
Abstract
Vanadium is the 21st most abundant element in the Earth’s crust and the 2nd-to-most abundant transition metal in sea water. The element is ubiquitous also in freshwater and nutrients. The average body load of a human individual amounts to 1 mg. The omnipresence of vanadium hampers checks directed towards its essentiality. However, since vanadate can be considered a close blueprint of phosphate with respect to its built-up, vanadate likely takes over a regulatory function in metabolic processes depending on phosphate. At common concentrations, vanadium is non-toxic. The main source for potentially toxic effects caused by vanadium is exposure to high loads of vanadium oxides in the breathing air of vanadium processing industrial enterprises. Vanadium can enter the body via the lungs or, more commonly, the stomach. Most of the dietary vanadium is excreted. The amount of vanadium resorbed in the gastrointestinal tract is a function of its oxidation state (VV or VIV) and the coordination environment. Vanadium compounds that enter the blood stream are subjected to speciation. The predominant vanadium species in blood are vanadate and vanadyl bound to transferrin. From the blood stream, vanadium becomes distributed to the body tissues and bones. Bones act as storage pool for vanadate. The aqueous chemistry of vanadium(V) at concentration <10 μM is dominated by vanadate. At higher concentrations, oligovanadates come in, decavanadate in particular, which is thermodynamically stable in the pH range 2.3–6.3, and can further be stabilized at higher pH by interaction with proteins.
The similarity between vanadate and phosphate accounts for the interplay between vanadate and phosphate-dependent enzymes: phosphatases can be inhibited, kinases activated. As far as medicinal applications of vanadium compounds are concerned, vanadium’s mode of action appears to be related to the phosphate-vanadate antagonism, to the direct interaction of vanadium compounds or fragments thereof with DNA, and to vanadium’s contribution to a balanced tissue level of reactive oxygen species. So far vanadium compounds have not yet found approval for medicinal applications. The antidiabetic (insulin-enhancing) effect, however, of a singular vanadium complex, bis(ethylmaltolato)oxidovanadium(IV) (BEOV), has revealed encouraging results in phase IIa clinical tests. In addition, in vitro studies with cell cultures and parasites, as well as in vivo studies with animals, have revealed a broad potential spectrum for the application of vanadium coordination compounds in the treatment of cardiac and neuronal disorders, malignant tumors, viral and bacterial infections (such as influenza, HIV, and tuberculosis), and tropical diseases caused by parasites, e.g., Chagas’ disease, leishmaniasis, and amoebiasis.
Please cite as: Met. Ions Life Sci. 13 (2013) 139–169
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
- antiparasitic vanadium compounds
- antiviral potential
- cardiovascular effects
- essentiality of vanadium
- insulin-enhancing action
- vanadate-phosphate antagonism
1 Introduction
Vanadium is a versatile and omnipresent element that can attain the oxidation states –III to +V. Low-valent vanadium is stabilized by strongly π-accepting ligands, carbon monoxide in particular, high valent vanadium by σ and π donors represented by hard, oxygen and nitrogen functional ligands. Soft ligands such as thio-functional ones are predominantly found in vanadium compounds with vanadium in intermediate oxidation states. Vanadium nitrogenase is an example for a naturally occurring vanadium compound where vanadium switches in-between the oxidation states +II and +IV: In vanadium nitrogenase from nitrogen fixing bacteria such as Azotobacter, vanadium – an integral constituent of the Fe7VS9 M-cluster – is coordinated to three sulfides, a histidine-N, and two oxygen functions of homocitrate. Vanadium(III) coordinated to water molecules is present in the vanadocytes of sea squirts. The +IV and +V oxidation states, which are the by far predominating ones in physiologically relevant vanadium systems, typically contain the VIVO2+, VVO3+ or VVO2 + ‘core’, although there are exceptions. An example for a ‘bare’ vanadium(IV) complex is the naturally occurring amavadin, where V4+ is coordinated to two tetradentate N-oxyimino-2,2′-dipropionate ligands. Amavadin is found in mushrooms belonging to the genus Amanita, such as the fly agaric. The oxidovanadium(V) core is present in vanadate-dependent haloperoxidases from, inter alia, marine algae, with vanadate H2VO −4 coordinatively linked to an active center histidine-N.
To date, vanadate-dependent haloperoxidases and vanadium nitrogenases have remained the only identified naturally occurring vanadium-based enzymes. Whether or not vanadium is an essential element for evolutionary younger organisms, including vertebrates, remains to be verified. A functional role of simple vanadium compounds (vanadate in particular) in vertebrates, and hence also in humans, is likely, an assumption which is based on the similarity between vanadate and phosphate. In this context, the vanadate-dependent haloperoxidases are of particular interest since they mimic, or model, enzymes involved in phosphate metabolism, where the protein binding domain for phosphate is blocked by vanadate.
The competitive behavior of vanadate with respect to phosphate is likely also the clue for the insulin-mimetic/insulin-enhancing potential of vanadium compounds, and hence the surge in the design of antidiabetic vanadium complexes during the past two decades. These auspicious developments have also initiated research towards the design of biologically active vanadium complexes in the search of pharmacological control of cancer, cardiovascular imbalances, and diseases caused by viruses, bacteria, amoebae, and flagellate protozoan parasites. In several cases, ligands have been employed that relate to original pharmacologically applied drugs, with the aim to increase the efficacy of the drug, and to widen the spectrum of therapeutic use by exploiting the cooperative effect of the metal and the ligand. The research into these medicinal applications includes functional alternatives to the phosphate-vanadate antagonism, e.g., the direct interaction of the vanadium compound with the DNA of a tumor cell or the pathogen.
The broad medicinal potential of vanadium will be extemporized in Section 4 of this chapter. Section 2.1 is dedicated to vanadium’s distribution and speciation in nature, and hence, its availability, potential inalienability, and occasional toxicity for humans. Section 2.2 addresses resorption, speciation in the blood stream and in the cytosol, tissue distribution, and excretion of vanadium within the human body, and hence, vanadium’s gateways commonly referred to as pharmacokinetics and pharmacodynamics. Given the importance of the similarities (and, to some extent, also the dissimilarities) between vanadate and phosphate for the potentiality of vanadium in toxic as well as in beneficial issues (such as regulatory function and medicinal applications), an extra section, Section 3, is devoted to the vanadate-phosphate antagonism.
2 Distribution and Cycling of Vanadium
2.1 Vanadium in Nature
The Earth’s crust, including the aqua- and atmosphere, contains ≈130 ppm (by mass) vanadium, making vanadium the 21st most abundant element in the outermost sphere of our home planet. The abundance of vanadium in the Earth’s crust thus exceeds its occurrence in the Universe (≈1 ppm) and in the Sun (≈ 0.4 ppm) by about two orders of magnitude [1]. Volcanic areas with basaltic layers are particularly rich in vanadium, as are hard coal (up to 0.34%) and some oil shales and crude oil. Venezuelan crude oil can contain up to 0.12% V, mostly in the form of oxidovanadium(IV) porphyrins (Figure 1a). VO2+ ions are strongly complexed by porphyrins and related chelators, and the enrichment of crude oil with vanadium is due to its extraction from vanadium-bearing rock by porphyrins present in oil that had passed through rocky layers. Natural release of vanadium mainly goes back to weathering of vanadium-containing rock and the erosion of soil.

(a) The porphinogenic vanadium compound oxidovanadium(IV)-deoxiphylloerythrin from crude oil. (b) Amavadin, present in the fly agaric (Amanita muscaria), is a non-oxido vanadium compound containing the ligand (S,S)-N-oxyimino-2,2′-diisopropionate(3–).
Vanadium concentrations in seawater and freshwater are around 30 nM. At the prevailing oxic conditions and about neutral to slightly alkaline pH, soluble vanadate(V), H2VO −4 , is the dominant species. The high Na+ concentration in seawater implies that ion pairs Na+[H2VO −4 ] are formed. Under non-oxic conditions, sparingly soluble oxidovanadium(IV) hydroxide ‘VO(OH)2’ is generated, transported in water in the form of colloids and absorbed to floating particulate sediment, or solubilized by complexing ligands.
Vanadium is the second-to-most abundant transition metal in seawater, outclassed only by molybdenum (MoO 2 −4 , c = 100 nM). The nanomolar concentration excludes the formation of vanadates of higher nuclearity otherwise typical for vanadates (see below). Vanadate concentrations in potable water are around 10 nM. In volcanic areas with basalt layers, the concentration of vanadium in underground water, and consequently also in drinking water, can rise to 2.5 μM [2]. Where drinking water is supplied through lead water pipe systems, vanadate is removed through the formation of deposits of sparingly soluble vanadinite, PbCl2⋅3Pb3[VO4]2. A decrease of pH and an increase of the phosphate concentration (e.g., by addition of phosphate-based corrosion inhibitors to drinking water) can re-mobilize vanadate (eqns 1 and 2) and thus eventually increase vanadate concentrations beyond a tolerable level [3].
Vanadinite is a common mineral containing vanadium in the oxidation state +V. Vanadium’s first discovery by the Spanish mineralogist Manuel del Rio y Fernández in1801 goes back to this mineral. Vanadium-based minerals are otherwise comparatively rare, i.e., most of the vanadium in the Earth’s crust is dissipated in other minerals, rocks, and sediments. Examples for defined minerals with vanadium in oxidation states other than +V are minasragrite, VOSO4⋅5H2O, and patronite, V(S2)2, with vanadium(IV), and karelianite, V2O3, with vanadium(III). Note that, under oxic conditions, only the oxidation states +V and, to some extent, also +IV are stable. Vanadium(II) has been found in forsterite (Mg2SiO4) and enstatite (MgSiO3) in chondrules of meteorites such as the Vigarano meteorite [4]. Here, V2+ can partially occupy Mg2+ and Ca2+ sites.
High contents of vanadium are found in various sea squirts (ascidians), in fan worms, and in Amanita mushrooms such as the fly agaric [5]. In ascidians, vanadium concentrations in specialized blood cells, where the predominant form of vanadium is [VIII(H2O)5HSO4]2+, can go up to 0.3 M. The concentration of vanadium in the fly agaric exceeds that in other plants by a factor of 102. In the fly agaric, also known as toad stool, vanadium is present as amavadin, a low molecular mass non-oxido VIV complex (Figure 1b).
Vanadium contents in food average 30 μg kg–1, the daily intake via food and beverages is 10 μg to 2 mg, only a minute proportion of which becomes resorbed. The body pool of an average human being (70 kg body mass) amounts to ca. 1 mg V, the average blood plasma concentration to 45 nM. Oral intake of vanadium is somewhat increased for sportsmen and bodybuilders resorting to preparations containing VOSO4. This ‘vanadyl fuel’ allegedly helps increasing the muscle mass. Since almost all of the vanadium is excreted in the form of insoluble VO(OH)2 prior to resorption, potential harm due to vanadium overload is not likely to occur.
Still another – and more critical – source of vanadium intake is breathable air in urban and industrialized areas. Vanadium, in the form of vanadium(IV) and vanadium(V) oxides, VOx, is present in air in particulate form or absorbed to tiny dust particles and aerosols, and thus enters the lungs and the pulmonary system, from where it becomes distributed in the body after solubilization. The main natural sources for vanadium loads in the atmosphere are continental dust, marine aerosols, and volcanic emissions [6]. In rural areas, the concentration of vanadium oxides is in the range of 50 ng m–3; pollution can go up to >103 ng m–3 in urban settings, and in industrial areas in particular [7], where combustion of petroleum and oil are the main contributors to aerial VOx. Potential toxic effects of vanadium overloads [8], in particular irritations of the respiratory tract in workers exposed to high loads of vanadium oxides at their working place, will briefly be addressed in the next section.
2.2 Pharmacokinetics and Pharmacodynamics
As noted, critical exposure to vanadium compounds, vanadium oxides in particular, is confined to inhalation in the frame of occupational exposure, including mining and milling of vanadium ores, metallurgical processing involving ferrovanadin, production of catalysts, batteries and glass melt additives based on vanadium oxides, and cleaning of oil-fired boilers. Inhaled vanadium oxides cause rhinitis, irritations of the respiratory tract and – commonly transient – pulmonary malfunctions such as bronchitis, pneumonia, and asthma. Whether or not vanadium oxides can promote lung cancer has yet to be shown. In any case, the maximum allowable concentration of V2O5 at the working place, the MAC value, has been set by the World Health Organisation to 0.05 mg m–3 (40-h week, 8-h time-weighted average).
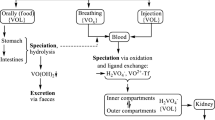
Vanadium oxides are readily absorbed in the lungs and enter the blood stream after solubilization in the form of vanadate, H2VO −4 . Skin does not appear to allow for an appreciable import of vanadium. The main ‘natural’ source for the body’s vanadium supply thus is dietary uptake, a comparatively ineffective process because up to 99% of the dietary vanadium is usually excreted with the feces. The main routes of vanadium uptake and distribution in the body are sketched in Figure 2. Dietary forms of vanadium are either vanadate, H2VO −4 , present mainly in drinking water, and oxidovanadium(IV) compounds {VOL}, where {VOL} represents any ligand-stabilized VO2+. Free VO2+ is, as noted, essentially unavailable, since it forms sparingly soluble oxidovanadium hydroxide, allowing for nanomolar concentration of ‘free’ VO2+ (actually [VO(OH)3]–) at the best. H2VO −4 is more easily taken up in the gastrointestinal tract. However, vanadate(V) is partially reduced in the stomach and precipitated in the form of VO(OH)2 in the slightly alkaline medium of the intestines. Vanadium can also enter the blood stream by injection or infusion, either intentionally when injected intravenously (or intraperitoneally), or accidentally when present as a ‘contaminant’ in infusion solutions [9].

Uptake, distribution and excretion of vanadium compounds. Uptake routes are indicated by broad arrows, excretion routes by broken arrows, and distribution routes by standard arrows and equilibrium arrows, respectively. Main vanadium compounds are indicated. Abbreviations: Tf = transferrin, L is any ligand provided by the nutritional matrix or in a medicinally applied vanadium compound, L′ is a low molecular mass ligand present in blood serum, and {L/L′VO} is the abbreviation for a VO2+ complex with L and/or L′.
Vanadium compounds ending up in the blood stream either after resorption in the gastrointestinal tract, or via the lungs, or by infusion/injection, are subjected to redox interconversion between VV and VIV, depending on the oxygen tension and the presence of redox-active agents. The main transporter for both anionic vanadate(V), cationic VO2+, and neutral or charged {LVIVO} is transferrin [10]. Transferrin (Tf) forms binary complexes {VO2+-Tf} and ternary complexes {VOL-Tf} and {VOL′-Tf}, where L is a ligand originally coordinated to VO2+, and L′ a low molecular mass (lmm) ligand provided by blood serum, such as lactate [11], the serum lmm compound with the highest concentration, i.e., 1.5 mM. The VO2+ ion binds into the same protein pocket as Fe3+, and hence to two tyrosinates, an aspartate, and the Nε of a histidine, plus a synergistic carbonate (Figure 3a). With increasing blood serum concentrations of vanadium, high-molecular mass transporters other than Tf come in, namely serum albumin (Ab) and immunoglobulin G (Ig) [12]. These proteins preferentially form ternary complexes, {VOL-Ab} and {VOL-Ig}. As shown in Figure 3b, the protein binds to the VOL moiety via a histidine residue.

Likely binding modes of VO2+ in (a) the ternary VO2+-transferrin complex [12b], and (b) in the ternary complexes LVO2+-albumin or LVO2+-immunoglobulin (L = ethylmaltol), coordinating through a histidine [12a]. In (a), the Tyr trans to the oxido ligands binds just weakly.
Plasma vanadium contents decline in three phases: The first phase is a rapid decline with a half-life t1/2 of 1 hour, followed by a second intermediate decline (t1/2 ≈ 26 hours) and a third slow decline with t1/2 ≈ 10 days. Vanadium contents in blood are thus reduced to about 30% within the first 24 hours [9]. Clearing occurs directly via urinary excretion, and after distribution over tissues of the inner compartment (heart, liver, kidney, spleen), the outer compartment (brain, muscle, adipose tissue), and bones. About 50% of the vanadium is recovered in urine after 12 days. The residence time of vanadium in bones, where it replaces phosphorus in hydroxyapatite, Ca5(PO4)3OH, is ca. 1 month [13], corresponding to a half-life of 4–5 days.
There are several alternative routes by which vanadium compounds can be transported from the blood plasma into blood and tissue cells. Vanadate is essentially present, at pH ≈ 7, in the form of dihydrogenvanadate, H2VO −4 (the pK a is 8.2), and may use phosphate and sulfate channels: Vanadate and phosphate HPO 2 −4 /H2PO −4 (pK a = 7.2) are structurally very similar (see also the next section). Vanadate and oxidovanadium(IV) bound to transferrin can enter the intracellular space by endocytosis – analogously to Fe3+, the main target ion for transferrin. An additional conceivable path – for a stable vanadium coordination compound with a sufficiently lipophilic coordination sphere – is diffusion across the cell membrane. The feasibility of this latter route of entry has been demonstrated for the uptake of the complex [VO(pyridinone)2H2O] by erythrocytes [14].
The low absorption rate of dietary vanadium and the rather efficient desorption of excess vanadium that has entered the blood and body tissues diminish toxic effects that contemporarily can emerge, such as irritations of the conjunctivae and the respiratory system on exposure to vanadium oxides in the breathing air (see above), or (mild) gastrointestinal and renal problems in the course of medicinal applications of vanadium compounds. The no-effect level has been set to a daily intake of 10 mg V per kg body mass. The respective limit values for intravenous application is 7 mg kg–1, for breathing air 35 mg m–3. Acute poisoning in animals fed an about tenfold excess of vanadium compounds causes paralysis, convulsion, and eventually death [5,15]. Vanadium compounds are considered potentially genotoxic and thus mutagenic, teratogenic, and ‘suspected carcinogenic’. Classification as a carcinogen is based on the fact that vanadium induces the formation of tumor-associated antigens, and that it can directly and indirectly damage DNA and affect DNA repair [1b,16]. ‘Indirectly’ here refers to the potentiality of VO2+ to effect the formation of reactive oxygen species (ROS) such as the OH radical in a Fenton-like reaction (eqn. 3a), and superoxide when directly interacting with O2 (eqn. 3b).
Superoxide in turn can cause the release of iron from the iron storage protein ferritin [17] and thus contributes to the disruption of iron homeostasis. In rat models, vanadium provokes neuro-toxicological effects in the brain, such as demyelination, i.e., damage of the myelin sheet of neurons. Myelin is a lipid-rich membrane of the nerves, and vanadium apparently promotes its peroxidative destruction [8].
While carcinogenic properties of vanadium compounds via ROS formation and interference with phosphokinases [18] are well in the realm of possibility, it should also be noted that vanadium compounds damage DNA in tumor cells more effectively than in healthy cells. As elaborated in Section 4.2.1, many vanadium compounds have an antitumor activity. Vanadium species can also annihilate reactive oxygen species. This is demonstrated by the sequence in equation (4) for the oxidation of peroxide to superoxide and further to O2 [19].
In reference to Paracelsus, who noted that “All substances are poisons; it is solely the dose that differentiates between a poison and a remedy” (“Alle Ding’ sind Gift, und nichts ohn’ Gift; allein die Dosis macht, daß ein Ding kein Gift ist”), there is so far no solid basis for categorizing vanadium compounds as harmful when administered in sensible amounts. Rather, as discussed in more detail in the oncoming sections, vanadium is likely an essential element in as far as vanadate can interfere with phosphatases, phosphorylases, and kinases and, more generally, is involved in regulating the phosphate metabolism and phosphate-dependent energetic processes. In addition, the participation of VIV and VV in levelling ROS suggests that vanadium can be beneficial in the treatment of several diseases and malfunctions related to ROS imbalances.
Generally, vanadium(V), in particular when present as vanadate, is more toxic than vanadium(IV). As noted, VO2+ either forms a sparingly soluble hydroxide or is ‘masked’, through coordination, by a variety of physiologically available ligand systems. Biological detoxification of vanadate occurs via integration into the hydroxyapatite structure of the bones (vide supra) and by reduction to vanadium(IV) [20]. Glutathione, ascorbate, NADH, and NADPH are examples for agents that can reduce vanadate. In ascidians, reduction equivalents for the reduction of H2VO −4 to VO2+ are supposedly delivered by NADPH (generated in the pentose phosphate pathway) via the redox couple 2GSH ⇌ GSSG + 2H+ + 2e–, where GSH and GSSG are the reduced and oxidized forms of glutathione, respectively [21]. Vanadium(III) plays, if any, a minor role only, since vanadium(IV) is not easily reduced to vanadium(III) at physiological conditions – and if so, rapidly re-oxidized to vanadium(IV).
3 The Aqueous Chemistry of Vanadium and the Vanadate-Phosphate Antagonism
At strongly acidic conditions (pH <2), cationic octahedral aqua complexes of VIII, VIVO2+, and VVO +2 can be present in aqueous media, i.e., [V(H2O)6]3+, [VO(H2O)5]2+, and [VO2(H2O)4]+. While [V(H2O)6]3+ has O h symmetry, the structures of the aqua complexes of VO2+ and VO2 + are somewhat distorted due to the trans influence exerted by the doubly bonded oxido ligand(s), giving rise to comparatively weakly bonded water trans to the oxo group(s). In addition, there are distortions in the octahedral arrangement as shown in Figure 4, reducing the local symmetry to C 4v in [VO(H2O)5]2+ and C 2v in [VO2(H2O)4]+ [22] (for the respective distances d(V-O) see Figure 4). Strongly acidic conditions are provided in the stomach, but otherwise physiologically irrelevant – with the exception of the vanadium sequestering blood cells of ascidians, where V3+ mainly exists in the form of [V(H2O)5HSO4]2+. In the presence of a ligand L, partial or complete replacement of H2O provides stability of the resulting complex(es) also in the less acidic, neutral, and slightly alkaline regimes. If L is a bidentate, singly negatively charged ligand, such as lactate, the composition of the resulting mono-ligand complexes is [VIII(H2O)4L]2+, [VIVO(H2O)3L]+, and [VVO2(H2O)2L]. Depending on the pH, mixed aqua-hydroxido complexes can form, such as [VIII(OH)(H2O)3L]+, [VIVO(OH)(H2O)2L], and [VVO2(OH)(H2O)L]–. Further, monooxidovanadium(V) complexes come in, for instance [VVO(H2O)3L]2+ and [VVO(OH)(H2O)2L]+. As the denticity and charge of the ligands increases – an example is citrate [23] – the diversity of potential vanadium species present around pH 7 rises substantially.

Cationic aqua complexes of vanadium(III, IV, and V) that can exist in strongly acidic aqueous media. For structure data see ref. [22].
Vanadium(IV) and vanadium(V) are easily interconverted by physiologically relevant redox agents such as NAD+/NADH, NADP+/NADPH, FAD2+/FADH2, glutathione, and ascorbate. Further, VIV and VV redox-interact with reactive oxygen species. The redox potential for the couple H2VO −4 /VO2+ (eqn. 5) at pH 7 is −0.34 V, which compares to −0.32 V for the couple NAD+/NADH.
The most prominent inorganic vanadium species present at micromolar concentrations and pH 7 is dihydrogenvanadate, H2VO −4 : At the physiological ionic strength of I ≈ 0.15 M, the pK a for the protonation/deprotonation equilibrium (eqn. 6), is 8.2 [24].
With increasing concentration, condensed vanadate species form. In the slightly acidic to alkaline regime (ca. pH 5–9), these are predominantly divanadate [H2V2O7]2–, cyclic tetravanadate [V4O12]4–, and cyclic pentavanadate [V5O15]5–. In the pH range 2.3–6.3, decavanadate [HnV10O28](6–n)– (n = 3–0) comes in. For the structural formulae of these oligovanadates see Figure 5. Commonly, physiological vanadate concentrations at subtoxic levels are too low to allow for the formation of oligovanadates. Locally, however, concentration enhancement or template-directed nucleation [25] may occur, and the oligovanadate(s) then formed can interact with proteins and DNA. An example is the incorporation of tetravanadate into Cu,Zn superoxide dismutase [26a], and the promotion of the hydrolytic cleavage of the ester bond in a phosphodiester RNA model by tetravanadate [26b]. Decavanadate is thermodynamically unstable at pH values exceeding 6.3; its decay into vanadates of lower nuclearity (including monovanadate) is, however, kinetically hampered, allowing for half-lives at pH 7, and even somewhat above pH 7, of several hours. Additional stabilization takes place by close interaction with proteins such as tubulins [27] and myosin [28] as well as with mitochondria [29]. Tubulins are proteins that assemble to structural elements (termed microtubules) of cytoskeletons. Myosin is involved in muscle contraction. Myosin binds decavanadate with high affinity, forming a myosin-MgATP-decavanadate complex that blocks the contractile cycle. Decavanadate that targets mitochondrial proteins inhibits mitochondrial oxygen consumption. Stabilization of decavanadate can also be achieved by its incorporation into tiny water pools of intracellular compartments – as suggested by model experiments with reverse micelles with nanoscopically confined water in the micelles’ cavities [30].

Vanadate(V) species that can be present in aqueous media, depending on concentration, pH, and stabilization by electrostatic interaction with, e.g., proteins. The protonation grade of decavanadate shown here corresponds to that at pH ≈ 2.7.
Vanadate and phosphate are structural analogues, and vanadate is only slightly larger than phosphate: The geometrical diameters are 6.2 Å for vanadate and 5.8 Å for phosphate, the volumes of the circumscribing spheres amount to 125 Å3 for vanadate and 102 Å3 for phosphate. These values are based on the following structure parameters: r(O2–) = 1.36 Å, d(V-O) = 1.72 Å, d(P-O) = 1.54 Å; V-O and P-O distances for tetrahedral coordination geometry, where r(VV) = 0.36 and r(PV) = 0.17 Å. From a geometrical point of view, the two anions are thus essentially indistinguishable, making vanadate an efficient competitor for phosphate in binding sites commonly targeted by phosphate. There are, however, also substantial differences: At pH ≈ 7 and physiological ionic strength, vanadate is almost exclusively present in the form of dihydrogenvanadate, H2VO −4 , while phosphate exists in approximately equal amounts of mono- and dihydrogenphosphate, HPO 2 −4 and H2PO −4 . The higher average charge enables phosphate to interact more efficiently than vanadate with dipoles (such as water) and anions (e.g., anionic amino acid residues in protein matrices). On the other hand, phosphorus can attain the coordination number 5 in transitional states only, while the d-block element vanadium easily extends its coordination number by forming stable penta- and hexa-coordinate complexes. Hence, once incorporated in lieu of phosphate into the active site of a phosphate-dependent enzyme, this enzyme is commonly deactivated with respect to its original function.
Naturally occurring enzymes relying on vanadate in a penta-coordinate trigonal-bipyramidal environment are the haloperoxidases in fungi, lichen, marine algae [5,31a], and Streptomyces [31b]; an example for a vanadate-inhibited phosphate-dependent enzyme is the vanadate variant of rat acid phosphatase [32] (Figure 6, top left), with the same first coordination sphere environment for vanadium as in vanadate-dependent haloperoxidases. Interestingly, vanadate-inhibited phosphatases do have some haloperoxidase activity [33], while vanadate-dependent haloperoxidases can exhibit phosphatase activity [34]. In the lower part of Figure 6, the mechanistic sequence of the phosphoester hydrolysis as catalyzed by phosphatases is illustrated. Apart from vanadate, many complexes of VIII, VIV, and VV with organic ligands that are related to or mimic physiologically available ligands, effectively inhibit phosphatases [35], likely after off-dissociation of all or part of the ligands and thus formation of vanadate or {VOxL} intermediates with an ‘open’ coordination site capable of interacting with, and thus blocking, the active site of the enzyme.

Top: The active sites of vanadate-substituted (and thus inhibited) rat acid phosphatase (left) [32a], the Cys215Ser mutant of protein tyrosine phosphatase 1B (center) [32b], and bovine phosphotyrosyl phosphatase (right) [32c]. Bottom: The mechanism of phosphate ester hydrolysis as catalyzed by a phosphatase. The transition state {} is ‘fixed’ as vanadate becomes coordinated into the active center.
Vanadates can also activate biochemical processes. An example for the activation of an otherwise phosphate-dependent process is the linkage of divanadate (‘pyrovanadate’, H2V2O 2 −7 ) to the 3′-end of the primer nucleotide sequence of DNA [DNA primers serve as a starting point for DNA replication (and hence DNA synthesis) through DNA polymerase]. This activates degradation (‘pyrovanadolysis’) of the primer, making available nucleotides for DNA replication by DNA polymerase [36]. The proposed mechanism for pyrovanadolysis is shown in Figure 7. Activation of kinases will be addressed in the context of the antidiabetic action of vanadium compounds in Section 4.1.

Proposed mechanism for the pyrovanadolysis of the DNA primer [36]: Divanadate attacks the primer at the 3′ position, a process which affords mediation by Mn2+. The transiently formed 3′-divanadophospho-nucleotide is hydrolytically split into divanadate and the phosphonucleotide to which DNA polymerase falls back.
In solutions containing vanadate and phosphate, phosphovanadates, [HVPO7]3– and [H2VPO7]2–, are present at pH 7 (the pK a is 7.2 at physiological ionic strength) [37] (eqn. 7). This mixed anhydride is labile; the formation constant at pH 7 is about 20 M–1, as compared to the formation constant of 350 M–1 for divanadate. Phosphovanadate hence is less stable against hydrolysis than divanadate by an order of magnitude, but more stable than diphosphate, [H2P2O7]2–, by six orders of magnitude. Phosphate can also form mixed species with VO2+ [38]: The dominant species in the slightly acidic regime is ‘VO(HPO4)’, where hydrogenphosphate is a ligand for VO2+ rather than a counter-ion. Stabilization against hydrolysis in the neutral to slightly alkaline range is again achieved by nutritionally or physiologically available organic ligands.
In view of the omnipresence of hydrogen peroxide which, at low concentration levels, is an important signalling and regulatory molecule in many biological processes, the potential formation of peroxidovanadates [39] and peroxidovanadophosphates [40] such as HVO3(O2)2– and HVPO6(O2)3– is also to be considered, as is the generation of vanadate esters according to equation (8). The monoesters of vanadate and divanadate – analogues of the physiologically important phosphate and diphosphate esters – are, however, hydrolytically labile. Formation constants in the case of aliphatic residues R are in the order of 0.1 M–1, for aromatic residues (phenyl esters) around 1 M–1 [41]. Figure 8 contains peroxidovanadates and vanadate esters of potential importance.

Examples for physiologically potentially relevant peroxidovanadates, peroxidovanadophosphates, and vanadate esters (from left to right).
4 The Medicinal Potential of Vanadium
4.1 Diabetes Mellitus
Worldwide, about 10% of the population are suffering from diabetes mellitus – knowingly and unknowingly. Approximately 90% of all diabetic cases are ascribed to type 2 diabetes, the remaining 10% to type 1 diabetes [42a]. Type 1 diabetes (“juvenile diabetes”) goes along with absent or only residual insulin supply by the β cells in the Langerhans islets of the pancreas, commonly caused by degeneration of the β cells in the frame of autoimmune reactions, or by accidental dysfunction or loss of the pancreas. Type 2 diabetes (“adult onset diabetes”) is related, in its initial stage, to insufficient response of the cellular insulin receptors to insulin. In its advanced stage, a feed-back mechanism comes in, provoking β cell failure caused by de-differentiation of the β cells [43]. Type 2 diabetes typically concerns people beyond the age of 50; its onset can be kept in check by physical exercise and reasonable nutritional behavior. However, diabetes type 2 is nowadays increasingly diagnosed also with young people and even children; this juvenile onset type 2 diabetes appears to be correlated to obesity [42b].
Intact β cells produce proinsulin, a peptide hormone consisting of an A-chain with 21 amino acids (aa), a B-chain (30 aa), and a C-chain (31 aa) (for the role of the C peptide see [86]), connecting the A- and B-chains. The C-chain is detached in the final step of insulin synthesis; in genuine insulin, the A- and B-chains are linked through two cystines. Insulin is stored as a C 3-symmetric hexamer, with the monomers linked through histidines via zinc ions [44]. The discharge of the active, monomeric form of insulin is initiated by elevated blood glucose levels. Insulin targets the cellular insulin receptor, triggering a complex mechanism by which glucose becomes internalized into the cytosol, followed by glucose metabolism. Further, insulin is involved in the inhibition of gluconeogenesis (the synthesis of glucose from smaller building blocks, for example amino acids), and in glycogenesis (the synthesis of glycogen). Insulin is thus strongly involved in glucose homeostasis. In addition, insulin stimulates lipogenesis and inhibits lipolysis, and thus prevents ketoacidosis caused by the accumulation of ketonic bodies such as acetyl acetic acid in the blood. Ketonic bodies are causative for the severe disease patterns accompanied with progressive states of diabetes, such as retinopathy and dying off of limbs.
Many inorganic (vanadate, vanadyl sulfate, peroxidovanadates) and organic vanadium compounds have been tested positive with respect to effectuating cellular glucose uptake and controlling free fatty acid levels. A selection of vanadium complexes of the general composition {VOL}, where L represents an organic ligand (system) in the coordination sphere, is provided in Figure 9; test conditions and results are summarized in Table 1 [45–52]. The compounds have been successfully tested in vitro (i.e., with cell cultures) and/or in vivo with diabetic rats or mice and, in the case of the maltolato complex 1b, with human individuals. Complex 1b (bis(ethylmaltolato)oxidovanadium(IV), BEOV), has passed clinical trials phase I and IIa with type 2 diabetic volunteers, essentially with encouraging response [13].

A selection of vanadium coordination compounds with antidiabetic in vitro and/or in vivo potential. Compounds 2 and 8 contain VV, the other complexes VIV. See Table 1 and text for details and references.
The ligand L in {VOL} largely influences the efficacy of a vanadium compound by steering resorption, transport, and stability of the complex, and thus the availability of the actual antidiabetic species, i.e., vanadate, at the locus operandi. Commonly, vanadium complexes are clearly more effective than inorganic vanadium compounds, underlining the advantageous bioavailability and pharmaceutical efficacy of organic vanadium compounds [13]. Where inorganic vanadium compounds, vanadate (H2VO −4 ) in particular, induce hypoglycemic effects, such as tea/vanadate decoctions (last entry in Table 1) [52], this effect is likely due to the intermittent formation of a coordination compound with tea ingredients.
Normal insulin supply and insulin ‘sensing’ provided, insulin docks to the carboxyterminal segment of the extracellular α-subunits, IRα, of the trans-membrane insulin receptor (a tyrosine kinase), de-repressing the tyrosine kinase activity of the intracellular β subunit, IRβ. In such a way, phosphorylation of the tyrosine residues of IRβ is promoted and the cytosolic signal cascade initiated, ending up in the activation of the glucose transporter (GLUT4). Once activated, GLUT 4 becomes translocated to the cell membrane where it picks up glucose for delivery into, and metabolism within, the cytosol. The basic signal transduction is illustrated in Figure 10. Among the various interposed and branching steps [45], insulin receptor substrates (IRS), phosphatidylinositol 3-kinase (PI3K, also known as Akt), and protein kinase B (PKB), have been considered.

Signal cascade for the internalization of glucose by the glucose transporter GLUT4, as triggered by the phosphorylated insulin receptor (IR). In the absence of insulin (diabetes 1) or insufficient insulin response (diabetes 2), protein tyrosine phosphatase 1B (PTP) dephosphorylates the IR, and the glucose intake is annulled. Vanadate can block PTP and thus restore the signalling path. Several of the steps of the signal cascade are shown: IRS = insulin receptor substrate, PI3K = phosphatidylinositol 3-kinase (which activates protein kinase B, also known as Akt), PKB = protein kinase B.
IRS are cytosolic proteins with tyrosine residues, and PKBs are kinases which have available serine and/or tyrosine residues for phosphorylation. PKB directly targets the glucose transporter. In the case of missing insulin supply (type 1 diabetes), or insufficient receptivity of the insulin receptor for insulin (type 2 diabetes), IRβ is dephosphorylated through the action of a protein tyrosine phosphatase (PTPase, PTP-1B), annulling signalling for glucose intake by GLUT4 and thus provoking hyperglycemia. A possible mechanism of action of antidiabetic vanadium compounds in stimulating cellular glucose uptake in the case of hyperglycemia is included in Figure 10. Accordingly, any vanadium compound {VOL} will be broken down in the extra- and/or intracellular space to generate vanadate. As noted in Section 3, vanadate is an efficient phosphatase inhibitor. Consequently, inhibition of PTP-1B by vanadate [53] prevents dephosphorylation of IRβ and thus restores the signalling path.
4.2 Activity in Health Hazards Other than Diabetes
4.2.1 Treatment of Cancer
Several animal and in vitro studies have revealed the virtue of inorganic and, more pronounced, organic vanadium compounds in reducing or preventing neoplasia (the malignant proliferation of cells), and thus tumors, including cancer and its metastatic potential, in various target tissues (see [54] and [55] for comprehensive reviews on cancer prevention and treatment with vanadium compounds). Selected examples are collated in Figure 11, and specific test results are provided in Table 2 [56–59].

Vanadium(V) and vanadium(IV) coordination compounds with anticancer potential. Simplistic names of the ligands are provided. 9b is a hydrolysis product of 9a. For details of the mode of action see Table 2 and text.
The vanadocene derivative 10 is a recent advancement of the more basic vanadocene (η5-C5H5)2VCl2 (= Cp2VCl2; Cp = cyclopentadienyl), introduced a quarter of a century ago by Köpf and Köpf-Maier, who demonstrated that the compound effectively degenerates and kills Ehrlich ascites tumor cells [60]. Ehrlich ascites are derived from breast tumors of female mice. The o-phenanthroline complex 11, dubbed “metvan”, stands for another group of closely related ‘classical’ vanadium coordination compounds that turned out to be particularly operant against various cancer cell lines, including cisplatin-resistant ovarian and testicular cancer [58]. The oxidovanadium(V) pyrimidinone complex 9 and the oxidovanadium(IV) complex 12 are examples for more recent developments in the search of vanadium-based anticancer compounds.
The operating mode of anticancer vanadium compounds is still elusive. Modes of action that have been proposed include
-
(i)
Inhibition of protein tyrosine phosphatases and activation of protein kinases;
-
(ii)
Activation of tyrosine phosphorylases, with concomitant activation of signal transduction pathways, followed by apoptosis and/or activation of tumor suppressor genes;
-
(iii)
Cleavage of, or intercalation into, DNA, resulting in cell cycle arrest;
-
(iv)
Enhanced formation of reactive oxygen species (ROS);
-
(v)
Down-regulation of ferritin expression and disruption of ferritin with concomitant, iron-induced mediation of ROS.
As in the case of antidiabetic vanadium species, the ligand system mediates the resorption and pharmacokinetics of the anticancer compound. In many cases, the active species again is likely vanadate, formed by (partial) degradation of the original drug: Speciation studies of the anionic pyrimidinone (L) complex, [VO2L2]– (9a), have demonstrated that at pH 7 the complex 9a coexists with the hydrolysis products, [VO2(OH)L]– (9b), and mono-, di-, and tetravanadate [56]. Intervention of vanadate with phosphatases, phosphorylases, and kinases will alter and eventually disrupt or enforce signalling paths involved in the regulation of the proliferation of malignant cells. Inorganic vanadate generated under physiological conditions from, for example, [VO(maltol)2] (1a in Figure 9) has been shown to discriminate between hepatocytes and hepatoma cells in as far as vanadate significantly increases the generation of ROS (superoxide and hydrogen peroxide), and concomitantly causes cell cycle arrest in hepatoma but not in normal liver cells (hepatocytes) [61]. Similarly, the formation of ROS by Fe2+ – after vanadium-induced disintegration of ferritin – may be responsible for the vulnerability of astrocytoma cells, while astrocytes (cells of the brain and spinal chord) remain unaffected [62].
Vanadocenes, Cp′2VCl2 (Cp′ stands for substituted Cp), such as complex 10 in Figure 11 will become partially hydrolyzed under physiological conditions to form [Cp′2V(H2O)x(OH)2–x]x+ (x = 0, 1, 2), and hence in a fashion comparable to the hydrolysis of cisplatin, cis-[(NH3)2PtCl2]. The {Pt(NH3) 2 +2 } moiety of cisplatin can directly interact with DNA by coordinating to the N-bases of DNA. The harder (with respect to Pt2+) V4+ in the {Cp′2V2+} moiety supposedly prefers coordination to oxo functionalities of the phosphoester linkages (Figure 12a). Alternatively, Cp′2V(OH)2 can interact with the phosphates via hydrogen bonds. In any case, the resulting ‘kink’ in the DNA will counteract DNA’s replication. Compounds such as metvan (11 in Figure 11), having an aromatic system strongly coordinated to vanadium, may interact with DNA, and thus deactivate DNA and cell proliferation, via intercalation (π−π interaction) (Figure 12b).

Possible interactions of stable (fragments of) vanadium compounds with DNA. (a) The Cp2V2+ moiety of vanadocene (e.g., compound 10) binds to two adjacent phosphates. (b) The o-phenanthroline unit of compound 11 intercalates in-between two nucleobases. X can be OH or H2O. The π stacking is indicated by broken lines.
4.2.2 Cardiovascular Effects; Bacterial and Viral Diseases
Interference of vanadium with protein tyrosine phosphatase (PTPase), in particular the inhibition of PTPase discussed in the context of the insulin-enhancing properties of vanadium compounds in the preceding section, appears to be a major mode of action also in beneficial effects for the vascular system in general, and vanadium’s cardio-protective effects in particular. The inhibition of PTPase by coordination of vanadate to the active site cysteine, as well as oxidative inhibition (cysteine → cystine) by vanadium-induced production of peroxide, result in an up-regulation of protein kinase B (Akt). Akt plays an important role in the regulation of cardiac hypertrophy and angiogenesis (the formation of new blood vessels from preformed ones), and thus in the prevention of, and recovery after, myocardial infarction. The up-regulation of Akt in turn enhances the expression of epithelial nitrogen oxide synthase (NO synthase) and thus the release of the vasodilator NO [63]. The maltolato complex 1a in Figure 9 [64], the pyridinethiolato complex 13, and the picolinato-bis(peroxido)vanadium(V) complex 14 in Figure 13 [65] perform significant cardio-protection in test animals (Table 3) [63–71].

Vanadium complexes with cardio-protective effects (13 and 14) or operant against viral (15, 16, and 17) and bacterial infections (17 and 18). The cut-out 17 is the active center of vanadate-dependent haloperoxidase, e.g., from Corallina inaequalis (see text and Table 3).
Several studies on vanadium’s potentiality in deactivating viral and bacterial infections have appeared during the last decade, and selected compounds and results are provided in Figure 13 and Table 3. The vanadium-substituted polyoxidotungstate [(VIVO)2(VVO)(SbW9O33)2]11– exhibits antiviral activity against viruses causing influenza and Dengue fever, and is also active against HIV-1 (human immune deficiency virus) and SARS (severe acute respiratory syndrome) in vitro [67a]. The cluster contains a linear {O=VO4}3 core (with the vanadium centers in a tetragonal-pyramidal environment), sandwiched by two {SbW9O33} halves in which the tungsten ions are octahedrally coordinated [67b]. At physiological conditions, this so-called ‘Keggin sandwich’ likely decomposes into the genuinely active species vanadate(V) and tungstate(VI). Anti-HIV activity has also been reported for the physiologically stable, water-soluble oxidovanadium(IV)-porphyrin 15 [68]. Water solubility of 15 is provided by the aminosulfonyl substituent at the porphyrin skeleton. The complex inhibits HIV-1 replication in virus infected Hut/CCR5 cells, possibly by targeting and deactivating the viral reverse transcriptase [Hut/CCR5 cells are derived from Hut78 cells, a human T cell line. Hut/CCR5 cells express the chemokine receptor CCR5, a protein on the surface of T lymphocytes (a group of white blood cells)].
The oxidovanadium(IV) xylyl-bicyclam complex 16 is a further example for an agent that is highly effective against strains of HIV-1 and HIV-2 [69], with IC50 values of 0.1–0.3 μM (IC50 values denote the concentration of a drug or prodrug at which the life function of 50% of the viable cells is inhibited). A possible mode of action is through binding of the complex to the chemokine receptor type 4 (CXCR-4), a receptor protein that HIV can use to infect T lymphocytes. Docking of compound 16 to CXCR-4 can occur via direct coordination of the metal center to aspartate and glutamate residues of the protein, or via weak interaction of the oxidovanadium moiety with tryptophan residues, and/or by hydrophobic interaction between tryptophan residues and the bicyclam rings.
Antiviral and antibacterial activity has also been noted for vanadate-dependent haloperoxidase isolated from the marine alga Corallina inaequalis, and its alkalophilic mutant Pro395Asp/Leu241Val/Thr343Ala in particular [70]. The enzyme contains vanadate (H2VO −4 ) coordinated to the Nε of a histidine residue in the active center (17 in Figure 13). The native peroxidase catalyzes the oxidation of halides, for example, bromide to hypobromous acid, HOBr (eq. 9a), and – in a side-reaction – generates singlet oxygen 1O2 (eq. 9b), commonly at slightly acidic conditions. The mutant is active at pH ~ 8, allowing for the disinfection, through HOBr and 1O2, of medicinal equipment under particularly mild and thus moderate conditions. Strong microbial abatement was observed for both enveloped (Herpes simplex) and non-enveloped viruses (Coxsackievirus B4), as well as for Gram-positive (Staphylococcus aureus) and Gram-negative bacteria (Pseudomonas aeruginosa).
Antibacterial action has also been reported for complex 18 in Figure 13. The VV thiosemicarbazone (tsc) complex [VO2(tsc)] and its VIV precursor [VO(acac)tsc] (acac = acetylacetonate(1–)) inhibit the pathogen of tuberculosis, Mycobacterium tuberculosis [71]. Minimal inhibitory concentrations (MIC values) of these vanadium complexes are lower than for the free tsc ligand, supporting a role of the vanadium center in these antituberculosis drugs.
4.2.3 Diseases Caused by Parasites
Vanadium coordination compounds have also been shown to exhibit potential in the abatement of epidemic diseases caused by parasites (amoebae and flagellates) predominantly in tropical and subtropical countries. Examples are amoebiasis, Chagas’ disease (American trypanosomiasis), and leishmaniasis. As in the case of antiviral and antibacterial vanadium compounds, none of these compounds has so far achieved the status of clinical tests, and studies have thus been restricted to in vitro tests with cultures of the parasites. In many cases, these studies reveal antiparasitic properties of the vanadium species, which are about comparable to or even more effective than established medications. In this section, selected examples of antiparasitic vanadium complexes are briefly described. For a recent overview see also the review by D. Gambino [72]. Respective vanadium compounds are illustrated in Figure 14; selected results are summarized in Table 4 [73–77].

Complexes and formulations that have been shown to exert antiparasitic potential against parasites causing amoebiasis (19), Chagas’ disease (20), and leishmaniasis (20, 21, 22). For details see text and Table 4.
According to the World Health Organisation, about 50 million people are affected worldwide by amoebiasis, with infections clearly cumulating in tropical countries. The etiologic agent of the disease is the amoeba Entamoeba histolytica, present in contaminated food and water in particular in areas of low sanitary and hygiene standards. Transmission occurs mainly by the fecal-oral route and through direct contact. 90% of the infected people are asymptomatic. For the remaining 10% the symptoms include diarrhea and, in more serious cases, dysentery (an inflammatory disorder mainly of the colon) with mucus and blood, the latter stemming from amoebae that succeed to overcome the epithelium of the intestines and thus travel to other organs, the liver in particular, where they cause deadly abscesses. The death toll amounts to ca. 70,000 per year. The hydrazone complex 19 in Figure 14 is an example for an efficient amoebocidal (pro)drug [73]. With an IC50 = 0.36 μM, the compound is more efficient than the standard drug Metronidazole (IC50 = 1.89 μM), and somewhat more toxic than Metronidazole against human cervical HeLa cancer cell lines.
Chagas’ disease is caused by the flagellate protozoan parasite Trypanosomas cruzi, transmitted by the feces of “kissing bugs”, blood-sucking bugs belonging to the subfamily Triatominae. About 10 million people are infected, with ca. 10,000 deaths per year. The disease is mainly distributed in Latin America, but also increasingly spreading into North America and Europe. Primary symptoms are skin lesions and swelling of the eye lids; secondary symptoms include digestive and neurological alteration, and cardiac disorder up to heart failure. The oxidovanadium complex 20 [74] contains a salicylidene semicarbazone ligand and hence a ligand that has also been shown to convey anticancer (compound 12) and antiamoebiasis activity (19). The additional ligand in 20 is phenanthroline, capable of intercalating DNA (Figure 12b). The metabolic pathways of parasites such as Trypanosoma and Leishmania are similar to those in tumor cells, suggesting a similar mode of action of anti-parasitic and anti-tumor drugs. Complexes such as compound 20, which are about as trypanocidal against epimastigotes (a developmental state of the parasite in the bug) of T. cruzi as the reference drug Nifurtimox, are in fact cytotoxic against leukemia cells [74] and cause conformational changes in plasmid DNA [75].
Complex 20 is also active on the promastigote and amastigote forms (the flagellate and non-flagellate stages, respectively) of Leishmania parasites, responsible for the tropical and subtropical disease leishmaniasis. The vectors for this disease, which is associated with malnutrition and weakness of the immune system, are sandflies of the subfamily Phlebotomina. About 12 million people are affected worldwide. The most serious form of this disease is visceral leishmaniasis, which goes along with high fever, weight loss, swelling of the spleen and liver, and anemia. Visceral leishmaniasis is mainly distributed in Brazil, India, Bangladesh, and Sudan, and accounts for 60,000 deaths each year.
Oxidovanadium complexes of galactomannan (21 in Figure 14) have been shown to be leishmanicidal on amastigotes of L. amazonensis, and to inhibit the growth of the promastigotes of this parasite [76]. Galactomannan is a polysaccharide with a mannose backbone and galactose side groups, isolated from the lichen Ramalina celastri, which is abundant in South Brazil. Antileishmanial effects have further been noted, with infected mice, for bis(peroxido)vanadate, [HVO2(O2)2]2–, and combinations of glutaminate and [HVO2(O2)2]2– (see 22 in Figure 14 for a tentative formulation of a formula unit) [77]. Finally, decavanadate deters the growth of the leishmania parasite, likely by interaction of decavanadate’s hydrolysis product [H2VO4]– with phosphatases [78], hence a mechanistic aspect which is again reminiscent of the antidiabetic, insulin-enhancing action of vanadate and (hydrolytically labile) vanadium complexes. For the molecular built-up of decavanadate see Figure 5.
5 Concluding Remarks and Prospects
The element vanadium was discovered in 1801 by Andrés Manuel del Rio y Fernández in vanadinite from the district Zimapán in Mexico [5]. It was rediscovered by Nils Gabriel Sefström in iron ore from the Taberg in Småland (Sweden) in 1830 by treating a “black powder obtained from the manufacturing of bar iron”, and in 1869/70 Sir Henry Enfield Roscoe in England was the first one who succeeded to isolate (an impure form of) metallic vanadium by reduction of VCl2 with H2 [79,87], nowadays widely employed in particularly tough and hard chromium-vanadium-iron alloys. Along with metallic vanadium, vanadium oxides (“VOx”) are in use as catalysts in oxidation reactions (an example is the production of sulfuric acid from SO2), as an UV-absorbing additive in glass ware, and in the form of lithium and silver vanadates in lithium batteries [80]. The first indication for the presence of vanadium in a living organism, i.e., the sugar beet, goes back to 1888 [81]; the essentiality of vanadium for some life forms became established in 1982/83 by the discovery of vanadate-dependent bromoperoxidase in the marine macro-alga Ascophyllum nodosum [82].
Toxic effects of vanadium were first reported by John Priestley [15], who applied large doses of sodium vanadate to animals, tediously describing their ailment and final death. Exploratory medicinal applications of vanadium, again in the form of vanadate, were carried out in Lyon (France) back in 1897–98 [83] on probands with health problems as diverse as anemia, tuberculosis, rheumatism, neurasthenia, and diabetes where, in the latter case, some decrease of blood glucose was observed. The prospective use of vanadium compounds in the treatment of diabetes thus has a long-standing tradition. The presently most advanced study of vanadium’s potential in the treatment of human diabetics are clinical tests phase IIa, carried out in 2007/8 with bis(ethylmaltolato)oxidovanadium(IV), BEOV [13], a compound developed in the group of Chris Orvig in Vancouver, Canada.
The successful application of vanadium compounds in low doses with diabetic animals (streptozotozin-induced rats in particular) and with type 2 diabetic humans has initiated research into medicinally active vanadium compounds for other medicinal applications, among these tropical and subtropical diseases caused by infectious parasites responsible for leishmaniasis, Chagas’ disease, and amoebiasis, just as diseases caused by viruses (HIV, influenza), and bacteria (tuberculosis). In all cases, in vitro tests with cell cultures or cultured parasites, as well as sporadic in vivo tests with infected test animals, have provided results that should encourage further investigations into the field of vanadium-based medications. Given that classical pharmaceuticals tend to have, sometimes severe, side effects, and parasites can develop resistance, the development of potential (pro-)drugs based on the essentially non-toxic element vanadium certainly is a future challenge.
The possibly most important aspect in choosing vanadium as the central metal in these coordination compounds is their (partial) rebuilt to vanadate(V) and oxidovanadium(IV) in the physiological broth. Vanadate is an antagonist of phosphate – as demonstrated for the first time by Cantley in 1977 for the inhibition of the Na,K-ATPase, the sodium-potassium pump [84]. Based on the interference of vanadate with metabolic processes depending on phosphate in general, and on phosphate-regulated enzymatic activity in particular, vanadate has also been introduced into the amelioration of cardiac dysfunctions, and in fighting malignant tumors. The latter application, first investigated systematically by Köpf and Köpf-Maier [60], is based, at least in part, on the direct interaction of stable vanadium-ligand fragments with the DNA of cancer cells – and is thus reminiscent of the action of the well established anticancer drug cisplatin. Stable fragments in vanadium compounds are represented by the cyclopentadienide ligands in vanadocenes, and by N-functional, oligodentate aromatic ligands such as ortho-phenanthroline and derivatives thereof. Last but not least, yet another functional aspect of vanadium compounds is the ability of VO2+ to interfere – in a Fenton-like reaction – with the tissue concentrations of reactive oxygen species [85].
- Ab:
-
albumin
- acac:
-
acetonylacetonate(1–)
- Akt:
-
protein kinase
- ATPase:
-
adenosine triphosphate cleaving enzyme
- BEOV:
-
bis(ethylmaltolato)oxidovanadium
- CAKI:
-
human renal carcinoma cell line
- CCR5:
-
chemokine receptor of T lymphocytes
- Cp:
-
cyclopentadienide
- CXCR-4:
-
chemokine receptor type 4
- FAD:
-
flavin adenine dinucleotide (oxidized form)
- FADH2 :
-
flavin adenine dinucleotide (reduced form)
- GLUT:
-
glucose transporter
- GSH:
-
glutathione
- GSSG:
-
oxidized form of glutathione
- HeLa cells:
-
cervical cancer cell line derived from Henrietta Lacks
- HIV:
-
human immune deficiency virus
- IC50 :
-
50% inhibitory concentration; the values denote the concentration of a drug or prodrug at which the life function of 50% of the viable cells are inhibited
- Ig:
-
immunoglobulin
- IR:
-
insulin receptor
- IRS:
-
insulin receptor substrate
- lmm:
-
low molecular mass (ligand)
- MAC:
-
maximum allowable concentration at the working place
- MIC:
-
minimal inhibitory concentration
- NAD:
-
nicotinamide adenine dinucleotide (oxidized form)
- NADH:
-
nicotinamide adenine dinucleotide (reduced form)
- NADP:
-
nicotinamide adenine dinucleotide phosphate (oxidized form)
- NADPH:
-
nicotinamide adenine dinucleotide phosphate (reduced form)
- PIK:
-
phosphatidyl-inositol kinase
- PKB:
-
protein kinase B
- PTPase:
-
protein tyrosine phosphatase
- ROS:
-
reactive oxygen species
- SARS:
-
severe acute respiratory syndrome
- STZ:
-
streptozotocin
- SV:
-
Simian virus
- Tf:
-
transferrin
- tsc:
-
thiosemicarbazone
References
(a) D. Rehder, Future Med. Chem. 2012, 4, 1823–1837. (b) D. Rehder, in Detoxification of Heavy Metals, Eds I. Sherameti, A. Varma, Soil Biology 2011, 30, ch. 11.
T. Shimizu, K. Ichikawa, M. Masayuki, Y. Shijo, Bunseki Kagaku 1989, 38, 201–203.
T. L. Gerke, K. G. Scheckel, M. R Schock, Environ. Sci. Technol. 2009, 43, 4412–4418.
G. Giuli, G. Pratesi, S. G. Eeckhout, E. Paris, 68th Ann. Meteoritic. Soc. Meeting, Zurich 2006, p. 5148.
D. Rehder, Bioinorganic Vanadium Chemistry, John Wiley & Sons, Chichester, 2008, pp 87–155.
D. G. Barcelaux, J. Toxicol. Clinic. Toxicol. 1999, 37, 265–278.
M. D. Cohen, ACS Symposium Series 2007, 974, 217–239. (b) T. Scior, A. Guevara-García, P. Bernard, Q.-T. Do, D. Domeyer, S. Laufer, Mini Rev. Med. Chem. 2005, 5, 995–1008.
J. O. Olopade, J. R. Condor, Curr. Topics Toxicol. 2011, 7, 33–39.
G. Heinemann, B. Fichti, W. Vogt, Clin. Pharmacol. 2003, 55, 241–245.
A. Gorzsás, I. Andersson, L. Pettersson, Eur. J. Inorg. Chem. 2006, 3559–3565.
D. Sanna, P. Buglyó, G. Micera, E. Garribba, J. Biol. Inorg. Chem. 2010, 15, 825–839.
(a) D. Sanna, L. Bíro, P. Buglyó, G. Micera, E. Garribba, J. Inorg. Biochem. 2012, 115, 87–99. (b) G. C. Justino, E. Garribba, S. Mehtab, J. Costa Pessoa, J. Biol. Inorg. Chem. 2013, 18, 803–813.
K. H. Thomson, J. Lichter, C. LeBel, M. C. Scaife, J. H. McNeill, C. Orvig, J. Inorg. Biochem. 2009, 103, 554–558.
T. C. Delgado, A. I. Tomaz, I. Corriera, J. Costa Pessoa, J. G. Jones, C. F. G. C. Geraldes, M. M. C. A. Castro, J. Inorg. Biochem. 2005, 99, 2328–2339.
J. Priestley, Phil. Trans. Roy. Soc. London 1876, 166, 495–556.
V. A. Ehrlich, A. K. Nersesyan, K. Alefie, C. Hoelzl, F. Ferk, J. Bichler, E. Valic, A. Schaffer, R. Schulte-Herrmann, M. Fenech, K.-H. Wagner, S. Knasmüller, Environ. Health Perspect. 2008, 116, 1689–1693.
H. P. Monteiro, C. C. Winterburn, A. Stern, Free Radic. Res. Commun. 1991, 12–13, 125–129.
N. Gao, M. Ding, J. Z. Zheng, Z. Zhang, S. S. Leonard, K. J. Liu, X. Shi, B.-H. Jiang, J. Biol. Chem. 2002, 277, 31963–31971.
H. Kelm, H.-J. Krüger, Angew. Chem. Int. Ed. 2001, 40, 2344–2348.
E. J. Baran, Chem. Biodivers. 2008, 5, 1475–1484.
(a) J. R. Treberg, J. E. Stacey, W. R. Driedzic, Comp. Biochem. Physiol. B 2012, 161, 323–330. (b) H. Michibata,T. Ueki, Biomol. Concepts 2010, 1, 97–107.
J. Krakowiak, D. Lundberg, I. Persson, Inorg. Chem. 2012, 51, 9598–9609.
M. Kaliva, E. Kyriakakis, A. Salifoglou, Inorg. Chem. 2002, 41, 7015–7023.
H. Schmidt, I. Andersson, D. Rehder, L. Pettersson, Chem. Eur. J. 2001, 7, 251–257.
J. Schemberg, K. Schneider, U. Demmer, E. Warkentin, A. Müller, U. Ermler, Angew. Chem. Int. Ed. 2007, 46, 2409–2413.
(a) L. Wittenkeller, A. Abraha, R. Ramasamy, D. Mota de Freitas, L. A. Theisen, D. C. Crans, J. Am. Chem. Soc. 1991, 113, 7872–7881. (b) N. Steens, A. M. Ramadan, T. N. Parac-Vogt, Chem. Commun. 2009, 965–967.
S. Lobert, N. Isern, B. S. Hennington, J. J. Correia, Biochemistry 1994, 33, 6244–6252.
M. Aureliano, Dalton Trans. 2009, 9093–9100.
S. S. Soares, C. Gutiérrez-Merino, M. Aureliano, J. Inorg. Biochem. 2007, 101, 789–796.
(a) N. E. Levinger, L. C. Rubenstrunk, B. Baruah, D. C. Crans, J. Am. Chem. Soc., 2011, 133, 7205–7214. (b) D. C. Crans, N. E. Levinger, Acc. Chem. Res., 2012, 45, 1637–1645.
(a) M. Weyand, H.-J. Hecht, M. Kieß, M.-F. Liaud, H. Vilter, D. Schomburg, J. Mol. Biol. 1999, 293, 595–611. (b) L. Kaysser, P. Bernhardt, S.-J. Nam, S. Loesgen, J. G. Ruby, P. Skewes-Cox, P. R. Jensen, W. Fenical, B. S. Moore, J. Am. Chem. Soc. 2012, 134, 11988–11991.
(a) Y. Lindquist, G. Schneider, P. Vihko, Eur. J. Biochem. 1994, 221, 139–142. (b) M. Z. Mehdi, A. K. Srivastava, Arch. Biochem. Biophys. 2005, 440, 158–164. (c) M. Zhang, M. Zhou, R. L. Van Etten, C. V. Stauffacher, Biochemistry 1997, 36, 15–23.
N. Tanaka, V. Dumay, Q. Liao, A. J. Lange, R. Wever, Eur. J. Biochem. 2002, 269, 2162–2167.
R. Renirie, W. Hemrika, R. Wever, J. Biol. Chem. 2000, 16, 11650–11667.
C. C. McLauchlan, J. D. Hooker, M. A. Jones, Z. Dymon, E. A. Backhus, B. A. Greiner, N. A. Dorner, M. A. Youkhana, L. M. Manus, J. Inorg. Biochem. 2010, 104, 274–281.
B. Akabayov, A. W. Kulczyk, S. R. Akabayov, C. Theile, L. W. McLaughlin, B. Beauchamp, A. M. van Oijen, C. C. Richardson, J. Biol. Chem., 2011, 286, 29146–29157.
M. J. Gresser, A. S. Tracey, K. M. Parkinson, J. Am. Chem. Soc. 1986, 108, 6229–62343.
T. Kiss, E. Kiss, G. Micera, D. Sanna, Inorg. Chim. Acta 1998, 283, 202–210.
I. Andersson, A. Gorzsás, C. Kerezsi, I. Tóth, L. Pettersson, Dalton Trans. 2005, 3658–3666.
I. Andersson, S. Angus-Dunge, O. W. Howarth, L. Pettersson, J. Inorg. Biochem. 2000, 80, 51–58.
(a) A. S. Tracey, M. J. Gresser, Proc. Natl. Acad. Sci. USA 1986, 83, 609–613. (b) A. S. Tracey, M. J. Gresser, Can. J. Chem. 1988, 66, 2570–2574.
(a) T. Scully, Nature 2012, 485, S2–S3. (b) L. Chen, D. J. Magliano, P. L. Zimmer, Nat. Rev. Endocrinol. 2012, 8, 228–236.
C. Talchai, S. Xuan, H. V. Lin, L. Sussel, D. Accili, Cell 2012, 150, 1223–1234.
M. F. Dunn, Biometals 2005, 18, 295–303.
M. Hiromura, Y. Adachi, M. Machida, M. Hattori, H. Sakurai, Metallomics 2009, 1, 92–100.
G. R. Willsky, L.-H. Chi, M. Godzala III, P. J. Kostyniak, J. J. Smee, A. M. Trujillo, J. A. Alfano, W. Ding, Z. Hu, D. C. Crans, Coord. Chem. Rev. 2011, 255, 2258–2269.
J. Gätjens, B. Meier, Y. Adachi, H. Sakurai, D. Rehder, Eur. J. Inorg. Chem. 2006, 3575–3585.
J. Nilsson, A. A. Shteinman, E. Degerman, E. A. Enyedy, T. Kiss, U. Behrens, D. Rehder, E. Nordlander, J. Inorg. Biochem. 2011, 105, 1795–1800.
H. Ou, L. Yan, D. Mustafi, M. W. Makinen, M. J. Brady, J. Biol. Inorg. Chem. 2005, 10, 874–886.
S. Karmaker, T. K. Saha, Y. Yoshikawa, H. Sakurai, ChemMedChem 2007, 2, 1607–1612.
R. Mukherjee, E. G. Donnay, M. A. Radomski, C. Miller, D. A. Redfern, A. Gericke, D. S. Damron, N. E. Brasch, Chem. Commun. 2008, 3783–3785.
T. A. Clark, C. E. Heylinger, M. Kopilas, A. L. Edel, A. Junaid, F. Aguilar, D. D. Smyth, J. A. Thliveris, M. Merchant, H. K. Kim, G. N. Pierce, Metabolism 2012, 61, 742–753.
K. G. Peters, M. G. Davis, B. W. Howard, M. Pokross, V. Rastogi, C. Diven, K. D. Greis, E. Eby-Wilkens, M. Maier, A. Evdokimov, S. Soper, F. Genbauffe, J. Inorg. Biochem. 2003, 96. 321–330.
A. Bishayee, A. Waghray, M. P. Patel, M. Chatterjee, Cancer Lett. 2010, 294, 1–12.
M. M. Evangelou, Oncol. Hematol. 2002, 42, 249–265.
H. Faneca, V. A. Figueiredo, I. Tomaz, G. Gonçalves, F. Avecilla, M. C. Pedroso de Lima, C. F. G. C. Geraldes, J. Costa Pessoa, M. M. C. A. Castro, J. Inorg. Biochem. 2009, 103, 601–608.
I. Fichtner, J. Claffey, A. Deally, B. Gleeson, M. Hogan, M. R. Markelova, H. Müller-Bunz, H. Weber, M. Tacke, J. Organomet. Chem. 2010, 695, 1175–1181.
O. J. D’Cruz, F. M. Uckun, Expert Opin. Investig. Drugs 2002, 11, 1829–1836.
N. A. Lewis, F. Liu, L. Seymour, A. Magnuse, T. R. Erves, J. Faye Arca, F. A. Beckford, R. Venkatraman, A. Gonzáles-Sarrías, F. R. Fronczkek, D. G. VanDerveer, N. P. Seeram, A. Liu, W. L. Jarrett, A. A. Holder, Eur. J. Inorg. Chem. 2012, 664–677.
P. Köpf-Maier, H. Köpf, Drugs Future 1986, 11, 297–319.
(a) Q. Wang, T.-T. Liu, Y. Fu, K. Wang, X.-G. Yang, J. Biol. Inorg. Chem. 2010, 15, 1087–1097. (b) T.-T. Liu, Y.-J. Liu, Q. Wang, X.-G. Yang, K. Wang, J. Biol. Inorg. Chem. 2012, 17, 311–320.
J. O. Olopade, A. B. Madhan Kumar, A. Das, X. Liu, B. Todorich, J. L. Liang, B. Slagle-Webb, J. R. Connor, Proc. Am. Ass. Cancer Res. 2009, 50, 1344 (#5575).
(a) Md. S. Bhuiyan, K. Fukunaga, J. Pharmacol. Sci. 2009, 110, 1–13. (b) K. Fukunaga, Yakugaku Zasshi 2012, 132, 279–284.
D. A. Liem, C. C. Gho, B. C. Gho, S. Kazim, O. C. Manintveld, P. D. Verdouw, D. J. Duncker, J. Pharmacol. Exp. Therapeut. 2004, 309, 1256–1262.
C. L. Walker, M. J. Walker, N.-K. Liu, E. C. Risberg, X. Gao, J. Chen, X,-M. Xu, PLoS ONE 2012, 7, e30012.
K. T. Keyes, J. Xu, B. Long, C. Zhang, Z. Hu, Y. Ye, Am. J. Phys. Heart Circ. Physiol. 2010, 298, H1198–H1208.
(a) S. Shigeta, S. Mori, T. Yamase, N. Yamamoto, N. Yamamoto, Biomed. Pharmacother. 2006, 60, 211–219. (b) T. Yamase, E. Ishikawa, K. Fukaya, H. Nojiri, T. Taniguchi, T. Atake, Inorg. Chem. 2004, 43, 8150–8157.
S.-Y. Wong, R. W.-Y. Sun, N. P.-Y. Chung, C.-L. Lin, C.-M. Che, Chem. Commun. 2005, 3544–3546.
A. Ross, D. C. Soares, D. Covelli, C. Pannecouque, L. Budd, A. Collins, N. Robertson, S. Parsons, E. De Clercq, P. Kennepohl, P. J. Sadler, Inorg. Chem. 2010, 49, 1122–1132.
R. Renirie, A. Dewilde, C. Pierlot, R. Wever, D. Hober, J.-M. Aubry, J. Appl. Microbiol. 2008, 105, 264–270.
P. I. da S. Maia, F. R. Pavan, C. Q. F. Leite, S. S. Lemos, G. F. de Sousa, A. A. Batista, O. R. Nascimento, J. Ellena, E. E. Castellano, E. Niquet, V. M. Deflon, Polyhedron 2009, 28, 398–406.
D. Gambino, Coord. Chem. Rev. 2011, 255, 2193–2203.
M. R. Maurya, A. A. Khan, A. Azam, S. Ranjan, N. Mondal, A. Kumar, F. Avecilla, J. Costa Pessoa, Dalton Trans. 2010, 39, 1345–1360.
J. Benítez, L. Becco, I. Correira, S. M. Leal, H. Guiset, J. Costa Pessoa, J. Lorenzo, S. Tanco, P. Escobar, V. Moreno, B. Garat, D. Gambino, J. Inorg. Biochem. 2011, 105, 303–312.
J. Benítez, L. Guggeri, I. Tomaz, G. Arrambide, M. Navarro, J. Costa Pessoa, B. Garat, D. Gambino, J. Inorg. Biochem. 2009, 103, 609–616.
G. R. Noleto, A. L. R. Mercê, M. Iacomini, P. A. J. Gorin, V. T. Soccol, M. B. M. Oloveira, Molec. Cellul. Biochem. 2002, 233, 73–83.
A. K. Haldar, S. Banerjee, K. Naskar, D. Kalita, N. S. Islam, S. Roy, Experim. Parasitol. 2009, 122, 145–154.
T. L. Turner, V. H. Nguyen, C. C. McLauchlan, Z. Dymon, B. M. Dorsey, J. D. Hooker, M. A. Jones, J. Inorg. Biochem. 2012, 108, 96–104.
H. E. Roscoe, Phil. Trans. Roy. Soc. A 1870, 160, 317–331.
Y. J. Kim, A. C. Marschilok, K. J. Takeuchi, E. S. Takeuchi, J. Power Sources 2011, 196, 6781–6787.
E. O. von Lippmann, Ber. Dtsch. Chem. Ges. 1888, 21, 3492–3493.
H. Vilter, K.-W. Glombitza, A. Grawe, Bot. Mar. 1983, 26, 331–340.
B. Lyonnet, X. Martz, E. Martin, La Presse Médicale 1899, 32, 191–192.
L. C. Cantley, Jr., L. Josephson, R. Warner, M. Yanagisawa, C. Lechene, G. Guidotti, J. Biol. Chem. 1977, 252, 7421–7423.
D. C. Crans, A. M. Trujillo, P. S. Pharazyn, M. D. Cohen, Coord. Chem. Rev. 2011, 255, 2178–2192.
L. Nordquist, M. Johansson, Vasc. Health Risk Manag. 2008, 4, 1283–1288.
H. E. Roscoe, Philos. Mag. 1870, 39, 146–150.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Rehder, D. (2013). Vanadium. Its Role for Humans. In: Sigel, A., Sigel, H., Sigel, R. (eds) Interrelations between Essential Metal Ions and Human Diseases. Metal Ions in Life Sciences, vol 13. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-7500-8_5
Download citation
DOI: https://doi.org/10.1007/978-94-007-7500-8_5
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-7499-5
Online ISBN: 978-94-007-7500-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)