
Abstract
The use of multiparental populations for QTL discovery has been recently highlighted by different theoretical and experimental developments. Here, we explored the interest of French populations using heterogeneous genetic stocks of cultivated wheat, maintained in situ over 12 sites since 1984 with an outcrossing mating system. We studied one of these populations (Le Moulon, 48.4°N, 21°E), derived from 12 cycles of random crosses between 60 founders, selected to maximize genetic diversity. Outcrossing was allowed by the integration of a nuclear male sterility allele (ms1b, Probus donor) in the population. We analyzed 1,000 Single Seed Descent lines (SSD) derived from the 12th generation of cultivation. This population was genotyped using the 9 K i-select SNPs (Single Nucleotide Polymorphisms) array, covering the whole genome. Polymorphism and quality checks resulted in the selection of around 6,500 SNPs. First, the evolution of genetic diversity was explored through the comparison of SSD lines and the inferred initial population. The low population structure and the strong decay in linkage disequilibrium between SSD lines and the inferred initial population confirmed the efficiency of the 12 cycles of the random outcrossing in producing a highly diverse and recombined population. Two years of observations of population earliness under different environments were used to show the complementarity of association genetics, which allowed the detection of already known Vrn major genes, and evolutionary approach, which, lead to the discovery of two new minor effect QTLs.
You have full access to this open access chapter, Download conference paper PDF
Similar content being viewed by others

Keywords
Introduction
Dynamic management (DM) aims at maintaining crop genetic diversity through in situ conservation of genetic resources. Genetically diverse populations are grown year after year, in various sites, differing for climate conditions, pathogen pressures and/or agricultural practices (Allard 1988; Henry et al. 1991; Porcher et al. 2004). In France, dynamic management has been experimented on bread wheat (Triticum aestivum L.) since 1984 (Henry et al. 1991), using three gene pools: two selfing populations (based on a pyramidal cross of 16 parents) and one outcrossing population. Samples of each of the three initial populations were sent to 7–12 sites in France and cultivated year after year in the same sites under the same conditions. Thus, these three “meta-populations” evolved over 10 to more than 20 generations without migration or conscious human selection (Enjalbert et al. 2011).
Studies on the selfing populations showed a good maintenance of global diversity at the network level (Raquin et al. 2008), both at phenotypic and molecular levels. A fast evolution of flowering time was observed, both over time and space (Rhoné et al. 2008): all populations flowered later than the initial population; and populations from Northern French sites flowered much later than Southern ones. Association genetics and spatio-temporal shifts in allelic frequencies revealed polymorphisms located in major genes controlling vernalization requirement or photoperiod sensitivity, partially explained climatic adaptation (Rhoné et al. 2008). Therefore, in addition to genetic resource preservation, DM populations can be an appropriate material to detect genes involved in local adaptation (Goldringer et al. 2001).
In the present study, we analyzed one outcrossing DM population, which is characterized by a high number of parents (60 lines), and numerous panmictic generations. In this population, wheat natural selfing habit was turned to an outcrossing mating system, using a recessive male sterility gene (ms1b), and harvesting solely open pollinated male-sterile plants. Taking the parental lines as reference, we studied an evolved population (12th generation) and tested for possible markers selection, trying to link detected markers to the observed evolution in vernalization requirement.
Materials and Methods
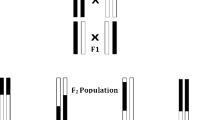
The population studied is derived from the cross of Probus, a mutant carrying a male sterile allele (ms1b), with 59 lines covering a large genetic diversity. Resulting F1 progenies were alternatively selfed (F2) or back-crossed (BC1) with the 59 parents to reduce the Probus genome contribution to the population. These two progenies (F2 & BC1) were sown together in an isolated field surrounded by rye. Male sterile spikes were tagged during flowering and harvested at maturity. Then, and over 12 generations, a random sample of harvested seeds on male sterile plants was drawn each year, and resown in fall, in order to reach between 5,000 and 10,000 adult plants (2,500–5,000 male-sterile plants). After 12 generations we derived 1,000 SSD lines (F5, Fig. 38.1).

Population creation scheme
First we inferred allelic frequencies in the initial population on the basis of the 56 parental lines (including Probus) still available in seed banks (four missing ones), estimating their contributions to the global pool using a Bayesian method (Thépot et al. 2015). The evolved population was studied through a subset of 380 SSD lines, representative of the phenotypic diversity of the 1,000 lines.
Vernalization requirement was assessed in field trial at Le Moulon over two seasons (2010–2011 and 2011–2012), with a spring sowing (April), on a single row of 20 seeds per genotype. For each row, the heading date was scored when half of the plants had half of the main ear emerged from the flag leaf. The heading date was transformed into sums of degree-days (dd) (sums of the mean temperature per day) from sowing to heading. On the basis of the bimodal distribution of the heading date (Fig. 38.2), SSD lines were classified as spring type (heading before 2,000dd), or winter type for the others. Genotypes with inconsistent behavior between both years were discarded (eight SSD lines).

Heading date distribution of all genotypes for spring sowing in 2011, the condition with the least vernalization to determine winter and spring type
Genotyping was performed using the 9 K i-select SNP array. Only SNPs unambiguously scored as biallelic after a visual inspection using Genome Studio software, were kept. Using KASPar SNP genotyping system (KBioscience), 14 additional polymorphisms located in candidate genes (earliness pathway such as PPD or VRN families) were genotyped.
The diversity detected by SNPs in both populations (parental lines and SSD lines) was compared using Minor Allele Frequency (MAF) and expected heterozygosity (He, Nei diversity). Evolution of growth habit was tested through a comparison of spring/winter ratio in the initial and the evolved populations (Chi square test). Strong shifts in allelic frequencies were used to detect markers under selection, using a new method (Thépot et al. 2015). Q-values were estimated to cope with the multiple tests (Storey and Tibshirani 2003). Each marker under selection was also tested for association with growth habit using a Logit model.
Results and Discussion
The genotyping of 436 lines (56 parents + 380 F5 lines) with the 9 K i-select SNP assay resulted in 7,270 SNPs with high scoring quality. Among these SNPs, 88.4 % were polymorphic in the initial population and 85.8 % in the evolved population. This slightly higher diversity in the initial population was also observed on allelic frequencies of polymorphic SNPs (mean MAF: 0.18 vs. 0.17 and He: 0.25 vs. 0.24). The MAF distribution (Fig. 38.3) showed a globally high frequency of SNPs with a MAF inferior to 0.05, rare alleles being more frequent in the evolved population. This distribution contrasts with the one observed on a worldwide panel, using the same SNP array, which demonstrates a deficit of SNPs with a low MAF (Cavanagh et al. 2013). This deficit might be due to the fact that lines were chosen to maximize the genetic diversity, and SNPs were intentionally selected to favor common allele in a panel of 26 cultivars from mainly USA and Australia (Cavanagh et al. 2013).

Distribution of minor allele frequency in the initial and evolved populations
Among the 6,476 polymorphic markers, 57 were detected under selection, representing 26 independent genomic areas. When assessing phenotypic evolution for flowering time, we observed a significant shift from 20 % of spring type in the initial population to 47 % in the evolved population. Among markers under selection, three were associated with the growth habit (p-value <0.05). These markers, located on the 5D and 4A chromosomes, only explained a rather limited part of the phenotypic variation (2.8 % with a global model included the three markers). Yet, they all have, experienced an increase of the spring allele frequency in the evolved population which explained a raise of spring type ranging from 3.5 % to 5.4 %. The 5D markers are located on the same chromosome as Vrn-D1 but did not present linkage disequilibrium with the marker located in this gene (r2 <0.009). Surprisingly polymorphisms in candidate genes like Vrn families (Vrn-A1-Prom, Vrn-A1-ex7 and Vrn-D1), although strongly associated to the growth habit (p-value <10−10, r2 = 28 %) have not been detected as targeted by selection. To take into account the complexity of interaction between these three markers, we assumed that spring alleles are both dominant and epistatic (Rousset et al. 2011). Thus as soon as there is at least one spring allele, the haplotype was classified as spring type or winter type otherwise. For parental lines, these Vrn haplotypes are almost completely explaining phenotype (97.5 % of correspondence, r2 = 0.85). However for the evolved population 30 % of SSD lines with winter Vrn haplotype exhibited a spring phenotype (Table 38.1) (r2 = 0.3). This evolution might be explained by (i) high level of recombination that broke the initial full linkage disequilibrium between causal mutations and the three SNPs genotyped, and/or (ii) the increase of spring alleles at one (or several) non-genotyped Vrn genes, such as Vrn-B1. As SNPs, Vrn haplotypes did not present a significant shift (p-value = 0.29), although a 6 % increase of spring haplotypes was observed between the initial and the evolved population (Table 38.1). One hypothesis to explain this absence of significant shift at these candidate genes could be their strong effect: a little variation in frequency at these major genes may have a strong effect on growth habit.
Association genetics and evolutionary approach provided complementary results. The first method detected QTLs with major effects while the second detected QTLs with lower effect but contributing to the evolution of phenotypes. Joint study of phenotypic and genetic evolutions allowed to detect new markers involved in the control of the growth habit on the 5D and 4A chromosomes.
With its high diversity, absence of structure and low LD (Thépot et al. 2015), this population appears as a new QTL mapping resource, allowing the discovery of original genomic regions controlling traits of interest. Ongoing studies will better explore the potential of this population for detection, using the 1,000 SSD lines.
References
Allard RW (1988) Genetic changes associated with the evolution of adaptedness in cultivated plants and their wild progenitors. J Hered 79:225–238
Cavanagh CR, Chao S, Wang S et al (2013) Genome-wide comparative diversity uncovers multiple targets of selection for improvement in hexaploid wheat landraces and cultivars. Proc Natl Acad Sci. doi:10.1073/pnas.1217133110
Enjalbert J, Dawson JC, Paillard S et al (2011) Dynamic management of crop diversity: from an experimental approach to on-farm conservation. C R Biol 334:458–468. doi:10.1016/j.crvi.2011.03.005
Goldringer I, Enjalbert J, Raquin A-L, Brabant P (2001) Strong selection in wheat populations during ten generations of dynamic management. Genet Sel Evol 33:S441–S463
Henry JP, Pontis C, David J, Gouyon PH (1991) An experiment on dynamic conservation of genetic resources with metapopulation. Species Conserv Popul Biol Approach 185–198. Birkhäuser Basel
Porcher E, Gouyon P-H, Lavigne C (2004) Dynamic management of genetic resources: maintenance of outcrossing in experimental metapopulations of a predominantly inbreeding species. Conserv Genet 5:259–269
Raquin A-L, Depaulis F, Lambert A et al (2008) Experimental estimation of mutation rates in a wheat population with a gene genealogy approach. Genetics 179:2195–2211
Rhoné B, Remoué C, Galic N et al (2008) Insight into the genetic bases of climatic adaptation in experimentally evolving wheat populations. Mol Ecol 17:930–943
Rousset M, Bonnin I, Remoué C et al (2011) Deciphering the genetics of flowering time by an association study on candidate genes in bread wheat (Triticum aestivum L.). Theor Appl Genet. doi:10.1007/s00122-011-1636-2
Storey JD, Tibshirani R (2003) Statistical significance for genomewide studies. Proc Natl Acad Sci 100:9440–9445. doi:10.1073/pnas.1530509100
Thépot S, Restoux G, Goldringer I et al (2015) Efficiently tracking selection in a multiparental population: the case of earliness in wheat. Genetics 199:609–623.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is distributed under the terms of the Creative Commons Attribution Noncommercial License, which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Copyright information
© 2015 The Author(s)
About this paper
Cite this paper
Thépot, S. et al. (2015). Interest of a Multiparental and Outcrossing Wheat Population for Fine Mapping. In: Ogihara, Y., Takumi, S., Handa, H. (eds) Advances in Wheat Genetics: From Genome to Field. Springer, Tokyo. https://doi.org/10.1007/978-4-431-55675-6_38
Download citation
DOI: https://doi.org/10.1007/978-4-431-55675-6_38
Publisher Name: Springer, Tokyo
Print ISBN: 978-4-431-55674-9
Online ISBN: 978-4-431-55675-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)