
Abstract
Over the last 50 years, restoring a durable and physiological performance to the aortic valve has been largely accomplished by surgical aortic valve replacement (AVR). The restoration of normal ventricular geometry, mass volume and function is a goal yet to be achieved. More robust management of hypertension, atrial fibrillation and early intervention in asymptomatic aortic stenosis (AS) or aortic regurgitation (AR) may offer a better outcome. This requires a comprehensive physiological consideration that combines classical force-velocity relationship and the time course of force development. Left ventricular hypertrophy (LVH) and myocardial fibrosis remain structural and morphological focus points in this process. Classical ECG strain pattern, CMR mapping of fibrosis and 3D speckle tracking echocardiography along with functional genomics are creating a new platform to yield more in-depth understanding of the nature of myocardial adaptation to pressure and volume overloading in heart valve diseases and myocardial response to valve surgery or intervention. Ultimately the physiological principles, the new knowledge and novel valve prostheses will work in synergy to improve quality of life in an ageing population.
Similar content being viewed by others


Keywords
- Aortic valve replacement
- TAVI
- Left ventricular function
- Left ventricular hypertrophy regression
- Myocardial force-velocity relationship
- Myocardial power
- Ventricular incoordination
- Cardiac genomic physiology
1 Introduction
Aortic valve disease involves the basic mechanical problems of obstruction to left ventricular ejection during systole or of regurgitation during diastole. These apparently simple mechanical abnormalities lead to profound disturbances of left ventricular physiology with progressive myocardial disease and consequent morbidity and mortality. The clinical solution is a simple mechanical surgical procedure, i.e. aortic valve replacement (AVR), or occasionally repair or recently percutaneous aortic valve implantation (TAVI). AVR or TAVI sets in train a series of adaptive changes in left ventricular structure and function [1,2,3,4,5] through a complex biological process that depends on many clinical factors. These include preoperative variables, such as the nature and extent of valve pathology and related pre-existing myocardial disease, the myocardial injury that occurred during the surgery or TAVI intervention [6, 7], changes in ventricular loading conditions, the extent to which the valve diseases have been corrected by valve prosthesis or valve reconstruction and the potential new pathology that may be introduced, such as significant prosthesis-patient mismatch, para-valvular aortic regurgitation (AR) and major ventricular activation abnormalities. In addition, concomitant cardiovascular and metabolic diseases, such as hypertension, atrial fibrillation, coronary artery disease and diabetes, also play an important role in LV remodelling before and after AVR in particular in the ageing population.
The comprehensive approach to and knowledge of the elucidation of LV function and hypertrophy in aortic valve disease as well as observing the effects of intervention has become ever more important in today’s clinical practice. Aortic valve surgery offers unique opportunities for research into fundamental cardiac physiology in a clinical setting. The derived knowledge of LV function and remodelling has guided clinical practice. Not only can this facilitate a better timing of intervention in symptomatic patients, thus retaining or recovering LV function and improving patient life quality and survival, but also increasingly forms a central stage for the decision of intervention in asymptomatic patients with severe aortic valve diseases. It also serves as the intermediate clinical end point when a new aortic valve prosthesis and its implantation approaches are introduced and the cardiac team-based practice evolves.
2 Overall Haemodynamic Response to Aortic Valve Replacement
In the 1960s and 1970s, following the wide acceptance of AVR, the basic haemodynamic response of the left ventricle to the operation was established. By relieving aortic stenosis, it was found that left ventricular systolic pressure and stroke work both fell, while stroke volume, cardiac index and systemic blood pressure usually increased particularly when their preoperative values were low [8,9,10]. After correcting aortic regurgitation, there is a fall in left ventricular stroke volume and stroke work and an increase in diastolic systemic blood pressure [8, 11]. The forward systemic flow may remain unaltered. Over the long term, exercise capacity was found to have increased significantly with respect to the preoperative level [2]. In spite of many changes in operative technique and valve prosthesis, these basic responses remain characteristically seen in today’s practice of AVR or TAVI. More recently, a small left ventricular stroke volume (LVSV) has been identified as an important marker of adverse long-term patient prognosis [12, 13] after AVR or TAVI, emphasizing the importance of basic haemodynamics in determining clinical outcome.
3 Left Ventricular Force-Velocity Relationship and Incoordination in AVR
It has been well recognized that increased LV ejection resistance due to aortic stenosis or ejection volume due to aortic regurgitation leads to an increase in the duration of ventricular systole relative to diastole and ventricular muscle mass. The classical physiology teaching believes that the systolic wall stress can be normalized by increasing or maintaining relative wall thickness in AS or AR, thus systemic cardiac output is maintained [14]. If hypertrophy does not develop or if the cavity dilates, wall stress will rise [15]. This state of affairs has been termed as “afterload mismatch” [16], and its presence in aortic valve disease is diagnosed when mean velocity of circumferential shortening and the fractional shortening of minor dimension or ejection fraction are lower than expected value when plotted against end-systolic wall stress [17]. However, the problem of differentiating patients with irreversible myocardial disease from those with afterload mismatch has not been fully solved [18,19,20]. Majority opinion about the transition of myocardial disease from an early and potentially reversible stage to a late and irreversible one remains confined to simple ideas based on loading and contractility [21]. It has, however, long been recognized that as ventricular cavity dilatation and hypertrophy develop, shortening velocity falls.
The main early adaptation of left ventricular function to relief of aortic stenosis has been interpreted on the basis of a fall in ventricular afterload [3, 18, 22, 23]. Following a successful aortic valve replacement, there is a consistent fall in systolic wall stress and a corresponding increase in the velocity of shortening [18]. With correction of aortic regurgitation, the corresponding changes are more complex, as the fall in systolic wall stress and the increase in the velocity of dimensional shortening are both inconsistent [19]. Early intraoperative studies using epicardial echocardiography have demonstrated that LV ejection fraction and the velocity of dimensional shortening may fall as the large stroke volume is corrected [23, 24]. In addition, considerable incoordination of LV contraction may also be present [25]. Impaired myocardial contractility has been implicated in a subgroup of patients whose preoperative force-velocity relationship was already depressed [18]. Recovery of systolic function after correction of aortic regurgitation appears to take a longer time and be less complete than with aortic stenosis [1, 2, 26], and thus its presence preoperatively predicts a possible poor outcome [27]. These early studies have formed the physiological basis of timing the indication for AVR in AR along with clinical symptoms in today’s guidelines by using 45 mm in end-systolic diameters and LVEF at 50% as the cut-off [28].
Clinical physiology based on force-velocity relationship provides an important framework in the assessment of myocardial contraction after AVR, but it also incurs significant limitations in common with the application of muscle mechanics to the intact heart, particularly when changes in loading conditions occur along with ventricular incoordination. In addition, measurements of regional power production do not give a complete picture of local ventricular function. Not only must the myocardium produce power, but this must be translated into useful work on the circulation. This step can be quantified in terms of regional cycle efficiency as the result of incoordination. This effect was found to be common in patients with aortic valve disease, in particular preoperatively (Fig. 22.1). As in other forms of ventricular disease, it occurred most commonly as the result of changes in ventricular cavity shape during the two isovolumic periods. It was commonly relieved within a few hours by AVR, suggesting that it was either directly or indirectly aggravated by the valve lesion itself and two other mechanisms: disturbances of ventricular activation, particularly loss of the septal q wave, and an increase in QRS duration [29].

(a) Left ventricular (LV) pressure-dimension loop constructed from simultaneous recordings of left ventricular cavity pressure and circumferential dimension. The loop area represents the external work done on the circulation by the segment studied. The area of the rectangle (A, B, C and D) that just encloses the loop represents the maximal possible work that could have been done by the ventricle over the same range of pressure and dimension. Cycle efficiency is defined as the ratio of loop area to that of the rectangle and reflects the efficiency of energy transfer. Loop (b), a typical example of the change in cycle efficiency (CE) before, and loop (c), 20 h after aortic valve replacement (AVR) from a patient with aortic stenosis. The lower cycle efficiency before AVR (52%) is due to abnormal dimension lengthening during isovolumic contraction and dimension shortening during isovolumic relaxation. These abnormalities are no longer present 20 h after AYR, and cycle efficiency has increased to 81%. Jin et al., Am J Cardiol 1994;74:1142–6 (with permission)
The presence of incoordination not only has effects on myocardial function but also has a major influence on the interpretation of measurements commonly used to assess it. When peak +dP/dt, either on its own or normalize to develop pressure, is used to assess LV inotropic state, it was demonstrated that it is affected to a major degree by incoordination. This is consistent with the notion by Wigglers nearly one century ago that the rate of rise of LV systolic pressure is the summation of its muscle contraction [30]. Thus, the classical approach to assess force-velocity relations in the intact ventricle by plotting wall stress against shortening velocity could be valid only in the absence of significant incoordination. In these circumstances, a low shortening rate reflects the direct effects of a high wall stress. When significant incoordination is present, peak shortening velocity relates directly to the degree of incoordination (Figs. 22.2 and 22.3). The assessment of LV contraction requires the incorporation of these factors, thus complex physiological process can be thoroughly examined. The recognition of such a complex interrelation has important therapeutic implications. The treatment of reduced inotropic state is a drug with a positive inotropic effect. However, such drugs are likely to act preferentially on normal myocardium thus making incoordination worse [31].

Peak velocity of circumferential fibre shortening (peak Vcf)
plotted against mean left ventricular (LV) systolic circumferential wall stress, before relief of aortic stenosis. Note that there is an inverse significant linear correlation between the two in coordinate ventricles (closed circles, r = −0.71, P < 0.01) but not in incoordinated ones (open circles, r = −0.45, P > 0.05). Demonstrating the importance of coordination in order to preserve the force-velocity relationship. Jin et al., Heart, 1996;76:495–501 (with permission)

Peak circumferential myocardial power plotted against mean LV systolic circumferential wall stress, before relief of aortic stenosis. Note that there is positive correlation between the two in those with a coordinate ventricle (closed circles, r = 0.83, P < 0.01) but not in those with incoordinated ventricles (open circles, r = 0.28, P > 0.05). Demonstrating the importance of coordination in order to preserve the force-power relationship. Jin et al., Heart, 1996;76:495–501 (with permission)
4 Left Ventricular Myocardial Power, Its Time Course and Implication for LV Hypertrophy
The alternative approach of studying LV physiology in AVR setting is to describe disturbances in simple physical terms. Their timing within the cardiac cycle and their position within the ventricle are specified. This approach has provided a framework for the many and complex disturbances of ventricular physiology that may be present in patients with aortic valve disease to be examined before and after valve replacement.
Myocardial power quantifies the effects of depressed force-velocity relations, loss of coordination and abnormal activation. But it has received less consideration in the 1970s and 1980s in comparison with more widely used load independent indexes, such as Emax or preload recruitable stroke work which unfortunately have yielded little insight into the exact nature of myocardial disease. Left ventricular power or rate of doing work is well defined in physical terms and, unlike contractility, has clear units (Watts). Measurement of local power production is a practical means of assessing LV systolic function. It can readily be measured from instantaneous values of wall stress and shortening rate, both of which are available from force-velocity relationship and can be derived using echocardiography with simultaneous LV pressure catheter. It reflects the inverse relation between force and shortening velocity; the condition of constant power implies a hyperbolic relation between the two, which is very close to that observed experimentally [32]. Power differs from variables such as Vmax, which have been used experimentally, in that it can be plotted continuously throughout systole, so that not only can the peak power value be obtained but also the pattern of the power generation with time can be observed. This proved to be very significant, since the duration of power generation in systole is prolonged in patients with aortic valve disease, a disturbance that would have been missed if peak values only had been recorded, and it cannot be derived by force-velocity relationship, per se [3]. Theoretically, external power production might be affected by input impedance, by the process known as matching [32, 33]. It was thus instructive to find that peak power was unchanged, even when aortic stenosis was corrected, which probably represents the largest change in ventricular afterload encountered in clinical practice [3]. Since neither end-diastolic volume nor pressure was consistently altered by aortic valve replacement, there was no concern about possible effects of alteration in preload on power production.
Altered loading conditions not only modify the amplitude of LV peak systolic stress and shortening velocity, and the interrelationship between them, but also change the entire time course of contraction. With aortic stenosis, there is a prolonged period of LV contraction due to prolongation of both isovolumic periods and of ejection [34, 35]. In any given cardiac cycle, therefore, the time available for diastolic filling and coronary perfusion is shortened. With aortic regurgitation, there is a similarly disproportionate increase in the duration of systole, particularly in ejection time and a corresponding decrease in diastolic time [36].
The display of myocardial power profile and its time course during LV contraction has offered a unique opportunity for the elucidation of fundamental physiology in aortic valve stenosis or regurgitation and after its correction. In clinical setting, this can be achieved using trans-esophageal echocardiogram combined with LV pressure recording. Jin et al. studied the very early changes in LV contraction following AVR for aortic stenosis or regurgitation by describing the timing and intensity of myocardial contraction. Simultaneous measurement of LV wall stress, circumferential shortening velocity and myocardial power along with their time course in a cardiac cycle, it was demonstrated that the abrupt decline of circumferential wall stress as LV pressure falls following AVR for AS, which resulted in an immediately reciprocal increase in peak circumferential shortening velocity (Vcf). Peak myocardial power thus remained constant; its peak was however registered much earlier in systole. Myocardial stroke work fell significantly due to the shortening of LV systolic duration. At the same time, ventricular diastolic time increased towards normal level [3]. When aortic regurgitation is corrected by AVR, however, neither peak value nor the timing of systolic wall stress, Vcf and myocardial power production changed immediately. Myocardial stroke work however also fell significantly, due similarly to the shortening of LV systolic duration. And diastolic time thus increased accordingly [36]. It is of more interesting that in the subsequent 4-year follow-up, LV systolic time remains unchanged and myocardial stroke work recovers and exceeds the baseline, while LV mass index regresses towards normal (Fig. 22.4). These data demonstrated that the physiological benefits of AVR to myocardial contraction is by restoring the normal time relationship between systole and diastole, thus the balance between oxygen demand and supply, and ultimately prevents or reverses myocardial disease, while that reducing the intensity (peak myocardial power) or the quantity (myocardial stroke work) of energy output of per unit volume of myocardium does not appear to be essential.

Percentage changes in LV ejection time (indexed to heart rate), LV mass index (g/m2) and myocardial stroke work (mW/cm3) in the first 4 years after AVR. Note that the 30% fall in LV ejection time is paralleled by the 40% fall in LV mass index, while the myocardial stroke work increased by 10–30% from before AVR, thus demonstrating that the reciprocal changes in LVH and myocardial stroke work is facilitated by a fall in LV systolic time. Adapted from Jin et al. 1994, 1996, 1999, 2004 (with permission)
We further consider that the disruption of normal time relationship between LV systole and diastole by aortic valve diseases can be one of the key underlying mechanisms leading to the development of LV hypertrophy and eventual heart failure. While the increase in muscle mass can normalize the value of LV systolic wall stress, it does not, however, rectify the prolonged duration of systolic wall stress. It is the late one that continuously serves as a positive feedback signal for myocardial remodelling. We have previously demonstrated that the development of excessive LVH in aortic stenosis was associated with major decline of myocardial contraction and its altered time course [37]. In addition, the duration of LV tension development rather than the wall stress value was identified as the positive predictors of LV mass in heart valve diseases [38]. The mechanism by which LV systolic tension development and ejection duration regulate the myocardial responses to mechanical stimulator remains to be fully elucidated. Nevertheless, the tension-time-based approach has added an important dimension to the conventional wall stress-based theory. Forty years after his initial publication, Grossman has recently noted that the process from valve stenosis to LVH involves complex interactions of biophysics and cardiac remodelling [14, 39]. Along with this line, our latest data on LV myocardial gene expression profiling of concentric versus eccentric LVH lends further support to such a notion. The LV tension-time-related parameters were found to have greater correlations with the gene expression changes than those based on pressure or volume measurement [40], thus emphasizing the critical role of time in the signal transduction and biological translation of mechanical stimulus into myocardial growth and remodelling. Translating these physiological principles into clinical practice, we ought to assess to what extent the systolic and diastolic durations are normalized for a given AVR patient, in particular when AVR or TAVI is complicated with a residual valve gradient, prosthesis regurgitation or LV activation abnormalities. Further discussions will be in the later part of this chapter.
5 Left Ventricular Diastolic Dysfunction and AVR
Left ventricular diastolic dysfunction has long been recognized in aortic stenosis along with LV hypertrophy. The characteristic patterns of dimensional changes and wall thinning during isovolumic relaxation and early diastolic filling have been well established in 1970s, based on M-mode echocardiography [41]. In the 1990s, trans-mitral flow velocity was used to assess global diastolic filling patterns. The interpretation of these Doppler measurements depends on the relationship of phasic pressure difference between left atrial and left ventricle. In isolated aortic stenosis, it was noted that patients with a normal ejection fraction and normal capillary wedge pressure also had a normal rate of isovolumic left ventricular pressure decay although the filling occurred primarily during atrial systole. In contrast, in patients with systolic dysfunction and elevated mean capillary wedge pressure, isovolumic pressure decay of left ventricle was prolonged, while filling occurred mainly during early diastole, with a reduced atrial contribution and a short isovolumic relaxation period. The single most important predictor of trans-mitral filling pattern was therefore the pulmonary capillary wedge pressure, while the left ventricular mass was the most powerful predictor of the rate of left ventricular pressure fall. Peak filling rate alone was not helpful in detecting diastolic dysfunction in patients with aortic valve disease [42]. From a series of invasive studies, it was demonstrated that left ventricular relaxation, filling and end-diastolic stiffness were all disturbed by LV hypertrophy due to aortic stenosis [43]. Non-invasively, pulmonary venous flow pattern and tissue Doppler velocity of mitral annulus motion when combined with mitral valve E wave velocity provide more useful estimation of PWP and the degree of LV diastolic dysfunction with wide clinical acceptance in the echocardiography guidelines. In recent years, speckle tracking imaging has provided more robust regional myocardial deformation data, and its global long-axis strain has become a sensitive early marker of LV impairment in contraction and relaxation.
In contrast to extensive reports on pre- and postoperative assessment of left ventricular diastolic function in AVR patients, the very acute changes in myocardial thinning, ventricular relaxation and filling during aortic valve replacement have been less frequently studied. Perioperative transoesophageal echocardiography study by quantifying the reduction in early diastolic lengthening rate of the transverse left ventricular dimension and a reduction in posterior wall thinning rate demonstrated that these abnormalities were not immediately corrected by valve replacement, particularly in patients with aortic stenosis, but improved over several hours with a time course similar to that of the regression of incoordination and increase in stroke volume [3]. The exact nature of these changes remains uncertain, but they do not appear to be the direct reduction of a low systolic shortening rate, while their partial normalization over a few hours in the postoperative period would seem to exclude irreversible changes such as fibrosis. A second and expected diastolic effect became apparent, namely, an increase in the duration of diastole secondary to shortening of systole. This is likely to have had physiological advantages in increasing the time available for ventricular filling and coronary flow after operation. In AR patients, it is likely to have interacted with the effects of aortic regurgitation in increasing left ventricular loading and at the same time reducing coronary perfusion pressure as aortic diastolic pressures fall and those in the left ventricle rise. These findings illustrate the common interaction between systolic and diastolic function and further stress the potential advantages in considering the time course as well as the intensity of ventricular contraction, which has been discussed in detail in the proceeding sections.
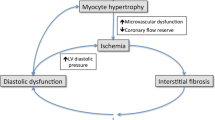
Regression of hypertrophy after AVR for AS was found to account for the long-term improvement in diastolic function along with longitudinal LV contraction [43]. However, the relative increase in interstitial fibrosis early after AVR when the myocyte hypertrophy has largely regressed was associated with elevated chamber stiffness, which took longer to regress than changes in the myocytes [43]. Indeed, the normalization of LV diastolic function and non-uniformity can take 5 years to establish. For AR patients LV diastolic dysfunction remains 7 years after AVR [44]. In today’s practice, the age of AVR and TAVI patients is continuously rising perhaps with more myocardial fibrosis. As such, the remodelling of LV diastolic function has been shown to be less satisfactory. The incidence of moderate to severe diastolic dysfunction increases after AVR partly due to lack of LV geometry remodelling and relative increase in myocardial stiffness [45]. Preop LA dilatation or diastolic dysfunction was another marker that is positively related with LVH degree and predicts worse long-term outcome [46].
6 LVH Regression After AVR
In the last 25 years, LVH regression has emerged as one of the hallmarks in the evaluation of AVR [5, 47,48,49,50]. Not only has this approach improved our understanding in myocardial disease process but also become important consideration in managing the timing of AVR in asymptomatic aortic stenosis patients. Preop severe LVH has been repeatedly demonstrated in large cohort studies as one of the key factors responsible for long-term adverse outcome in patients. The regression of LVH is widely accepted as the intermediary endpoint in assessing the effectiveness of aortic valve intervention [48]. Nearly half of AVR patients will have incomplete LVH regression which has been recognized for decades as an indicator of adverse outcome and thus an argument for earlier surgery [49,50,51,52,53].
From the pathological point of view, cardiac hypertrophy should only refer to myocyte enlargement in contrast to hyperplasia which occurs primarily in non-myocytes in the extracellular matrix and vasculature [54]. In clinical practice LVH is often defined by an increase in mass volume or ECG voltage along with strain pattern. Nevertheless, the recognition of two distinctive biological processes, the myocyte hypertrophy and extracellular matrix hyperplasia and fibrosis has important clinical implications. Vliegen and colleagues demonstrated that with an increase of LV mass, the increase of connective tissue cells are far greater than that in myocytes at 140% versus 20% [55]. By the time of undergoing AVR, 90% of the patient has developed a varying degree of LVH, but only in 50–60% will have LVH regressed to normal level after the surgery. Studies of preoperative myocardial pathology in AVR patients have long established that severe LVH and myocardial fibrosis predict adverse early and late outcome [49, 56]. In patients who developed cardiac dysfunction, the interstitial fibrosis becomes much more significant compared with those of normal function. Severe concentric LVH or septal hypertrophy proved to be associated with an increased surgical mortality [57, 58] or late mortality in AVR patients [59, 60]. In today’s practice, patients with degenerative aortic valve pathology are much older with additional ageing-related myocardial remodelling. Olivetti and colleagues demonstrated that LV myocyte number reduced significantly from age of 17 to 90, with a loss of 38 million myocytes per year in the left ventricle, which is partially replaced by myocyte enlargement by 110 mcg per year. The cellular enlargement of the remaining myocytes represents a structural basis with a reduced compensatory capacity of the ageing heart [61]. This is particularly relevant for patients undergoing AVR or TAVI.
6.1 Incomplete LVH Regression After AVR
Following AVR, LVH regression occurs most rapidly in the first 6 months [4, 5, 65, 72]. Earlier studies showed that left ventricular hypertrophy regression followed the time course of myocyte fibre diameter regression [47], but the regression of interstitial fibrosis took a much longer period; therefore, the early postoperative relative percentage of volume occupied by interstitial fibrosis could have increased as the myocyte volume decreased rapidly and myocardial stiffness increased [43].
Repeated studies have emphasized the incompleteness of left ventricular hypertrophy regression in over 40% of AVR cases despite apparently successful AVR surgery [53, 62] which is associated with worse clinical symptoms, cardiac dysfunction or long-term survival. The underlying mechanism of residual left ventricular hypertrophy is complex but can be largely considered into two categories, i.e. aortic valve disease and aortic valve replacement related or vascular diseases and heart rhythm related. Preoperative LVH quantified by echocardiography has been shown as one of the most important determinants of the degree of LVH regression as well as the level of residual LVH [50, 51, 53]. This is consistent with the myocardial pathology studies of profound extracellular matrix remodelling and interstitial fibrosis in severe LVH. Indeed, even in patients after stentless AVR with superior flow dynamics, the medium-term LV mass index remains dependent on baseline LVH, NYHA class, blood pressure (HBP) and the presence or absence of atrial fibrillation [63, 64].
6.2 The Impact of Aortic Prosthetic Haemodynamics on LVH Regression
The residual pressure drop across aortic valve prostheses and non-physiological aortic flow profiles can adversely affect long-term clinical outcome, particularly in those having a small-sized prosthesis and impaired systolic function [65, 66]. Indeed, prosthesis-related LV pressure increase has been demonstrated to cause incomplete regression of ventricular and cellular hypertrophy, as well as for increased interstitial fibrosis after AVR [56]. These findings have facilitated the research in the last 20 years regarding whether stentless AVR can improve LVH regression and survival and whether prosthesis-patient mismatch could compromise patient outcome.
The removal of the stent has brought several advantages. Not only can a larger valve be implanted into a given size of aortic annulus, thus maximizing the available cross-sectional area, but also is the native dynamics of aortic annulus well preserved. The residual obstruction of the stentless valves to trans-aortic flow is significantly reduced. Indeed, the in vivo haemodynamic results of stentless aortic bioprostheses have clearly lived up to these expectations and outperformed stented or mechanical ones. The lower pressure gradients seen with stentless valves during exercise have been attributed in part to a flow-related increase in EOA [67]. This is due to the presence of a predominant inertial flow rather than a resistive flow and a more uniform flow velocity profile when mean trans-valvular pressure gradient of stentless valves is below 5 mmHg. These haemodynamic characteristics distinguish clearly from their stented counterparts in which flow characteristics are predominantly resistive and turbulent in nature. From a historical point of view, the renaissance of stentless AVR in 1990s has provided the key intellectual and technical basis for the subsequent development of sutureless AVR and TAVI. Those using bovine pericardium such as Sorin Freedom stentless aortic valve [68] has evolved into the Perceval S sutureless AVR, while the tubular aortic cusp design using equine pericardium, such as ATS 3F stentless aortic valve [69], has been adopted by the TAVI prosthesis.
Despite studies of hypertrophy regression after AVR in the 1980s, the recognition of LVH as a marker for the comparison of bioprostheses was not widely adopted until early 1990s when stentless AVR come into surgical practice. In 1996, Jin and colleagues from the Brompton group published one of the very first large series that demonstrated aortic homograft or stentless bioprosthesis could offer more rapid and great regression of LVH [5]. This was supported by a large body of literature that superior haemodynamics of stentless aortic valves can facilitate a more rapid and greater resolution of LVH, and therefore better LV function early after AVR. This was expected to reduce the risk of sudden death and congestive heart failure after AVR and improve long-term outcome [70]. Some of subsequent reports, however, voiced different views by showing lack of difference in LVH regression between stentless and stented aortic bioprostheses. In order to analyse these discrepancies, a meta-analysis [71] confirmed that stentless AVR did provide an advantage of LVH regression in the first 6 months by offering more orifice area and a lower gradient. The accompanying editorial [72] reiterated the importance of having a significant baseline LVH as the essential requirement when comparing its regression after different prostheses. Therefore, only in patients with significant hypertrophy could the advantage of a stentless bioprosthesis be demonstrated. In the longer-term follow-up data, the LVH regression appears to be more affected by preop LV mass and the nonvalve-related factors, such as hypertension and atrial fibrillation [63, 64, 73].
In parallel to the studies of stentless AVR and LVH regression, investigations into the prosthesis-patient mismatch (PPM) have served the other side of the coin regarding the clinical importance of aortic prosthesis haemodynamics. After 20 years of continuous active research, the latest large series and long-term follow-up data [65, 66, 74] concluded that a significant residual gradient due to PPM defined as aortic EOA less than 0.65 cm2/m2 has prognostic value. The clinical impact of severe PPM not only has an adverse effect on the early postop surgical mortality but also on medium- to long-term patient outcome and LVH regression. It is worth noting that patients who have impaired baseline LV function and co-existence of coronary artery disease are the most vulnerable group to suffer from PPM.
The decades-long research into stentless valve haemodynamics and the PPM impact has independently pointed to the same principle that aortic valve replacement, particularly in impaired cardiac function and/or severe hypertrophy, should aim for achieving the most physiological valve haemodynamics, thereby a better opportunity for left ventricle to recovery, hypertrophy to regress and interstitial fibrosis to remodel.
6.3 Systemic Hypertension Diminishing LVH Regression After AVR
Hypertension occurs in 50–60% of aortic stenosis and plays a very important role in the development and regression of LVH in patients with aortic valve diseases. Not only does it cause baseline LVH disproportional to the degree of aortic valve disease, but it continuously diminishes long-term LVH regression after surgery. An early report [75] elegantly demonstrated two different patterns of LVH regression after AVR, the significant and sustained LVH regression in those without hypertension versus a slow and insignificant regression (Fig. 22.5). This pattern was also demonstrated in patients undergoing stentless AVR in that the hypertension was a major factor causing residual hypertrophy with late upswings at medium term [63, 76]. Furthermore in patients receiving stentless AVR, systemic hypertension has also been shown to reduce long-term survival by causing residual LVH [73, 77]. These findings have led to demands for more effective and prospective control of hypertension in AVR patients [78, 79].

LVH regression after AVR is significantly diminished by hypertension. Wilcoxon signed-rank test was employed for evaluation of significance. Gaudino et al. Euro Heart J 2005;26:51–57 (with permission)
6.4 The Effects LBBB, PMI and AF on LVH Regression and Remodelling
The recent introduction of TAVI benefited many high-risk patients who otherwise would be unsuitable for surgical AVR. Physiological benefits are evident after uneventful TAVI regarding valve haemodynamics, LVH regression and functional improvement [80,81,82]. It has however also faced new challenges for achieving these objectives when incurring complications of bundle branch block and/or permanent pacemaker insertion. Conventional aortic valve surgery has an incidence of a requirement for permanent pacemaker of 3–5% depending on the age group, but left bundle branch block occurs in less than 5% of all patients. Therefore, the impact of ventricular activation abnormalities on LVH regression after AVR has not been a significant issue in surgical practice. With the wider acceptance of TAVI in the elderly population, 15–30% incidence of TAVI-induced LBBB and/or PMI has prompt the recognition of their physiological implications for ventricular remodelling and LVH regression. In the last 5 years, consistent data from a variety of TAVI trials has been published indicating that with intraventricular conduction abnormalities the patient experienced an insignificant improvement in cardiac function and little or no change in left ventricular hypertrophy [83,84,85] and even with reverse LV remodelling [86]. The combination of LVH with new LBBB in elderly patients appears to be a worst possible case from cardiac physiological point of view. The underlying physiological mechanism is likely due to incoordinated LV contraction and prolonged systolic tension development duration, commonly caused by ventricular activation abnormality [29, 87].
Apart from the ventricular activation abnormalities, atrial fibrillation also has a major adverse impact on left ventricular remodelling and hypertrophy regression after AVR. The mechanism is intuitively simple in that the loss of atrial contraction leads to LV longitudinal functional recovery much compromised after AVR. Our data from stentless AVR follow-up demonstrated that those who have non-sinus rhythm due to AF or pacemaker have different ventricular geometry, with increased diameter in LV short axis and wall thickness thus associated with a more spherical ventricular geometry and a greater LV mass. This explains the potential physiological mechanism for long-established clinical findings that AF is an independent risk factor for worse survival after AVR (Fig. 22.6) [88].
6.5 TAVI and Para-valvular AR
In modern cardiac surgery, intraoperative transoesophageal echocardiography has become integral part of the procedure. Moderate or severe para-valvular aortic prosthesis regurgitation has been largely diagnosed and resolved in the operating theatre. Thus, its incidence is below 1% per patient year in the long-term follow-up after AVR. Following TAVI implantation, the incidence of moderate or greater para-valvular AR can occur in 10–15% of patients. To manage significant new AR, the LV has to suddenly remodel from pressure-overloaded concentric hypertrophy to volume-overloaded eccentric hypertrophy [89]. From the cellular and molecular biology point of view, the myocyte and extracellular matrix requires a very complex and time-consuming process, which is often even more difficult to achieve in elderly patients for the reasons discussed earlier. This has inevitably rendered a higher LV end-diastolic pressure, failure of LVH regression and systolic and diastolic dysfunction and ultimately a worse early and medium term survival [89, 90]. With the improvement of TAVI device design and implantation experience, para-valvular has been on the way down more recently. Nevertheless, the current incidence in para-valvular AR and significant activation abnormality does raise clinical concerns when TAVI started to be considered for moderate risk AS with younger age patients. The debate is vigorous and ongoing (Fig. 22.5) [91].

Relative survival after primary AVR is significantly reduced in patients with preoperative atrial fibrillations who survived the first postoperative month. Kvidal et al. JACC 2000;35:747–56 (with permission)
7 Future Perspectives
7.1 LVH as an Indicator for Early AVR in Asymptomatic AS
Defining the optimal timing of aortic valve replacement remains to be a difficult task in the management of the individual patient, particularly in asymptomatic with severe aortic stenosis or regurgitation. While the role of LV function and cavity size has been more widely accepted in timing AR patients for AVR [92,93,94], it is yet to be established in AS patients with respect to LVH. Given the current indications for AVR remain based on patient’s symptoms rather that LV hypertrophy or function, inevitably a significant amount of AVR patients are operated on with advanced LVH with the risk of developing heart failure [12, 53]. Even with the best stentless AVR haemodynamics, the existing myocardial disease may not be fully reversed, and the outcome remains to be improved [63], thus emphasizing the importance of early intervention. The leading opinions on early AVR for asymptomatic AS, however, remain divergent [95, 96], but the gap is narrowing following more recent compelling clinical studies that demonstrated much improved outcome if AVR were undertaken [97, 98]. In the light of ensuring best risk and benefit ratio, a range of diagnostic approaches has been deployed. This include using ECG strain pattern and CMR T1 mapping for picking up those with significant myocardial fibrosis [99] and echocardiography strain image to identify an important prognostic marker based on myocardial deformation [100, 101]. The extensive study of hypertrophy and its underlying mechanism as well as the impact on AVR outcome has also led to the questions, should LVH be introduced as one of the indictors for aortic valve replacement despite the patient remaining asymptomatic? Our personal experiences suggest that if severe LVH is combined with severe AS, AVR is likely to be warranted. However, if severe LVH is associated with only mild to moderate AS but significant hypertension, the benefits of early AVR could be less certain.
7.2 Genomics in the Elucidation of LV Function and Hypertrophy
Genomics is the study of genome-wide data set at the DNA or RNA level. When combined with studies of physiological traits or disease phenotypes, the functional genomics can be used to infer the molecular insights. Since the early genomic profiling analyses of cardiac remodelling in the animal model [102] and human failing heart [103], much progress has been made in the study of the molecular process concerning the left ventricular remodelling, hypertrophy and overt heart failure following various disease aetiology including myocardium infarction [104], DCM or ICM [105], hypertension [106] and valvular disease [107,108,109].
Functional genomic approaches can play the following roles in the context of concentric remodelling with aortic stenosis and subsequently the planning and the management of AVR. Firstly, it can uncover the biological mechanisms underlying the process of LVH due to aortic stenosis, therefore, to understand why similar patients could have different outcomes after AVR irrespective of the aetiologies of AS. For an example, ECG strain pattern is frequently presented in AS patients as a marker of LV hypertrophy and is emerging as one of the predictors for AVR [110, 111]. We have observed the different time course of normalization between ST segment depression and T-wave inversion after AVR. For ST depression, it recovered within first 24 h after AVR, while that of inverted T-wave took 6–12 months to improve in parallel to LVH regression [112, 113]. To elucidate the underlying molecular mechanism for such a different time course, we studied the myocardial gene expression profiling using oligonucleotide array and found that the molecular pathways and their network connections underlining the T-wave inversion are much more complex than those in ST depression. Furthermore, the different relationships to LVH regression after AVR between T-wave inversion and ST depression patients were indeed reflected by their different molecular signatures and pathways [114]. Another example of the study of molecular mechanisms of LVH is to approach the response to the stimulus and/or mechanical regulators from pressure overloaded versus volume overloaded during cardiac cycle. Again using gene expression profiling, we compared the pressure-overloaded human LV due to aortic stenosis with volume-overloaded LV from mitral regurgitation. We are able to identify a group of molecular clusters that can fully distinguish these two LV remodelling phenotypes. Moreover, employing stepwise regression analysis of each gene’s expression level against the mechanical stimulus, we found that the time-based parameters have more abundant correlations with the gene expression changes than the parameters based on pressure, volume or stroke work [40]. We therefore hypothesize that the time-based regulators play the critical roles in the signal transduction and biological conversion of mechanical stimulus into myocardial growth and interstitials remodelling. This adds an important dimension to the conventional theory based on mechanical workload and wall stress in the elucidation of LVH mechanisms.
The second important application of genomic approaches is to provide the decision-making strategy concerning the timing of AVR and predict the prognosis of AVR. It is evident that the consideration for AVR prior to the onset of adverse myocardial ECM sequelae may prove valuable in patients with already severe but asymptomatic AS. The hallmarks of maladaptive LV remodelling are represented by increased myocardial fibrosis and increased myocardial stiffness. It has taken a long time to notice whether or not AVR can reverse AS-induced maladaptive LV remodelling, largely determined by the timing of the AVR. It is imperative to identify such biomarkers that are able to differentiate the transition from adaptive to maladaptive LV remodelling in order to more accurately time AVR. Because myocyte hypertrophy represents an early and relatively reversible event in the progression of pressure overload-induced myocardial remodelling, over the past decades, it seems appropriate to focus on the identification of biomarkers that are more closely associated with the myocardial ECM remodelling. Such biomarkers include transforming growth factor-β1 [115], cardiotrophin-1 [116, 117], collagen-derived peptides [118], the matrix metalloproteinase [119, 120] and the tissue inhibitors of the matrix metalloproteinases [121], and such biomarker lists are under tirelessly development [122].
Thirdly, it is to provide the possibility of combined therapy including using the molecular agent along with the surgical procedure. Due to the ageing population, AS is becoming increasingly common. Despite the pressing need for the development of a molecular therapeutic strategy, the promising molecular targets to slow down the progression of the disease in all age group are still lacking. Dahl et al. reported in their prospective randomized investigation that the modulation of the renin-angiotensin system utilizing angiotensin receptor blockade (ARB) achieved greater LV mass regression than the conventional beta-blockade therapy in patients with AS undergoing AVR [123]. Benedetto et al. also demonstrated the effectiveness of using both ACEI and ARB II blockade to enhance LVH regression in AVR patients with PPM [124]. While these anecdote reports are very encouraging, no established molecular strategies exist to systematically treat adverse myocardial ECM remodelling following AVR. miRNAs are small, non-coding RNAs that act to inhibit translation of messenger RNA (mRNA) into proteins and are involved in many cardiovascular diseases including LV hypertrophy. Most recently, Coffey et al. reported integrated mRNA expression and microRNA analysis in the comparison of patients with and without AS and showed that pathways involved in extracellular matrix function are the most significant in aortic stenosis. They subsequently identified two specific miRNAs, miR-122-5p and miR-21-5p, as being the potential important targets [125]. The application of a system biology approach has a great potential for paving the way to identify potential therapeutic targets for more effectively regress LVH in the future.
8 Summary
Over the last 50 years, restoring a durable and physiological performance to the aortic valve has been largely accomplished by surgical AVR. There remains scope for improvement in the normalization of ventricular geometry, hypertrophy and function. More robust management of hypertension, atrial fibrillation and early intervention in asymptomatic AS or AR may offer better outcome. This requires a comprehensive physiological consideration that combines classical force-velocity relationship and the time course of force development. LVH and myocardial fibrosis remain a structural and morphological focus point in this process. Classical ECG strain pattern, CMR mapping of fibrosis and 3D speckle tracking echocardiography along with functional genomics are creating a new platform to yield more in-depth understanding of the nature of myocardial adaptation to pressure and volume overloading in heart valve diseases, and myocardial response to valve surgery or intervention. Ultimately the physiological principles, the new knowledge and novel valve prosthesis will work in synergy to improve quality of life in an ageing population.
References
Gaasch W, Andrias W, Levine HJ. Chronic aortic regurgitation: the effect of aortic valve replacement on left ventricular volume, mass and function. Circulation. 1978;58:825–36.
Borer JS, Herrold EM, Hochreither C, Roma M, Supino P, Devereux RB, Kligfield P, Nawaz H, Chlouverakis G. Natural history of left ventricular performance at rest and during exercise after aortic valve replacement for aortic regurgitation. Circulation. 1991;84:S133–9.
Jin XY, Pepper JR, Brecker SJ, Carey JA, Gibson DG. Early changes in left ventricular function after aortic valve replacement for isolated aortic stenosis. Am J Cardiol. 1994;74:1142–6.
Jin XY, Westaby S, Gibson DG, Pillai R, Taggart D. Left ventricular remodelling and improvement in freestyle stentless valve haemodynamics. Eur J Cardiothorac Surg. 1997;12:63–9.
Jin XY, Zhang ZM, Gibson DG, Yacoub MH, Pepper JR. Effects of valve substitute on changes in left ventricular function and hypertrophy after aortic valve replacement. Ann Thorac Surg. 1996;62:683–90.
Dworakowski R, Wendler O, Bhan A, Smith L, Pearson P, Alcock E, Rajagopal K, Brickham B, Dew T, Byrne J, Monaghan MJ, Sherwood R, Shah AM. Successful transcatheter aortic valve implantation (TAVI) is associated with transient left ventricular dysfunction. Heart. 2012;98:1641–6.
Ribeiro HB, Nombela-Franco L, Munoz-Garcia AJ, Lemos PA, Amat-Santos I, Serra V, et al. Late incidence and predictors of persistent or recurrent heart failure in patients with aortic prosthetic valves. J Thorac Cardiovasc Surg. 2004;127:149–59.
Austen WG, Corning HB, Moran JM, Sanders CA, Scannell JG. Cardiac hemodynamics immediately following aortic valve surgery. J Thorac Cardiovasc Surg. 1966;51:461–7.
Carey JS, Plested WG. Immediate hemodynamic response to correction of cardiac valvular lesions. Ann Thorac Surg. 1972;13:311–23.
Harpole DH, Jones RH. Serial assessment of ventricular performance after valve replacement for aortic stenosis. J Thorac Cardiovasc Surg. 1990;99:645–50.
Pantely G, Morton M, Rahimtoola SH. Effects of successful, uncomplicated valve replacement on ventricular hypertrophy, volume, and performance in aortic stenosis and in aortic incompetence. J Thorac Cardiovasc Surg. 1978;75:383–91.
Parikh R, Goodman AL, Barr T, Sabik JF, Svensson LG, Rodriguez LL, Lytle BW, et al. Outcomes of surgical aortic valve replacement for severe aortic stenosis: incorporation of left ventricular systolic function and stroke volume index. J Thorac Cardiovasc Surg. 2015;149:1558–66.
Eleid MF, Goel K, Murad MH, Erwin PJ, Suri RM, Greason KL, et al. Meta-analysis of the prognostic impact of stroke volume, gradient, and ejection fraction after transcatheter aortic valve implantation. Am J Cardiol. 2015;116:989–94.
Grossman W, Jones D, McLaurin LP. Wall stress and patterns of hypertrophy in the human left ventricle. J Clin Invest. 1975;56:56–64.
Quinones MA, Gaasch WH, Cole JS, Alexander JK. Echocardiographic determination of left ventricular stress-velocity relations in man. With reference to the effects of loading and contractility. Circulation. 1975;51:689–700.
Ross J Jr. Afterload mismatch and preload reserve: a conceptual framework for the analysis of ventricular function. Prog Cardiovasc Dis. 1976;18:255–64.
Gunther S, Grossman W. Determinants of ventricular function in pressure-overload hypertrophy in man. Circulation. 1979;59:679–88.
Henry WL, Bonow RO, Borer JS, Kent KM, Ware JH, Redwood DR, Itscoitz SB, McIntosh CL, Morrow AG. Evaluation of aortic valve replacement in patients with valve aortic stenosis. Circulation. 1980;61:814–25.
Starling MR, Kirsh MM, Montgomery DG, Gross MD. Mechanisms for left ventricular systolic dysfunction in aortic regurgitation: importance for predicting the functional response to aortic valve replacement. J Am Coll Cardiol. 1991;17:887–97.
Pirwitz MJ, Lange RA, Willard JE, Landau C, Glamann B, Hillis LD. Use of the left ventricular peak systolic pressure/end-systolic volume ratio to predict symptomatic improvement with valve replacement in patients with aortic regurgitation and enlarged end-systolic volume. J Am Coll Cardiol. 1994;24:1672–7.
Taniguchi K, Nakano S, Kawashima Y, Sakai K, Kawamoto T, Sakaki S, Kobayashi J, Morimoto S, Matsuda H. Left ventricular ejection performance, wall stress, and contractile state in aortic regurgitation before and after aortic valve replacement. Circulation. 1990;82:798–807.
Smith N, McAnulty JH, Rahimtoola SH. Severe aortic stenosis with impaired left ventricular function and clinical heart failure: results of valve replacement. Circulation. 1978;58:255–64.
St J, Sutton M, Plappert T, Spiegel A, Raichlen J, Douglas P, Reichek N, Edmunds L. Early postoperative changes in left ventricular chamber size, architecture, and function in aortic stenosis and aortic regurgitation and their relation to intraoperative changes in afterload: a prospective two-dimensional echocardiographic study. Circulation. 1987;76:77–89.
Ren JF, Panidis IP, Kotler MN, Mintz GS, Goel I, Ross J. Effect of coronary bypass surgery and valve replacement on left ventricular function: assessment by intraoperative two-dimensional echocardiography. Am Heart J. 1985;109:281–9.
Venco A, St J, Sutton MG, Gibson DG, Brown DJ. Non-invasive assessment of left ventricular function after correction of severe aortic regurgitation. Br Heart J. 1976;38:1324–31.
Roman MJ, Klein L, Devereux RB, Kligfield P, Niles NW, Hochreiter C, Isom W, Borer JS. Reversal of left ventricular dilatation, hypertrophy, and dysfunction by valve replacement in aortic regurgitation. Am Heart J. 1989;118:553–63.
Bonow RO. Regional left ventricular nonuniformity. Effects on left ventricular diastolic function in ischemic heart disease, hypertrophic cardiomyopathy, and the normal heart. Circulation. 1990;81:S54–65.
Nishimura RA, et al. 2014 AHA/ACC guideline for the management of patients with valvular heart disease: executive summary. J Am Coll Cardiol. 2014;22:2438–88.
Jin XY, Pepper JR, Xiao HB, Gibson DG. Effect of abnormal activation on the development of left ventricular incoordination in aortic valve disease (abstract). Circulation. 1996;94:I-668.
Wiggers CJ. The interpretation of the intraventricular pressure curve on the basis of rapid summated fractionate contractions. Am J Phys. 1927;80:1–30.
Heyndrickx G, Vilaine JP, Knight DR, Vatner SF. Effects of altered site of electrical activation on myocardial performance during inotropic stimulation. Circulation. 1985;71:1010–6.
Ford LE. Effect of afterload reduction on myocardial energetics. Circ Res. 1980;46:161–6.
Chiu YC, Walley KR, Ford LE. Comparison of the effects of different inotropic interventions on force, velocity, and power in rabbit myocardium. Circ Res. 1989;65:1161–71.
Katz LN, Feil HS. Clinical observations on the dynamics of ventricular systole: III. Aortic stenosis and aortic insufficiency. Heart. 1926;12:171–80.
Moskowitz RL, Wechsler BM. Left ventricular ejection time in aortic and mitral valve disease. Am J Cardiol. 1965;15:809–14.
Jin XY, Pepper JR, Gibson DG, Yacoub MY. Early changes in the time course of myocardial contraction after correcting aortic regurgitation. Ann Thorac Surg. 1999;67:139–45.
Jin XY, Pepper J. Severe left ventricular hypertrophy(LVH) indicates the failure of myocardial adaptation in aortic stenosis. Heart. 2005;91:A71.
Jin XY, Pepper J. Mechanical determinants of left ventricular hypertrophy in patients with heart valve diseases. Heart. 2015;101:A28–9.
Grossman W, Paulus WJ. Myocardial stress and hypertrophy: a complex interface between biophysics and cardiac remodeling. J Clin Invest. 2013;123:3701–3.
Jin XY, Hu J, Pezzella F, Pepper JR. The critical role of ERBB2 in human left ventricular remodelling due to pressure or volume overload. Heart. 2007;93:A18–9.
Gibson DG, Traill TA, Hall RJC, Brown DJ. Echocardiographic features of secondary left ventricular hypertrophy. Br Heart J. 1979;41:54–9.
Vanoverschelde JLJ, Essamri B, Michel X, Hanet C, Cosyns JR, Detry JMR, Wijns W. Hemodynamic and volume correlates of left ventricular diastolic relaxation and filling in patients with aortic stenosis. J Am Coll Cardiol. 1992;20:813–21.
Hess OM, Villari B, Krayenbuehl HP. Diastolic dysfunction in aortic stenosis. Circulation. 1993;87:S73–6.
Villari B, Sossalla S, Ciampi Q, Petruzziello B, Turina J, Schneider J, Turina M, Hess OM. Persistent diastolic dysfunction late after valve replacement in severe aortic regurgitation. Circulation. 2009;120:2386–92.
Gjertsson P, Caidahl K, Bech-Hanssen O. Left ventricular diastolic dysfunction late after aortic valve replacement in patients with aortic stenosis. Am J Cardiol. 2005;9:722–7.
Gjertsson P, Caidahl K, Farasati M, Odén A, Bech-Hanssen O. Preoperative moderate to severe diastolic dysfunction: a novel Doppler echocardiographic long-term prognostic factor in patients with severe aortic stenosis. J Thorac Cardiovasc Surg. 2005;129:890–6.
Krayenbuehl HP, Hess OM, Monrad ES, Schneider J, Mall G, Turina M. Left ventricular myocardial structure in aortic valve disease before, intermediate and late after aortic valve replacement. Acta Cardiol. 1992;47:145–56.
Jin XY. Elucidation of cardiac physiology in aortic valve replacement: what should we know? J Heart Valve Dis. 2004;13:S70–5.
Lund O. Valve replacement for aortic stenosis: the curative potential of early operation. Scand J Thorac Cardiovasc Surg. 1993;40:S1–137.
Fuster RG, Argudo JA, Albarova OG, Sos FH, López SC, Sorlí MJ, Codoñer MB, Miñano JA. Left ventricular mass index in aortic valve surgery: a new index for early valve replacement? Eur J Cardiothorac Surg. 2003;23:696–702.
Ruel M, Rubens FD, Masters RG, Pipe AL, Bédard P, Hendry PJ, Lam BK, Burwash IG, Goldstein WG, Brais MP, Keon WJ, Mesana TG. Late incidence and predictors of persistent or recurrent heart failure in patients with aortic prosthetic valves. J Thorac Cardiovasc Surg. 2004;127:149–59.
Brown ML, Pellikka PA, Schaff HV, Scott CG, Mullany CJ, Sundt TM, Dearani JA, Daly RC, Orszulak TA. The benefits of early valve replacement in asymptomatic patients with severe aortic stenosis. J Thorac Cardiovasc Surg. 2008;135:308–15.
Beach JM, Mihaljevic T, Rajeswaran J, Marwick T, Edwards ST, Nowicki ER, Thomas J, Svensson LG, Griffin B, Gillinov AM, Blackstone EH. Ventricular hypertrophy and left atrial dilatation persist and are associated with reduced survival after valve replacement for aortic stenosis. J Thorac Cardiovasc Surg. 2014;147:362–9.
Knöll R, Iaccarino G, Tarone G, Hilfiker-Kleiner D, Bauersachs J, Leite-Moreira AF, Sugden PH, Balligand JL. Towards a re-definition of ‘cardiac hypertrophy’ through a rational characterization of left ventricular phenotypes: a position paper of the Working Group ‘Myocardial Function’ of the ESC. Eur J Heart Fail. 2011;13:811–9.
Vliegen HW, van der Laarse A, Cornelisse CJ, Eulderink F. Myocardial changes in pressure overload-induced left ventricular hypertrophy. A study on tissue composition, polyploidization and multinucleation. Eur Heart J. 1991;12:488–94.
Lund O, Emmertsen K, Dørup I, Jensen FT, Flø C. Regression of left ventricular hypertrophy during 10 years after valve replacement for aortic stenosis is related to the preoperative risk profile. Eur Heart J. 2003;24:1437–46.
Mehta RH, Bruckman D, Das S, Tsai T, Russman P, Karavite D, Monaghan H, Sonnad S, Shea MJ, Eagle KA, Deeb GM. Implications of increased left ventricular mass index on in-hospital outcomes in patients undergoing aortic valve surgery. J Thorac Cardiovasc Surg. 2001;122:919–28.
Duncan AI, Lowe BS, Garcia MJ, Xu M, Gillinov AM, Mihaljevic T, Koch CG. Influence of concentric left ventricular remodeling on early mortality after aortic valve replacement. Ann Thorac Surg. 2008;85:2030–9.
van Straten AH, Soliman Hamad MA, Peels KC, van den Broek KC, ter Woorst JF, Elenbaas TW, van Dantzig JM. Increased septum wall thickness in patients undergoing aortic valve replacement predicts worse late survival. Ann Thorac Surg. 2012;94:66–71.
Milano AD, Faggian G, Dodonov M, Golia G, Tomezzoli A, Bortolotti U, Mazzucco A. Prognostic value of myocardial fibrosis in patients with severe aortic valve stenosis. J Thorac Cardiovasc Surg. 2012;144:830–7.
Olivetti G, Melissari M, Capasso JM, Anversa P. Cardiomyopathy of the aging human heart. Myocyte loss and reactive cellular hypertrophy. Circ Res. 1991;68:1560–8.
Mihaljevic T, Nowicki ER, Rajeswaran J, Blackstone EH, Lagazzi L, Thomas J, Lytle BW, Cosgrove DM. Survival after valve replacement for aortic stenosis: implications for decision making. J Thorac Cardiovasc Surg. 2008;135:1270–8.
Jin XY, Pillai R, Westaby S. Medium-term determinants of left ventricular mass index after stentless aortic valve replacement. Ann Thorac Surg. 1999;67:411–6.
Del Rizzo DF, Abdoh A, Cartier P, Doty D, Westaby S. Factors affecting left ventricular mass regression after aortic valve replacement with stentless valves. Semin Thorac Cardiovasc Surg. 1999;11:S114–20.
Mohty D, Dumesnil JG, Echahidi N, Mathieu P, Dagenais F, Voisine P, Pibarot P. Impact of prosthesis-patient mismatch on long-term survival after aortic valve replacement: influence of age, obesity, and left ventricular dysfunction. J Am Coll Cardiol. 2009;53:39–47.
Head SJ, Mokhles MM, Osnabrugge RL, Pibarot P, Mack MJ, Takkenberg JJ, Bogers AJ, Kappetein AP. The impact of prosthesis-patient mismatch on long-term survival after aortic valve replacement: a systematic review and meta-analysis of 34 observational studies comprising 27 186 patients with 133 141 patient-years. Eur Heart J. 2012;33:1518–29.
Jin XY, Westaby S. In vivo haemodynamic characteristics of porcine stentless aortic valves. Semin Thorac Cardiovasc Surg. 2001;13:S67–74.
Jin XY, Westaby S. Pericardial and porcine stentless aortic valves: are they hemodynamically different? Ann Thorac Surg. 2001;71:S311–4.
Pillai R, Kattach H, Soon JL, Ratnatunga C, Jin XY. 3F prosthesis aortic cusp replacement: implantation technique and early results. Asian Ann Cardiovasc Med Surg. 2010;18:13–6.
Westaby S, Horton M, Jin XY, Katsumata T, Li H, Grunkemeir G. Survival advantage of stentless aortic bioprosthesis. Ann Thorac Surg. 2000;70:785–90.
Kunadian B, Vijayalakshmi K, Thornley AR, de Belder MA, Hunter S, Kendall S, Graham R, Stewart M, Thambyrajah J, Dunning J. Meta-analysis of valve hemodynamics and left ventricular mass regression for stentless versus stented aortic valves. Ann Thorac Surg. 2007;84:73–8.
Jin XY, Ratnatunga C. Invited commentary on “A meta-analysis of valve hemodynamics and LV mass regression for stentless versus stented aortic valves”. Ann Thorac Surg. 2007;84:78–9.
Cohen G, Zagorski B, Christakis GT, Joyner CD, Vincent J, Sever J, Harbi S, Feder-Elituv R, Moussa F, Goldman BS, Fremes SE. Are stentless valves hemodynamically superior to stented valves? Long-term follow-up of a randomized trial comparing Carpentier-Edwards pericardial valve with the Toronto Stentless Porcine Valve. J Thorac Cardiovasc Surg. 2010;139:848–59.
Dayan V, Vignolo G, Soca G, Paganini JJ, Brusich D, Pibarot P. Predictors and outcomes of prosthesis-patient mismatch after aortic valve replacement. JACC Cardiovasc Imaging. 2016;9:924–33.
Gaudino M, Alessandrini F, Glieca F, Luciani N, Cellini C, Pragliola C, Morelli M, Canosa C, Nasso G, Possati G. Survival after aortic valve replacement for aortic stenosis: does left ventricular mass regression have a clinical correlate? Eur Heart J. 2005;26:51–7.
Westaby S, Jin XY, Katsumata T, Arifi A, Braidley P. Valve replacement with a stentless bioprosthesis: versatility of the porcine aortic root. J Thorac Cardiovasc Surg. 1998;116:477–84.
Bové T, Van Belleghem Y, François K, Caes F, Van Overbeke H, Van Nooten G. Stentless and stented aortic valve replacement in elderly patients: factors affecting midterm clinical and hemodynamical outcome. Eur J Cardiothorac Surg. 2006;30:706–13.
Imanaka K, Kohmoto O, Nishimura S, Yokote Y, Kyo S. Impact of postoperative blood pressure control on regression of left ventricular mass following valve replacement for aortic stenosis. Eur J Cardiothorac Surg. 2005;27:994–9.
Tjang YS, van Hees Y, Körfer R, Grobbee DE, van der Heijden GJ. Predictors of mortality after aortic valve replacement. Eur J Cardiothorac Surg. 2007;32:469–74.
Tzikas A, Geleijnse ML, Van Mieghem NM, Schultz CJ, Nuis RJ, van Dalen BM, Sarno G, van Domburg RT, Serruys PW, de Jaegere PP. Left ventricular mass regression one year after transcatheter aortic valve implantation. Ann Thorac Surg. 2011;91:685–91.
Schueler R, Sinning JM, Momcilovic D, Weber M, Ghanem A, Werner N, Nickenig G, Grube E, Hammerstingl C. Three-dimensional speckle-tracking analysis of left ventricular function after transcatheter aortic valve implantation. J Am Soc Echocardiogr. 2012;25:827–34.
Hahn RT, Pibarot P, Stewart WJ, Weissman NJ, Gopalakrishnan D, Keane MG, Anwaruddin S, et al. Comparison of transcatheter and surgical aortic valve replacement in severe aortic stenosis: a longitudinal study of echocardiography parameters in cohort a of the PARTNER trial (placement of aortic transcatheter valves). J Am Coll Cardiol. 2013;61:2514–21.
Houthuizen P, Van Garsse LA, Poels TT, de Jaegere P, van der Boon RM, Swinkels BM, Ten Berg JM, et al. Left bundle-branch block induced by transcatheter aortic valve implantation increases risk of death. Circulation. 2012;126:720–8.
Dizon JM, Nazif TM, Hess PL, Biviano A, Garan H, Douglas PS, Kapadia S, Babaliaros V, et al. PARTNER Publications Office. Chronic pacing and adverse outcomes after transcatheter aortic valve implantation. Heart. 2015;101:1665–71.
Massoullié G, Bordachar P, Ellenbogen KA, Souteyrand G, Jean F, Combaret N, Vorilhon C, Clerfond G, Farhat M, Ritter P, Citron B, Lusson JR, Motreff P, Ploux S, Eschalier R. New-onset left bundle branch block induced by transcutaneous aortic valve implantation. Am J Cardiol. 2016;117:867–73.
Magalhaes MA, Koifman E, Torguson R, Minha S, Gai J, Kiramijyan S, Escarcega RO, Baker NC, Wang Z, Goldstein S, Asch F, Satler LF, Pichard AD, Waksman R. Outcome of left-sided cardiac remodeling in severe aortic stenosis patients undergoing transcatheter aortic valve implantation. Am J Cardiol. 2015;116:595–603.
Jin XY, Pepper JR, Gibson DG. Effects of incoordination on left ventricular force-velocity relation in aortic stenosis. Heart. 1996;76:495–501.
Kvidal P, Bergström R, Hörte LG, Ståhle E. Observed and relative survival after aortic valve replacement. J Am Coll Cardiol. 2000;35:747–56.
Gotzmann M, Lindstaedt M, Mügge A. From pressure overload to volume overload: aortic regurgitation after transcatheter aortic valve implantation. Am Heart J. 2012;163:903–11.
Poulin F, Carasso S, Horlick EM, Rakowski H, Lim KD, Finn H, Feindel CM, Greutmann M, Osten MD, Cusimano RJ, Woo A. Recovery of left ventricular mechanics after transcatheter aortic valve implantation: effects of baseline ventricular function and postprocedural aortic regurgitation. J Am Soc Echocardiogr. 2014;27:1133–42.
Falk V. Transcatheter aortic valve replacement indications should not be expanded to lower-risk and younger patients. Circulation. 2014;130:2332–42.
Carabello BA, Usher BW, Hendrix GH, Assey ME, Crawford FA, Leman RB. Predictors of outcome for aortic valve replacement in patients with aortic regurgitation and left ventricular dysfunction: a change in the measuring stick. J Am Coll Cardiol. 1987;10:991–7.
Borer JS. Aortic valve replacement for the asymptomatic patient with aortic regurgitation: a new piece of the strategic puzzle. Circulation. 2002;106:2637–9.
Brown ML, Schaff HV, Suri RM, Li Z, Sundt TM, Dearani JA, Daly RC, Orszulak TA. Indexed left ventricular dimensions best predict survival after aortic valve replacement in patients with aortic valve regurgitation. Ann Thorac Surg. 2009;87:1170–5.
Shah PK. Should severe aortic stenosis be operated on before symptom onset? Severe aortic stenosis should not be operated on before symptom onset. Circulation. 2012;126:118–25.
Carabello BA. Should severe aortic stenosis be operated on before symptom onset? Aortic valve replacement should be operated on before symptom onset. Circulation. 2012;126:112–7.
Taniguchi T, Morimoto T, Shiomi H, Ando K, Kanamori N, Murata K, et al; CURRENT AS Registry Investigators. Initial surgical versus conservative strategies in patients with asymptomatic severe aortic stenosis. J Am Coll Cardiol. 2015;66:2827–38.
Zilberszac R, Gabriel H, Schemper M, Laufer G, Maurer G, Rosenhek R. Asymptomatic severe aortic stenosis in the elderly. JACC Cardiovasc Imaging. 2017;10:43–50.
Chin CW, Messika-Zeitoun D, Shah AS, Lefevre G, Bailleul S, Yeung EN, Koo M, Mirsadraee S, Mathieu T, Semple SI, Mills NL, Vahanian A, Newby DE, Dweck MR. A clinical risk score of myocardial fibrosis predicts adverse outcomes in aortic stenosis. Eur Heart J. 2016;37:713–23.
Kearney LG, Lu K, Ord M, Patel SK, Profitis K, Matalanis G, Burrell LM, Srivastava PM. Global longitudinal strain is a strong independent predictor of all-cause mortality in patients with aortic stenosis. Eur Heart J Cardiovasc Imaging. 2012;13:827–33.
Yingchoncharoen T, Gibby C, Rodriguez LL, Grimm RA, Marwick TH. Association of myocardial deformation with outcome in asymptomatic aortic stenosis with normal ejection fraction. Circ Cardiovasc Imaging. 2012;5:719–25.
Stanton LW, Garrard LJ, Damm D, Garrick BL, Lam A, Kapoun AM, Zheng Q, Protter AA, Schreiner GF, White RT. Altered patterns of gene expression in response to myocardial infarction. Circ Res. 2000;86:939–45.
Yang J, Moravec CS, Sussman MA, DiPaola NR, Fu D, Hawthorn L, Mitchell CA, Young JB, Francis GS, McCarthy PM, Bond M. Decreased SLIM1 expression and increased gelsolin expression in failing human hearts measured by high-density oligonucleotide arrays. Circulation. 2000;102:3046–52.
Yang J, Brown ME, Zhang H, Martinez M, Zhao Z, Bhutani S, Yin S, Trac D, Xi JJ, Davis ME. High-throughput screening identifies microRNAs that target Nox2 and improve function after acute myocardial infarction. Am J Physiol Heart Circ Physiol. 2017;312:H1002–H12.
Qiao A, Zhao Z, Zhang H, Sun Z, Cui X. Gene expression profiling reveals genes and transcription factors associated with dilated and ischemic cardiomyopathies. Pathol Res Pract. 2017;213:548–57.
Wang H, Kwak D, Fassett J, Hou L, Xu X, Burbach BJ, Thenappan T, Xu Y, Ge JB, Shimizu Y, Bache RJ, Chen Y. CD28/B7 deficiency attenuates systolic overload-induced congestive heart failure, myocardial and pulmonary inflammation, and activated T cell accumulation in the heart and lungs. Hypertension. 2016;68:688–96.
Balakumar P, Jagadeesh G. Multifarious molecular signaling cascades of cardiac hypertrophy: can the muddy waters be cleared? Pharmacol Res. 2010;62:365–83.
Pasipoularides A. Calcific aortic valve disease: Part 1—molecular pathogenetic aspects, hemodynamics, and adaptive feedbacks. J Cardiovasc Transl Res. 2016;9:102–18.
Pasipoularides A. Calcific aortic valve disease: Part 2—morphomechanical abnormalities, gene reexpression, and gender effects on ventricular hypertrophy and its reversibility. J Cardiovasc Transl Res. 2016;9:374–99.
Greve AM, Boman K, Gohlke-Baerwolf C, Kesäniemi YA, Nienaber C, Ray S, Egstrup K, Rossebø AB, Devereux RB, Køber L, Willenheimer R, Wachtell K. Clinical implications of electrocardiographic left ventricular strain and hypertrophy in asymptomatic patients with aortic stenosis: the Simvastatin and Ezetimibe in Aortic Stenosis study. Circulation. 2012;125:346–53.
Shah AS, Chin CW, Vassiliou V, Cowell SJ, Doris M, Kwok TC, Semple S, Zamvar V, White AC, McKillop G, Boon NA, Prasad SK, Mills NL, Newby DE, Dweck MR. Left ventricular hypertrophy with strain and aortic stenosis. Circulation. 2014;130:1607–16.
Du X, Jin XY. The time course of improvement in ECG strain pattern after AVR for aortic stenosis and its underlying mechanisms. Heart. 2007;93:A39.
Jin XY, Yuan L, Hu JT, Pepper JR. The simultaneous changes in ECG strain pattern and left ventricular force-velocity relationship immediately after valve replacement for aortic stenosis. Heart. 2013;99:A87.
Hu JT, Pepper J, Pezzella F, Gatter K, Jin XY. Genomic insights of ECG strain patten in aortic stenosis: T wave inversion and ST-segment depression are underlined by different molecular pathways. Heart. 2017;103:A97.
Kuwahara F, Kai H, Tokuda K, Kai M, Takeshita A, Egashira K, Imaizumi T. Transforming growth factor-beta function blocking prevents myocardial fibrosis and diastolic dysfunction in pressure-overloaded rats. Circulation. 2002;106:130–5.
Pennica D, Shaw KJ, Swanson TA, Moore MW, Shelton DL, Zioncheck KA, Rosenthal A, Taga T, Paoni NF, Wood WI. Cardiotrophin-1. Biological activities and binding to the leukemia inhibitory factor receptor/gp130 signaling complex. J Biol Chem. 1995;270:10915–22.
López B, González A, Querejeta R, Barba J, Díez J. Association of plasma cardiotrophin-1 with stage C heart failure in hypertensive patients: potential diagnostic implications. J Hypertens. 2009;27:418–24.
Huang-Lee LL, Nimni ME. Fibroblast contraction of collagen matrices with and without covalently bound hyaluronan. J Biomater Sci Polym Ed. 1993;5:99–109.
Ahmed SH, Clark LL, Pennington WR, Webb CS, Bonnema DD, Leonardi AH, McClure CD, Spinale FG, Zile MR. Matrix metalloproteinases/tissue inhibitors of metalloproteinases: relationship between changes in proteolytic determinants of matrix composition and structural, functional, and clinical manifestations of hypertensive heart disease. Circulation. 2006;113:2089–96.
González A, López B, Querejeta R, Zubillaga E, Echeverría T, Díez J. Filling pressures and collagen metabolism in hypertensive patients with heart failure and normal ejection fraction. Hypertension. 2010;55:1418–24.
Melendez-Zajgla J, Del Pozo L, Ceballos G, Maldonado V. Tissue inhibitor of metalloproteinases-4. The road less traveled. Mol Cancer. 2008;7:85.
Yarbrough WM, Mukherjee R, Ikonomidis JS, Zile MR, Spinale FG. Myocardial remodeling with aortic stenosis and after aortic valve replacement: mechanisms and future prognostic implications. J Thorac Cardiovasc Surg. 2012;143:656–64.
Dahl JS, Videbaek L, Poulsen MK, Pellikka PA, Veien K, Andersen LI, Haghfelt T, Møller JE. Effect of candesartan treatment on left ventricular remodeling after aortic valve replacement for aortic stenosis. Am J Cardiol. 2010;106:713–9.
Benedetto U, Melina G, Refice S, di Bartolomeo R, Roscitano A, Angeloni E, Sinatra R. Dual renin-angiotensin system blockade for patients with prosthesis-patient mismatch. Ann Thorac Surg. 2010;90:1899–903.
Coffey S, Williams MJ, Phillips LV, Galvin IF, Bunton RW, Jones GT. Integrated microRNA and messenger RNA analysis in aortic stenosis. Sci Rep. 2016;6:36904.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer-Verlag GmbH Austria, part of Springer Nature
About this chapter

Cite this chapter
Jin, X.Y., Hu, J.T., Pepper, J.R. (2019). Left Ventricular Function and Aortic Valve Replacement. In: Stanger, O., Pepper, J., Svensson, L. (eds) Surgical Management of Aortic Pathology. Springer, Vienna. https://doi.org/10.1007/978-3-7091-4874-7_22
Download citation
DOI: https://doi.org/10.1007/978-3-7091-4874-7_22
Published:
Publisher Name: Springer, Vienna
Print ISBN: 978-3-7091-4872-3
Online ISBN: 978-3-7091-4874-7
eBook Packages: MedicineMedicine (R0)