
Abstract
International Wheat Genome Sequencing Consortium (IWGSC) decided to adopt the strategy of chromosome sorting and short read assembly to overcome difficulties of wheat genome sequencing derived from the hexaploid status, the large genome size (about 17 Gb) and high repeat contents (more than 80 %). Our Japanese group was responsible for the sequencing of wheat chromosome 6B. Using DNAs from the flow-sorted chromosome arms, we conducted whole-chromosome shotgun sequencing of chromosome 6B. We sequenced more than 12 million reads obtained from the short and long arms by GS-FLX Titanium, and assembled contigs of 235 Mb for 6BS and 273 Mb for 6BL were generated by GS assembler 2.7 (Roche). These assemblies cover 56.6 % and 54.9 % of estimated sizes of 6BS (415 Mb) and 6BL (498 Mb), respectively. We annotated repetitive regions covering more than 80 % of contigs, 4,798 possible expressed loci, and various kinds of RNA genes using our annotation pipeline. We also found the evolutionary conserved regions among syntenic chromosomes from four grass genomes. For application of the 6B sequences to wheat genomics, various kinds of markers, such as simple sequence repeat (SSR) and insertion site-based polymorphism (ISBP) markers were constructed. Combination of the marker data with the comparative genome analysis will lay a strong foundation of functional genomics of the group-6 chromosomes in wheat.
You have full access to this open access chapter, Download conference paper PDF
Similar content being viewed by others


Keywords
Chromosome by Chromosome Sequencing
Completed rice genome sequence in 2004 by International Rice Genome Sequencing Project was the first cereal genome sequence (IRGSP 2005), and then sorghum and maize genome sequences were followed (Paterson et al. 2009; Schnable et al. 2009). For the rice genome sequencing, because BAC by BAC sequencing method using Sanger sequencing was adopted, the sequencing accuracy was less than one error in 10 kb. This accuracy was validated by genome resequencing by Next Generation Sequence (NGS) data (Kawahara et al. 2013), and it showed that the rice genome sequence is the most accurate one in the cereal genomes sequenced so far. Sorghum genome was determined by whole-genome shotgun sequencing method and maize genome sequence was achieved by the combination of the minimum tiling path (MTP) method and BAC by BAC sequencing. However, their genome sequences were less accurate than rice genome and were still fragmented into many scaffolds.
In Pooideae, Brachypodium distachyon Bd21 genome was sequenced in 2010 by whole-genome shotgun sequencing method, because of its small genome size (272 Mb) (The International Brachypodium Initiative 2010). However, compared with the B. distachyon genome sequencing, the sequencing of other Pooideae genomes, such as wheat (Triticum aestivum L.) and barley (Hordeum vulgare L.) has fallen behind due to the complexity of their genome structures. First, the wheat and barley genome size was 17 and 5.1 Gb, respectively. They are more than 40-times and 13-times larger than rice genome. Second, repeat regions occupied more than 80 % of the genome hamper their genome assembly. Third, in particular, since wheat is a hexaploidy, it is quite hard to distinguish homoeologous sequences from A, B and D genomes.
To overcome these problems, the various new methods were applied. NGS technology enables us to assemble large sized genome with the low cost. Even if the NGS read length is several hundred bp, millions of NGS reads can be used in one analysis (the total read length is up to several Gb) so that assembly of large genomes can be conducted. For the barley genome sequencing, BAC by BAC sequencing and NGS sequencing methods were combined, and then 1.9 Gbp of the sequences were released in 2012 (The International Barley Genome Sequencing Consortium 2012). In 2013, the genome sequences of Aegilops tauschii and T. urartu were determined (Jia et al. 2013; Ling et al. 2013; Luo et al. 2013). Wheat genome was also sequenced by whole-genome shotgun technology with NGS data (Brenchley et al. 2012). However, because of the hexaploidy, whole genome assembly was not achieved as same as other diploid genomes of Triticeae.
To solve the genome complexity, chromosome sorting by flow cytometry was developed in cereal genomics (Doležel et al. 2007). This method can reduce sample complexity, such as the hexaploid status of the wheat genome, therefore International Wheat Genome Sequencing Consortium (IWGSC) decided to apply this technologies to their activity. Single chromosomes or chromosome arms were sorted by the flow cytometric analysis and chromosome (arm)-specific BAC libraries were constructed. Progress of physical map construction and genome sequencing of each chromosome and chromosome arms can be seen on the IWGSC website (http://www.wheatgenome.org/) and URGI wheat portal site (http://wheat-urgi.versailles.inra.fr/).
Survey Sequencing and Annotation of Chromosome 6B
Under the framework of IWGSC, sequencing project of chromosome 6B was started in Japan in 2011 and the first survey sequences of 6B was released in 2013 (Tanaka et al. 2014). In this analysis, the DNA libraries of sorted 6B chromosome arms were constructed and sequenced independently using the 454 GS-FLX Titanium (Roche, CT, USA). The sequence reads (454 reads) from each arm were assembled by GS assembler 2.7 (Roche). From more than 12 million reads for each arm, 234 and 273 Mbp were assembled comprising 262,375 and 173,655 contigs for 6BS and 6BL, respectively. They correspond to 56.6 % and 54.9 % of the estimated lengths of both arms (415 Mbp for 6BS and 498 Mbp for 6BL).
As described before, the wheat genome is composed of abundant repetitive elements. Known classes of repeat elements were detected using the repeat libraries, such as TREP and MIPS repeat libraries. In addition, to detect novel repeat elements, we constructed a new repeat library by RepeatModeler (http://www.repeatmasker.org/RepeatModeler.html). Using a repeat masking program, censor (http://www.girinst.org/censor/index.php) with TREP and the new repeat library (Jurka et al. 1996), 76.6 % and 85.5 % of 6BS and 6BL assembly were masked, respectively. Since 63.6 % and 72.2 % of 6BS and 6BL assemblies were masked by TREP library, around 13 % of repetitive regions may be novel repeat elements detected only by the new library.
After repeat detection, we identified transcribed regions by mapping many transcripts in public domains. In addition to mRNA and millions of ESTs in DDBJ/EMBL/GenBank, wheat full-length cDNAs (FLcDNAs) were available from TriFLDB (http://trifldb.psc.riken.jp/index.pl) (Mochida et al. 2009). In combination with transcriptome mapping and an ab initio gene prediction program, 4,798 transcribed regions were determined. We found several genes that were known to locate on chromosome 6B, such as α-gliadin gene, the stripe rust resistance gene Yr36, the grain protein content gene Gpc-B1, α-amylase gene, the genes for three low-temperature-responsive dehydrins, Wcs120, Wcs66 and Wcor410, the flowering time gene TaHd1-2 and the gene involved in vernalization TmVIL2.
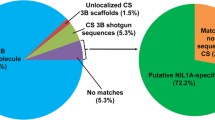
Our assemblies also showed the conservation of syntenic genes between monocots. First, 2,399 of 2,573 high-confidence barley genes on chromosome 6H could be mapped on our assemblies (E value <10−5). Second, 3,772 syntenic loci were detected from homology search of syntenic genes from chromosome 2 of O. sativa, chromosome 3 of B. distachyon and chromosome 4 of S. bicolor. Since 57.4 % of the syntenic regions had wheat transcriptome evidence, which was significant higher than that of non-syntenic regions (32.7 %), we concluded that wheat 6B has a conserved synteny with the chromosomes of other grass species.
Our annotation pipeline included detection of RNA genes, rRNAs, tRNAs, and miRNAs. It is known that chromosome 6B has a locus for ribosomal DNA (rDNA) containing approximately 5,500 rRNA genes. Moreover, non-protein coding RNAs, such as microRNAs (miRNAs) are currently recognized as biologically important genetic components. We found that some RNA genes were associated to a particular repetitive element. For example, 83 of 131 tRNALys were located in an LTR retrotransposon, Gypsy, and de novo repeats. Almost predicted miRNAs were also located in repeat-masking regions, especially DNA transposons, Mariner and CACTA. In case of rRNA genes, the quite small number of contigs with rRNA genes could be explained by high read depth of contigs. Because of the high sequence similarity, rRNA regions were degenerated during the assembly so that a few contigs with high depth reads existed in our data. This result is quite similar to that of repetitive regions. These results suggested that RNA genes were distributed in the wheat genome with the diffusion of transposons and repetitive elements
Application of Chromosome 6B Sequences to Wheat Genomics
Decipher of genome sequences enables us not only to know representative gene set containing many novel genes, but also to prepare resources for genomics and breeding, such as maker information. In case of wheat, chromosome information is quite useful to distinguish homoeologous genes. For example, there are three homoeologs of flowering time genes, TaHd1-1, TaHd1-2 and TaHd1-3. Our 6B assembly can distinguish TaHd1-2 transcribed from 6B and other two homoeologs from 6A and 6D in the sequence similarity level. In addition, since exon-intron structures are determined on wheat genomes, constructions of transcript-based markers, such as PLUG markers, are easier and more accurate than the previous situation using rice genome data.
Insertion site-based polymorphism (ISBP) marker can be constructed using genome sequences (Paux et al. 2010). Genome wide survey of simple sequence repeat (SSR) is applied to construct SSR markers on non-genic regions that have not been focused by the transcript-based marker constructions. As same as the genome zipper analysis (Mayer et al. 2009, 2011), virtual order of the markers would be speculated by sequence homology of the flanking regions of the markers to closely related species, such as barley and Brachypodium. In fact, we found 16,728 SSRs on non-repetitive regions of 6B and at least 1,354 SSRs of them were positioned on barley chromosome 6H. Since more than 80 % of the SSRs were located in intergenic regions of 6H, the new SSR markers can be efficiently used for the gap filling between known markers.
Survey sequences of wheat chromosome 6B provided the various types of novel information, e.g. repeat information, genome annotation including genes and RNA genes, and marker information. However the current genomic sequences of the chromosome 6B are fragmented and not completely covered so that improvement of genome assembling should be needed. Sequencing of chromosome 6B is ongoing with MTP method and BAC by BAC sequencing using Roche 454, and more accurate and physical positioned sequences will be available in near future.
References
Brenchley R, Spannagl M, Pfeifer M et al (2012) Analysis of the bread wheat genome using whole-genome shotgun sequencing. Nature 491:705–710
Doležel J, Kubaláková M, Paux E et al (2007) Chromosome-based genomics in the cereals. Chromosome Res 15:51–66
International Rice Genome Sequencing Project (2005) The map-based sequence of the rice genome. Nature 436:793–800
Jia J, Zhao S, Kong X et al (2013) Aegilops tauschii draft genome sequence reveals a gene repertoire for wheat adaptation. Nature 496:91–95
Jurka J, Klonowski P, Dagman V et al (1996) CENSOR-a program for identification and elimination of repetitive elements from DNA sequences. Comput Chem 20:119–121
Kawahara Y, de la Bastide M, Hamilton JP et al (2013) Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice 6:4
Ling HQ, Zhao S, Liu D et al (2013) Draft genome of the wheat A-genome progenitor Triticum urartu. Nature 496:87–90
Luo MC, Gu YQ, You FM et al (2013) A 4-gigabase physical map unlocks the structure and evolution of the complex genome of Aegilops tauschii, the wheat D-genome progenitor. Proc Natl Acad Sci U S A 110:7940–7945
Mayer KF, Taudien S, Martis M et al (2009) Gene content and virtual gene order of barley chromosome 1H. Plant Physiol 151:496–505
Mayer KF, Martis M, Hedley PE et al (2011) Unlocking the barley genome by chromosomal and comparative genomics. Plant Cell 23:1249–1263
Mochida K, Yoshida T, Sakurai T et al (2009) TriFLDB: a database of clustered full-length coding sequences from Triticeae with applications to comparative grass genomics. Plant Physiol 150:1135–1146
Paterson AH, Bowers JE, Bruggmann R et al (2009) The Sorghum bicolor genome and the diversification of brasses. Nature 457:551–556
Paux E, Faure S, Choulet F et al (2010) Insertion site-based polymorphism markers open new perspectives for genome saturation and marker-assisted selection in wheat. Plant Biotechnol J 8:196–210
Schnable PS, Ware D, Fulton RS et al (2009) The B73 maize genome: complexity, diversity, and dynamics. Science 326:1112–1115
Tanaka T, Kobayashi F, Joshi GP et al (2014) Next-generation survey sequencing and the molecular organization of wheat chromosome 6B. DNA Res 21:103–114
The International Barley Genome Sequencing Consortium (2012) A physical, genetic and functional sequence assembly of the barley genome. Nature 491:711–716
The International Brachypodium Initiative (2010) Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature 463:763–768
Acknowledgments
This work was supported by grants from the Ministry of Agriculture, Forestry and Fisheries of Japan (KGS1001, KGS1003, KGS1004, and NGB1003) and funding from Nisshin Flour Milling Inc.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is distributed under the terms of the Creative Commons Attribution Noncommercial License, which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Copyright information
© 2015 The Author(s)
About this paper
Cite this paper
Tanaka, T. et al. (2015). Sequencing of Wheat Chromosome 6B: Toward Functional Genomics. In: Ogihara, Y., Takumi, S., Handa, H. (eds) Advances in Wheat Genetics: From Genome to Field. Springer, Tokyo. https://doi.org/10.1007/978-4-431-55675-6_12
Download citation
DOI: https://doi.org/10.1007/978-4-431-55675-6_12
Publisher Name: Springer, Tokyo
Print ISBN: 978-4-431-55674-9
Online ISBN: 978-4-431-55675-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)