Aggregation of scaffolding protein DISC1 dysregulates phophodiesterase 4 in Huntington’s disease. J Clin Invest 127:1438–1450
Tsuboi D et al (2015) Disrupted-in-schizophrenia 1 regulates transport of ITPR1 mRNA for synaptic plasticity. Nat Neurosci 18:698–707
Wang IF, Wu LS, Shen CK (2008) TDP-43, the signature protein of FTLD-U, is a neuronal activity-responsive factor. J Neurochem 105:797–806
Acknowledgments
We thank Akira Sawa, Yoko Machida, Yusuke Komi, Shigeo Murayama, Masaki Takao, Hiroyasu Akatsu, Koko Ishizuka, Miles Houslay and the members of Tanaka Laboratory for helpful discussion. We are grateful to the Brain Bank for Aging Research at Tokyo Metropolitan Institute of Gerontology and Fukushimura Brain Bank, and the families of the patients for providing brain samples used in this study. DNA sequencing and preparation of polyclonal antibodies were performed at the Research Resource Center facility in RIKEN Brain Science Institute. Animals were maintained by RIKEN Brain Science Institute animal facility. This work was supported by The Uehara Memorial Foundation (M.T.), Grant-in-Aid for Scientific Research on Innovative Areas from Ministry of Education, Culture, Sports, Science and Technology, Japan (26116004 to M.T.) and RIKEN Aging Project (MT).
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2020 The Author(s)
About this paper

Cite this paper
Endo, R., Takashima, N., Tanaka, M. (2020). Abnormal Local Translation in Dendrites Impairs Cognitive Functions in Neuropsychiatric Disorders. In: Toyama, Y., Miyawaki, A., Nakamura, M., Jinzaki, M. (eds) Make Life Visible. Springer, Singapore. https://doi.org/10.1007/978-981-13-7908-6_18
Download citation
DOI: https://doi.org/10.1007/978-981-13-7908-6_18
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-7907-9
Online ISBN: 978-981-13-7908-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)