
Zusammenfassung
Die Pumpleistung des Herzens nimmt mit steigendem venösem Zufluss durch folgende Mechanismen automatisch zu: - Frank-Starlin-Mechanismus: Vergrößerung des venösen Angebotes → stärkere diastolische Kammerfüllung unter Zunahme der Faserdehnung → Steigerung der Kontraktionskraft und des Schlagvolumens. - Mechanische Steigerung der Herzfrequenz: Stärkere Füllung des rechten Vorhofes → Stimulation des Sinusknotens durch Dehnung → Anstieg der Schlagfrequenz (mit frequenzbedingter Kontraktilitätssteigerung).
We’re sorry, something doesn't seem to be working properly.
Please try refreshing the page. If that doesn't work, please contact support so we can address the problem.
1.1 Mechanismen der Kreislaufregulation
Mechanismen der Herz-Kreislauf-Regulation
-
Intrinsic-Regulation
-
Nervale Regulation
-
Hormonale Regulation
-
Physikalische Regulation
-
Gesamtregulationsmechanismen
1.1.1 Intrinsic-Regulation
Intrinsic-Regulation
Basale Regulationsmechanismen des Kreislaufs, die von der übergeordneten nervalen und humoralen Steuerung unabhängig sind.
1.1.1.1 Autoregulation der kardialen Pumpleistung
Die Pumpleistung des Herzens nimmt mit steigendem venösem Zufluss durch folgende Mechanismen automatisch zu:
-
Frank-Starlin-Mechanismus: Vergrößerung des venösen Angebotes → stärkere diastolische Kammerfüllung unter Zunahme der Faserdehnung → Steigerung der Kontraktionskraft und des Schlagvolumens.
-
Mechanische Steigerung der Herzfrequenz: Stärkere Füllung des rechten Vorhofes → Stimulation des Sinusknotens durch Dehnung → Anstieg der Schlagfrequenz (mit frequenzbedingter Kontraktilitätssteigerung).
1.1.1.2 Autoregulation der Gewebe- und Organdurchblutung
Sie passt die Blutversorgung der Organe und Gewebe genau ihrer Stoffwechselaktivität an, verhindert somit Schädigungen durch Über- und Unterperfusion und trägt maßgeblich zur Ökonomie der Herztätigkeit bei. Gesteuert wird der lokale Blutfluss durch schnelle Änderungen des Kontraktionszustandes der Arteriolen, Metarteriolen (Endarteriolen) und präkapillären Sphinkter. Die letzteren werden abwechselnd geschlossen und wieder ganz geöffnet, so dass die Durchblutung von der Zahl der geöffneten Sphinkter abhängt. Die Steuerungsmechanismen sind:
-
Vasokonstriktion: Bei akutem Blutdruckanstieg bewirkt die plötzliche Zunahme des Blutflusses nach der metabolischen Theorie ein zu großes Angebot von O2 und Nährstoffen im Gewebe, das den Tonus der Arteriolen und der präkapillären Sphinkter erhöht. Nach der myogenen Theorie steigt der Tonus dieser Gefäße reflektorisch, wenn eine plötzliche Wanddehnung erfolgt. Zur Vasokonstriktion trägt das vom Gefäßendothel sezernierte Endothelin 1 bei, ein Peptid aus 21 Aminosäuren, für das auch spezifische Rezeptoren existieren. In jedem Fall wird die Gewebeperfusion auch bei größeren Blutdruckausschlägen in normalen Grenzen gehalten.
-
Vasodilatation: Erfolgt durch vasodilatierende Substanzen, die bei Stoffwechselsteigerung bzw. O2-Mangel im Gewebe vermehrt anfallen: Adenosinphosphat (aus ATP), CO2, Laktat, Histamin, Kalium- und Wasserstoffionen. Das Absinken der O2-Konzentration im Gewebe vermindert auch direkt den Tonus der Arteriolen und öffnet die präkapillären Sphinkter. Das verstärkt einschießende Blut setzt durch Scherstress in den Endothelzellen der Arteriolen und kleinen Arterien Nitroxid (NO) frei, das vasodilatierend auf die stromaufwärts gelegenen intermediären und größeren Arterien wirkt. Das Radikal NO entsteht nach Aktivierung der NO-Synthase aus Arginin, das dabei in Citrullin übergeht.
-
Vaskularisierung: Langfristigen Änderungen der lokalen Durchblutung wird durch Zunahme bzw. Abnahme der Vaskularisierung (Kapillarisierung) begegnet. Es können Umgehungskreisläufe entstehen. Auf die Dauer passt sich der Vaskularisierungsgrad genau der Größe des Stoffwechsels an. Die wichtigsten Induktoren der Angiogenese sind FGF-1 (fibroblast growth factor 1), VEGF (vascular endothelial growth factor) und PDGFs (platelet derived growth factors), die alle von hypoxieinduzierenden Genen gebildet werden und auch bei der Vaskularisierung expandierender Karzinome mitwirken.
Besonderen Regulationsmechanismen unterliegen Gehirn- und Nierenkreislauf. Im Gehirn wirkt CO2 stark vasodilatierend. Die überwiegend nicht nutritiven Zwecken dienende Nierendurchblutung wird durch den juxtaglomerulären Apparat reguliert.
1.1.1.3 Hormonunabhängige renale Regulation des Blutvolumens
Durch das Phänomen der Druckdiurese (pressure diuresis) trägt die Niere zur Kontrolle des Blutvolumens und damit auch des Blutdrucks bei:
-
Reaktion auf Verminderung des Blutvolumens: → Abnahme des Herzzeitvolumen s (cardiac output) → Absinken des Blutdrucks → Verminderung der renalen Wasser- und Salzausscheidung (durch Abnahme der glomerulären Filtrationsrate und des peritubulären Kapillardrucks) → Zunahme der extrazellulären Flüssigkeit (Blutplasma + interstitielle Flüssigkeit) → Anstieg des Blutvolumens und des Blutdrucks zur Norm.
-
Reaktion auf Vermehrung des Blutvolumens: → Zunahme des Herzzeitvolumen s → autoregulatorische Vasokonstriktion in der zu stark durchbluteten Kreislaufperipherie → Anstieg des Blutdrucks → Zunahme der renalen Wasser- und Salzausscheidung („pressure-diuresis“ durch Erhöhung der glomerulären Filtrationsrate und des peritubulären Kapillardrucks) → Absinken des Blutvolumens und des Blutdrucks zur Norm. Voraussetzung ist eine normale Nierenfunktion.
1.1.2 Nervale Regulation
Nervale Regulation
Kreislaufregulation durch das autonome Nervensystem, das den Intrinsic-Mechanismen zum Zweck integrierter Gesamtregulation übergeordnet ist, schneller reagiert und die Funktionsreserven weiter ausschöpft.
1.1.2.1 Medulläre autonome Zentren der Kreislaufregulation
1.1.2.1.1 Vasokonstriktorisches Areal des Vasomotorenzentrum s
Bilateral, im anterolateralen Bereich der oberen Medulla gelegene Neuronengruppe, die über den Sympathikus spontan und kontinuierlich vasokonstriktorische Impulse aussendet. Diese fortlaufende Stimulation wird als Vasomotorentonus bezeichnet. Von demselben Areal gehen via Sympathikus auch exzitatorische Impulse zum Herzen aus.
1.1.2.1.2 Vasodilatatorisches Areal des Vasomotorenzentrums
Bilateral, in der unteren Hälfte der Medulla, dicht neben dem motorischen Vaguskern gelegene Neuronengruppe. Ihre Fasern ziehen aufwärts in das vasokonstriktorische Areal, hemmen dessen vasokonstriktorische Aktivität und bewirken eine Vasodilatation. Zusätzlich stimulieren sie über den motorischen Vaguskern parasympathische kardioinhibitorische Impulse zum Herzen.
1.1.2.1.3 Sensorisches Areal
Beiderseits im Tractus solitarius lokalisiert, verläuft posterolateral in der Medulla oblongata und erstreckt sich bis in den unteren Abschnitt der Pons. Die Neuronen dieses Areals empfangen sensorische Nervensignale vom Vagus und Glossopharyngeus und senden Signale zum vasokonstriktorischen und zum vasodilatatorischen Areal aus, die eine reflektorische Kontrolle des Blutdrucks ermöglichen.
1.1.2.2 Supramedulläre Kontrolle des Vasomotorenzentrums
Das selbständig arbeitende medulläre Vasomotorenzentrum kann durch Impulse aus höheren Zentren beeinflusst werden:
-
Hypothalamus: Stimulation des vorderen Hypothalamus erhöht den Vagus- oder Parasympathikustonus. Stimulation des hinteren Hypothalamus steigert den Sympathikustonus und setzt den Parasympathikustonus herab.
-
Cortex: Verschiedene Regionen der Hirnrinde (motorische Rinde, vorderer Temporallappen, Hippokampus u. a.) können die hypothalamischen und medullären Zentren stimulierend oder hemmend beeinflussen. Damit werden die vielfältigen emotionalen, psychomotorischen und sensorischen Effekte auf den Kreislauf verständlich.
1.1.2.3 Afferente und direkte Stimulation der medullären Zentren
1.1.2.3.1 Arterielle Barorezeptoren (high pressure receptors)
Dehnungsrezeptoren in der Arterienwand (Karotissinus, Aorta, große Arterien), deren Impulse über den N. vagus und N. glossopharyngeus zum sensorischen Areal (Tractus solitarius) geleitet werden. Von dort gehen dann inhibitorische Impulse zum vasokonstriktorischen Areal des Vasomotorenzentrums und exzitatorische Impulse zum Vaguskern aus. Die permanente Entladungsfrequenz der Rezeptoren steigt bei Blutdruckanstieg und sinkt bei plötzlichem Blutdruckabfall. Auf diese Weise wirkt der Barorezeptorreflex vor allem plötzlichen Änderungen des Blutdrucks entgegen und zwar nach beiden Seiten. Auf langfristige Blutdruckerhöhung sprechen die Barorezeptoren nicht an, sondern verändern ihren Stellwert. Mechanischer Druck auf den Karotissinus stimuliert die Barorezeptoren und bewirkt Blutdruckabfall und Bradykardie. Bei einem durch Wandveränderungen (Arterioklerose, Narben, Lymphome) hypersensitiven Karotissinus ist die Reaktion auf den Karotisdruck verstärkt. Durch massiven Blutdruckabfall und extreme Bradykardie kann es zum Ohnmachtsanfall kommen (Karotis-Sinus-Synkope).
1.1.2.3.2 Venöse Barorezeptoren (low pressure receptors)
Dehnungsrezeptoren in den Vorhöfen und Pulmonalarterien, die einem Blutdruckanstieg bei Zunahme des Blutvolumens entgegenwirken. Wenn bei einem Volumenanstieg der Druck in den Vorhöfen und den Pulmonalarterien zunimmt, senden die Dehnungsrezeptoren vermehrt Impulse zum vasodilatatorischen Areal des Vasomotorenzentrums aus, die durch Vasodilatation den Blutdruck senken. Besonders intensiv ist die Dilatation der afferenten Nierenarteriolen, die über eine Steigerung der glomerulären Filtrationsrate die Urinausscheidung steigert. Die atrialen Rezeptoren hemmen bei Druckanstieg reflektorisch den Vagustonus und bewirken dadurch einen Frequenzanstieg des Herzens (Bainbridge-Reflex ).
1.1.2.4 Direkte Stimulation des Vasomotorenzentrums
1.1.2.4.1 Ischämiereaktion des ZNS
Erfolgt bei systolischem Blutdruckabfall unter 50 mmHg oder bei lokaler Durchblutungsstörung der Medulla oblongata: Maximale Stimulation des vasokonstriktorischen Areals des Vasomotorenzentrums mit intensiver generalisierter Vasokonstriktion und extremem Blutdruckanstieg.
1.1.2.4.2 Cushing-Reaktion
Spezieller Typ der Ischämiereaktion bei Kompression zerebraler Arterien durch überhöhten Liquordruck. Der reflektorische Blutdruckanstieg lässt den Blutdruck wieder über den Liquordruck steigen.
1.1.2.5 Sympathikus
1.1.2.5.1 Anatomische Gliederung und Überträgerstoffe
Vom Sympathikus gehen alle efferenten adrenergen Impulse des autonomen Nervensystems aus. Sie bewirken an den innervierten Endorganen die Freisetzung von Noradrenalin und im Nebennierenmark die Ausschüttung von Adrenalin, das auf dem Blutweg seine Wirkung entfaltet.
Die Sympathikusbahn ist in 2 Neurone gegliedert:
-
das pränglionäre Neuron mit dem Zellkörper im Seitenhorn des Rückenmarkgrau und
-
das postganglionäre Neuron mit dem Zellkörper in den paravertebralen oder prävertebralen sympathischen Ganglien.
Die präganglionären Sympathikusfasern sind cholinergisch, d. h. sie setzen an der Synapse in den Ganglien als Überträgerstoff Acetylcholin frei.
1.1.2.5.1.1 Adrenerge Rezeptoren
Die Wirkung von Adrenalin und Noradrenalin auf die sympathischen Effektorzellen wird durch den Rezeptortyp bestimmt, den diese Zellen tragen. Es kann eine exzitatorische oder inhibitorische Wirkung sein. Zu unterscheiden sind die Rezeptoren α1, α2, β1, β2 und β3. Vom α1- und α2-Rezeptor konnten je 3 Subtypen geklont werden, die zwar in ihrer Gewebeverteilung, aber nicht in ihren funktionellen Eigenschaften differieren. Ein β3-Rezeptor ist hauptsächlich im Fettgewebe lokalisiert. Seine Bedeutung für die Lipolyse ist ungeklärt. Noradrenalin und Adrenalin wirken auf α1-, α2- und β1-Rezeptoren annähern gleich stark. Auf β2-Rezeptoren, deren Stimulation zur Vasodilatation führt, wirkt Noradrenalin erheblich schwächer als Adrenalin.
1.1.2.5.2 Innervation und Wirkung am Kreislaufsystem
1.1.2.5.2.1 Periphere Gefäße
Vasokonstriktorische Sympathikusfasern innervieren die mit α1-Rezeptoren ausgestatteten kleinen Arterien, Arteriolen, Venolen und Venen, nicht aber Kapillaren, präkapilläre Sphinkter und Metarteriolen (◘ Abb. 1.1). Die Konstriktion der Arteriolen und Arterien bewirkt eine Erhöhung des peripheren Gesamtwiderstandes und des Blutdrucks. Durch die Konstriktion der Venolen und Venen wird die Füllungskapazität des Venensystems reduziert. Die Folge ist eine Zunahme des venösen Rückflusses zum Herzen und eine volumeninduzierte Steigerung der Pumpleistung. α2-Rezeptoren sind an den sympathischen Endausbreitungen und in der pontomedullären Region des ZNS lokalisiert. Ihre Stimulation durch Katecholamine hemmt die Freisetzung von Noradrenalin in den vom Sympathikus innervierten Gefäßen und den zentralen Sympathikustonus.


Sympathikus-Innervation des Gefäßsystems
1.1.2.5.2.2 Skelettmuskelgefäße
In der Skelettmuskulatur ist der vasokonstriktorische Sympathikuseffekt relativ schwach. Hier sind die Gefäßmuskelzellen mit α1- und β2-Rezeptoren besetzt. Bei körperlicher Arbeit erfolgt eine intensive Vasodilatation durch Autoregulation, und den vasodilatatorischen Effekt des vom Nebennierenmark ausgeschütteten Adrenalins auf die α2-Rezeptoren.
1.1.2.5.2.3 Herzkranzgefäße
Die epikardialen Gefäße haben überwiegend α1- und α2-Rezeptoren, die intramyokardialen überwiegend β2-Rezeptoren. Unter Ruhebedingungen resultiert ein schwacher vasokonstriktorischer Sympathikustonus. Bei Belastung erfolgt durch metabolische Autoregulation eine dem O2-Verbrauch proportionale Vasodilatation. Daran ist der Sympathikus indirekt beteiligt, weil er an seinen myokardialen Angriffspunkten die Kontraktilität und damit den Stoffwechsel steigert.
1.1.2.5.2.4 Herz
Sinusknoten, Vorhöfe, AV-Knoten, His-Purkinje-Fasern und Ventrikel haben Adrenorezeptoren der Typen β1, β2 und α1. Im normalen Myokard beträgt das Verhältnis 70–80:20–30. Die adrenergen Rezeptoren im Sarkolemm der Myozyten gehören zu den Protein-G-gekoppelten Rezeptoren . Nach Stimulation der β-Rezeptoren durch Katecholamine verbindet sich der Rezeptor mit dem stimulatorischen G-Protein (Gs), das aus den Untereinheiten α, β und γ besteht. Danach löst sich das an die α-Untereinheit gebundene GDP und wird durch GPT ersetzt. Die damit verbundene Aktivierung des G-Proteins führt zur Abspaltung der GPT tragenden α-Untereinheit. Der GTP-α-Komplex aktiviert nun die in der Membran lokalisierte Adenylatzyklase, die als Second Messenger cAMP bildet. Durch cAMP wird die Proteinkinase A aktiviert, die über eine Phosphorylierung der Calciumkanäle vom Typ L den Calciuminflux und damit die Inotropie steigert. β-Rezeptoren können sich auch mit inhibierendem G-Protein (Gi) verbinden. Daraus resultiert eine Hemmung der Adenylatzyklase. Die adrenergen α1-Rezeptoren verbinden sich nach der Stimulation mit dem G-Protein Gq, das in der Zellmembran die Phospholipase Cβ aktiviert. Diese spaltet Phosphoinositide in 2 Second Messenger, Inositoltriphosphat (IP3) und Diacylglycerol (DAG). IP3 stimuliert die Freisetzung von Ca++ aus intrazellulären Speichern, während DAG ein potenter Aktivator der Phospholipase Cβ ist.
Insgesamt nehmen bei gesteigerter Sympathikusaktivität im Sinusknoten die Impulsfrequenz, im AV-Knoten und den His-Purkinje-Fasern die Leitungsgeschwindigkeit und im Arbeitsmyokard die Inotropie zu.
1.1.2.6 Parasympathikus
1.1.2.6.1 Anatomische Gliederung, Überträgerstoff und Rezeptoren
Der Parasympathikus ist in prä- und postganglionäre Neurone gegliedert. Der Zellkörper der präganglionären Neurone liegt in den Kernen der Hirnnerven III (N. oculomotorius), VII (N. facialis), IX (N. glossopharyngeus) und X (N. vagus), sowie im 2.–4. Sakralsegment des Rückenmarks. Die Neuriten der präganglionären Neurone werden erst in den Erfolgsorganen auf die postganglionären Neurone umgeschaltet und sind deshalb erheblich länger als die der postganglionären. Etwa 75 % aller parasympathischen Nervenfasern, darunter die kreislaufwirksamen, verlaufen im N. vagus. Überträgerstoff sowohl an den Synapsen zwischen prä- und postganglionären Neuronen als auch an den Endausbreitungen der postganglionären Neuriten ist das Acetylcholin . Man spricht deshalb auch vom cholinergen autonomen Nervensystem. Das Acetylcholin reagiert mit 2 Rezeptortypen. An den Synapsen zwischen prä- und postganglionären parasympathischen (und sympathischen) Neuronen befinden sich Rezeptoren vom Nikotintyp (selektive Aktivierung durch Nikotin). Alle von postganglionären parasympathischen Neuronen stimulierten Effektorzellen tragen Rezeptoren vom Muskarintyp (selektive Aktivierung durch Muskarin).
1.1.2.6.2 Innervation und Wirkung am Kreislaufsystem
1.1.2.6.2.1 Periphere und Skelettmuskelgefäße
Sie haben keine parasympathische Innervation, sondern werden nur vom Sympathikus gesteuert.
1.1.2.6.2.2 Herz
Vom Vagus innerviert werden der Sinusknoten, das Vorhofmyokard und der AV-Knoten. Nur wenige, funktionell unbedeutende Vagusfasern erreichen das Kammermyokard. Eine Vagusstimulation verlangsamt die Sinusknotenimpulse und die Erregungsleitung in den Verbindungsfasern zwischen Vorhofmyokard und AV-Knoten. Außerdem setzt sie die Kontraktilität der Vorhöfe herab. Es resultiert eine Abnahme der Herzfrequenz und der kardialen Pumpleistung.
Extreme Vagusstimulation kann zum Sinusstillstand und zum AV-Block führen.
1.1.3 Hormonale Regulation
1.1.3.1 Renin-Angiotensin-Aldosteron-System
Das System dient der kurz- und langfristigen Blutdruckkontrolle, die durch Einwirkung auf den Gefäßtonus und das Blutvolumen erreicht wird.
1.1.3.1.1 Renin
Aktiviert wird das System durch das Renin, eine Protease, die in den juxtaglomerulären Zellen (JG-Zellen) des Vas afferens der Glomeruli aus Prorenin gebildet und zusammen mit seiner Vorstufe gespeichert wird. Zur Reninsekretion in die Blutbahn kommt es durch:
-
Stimulation der Barorezeptoren im Vas afferens als Reaktion auf Blutdruckabfall oder lokale Ischämie,
-
Stimulation der JG-Zellen über die Macula densa des juxtaglomerulären Apparates bei Absinken der Na+- und Cl–-Konzentration im distalen Tubulus,
-
Sympathikusstimulation der β1-Rezeptoren an den JG-Zellen.
1.1.3.1.2 Angiotensin
Das Renin spaltet im Blutplasma aus den Angiotensinogen, einem α2-Globulin, das weitgehend kreislaufinaktive Dekapeptid Angiotensin I ab. Daraus entsteht durch Einwirkung des am Gefäßendothel (hauptsächlich in der Lunge) lokalisierten Angiotensin-Converting-Enzym (ACE) das aktive Oktapeptid Angiotensin II. Ebenfalls wirksam, wenn auch viel schwächer ist das Heptapeptid Angiotensin III, das beim Abbau des Angiotensin II durch eine Aminopeptidase gebildet wird. Das Angiotensin II ist die Schlüsselsubstanz des Systems. Mit 2 unterschiedlichen Wirkungsmechanismen trägt es maßgeblich zur Blutdruckstabilisierung bei:
-
Vasokonstriktion durch:
-
hauptsächlich intensive direkte Kontraktion der Arteriolen, weniger der postkapillären Venolen
-
Verstärkung des peripheren Symathikustonus über eine Hemmung des Noradrenalin-Reuptake in die Nervenenden
-
Stimulation des Vasomotorenzentrums.
-
Die Renin-Angiotensin-Vasokonstriktion wird erst 20 Minuten nach Stimulation des Systems voll wirksam, also deutlich langsamer als die Sympathikusaktivierung.
-
Expansion des Blutvolumens durch:
-
Stimulation der Rückresorption von Na+, Cl- und Bikarbonat im proximalen Tubulus, die zur renalen Wasser- und Salzretention führt
-
Konstriktion der renalen Blutgefäße, die den renalen Blutfluss und die Harnausscheidung drosselt
-
Stimulation der Aldosteronsekretion in der Nebennierenrinde. Aldosteron steigert im distalen Tubulus die renale Na+-Rückresorption und expandiert damit das extrazelluläre Flüssigkeitsvolumen. Die direkten Effekte des Angiotensin II auf die Niere sind dreimal stärker als der indirekte Effekt über das Aldosteron.
-
Die Expansion des Blutvolumens beginnt einige Stunden nach der Stimulation des Renin-Angiotensin-Aldosteron-Systems, dauert aber so lange, bis die Ursache der Stimulation entfällt.
Alle kreislaufwirksamen Effekte des Angiotensin II werden an die Effektorzellen durch AT1-Rezeptoren vermittelt. AT2-Rezeptoren sind weit verstreut in fetalen Geweben anzutreffen, weniger bei Erwachsenen. Ihre Funktion ist noch nicht genau bekannt.
1.1.3.1.2.1 Anmerkung
Mehrere Gewebe haben ein lokales Renin-Angiotensin-System, das Angiotensin II produzieren kann. Dazu gehören Gefäßwand, Herz, Gehirn, Nebennierenrinde, Nieren, Uterus und Plazenta. In Tierexperimenten zeigte Angiotensin II an kardiovaskulären Strukturen folgende, wahrscheinlich parakrine Effekte:
-
Stimulation von Migration, Proliferation und Hypertrophie glatter Muskelzellen
-
Steigerung der Matrixproduktion glatter Muskelzellen
-
Hypertrophie kardialer Myozyten
-
Stimulation der Fibroblasten zu vermehrter Bildung von extrazelllärer Matrix
In analoger Weise scheint Angiotensin II auch beim Menschen wirksam zu sein. Denn die Behandlung mit ACE-Blockern wirkt der Hypertrophie und Fibrosierung des Herzmuskels sowie dem Remodeling des linken Ventrikels nach Myokardinfarkt entgegen.
1.1.3.2 Antidiuretisches Hormon (ADH, Vasopressin )
Das im Hypothalamus gebildete und im Hypophysenhinterlappen sezernierte Nonapeptid ADH hat 2 kreislaufstabilisierende Wirkungen:
-
Durch Stimulation der Wasserrückresorption im distalen Nephron stabilisiert es das Blutvolumen.
-
Durch generalisierte Arteriolenkonstriktion, auch in der Skelettmuskulatur, erhöht es den peripheren Gesamtwiderstand und damit den Blutdruck. Die Vasokonstriktion wird durch V1-Rezeptoren an den Gefäßen vermittelt, die Wasserrückresorption durch V2-Rezeptoren an den Sammelrohren.
Die Stimulation der ADH-Sekretion durch das Kreislaufsystem erfolgt auf nervalem Weg über die Barorezeptoren im Karotissinus (bei Blutdruckabfall) und in den Vorhöfen (bei Volumenmangel). Während die ADH-Sekretion auf einen von zentralnervösen Osmorezeptoren signalisierten Anstieg der Plasmaosmolarität höchst empfindlich reagiert, springt sie erst auf starken Blutdruckabfall und schwere Volumenverluste an.
1.1.3.3 Natriuretische Peptide
Zwei im Herzen gebildete Peptide wirken vasodilatatorisch und natriuretisch und vermindern dadurch das Blutvolumen. Sie supprimieren außerdem die Aktivität des Renin-Angiotensin-Aldosteron- und des adrenergen Systems. Ein drittes natriuretisches Peptid (CNP) wird im Gefäßsystem gebildet. Seine physiologische Bedeutung ist noch nicht genau geklärt. Der natriuretische und vasodilatatorische Effekt wird durch Rezeptoren der Typen A und B vermittelt. Ein Rezeptor vom Typ C trägt zusammen mit einer neutralen Endopeptidase zur Elimination bzw. Regulation der Peptide bei.
1.1.3.3.1 ANP (atriales natriuretisches Peptid )
Das Peptid aus 28 Aminosäuren wird in den Myokardzellen beider Vorhöfe gebildet und bei Vorhofdehnung vermehrt sezerniert. Bei seiner Freisetzung wird es vom Pro-ANP, einem in Granula gespeicherten Vorläuferprotein, abgespalten. Die normalen Plasmaspiegel betragen 2–80 pmol/l. Der natriuretische Effekt kommt hauptsächlich durch Dilatation des Vas afferens und die damit verbundene Steigerung der glomerulären Filtrationsrate zustande. Ergänzt wird er durch die Hemmung der Aldosteronsekretion. Per saldo wirkt ANP einer Stauung in den Vorhöfen durch Herzinsuffizienz, Niereninsuffizienz, Hypervolämie und Hypertonie entgegen. Das ANP im Plasma steigt auch beim paroxysmalen Vorhofflimmern an.
1.1.3.3.2 BNP (brain natriuretic peptide )
Die Bezeichnung beruht auf der erstmaligen Isolierung des Peptids aus Schweinegehirn. Es hat 32 Aminosäuren und wird hauptsächlich von den Zellen des Kammermyokards gebildet und bei Zunahme des ventrikulären Füllungsdrucks vermehrt freigesetzt. Der Wirkungsmechanismus entspricht dem des ANP, denn es greift an den gleichen Rezeptoren an. BNP entsteht aus proBNP und wird daraus bei der Sekretion abgespalten. In äquimolaren Mengen sezerniert wird auch das inaktive N-terminale Fragment des proBNP (NT-proBNP). Da es mit > 120 min eine höhere Plasmahalbwertszeit hat als BNP (< 20 min), wird die Bestimmung des NT-proBNP bevorzugt zur Messung der BNP-Sekretion verwendet. In ◘ Abb. 1.2 sind die Normalwerte und ihre Altersabhängigkeit wiedergegeben. Inzwischen steht ein Schnelltest für Bedside-Bestimmungen zur Verfügung (◘ Abb. 1.2).


Erwartete NT-proBNP-Werte bei gesunden Individuen in Bezug auf das Alter. Mit zunehmendem Alter werden erhöhte NT-proBNP-Werte immer häufiger auch bei scheinbar Gesunden gefunden. Diese erhöhten Werte zeigen wahrscheinlich ein frühes Stadium und/oder milde Formen einer asymptomatischen kardialen Dysfunktion an (Roche Diagnostics)


Röntgenthorax (liegend) eines 72-jährigen beatmeten Patienten im kardiogenen Schock mit liegender intraaortaler Gegenpulsation (IABP) zur Kreislaufunterstützung und invasivem Kreislaufmonitoring mittels zentralem Pulmonaliskatheter
1.1.4 Physikalische Mechanismen
1.1.4.1 Kapillare Flüssigkeitsverschiebung
-
Blutdruckanstieg: Anstieg des Kapillardrucks → Zunahme der Filtration von Flüssigkeit ins Interstitium → Abnahme des Blutvolumens → Absinken des Blutdrucks.
-
Blutdruckabfall: Absinken des Kapillardrucks → Flüssigkeitseinstrom aus dem Interstitium in die Kapillaren → Zunahme des Blutvolumens → Blutdruckanstieg.
1.1.4.2 Stressrelaxation der Gefäße
Spontane Anpassung der Wandspannung, vor allem der Venen, an Änderungen der Druckbelastung.
-
Blutdruckanstieg (z. B. durch Hypervolämie): Zunahme der Gefäßwandspannung → Relaxation der glatten Muskelzellen in der Gefäßwand → Gefäßerweiterung → Blutdruckabfall.
-
Blutdruckabfall (z. B. durch Blutverlust): Abnahme der Gefäßwandspannung → Tonisierung der glatten Muskelzellen (umgekehrte Stressrelaxation) → Gefäßverengung → Blutdruckanstieg.
Durch die Adaptation der Wandspannung können akute Veränderungen des Blutvolumens von +30 % bis –15 % befristet kompensiert werden.
1.1.5 Gesamtregulation
Die adäquate Blutversorgung der Organe und Gewebe hängt von einer bedarfsgerechten Förderleistung des Herzens ab. Bestimmt wird das Herzminutenvolumen durch den venösen Rückfluss, der die Summe aller örtlich regulierten Flussraten im Organismus darstellt.
Die treibende Kraft für den Blutumlauf ist der arterielle Blutdruck, für den die Formel gilt: Arterieller Blutdruck = Herzminutenvolumen × peripherer Gesamtwiderstand
Die dargestellten Regulationsmechanismen gewährleisten durch ihr Zusammenwirken, dass in Ruhe und bei Belastung das benötigte Blutvolumen gefördert wird und der Blutdruck die erforderliche Höhe erreicht.
1.1.5.1 Adaptation an erhöhten Blutbedarf
Basisregulation durch Intrinsic-Mechanismen: Erhöhter peripherer Blutbedarf → autoregulatorische Vasodilatation → Zunahme des venösen Rückflusses → Zunahme der kardialen Pumpleistung → Anstieg des Herzminutenvolumens und des Blutdrucks. Bei starker Belastung zusätzliche Sympathikusaktivierung (bei gleichzeitiger Herabsetzung des Parasympathikustonus) mit Steigerung der Schlagfrequenz und Inotropie des Herzens, Vasokonstriktion (Haut, Splanchnikus, Niere) mit Änderung der Blutverteilung zugunsten der Organe mit erhöhtem Bedarf (Muskulatur) und Steigerung des venösen Rückflusses durch Venenkonstriktion.
1.1.5.2 Adaptation an Blutdruckabfall
1.1.5.2.1 Vorkommen
Orthostase, generalisierte Vasodilatation, Hypovolämie, Herzschwäche.
1.1.5.2.2 Sofortkompensation
Sympathikusstimulation durch Barorezeptoren, evtl. durch zerebrale Ischämiereaktion. Wirkungseintritt in Sekunden. Große Wirkungsintensität, Wirkungsdauer einige Stunden bis Tage.
1.1.5.2.3 Mittelfristige Kompensation
Vasokonstriktion durch Angiotensin II und Vasopressin, umgekehrte Stressrelaxation der Gefäße, kapillare Flüssigkeitsverschiebung aus dem Interstitium in die Blutbahn. Wirkungseintritt nach Minuten. Wirkungsintensität schwächer als bei der Sofortkompensation, Wirkungsdauer Stunden, Tage, Wochen.
1.1.5.2.4 Langfristige Kompensation
Renale Wasser- und Salzretention mit Anstieg des Blutvolumens durch Aktivierung des Renin-Angiotensin-Aldosteron-Systems. Mit dem Blutvolumen steigen venöser Rückfluss, Herzminutenvolumen und arterieller Blutdruck. Wirkungseintritt nach Stunden, Wirkungsintensität groß, Wirkungsdauer unbefristet.
1.1.5.3 Adaptation an Blutdruckanstieg
1.1.5.3.1 Vorkommen
Hypervolämie verschiedener Genese mit peripherer Vasokonstriktion. Pharmaka-Überdosierung.
1.1.5.3.2 Sofortkompensation
Sympathikushemmung und Vagusstimulation durch den Barorezeptorreflex. Wirkungseintritt in Sekunden, Wirkungsintensität groß, Wirkungsdauer Stunden bis einige Tage.
1.1.5.3.3 Mittelfristige Kompensation
Stressrelaxation der Gefäße, kapillare Flüssigkeitsverschiebung aus der Blutbahn ins Interstitium. Drosselung der Freisetzung von Angiotensin II und Vasopressin. Abnahme des Blutvolumens, des venösen Rückflusses, des Herzminutenvolumens und des Blutdrucks. Wirkungseintritt nach Minuten, Wirkungsintensität mittelstark, Wirkungsdauer Stunden, Tage, Wochen.
1.1.5.3.4 Langfristige Kompensation
Steigerung der renalen Wasserausscheidung durch „pressure-diuresis“, Ausschüttung von ANP und Inaktivierung des Renin-Angiotensin-Aldosteron-Systems. Dadurch Beseitigung der Hypervolämie und Normalisierung des Blutdrucks, Wirkungseintritt nach Stunden, Wirkungsintensität groß, Wirkungsdauer unbefristet. Voraussetzung ist eine normale Nierenfunktion.
1.2 Kreislaufschock
1.2.1 Definition
Akute Minderdurchblutung ausgedehnter Körperregionen, die zu schweren Zellschäden und zum irreversiblen Kreislaufversagen führen kann.
1.2.2 Pathogenetische Klassifizierung
1.2.3 Schock durch primäres Herzversagen (kardiogener Schock )
Kritische Verminderung des Herzminutenvolumens infolge Pumpschwäche des Herzens bei ausreichendem venösem Blutrückfluss. Ätiologie: Herzinfarkt, Kardiomyopathie, dekompensierte Vitien, tachy- und bradykarde Rhythmusstörungen, Perikarditis constrictiva, Herztamponade.
1.2.4 Schock durch Verminderung des venösen Rückflusses
Kritische Herabsetzung des Herzminutenvolumens infolge Verminderung des venösen Blutangebotes bei intaktem Herzen. Ätiologie:
-
Hypovolämie: Blutverluste, Plasmaverluste (Verbrennungen, entzündliche oder traumatische Kapillarschädigung), Dehydratation (renale oder extrarenale Flüssigkeitsverluste).
-
Mechanische Rückflussverminderung: Verlegung großer Venen, Lungenembolie.
-
Periphere Vasodilatation: neurogener Schock (Schädigung des Vasomotorenzentrums durch Ischämie, Hirnödem oder Traumen), reflektorischer Abfall des Vasomotorentonus (Schmerz, Schreck), toxische periphere Vasodilatation (Anaphylaxie, Narkotika), regulatorische Vasodilatation der Haut (Fieber, Hitze) oder der Muskulatur (Arbeit). Orthostatische Regulationsstörungen bezeichnet man nicht als Schock, da sie im Liegen aufhören.
1.2.5 Schock durch primäre Mikrozirkulationsstörungen
Inadäquate Gewebeperfusion infolge Zirkulationssperre in der Endstrombahn mit arteriovenösen Kurzschlüssen. Vorkommen bei gramnegativen Infektionen (Endotoxin-Intoxikation, generalisierte intravaskuläre Gerinnung).
1.2.6 Hämodynamik und Kompensationsmechanismen
1.2.7 Primärer Abfall des Herzminutenvolumen s
-
Frühphase: Blutdruckabfall → Reaktion der Barorezeptoren → reflektorische Steigerung des Sympathikustonus, Abnahme des Vagustonus, Freisetzung von → Angiotensin II und Vasopressin → intensive periphere Vasokonstriktion unter Aussparung des Koronar- und Hirnkreislaufs (Zentralisation) → Erhöhung des peripheren Gesamtwiderstandes → Wiederanstieg des Blutdrucks auf ein mäßig erniedrigtes Niveau.
-
Spätphase: Anhaltende Drosselung der Gewebedurchblutung → Gewebehypoxie und Azidose → Steigerung der Kapillarpermeabilität → Plasmaübertritt ins Interstitium → Bluteindickung (sekundäre Hypovolämie), Erythrozytenaggregation, zuletzt auch intravaskuläre Gerinnung (durch gerinnungsaktive Substanzen aus Endothelzellen, Thrombozyten und Leukozyten) → Behinderung der Mikrozirkulation, Abnahme des venösen Rückflusses → weitere Abnahme des Herzminutenvolumens mit Blutdruckabfall → Zunahme der ischämischen Gewebeschädigung (Circulus vitiosus).
1.2.8 Primäre Störung der Mikrozirkulation (septischer Schock)
-
Frühphase: Endotoxineinwirkung auf die kleinen Blutgefäße → Spasmus der Arteriolen und Venolen, Stagnation des Blutes in den Kapillaren → Eröffnung arteriovenöser Anastomosen mit Shunteffekt → Herabsetzung des peripheren Gesamtwiderstandes trotz Perfusionsstörung der Gewebe → Zunahme des venösen Rückflusses und des Herzminutenvolumens bei mäßigem Blutdruckabfall.
-
Spätphase: Stase im Kapillargebiet → O2-Mangel und Azidose im Gewebe → Lösung des Arteriolenspasmus bei weiterbestehender Venolenkonstriktion, Freisetzung von Bradykinin, Aktivierung des Hagemann-Faktors → Blutpooling im Kapillargebiet mit Erhöhung der Kapillardrucks, Permeabilitätssteigerung, Plasmaübertritt ins Interstitium → Bluteindickung (roter Sludge), intravaskuläre Gerinnung → Verminderung des effektiven Blutvolumens und des venösen Rückflusses → Absinken des Herzminutenvolumens und des Blutdrucks → reaktive periphere Vasokonstriktion mit Perfusionsstörung im Gewebe (Circulus vitiosus).
1.2.9 Organschäden und Funktionsstörungen
-
Haut: Im Schock mit herabgesetztem Herzminutenvolumen und Zentralisation des Kreislaufs feucht-kalt und extrem blass. In der Frühphase des septischen Schocks warm und trocken. Bei intravaskulärer Gerinnung und Verbrauchskoagulopathie Blutungen und Nekrosen.
-
Nieren: Blutdruckabfall und Vasokonstriktion → Abnahme der glomerulären Filtrationsrate → Oligurie bis zur Anurie, Anstieg von Harnstoff und Kreatinin im Serum. Bei anhaltender schwerer Ischämie akutes Nierenversagen infolge tubulärer Nekrose. Bei intravaskulärer Gerinnung (septischer Schock) droht irreversible Rindennekrose beider Nieren.
-
Gastrointestinaltrakt: Reflektorische Vasokonstriktion oder intravaskuläre Gerinnung mit Mikrothromben → Sugillationen, blutende Erosionen und Ulzera der Magen- und Darmschleimhaut.
-
Leber: Ischämie durch Vasokonstriktion → Einzelzellnekrosen bis zu ausgedehnten läppchenzentralen Nekrosen.
-
Pankreas: Ischämie durch Vasokonstriktion → Drosselung der Insulinsekretion mit Hyperglykämie.
-
Lunge: Bei kardiogenem Schock Lungenödem durch pulmonale Stauung (pulmonaler Kapillardruck erhöht), besonders nach zu starker Infusionsbehandlung. Bei septischem Schock durch Endotoxin oft irreversibles Lungenödem mit normalem intrapulmonalen Kapillardruck (ARDS: acute respiratory distress syndrome). Ursache ist eine Kapillarschädigung mit Permeabilitätssteigerung durch Zytokine aus Blutzellen und eine Störung der Surfactantproduktion.
-
Herz: Die Ventrikelfunktion bleibt im Schock relativ lange intakt (Ausnahme kardiogener Schock). Erst ein kritischer Blutdruckabfall führt über einen Circulus vitiosus zum Herzversagen: Abnahme der Koronarperfusion → myokardialer O2-Mangel (beschleunigt durch Sympathikusstimulation, die den O2-Verbrauch steigert) → Störung des oxidativen Myokardstoffwechsels (Glykolyse, Laktatbildung, ATP-Abfall) → Verminderung der Kontraktilität → Abnahme von Herzminutenvolumen und Koronarperfusion → weiterer Blutdruckabfall. Zusätzliche Komplikationen: Supraventrikuläre und ventrikuläre Rhythmusstörungen, bei septischem Schock Freisetzung myokarddepressorischer Substanzen.
-
Gehirn: Perfusionsstörung bei Abfall des arteriellen Mitteldrucks unter 60 mmHg. Zunächst psychomotorische Unruhe (Adrenalineffekt), Angstzustände, Verwirrtheit. Bei arteriellem Mitteldruck von 35 mmHg Übergang in Bewusstlosigkeit. Im Terminalstadium ischämische Schädigung des Vasomotorenzentrums mit Absinken des Vasomotorentonus bis zur Aufhebung der zentralen Kreislaufregulation.
-
Stoffwechsel: Laktatazidose durch O2-Mangel im Gewebe. Hyperglykämie durch Glykogenolyse und Hemmung der Insulinsekretion. Nach Erschöpfung der Glykogenvorräte Tendenz zur Hypoglykämie.
1.2.10 Stadien des Schock s
1.2.11 Kompensierter (nicht progredienter) Schock
Kompensationsmechanismen erhalten die Perfusion der lebenswichtigen Organe aufrecht. Nur relativ geringer Blutdruckabfall. Volle Reversibilität durch Beseitigung der Schockursache (z. B. Blutersatz bei hämorrhagischem Schock).
1.2.12 Progredienter Schock
Kompensationsmechanismen so weit ausgeschöpft, dass ein Circulus vitiosus in Gang gesetzt wird: Die schockbedingten Funktionsstörungen verschlechtern die Perfusion der Organe und Gewebe weiter und verstärken den Schock. Kritische Situation besonders für das Herz. Lebensrettung nur durch intensive Sofortbehandlung möglich.
1.2.13 Irreversibler Schock
Exitus durch keine Therapie mehr abzuwenden. Die Irreversibilität ist klinisch nicht zu diagnostizieren, sondern ergibt sich aus dem Verlauf. Nicht das Ausmaß des Blutdruckabfalls, sondern das der Perfusionsstörung ist für den Schweregrad des Schocks maßgebend. Die Irreversibilität des Schocks resultiert aus der Zellzerstörung in den vitalen Organen (durch Unterschreitung des kritischen O2-Defizits und zirkulierende toxische Substanzen). Todesursache ist meisten die irreparable Schädigung des Herzens.
1.2.14 Klinik
1.2.15 Symptome des verkleinerten Herzminutenvolumen s
-
kleiner Puls: durch verkleinertes Schlagvolumen
-
niedriger Blutdruck, Verkleinerung der Blutdruckamplitude
-
Muskelschwäche: durch reflektorische Ischämie
-
Temperaturabfall: durch verminderte Stoffwechselaktivität
-
Bewusstseinstrübung, Stupor, Bewusstlosigkeit: durch zerebrale Ischämie
1.2.16 Symptome der sympathikoadrenalen Stimulation
-
Tachykardie
-
motorische Unruhe
-
feucht-kalte Haut, Akrozyanose
-
Oligurie, Anurie
1.2.17 Ursachenabhängige Symptome
-
kardiogener Schock: Zeichen der Herzkrankheit (Infarkt, Lungenembolie etc.)
-
septischer Schock: Schüttelfrost, Fieber, intravaskuläre Gerinnung mit Verbrauchskoagulopathie
-
Schocklunge: Hyperventilation mit Hypokapnie, Hypoxie; initial respiratorische Alkalose, final metabolische Azidose
-
hämorrhagischer Schock: Hämatokritabfall nach 2–6 h
-
hypovolämischer, nichthämorrhagischer Schock (z. B. bei Cholera): Hämokonzentration mit erhöhtem Hämatokrit
-
anaphylaktischer Schock: Dyspnoe, Bronchospastik, Urticaria nach Antigenkontakt
1.2.18 Symptomatische Therapie
1.2.19 Allgemeinmaßnahmen
Patient warmhalten, Beine etwas hochlagern, um den venösen Rückfluss zu verbessern. Beim kardiogenen Schock Oberkörper hochlagern. Großzügige Schmerzlinderung mit Morphin i.v. (5 mg in 2 min). O2-Applikation durch Nasensonde, Infusion anlegen. Transport zur nächsten Intensivstation. Dort fortlaufende Kontrolle von Temperatur, Atmung, Urinausscheidung (Blasenkatheter), EKG, Blutdruck (möglichst intraarteriell), mit zentralem Venenkatheter Druck im rechten Vorhof (CVP) und in den Lungenkapillaren (PCWP: pulmonary capillary wedge pressure). Messung des Herzminutenvolumens mit der Thermodilutionsmethode. Bestimmung des peripheren Gefäßwiderstandes. Zu überwachen sind ferner pO2, pCO2 und pH, Elektrolyte, Harnstoff, Kreatinin, Blutbild und Gerinnungsparameter. Bei Herzkreislaufstillstand kardiopulmonale Reanimation.
1.2.20 Volumensubstitution
Wichtigste Sofortmaßnahme zur Steigerung von venösem Rückfluss, Herzminutenvolumen und Blutdruck. Erreicht wird damit ein Nachlassen der kompensatorischen Vasokonstriktion und der Tachykardie. Abgesehen vom kardiogenen Schock mit Lungenstauung und einigen Sonderfällen (Herztamponade, Lungenembolie) liegt beim Schock immer eine absolute Hypovolämie oder eine Verminderung des effektiven vaskulären Volumens (infolge venösen Poolings) vor.
Infundiert wird bis zum normalen bzw. erhöhtem CVP, beim kardiogenen Schock bis zum erhöhten PCWP, der dem erhöhten diastolischen Füllungsdruck des linken Ventrikels entspricht.
Aufgefüllt wird der Kreislauf initial mit 0,9%iger Kochsalzlösung oder mit Ringerlösung, die durch Kolloidlösungen (Dextran 60) ergänzt werden können. Im hämorrhagischen Schock sind Erythrozyten- oder Vollblutkonserven angezeigt, bis zu einem Hb-Wert von mindestens 10 g/dl.
1.2.21 Azidoseausgleich
Gegen die metabolische Azidose werden 50–100 ml 8,4%ige Natriumbikarbonatlösung (1 mEq/ml) unter Kontrolle des Bikarbonatspiegels im Blut langsam infundiert. Wichtiger zur Beseitigung der Azidose ist die Behandlung der Schockursache.
1.2.22 Katecholamine
Bei ungenügendem Blutdruckanstieg nach Volumensubstitution oder primär normalem CVP ist Dopamin i. v. indiziert: 4–20 µg/kg/min. Es steigert die Freisetzung von Noradrenalin an den sympathischen Nerven im Herzen und an den peripheren Gefäßen und besitzt dadurch einen positiv inotropen und vasokonstriktorischen Effekt. Zugleich wirkt es über Dopaminrezeptoren vasodilatatorisch auf Nieren- und Splanchnikusgefäße. Reicht Dopamin nicht aus, wird Noradrenalin infundiert (2–20 µg/min). Nach Anhebung des Blutdrucks kann zur Steigerung der kardialen Kontraktilität die Infusion von Dobutamin indiziert sein (2,5–15 µg/kg/min). Dobutamin stimmuliert nur die β1- und β2- adrenergen Rezeptoren und steigert die Vasokonstriktion nicht. Bei mangelhafter Wirksamkeit kommen zusätzlich die Phosphodiesterase-Inhibitoren Milrinon und Enoximon in Betracht. Beide Substanzen greifen nicht an den β-Rezeptoren an und wirken positiv inotrop und vasodilatatorisch. Bei infauster Prognose ggf. Assist Device oder Herztransplantation. Beim anaphylaktischen Schock ist die sofortige i. v. Injektion von Adrenalin indiziert. Starke Bradykardien werden mit Atropin behandelt.
1.2.23 Apparative Maßnahmen
1.2.24 Endotracheale Intubation
Zur Freihaltung der Atemwege, Sekretentfernung und Sauerstoffzufuhr.
1.2.25 Maschinelle Beatmung
Bei respiratorischer Insuffizienz und beginnender Schocklunge. Mit positivem endexspiratorischem Druck werden Mikroatelektasen beseitigt und die O2-Sättigung des Blutes gesteigert.
1.2.26 Manuelle Herzdruckmassage
Bei systolischem RR <50 mmHg und Herzstillstand.
1.2.27 Intraaortale Ballongegenpulsation (◘ Abb. 1.3)
Platzierung des über die A. femoralis eingeführten Ballonkatheters in der Aorta descendens unterhalb des Subklaviaabganges. EKG-gesteuertes Aufblasen in der frühen Diastole steigert die Koronarperfusion. Deflation in der frühen Systole senkt die Nachlast und steigert das Schlagvolumen des Herzens. Indiziert im kardiogenen Schock (Kontraindikation bei Aorteninsuffizienz).
1.2.28 Passagerer transvenöser elektrischer Herzschrittmacher
Bei SA- und AV-Block 3. Grades.
1.2.29 Elektrische Defibrillation
Bei tachykarden Rhythmusstörungen.
1.2.30 Chronische venovenöse Hämofiltration
Bei prärenalem Nierenversagen.
1.2.31 Kausale Therapie
1.2.32 Kardiogener Schock
Bei akutem Infarkt Revaskularisierung durch Thrombolyse mit anschließender Antikoagulation (Heparin) falls kein kardiologisches Zentrum zur Verfügung steht. Wenn vorhanden oder in 1–2 h erreichbar, sofortige Verlegung (mit ärztlicher Begleitung) zur primären Revaskularisierung durch perkutane transluminale koronare Angioplastie (PTCA), möglichst mit Stentimplantation. Beim Misslingen der PTCA auch Frühbypassoperation. Besonders erfolgversprechend sind Revaskularisierungsversuche in den ersten 12 h nach Beginn der Infarktsymptome. Der günstigste Abstand beträgt 1–3 h. Perikarddrainage bei Herztamponade. Klappenoperation bei akuter Klappeninsuffizienz oder dekompensierter Aortenstenose.
1.2.33 Lungenembolie
Thrombolyse, besser kathetergesteuerte Thrombusfragmentation plus Thrombolyse oder Embolektomie.
1.2.34 Hämorrhagischer Schock
Chirurgische oder endoskopische Blutstillung.
1.2.35 Septischer Schock
Bakterizide Antibiotika, die gegen grampositive und gramnegative Erreger wirksam sind (z. B. Ceftriaxon 2 g/Tag plus Gentamycin 3 mg/kg/Tag oder Levofloxacin 500–700 mg alle 12 h plus Clindamycin 600 mg alle 8 h). Ausschaltung des Sepsisherdes. Bei partieller NNR-Insuffizienz (Plasmacortisol <9 µg/dl nach 25 µg ACTH) 50 mg Hydrocortison alle 6 h Antikoagulation mit rekombinantem aktiviertem Protein C (aPC). Antimediatoren-Wirkstoffe (Ibuprofen, lösliche Rezeptoren für TNF-α und monoklonale Antikörper gegen TNF-α) haben den Tod am septischen Schock nicht verhindert. Trotz aller Maßnahmen sterben 40–60 % der Patienten.
1.2.36 Schock bei Intoxikationen
Spezifisches Antidot, Hämoperfusion, Hämodialyse, Hämofiltration.
1.3 Arterielle Hypotonie und Synkopen
1.3.1 Essenzielle Hypotonie
1.3.1.1 Definition
Niedriger Blutdruck, bei Männern dauernd unter 110/60, bei Frauen unter 100/60 mmHg, ohne organische Ursache.
1.3.1.2 Klinik
Konstitutionell niedrig eingestellter Blutdruck mit intakter Blutdruckregulation ohne klinische Symptome und ohne Krankheitswert, ja sogar mit erhöhter Lebenserwartung. Die nach unten eingeschränkte Reaktionsbreite des Blutdrucks bedingt aber eine erhöhte Anfälligkeit gegenüber hypotensiv wirkenden Faktoren.
1.3.2 Sekundäre Hypotonie
1.3.2.1 Ätiologie und Pathogenese
Symptomatische Hypotonien entstehen durch Herabsetzung des Herzminutenvolumens, des peripheren Gefäßwiderstandes, des Blutvolumens oder des venösen Rückflusses. Diese pathogenetischen Faktoren kommen auch in Kombinationen vor. Die wichtigsten Ursachen sind:
-
kardiale Funktionsstörungen: schwere Herzinsuffizienz, Stenose der Aorten,- Mitral-, Pulmonal- oder Trikuspidalklappe, hypertrophisch-obstruktive Kardiomyopathie, dilatative Kardiomyopathie, Herzinfarkt, Lungenembolie, paroxysmale Tachykardien und Tachyarrhythmien, extreme Bradykardien,
-
nervale und vaskuläre Funktionsstörungen: orthostatische Hypotonie und Synkopen.
-
metabolische und endokrine Störungen: Morbus Addison, Hypaldosteronismus, Hypothyreose, Anorexia nervosa, Hyperparathyreoidismus (polyurisch bedingte Hypovolämie), Flüssigkeitsverluste (Blutungen, Verbrennungen, Durchfälle etc.).
-
blutdrucksenkende Pharmaka: Vasodilatatoren, α- und β-Rezeptorenblocker, ACE-Hemmer, Angiotensin-II-Rezeptor-Antagonisten, zentral wirksame Sympathikolytika, Ganglienblocker, Reserpin, negativ-inotrop wirkende Pharmaka.
1.3.2.2 Klinik
Durch Kompensationsmechanismen können die sekundären Hypotonien symptomlos bleiben. Sie manifestieren sich am häufigsten im Stehen mit Schwindelgefühl, Herzklopfen und Schwächegefühl. Nicht selten wird über rasche Erschöpfung bei körperlicher Belastung geklagt, über Kopfschmerzen Konzentrationsschwäche und kalte Füße. Klinische Zeichen sind neben den Symptomen der Grundkrankheit Hautblässe und ein kleiner, leicht unterdrückbarer Puls.
1.3.2.3 Therapie
Behandlung der Grundkrankheit. Verbesserung des venösen Rückflusses durch Erhöhung des Blutvolumens (mehr trinken und mehr Salz konsumieren, Mineralokortikoide), vasokonstriktorische Substanzen.
1.3.3 Orthostatische Kreislaufstörungen
1.3.3.1 Normale Orthostasereaktion
Stabilisiert den Kreislauf in aufrechter Körperhaltung durch folgende Mechanismen: Aufstehen → Blutpooling in den Beinen (300–800 ml) → Abnahme des venösen Rückflusses → Abfall der kardialen Förderleistung → Blutdruckabfall → Reaktion der arteriellen Pressorezeptoren (Abnahme der inhibitorischen Impulse zum Vasomotorenzentrum (▶ Abschn. 1.1.2) → Zunahme des Sympathikustonus und Abnahme des Vagustonus → Anstieg des peripheren Gefäßwiderstandes, Steigerung des venösen Rückflusses durch Tonisierung der venösen Kapazitätsgefäße, Zunahme der Herzfrequenz → Wiederanstieg des Blutdrucks. Bei fortdauerndem Stehen werden auch die ADH-Sekretion und das Renin-Angiotensin-System aktiviert. Die kontrahierte Beinmuskulatur steigert den venösen Rückfluss.
1.3.3.2 Orthostatische Hypotonie
1.3.3.2.1 Definition
Abfall des systolischen Blutdrucks um mindestens 20 mmHg, oder des diastolischen um mindestens 10 mmHg nach dem Aufstehen aus liegender Position über mindestens 3 Minuten.
1.3.3.2.2 Klassifikation
1.3.3.2.3 Asympathikotone orthostatische Hypotonien durch partiellen oder totalen Ausfall des Barorezeptorreflexes
Blutdruckabfall ohne oder mit ungenügender reflektorischer Sympathikusaktivierung. Herzfrequenz und peripherer Gefäßtonus steigen nicht oder nur inadäquat an.
-
Bradbury-Eggleston-Syndrom (PAF: pure autonomic failure): Zugrunde liegt der orthostatischen Hypotonie eine chronische postganglionäre autonome Insuffizienz unklarer Ursache mit herabgesetzter Neuronendichte in den sympathischen Ganglien und erniedrigtem, nicht stimulierbarem Plasmanoradrenalinspiegel. Kommt sehr selten vor und betrifft überwiegend Männer nach dem 60. Lebensjahr. Die Beeinträchtigung ist erheblich, die Lebenserwartung aber nicht verkürzt. Im Verlauf können Impotenz, Blasenstörungen, Verlust des Schwitzens und verminderte Tränen- und Speichelsekretion hinzukommen.
-
Shy-Drager-Syndrom: Ätiologisch ungeklärte Degeneration der präganglionären sympathischen Neurone in der Columna mediolateralis des Rückenmarks. Der Prozess bleibt aber nicht auf das autonome Nervensystem beschränkt, sondern greift auf viele andere Hirnregionen über. Vorkommen sehr selten. Krankheitsbeginn zwischen dem 45. und 55. Lebensjahr. Im Frühstadium nur Symptome der autonomen Dysfunktion: Schwere orthostatische Hypotonie mit normalem, aber nicht steigerungsfähigen Plasmanoradrenalinspiegel, Hypertonie im Liegen, Impotenz, Blasenentleerungsstörungen, Obstipation und verminderte Schweißbildung. Im Laufe von 5 Jahren treten parkinsonähnliche Symptome (Rigor, Tremor, Bradykinesie, Dysarthrie, Psycholabilität) und Pyramidenzeichen hinzu. Die Patienten sterben nach 7–10 Jahren, meistens an pulmonalen Komplikationen der Immobilität.
-
Weitere zentralnervöse Erkrankungen: Eine orthostatische Hypotonie tritt relativ häufig im fortgeschrittenen Stadium des Morbus Parkinson auf. Sie kommt auch bei Tumoren der hinteren Schädelgrube und bei Syringobulbie vor. Zugrunde liegt ihr offenbar eine zentrale Blockierung des Barorezeptorreflexes.
-
Periphere autonome Neuropathien: Infolge Schädigung der Neuriten verminderte Speicherung und Freisetzung von Noradrenalin an den sympathischen Endausbreitungen in den Arteriolen und venösen Kapazitätsgefäßen. Vorkommen bei Diabetes mellitus, Amyloidose, Porphyrie, chronische Alkoholintoxikation und Guillain-Barr-Syndrom (akute entzündliche demyelinisierende Radikulitis).
1.3.3.2.4 Orthostatische Hypotonie durch venöse Rückflussstörung bei intaktem Barorezeptorreflex (sympathikotone orthostatische Hypotonie)
Trotz Sympathikusaktivierung mit Tachykardie und Vasokonstriktion sinkt der Blutdruck, weil der venöse Rückfluss zum Herzen abnimmt. Ursachen sind:
-
Hypovolämie: Flüssigkeitsverluste nach außen (Blutungen, Saluretika, Polyurien, intestinale Flüssigkeitsverluste, Verbrennungen), Flüssigkeitsverluste nach innen (Aszites, nephrotisches Syndrom), ungenügende Flüssigkeitszufuhr. Blutdruck meistens schon im Liegen niedrig. Nach dem Aufstehen stärkere Herzbeschleunigung, Vasokonstriktion und stärkere Erhöhung des Plasmanoradrenalins als bei normaler Orthostasereaktion – dennoch Blutdruckabfall. Seltenes Vorkommen beim chronischen Phäochromozytom (▶ dort).
-
Venöses Pooling: Versacken des Blutes in der unteren Körperhälfte bei Varikosis und mangelhaftem Muskeltonus (Wegfall der Venenkompression durch Bein- und Bauchmuskeln) nach längerer Bettlägerigkeit, bei geschwächten und alten Menschen. Vasokonstriktion und Tachykardie genügen zur Normalisierung des venösen Rückflusses nicht.
-
Vasodilatation:
-
Durch blutdrucksenkende Pharmaka: Alpharezeptorenblocker (Urapidil, Prazosin, Chlorpromazin), Antidepressiva, Clonidin, ACE-Blocker, AT1-Rezeptorenblocker, Nitrate, Alkohol etc. Im Stehen reflektorische Tachykardie, die bei Substanzen mit blockierendem Effekt auf Betarezeptoren ausbleibt (Carvedilol).
-
Postprandiale Vasodilatation im Splachnikusgebiet kann bei alten Personen eine ausreichende periphere Vasokonstriktion nach dem Aufstehen verhindern (postprandiale orthostatische Hypotonie).
-
Durch wämeregulatorische Vasodilatation der Haut in hoher Umgebungstemperatur kann im Stehen ebenfalls eine ausreichende periphere Vasokonstriktion ausbleiben.
-
1.3.3.2.5 Orthostatische Hypotonie aus kardialer Ursache
Die Förderleistung des Herzens kann trotz Sympathikusaktivierung nicht adäquat gesteigert werden: Vorkommen bei akutem Herzinfarkt, Tachyarrhythmien, hochgradiger Aortenstenose und schwerer Kardiomyopathie.
1.3.3.2.6 Orthostatische Hypotonie durch Cortisolmangel
Cortisol steigert die Reaktivität der glatten Gefäßmuskulatur gegenüber Noradrenalin, stimuliert die Bildung adrenerger Rezeptoren in der Gefäßwand und hat einen mineralokortikoiden Effekt auf das Blutvolumen. Bei Nebennierenrindeninsuffizienz und plötzlichem Absetzen einer langdauernden Glukokortikoidtherapie resultiert eine Hypotonie mit ausgeprägter orthostatischer Komponente.
1.3.3.2.7 Klinik
Die Symptome der orthostatischen Hypotonie beruhen auf mangelhafter Durchblutung des Gehirns und der Muskulatur und – soweit vorhanden – auf der Symathikusstimulation. Sie bilden sich im Liegen mit der Verbesserung des venösen Rückflusses schnell zurück. Bei intakten zerebralen Arterien mit guter Compliance kann die orthostatische Hypotonie symptomlos bleiben.
1.3.3.2.8 Hypotone Regulationsstörung (faintness)
Müdigkeit, Mattigkeit, Leeregefühl im Kopf, Schwindel, Herzklopfen, Hautblässe, Empfindung von Ohnmachtsnähe mit Übelkeit und Schwäche. Die Patienten können sich aber aufrecht halten.
1.3.3.2.9 Orthostatischer Kollaps
Plötzliches Einsetzen von generalisierter Muskelschwäche, Schweißausbrüchen, Flimmern oder Schwarzwerden vor den Augen mit nachfolgendem Bewusstseinsverlust, durch den die Patienten umsinken. Übelkeit und Erbrechen können nachfolgen. Man beobachtet hochgradige Hautblässe, kalten Schweiß auf der Stirn und einen kleinen frequenten Puls. Bei asympathikotonem Kollaps fehlen Tachykardie und Hautschweiß.
1.3.3.2.10 Diagnostik
1.3.3.2.11 Stehversuch
Nach 10 Minuten Ruhelage 5 Minuten frei stehen. Puls- und Blutdruckmessung jede Minute.
-
Normale Regulation: Nach dem Aufstehen geringer Abfall des systolischen Drucks (0–15 mmHg) und leichter Anstieg des diastolischen Drucks (0–5 mmHg). Der Pulsdruck (Blutdruckamplitude) sinkt nicht unter 30 mmHg. Anstieg der Pulsfrequenz um 10–20 %. Im Liegen werden nach einer Minute die Ausgangswerte erreicht.
-
Labile Regulation: Systolischer Druckabfall und diastolischer Druckanstieg etwas stärker. Absinken des Pulsdrucks auf 10–15 mmHg. Zeichen der gesteigerten Sympathikusstimulation. Im Liegen nach einer Minute Ausgangswerte.
-
Insuffiziente Regulation: Absinken des Pulsdrucks auf Werte unter 10 mmHg bei starkem Abfall des systolischen und entsprechendem Anstieg des diastolischen Drucks. Pulsfrequenz über 120 mmHg. Häufig Übergang in Kollaps. Trotz maximaler Stimulation reicht der Sympathikustonus zur Kompensation nicht aus. Es muss eine nichtneurogene Störung des venösen Rückflusses vorliegen.
-
Asympathikotone Regulation: Nach dem Aufstehen sofortiges Absinken des systolischen und diastolischen Drucks ohne Pulsbeschleunigung, Schwächeanfall, Kollaps.
1.3.3.2.12 Steh-EKG
Erfasst normale und gesteigerte reflektorische Sympathikotonie.
-
Normal: Zunahme der Herzfrequenz. P-Vergrößerung in II, aVF und III. Rechtsdrehung des QRS-Vektors. Geringe aszendierende ST-Senkung und T-Abflachung in V4–V6.
-
Gesteigerte Sympathikotonie: Erhebliche Tachykardie. Hohe P-Zacken in II, aVF und III. Erhebliche ST-Senkung und biphasisches T in II, aVF, III und V4–V6.
1.3.3.2.13 Plasma-Noradrenalin
Gradmesser für die Noradrenalinfreisetzung an den sympathischen Endausbreitungen.
-
Normal: Im Stehen nach 5 Minuten Anstieg auf das Doppelte.
-
Hypovolämie: Im Stehen nach 5 Minuten Anstieg auf das Dreifache.
-
Neurogene orthostatische Hypotonie:
-
periphere Form: erniedrigter Ruhewert, kein Anstieg im Stehen und nach Tyramingabe
-
zentrale Form: normaler Ruhewert, kein Anstieg im Stehen, normaler Anstieg nach Tyramingabe
-
1.3.3.2.14 Therapie
Die Therapie der orthostatischen Hypotonie erfolgt symptomatisch:
-
Volumenauffüllung: Langfristig am wirksamsten ist die Steigerung bzw. Stabilisierung des Blutvolumens durch reichlichen Flüssigkeits- und Kochsalzkonsum, im Bedarfsfall ergänzt durch das Mineralokortikoid Fludrocortison (Astonin HR) in einer Tagesdosis von 0,05–0,1 mg per os. Mit dem Füllungszustand des Gefäßsystems nimmt der venöse Rückfluss zu. Der systolische Druck wird um 10–20 mmHg angehoben. Kontraindikationen: Herzinsuffizienz und Ödeme.
-
Verminderung des venösen Pooling: Varizenbehandlung, Steigerung des Muskeltonus (Kräftigung der Bein- und Bauchmuskulatur, Stütz- oder Kompressionsstrümpfe, bei asympathikotoner Hypotonie Kompressionsanzug. Bei postprandialer Hypotonie kleine Mahlzeiten. Bei Hypotonie im Alter langsame Lagewechsel.
-
Verbesserung der Herzfunktion: Behandlung von Herzinsuffizienz, Rhythmusstörungen und Vitien.
-
Steigerung des peripheren Gefäßwiderstands: Sympathikomimetika (Etilefrin, Norfenefrin, Dihydroergotamin) sind bei normalem Barorezeptorreflex nur vorübergehend indiziert, bei Ödemneigung hilfreich. Betarezeptorenblocker schalten die vasodilatierende Komponente des Sympathikuseffektes aus. Bei asympathikotoner Hypotonie kommt der selektive α1-Rezeptoragonist Midodrin (3×2,5–10 mg/Tag) in Betracht. Octreotid, ein Somatostatin-Analogon, das im Darm die Freisetzung vasodilatierender Peptide hemmt, wirkt der postprandialen Hypotonie entgegen. Selbstverständlich sind alle Medikamente mit blutdrucksenkender Wirkung zu vermeiden.
Zur Therapie des orthostatischen Kollapses Anheben der Beine zur Verbesserung des venösen Rückflusses. Prednisolon intravenös (25–50 mg) verstärkt den Sympathikuseffekt auf die Gefäße. Alternativ kommen Noradrenalin oder Norfenefrin parenteral in Betracht. Volumengabe.
1.3.4 Synkope n
Synkopen
-
Reflektorische Synkopen
-
Neurokardiogene Synkopen
-
Kardiale Synkopen
-
Adam-Stoke-Syndrom
-
Aortenstenose und hypertrophisch-obstruktive Kardiomyopathie
-
Primäre pulmonale Hypertonie
-
Akutes Herzversagen
-
1.3.4.1 Definition
Bewusstseinsverlust durch plötzliche, meistens schnell vorübergehende zerebrale Perfusionsstörung. Im Gegensatz dazu sind epileptische Ohnmachtsanfälle durch ein zerebrales Krampfleiden bedingt. Der orthostatische Kollaps (▶ Abschn. 1.3.3) kann auch zu den Synkopen gezählt werden, tritt aber gewöhnlich weniger abrupt auf.
-
Präsynkopen: Plötzliches Schwächegefühl, Angst, Hyperventilation, verschwommenes Sehen, Gefühl der Ohnmachtnähe. Nicht selten fehlt dieses warnende Vorstadium.
-
Synkopen: Bewusstseinsverlust im Stehen oder Sitzen (bei kardialen Synkopen auch im Liegen), Sturz oder Umsinken, Bewegungslosigkeit, Hautblässe, Schweißausbruch, flache Atmung, meistens langsamer Puls, Blutdruckabfall. Pupillen reagieren, kein unwillkürlicher Stuhl- oder Urinabgang, Krämpfe nur bei länger dauernder Ischämie.
1.3.4.2 Reflektorische Synkope n
1.3.4.2.1 Hypersensitiver Karotissinus
1.3.4.2.1.1 Definition
Überschießende Reaktion der Barorezeptoren in den Karotiden bei älteren Menschen, insbesondere Männern. Auslösung durch Kopfdrehung (Rasieren), engen Kragen, leichten Druck oder Stoß, aber auch spontan. Zugrunde liegt meistens eine Arteriosklerose, seltener eine Narbenbildung oder Halslymphknotenschwellung. Die Verlangsamung oder Unterbrechung der inhibitorischen Barorezeptorimpulse zum Vasomotorenzentrum hemmen den Sympathikus und stimulieren den Vagus.
1.3.4.2.1.2 Klinik
Klinisch lassen sich Reaktionsformen abgrenzen, die auch kombiniert vorkommen:
-
vagaler (kardioinhibitorischer) Typ: Sinusbradykardie, Sinusstillstand, AV-Block, Asystolie
-
vasopressorischer Typ: Ausgeprägte Hypotonie ohne signifikante Bradykardie. Diagnose durch Karotisdruckversuch: Vorsichtige einseitige manuelle Karotiskompression im Liegen unter laufender EKG- und Blutdruckkontrolle.
1.3.4.2.1.3 Therapie
Vermeidung extremer Halsbewegungen, offene Kragen. In schweren Fällen mit Bradykardie Implantation eines Herzschrittmachers.
1.3.4.2.2 Weitere reflektorische Synkopen
Induziert durch afferente Impulse aus dem Vagus oder Glossopharyngeus:
-
Glossopharyngeusneuralgie : Läsionen im Ohr, Pharynx oder Larynx
-
Schlucksynkope : Bei Divertikeln oder Tumoren des Ösophagus
-
Hustensynkope : Ohnmachtsanfall bei schwerer Hustenattacke
-
Miktionssynkope : Ohnmachtsanfall nach dem Wasserlassen im Stehen, hauptsächlich während der Nacht
-
Synkopen nach schmerzhaften Punktionen: Venen-, Pleura- oder Bauchpunktion
1.3.4.3 Neurokardiogene (vasovagale) Synkope n
1.3.4.3.1 Definition
Reflektorische Ohnmachtsanfälle mit hochgradiger Bradykardie infolge intensiver Vagusstimulation.
1.3.4.3.2 Vorkommen und Pathogenese
Auslösung durch langes Stillstehen in Menschenansammlungen, begünstigt durch Hitze und schlechte Luft. Aber auch psychogen durch Schreck, Angst oder Schmerzen.
Eine Variante, die bei empfindlichen Menschen am Kipptisch auszulösen ist, hat folgende Kausalkette: Venöses Pooling → Abnahme des venösen Rückflusses → Sympathikusstimulation → intensive Kontraktion des gering gefüllten linken Ventrikels → Stimulation myokardialer Mechanorezeptoren, die über afferente Nervenfasern den Vagus aktivieren → vagotone Bradykardie → Blutdruckabfall bis zur Ohnmacht.
Da vasovagale Synkopen auch bei Herztransplantierten vorkommen, kann eine kardiogene Vagusstimulation nicht der einzige Pathomechanismus sein, denn das transplantierte Herz ist denerviert. Neben der kardiogenen dürfte eine zentralnervöse Variante anzunehmen sein: Psychogene Erregung → zentrale Herabsetzung des Sympathikustonus mit peripherer Vasodilatation und simultane intensive Vagusstimulation mit Bradykardie. Für diese Reaktionsweise besteht eine individuelle Disposition.
1.3.4.3.3 Klinik
Vorstadien der Ohnmacht (Übelkeit, Hyperventilation, Schwindelgefühl, Schwäche) fehlen oder sind kürzer als beim orthostatischen Kollaps. Sturzverletzungen kommen vor, gelegentlich auch Inkontinenz.
1.3.4.3.4 Therapie
Spontanremission im Liegen nach einigen Sekungen oder Minuten. Prophylaktisch kommen β1-selektive β-Blocker in Betracht (Metoprolol). Sie hemmen die linksventrikulären Mechanorezeptoren. Schrittmacher helfen nicht, weil sie die Vasodilatation nicht verhindern. In der Aura sofort Beine und Arme verschränken, um den peripheren Widerstand zu erhöhen. Gegen die Vasodilatation hilft Midodrin 3×2,5 mg/Tag. Zur Prophylaxe eignen sich selektive β1-Blocker, z. B. Metoprolol. Sie hemmen die linksventrikulären Mechanorezeptoren.
1.3.4.4 Kardiale Synkope n
1.3.4.4.1 Adams-Stokes-Syndrom
Ohnmachtsanfall infolge eines akuten passageren Kreislaufstillstands bei:
-
Komplettem SA- oder AV-Block: wenn das subsidiäre Automatie-Ersatzzentrum verzögert einsetzt
-
Sinusknoten-Syndrom: Sinusstillstand, extreme Bradykardie nach Beendigung eines Vorhofflimmerns (verzögerte Reaktivierung des Sinusknotens)
-
Tachykarde Rhythmusstörungen: paroxysmale Kammertachykardie, Kammerflattern oder extreme Tachyarrhythmie (>300/min) bei WPW-Syndrom mit Vorhofflimmern.
1.3.4.4.1.1 Klinik
Plötzliches Einsetzen der Ohnmacht, unabhängig von der Position des Körpers. Pulslosigkeit, Verdrehung der Augen, Muskelerschlaffung, retrograde Amnesie. Bei Kreislaufstillstand über 20 Sekunden Zyanose, schnarchende Atmung, krampfartige (Zuckungen ohne Zungenbiss), Sphinkterinsuffizienz. Verzögertes Erwachen, anschließend noch persistierende Schwäche.
1.3.4.4.2 Aortenstenose und hypertrophisch-obstruktive Kardiomyopathie
Typisch ist das Auftreten der Synkopen unmittelbar nach körperlicher Belastung. Bei dem kleinen fixierten Herzminutenvolumen führt der Blutabfluss in die Muskulatur zur zerebralen Ischämie.
1.3.4.4.3 Primäre pulmonale Hypertonie
Synkope durch fixiertes Herzminutenvolumen und arterielle Hypoxie bei körperlicher Belastung. Synkopen auch bei großen Lungenembolien.
1.3.4.4.4 Akutes Herzversagen
Plötzlicher Abfall der kardialen Förderleistung bei großem Myokardinfarkt oder großen inneren Blutungen (Magen-Darm-Trakt, tubarer Abort).
1.3.5 Episodische Schwächezustände ohne Synkopen
1.3.5.1 Angstzustände mit Hyperventilationstetanie
1.3.5.1.1 Definition
Manifestationen einer durch bewusste oder unbewusste Konflikte begünstigten Neurose, worunter nach Freud der Übergang belastender seelischer Inhalte in körperliche Symptome zu verstehen ist. Eine typische Attacke läuft wie folgt ab: Akuter Angstanfall → Gefühl, keine Luft zu bekommen → Hyperventilation → respiratorische Alkalose → Parästhesie an Händen und Füßen, Spannungsgefühl um den Mund → Steigerung der Hyperventilation, starker CO2-Abfall im Blut → Karpopedalspasmen, Drosselung der zerebralen Durchblutung, Schwindelgefühl und Benommenheit, die letztlich zur Senkung der Atemfrequenz führt. Die begleitende Adrenalinausschüttung führt zur Tachykardie, zu Herzklopfen und Schwitzen und kann die Angst zum Vernichtungsgefühl steigern. Das Anfallerlebnis ist sehr belastend.
1.3.5.1.2 Therapie
Im Anfall Calcium i. v. (obwohl das Plasmacalcium nicht herabgesetzt ist) und Diazepam i. v. oder per os.
1.3.5.1.3 Prophylaxe
Erklärung des Anfallmechanismus, bewusstes Vermeiden der Hyperventilation, Psychotherapie, kleine Dosen Diazepam bei den ersten Vorboten des Anfalls.
1.3.5.2 Neuropathisches posturales Tachykardie-Syndrom
Orthostatische Intoleranz ohne Hypotonie mit Tachykardie (>100/min) im Stehen, Leeregefühl im Kopf, verminderter körperlicher Belastbarkeit, Schwäche, Missempfindungen in der Brust, Herzklopfen, Angstgefühl, Übelkeit und blauroter Verfärbung der Beine. Es kann auch zu Synkopen kommen. Im Sitzen und Liegen verschwinden alle Symptome. Das im Liegen normale Plasma-Noradrenalin steigt im Stehen doppelt so hoch an als bei Normalpersonen. Es handelt sich um eine partielle autonome Dysregulation, bei der die Speicherung und Freisetzung von Noradrenalin an den sympathischen Endausbreitungen in den Beinen herabgesetzt ist. Daraus folgt ein Versacken des Blutes im Stehen. Die reflektorische Tachykardie verhindert jedoch einen Blutdruckabfall. Die ätiologisch unklare Anomalie betrifft überwiegend Frauen jüngeren und mittleren Alters. Therapie: Volumenauffüllung durch reichliche Flüssigkeits- und Kochsalzaufnahme, ergänzt durch Fludrocortison, Kompressionsstrümpfe, α1-Rezeptoragonisten (Midodrin), körperliches Training.
1.3.5.3 Hypoglykämie
1.3.5.3.1 Pathogenese
Das Absinken des Blutzuckers unter 60 mg/dl (3,3 mmol/l) hat zerebrale Funktionsstörungen (Neuroglukopenie) und eine gegenregulatorische Adrenalinausschüttung zur Folge.
1.3.5.3.2 Klinik
Schwindelgefühl, Kopfschmerz, verschwommenes Sehen, Verwirrtheit, Verhaltensstörungen, schließlich Konvulsionen und Bewusstseinsverlust. Die adrenergen Begleiterscheinungen sind Tachykardie, Tremor, Schwitzen, Hungergefühl und Angst.
1.3.5.3.3 Postprandiale Hypoglykämien
Meistens funktioneller Natur, induziert durch Zucker oder Süßspeisen (besonders bei Magenoperierten). Führt nicht zu Bewusstseinsstörungen. Durch wasserlösliche Ballaststoffe oder Fett-Eiweiß-Diät vermeidbar.
1.3.5.3.4 Nüchternhypoglykämien
Bei Insulinomen, extrapankreatischen Tumoren mit abnorm starker Glukoseutilisation, Leberkrankheiten und endokrinen Störungen (Morbus Addison).
1.3.5.3.5 Exogene Hypoglykämie n
Bei Diabetikern durch Insulin und orale Antidiabetika.
1.4 Arterielle Hypertonie
1.4.1 Definition der Hypertonie
In ihrer Leitlinie zur Diagnostik und Behandlung der arteriellen Hypertonie (2006) hat die Deutsche Hochdruckliga Definition und Klassifikation der Blutdruckstufen vorgeschlagen (◘ Tab. 1.1). Es basiert auf der epidemiologischen Erkenntnis, dass Sterberate und Mortalität an koronarer Herzkrankheit vom niedrigsten noch physiologischen Blutdruck aus kontinuierlich zunehmen.
Mit transportablen Blutdruckautomaten gelingt eine objektive Erfassung des Blutdrucktagesprofils, das zur Beurteilung des Schweregrades der Hypertonie und des Therapieeffektes von großem Nutzen ist. Es schützt auch davor, einen erregungsbedingten „Sprechstundenhochdruck“ zu missdeuten und falsch zu behandeln. Beim ambulanten Blutdruckmonitoring (ABDM) wird der Blutdruck in halb- bis einstündigen Intervallen über 24 h gemessen. In der Nacht sinkt der Blutdruck normalerweise deutlich ab. In ◘ Abb. 1.4 ein normales, in ◘ Abb. 1.5 ein pathologisches ABDM wiedergegeben.


Unauffällige Langzeit-Blutdruck-Registrierung über 24 h


Pathologische Langzeit-Blutdruck-Registrierung über 24 h
Für das ABDM gelten folgende Normalwerte:
-
24-h-Mittelwert: <130/80 mmHg
-
Tagesmittelwert: <135/85 mmHg
-
Nachtabsenkung:
-
systolisch 10–15 mmHg
-
diastolisch 15–20 mmHg
-
-
Nachtmittelwert: <120/70 mmHg
1.4.2 Prävalenz und Altersabhängigkeit der Hypertonie
In der erwachsenen Allgemeinbevölkerung beträgt die Prävalenz der Hypertonie 23–25 %. Sie ist für Frauen etwas niedriger als für Männer.
1.4.3 Hypertonie als Risikofaktor
Durch Druckbelastung schädigt erhöhter Blutdruck die Arterien und das Herz. Hypertoniker haben deshalb ein zur Blutdruckhöhe proportionales Risiko an Herzinfarkt, Herzinsuffizienz, Schlaganfall und terminaler Niereninsuffizienz zu erkranken und zu sterben. Die Risiken steigen, wenn die Hypertonie mit weiteren Risikofaktoren der Arteriosklerose zusammentrifft: Zigarettenrauchen, Diabetes mellitus, Hypercholesterinämie und Hyperhomocysteinämie.
1.4.4 Hämodynamische Typen der Hypertonie
Hämodynamisch liegt eine Hypertonie bei Erhöhung des für die Gewebeperfusion maßgeblichen mittleren arteriellen Drucks vor. Das ist der durchschnittliche arterielle Druck während eines Pulszyklus. Er ist nicht dem Mittelwert aus systolischem und diastolischen Druck gleichzusetzen, sondern liegt etwas niedriger. Bei erhöhtem diastolischen Druck besteht immer eine Hypertonie, bei erhöhtem systolischen Druck nur, wenn der diastolische Druck normal oder nicht stärker gesenkt ist.
Aus der Formel:
arterieller Mitteldruck = Herzminutenvolumen (bzw. Schlagvolumen) × peripherer Gesamtwiderstand
ergeben sich für die Hypertonie folgende hämodynamische Konstellationen:
-
Widerstandshochdruck: Peripherer Gesamtwiderstand erhöht, Herzminutenvolumen im Normbereich oder herabgesetzt. Die Höhe des diastolischen Drucks geht mit der Widerstandszunahme parallel. Hauptwiderstand im Arteriolenbereich. Kommt bei den meisten Formen der Hypertonie vor. Mechanismen der Widerstandserhöhung:
-
Vasokonstriktion: durch vasokonstriktorische Substanzen (Adrenalin, Noradrenalin, Angiotensin II)
-
generalisierte autoregulatorische Vasokonstriktion: Induziert durch eine Überperfusion der Gewebe bei erhöhtem Herzminutenvolumen.
-
-
Minutenvolumenhochdruck: Herzminutenvolumen gesteigert, Herzfrequenz erhöht, peripherer Gesamtwiderstand herabgesetzt oder normal. Systolischer Druck erhöht, diastolischer Druck mäßig erhöht oder normal. Vorkommen: Hyperthyreose, Fieber, Anämie, hyperkinetisches Herzsyndrom.
-
Erhöhter systolischer Druck bei normalem arteriellen Mitteldruck: Herzminutenvolumen normal. Systolischer Druck erhöht, diastolischer Druck erniedrigt. Peripherer Gesamtwiderstand normal. Ursachen: Abnorm großes Schlagvolumen (Bradykardie, Aorteninsuffizienz) oder altersbedingter Elastizitätsverlust der Aorta.
1.4.5 Ätiologische Klassifikation
1.4.6 Primäre oder essenzielle Hypertonie
Hochdruck ungeklärter Ursache mit einem Häufigkeitsanteil von 90 %, in klinischen Statistiken von 80 %. Gehäuftes familiäres Vorkommen ohne definierten Erbgang. Manifestation zwischen dem 35. und 55. Lebensjahr.
1.4.7 Sekundäre Hypertonie n
Hochdruck mit bekannter Ursache, durch bestimmte Grundkrankheiten oder exogene Faktoren entstehend. Häufigkeitsanteil etwa 10 %, in klinischen Statistiken 20 %. Die wichtigsten Formen sind in ◘ Tab. 1.2 aufgeführt.
1.4.8 Pathogenese
1.4.9 Essenzielle Hypertonie
Am plausibelsten ist die Hypothese, dass dem Hochdruck eine Störung der renalen Wasser- und Salzausscheidung zugrunde liegt, aus der folgende Kausalkette resultiert: Zunahme der extrazellulären Flüssigkeit → Hypervolämie → Steigerung des venösen Rückflusses → Zunahme der kardialen Förderleistung → Überperfusion der Organe und Gewebe → generalisierte autoregulatorische Vasokonstriktion (▶ Abschn. 1.1.1) → Zunahme des peripheren Gesamtwiderstandes → Anstieg des Blutdrucks → Steigerung der renalen Wasser- und Salzausscheidung (pressure diuresis) → auf dem erhöhten Blutdruckniveau bleibt künftig eine Salz- und Flüssigkeitsretention aus.
Durch medikamentöse Steigerung der Saliurese kann die täglich aufgenommene Flüssigkeits- und Salzmenge ohne kompensatorische Hypertonie von den Nieren ausgeschieden werden. Bei langdauernder Hypertonie drohen durch Hyalinose der kleinen Arterien und Arteriolen eine Fixierung der peripheren Widerstandserhöhung und eine progrediente Nierenschädigung.
1.4.10 Hypertonie bei renalen Parenchymerkrankungen
Widerstandshochdruck durch renale Salz- und Flüssigkeitsretention nach dem für die essenzielle Hypertonie dargelegten Mechanismus. Mit fortschreitender Niereninsuffizienz wird die Abhängigkeit des Blutdruckniveaus von der Wasser- und Salzzufuhr immer deutlicher. Einseitige Nephrektomie führt bei intakter Restniere und normalem Salzkonsum nicht zur Hypertonie.
1.4.11 Renovaskuläre Hypertonie
Widerstandshochdruck durch Nierenarterienstenose, die in 80 % der Fälle durch Atherome, in 20 % durch fibromuskuläre Dysplasie, selten durch Arteriitis oder Kompression von außen bedingt ist. Mechanismus: Druckabfall distal der Stenose → Stimulation der Barorezeptoren in den Vasa afferentia → Steigerung der Reninsekretion der juxtaglomerulären Zellen → Erhöhung der lokalen und systemischen Konzentration von Angiotensin II → (a) generalisierte Vasokonstriktion, (b) renale Flüssigkeitsretention durch Aldosteronausschüttung und direkte Stimulation der tubulären Na+-Rückresorption.
Die Erhöhung des peripheren Gesamtwiderstandes durch Angiotensin II erfolgt teils direkt, teils indirekt über eine Hypervolämie, die zur autoregulatorischen Vasokonstriktion führt. Bei einseitiger Nierenarterienstenose drosselt das zirkulierende Angiotensin II auch die Wasser- und Salzausscheidung der gesunden Niere.
1.4.12 Aortenisthmusstenose
Angeborene hochgradige, meist kurze Einengung der Aorta descendens distal der linken A. subclavia vor, gegenüber oder nach Abgang des Lig. bzw. Ductus arteriosus Botalli (postduktale Form) mit einem Umgehungskreislauf über die Interkostalarterien und die A. epigastrica superficialis (◘ Abb. 1.10). Hypertonie in der oberen, Normotonie in der unteren Körperhälfte. Mechanismus: Primärer Druckabfall hinter der Stenose, auch in beiden Nierenarterien → Steigerung der Reninsekretion und der Bildung von Angiotensin II → Vasokonstriktion und Drosselung der renalen Wasser- und Salzausscheidung → Hypervolämie → Zunahme der kardialen Förderleistung → Blutdruckanstieg in der oberen, Normalisierung des Blutdrucks in der unteren Körperhälfte → Normalisierung der renalen Wasser- und Salzausscheidung und weitgehend auch der Reninsekretion → Überperfusion der oberen Körperhälfte → autoregulatorische Vasokonstriktion in der oberen Körperhälfte → Die Perfusionsraten werden im ganzen Körper gleich groß. Der Hypertonus in der oberen Körperhälfte bewirkt eine ausgeglichene Flüssigkeitsbilanz.
1.4.13 Primärer Aldosteronismus
Gesteigerte Aldosteronproduktion durch solitäre Adenome (selten Karzinome), in 30 % der Fälle doppelseitige Nebennierenrindenhyperplasie. Extrem selten: Aldosteronbildende Ovarialkarzinome und der Glukokortikoid-supprimierbare Aldosteronismus, der durch eine Genanomalie bedingt ist.
Der Hochdruck entsteht durch renale Salz- und Flüssigkeitsretention, die eine Hypervolämie mit Blutdrucksteigerung induziert und als Reaktion darauf eine systemische autoregulatorische Vasokonstriktion. Der Mineralokortikoidexzess manifestiert sich zugleich in einer Hypokaliämie und einer Suppression des Plasmareninspiegels.
1.4.14 Cushing-Syndrom und exogene Glukokortikoid-Hypertonie
Der Hochdruck entsteht hauptsächlich durch renale Salz- und Flüssigkeitsretention, die vom Cortisol über 3 Angriffspunkte gesteigert wird:
-
Stimulation des Glukokortikoidrezeptors
-
Stimulation des Mineralokortikoidrezeptors
-
eine vom Cortisol induzierte (wahrscheinlich von beiden Rezeptoren unabhängige) Na+-Retention
Am stärksten sind Hypertonie und mineralokortikoider Effekt (Hypokaliämie) bei Tumoren mit ektopischer ACTH-Sekretion. Synthetische Glukokortikoide steigern den Blutdruck nur moderat. Ihre mineralokortikoide Wirkung ist gering. Zur Blutdrucksteigerung durch Glukokortikoide könnten weitere Effekte der Hormone beitragen: Erhöhung der Plasmakonzentrationen von Angiotensinogen und Prorenin, Hemmung der Prostaglandinsynthese, Steigerung der Gefäßsensitivität gegen Pressorsubstanzen.
1.4.15 Hypertonus bei Akromegalie
Ein Hochdruck besteht in einem Drittel der Fälle. Die Pathogenese ist ungeklärt. Im Plasma ist der Reninspiegel niedrig und die Aldosteronkonzentration nicht erhöht. Von Bedeutung könnte der antinatriuretische Effekt des Wachstumhormons sein.
1.4.16 Hypertonus bei Schilddrüsenkrankheiten
-
Hyperthyreose: Minutenvolumenhochdruck mit herabgesetztem peripheren Gesamtwiderstand. Die hormoninduzierte Stoffwechselsteigerung führt zur generalisierten Vasodilatation und Hyperzirkulation des Blutes.
-
Hypothyreose: Die Drosselung des Stoffwechsels führt zur generalisierten autoregulatorischen Vasokonstriktion und damit zur Erhöhung des peripheren Gesamtwiderstandes. Die obligatorische Bradykardie vergrößert das Schlagvolumen. Ein Hochdruck wird in 20–30 % der Fälle manifest.
1.4.17 Schwangerschaftshypertonie
-
Präeklampsie: Blutdruckanstieg nach der 20. Schwangerschaftswoche um >30 mmHg systolisch oder um >15 mmHg diastolisch. Vorkommen in 5–8 % aller Schwangerschaften, am häufigsten in der ersten. Zugrunde liegt ein generalisierter Vasospasmus unklarer Genese mit Erhöhung des peripheren Gesamtwiderstandes und herabgesetztem Blutvolumen. In schweren Fällen starke Albuminurie, Ödeme und Bauchkrämpfe.
-
Eklampsie: Entwickelt sich in 1 % der Fälle von Präeklampsie und ist durch das Hinzutreten von Bewusstseinsstörungen und zerebralen Krampfanfällen gekennzeichnet, die durch Hirnödem und fokale Ischämien entstehen.
1.4.18 Phäochromozytom
Tumor aus chromaffinen Zellen, der Katecholamine sezerniert. Häufigkeit unter Hypertoniepatienten nur 0,1 %. Lokalisation: Nebennierenmark 85 %, Paraganglien und Zuckerkandl-Organ 10 %, hinteres Mediastinum 5 %, selten Glomus caroticum. Multizentrische Tumoren bei Erwachsenen unter 10 %, bei Kindern 30 %. Familiäres Vorkommen: Multiple endokrine Neoplasie Typ II (kombiniert mit Schilddrüsenkarzinom, das Calcitonin bildet, Nebenschilddrüsenadenom und Hypophysenadenom) oder Typ III (kombiniert mit multiplen Neurinomen an Lippen und Zunge). Anteil der malignen Tumoren unter 10 %.
Inzwischen sind mehrere Gene identifiziert worden, die ein Phäochromozytom verursachen (VHL, RET, SDHB, SDHD, NF1).
Die meisten Phäochromozytome des Nebennierenmarks bilden Noradrenalin und Adrenalin (mit höherem Noradrenalinanteil als das normale Nebennierenmark), einige nur Adrenalin. Die extraadrenalen Phäochromozytome sezernieren ausschließlich Noradrenalin. Die Sekretion erfolgt kontinuierlich oder diskontinuierlich, nicht durch Sympathikusstimulation.
Die Hypertonie ist vasokonstriktorisch mit stark erhöhtem peripheren Widerstand und herabgesetztem Blutvolumen (Tonisierung der venösen Kapazitätsgefäße, Druckdiurese, Hemmung des Renin-Angiotensin-Aldosteron-Systems). In 60 % der Fälle Dauerhochdruck, dem sich bei der Hälfte der Patienten Blutdruckkrisen aufpfropfen. In 40 % der Fälle nur Blutdruckkrisen bei normalem Blutdruck im Intervall. Im Verlauf kann es zur orthostatischen Hypotension mit Tachykardie und Schwindelanfällen kommen, die zum führenden Symptom werden. Zur orthostatische Intoleranz tragen wahrscheinlich eine Down-Regulation der peripheren α1-Rezeptoren, eine herabgesetzte Reninfreisetzung und die Hypovolämie bei. Im Anfall Herzklopfen, Angstgefühl, Tremor, Kopfschmerz, Übelkeit und Erbrechen, Schweißausbruch, Sehstörungen, auch anginöse und abdominale Schmerzen. Hypertonische Krisen können durch zerebralen Insult, Herzinfarkt oder Multiorganversagen zum Tode führen.
1.4.19 Neurogene Hypertonie
Entsteht durch Überaktivität des sympathischen Nervensystems aus folgenden Ursachen:
-
Direkte Stimulation des Vasomotorenzentrums: Bei Hirndrucksteigerung, Ischämie und Hirnstammerkrankungen. Nur kurzdauernde Hypertonie bei Ausschaltung der Barorezeptoren. Das Vasomotorenzentrum adaptiert sich dann an den Wegfall der hemmenden Impulse aus dem Karotissinus.
-
Zentrale Hyperreaktivität auf emotionale Belastungen: Etwa 20 % der Patienten mit einer als essenziell eingestuften Hypertonie sind hyperadrenergisch: Zirkulierende Katecholamine im Plasma erhöht, Herzfrequenz gesteigert, auf α1- und β-Rezeptorenblocker und kombinierte adrenerge Blockade stärkerer Blutdruckabfall als bei nichthyperadrenergen Hypertonikern.
1.4.20 Organschäden durch Hypertonie
1.4.21 Herz
Linksherzhypertrophie, Schädigung des linken Ventrikels durch subendokardiale Ischämie, Linksinsuffizienz mit Lungenödem, oft auch auf dem Boden einer diastolischen Funktionsstörung infolge der Hypertrophie des linken Ventrikels, Rhythmusstörungen, Arteriosklerose der Koronararterien, Angina pectoris, Infarkt.
1.4.22 Arterien
-
Aortenaneurysma: Hypertonische Schädigung der Media (besonders bei Dissektion), akzelerierte Arteriosklerose.
-
Periphere arterielle Verschlusskrankheit: Akzelerierte Arteriosklerose.
-
Arteriolosklerose: Intimaverdickung und hyaline Degeneration der Widerstandsgefäße (kleine Arterien, Arteriolen), im Frühstadium nach Drucksenkung reversibel.
1.4.23 Gehirn
-
Hypertonische intrazerebrale Blutung: Aus rupturierten Mikroaneurysmen kleiner penetrierender Arterienäste, überwiegend im Bereich der inneren Kapsel (◘ Abb. 1.6). Unterteilung in große Blutung, meist mit Ventrikeleinbruch, kleine Blutung, schlitzförmige Blutung und petechiale Blutung.
Abb. 1.6 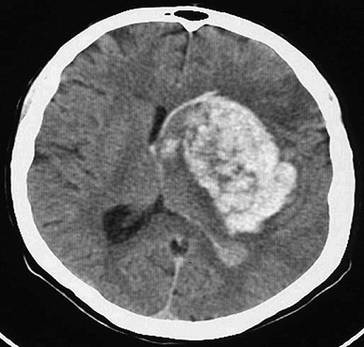
Natives CCT einer schweren linksseitigen Stammganglienblutung mit Mittellinienverlagerung, Kompression und Einblutung des linken Seitenventrikels bei einer 65-jährigen Patientin
-
Zerebrale Infarkte: Arteriosklerotische Thrombose der Hirnarterien, lakunäre Infarkte durch segmentale Lipohyalinose kleiner penetrierender Äste der A. cerebri media und der A. basilaris (◘ Abb. 1.7). Mikroembolien aus Atheromen extrakranieller Hirnarterien. Transitorische ischämische Attacken.
Abb. 1.7 
MRT mit lakunärem Insult im rechten Nucleus caudatus bei einem 72-jährigen Patienten mit langjährigem arteriellen Hypertonus und Zustand nach Aortenklappenersatz
-
Hypertonische Enzephalopathie: Hirnödem durch akuten Blutdruckanstieg mit Hyperperfusion des Gehirns. Symptome: Kopfschmerz, Erbrechen, Sehstörungen, Benommenheit, Stupor, Koma, auch generalisierte Krämpfe und fokale neurologische Ausfallserscheinungen.




1.4.24 Retina
-
Retinopathie der Sklerose: Altersveränderungen (Hyalinisierung), durch chronische Hypertonie verstärkt, aber auch ohne Hypertonie vorkommend:
-
Kreuzungszeichen (Gunn): Venen an den Kreuzungsstellen nicht mehr hinter den verdickten Arterien sichtbar, ihr Verlauf erscheint unterbrochen. Venenerweiterung distal der Kreuzung, an der Arterie und Vene eine gemeinsame Adventitia haben.
-
Verbreiterung der vaskulären Lichtreflexe: Der normale schmale helle Reflexstreifen (der Blutsäule bei durchsichtiger Gefäßwand) wird mit zunehmender Sklerosierung diffuser und breiter. Leichter Grad: Kupferdraht-Arterien; stärkerer Grad: Silberdraht-Arterien.
-
-
Retinopathie der akuten Hypertonie:
-
Veränderungen an den Arterien: Generalisierte oder fokale Engstellung der Arterien und Arteriolen, die blass erscheinen, gestreckt verlaufen und sich spitzwinklig verzweigen. Potenziell reversibel, bei Persistenz des Hochdrucks in Sklerosierung übergehend.
-
Blutungen: Durch Diapedese oder Nekrose von Kapillaren und Präkapillaren. Flammenförmige Blutungen der oberen, punkt- und fleckförmige Blutungen der tieferen Netzhautschichten.
-
Retinaödem und fettige Degenerationsherde: Degeneration von Nervenzellen durch das Retinaödem. Dabei entstehen gelbliche Lipid-Protein-Residuen („harte Exsudate“), die innerhalb einiger Wochen von Makrophagen abgeräumt werden können.
-
Cotton-Wool-Exsudate: Weiß-graue Herde, die durch ischämische Netzhautinfarkte entstehen. Verschluss der zuführenden Arteriole, dilatierte, z. T. aneurysmatisch erweiterte Kapillaren im Randgebiet. Rückbildungsdauer 4–12 Wochen.
-
Papillenödem: Beginnt mit grauer Verfärbung am temporalen Papillenrand. Arterien extrem enggestellt, Venen gestaut. Entstehung durch lokale ischämische Kapillarschädigung und durch Anstieg des intrakraniellen Drucks.
-
-
Retinopathie der chronischen Hypertonie: Mischbild aus sklerotischer und akuter Retinopathie. Verstärkt gewundene Kupfer- und Silberdraht-Arterien. Terminale Arteriolen verengt und gestreckt. Kreuzungszeichen mit gestauten Venen, Blutungen, in schweren Fällen Cotton-Wool-Exsudate. Bei maligner Hypertonie Papillenödem.
1.4.25 Nieren
-
Chronische Hypertonie: Arteriolosklerose, die zur Progredienz der Hypertonie, zu allmählicher Atrophie und zur Einschränkung der Nierenfunktion führt.
-
Maligne Hypertonie: Akute schwere Wandläsionen der interlobulären Arterien und Arteriolen, mit progredienter ischämischer Atrophie und Niereninsuffizienz, die in terminales Nierenversagen übergehen kann. Verschlimmerung der Hypertonie im Sinne eines Circulus vitiosus.
1.4.26 Diagnostik
1.4.27 Leitsymptome
Hinterkopfschmerzen beim Erwachen, Schwindelgefühl, Herzklopfen und gesteigerte Erschöpfbarkeit können auf eine Hypertonie hinweisen. Es gibt aber keine hochdruckspezifischen Beschwerden. Die meisten Hypertoniker sind asymptomatisch, so lange keine kardiovaskulären oder zerebrovaskulären Komplikationen auftreten. Die Hypertonie ist daher sehr häufig eine Zufallsdiagnose. Man muss sie durch mehrmalige Blutdruckmessung sichern. Am zuverlässigsten ist die ambulante 24-Stunden-Registrierung (ABDM).
1.4.28 Erfassung von Schweregrad und sekundären Organschäden
-
Klinisch: Herzverbreiterung? Hebender Spitzenstoß? Lungenstauung? Ödeme? Seitendifferenz des Blutdrucks? Stenosegeräusche über den großen Arterien? Neurologische Defizite?
-
Elektrokardiogramm: Zeichen der Linksherzhypertrophie und Linksschädigung? Infarktnarben? Ischämiezeichen unter Belastung? Herzrhythmusstörungen (Vorhofflimmern)?
-
Echokardiogramm: Hypertrophie oder Dilatation des linken Ventrikels? Normale Ejektionsfraktion? Normale diastolische Funktion? Dilatation des linken Vorhofs? Ektasie der Aorta ascendens? Eine diastolische Funktionsstörung ist frühzeitig mittels PW-Doppler-Echokardiographie am transmitralen Einstromprofil zu erkennen (◘ Abb. 1.8): verminderte frühe linksventrikuläre Füllungsgeschwindigkeit (E) bei erhöhter Einflussgeschwindigkeit während der Vorhofkontraktion (A).
Abb. 1.8a–c 
PW-Doppler-Echokardiographie: a Normales transmitrales Einstromprofil eines Herzgesunden mit regelrechtem E/A-Verhältnis, b Umgekehrtes E/A-Verhältnis bei einem Patienten mit diastolischer Relaxationsstörung auf der Basis eines Hypertonus, c Restriktives Füllungsmuster mit hoher schmaler E-Welle und sehr kleiner A-Welle bei einem Patienten mit kardialer Amyloidose und schwerer restriktiver diastolischer Funktionsstörung


1.4.29 Röntgenaufnahme des Thorax
Abnorme Größe und Form des Herzens? Pulmonale Stauung? Verbreiterung der Aorta?
1.4.30 Augenhintergrund
Klassifikation nach Keith-Wagner:
-
I: geringe Einengung und Sklerose der Arterien
-
II: wie I, aber stärker plus Kreuzungszeichen
-
III: Netzhautödem, Exsudate, Blutungen
-
IV: wie III plus Papillenödem
1.4.31 Labordiagnostik
Kreatinin und Harnstoff im Serum erhöht?
1.4.32 Zusätzliche koronare Risikofaktoren
Diabetes mellitus? Hypercholesterinämie? Hyperhomocysteinämie? Zigarettenrauchen?
1.4.33 Differenzialdiagnosen
1.4.34 Essenzielle Hypertonie
Familiäres Vorkommen, Manifestationsalter zwischen dem 35. und 55. Lebensjahr. Ausschluss sekundärer Hypertonien.
1.4.35 Renoparenchymatöse Hypertonie
Nierenerkrankung in der Anamnese: Eingeschränkte Nierenfunktion, Albuminurie, entzündliches Harnsediment, evtl. Bakteriurie. Verkleinerung der Nieren im Sonogramm.
1.4.36 Renovaskuläre Hypertonie
Alter unter 30 oder über 50 Jahre. Einseitig kleine Niere im Sonogramm (Differenz >1,5 cm). Schlechtes Ansprechen auf antihypertensive Therapie. Hypokaliämie. Plasmarenin nicht zuverlässig. Direkter Stenosenachweis durch Farbduplexsonographie (70–90 % bei erfahrenem Untersucher). Captopril-Nierensequenzszintigraphie, MR-Angiographie (◘ Abb. 1.9) oder konventionelle Angiographie.


MRT (nach KM-Gabe) einer hochgradigen rechtsseitigen Nierenarterienstenose mit Niereninfarkt (Sammlung Dr. Esdorn, Bad Oeynhausen)
1.4.37 Phäochromozytom
-
Vorkommen: Bei 0,1 % aller Hypertonien. Unter 1 Mio. Einwohnern 2–8 Fälle.
-
Klinik: Paroxysmen oder Dauerhochdruck. Trias: Anfälle von Kopfschmerz, Herzklopfen und Schweißausbruch. Flush oder Blässe. Tachykardie oder Bradykardie, auch Vorhofflimmern und Angina pectoris. Kohlenhydratintoleranz (hepatische Glukogenese und Suppression der Insulinsekretion). Resistenz gegen übliche Antihypertensiva.
-
Diagnostik: Erhöhte freie Metanephrine im Plasma sicherster Suchtest). Im 24-h-Harn sind Vanillinmandelsäure, Katecholamine und totale Metanephrine erhöht, im Plasma auch Katecholamine und Chromogranin A. Direkter Tumornachweis mittels CT, MRT oder MIBG-Scan.
-
Therapie: ▶ unten.
1.4.38 Aortenisthmusstenose
Jugendliches Alter. Schwache Fußpulse. Mesosystolikum (p.m. interskapulär am Rücken). Blutdruckdifferenz zwischen oberer und unterer Extremität (simultane Dopplermessung in Ruhe und nach 10 Kniebeugen). Echo: Darstellung der Stenose und Bestimmung des Druckgradienten in Ruhe und unter Valsalva-Manöver. Röntgenbild des Thorax: Rippenusuren. Darstellung der Stenose im MR-Angiogramm (◘ Abb. 1.10).


MRT (nach KM-Gabe) einer Aortenisthmusstenose (Sammlung Dr. Esdorn, Bad Oeynhausen)
1.4.39 Endokrine Hypertonie
Primärer Aldosteronismus, Cushing-Syndrom, Akromegalie, Hyperthyreose, Hypothyreose (▶ Kap. 6).
1.4.40 Allgemeine Therapie
1.4.41 Nichtmedikamentöse Maßnahmen
Gründliche Aufklärung des Patienten über die Hochdruckkrankheit. Belehrung über den blutdrucksenkenden Effekt von Gewichtsreduktion, Kochsalz- und Alkoholbeschränkung, Nikotinverzicht und körperlichem Training. Zur Verbesserung von Therapiekontrolle und Kooperation Selbstmessung empfehlen. Vor Beginn der symptomatischen Behandlung heilbare sekundäre Hypertonien ausschließen.
1.4.42 Diuretika
Durch Steigerung der Natriurese senken sie extrazelluläres Flüssigkeitsvolumen und Herzminutenvolumen. Dadurch werden autoregulatorische periphere Vasokonstriktion und Blutdruck herabgesetzt (▶ Abschn. 1.1). Ein direkter antihypertensiver Effekt ist nicht gesichert. Da die Diuretika die Wirkung fast aller Antihypertensiva verstärken, sollten sie in jede Kombination aufgenommen werden.
-
Thiazide: In der 2002 publizierten großen amerikanischen ALLHAT-Studie an 42418 hypertonen Patienten wurde das Thiazid-Diuretikum Chlorthalidon (12,5–25 mg/Tag) mit dem Ca-Antagonisten Amlodipin (2,5–10 mg/Tag) und dem ACE-Hemmer Lisionopril (10–40 mg/Tag) verglichen. Die Autoren resümierten, dass Chlorthalidon den beiden untersuchten modernen Antihypertonika in Bezug auf kardiovaskuläre Protektion überlegen war und empfahlen. die kostengünstigen Thiazid-Diuretika bei der Initialtherapie der Hypertonie zu bevorzugen. Die Studie blieb nicht unwidersprochen, doch lässt sich eine essenzielle Hypertonie bei vielen, vor allem älteren Patienten gut mit Thiaziden kontrollieren. Einzuhalten sind niedrige Dosierungen (z. B. nicht >25 mg Hydrochlorothiazid/Tag), weil dann die metabolischen Nebenwirkungen (Hypokaliämie, Hyperurikämie, Abnahme der Glukosetoleranz, Hyperlipämie) gering sind. Außerdem wird eine übermäßige Volumenkontraktion mit Aktivierung des Renin-Angiotensin-Aldosteron-Systems vermieden. Vorteilhaft ist die Kombination mit einem Kalium-sparenden Diuretikum.
Die Gruppe der Thiazide mit Wirkungsmechanismus und Dosierungen wird im Abschnitt über Ödeme (▶ Kap. 3.2) abgehandelt. Falls bei der Hypertonie eine niedrige Initialdosis nicht ausreicht, ist zusätzlich ein weiteres Mittel einzusetzen.
-
Schleifendiuretika: Ihr Einsatz erfolgt bei schweren, vor allem renalen Hypertonien mit eingeschränkter Nierenfunktion, ferner bei Herzinsuffizienz. Bezüglich Wirkungsmechanismus und Dosierung der einzelnen Pharmaka wird auf ▶ Kap. 3.2 verwiesen.
-
Aldosteronantagonisten: Verfügbar sind Spironolacton und neuerdings Eplerenon. Indiziert ist die Anwendung bei Hypertonie mit Herzinsuffizienz und beim sekundären Aldosteronismus mit Hypokaliämie unter Diuretikatherapie, ferner bei Hypertonie unter Glukokortikoidtherapie.
1.4.43 Inhibitoren des Renin-Angiotensin-Systems
-
ACE (Angiotensin-Converting-Enzyme)-Blocker: Sie senken den Blutdruck, indem sie die Bildung des vasokonstriktorischen Angiotensin II und den Abbau des vasodilatierenden Bradykinin hemmen. Dabei bleiben Schlagfrequenz und Förderleistung des Herzens unbeeinflusst. Zusätzlich unterdrücken ACE-Blocker die Stimulation der Aldosteronsekretion durch Angiotensin II und wirken damit einer Salz- und Flüssigkeitsretention entgegen. Hinzu kommen wichtige protektive Wirkungen auf kardiovaskuläre Strukturen durch Ausschaltung der lokalen Bildung von Angiotensin II. Besonders effektiv sind ACE-Blocker deshalb bei koronarer Herzkrankheit und Herzinsuffizienz. Als weitere Domäne haben sich chronische Nierenkrankheiten mit und ohne Diabetes mellitus erwiesen. Eine häufige Nebenwirkung (bei 10–15 % der Patienten) der ACE-Blocker, der zum Absetzen zwingt, ist ein anhaltender heftiger Reizhusten.
-
Kontraindikationen: Hyperkaliämie, Kreatininwerte >2,5 mg/dl und Schwangerschaft. Aspirin und nichtsteroidale Antiphlogistika können die Wirkung der ACE-Blocker einschränken.
-
Repräsentative Präparate: Captopril (2-mal 12,5–50 mg/Tag), Enalapril (1-mal 5–40 mg/Tag), Lisinopril (1-mal 5–40 mg/Tag). Nicht mit kaliumsparenden Diuretika kombinieren.
-
-
Angiotensin-II-Rezeptor-Blocker (ARB): Sie verdrängen Angiotensin II kompetitiv von seinem spezifischen Rezeptor AT1 und schalten bei entsprechender Dosierung alle Effekte des Angiotensin II aus. Sie heißen deshalb auch AT1-Antagonisten und sind mit den ACE-Blockern praktisch wirkungsgleich. Hustenreiz lösen sie aber nicht aus. Der durch die Rezeptorblockade bewirkte Anstieg des zirkulierenden Angiotensin II hat keine nachteiligen Konsequenzen.
-
Repräsentative Präparate: Lorsartan (1-mal 50–100 mg/Tag), Irbesartan (1-mal 150–300 mg/Tag), Valsartan (1-mal 80–160 m/Tag), Candesartan (1-mal 8–16 mg/Tag).
-
-
Direkte Renin-Inhibitoren: Die einzige zugelassene Substanz ist Aliskiren, ein Non-Peptid mit niedrigem Molekulargewicht. Durch Bindung an die aktive Seite des Renins blockiert Aliskiren die Umwandlung von Angiotensinogen in Angiotensin I. Im Plasma sinken beide Angiotensine, während Renin stark ansteigt. Die Aldosteronproduktion sinkt, die Natriurese nimmt zu. Dosisabhängig senkt Aliskiren den Blutdruck genau so stark wie die ACE-Blocker. Die Kosten sind aber deutlich höher.
-
Nebenwirkungen: Diarrhö, abdominale Schmerzen, Nasopharyngitis, Angioödem.
-
Dosierung: 1×150 oder 300 mg/Tag.
-
1.4.44 Calciumantagonisten (Calciumkanal-Blocker)
Zu unterscheiden sind 3 Hauptgruppen: Dihydropyridine (Nifedipin, Amlodipin), Phenylalkylamine (Verapamil) und Benzothiazepine (Diltiazem).
-
Wirkungsmechanismus: Die relativ starke blutdrucksenkende Wirkung der Calciumantagonisten beruht auf Relaxation der Arteriolen und damit verbundenen Senkung des peripheren Kreislaufwiderstands. Durch Bindung an die spannungsabhängigen Calciumkanäle vom Typ L wird der Ca++-Einstrom in die glatten Muskelzellen reduziert, deren Kontraktilität von der freien intrazellulären Ca++-Konzentration abhängt. Der Venentonus wird nicht herabgesetzt, so dass ein orthostatischer Blutdruckabfall ausbleibt. Durch unterschiedliche Bindungsstellen an den Calciumkanälen wird das Wirkungsprofil der Calciumantagonisten modifiziert.
Auf den Herzmuskel wirken Dihydropyridine nur schwach, Verapamil und Diltiazem etwas stärker negativ inotrop. Auf den Sinusknoten und den AV-Knoten, die vom langsamen Ca++-Einstrom depolarisiert werden, haben nur Verapamil einen deutlichen Hemmeffekt, der die Herzfrequenz und die AV-Überleitung verlangsamt, letztere am stärksten bei Vorhofflimmern. Bei raschem Blutdruckabfall durch Dihydropyridine kommt es via Barorezeptoren zur reflektorischen Sympathikusstimulation mit Tachykardie und Steigerung des Herzzeitvolumens. Verapamil und Diltiazem senken den Blutdruck protrahierter und deutlich schwächer.
-
Anwendung und Kontraindikationen:
-
Dihydropyridine: Indiziert bei Hypertonie und vasospastischer Angina pectoris (Prinzmetal-Angina), weil es den Blutdruck senkt und Koronarspasmen vorbeugt. Gute Wirkung bei systolischer Hypertonie im Alter (Senkung des Schlaganfallrisiko). Verstärkt bei renaler Hypertonie ungenügend wirksame Zweierkombinationen aus Diuretikum und ACE-Blocker. In der ALLHAT-Studie schnitt Amlodipin nicht schlechter ab als der ACE-Blocker Lisinopril. In hypertensiven Notfällen ist Nifedipin schnell wirksam (5–10 mg Kapsel zerbeißen). In der Langzeittherapie sind Nifedipin in Retardform (2-mal 20 mg/Tag) und protrahierter wirkende DHP-Calciumantagonisten, z. B. Amlodipin (1-mal 5–10 mg/Tag), Nitrendipin (2-mal 10–20 mg/Tag) und Isradipin (2-mal 2,5 mg oder 5 mg in Retardform/Tag einzusetzen.
-
Kontraindikationen: Koronare Herzkrankheit, instabile Angina pectoris und frischer Myokardinfarkt wegen reflektorischer Sympathikusaktivierung.
-
Nebenwirkungen: Kopfschmerzen, Flush, Knöchelödeme.
-
-
Verapamil und Diltiazem: Beide Substanzen sind bei leichter bis mittelschwerer Hypertonie einsetzbar (Verapamil retard 120–480 mg/Tag, Diltiazem retard 180–300 mg/Tag).
-
Kontraindikationen: Bradykardie und Erregungsleitungsstörungen, Kombination mit β-Blockern, manifeste Herzinsuffizienz.
-
Nebenwirkungen: Obstipation, Unterschenkelödeme, Gingivahyperplasie bei Diltiazem.
-
-
1.4.45 β-Adrenorezeptor-Antagonisten (β-Blocker )
-
Klassifizierung:
-
nichtselektive: Propranolol, Nadolol, Timolol, Sotalol u. a.
-
nichtselektive mit intrinsischer sympathikomimetischer Aktivität: Pindolol, Penbutolol u. a.
-
β1-selektive oder kardioselektive: Metoprolol, Atenolol, Bisoprolol, Nebivolol u. a.
-
β1-selektive mit intrinsischer sympathikomimetischer Aktivität: Acebutolol, Celilprolol
-
-
Antihypertensive Wirkungsmechanismen: Der Blutdruck sinkt, weil die Förderleistung des Herzens durch Herabsetzung der myokardialen Kontraktilität und der Herzfrequenz nachlässt. Dem wirkt zunächst eine Zunahme des peripheren Kreislaufwiderstands entgegen, denn auch die vasodilatatorischen peripheren β-Rezeptoren werden blockiert. Die vaskuläre Resistenz tendiert jedoch allmählich wieder zur Norm. Dazu trägt bei, dass die β-Blocker die renale Reninsekretion und damit die Bildung des Angiotensin II hemmen.
-
Anwendung: Am besten spricht die hyperkinetische, mit Tachykardie verbundene Hypertonie auf β-Blocker an (z. B. Nadolol 1-mal 30–120 mg/Tag). Weitere Indikationen sind renale Hypertonien mit gesteigerter Sympathikusaktivität, Tachyarrhythmien, hypertensive koronare Herzkrankheit, Zustand nach Infarkt und Herzinsuffizienz. Bei koronarer Herzkrankheit schützen β-Blocker zugleich vor Rhythmusstörungen und akutem Herztod. Bei Herzinsuffizienz sind nur β1-Blocker (in langsam steigender Dosis!) erlaubt. Sie verbessern die Herzleistung und wirken protektiv auf das Myokard (▶ Abschn. 1.9). Bei Bronchospastik sind nur die kardioselektiven β1-Blocker anzuwenden. Sie beeinträchtigen auch den Lipid- und Kohlenhydratstoffwechsel weniger als die nichtselektiven.
-
Kontraindikationen: Verdacht auf Phäochromozytom, Schwangerschaft, hydropische Herzinsuffizienz, schweres Bronchialasthma, Bradykardie und AV-Leitungsstörungen, periphere arterielle Verschlusskrankheit, insulinabhängiger Diabetes (hemmt Wahrnehmung und Gegenregulation bei Hypoglykämie).
-
Nebenwirkungen: Müdigkeit, Schwäche, Herabsetzung der maximalen körperlichen Leistungsfähigkeit, Schlafstörungen, Depression, Potenzschwäche, Haarausfall, Kältegefühl in den Extremitäten, Bradykardie, AV-Leitungsstörungen.
-
Repräsentative Präparate: Propranolol (2- bis 4-mal 40‒80 mg/Tag), Bisoprolol (1-mal 1,25–10 mg/Tag), Metoprolol (2-mal 20–150 mg/ Tag), Atenolol (1-mal 25‒100 mg/Tag).
1.4.46 Kombinierte α1- und β-Adrenorezeptoren-Antagonisten
-
Substanzen: Zugelassen sind Carvedilol, in den USA auch Labetalol.
-
Wirkungsmechanismus: Die Förderleistung des Herzens wird weniger reduziert als durch reine β-Blocker. Die Blutdrucksenkung kommt hauptsächlich durch die Blockade der α1-Rezeptoren zustande, die den peripheren Widerstand und damit das Afterload herabsetzen. Zugleich resultiert eine Tendenz zu orthostatischem Blutdruckabfall. Die Bradykardie kann ausgeprägt sein.
-
Anwendung: Bei mittelschwerer Hypertonie effektiv, senkt den diastolischen Druck deutlich. Besonders indiziert bei Herzinsuffizienz. Dosierung (Carvedilol): Bei essenzieller Hypertonie 1-mal 12,5 mg/Tag für 2 Tage, danach 1-mal 25 mg/Tag, maximal 2-mal 25 mg. Bei Herzinsuffizienz 2-mal 3,125 mg/Tag, Dosisverdopplung alle 2 Wochen auf maximal 2-mal 50 mg/Tag bei Verträglichkeit.
-
Kontraindikationen: Dekompensierte Herzinsuffizienz, Herzfrequenz <60/min, Schenkelblockbilder, Phäochromozytom, Prinzmetal-Angina, akute Lungenembolie, Aortenstenose, Niereninsuffizienz, Bronchialasthma.
-
Nebenwirkungen: Bradykardie, Dyspnoe, Gliederschmerzen, Verschlimmerung peripherer Durchblutungsstörungen, Sehstörungen, Verwirrtheit, Psychose, Ödeme. Dazu Thrombopenie, Leukopenie und Transaminasenanstieg.
1.4.47 Selektive α1-Adrenorezeptor-Antagonisten (α-Blocker )
-
Substanzen: Bunazosin , Doxazosin , Prazosin , Terazosin , Urapidil .
-
Antihypertensiver Wirkungsmechanismus: Die selektive Hemmung der α1-adrenergen Rezeptoren führt zur peripheren Vasodilatation und damit zur deutlichen Senkung des systolischen und diastolischen Blutdrucks. Auch die venösen Kapazitätsgefäße werden erweitert, so dass gleichzeitig der venöse Rückfluss abnimmt. Auf diese Weise sinkt der Blutdruck im Stehen stärker ab als im Liegen. Außerdem entsteht eine Tendenz zur Flüssigkeitsretention, die zu Ödemen führen kann.
-
Anwendung: Zur Monotherapie des Hochdrucks sind die α1-Adrenorezeptor-Antagonisten nicht geeignet. Denn in der ALLHAT-Studie erhöhte sich bei den mit Doxazosin Behandelten das Risiko einer kardiovaskulären Erkrankung um 25 % und das der Herzinsuffizienz auf das Doppelte. In jedem Fall ist die Kombination mit einem Diuretikum erforderlich. Wenn Dreierkombinationen ungenügend wirksam sind, können α1-Blocker gegen eine der Komponenten ausgetauscht oder zusätzlich gegeben werden.
Die erste Dosis kann zu einem unerwartet intensiven Blutdruckabfall führen und sollte deshalb kurz vor dem Schlafengehen eingenommen werden.
-
Dosierungen:
-
Bunazosin: 1-mal 6 mg/Tag
-
Doxazosin: Anfangsdosis 1-mal 1 mg/Tag, langsam erhöhen auf 2–4 mg, selten auf maximal 16 mg/Tag
-
Prazosin: Anfangsdosis 1 mg/Tag, in wöchentlichen Abständen steigern auf max. 2-mal 10 mg/Tag
-
Terazoin: Anfangsdosis 1-mal 1 mg, langsam erhöhen auf 4–5, max. auf 10 mg/Tag
-
Urapidil: Anfangsdosis 2-mal 60 mg/Tag, maximal 180 mg/Tag
-
-
Kontraindikationen: Für Prazosin, Doxazosin und Terazosin Überempfindlichkeit gegen Chinazoline. Für Prazosin und Urapidil Aorten- und Mitralstenose im Dekompensationsstadium, Linksinsuffizienz mit niedrigem Füllungsdruck, Herzinsuffizienz mit pulmonaler Hypertonie.
1.4.48 Zentral und peripher wirkende Antisympathikotonika : Sympathikolytika
-
Substanzen: Clonidin, α-Methyldopa, Urapidil, Moxonidin, Reserpin.
-
Clonidin: Stimuliert die α2-Adrenorezeptoren im Vasomotorenzentrum und setzt dadurch die periphere Sympathikusaktivität herab. Es senkt den Blutdruck kräftig, erzeugt aber 12–18 h nach Absetzen ein Rebound-Phänomen. Clonidin eignet sich zur Kombination mit Medikamenten, die eine reaktive Sympathikusstimulation bewirken (Diuretika, Calciumantagonisten), auch als dritte Komponente zu ungenügend wirksamen Zweierkombinationen. Wegen additiver negativ-inotroper Wirkung soll es nicht mit β-Blockern kombiniert werden. Dosis: 2-mal 0,038–0,0075 mg/Tag bei leichter, 2-mal 0,150–0,300 mg/Tag bei schwerer Hypertonie. Nebenwirkungen: Sedierung, Mundtrockenheit.
-
α-Methyldopa: Es wird enzymatisch zu Methylnoradrenalin umgewandelt, das die zentralen α2-Rezeptoren stimuliert und damit den Sympathikustonus herabsetzt. Wegen seiner Nebenwirkungen (Sedierung, Nasenkongestion, immunhämolytische Anämie, Hepatitis) wird es kaum mehr eingesetzt. Noch ist es aber Mittel der Wahl bei Schwangerschaftshypertonie, da es den Feten nicht schädigt. Dosis: 2-mal 250 mg/Tag.
-
Moxonidin: Die Substanz führt durch Bindung an zentrale I1-Imidazolidin-Rezeptoren und die Stimulation zentraler α2-Rezeptoren zur Hemmung der Sympathikusaktivität. Die Blutdrucksenkung ist schwächer als die durch Clonidin. Dafür kommt es nicht zum Rebound-Phänomen und weniger zu Sedierung und Mundtrockenheit. Moxonidin wird in Mehrfachkombinationen eingesetzt, doch fehlen Langzeitstudien über seinen Effekt auf Morbidität und Mortalität durch Hochdruckkomplikationen. Dosis: 1-mal 0,2 bis maximal 0,6 m/Tag.
-
Reserpin: Das Rauwolfia-Alkaloid senkt den Blutdruck durch Aktivitätsminderung zentraler und peripherer noradrenerger Neurone. Das geschieht durch Hemmung der Wiederaufnahme von Noradrenalin und Dopamin in die präsynaptischen Speichergranula. Herzfrequenz, Herzzeitvolumen und peripherer Widerstand werden herabgesetzt. Neben der sympathikolytischen Wirkung findet eine Vagusstimulation statt. Früher war Reserpin ein häufig eingesetztes Hochdruckmittel. Wegen seiner Nebenwirkungen (Sedierung, Depression, Kongestion der Nasenschleimhaut, Mundtrockenheit, Magenkrämpfe, Diarrhö und eventueller Erhöhung des Brustkrebsrisikos) wird es nur noch sehr selten verordnet. Dosis: 0,075–0,15 mg/Tag.
1.4.49 Direkte Vasodilatatoren
Bewirken auf unterschiedliche Weise eine Weitstellung der Arteriolen mit starkem Blutdruckabfall und eine intensive reflektorische Sympathikusstimulation.
-
Substanzen: Hydralazin, Dihydralazin, Minoxidil, Diazoxid, Nitroprussidnatrium.
-
Hydralazin, Dihydralazin: Relaxieren selektiv die glatten Muskelzellen der Arteriolen, wobei der Wirkungsmechanismus unbekannt ist. Der Blutdruckabfall induziert eine intensive kompensatorische Sympathikusstimulation, einen Anstieg der Plasmareninaktivität und eine renale Salz- und Flüssigkeitsretention. Folglich ist die Kombination mit einem β-Blocker und einem Diuretikum erforderlich. Nebenwirkungen: Kopfschmerz, Übelkeit, Herzklopfen, Ödeme. Bei längerer Anwendung treten in 5–15 % der Fälle antinukleäre Autoantikörper und bei 5–10 % ein Lupus-erythematodes-Syndrom auf, seltener eine immunhämolytische Anämie oder Glomerulonephritis. Anwendung nur bei schwerer, sonst refraktärer Hypertonie als Komponente in Mehrfachkombinationen.
-
Minoxidil: Dilatiert selektiv die Arteriolen, indem es über eine Öffnung ATP-modulierter K+-Kanäle spannungsabhängige Calciumkanäle blockiert. Durch den Blutdruckabfall kommt es wie durch Hydralazin zur Sympathikusstimulation des Herzens und zur Aktivierung des Renin-Angiotensin-Aldosteron-Systems mit Salz- und Flüssigkeitsretention. Nebenwirkungen: Gewichtszunahme und Herzinsuffizienz. Anwendung nur in Mehrfachkombinationen bei refraktärer Hypertonie. Kontraindikation: koronare Herzkrankheit.
-
Diazoxid: Dilatiert die Arteriolen auf die gleiche Weise wie Minoxidil. Es resultieren Sympathikusaktivierung mit Zunahme von Herzfrequenz und Herzzeitvolumen und Anstieg der extrazellulären Flüssigkeit. Nebenwirkungen: Kopfschmerz, Übelkeit, Erbrechen, Hemmung der Insulinsekretion mit Hyperglykämie, Hypertrichose. Anwendung nur noch selten bei hypertensiven Notfällen und maligner Hypertonie. Applikation als Infusion in Boli von 60 mg alle 2–3 Minuten bis zur erwünschten Wirkung. Zur Begleittherapie sind Schleifendiuretika indiziert. Durch die gleichzeitige Gabe von β-Blockern wird die Wirkung verstärkt.
-
Nitroprussidnatrium: Gilt als stärkster Vasodilatator. Relaxiert arterielle Widerstandsgefäße und venöse Kapazitätsgefäße durch nichtenzymatische Freisetzung von Nitroxid. Vor- und Nachlast nehmen ab, was sich besonders günstig bei Linksinsuffizienz auswirkt. Anwendung bei hypertensiven Notfällen per Infusionspumpe in 5%iger Glukoselösung über einen zentralen Venenkatheter. Der Blutdruck fällt nach einigen Sekunden und steigt nach Infusionsstop innerhalb von 10 Minuten wieder an. Dadurch lässt sich das Infusionstempo gut steuern. Anfangsdosis 0,5 µg/kg/min. Anwendung nur unter intensivmedizinischem Kreislaufmonitoring. Bei längerer Anwendung und gestörter Nierenfunktion kann es zur Thiozyanat-Intoxikation kommen.
1.4.50 Praktisches Vorgehen
Als Zielwert für die Behandlung der Hypertonie gilt allgemein ein Blutdruck von <140/90 mmHg. Bei Patienten mit Diabetes mellitus und bei niereninsuffizienten Patienten ist ein systolischer Druck von <130 mmHg und ein diastolischer Druck von <85 mmHg erforderlich. Bei Beginn der Hypertoniebehandlung sollte zur Vermeidung von Beschwerden durch abrupte Blutdrucksenkung niedrig dosiert und langsam gesteigert werden. Dabei bewährt sich das ambulante Blutdruckmonitoring.
Für die medikamentöse Hypertoniebehandlung gibt es keine allgemein anerkannten Leitlinien. Die Deutsche Hochdruckliga hat in Übereinstimmung mit der European Society of Hypertension (ESH) für den Beginn der Hochdruckbehandlung 5 Medikamentengruppen als Mittel der ersten Wahl benannt: Diuretika, β-Blocker, ACE-Hemmer, AT1-Antagonisten und Calciumantagonisten. Das US Joint National Committee (JNC) weicht von dieser Leitlinie insofern ab, als zur Einleitung der Therapie ein niedrig dosiertes Thiazid-Diuretikum empfohlen wird, das zur Vorbeugung einer Hypokaliämie mit einem kaliumsparenden Diuretikum (Amilorid oder Triampteren) kombiniert werden kann. Eine Hypertonie 1. Grades würde sich in etwa 30 % der Fälle damit einstellen lassen. Diese Basistherapie erscheint insofern plausibel, als sie den seit hundert Jahren verordneten, aber selten eingehaltenen diätetischen Kochsalzentzug mit anderen Mitteln herbeiführt.
Da Diuretika durch Natriurese die Wirkung aller anderen antihypertensiven Mittel verstärken, wird allgemein empfohlen, sie in alle Kombinationen aufzunehmen. Bei unkomplizierter Hypertonie kann das Thiazid im Fall ungenügender Wirksamkeit in niedriger Dosierung mit jedem der angegebenen Mittel kombiniert werden. Für bestimmte Begleiterkrankungen ist eine besondere Auswahl vorzuziehen:
-
Thiazid + β-Blocker: koronare Herzkrankheit, Tachyarrhythmie, Herzinsuffizienz
-
Thiazid + ACE-Blocker oder AT1-Antagonist: Herzinsuffizienz, systolische Dysfunktion, koronare Herzkrankheit, diabetische Nephropathie, Proteinurie
-
Calciumantagonisten: systolische Hypertonie im Alter, stabile Angina pectoris, periphere Gefäßkrankheit
-
dritte Komponente: ACE-Blocker oder AT1-Antagonist, Calciumantagonist oder β-Blocker, wenn diese in der Zweierkombination nicht enthalten sind.
1.4.51 Nierennervenablation
Bei dieser ganz neuen Methode werden die in den Nierenarterien verlaufenden Sympathikusfasern per Ablationskatheter mit Radiofrequenzenergie (5 – -8 Watt) ausgeschaltet. Indiziert und sehr erfolgreich ist der Eingriff bei Patienten mit therapieresistenter Hypertonie unter 3 Medikamenten. Der Katheter wird über die Leistenarterie eingeführt und unter Röntgenkontrolle in die Nierenarterie vorgeschoben. Danach erfolgt beiderseits an 4–6 Stellen nach Passage der Intima die Hitzekoagulation. Die Prozedur dauert ca. 1 h. Die Patienten erhalten eine Sedierung und ein Schmerzmittel. Sie vertragen den Eingriff gut. Medikamente sind anschließend weiter einzunehmen, doch sinkt der Blutdruck jetzt durchschnittlich um 30/15 mmHg. Nach 2 Jahren dauerte die Wirkung an. Langzeitbeobachtungen stehen noch aus.
1.4.52 Spezielle Therapie
1.4.53 Phäochromozytom
-
Medikamentöse Therapie: Notwendig ist eine 10- bis 14-tägige Operationsvorbereitung mit Phenoxybenzamin zur anhaltenden α-Rezeptorenblockade. Im Gegensatz zu den kompetitiven selektiven α1-Rezeptor-Antagonisten (▶ oben) blockiert Phenoxybenzamin die adrenergen α-Rezeptoren irreversibel. Es dauert einige Tage, bis sich neue Rezeptoren gebildet haben. Man beginnt mit 10 mg p. o. alle 12 h und steigert die Dosis um 10–20 mg/Tag. Blutdruckkrisen können mit Diazoxid oder Nitroprussidnatrium abgefangen werden. β-Rezeptorenblocker sind initial streng kontraindiziert. Nach Eintritt der α-Blockade sind bei Tachykardien niedrige Dosen indiziert. Präoperataiv bedürfen die Patienten auch gesteigerter Salz- und Flüssigkeitszufuhr, um die meistens bestehende Hypovolämie (mit Orthostasetendenz) auszugleichen.
Inoperable Phäochromozytome werden langfristig mit α-Blockade und wenn nötig mit Metyrosin behandelt, das die Katecholaminsynthese im Tumorgewebe hemmt. Eine erfolgversprechende Chemotherapie ist nicht bekannt.
-
Chirurgische Therapie: Tumorresektion laparoskopisch oder konventionell nach Unterbindung der Nebennierenvene. Postoperativ sind bei benignem Tumor 95 % der Patienten mit Blutdruckkrisen und 70 % der Patienten mit kontinuierlicher Hypertonie normotensiv.
1.4.54 Aortenisthmusstenose
Standard ist die operative Therapie im Vorschulalter mit Resektion der Isthmusstenose und End-zu-End-Anastomose der Aorta. Eine Alternative zur Operation ist die Ballondilatation mit Implantation einer Gefäßstütze (◘ Abb. 1.11).


Angiographie einer hochgradigen postduktalen Aortenisthmusstenose eines 26-jährigen Patienten vor und nach Implantation einer Gefäßstütze (Stent) mit Reduktion des Druckgradienten von 40 auf 1 mmHg
1.4.55 Renovaskuläre Hypertonie
-
Medikamentöse Therapie: Bei einseitiger Nierenarterienstenose initial ACE-Blocker oder Angiotensin-II-Rezeptor-Antagonisten unter Kontrolle des Serumkreatinins. Viele Fälle lassen sich damit befriedigend einstellen. Bei arteriosklerotischen Stenosen rigorose Behandlung der atherogenen Risikofaktoren. Bei doppelseitiger Nierenarterienstenose sind ACE-Blocker streng kontraindiziert, da sie zur Niereninsuffizienz führen.
-
Perkutane Revaskularisierung: PTA mittels Ballondilatation und nötigenfalls Stentimplantation (◘ Abb. 1.12). Indikationen: Fibromuskuläre Dysplasie mit asymmetrischem Perfusionsdefekt, arteriosklerotische Ostiumstenosen mit refraktärer Hypertonie oder drohendem Nierenfunktionsverlust infolge ischämischer Nephropathie (pathologische Captopril-Nierensequenzszintigraphie). Erfolgsquote bei fibromuskulärer Dysplasie 80–100 % mit 10–11 % Rezidiven, bei arteriosklerotischer Stenose 60–80 % mit 10–47 % Rezidiven. (Solange die Hypertonie medikamentös zu beherrschen und die Nierenfunktion normal ist, besteht bei arteriosklerotischen Stenosen keine zwingende Notwendigkeit zur Revaskularisierung, zumal das Komplikationsrisiko mit dem Alter zunimmt).
Abb. 1.12 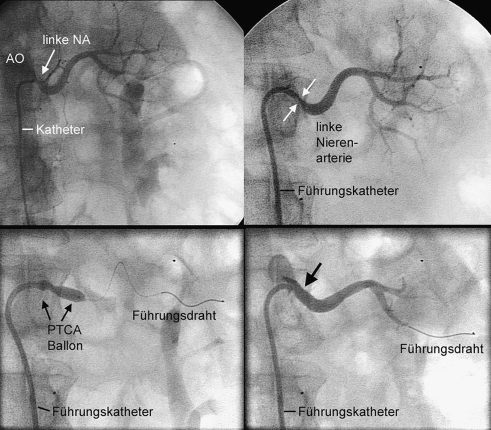
Ballondilatation (PTA) einer symptomatischen hochgradigen linksseitigen Nierenarterienstenose bei einem 65-jährigen Patienten mit schwerer Hypertonie
-
Operative Revaskularisierung: Indiziert bei langstreckigen Stenosen, Nierenarterienaneurysmen und bei Versagen der PTA.
-
Methoden: Transaortale TEA, aortorenaler Bypass oder Bypass zu einer Viszeralarterie. Operationsletalität 1–2 %. Normalisierung der Hypertonie in 59 %, Verbesserung in 33 % der Fälle. Die Niere kann bei 90 % in guter Funktion erhalten werden.


1.4.56 Schwangerschaftshypertonie
-
Vorbestehende Hypertonie: Erhöht das Risiko einer Präeklampsie. Bei Blutdruckwerten bis 150/100 mmHg Therapie absetzen, körperliche Aktivität drastisch reduzieren. Bei Blutdruck >150/100 mmHg im 2. Trimester Umstellung der Therapie auf α-Methyldopa, nötigenfalls zusätzlich Dihydralazin. Beide Medikamente schädigen den Feten nachweislich nicht. β1-Rezeptorblocker weniger unbedenklich, sollen vor der Entbindung abgesetzt werden. Keine Diuretika, da sie das effektive Blutvolumen der Mutter reduzieren und das Wachstum des Feten verzögern. ACE-Blocker sind im 2. und 3. Trimester kontraindiziert, weil im Tierversuch Fehlbildungen auftraten. Nifedipin im ersten Trimenon kontraindiziert, in der Spätschwangerschaft für kurzfristigen Einsatz bei Blutdruckspitzen zulässig.
-
Präeklampsie und Eklampsie: Klinikeinweisung. Strenge Bettruhe, normale Salz- und gesteigerte Flüssigkeitszufuhr, Linksseitenlage zur Steigerung der Diurese. In schweren Fällen Infusionen mit Ringer-Lösung und Magnesiumsulfat (4 g langsam i. v. in 15 Minuten, danach 1–3 g/h per Infusionspumpe) zur Dämpfung der Hyperreflexie und Blutdrucksenkung. Nötigenfalls Dihydralazin infundieren. Bei ausbleibender Diurese 10–20 mg Furosemid i. v. Bei Eklampsie die gleiche Therapie, ergänzt durch Valium gegen die Krämpfe. Nach Stabilisierung der Patientin unverzügliche Entbindung. Anschließend 1–2-wöchige stationäre Nachbehandlung. Etwa 25 % der Fälle von Eklampsie treten in den ersten Stunden post partum auf.
1.4.57 Hypertonische Notfälle
-
Blutdruckspitzen bei ambulanter Hypertoniebehandlung: Nifedipin-Kapsel 5–10 mg zerbeißen. Wegen reflektorischer Sympathikusaktivierung nicht bei Patienten mit koronarer Herzkrankheit anwenden. Oft genügt hier eine Kapsel oder ein Spray mit Nitroglycerin.
-
Blutdruckkrisen bei maligner Hypertonie, hypertonischer Enzephalopathie und Phäochromozytom: Sofortige Blutdrucksenkung in der Praxis mit Diazoxid (50–100 mg in 10–15 Minuten i. v.) plus Furosemid 40–80 mg i. v., in der Klinik mit Nitroprussidnatrium-Infusion bis zu einer Thiocyanatkonzentration von maximal 12 mg/dl unter intensivmedizinischem Monitoring.
-
Blutdruckkrisen bei Linksherzinsuffizienz mit Lungenödem, instabiler Angina pectoris oder Herzinfarkt: Bei Linksherzinsuffizienz sind Furosemid i. v., Nitroglycerin i. v., ACE-Blocker i. v. und Sedierung einzusetzen, nur selten ist eine Infusion mit Nitroprussidnatrium erforderlich.
1.5 Krankheiten der peripheren Arterien
1.5.1 Klassifizierung
Klassifizierung der Krankheiten der peripheren Arterien
-
Degenerative Arterienerkrankungen
-
atheromatöse Arteriosklerose (Atherosklerose)
-
nichtatheromatöse Arteriosklerose
-
Altersveränderungen der Arterien
-
Mediaverkalkung (Mönckeberg-Sklerose)
-
Arteriolosklerose
-
-
-
Entzündliche Arterienerkrankungen
-
Thrombangiitis obliterans (Morbus Winiwarter-Buerger)
-
Aortoarteriitis (Morbus Takayasu)
-
Periiarteriitis nodosa (Polyarteriitis nodosa)
-
Arteriitis temporalis (cranialis) – Polymyalgia rheumatica
-
-
Mechanische Arterienläsionen und -verschlüsse
-
Kompressionssyndrome
-
Kompressionssyndrom der A. poplitea
-
Kompressionssyndrom der A. brachialis
-
neurovaskuläre Schultergürtelsyndrome
-
-
Arterienverschlüsse durch stumpfe Traumen
-
intravasale Verschlüsse
-
arterielle Thrombose
-
Embolien
-
-
-
Funktionelle arterielle Durchblutungsstörungen
-
Raynaud-Krankheit (primäres Raynaud-Phänomen) und Digitus mortuus
-
Raynaud-Syndrom (sekundäres Raynaud-Phänomen)
-
Livedo reticularis
-
Akrozyanose
-
1.5.1.1 Degenerative Arterienerkrankungen
Unter dem Begriff Arteriosklerose zusammengefasste nichtentzündliche Arterienerkrankungen, die sich makroskopisch in Schlängelung, Verdickung, Verhärtung und Lumeneinengung, seltener in aneurysmatischer Erweiterung äußern. Man unterscheidet 2 Formen, die nebeneinander bestehen können.
1.5.1.1.1 Atheromatöse Arteriosklerose (Atherosklerose )
1.5.1.1.1.1 Erkrankung der mittleren und großen Arterien
Gekennzeichnet durch herdförmig auftretende fibröse Intimaplaques mit zentraler Nekrose und Cholesterinablagerungen.
1.5.1.1.1.2 Komplikationen
Einengung des Lumens bis zum Verschluss, Kalzifizierung, Ulzeration mit sekundärer Thrombose, von der Mikroembolien ausgehen können, Arterienruptur.
1.5.1.1.1.3 Pathogenese
Beteiligt sind 3 Zelltypen: Endothelzellen, glatte Muskelzellen und inflammatorische mononukleäre Zellen (Makrophagen, Lymphozyten). Initial erfolgt eine fokale Anheftung zirkulierender Monozyten an die Endotheloberfläche. Welche Faktoren das bewirken, ist nicht genau geklärt. In erster Linie werden oxidierte oder modifizierte Lipoproteine (LDL) verantwortlich gemacht, die sich infolge Hyperlipidämie subendothelial an Proteoglykanen abgelagert haben. Aber auch Areale mit Mikroläsionen des Endothels könnten Monozyten binden. Die Monozyten induzieren an der Endotheloberfläche die Expression von Adhäsionsmolekülen (VCAM-1), die es ihnen ermöglichen, zwischen den Endothelzellen in den subendothelialen Spalt einzuwandern. Dort differenzieren sie zu Makrophagen aus. Auch LDL und andere atherogene Lipoproteine dringen in diesen Spalt ein, wo sie von der Matrix festgehalten und oxidiert oder chemisch modifiziert werden. Die Lipide werden dann von den Makrophagen phagozytiert. Dabei gehen die Makrophagen in Schaumzellen über. Zugleich werden sie zur Sekretion von Wachstumsfaktoren (Mediatoren für Zellproliferation und Chemotaxis) und Zytokinen (Mediatoren für Entzündung und Immunität) aktiviert. Durch Wachstumsfaktoren werden die wenigen glatten Muskelzellen in der Intima zur Proliferation und Sekretion von Matrix (Kollagen, elastische Fasern) stimuliert. In der Elastica interna entstehen Defekte, durch die weitere glatte Muskelzellen in die Intima gelangen. Durch Proliferation und Matrixproduktion vergrößern sie die fibröse Läsion, die sich allmählich in das Gefäßlumen vorwölbt. Im weiteren Verlauf kommt es zur Nekrose von Schaumzellen und zum Untergang des Endothels über der Läsion, die damit thrombogen wird und wandständige Thromben entstehen lässt. In diesem fortgeschrittenen Stadium wird die Läsion von T-Lymphozyten infiltriert, die möglicherweise eine Autoimmunreaktion induzieren. Zuletzt rupturiert der atheromatöse Plaque, worauf an seiner Oberfläche ein frischer Thrombus entsteht, der das Gefäßlumen partiell oder total verschließt. Er kann auch als Embolus abgeschwemmt werden.
Die Hauptrisikofaktoren der Atherogenese sind Zigarettenrauchen, Diabetes mellitus, Hypertonie, Hypercholesterinämie und Hyperhomozysteinämie.
1.5.1.1.2 Nichtatheromatöse Arteriosklerose
1.5.1.1.2.1 Altersveränderungen der Arterien
Langsam progrediente, diffuse Zunahme des Bindegewebes und der glatten Muskelzellen in der Media, Elastizitätsverlust, Steigerung der Gefäßrigidität.
1.5.1.1.2.2 Mediaverkalkung (Mönckeberg-Sklerose)
Fokale Kalzifizierung der Media nach Degeneration glatter Muskelfasern ohne Einengung des Gefäßlumens. Alterserscheinung, akzeleriert bei Diabetes mellitus und Langzeitbehandlung mit Glukokortikoiden.
1.5.1.1.2.3 Arteriolosklerose
Erkrankung kleiner Arterien und Arteriolen, gekennzeichnet durch Hyalinisierung von Intima und Media, die zur Okklusion führen kann. Vorkommen bei Hypertonie. In schweren Fällen fibröse und elastische Hyperplasie. Bei maligner Hypertonie Nekrose der Intima und Media. Endothelproliferation bei diabetischer Mikroangiopathie.
1.5.1.2 Entzündliche Arterienerkrankungen
Die Entzündungsprozesse sind außer bei der Aortitis luetica immunologischer oder autoimmunologischer Natur. Einige Formen gehen mit Fieber und erheblichen Allgemeinerscheinungen einher.
-
Thrombangiitis obliterans (Morbus Winiwarter-Buerger ): Nur bei Rauchern vorkommende Entzündung kleiner und mittlerer Arterien mit sekundärer Thrombose und charakteristischer Mitbeteiligung oberflächlicher Venen.
-
Wegener-Granulomatose
-
Periarteriitis nodosa (Polyarteriitis nodosa)
-
Riesenzellarteiitis (temporale und aortale Form; ▶ Kap. 8.4.9 und 8.4.10)
-
Takayasu-Arteriitis (▶ Abschn. 1.5.7)
1.5.1.3 Mechanische Arterienläsionen und -verschlüsse
1.5.1.3.1 Kompressionssyndrome
Arterienkompression durch Muskeln oder Knochen, die durch Wandschädigung zur Stenosierung des Lumens und Thrombose führen kann.
1.5.1.3.1.1 Kompressionssyndrom der A. poplitea
Entsteht durch den Gastroknemius bei abnormem Gefäßverlauf oder Muskelansatz.
1.5.1.3.1.2 Kompressionssyndrom der A. brachialis
Quetschung durch einen kräftigen Lacertus fibrosus (Nebensehne des Bizeps).
1.5.1.3.1.3 Neurovaskuläre Schultergürtelsyndrom e
Intermittierende oder dauernde Kompression der A. subclavia und des Plexus brachialis:
-
Skalenus-anterior-Syndrom: Einengung der Skalenuslücke durch Muskelhypertrophie oder Ödem.
-
Halsrippe: In 5 % der Fälle komprimierend.
-
Kostoklavikular-Syndrom: Kompression zwischen Klavikula und erster Rippe
-
Pektoralis-minor-Syndrom: Kompression durch hypertrophierten M. pectoralis minor.
1.5.1.3.2 Arterienverschlüsse durch stumpfe Traumen
Durch Intimaruptur mit sekundärer Thrombose, oft nach längerem Intervall. Prädilektionsstelle A. iliaca externa (Traumen der Leistenregion). Hypothenar-Hammer-Syndrom: Schädigung des ulnaren Hohlhandarkus durch Hammerschläge mit der Hand. Akrale Verschlusskrankheit durch chronisches Vibrationstrauma der Hände.
1.5.1.3.3 Intravasale Verschlüsse
-
Arterielle Thrombose n
-
auf dem Boden einer Intimaschädigung (Atherom, Entzündung)
-
bei intravaskulärer Gerinnung oder Plättchenaggregation (Thrombozythämie)
-
bei Kälteagglutininkrankheit
-
-
Embolie n
-
durch verschleppte Thromben aus dem Herzen, aus großen Arterien, bei offenem Foramen ovale aus Venen
-
Mikroembolien aus Atheromen (Gerinnsel, Cholesterinkristalle)
-
1.5.1.4 Funktionelle arterielle Durchblutungsstörungen
Vasospastische Erkrankungen der kleinen Arterien und Arteriolen mit oder ohne organische Gefäßläsionen. Bei zunächst rein funktionellen Spasmen sekundäre Gefäßveränderungen im Verlauf möglich.
1.5.1.4.1 Raynaud-Krankheit (primäres Raynaud-Phänomen ) und Digitus mortuus
Intermittierende Spasmen der Digitalarterien auf Kältereiz ohne primäre Gefäßläsionen (◘ Abb. 1.13).


Primäres vasospastisches Raynaud-Phänomen. Typischerweise sind die Daumen nicht beteiligt. Es handelt sich um einen durch Kältereiz oder emotional ausgelösten Spasmus der Digitalarterien. Meist dreiphasiger („trikolorer“) Verlauf. a Weißfärbung (Entleerung des Kapillarbettes). b zyanotische Verfärbung der Akren (Kapillaren und Venolen erweitern sich). c Rotverfärbung/reaktive Hyperämie (Arterienspasmus löst sich)
1.5.1.4.2 Raynaud-Syndrom (sekundäres Raynaud-Phänomen )
Spasmen vorgeschädigter Digitalarterien, die in einen kompletten Gefäßverschluss übergehen können. Vorkommen: Thrombangiitis obliterans, Thrombozythämie, Kollagenosen, Mikroembolien, Erfrierungen, Traumen etc.
1.5.1.4.3 Livedo reticularis
Netzförmige livide Hautverfärbung durch spastische Verengung der Hautarteriolen. Livedoide Vaskulitis bei organischen Arteriolenverschlüssen.
1.5.1.4.4 Akrozyanose
Blaurote Verfärbung der Akren durch Engstellung der Arteriolen und Erweiterung von Kapillaren und Venolen mit erhöhter peripherer O2-Ausschöpfung.
1.5.2 Untersuchungsmethoden
Übersicht der Untersuchungsmethoden
-
Klinische Untersuchung
-
Hautfarbe und -temperatur
-
Trophische Störungen
-
Palpatation und Auskultation der Arterien
-
-
Einfache Funktionsprüfungen
-
Lagerungsprobe (Ratschow)
-
Faustschlussprobe
-
Gehprobe
-
Ergometrie
-
-
Oszillographie
-
Plethysmographie:
-
Venenverschlussplethysmographie
-
Quantitative Segmentplethysmographie
-
-
Sonographie (B-Bild-Darstellung)
-
Doppler- und Duplexsonographie
-
radiologische Gefäßdarstellung:
-
Intraarterielle digitale Substraktionsangiographie (DSA)
-
Computertomographische Angiographie (CTA)
-
-
Magnetresonanzangiographie (MRA)
1.5.2.1 Klinische Untersuchung
1.5.2.1.1 Hautfarbe
Blass, rot oder zyanotisch, bei herabgesetzter Hauttemperatur Zeichen der Ischämie. Warme zyanotische Haut bei Stauung.
1.5.2.1.2 Hauttemperatur
Orientierende Prüfung mit dem Handrücken und Seitenvergleich.
1.5.2.1.3 Trophische Störungen
Atrophie, Nekrosen, Ulzera, Gangrän.
1.5.2.1.4 Palpation und Auskultation der Arterien
Routinemäßige Ausführung an allen typischen Stellen. Pathologische Befunde auch beim Fehlen von Beschwerden möglich. Abschwächung oder Fehlen des Pulses bei Stenose bzw. Gefässverschluss. Verstärkte Pulsation bei Aneurysmen. Pulssynchrone Geräusche bei Arterienstenosen (Gefäße nicht mit dem Stethoskop komprimieren).
1.5.2.2 Einfache Funktionsprüfungen
1.5.2.2.1 Lagerungsprobe (Ratschow)
Patient hebt in Rückenlage die gestreckten Beine möglichst senkrecht hoch, stützt die Oberschenkel mit den Händen und führt mit den Füßen für 3–5 Minuten bzw. bis zum Schmerzeintritt Roll- oder Pedalbewegungen aus. Danach setzt er sich an den Bettrand und lässt die Füße frei herabhängen. Beobachtet werden Fußrücken und Fußsohlen. Ischämiezeichen: Abblassen der Haut nach Hochlagerung und während der Fußbewegungen, Bewegungsschmerz. Verzögerung der reaktiven Hyperämie (normal nach 3–5 s) und der Venenauffüllung (normal nach 20 s) im Sitzen. Bei Varikosis ist die Venenwiederauffüllungszeit nicht verwertbar.
1.5.2.2.2 Faustschlussprobe
30 kräftige Faustschlüsse in einer Minute mit erhobenen Armen. Danach Öffnen der hochgehaltenen Hände ohne Anspannung der Handflächen. Bei Durchblutungsinsuffizienz rasche Ermüdung und Abblassen von Fingern und Handteller sowie Verzögerung der reaktiven Hyperämie auf der betroffenen Seite.
1.5.2.2.3 Gehprobe
Der Untersucher begleitet den Patienten mit einer Geschwindigkeit von 120 Schritten/min auf einem längeren Korridor oder im Freien und registriert die Wegstrecke bis zum Auftreten des Claudicatio-Schmerzes.
1.5.2.2.4 Ergometrie
Definierte Belastung mit dem Fahrradergometer oder mit dem Laufbandergometer (Gehgeschwindigkeiten: 3,2 km/Std, 5 km/Std = 120 Schritte/min, 6 km/Std; Anstiegwinkel: 12,5°).
1.5.2.3 Oszillographie
1.5.2.3.1 Methode
Aufzeichnung der peripheren Arterienpulse zur Objektivierung und Erweiterung des Palpationsbefundes. Das in der Systole unter Druckanstieg in die proximale Aorta ausgeworfene Blut drängt das weiter distal gelegene Blut in das arterielle System, wodurch sich die Druckwelle schnell in die Peripherie fortpflanzt. (Die Fortleitungsgeschwindigkeit der Druckwelle in der Aorta ist mit 3–5 m/s 15-mal schneller als die Flussgeschwindigkeit des Blutes.) Der Oszillograph registriert die pulssynchronen Änderungen des Extremitätenumfanges (nicht Arteriendruck und Blutfluss), die distal von Stenosen (besonders nach Belastungen mit poststenotischem Druckabfall) abgeschwächt oder aufgehoben sind. Die Pulsabnehmer (mechanische oder elektronische) werden zur Verstärkung der Ausschläge unter dosiertem Druck (aufblasbare Gummimanschetten) angelegt. Registrierung unter stufenweiser Senkung des Manschettendrucks oder bei konstant niedrigem Manschettendruck (60 mmHg). Registrierstellen: Distaler Oberschenkel, proximaler und distaler Unterschenkel, Fuß; entsprechende Partien an den oberen Extremitäten. Akrale Oszillographie an Fingern und Zehen mit elektronischem Pulsabnehmer.
1.5.2.3.2 Normalbefunde
Seitendifferenz der Amplituden unter 30 %. An Ober- und Unterschenkeln etwa gleich große, an den Füßen wesentlich kleinere Ausschläge. Maximale Amplitude (oszillometrischer Index) nicht standardisierbar. Nach Arbeitsbelastung Amplitudenzunahme (positive Reaktion) oder gleichbleibende Amplitude. Die Pulskurve (nur im elektronischen Oszillogramm zu beurteilen) zeigt einen steil ansteigendem Schenkel und eine dikrote Welle (Eigenschwingung der Arterie) im langsam abfallenden Schenkel.
1.5.2.3.3 Pathologische Befunde
Seitendifferenz der Amplituden über 30 %. Abnorme Amplitudenverkleinerung im Längsschnittprofil. Nach Arbeitsbelastung (Zehenstand, Kniebeugen, Liegestütz) Amplitudenverkleinerung bis zum Verschwinden mit Erholungszeiten von einigen Minuten (je nach Schweregrad der Durchblutungsstörung). Pulskurve zeigt integrierten oder „Stenosepuls“ mit erniedrigtem abgerundeten Gipfel und Verlust der dikroten Nachwelle.
1.5.2.4 Plethysmographie
1.5.2.4.1 Venenverschlussplethysmographie
Dient der Messung des arteriellen Blutflusses (ml/min) in peripheren Gliedmassenabschnitten.
1.5.2.4.1.1 Prinzip
Blockierung des venösen Rückflusses mittels Blutdruckmanschette (subdiastolische Druckwerte); Registrierung der Volumenzunahme, die der ungehinderte arterielle Einstrom im gestauten Gliedmaßenabschnitt bewirkt, mittels luftgefüllter Manschetten oder Dehnungsmeßstreifen (Dauer der Blockierung 5–10 s).
1.5.2.4.1.2 Anwendung
Messung der Ruhedurchblutung und der reaktiven Hyperämie nach arterieller Drosselung zur Beurteilung der funktionellen Kapazität der peripheren Arterien.
1.5.2.4.2 Quantitative Segmentplethysmographie
Registrierung der Pulsvolumenkurven in verschiedenen Extremitätenabschnitten beider Seiten.
1.5.2.4.2.1 Prinzip
Luftgefüllte Manschetten (Druck 65 mmHg) übertragen die pulssynchronen Volumenschwankungen des darunter liegenden Gewebes auf das Messgerät (Pulsvolumen-Rekorder nach Raines). Messung in ml oder standardisierter Amplitudenhöhe.
1.5.2.4.2.2 Anwendung
Nachweis und Lokalisation von Stenosen und Verschlüssen, deutliche Abweichungen oft erst nach Belastung. Auch zur Druckmessung geeignet.
1.5.2.5 Sonographie
Das Verfahren beinhaltet die Gefäßdarstellung mit reflektiertem Ultraschall.
1.5.2.5.1 Prinzip
Ultraschallköpfe besitzen Keramikelemente, die nach Anlegung einer elektrischen Wechselspannung Druckwellen aussenden (piezoelektrischer Effekt) und reflektierte Druckwellen in elektrische Spannung transformieren. Die Reflexion erfolgt an Grenzflächen zwischen zwei Gewebsschichten unterschiedlicher akustischer Dichte. Verwendet werden Frequenzen von 2,0–15 MHz, die außerhalb des hörbaren Bereichs (0–20 kHz) liegen und deshalb als Ultraschall bezeichnet werden. Die Tiefe, aus der das Echo kommt, wird von einem Rechner aus der Zeit zwischen Aussendung und Rückkehr des Schallimpulses ermittelt.
1.5.2.5.2 B-Bild-Darstellung
1.5.2.5.2.1 Methode
Zur Gewinnung eines zweidimensionalen Schnittbildes wird der Schallstrahl von Impuls zu Impuls mit insgesamt 64–256 Schallinien durch einen Gewebesektor geschwenkt (5–20 Linien/cm). Die von den Echos produzierten elektrischen Impulse werden vom Gerät in punktförmige Flecke umgewandelt, deren Helligkeit (Brightness) der Intensität der Echos proportional ist und in 64–128 Graustufen wiedergegeben wird.
1.5.2.5.2.2 Befunde
Erlaubt die Messung der Intima-Media-Dicke, die Erkennung von Wandauflagerungen und Aneurysmen, die Feststellung von Stenosegraden aber nur ungenau.
1.5.2.6 Doppler - und Duplexsonographie
Es handelt sich um Methoden zur Strömungsanalyse des Blutes. Mit einer farbkodierten Duplexsonographie kann strömendes Blut und damit auch das durchströmte Gefäß farblich dargestellt werden.
1.5.2.6.1 Prinzip
Die Verfahren der Dopplersonographie basieren auf dem Phänomen, dass Schallwellen von einem bewegten Reflektor mit veränderter Frequenz reflektiert werden. Bewegung auf die Schallquelle zu erhöht die Frequenz des reflektierten Schalls, Bewegung von der Schallquelle weg erniedrigt sie (Doppler-Effekt). Bei der Untersuchung der Blutströmung sind die Erythrozyten die bewegten Reflektoren. Die vom Schallkopf ausgesandten Frequenzen betragen 4 oder 8 MHz. Die Frequenzdifferenz (Dopplerfrequenz) liegt im hörbaren Bereich und wird vom Dopplergerät akustisch wiedergegeben oder graphisch dargestellt. Dabei werden vom flektierten Schall nur die Dopplerfrequenzen ausgewertet. Deren Höhe gibt die Flussgeschwindigkeit wieder.
1.5.2.6.2 Dopplersysteme
1.5.2.6.2.1 Continuous-wave-Doppler (CW)
Der Liniendoppler hat einen Quarzkristall, der kontinuierlich sendet und einen zweiten Kristall, der die reflektierten Schallwellen kontinuierlich empfängt. Entlang des Schallstrahles werden alle Flussgeschwindigkeiten erfasst (◘ Abb. 1.14a).


Die drei Systeme der Doppler-Sonographie: a Linien-Doppler: Entlang eines Schallstrahles werden alle Flussgeschwindigkeiten erfasst. b Punkt-Doppler: Flussgeschwindigkeiten werden nur an einem Punkt gemessen (sample volume = Messvolumen). c Flächen-Doppler: Die durch viele Messvolumina (sample volume) bestimmten mittleren Flussgeschwindigkeiten werden im gewohnten 2-D-Sektor-Bild farbkodiert wiedergegeben und liefert in Echtzeit Informationen über Richtung und Qualität des Blutflusses sowie die Ausbreitung
1.5.2.6.2.2 Gepulster Doppler (PW)
Der Punktdoppler hat nur einen Kristall, der abwechselnd sendet und empfängt. In einem bestimmten zeitlichen Abstand vom gesendeten Impuls werden also selektiv Signale aus einem entsprechenden Tiefenbereich (sample volume) empfangen (◘ Abb. 1.14b). Korrekt registriert werden Dopplerfrequenzen nur bis zur halben Impulswiederholungsfrequenz (Nyquist-Grenze). Dopplerfrequenzen bzw. Flussgeschwindigkeiten oberhalb der Nyquist-Grenze werden in der Gegenrichtung dargestellt (Alias-Phänomen), sofern man die Nulllinie nicht verschiebt.
1.5.2.6.2.3 Farbdoppler
Flächendoppler (◘ Abb. 1.14c), der mit einem gepulsten Verfahren die Dopplersignale aus mehreren Messvolumina empfängt und als mittlere Flussgeschwindigkeit farbkodiert wiedergibt. Strömungen, die auf den Schallkopf zufließen, werden in der Regel rot, bei hohen Geschwindigkeiten hellrot bis gelb kodiert, Strömungen vom Schallkopf weg dagegen blau, bei höheren Geschwindigkeiten hellblau bis türkis. Wenn die Nyquist-Grenze überschritten wird, kommt es zum Alias-Phänomen mit Umschlag in den hellsten Farbton der Gegenrichtung, beispielsweise von orangegelb nach türkisblau. Ein Farbumschlag erfolgt auch bei Turbulenzen. Der Farbdoppler wird stets einem B-Bild zugeschaltet und macht darin die Gefäße sichtbar.
1.5.2.6.3 Dopplerdruckmessung en
Vergleichende Bestimmung des systolischen Drucks an den Oberarmen (Riva-Rocci-Methode) und an den Knöchelarterien (mittels Dopplersonde). Eine Dopplersonde (8 MHz) wird über der A. tibialis anterior auf dem Fußrücken im Winkel von 60° mit Kontaktgel angelegt, die Blutdruckmanschette (13 cm) am Unterschenkel, 10 cm oberhalb des Knöchels. Messpunkt für den systolischen Druck ist der Beginn des akustischen Strömungssignals. Normalbefund: Systolischer Knöchelarteriendruck 10-20 mmHg höher als systolischer Oberarmarteriendruck. Entsprechend beträgt der normale Dopplerindex (Quotient aus Knöchelarteriendruck und Oberarmarteriendruck) ≥1.
Dopplerindizes von 0,7–1,0 weisen auf Arterienstenosen oder gut kollateralisierte Verschlüsse hin. Ein Knöchelarteriendruck <60 mmHg zeigt einen schlecht kompensierten Arterienverschluss mit kritischer Ischämie an, ein Druckwert von 40 mmHg eine vitale Gefährdung des Beines. Arterienstenosen von 50–75 % lassen sich mit der Druckmessung in Ruhe schwer erfassen. Nach Belastung sinkt der Dopplerindex jedoch deutlich ab.
1.5.2.6.4 Strömungskurven mit der Doppler-Stiftsonde
Als Strömungssignale empfängt die Sonde aus der Arterie Dopplerfrequenzen, deren Höhen der Flussgeschwindigkeit des Blutes proportional sind. Wenn der Winkel der Sonde zur Gefäßachse bekannt ist (Duplexsonographie), zeigt das Gerät auf der Ordinate statt der Dopplerfrequenz die Flussgeschwindigkeit an. Normale Strömungskurve: Steiler systolischer Anstieg, systolischer Peak, frühdiastolische Rückflusskomponente, spätdiastolischer Vorwärtsfluss, präsystolischer Nullfluss.
1.5.2.6.4.1 Analogkurven
Bei der Aufzeichnung der Strömungssignale ermittelt das Gerät zu jedem Zeitpunkt die am häufigsten vorkommende Frequenz (Modal-Frequenz), die annähernd dem arithmetischen Mittelwert aller gemessenen Frequenzen entspricht (◘ Abb. 1.15).


Doppleranalogkurve der A. poplitea, die eine Etage unterhalb einer Arterienstenose abgeleitet wurde. Sie zeigt im Vergleich zur gesunden Seite eine Abnahme der systolischen Amplitude sowie die Zunahme der systolischen Signalbreite, der Dip fehlt. Aufgrund der guten Kompensation der Durchblutungsstörung liegt die enddiastolische Frequenz auf der Nulllinie
1.5.2.6.4.2 Frequenzspektrum
Die Frequenzanalyse ist mit dem CW- und PW-Doppler möglich. Zu jedem Zeitpunkt werden alle empfangenen Dopplerfrequenzen aufgezeichnet. Die Linie der Spektralkurve stellt also ein Frequenzband dar, das alle Flussgeschwindigkeiten wiedergibt (◘ Abb. 1.16). Es bleibt schmal, solange im Gefäßlumen laminare Strömung mit annähernd gleicher Flussgeschwindigkeit vorherrscht. Der frequenzfreie Bereich unter der Spektralkurve wird als systolisches Fenster bezeichnet. Bei höheren Flussgeschwindigkeiten durch Belastung oder im Bereich von Stenosen steigt die Spitzenfrequenz an. Da das laminare zunehmend in ein paraboles Strömungsprofil übergeht, wird das Frequenzband breiter und die Spektralkurve biphasisch, weil der spätdiastolische Vorwärtsfluss präsystolisch nicht auf Null abfällt. Das systolische Fenster bleibt zunächst noch offen. Im Bereich hochgradiger Stenosen ist die Spitzenfrequenz maximal. Unmittelbar hinter der Enge wird die Strömung turbulent. In Wandnähe entstehen Wirbel mit Flussumkehr, die das Frequenzband stark verbreitern und das systolische Fenster schließen. Eine Etage unterhalb von über 50%igen Stenosen kommt es zu monophasischer abgeflachter Kurvenform ohne Rückflusskomponente mit offenem systolischem Fenster.


Dopplerfrequenzspektren in Abhängigkeit vom Stenosegrad (intrastenotische Ableitung im Strömungsjet). a Normalbefund, b Stenosegrad <25 %, c Stenosegrad 25–50 %, d Stenosegrad 50–75 %, e Stenose >75 %
1.5.2.6.5 Duplexsonographie
Bei diesem Verfahren werden B-Bild und Doppler kombiniert.
1.5.2.6.5.1 Schwarz-weiß-Duplexsonographie
Kombination des B-Bildes mit einem CW- oder PW-Doppler. Erlaubt die gezielte Messung der Flussgeschwindigkeit in Gefäßen und im Herzen. Aber nur der PW-Doppler hat dabei Tiefenselektivität.
1.5.2.6.5.2 Farbkodierte Duplexsonographie (FKDS)
Bei der Kombination des B-Bildes mit dem Farbdoppler erscheint das Lumen des anvisierten Gefäßabschnittes bei der üblichen Farbzuordnung in der Systole rot, in der frühen Diastole blau, in der Spätdiastole wieder rot und erlischt während des präsystolischen Nullflusses (◘ Abb. 1.17). Die Farbmarkierung erleichtert das Auffinden der Gefäße im Gewebe sehr. Wandauflagerungen sind an Aussparungen der Farbkodierung zu erkennen. Die Färbung des Gefäßlumens erleichtert vor allem den Nachweis von Stenosen und die Positionierung des PW- oder CW-Dopplers, die zur Messung der Frequenzspektren bzw. Flussgeschwindigkeiten benötigt werden. Bei hohen intrastenotischen Flussbeschleunigungen wird die Pulsalität des Farbdopplersignals aufgehoben, die Farbe heller, und der diastolische Farbumschlag bleibt aus. Zur Quantifizierung des lokalen Stenosegrades kann im Querschnitt durch Planimetrie die Relation des intrastenotischen, farbkodierten Restlumens zum originären Gefäßquerschnitt als Berechnungsgrundlage ermittelt werden.


Farbkodierte Duplexsonographie: Längsschnitt einer normalen Arteria (rot) und Vena femoralis (blau)
1.5.2.7 Radiologische Gefäßdarstellung
1.5.2.7.1 Intraarterielle digitale Subtraktionsangiographie (DSA)
Vor der Kontrastmittelinjektion wird ein Leer- oder Maskenbild erstellt, das nach der Gefäßkontrastierung von einer identischen Aufnahme computergestützt subtrahiert wird. Optimales Verfahren zur Gefäßdarstellung, vor interventionellen Eingriffen meistens unverzichtbar. Nachteile: Invasives Verfahren mit Strahlenbelastung und Komplikationsmöglichkeiten. Stellt nur das perfundierte Gefäßlumen dar und gibt wenig Aufschluss über die Gefäßwand.
1.5.2.7.2 Computertomographische Angiographie (CTA)
Erfasst Lumenweite und Wandstrukturen. Moderne Rekonstruktionsverfahren ermöglichen eine 3-D-Darstellung größerer Gefäße. Das Kontrastmittel wird intravenös injiziert. Schonendes Verfahren zur Darstellung vor allem von Hirnarterien. Nachteile: Strahlenbelastung, Möglichkeit der Kontrastmittelunverträglichkeit.
1.5.2.8 Magnetresonanzangiograpie (MRA)
Die MRA oder Kernspinangiographie ist durch Einführung schneller Sequenzen mit oder ohne Kontrastmittel (Gadolinum) zu einer schonenden, sehr guten Methode der Gefäßdarstellung geworden. Räumliche und zeitliche Auflösung und die Kontrastauflösung sind ausgezeichnet. Perfundiertes Lumen und Gefäßwandveränderungen werden sicher dargestellt. Hinsichtlich Kontrastauflösung ist die MRA der CTA eindeutig überlegen. Arterien und Venen werden gleichzeitig erfasst, dazu noch Parenchymveränderungen. Eine Strahlenbelastung entfällt. Das Kontrastmittel ist nicht jodhaltig.
1.5.3 Extremitätenarterien
1.5.3.1 Akute Verschlüsse
1.5.3.1.1 Ätiologie
Arterielle Embolie, akute Thrombose im Bereich atheromatöser Läsionen (begünstigt durch Hämokonzentration und Hyperkoagulabilität), Plättchenthromben (Thrombozytose), Arterienspasmus bei Thrombose der Begleitvene (Phlegmasia coerulea dolens), Kompression von außen, Aortendissektion.
1.5.3.1.2 Lokalisationen
-
Embolien: In abnehmender Häufigkeit Femoralisgabel, Iliakagabel, A. poplitea, A. brachialis.
-
Thrombosen: Hauptsächlich in mittel- und kleinkalibrigen Arterien des Ober- und Unterschenkels, seltener in der Aorta und den Beckenarterien. Phlegmasia coerulea dolens: Strombahnblockade im Bereich des ganzen Beines.
1.5.3.1.3 Pathophysiologie
Akute Arterienverschlüsse führen je nach Sitz und Kollateralversorgung zum vollständigen oder unvollständigen Ischämiesyndrom. Ersteres ist meistens bei Embolien der Fall. Da arterielle Thrombosen den kompletten Verschluss einer bereits bestehenden arteriosklerotisch bedingten Stenose darstellen, hat sich meistens bereits ein mehr oder weniger gut ausgeprägter Kollateralkreislauf ausgebildet. Sitz der Verschlüsse bei vollständiger Ischämie in den Hauptarterien zentral der Kniekehle und Ellenbeuge, an Unterschenkeln und Unterarmen nur, wenn kein Gefäß offen bleibt. Progredienz durch Anlagerung von Stagnationsthromben proximal und distal des Verschlusses vor allem bei Embolien. Irreversible Schädigungen der Muskulatur 4–6 h, der Haut 2 h und der peripheren Nerven 24 h nach Kreislaufstillstand.
1.5.3.1.4 Klinik
Bei Zirkulationssperre mit Druckabfall unter 40 mmHg distal der Obstruktion resultieren: intensiver Schmerz, kalte Blässe, Gefühlsstörung, Pulsverlust und Bewegungsunfähigkeit in der Gliedmaßenperipherie, ferner Erschöpfung und allgemeine Schockzeichen. Zeichen der irreversiblen Gewebeschädigung: Spannungsblasen, Gangrän, totaler Sensibilitätsverlust, verspannte und verdickte Muskelgruppen, schlaffe Paresen (nach 43 h). Blasse Ischämie bei offenen Venen, blaue Ischämie bei sekundärer Stagnationsthrombose in Kapillaren und Venen. Bei unvollständiger Ischämie oft nur Blässe, Kälte und Taubheitsgefühl in der betroffenen Extremität.
1.5.3.1.5 Diagnostik
Etagenlokalisation des Verschlusses durch Pulspalpation. Distale Dopplerdruckmessung ergibt Aufschluss über den Ischämiegrad. Bei der FKDS fehlt im Verschlussbereich das Farbdopplersignal. Kollateralenfluss erzeugt poststenotisch ein monophasisches Frequenzspektrum. Fahndung nach der Emboliequelle im Herzen mittels Echokardiographie, in der Aorta durch Abdomensonographie. Zur definitiven Abklärung wird meistens prä- oder intraoperativ eine Angiographie durchgeführt.
1.5.3.1.6 Therapie
1.5.3.1.7 Sofortmaßnahmen
Tieflagerung der Extremität mit Wattepolsterung der Auflageflächen. Hochlagerung verstärkt die Ischämie. Weder Kälte- noch Wärmeapplikation. Schmerzbekämpfung mit Morphinpräparaten. Antikoagulation mit 10.000 E Heparin gegen Appositionsthromben.
1.5.3.1.8 Revaskularisierung
-
Bei Embolie: Embolektomie mit Ballonkatheter oder Ringstripper bzw. durch Arteriotomie in Höhe des Verschlusses. Wenig belastender Eingriff, so früh wie möglich durchführen. Spätembolektomie bei inkomplettem Ischämiesyndrom noch nach 3–6 Wochen erfolgversprechend
Nach Revaskularisation besteht die Gefahr eines postischämischen Ödems mit hypovolämischem Schock.
-
Bei Thrombose: Notfalloperation (Thrombektomie, TEA) nur bei komplettem Ischämiesyndrom der unteren Extremität, bei Befall der aortoiliakalen Strombahn und des Armes. Bei infrainguinalen Verschlüssen wenn möglich perkutane Angioplastie, sonst intraarterielle Thrombolyse.
1.5.3.1.9 Konservative Maßnahmen
Lokale Thrombolyse und anschließend Heparinisierung. Indiziert bei Embolien im Bereich peripherer kleiner Arterien, bei Thrombosen mit inkompletter Ischämie oder Inoperabilität wegen schweren Schocks oder Herzinsuffizienz.
1.5.3.1.10 Prognose
Amputationsrate bei einer Operation innerhalb 12 h 3 %, innerhalb von 48 h 10 %, für die Gesamtheit der Embolien 6 %.
1.5.3.2 Chronische Verschlüsse (periphere arterielle Verschlusskrankheit )
1.5.3.2.1 Häufigkeit
Etwa 2,2 % aller Männer und 1,8 % aller Frauen sind betroffen. In einer Basler Studie an 6400 Berufstätigen trat innerhalb von 5 Jahren bei 5 % der 40-jährigen und bei 18 % der 70-jährigen Männer eine frische periphere Verschlusskrankheit auf, wobei die asymptomatische Form 3-mal häufiger war als die symptomatische. Frauen erkrankten 5-mal seltener.
1.5.3.2.2 Ätiologie
In 80–90 % der Fälle atheromatöse Arteriosklerose mit den bereits genannten Risikofaktoren, unter denen das Rauchen an erster Stelle steht. Bei peripherer Verschlusslokalisation (besonders an Unterschenkeln und Füßen) liegt in 10–20 % der Fälle eine Endangiitis obliterans vor.
1.5.3.2.3 Pathophysiologie
Je weiter zentral die Verschlusslokalisation, desto besser die Kompensationsmöglichkeit über Kollateralen, desto später auch die klinischen Symptome. Nach den funktionellen Auswirkungen unterscheidet man folgende Schweregrade der Durchblutungsstörungen:
-
Stadium I: Stenosen oder Verschlüsse ohne klinische Symptomatik
-
Stadium II: Belastungsinsuffizienz (z. B. Claudicatio intermittens)
-
Stadium III: Dauerinsuffizienz mit Ruheschmerz
-
Stadium IV: manifester anoxischer Gewebeschaden (distale Nekrosen), mit oder ohne Ruheschmerz
1.5.3.2.4 Obliteration der Becken - und Beinarterien
Mit Abstand die häufigste chronische arterielle Verschlusskrankheit. Initialsymptom ist das intermittierende Hinken (Claudicatio intermittens ) durch ischämischen Muskelschmerz und Schwäche beim Gehen.
Im Gegensatz zu neuritischen Schmerzen klingt der Claudicatio-Schmerz nach Unterbrechung der Belastung innerhalb einiger Minuten ab. Progredienz manifestiert sich in zunehmender Verkürzung der Gehstrecke, Taubheitsgefühl und Parästhesien, Ruheschmerz, Atrophie und Nekrosen an Zehen und Vorfuß. Nach der Lokalisation unterscheidet man 3 Verschlusstypen:
-
Beckentyp (aortoiliakale Verschlüsse): Betrifft kaudale Aorta, Aortenbifurkation, A. iliaca communis, A. iliaca interna, A. iliaca externa (bis zum Leistenband). Ischämischer Schmerz bevorzugt in der Gesäß- und Oberschenkelmuskulatur. Bei 20 % erektile Potenzstörung. Ruheschmerz und Nekrosen fast nur bei akuten und subakuten Verschlüssen oder bei zusätzlichen Verschlüssen weiter peripher. Stenosegeräusche in der Leistenregion. Alle Beinpulse abgeschwächt. Schweregrad ergibt sich aus Dopplerdruckmessung und FKDS. Präoperativ DSA oder Magnetresonanzangiographie zur genauen Lokalisation und Beurteilung der peripheren Ausflussbahn.
-
Oberschenkeltyp (femoropopliteale Verschlüsse): Betrifft: A. femoralis communis (vom Leistenband bis zur Femoralisgabel), A. femoralis superficialis (Leitungsarterie von der Femoralisgabel bis zum Ende des Adduktorenkanals), A. profunda femoris (von der Femoralisgabel als Versorgungsarterie in die Oberschenkelmuskulatur mit Kollateralen zum distalen Segment der A. femoralis superficialis und zum 1. und 2. Segment der A. poplitea ‒ sog. Profundakreislauf) und A. poplitea (vom Ende des Adduktorenkanals über das Kniegelenk bis zum unteren Rand des M. popliteus). Etwa 50 % der Beinarterienverschlüsse entfallen auf die A. femoralis superficialis (◘ Abb. 1.18). Gute Kompensation bei offenem Profundaabgang und offener A. poplitea über den Profundakreislauf. Ischämischer Schmerz in den Waden, Parästhesien und Kältegefühl in den Zehen. Fehlen oder starke Abschwächung des Popliteapulses und der Fußpulse bei normalem Leistenpuls. Gefäßgeräusche distal des Verschlusses (nach Kniebeugen oder Zehenstand). Schweregrad durch FKDS, Dopplerdruckmessung und Oszillographie zu erfassen. Angiographie oder MRA nur bei Operationsindikation.
Abb. 1.18 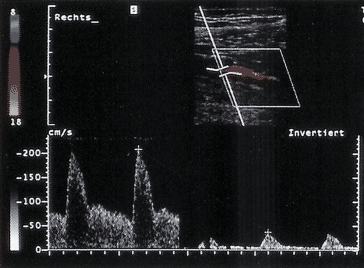
Duplexsonographie mit Farb- und CW-Doppler: Hochgradige Stenose der A. femoralis superficialis mit hohen systolischenund diastolischen Flüssen innerhalb sowie niedrigen Flüssen distal der Stenose
-
Unterschenkeltyp (Verschlüsse unterhalb der A. poplitea): Betrifft A. tibialis anterior, A. tibialis posterior und A. peronea (aus A. tibialis posterior). Obliteration von 2 Unterschenkelarterien kann durch die dritte ausgeglichen werden. Bei ungenügender Kompensation (2–3 Arterien verlegt) Belastungsschmerz der Fußgelenke oder Fußsohlen, Parästhesien und Ruheschmerz in den Zehen. Fehlender Puls der A. dorsalis pedis bzw. A. tibialis posterior bei normalem Popliteapuls. Lokalisation und Schweregrad durch FKDS, Oszillographie (auch der Zehen) und etagenweise Dopplerdruckmessung zu erfassen, ggf. durch MRA.


1.5.3.2.4.1 Kombinierte Verschlüsse
Becken-Oberschenkel-Typ und Oberschenkel-Unterschenkel-Typ kommen häufig vor. Becken-Unterschenkel-Typ und Verschlüsse aller 3 Etagen sind selten.
1.5.3.2.5 Obliteration der Armarterien
Nur etwa 10 % der chronischen Gliedmaßenarterienverschlüsse entfallen auf die oberen Extremitäten.
1.5.3.2.5.1 Schultergürteltyp (Subklavia- und Axillarisverschlüsse)
Prädilektionsstellen: Linke A. subclavia zwischen Abgang von der Aorta und Abzweigung der A. vertebralis, seltener rechte A. subclavia zwischen Abgang der A. carotis communis und A. vertebralis, noch seltener Verschlüsse distal des Vertebralisabgangs und in der A. axillaris. Bei Verschlüssen proximal des Vertebralisabgangs wenig Symptomatik, da kollateraler Zufluss über die Vertebralis erfolgt. Nachweis durch Blutdruck- und Pulsdifferenz an den Armen im Seitenvergleich. Claudicatio bei Belastung des Armes, zerebrale Ischämie bei proximalen Subklaviaverschlüssen durch Steal-Effekt (▶ Abschn. 1.5.7) mit Steal-Syndrom. Bedrohliche Ischämie nur bei akuten Verschlüssen und Kombination mit peripheren Verschlüssen. Lokalisation mittels FKDS. Präoperativ Aortoangiographie.
1.5.3.2.5.2 Oberarmtyp
Sehr selten, überwiegend entzündlich, traumatisch oder thrombotisch bedingt. Führt zur Claudicatio des Armes.
1.5.3.2.5.3 Unterarmtyp (Radialis- und Ulnarisverschluss)
Symptome nur bei Verschluss beider Unterarmarterien, am häufigsten durch Thrombangiitis obliterans bedingt. Faustschlussprobe positiv. Oszillogramm pathologisch, Pulse abgeschwächt oder fehlend. Verschlussnachweis durch FKDS und Angiographie.
1.5.3.2.5.4 Digitaler Verschlusstyp
Überwiegend bei entzündlichen Gefäßerkrankungen und Diabetes mellitus, Mikroembolien und Thrombozytose. Betroffen sind Digitalarterien, Hohlhandarkus, nicht selten auch die distalen Abschnitte von A. radialis und ulnaris.
1.5.3.2.5.5 Klinik
Anfallsweise Blass- bzw. Blauverfärbung der Finger mit ischämischen Schmerzen und Abkühlung, trophische Störungen an den Endgliedern, Entzündungen, Nekrosen, Narben, Wachstumsstörungen der Nägel, zyanotische Hautverfärbung, pathologische Faustschlussprobe.
1.5.3.2.5.6 Diagnostik
Verschlussnachweis durch MR-Angiographie mit Gadolinum, Fingeroszillogramm.
Die Gefäßspasmen auf dem Boden einer Verschlusskrankheit werden als sekundäres Raynaud-Syndrom bezeichnet und unterscheiden sich vom primären Raynaud-Syndrom (Raynaud-Krankheit) durch die in ◘ Tab. 1.3 dargestellten Kriterien.
1.5.3.2.5.7 Therapie
1.5.3.2.5.8 Konservative Therapie
Indiziert in den Stadien I und II:
-
Allgemeine Maßnahmen: Nikotinabstinenz und Ausschaltung weiterer Risikofaktoren der Arteriosklerose.
-
Gehtraining: Bis zu 75 % der Belastungstoleranz zur Verbesserung des Kollateralkreislaufs.
-
Antikoagulation: Thrombozytenaggregationshemmer, seltener Cumarine.
-
Beim Raynaud-Syndrom: Schutz vor Abkühlung, vasokonstriktorische Mittel absetzen (β-Blocker, Ergotaminderivate). Medikamente: Calciumantagonisten (Nifedipin, Diltiazem, Prazosin, Reserpin, Phenoxybenzamin).
-
Angiologische Interventionen peripherer Arterien: Häufig ist es möglich geworden, verengte periphere Arterien mit einem Ballonkatheter aufzudehnen und mit einem Stent offenzuhalten.
1.5.3.2.5.9 Chirurgische Gefäßrekonstruktion
Indiziert in den Stadien III und IV.
-
Beckentyp:
-
Langstreckige Stenosen: Aortobifemoraler Bypass (Offenheitsrate nach 5 Jahren 80–90 %), bei einseitiger Stenose iliofemoraler Crossover-Bypass.
-
Kurzstreckige Stenosen: Perkutane transluminale Angioplastie (PTA), im Bereich der A. iliaca com. mit Stenteinlage. Alternativ offene oder halbgeschlossene retrograde Iliaka-TEA. Entfernung embolisierender atheromatöser Plaques aus der Aorta abdominalis durch offene TEA mit Direktverschluss.
-
-
Oberschenkeltyp:
-
Isolierter Verschluss der A. femoralis superficialis mit offenem Profundakreislauf: Konservative Behandlung. Profundaplastik bei Abgangsstenose. Femoropoplitealer Bypass zum ersten Popliteasegment bei langstreckigem Verschluss. Kniegelenküberschreitende Bypässe in den Stadien III und IV bei Gehstrecke <20 m.
-
Kurzstreckige Stenosen oder Verschlüsse der A. femoralis superficialis: Versuch der PTA.
-
-
Unterschenkeltyp: Verschlüsse im femoropoplitealen Bereich: Überbrückung durch Bypass, möglichst auf 2 Unterschenkelarterien. Bei Beteiligung der Unterschenkelarterien Bypässe zu den Arterien in Sprunggelenkshöhe. Generell haben Venenbypässe eine bessere Prognose als Kunststoffbypässe.
1.5.3.2.5.10 Amputation en
Bei Nekrosen, die sich unter konservativer Behandlung (Reinigung mit hypertonischen und tryptischen Lösungen, Trockenföhnen, Antibiotika) nicht spontan demarkieren, bzw. die Gebrauchsfähigkeit der Gliedmaße stark einschränken. Grenzzonenamputation in der nekrobiotischen Zone mit sekundärer Stumpfheilung oder große Gliedmaßenamputation (Unterschenkel, Oberschenkel).
1.5.3.2.5.11 Schultergürtelsyndrom
Bei ausgeprägten vaskulären und nervalen Komplikationen je nach Ursache: Resektion einer Halsrippe, Entfernung narbiger Zügelungen von Gefäßen und Nerven.
1.5.3.2.5.12 Subklaviastenose
Nur in symptomatischen Fällen PTA, TEA oder Karotis-Subklavia-Bypass (◘ Abb. 1.19).


Dilatation und Stentimplantation bei symptomatischer linksseitiger Subklavia-Stenose
1.5.3.2.5.13 Sekundäres Raynaud-Syndrom
Nach Ausschöpfung der medikamentösen Therapie Sympathektomie.
1.5.4 Viszeralarterien
1.5.4.1 Akute Verschlüsse
1.5.4.1.1 Ätiologie
Arterielle Embolie und arterielle Thrombose dominieren (gleich häufig, zusammen 90 % aller Fälle). Selten: Aortenaneurysma, Aortendissektion, Kompression der Arterien von außen (Strangulation, Tumoren), Traumen.
1.5.4.1.2 Truncus coeliacus
Verlegung des Hauptstammes dank kollateraler Zuflüsse in der Regel symptomlos. Coeliaca-Ischämiesyndrom mit akutem Oberbauchschmerz, Übelkeit, sanguinolentem Erbrechen (infarzierte Magenschleimhaut) und Schock nur bei fehlendem kollateralem Strömungsausgleich. Bei Astverschlüssen partielle oder totale Organnekrosen (Leber- und Milzinfarkte). Diagnose: FKDS, Angiographie.
1.5.4.1.3 A. mesenterica superior
Hauptstammblockade bewirkt Darmnekrose im ganzen Versorgungsgebiet (Jejunum, Ileum, Kolon bis zur linken Flexur). Astverschlüsse 2. und 3. Ordnung wegen postokklusiver Brücken meistens symptomlos. Verschlüsse der terminalen Arkaden bzw. Vasa recta führen zu multiplen zirkumskripten Infarkten. Symptome des Hauptstammverschlusses:
-
Initialstadium (1.–6. Stunde): Abdominalschmerz. Zeichen des Schocks, Durchfälle, lebhafte Peristaltik.
-
Stilles Intervall (7.–12. Stunde): Erträglicher Dauerschmerz, kein auffälliger Lokalbefund, Verschlechterung des Allgemeinzustandes zuletzt Verschwinden der Darmperistaltik.
-
Endstadium (nach 12–48 h): Paralytischer Ileus, Peritonitis.
Begünstigung von embolischen Verschlüssen durch großes Gefäßkaliber und schrägen Abgang aus der Aorta. Akute Leibschmerzen von Herzkranken (Vorhofflimmern) stets embolieverdächtig. Thromboseverdacht bei protrahierter Symptomentwicklung und vorausgegangener Angina abdominalis (▶ unten).
Diagnostik: Laktatanstieg im Serum, FKDS, Angiographie, Laparotomie auch im Verdachtsfall. Ohne Operation keine Überlebenschance: Embolektomie, Thrombendarteriektomie oder Bypass zur Wiederherstellung der arteriellen Strombahn, Darmresektion im nötigen Umfang. Rapide Verschlechterung der Prognose jenseits der 6-Stunden-Grenze. Gesamtletalität 80–90 %.
1.5.4.1.4 A. mesenterica inferior
Akuter Verschluss meistens symptomlos. Bei Störungen der kollateralen Zirkulation (Verschlussprozesse der inneren Beckenarterien und der A. mesenterica superior) Nekrose des Sigma und oberen Rektum mit blutigen Stühlen und paralytischem Ileus. In weniger schweren Fällen ischämische Kolitis, narbige Stenosen im Spätstadium. Frühestmögliche Operation (Darmresektion, evtl. Revaskularisierung). Diagnostik: Mit FKDS selten möglich, in Regel durch Angiographie.
1.5.4.2 Chronische Verschlüsse
1.5.4.2.1 Angina abdominalis
Intermittierende Leibschmerzen wechselnder Lokalisation meistens nach dem Essen, in 15–20 % der Fälle auch Malabsorption mit Gewichtsverlust. Häufig Gefäßgeräusche bei der Auskultation des Abdomens. Sicherung der Diagnose durch FKDS oder Arteriographie. Erst wenn zwei der drei Viszeralarterien von Abgangsstenosen (Arteriosklerose, Endangiitis obliterans, fibromuskuläre Dysplasie) betroffen sind, treten Symptome auf. Langsame Progredienz begünstigt die Entwicklung der Kollateralen, dennoch Gefahr akuter Mesenterialinfarkte. Operative Korrektur durch TEA, Bypass oder Transsektion mit Reinsertion.
1.5.4.2.2 Kompressionssyndrom des Truncus coeliacus
Intermittierende Schmerzzustände im Oberbauch von unterschiedlicher Intensität und Dauer, selten mit Übelkeit und Erbrechen. Meistens bei jüngeren Patienten ohne Hinweis auf Gefäßleiden. Der Truncus wird vom Lig. arcuatum im Hiatus aorticus des Zwerchfells komprimiert. Nachweis mit FKDS oder Angiographie. Operative Dekompression durch Zwerchfellspaltung.
1.5.5 Krankheiten der Nierenarterien
1.5.5.1 Akute Verschlüsse
1.5.5.1.1 Ätiologie
Embolie, arterielle Thrombose, stumpfes Trauma oder Aneurysma dissecans.
1.5.5.1.2 Klinik
Es entstehen Infarkte unterschiedlicher Größe, die klinisch oft stumm verlaufen. Auch Hämaturie und Albuminurie können fehlen. Keine Retention harnpflichtiger Substanzen bei einseitigem Infarkt und intakter zweiter Niere. Klinische Symptome in etwa 50 % der Fälle: Heftiger Flanken- oder diffuser Leibschmerz, Übelkeit und Erbrechen, Leukozytose und Fieber. In allen Fällen Enzymanstieg: GOT sofort, LDH nach 1–2 Tagen, alkalische Phosphatase nach 3–5 Tagen. Niereninsuffizienz bei Doppelverschlüssen.
1.5.5.1.3 Diagnostik
Durch FKDS und Angiographie. Stumme Niere im i. v. Pyelogramm und Isotopennephrogramm. Ischämie-Toleranz-Zeit der Niere 40–50 Minuten.
1.5.5.1.4 Therapie
Bei normaler globaler Nierenfunktion komplikationsloser Verlauf unter konservativer Behandlung (Bettruhe, Analgetika, Antiemetika, Antikoagulation). Restkanal und Kollateralbahnen erklären, dass Revaskularisation innerhalb einer Woche häufig erfolgreich ist, gelegentlich noch später (15–43 Tage). Spätfolge inkompletter Verschlüsse renovaskuläre Hypertonie. Revaskulariserung in der Regel nur bei früh erkannten doppelseitigen Verschlüssen erfolgreich.
1.5.5.2 Chronische Verschlüsse
Die klinischen Manifestationen sind ischämische Nephropathie mit Nierenverkleinerung und renovaskuläre Hypertonie (▶ Abschn. 1.4 und ◘ Abb. 1.12).
1.5.6 Aorta
1.5.6.1 Aneurysmen
Diffuse (fusiforme) oder sackförmige Erweiterungen der Aorta infolge Wandschädigung durch Arteriosklerose, zystische Medianekrose, Traumen, Lues und andere Infektionen. Hypertonie, die systolischen Scherkräfte des Blutes, besonders bei hoher Blutdruckamplitude, und die mit dem Durchmesser steigende Wandspannung begünstigen die Aneurysmaentwicklung. Echte Aneurysmen haben alle 3 Wandschichten, falsche Aneurysmen eine Wand aus Adventitia und perivaskulärem Gewebe, da sie nach traumatischer Ruptur von Intima und Media entstehen.
1.5.6.1.1 Aorta ascendens
Hauptlokalisation der selten gewordenen luetischen Aneurysmen. Jetzt überwiegend durch zystische Medianekrose (Marfan-Syndrom , Hypertonie, Altersdegeneration) bedingt.
1.5.6.1.1.1 Klinik
Retrosternaler Schmerz, bei der häufigen Einbeziehung des Klappenringes Aorteninsuffizienz mit kardialer Dekompensation. Hustenreiz durch Verdränguung der Trachea nach rechts. Kompression der V. cava superior.
1.5.6.1.1.2 Diagnostik
Erkennbar auf der Thoraxaufnahme. Darstellung durch CT-Angiographie, MR-Angiographie oder konventionelle DSA.
1.5.6.1.1.3 Therapie
Operationsindikation bei einem Durchmesser von 5,5 cm oder rascher Progression (bei Marfan-Syndrom 4,5–5 cm). Ersatz des Aneurysmas durch eine Dacron-Prothese. Bei Aorteninsuffizienz Ersatz von Aszendens und Bulbus mit einer „Composite-Graft“, die eine künstliche Klappe beinhaltet. Anschließend werden beide Koronarostien reimplantiert.
1.5.6.1.2 Aortenbogen
Seltenste Lokalisation.
1.5.6.1.2.1 Klinik
Dysphagie, Husten, Heiserkeit (Rekurrensparese), Verlegung der linken A. subclavia, Schmerzen.
1.5.6.1.2.2 Therapie
Blutdrucksenkung, β-Rezeptoren. Operation in Hypothermie (18–20 °C) bei kritischer Größe. Ersatz des Aortenbogens mit Dacron-Prothese und Belassung einer Gefäßmanschette mit den Abgängen der Hals- und Armarterien.
1.5.6.1.3 Aorta descendens
Zweithäufigste Form des Aortenaneurysmas, meistens am Isthmus, dicht unter dem Abgang der A. subclavia lokalisiert. Echte Aneurysmen überwiegend arteriosklerotisch bedingt, falsche traumatische Aneurysmen durch Thoraxkompression bei Verkehrsunfällen. Operationsindikation für echte Aneurysmen bei Rupturgefahr (Durchmesser über 6,0–7,0 cm), bei traumatischen Aneurysmen sofort. Operationsrisiko 10 % und höher. Wesentlich schonender und risikoärmer ist die transluminale Applikation einer endovaskulären Stentprothese.
1.5.6.1.4 Aorta abdominalis
Häufigste Lokalisation (75 % aller Aortenaneurysmen). Die infrarenale Form (96 %) dominiert über die suprarenale (4 %).
1.5.6.1.4.1 Ätiologie
Fast immer Arteriosklerose.
1.5.6.1.4.2 Klinik
Nur bei 40 % der Kranken Bauchschmerzen und pulsierende Sensationen. Bei gedeckter retroperitonealer Ruptur zunehmender Dauerschmerz in der Lendenregion, bei Ruptur in die Bauchhöhle massive Blutung und schwerer Schock.
1.5.6.1.4.3 Diagnostik
In asymptomatischen Fällen Zufallsbefund bei Sonographie des Abdomens. Größere Aneurysmen verursachen tastbare Pulsationen. Ausmessung im sonographischen B-Bild, wird durch Farbkodierung erleichtert. Präoperativ DSA oder CT-Angiographie. Operationsindikation bei einem Durchmesser des Aneurysmas von >5,5 cm. Resektion des aneurysmatischen Aortenabschnitts und Ersatz durch Gefäßtransplantat, bei suprarenalen Aneurysmen mit Anschluss der Viszeralarterien. Operationsletalität vor Ruptur 5–10 %, nach Ruptur 50 %. Bei Risikopatienten wird wegen sehr geringer Letalität über die freigelegte Femoralarterie eine selbstexpandierende ummantelte Stentprothese in das Gefäßlumen eingebracht.
1.5.6.2 Dissektion
1.5.6.2.1 Definition
Eindringen des Blutes in die Aortenwand durch einen Intimariss mit Zerstörung der Media und Abtrennung der Intima von der Adventitia (dissezierendes Hämatom bzw. Aneurysma).
Das Hämatom (falsches Lumen) engt das wahre Lumen der Aorta ein, vergrößert den Aortendurchmesser, kann Gefäßabgänge stenosieren, die Aortenklappen dislozieren (Aorteninsuffizienz), und nach außen oder innen perforieren. Letzteres bedeutet eine Spontankorrektur.
Entstehung des Intimarisses auf dem Boden einer Mediaschädigung durch zystische Medianekrose, Hypertonie, Schwangerschaft, Aortenisthmusstenose, bikuspidale Aortenklappe. Zwei Prädilektionsstellen (95 % aller Dissektionen) für Intimarisse: 2–5 cm oberhalb der Aortenklappe (proximal) und Aorta descendens unmittelbar unter dem Abgang der A. subclavia (Einteilung nach Debakey, ◘ Abb. 1.20).


Typeneinteilung der Aortendissektion. a Typ I: Dissektion beginnend in der Aorta ascendens und sich über den gesamten Aortenverlauf erstreckend, b Typ II: Dissektion der Aorta ascendens, c Typ III: Beginn der Aortendissektion distal des Abgangs der A. subclavia sinistra und fortschreiten der Dissektion bis zur Aortenbifurkation bzw. bis in die Beckenstrombahn
1.5.6.2.2 Dissektion der Aorta ascendens
Antegrade Ausbreitung der Dissektion von der proximalen Aszendens über den Aortenbogen bis in die A. abdominalis (Typ I) oder nur im Bereich der Aszendens (Typ II, ◘ Abb. 1.21). Gelegentlich retrograde Ausbreitung einer distalen Dissektion.


Supraaortale Angiographie eines 24-jährigen Patienten mit einer Aortendissektion Typ de Bakey II bzw. Stanford-Typ A mit Ausbildung eines großen falschen Lumens zwischen rechter Koronararterie und Truncus brachiocephalicus sowie dissektionsbedingtem Verschluss der rechten Koronararterie
1.5.6.2.2.1 Klinik
Heftigster retrosternaler Schmerz, im Gegensatz zum Crescendoschmerz des Infarktes sofort maximal, oft in der Dissektionsrichtung zum Hals und zwischen die Schulterblätter ausstrahlend. Aorteninsuffizienz (>50 %), die zur kardialen Dekompensation führen kann. Pulsverlust oder -abschwächung an den Arm- und Halsgefäßen, zerebrale Ischämie mit Hemiparese, bei Typ I Abschwächung der Femoralispulse, auch Paraplegie durch ischämische Rückenmarkschädigung. Häufigste Todesursache: externe Ruptur in den Herzbeutel (Tamponade), in eine der Pleurahöhlen, ins Mediastinum, in die Bauchhöhle oder in den Gastrointestinaltrakt.
1.5.6.2.2.2 Diagnostik
Klinische Symptome, Röntgenbild des Thorax, transösophageale Echokardiographie, MR-Angiographie, CT-Angiographie, Spiralcomputertomographie. Die klassische DSA zeigt nur das eingeengte freie Lumen. CK und CK-MB normal, keine Infarktzeichen im EKG.
1.5.6.2.2.3 Therapie
Schmerzbekämpfung, Blutdrucksenkung auf 100–120 mmHg (Infusion von Nitraten und ACE-Blocker, selten von Natriumnitroprussid) und Propranolol i. v. außer bei schwerer Aorteninsuffizienz (zur Herabsetzung der Ejektionsgeschwindigkeit des Blutes) als Sofortmaßnahmen. Standard ist die sehr schnelle operative Versorgung (Resektion an der Stelle des Intimarisses, Nahtverschluss oder Gefäßprothese evtl. mit Aortenklappenprothese). Medikamentöse Langzeitnachbehandlung (Blutdrucksenkung, β-Rezeptorenblocker). Ohne Behandlung sterben 21 % der Patienten innerhalb 24 h, 60 % in den ersten 2 Wochen, 90 % innerhalb von 3 Monaten.
1.5.6.2.3 Dissektion der Aorta descendens
Antegrade Ausbreitung der Dissektion vom Lig. arteriosum zum Zwerchfell oder in die Aorta abdominalis (Typ III, ◘ Abb. 1.22). In der Regel keine retrograde Ausbreitung. Seltener als Typen I und II, überwiegend bei alten Hypertonikern.


MRT (nach KM-Gabe) eines 52-jährigen Patienten mit Aortendissektion Typ de Bakey III bzw. Stanford Typ B (Sammlung Dr. Esdorn, Bad Oeynhausen)
1.5.6.2.3.1 Klinik
Intensiver plötzlicher Schmerz zwischen den Schulterblättern, der nach vorn ausstrahlen kann, gelegentlich Stenosierung der linken A. subclavia und Abschwächung der Femoralispulse durch Stenosierung der Aortenbifurkation.
1.5.6.2.3.2 Diagnostik
Röntgenaufnahme des Thorax, transösophageale Echokardiographie, Spiralcomputertomographie, FKDS, Aortographie.
1.5.6.2.3.3 Therapie
In unkomplizierten Fällen konservative Behandlung (Blutdrucksenkung, β-Rezeptorenblocker). Bei alten und instabilen Patienten mit erhöhtem Operationsrisiko kommt eine Versorgung mit endoluminaler Stentprothese in Betracht. Bei drohender Ruptur, bedrohlicher Ischämie vitaler Organe (Nieren, Darm) oder anhaltenden Schmerzen operative Beseitigung des Intimarisses und Gefäßprothese. Unter konservativer Behandlung überleben 80 % das Akutstadium in der Klinik.
1.5.7 Kopfarterien
1.5.7.1 Akuter zerebrovaskulärer Insult (Schlaganfall , Apoplexie )
1.5.7.1.1 Häufigkeit
Die Inzidenz steigt von etwa 8 pro 100.000 in der Altersgruppe 25–34 Jahre auf über 2000 pro 100.000 im hohen Lebensalter (>85 Jahre). In den USA ereignen sich pro Jahr rund 750.000 Schlaganfälle mit einer Mortalitätsrate von über 150.000.
1.5.7.1.2 Ätiologie
-
Hirninfarkt:
-
arteriosklerotische Thrombose
-
Hirnembolie (◘ Abb. 1.23)
Abb. 1.23 
Das native CCT zeigt einen kleinen älteren frontoparietalen Territorialinfarkt bei einer 68-jährigen Patientin mit intermittierendem Vorhofflimmern
-
Infarzierung vom lakunären Typ (Obliteration kleiner perforierender Arterienäste an der Hirnbasis infolge hypertoniebedingter Hyalinisierung) (◘ Abb. 1.7)
-
-
intrazerebrale Blutung:
-
aus hypertoniegeschädigten perforierenden Arterien mit Mikroaneurysmen
-
bei Amyloidangiopathie (im Alter)
-
durch Einblutung in ischämische Infarkte
-
-
Subarachnoidalblutung: in den Subarachnoidalraum aus rupturierten sackförmigen Aneurysmen an den Teilungsstellen der Hirnbasisarterien (Entwicklungsdefekte der Media und Elastika), selten mykotische bei Endokarditis
-
andere Blutungsursachen: traumatisches epidurales und subdurales Hämatom. Intrakraniale Blutungen bei hämorrhagischen Diathesen und Antikoagulation.


1.5.7.1.3 Klinik
1.5.7.1.4 Kardinalsymptome
Plötzlich auftretende fokale neurologische Ausfallserscheinungen, die von Kopfschmerzen, Erbrechen und Bewusstseinsverlust mit zentralen Atmungs- und Kreislaufstörungen begleitet sein können.
1.5.7.1.5 Zeitliches Insultprofil
-
Thrombotischer Hirninfarkt: In 60 % der Fälle vorausgehende transitorische ischämische Attacken (s. unten). Neurologische Ausfallserscheinungen innerhalb von Minuten voll ausgebildet oder als Zeichen der Thrombusausdehnung bis zu 3 Tagen stufenweise zunehmend (progressing stroke). Remission je nach Infarktgröße nach Stunden, Tagen oder Wochen beginnend, selten komplett. Defekte, die sich innerhalb von 6 Monaten nicht zurückbilden, bleiben bestehen.
-
Embolie: Selten transitorische ischämische Attacken (Mikroembolien) als Vorboten. Blitzartig einsetzende neurologische Ausfallserscheinungen, die sofort ihr Maximum erreichen. Bei Fragmentation des Thrombus und Weitertransport von Thrombusteilen in periphere Gefäßäste Besserung der Symptome nach kurzer Zeit. Rekanalisierung der Arterien nach Infarkteintritt relativ häufig. Dauerschäden wie bei thrombotischen Infarkten.
-
Hypertonische Parenchymblutung: Keine Prodromi. Neurologische Defekte nach plötzlichem Beginn kontinuierlich zunehmend. Maximum nach 1–24 h (abhängig von der Stärke der Blutung). Spät einsetzende, langsame Remission mit gutem Endresultat bei kleinen Blutungen.
-
Subarachnoidalblutung: Selten Kopfschmerzen und neurologische Symptome als Vorboten (Sickerblutung). Nach der Ruptur blitzartige schwerste Kopfschmerzen mit sofort folgender Bewusstlosigkeit (50 % der Fälle) oder Bewusstlosigkeit ohne vorausgehende Beschwerden. In weniger schweren Fällen heftigster Kopfschmerz ohne Bewußtseinsverlust. Hemiplegie und andere Ausfälle in der Regel erst 4–9 Tage nach der Ruptur (Ischämie und Nekrosen infolge Spasmen der Hirnbasisarterien).
-
Posttraumatische Blutungen: Bewusstseinsverlust und Hirnstammsymptome nach einem freien Intervall.
1.5.7.1.6 Neurologische Leitsymptome der Hirninfarkte:
-
Karotissystem (Versorgungsgebiete der A. ophthalmica, A. cerebri anterior, A. cerebri media): Kontralaterale Hemiparese, Hemihypästhesie und homonyme Hemianopsie kombiniert mit sensomotorischer Aphasie (dominante Hemisphäre) oder Apraktagnosie (andere Hemisphäre). Reine motorische Hemiplegie bei lakunärem Infarkt der inneren Kapsel (A. lenticulostriata aus der A. cerebri media). Weitere lakunäre Syndrome im Versorgungsgebiet perforierender Mediaäste mit Dysarthrie, Hemichorea und Hemiballismus.
-
Vertebralis-Basilaris-System (Versorgungsgebiete der A. vertebralis, A. basilaris und der A. cerebri posterior): Bilaterale motorische und/oder sensorische Ausfallserscheinungen, kombiniert mit Störungen der Hirnnerven, des Kleinhirns und anderer Hirnstammstrukturen. Reine Hemianästhesie bei lakunärem Infarkt im ventralen hinteren Thalamuskern (perforierender Ast aus der A. cerebri posterior). Zahlreiche weitere lakunäre Hirnstammsyndrome (▶ Lehrbücher der Neurologie).
1.5.7.1.7 Neurologische Leitsymptome intrazerebraler Blutungen
-
Putamen und angrenzende innere Kapsel (A. lenticulostriata): Kontralaterale Hemiplegie, Blickwendung zur nicht gelähmten Seite, nach 5–30 Minuten Koma, im Verlauf obere Hirnstammkompression (Babinskireflex positiv, irreguläre Atmung, dilatierte fixierte Pupillen, finale Streckkrämpfe infolge Dezerebration). Häufigste Blutungslokalisation (50 %).
-
Thalamus: Kontralaterale Hemiparese oder -plegie durch Kompression der inneren Kapsel, Blick nach unten gerichtet, reaktionslose Pupillen. Zusätzlich sensorische Ausfallserscheinungen (Aphasie, Apraktognosie, homonyme Hemianopsie, Mutismus).
1.5.7.1.8 Diagnostik
Zu klären sind:
-
Ursache, Lokalisation und Ausdehnung des Gefäßverschlusses
-
Lokalisation des Hirninfarktes
-
das Vorliegen von intra- oder extrazerebralen Blutungen
1.5.7.1.9 Anamnese und klinische Untersuchung
Hinweise auf Thrombose: vorausgegangene TIA, Strömunsgeräusche über den Halsschlagadern, Zeitprofil. Hinweise auf Embolie: Vorhofflimmern, Atheromatose der Aorta ascendens, Zeitprofil. Hinweis auf Blutung: Trauma, schwere Hypertonie, Nackensteife, Zeitprofil.
1.5.7.1.10 Computertomographie
Sehr wichtig zur sofortigen Erkennung von Blutungen. Hirninfarkte sind erst nach 24–48 h eindeutig nachzuweisen. Ausschluss raumfordernder Tumoren als Insultursache.
1.5.7.1.11 MR-Tomographie
Infarktnachweis in allen Hirnregionen innerhalb einer Stunde. Auch Blutungen werden sofort erkannt. Wenn verfügbar, dem CT vorzuziehen.
1.5.7.1.12 MR-Angiographie
Erfasst Veränderungen an den extrakranialen und den großen intrakranialen Arterien. Keine sichere Unterscheidung zwischen komplettem und fast komplettem Karotisverschluss. Im Gegensatz zur konventionellen Arteriographie risikolos.
1.5.7.1.13 Farbkodierte Duplexsonographie (FKDS)
Ermöglicht schnell und zuverlässig den Nachweis von Stenosen und Verschlüssen der A. carotis interna. Schwieriger ist oft die Darstellung von Stenosen und Verschlüssen der Vertebralarterien im extrakranialen Bereich. Durch transkraniale Doppleruntersuchung sind Stenosen und Fluss in der A. cerebri media, der A. basilaris und den distalen Vertebralarterien zu erfassen. Kollateralkreisläufe lassen sich durch die Flussmessung an den intrakranialen Gefäßen und an der A. supratrochlearis erkennen, die eine Kollaterale zwischen dem Stromgebiet der A. carotis interna und A. carotis externa darstellt. Beim Internaverschluss fließt das Blut durch die A. supratrochlearis retrograd über die A. ophthalmica in den Hirnkreislauf.
1.5.7.1.14 Selektive zerebrale Arteriographie (DSA)
Das jodhaltige Kontrastmittel wird über einen transfemoral eingeführten Katheter selektiv in die extrakranialen Arterien injiziert. Genaueste Methode der zerebralen Gefäßdarstellung. Komplikationsrate bis 3 %. Indiziert vor intraarterieller Thrombolyse. Sonst weitgehend durch die Kombination von FKDS und MR-Angiographie zu ersetzen.
1.5.7.1.15 Differenzialdiagnosen
Hypoglykämie, epileptischer Anfall, Hirntumor, hypertensive Enzephalopathie, Migräne.
1.5.7.1.16 Therapie
Nach Untersuchungen mittels MRT und Positronen-Emissionstomographie entsteht im Ischämiebezirk schnell ein infarzierter Kern der von hypoxischem, jedoch potenziell erholungsfähigem Nervengewebe umgeben ist. Unter guter Therapie kann sich daher ein erheblicher Teil der neurologischen Ausfallserscheinungen zurückbilden.
1.5.7.1.17 Allgemeine Maßnahmen
Einweisung auf eine Intensivstation, wenn erreichbar in eine Spezialeinrichtung für Schlaganfälle (stroke unit). Dort Überwachung von Atmung und Kreislauf. Freihaltung der Atemwege, Bestimmung der Blutgase zum Ausschluss einer Hypoxie. Erhöhter Blutdruck verbessert die Perfusion. Daher auch Werte bis 220/120 mmHg nicht senken. Nur vor Thrombolyse und bei maligner Hypertonie langsames Absenken auf 180/105 mmHg. Bei Fieber Antipyretika.
1.5.7.1.18 Laboranalysen
Blutzucker, Elektrolyte, Kreatinin, Harnstoff, Gerinnungsfaktoren, Blutbild. Außerdem EKG und Röntgenaufnahme des Thorax.
1.5.7.1.19 Intravenöse Thrombolyse mit t-PA
Sicher und erfolgversprechend nur innerhalb der ersten 3 h nach Symptombeginn. Führt allerdings häufiger zu Einblutungen als in nicht behandelten Fällen.
1.5.7.1.20 Intraarterielle Thrombolyse mit t-PA
Dazu wird ein Mikrokatheter dicht vor oder im Thrombus platziert. Indikation bei gegebener Möglichkeit innerhalb der ersten 3 h bei Verschluss der A. cerebri media, vor allem bei Verschluss der A. basilaris, der ohne Wiederöffnung des Gefäßes eine sehr schlechte Prognose hat.
1.5.7.1.21 Antikoagulation
Low-Dose-Heparin reduziert das Risiko von Beinvenenthrombosen. Auf den Insultverlauf hat es keinen positiven Einfluss, erhöht sogar die Blutungsgefahr. Acetylsalicylsäure (300 mg) verringert in geringem Maße das Auftreten von Insultrezidiven.
1.5.7.1.22 Behandlung des Hirnödem s
In 5–10 % der Fälle kommt es proportional zur Infarktgröße zu klinischen Symptomen des Hirnödems (Nachlassen der Aufmerksamkeit, Benommenheit) und Raumforderung im CT. Beginn am 2. oder 3. Tag, bis zum 10. Tag andauernd. Therapieprinzip: Erhöhung der Osmolarität auf 320 mosmol/l durch strikte Beschränkung der Zufuhr freien Wassers und osmotische Diurese mittels Mannitolinfusionen (25–50 g alle 5 h bis maximal 2 g/kg/Tag). Unterstützend kann auch Furosemid injiziert werden. Keine Glukokortikoide! In schweren Fällen Intubation und mechanischer Hyperventilation zur Senkung des pCO2 auf 25–30 mmHg. Ultima ratio: Chirurgische Dekompressionstrepanation. Raumfordernde Hämatome werden mikrochirurgisch ausgeräumt.
1.5.7.1.23 Neuroprotektiva und Vasodilatanzien
Bei Hirninfarkt wegen erwiesener Unwirksamkeit nicht indiziert. Bei Subarachnoidalblutung drohen neurologische Ausfallserscheinungen durch Arterienspasmen an der Hirnbasis. Einziges wirksames Mittel dagegen ist der Calciumantagonist Nimodipin, zuerst als Infusion später per os.
1.5.7.1.24 Rehabilitation
Frühzeitige passive Bewegungen der gelähmten Extremitäten zur Verhütung von Gelenkkontrakturen und Periarthritiden. Aufsitzen im Stuhl nach einer Woche, aktive Bewegungstherapie, Gehübungen, Sprachübungen etc. Nach 3–6 Monaten können die meisten Hemiplegiker wenigstens etwas gehen.
1.5.7.1.25 Prognose
Nach dem ersten Hirninfarkt erreicht die Mortalität binnen 4 Wochen Werte bis 25 %. Von den Überlebenden des ersten Infarktes stirbt etwa jeder zweite innerhalb von 5 Jahren. Ungünstige Faktoren: Hohes Alter, großer neurologischer Defekt, anhaltende Bewusstlosigkeit, Herzkrankheiten, Hypertonie. Die Basilaristhrombose wird ohne arterielle Thrombolyse selten überlebt. Prognose der embolischen Infarkte schlechter als die der thrombotischen. Relativ gute Prognose der lakunären Infarkte. Mortalität der intrazerebralen Blutung 60–70 % innerhalb von 30 Tagen. Mortalität der Subarachnoidalblutung 27 % innerhalb einer Woche, bei Rezidivblutung 40–45 %.
1.5.7.2 Chronische Kopfarterienverschlüsse
1.5.7.2.1 Pathophysiologie
Die 4 hirnversorgenden Arterien (Aa. carotides, Aa. vertebrales → A. basilaris) kommunizieren an der Hirnbasis zum Circulus arteriosus cerebri. Sie sind außerdem auf mehreren Ebenen durch extra- und intrakraniale Anastomosen untereinander verbunden. Langsam entstehende Verschlüsse können deshalb durch kollateralen Blutfluss kompensiert werden. Blutdruckabfall, Hypoxie und Anämie gefährden diesen Kompensationsmechanismus. Abgangsstenosen der Aa. subclaviae bewirken einen Blutentzug aus dem Hirnkreislauf, der über die retrograd durchströmte A. vertebralis zum Umgehungskreislauf für den Arm wird (Subclavian-Steal-Syndrom).
1.5.7.2.2 Diagnostik
-
Magnetresonanzangiographie (MRA): Schonende Darstellung des Hirnkreislaufs, die multiple Stenosen aufdecken kann (◘ Abb. 1.24).
Abb. 1.24 
MRT (nach KM-Gabe) einer 70-jährigen Patientin mit höhergradigen Stenosen der linken A. carotis interna (ACI) und externa (ACE) sowie der rechten A. carotis interna (ACI) (Sammlung Dr. Esdorn, Bad Oeynhausen)
-
Duplexsonographie: Eignet sich zur Untersuchung der A. carotis (◘ Abb. 1.25).
Abb. 1.25a–c 
Duplexsonographie der A. carotis communis (ACC): a Intima-Media-Komplex leicht (1,1 mm) verbreitert, b Intima-Media-Komplex mässig (1,6 mm) verbreitert, c mittelgradige ACC-Stenose
-
Farbkodierte Duplexsonographie: Erfasst Stenosierungen der A. carotis interna unterschiedlicher Schweregrade (◘ Abb. 1.26 und ◘ Abb. 1.27).
Abb. 1.26 
Duplexsonographie einer leichtgradigen A.-carotis-interna-(ACI-)Stenose mit zugeschaltetem Farbdoppler
Abb. 1.27 
Duplexsonographie mit Farb- und CW-Doppler: hochgradige A.-carotis-interna-Stenose mit hohen systolischen und diastolischen Flussgschwindigkeiten








1.5.7.2.3 Asymptomatische Stenosen und Verschlüsse
Überwiegend extrakranial lokalisiert und durch Kollateralen kompensiert. Erfassung zur Insultprophylaxe äußerst wichtig: Seitendifferenz des Blutdrucks, Stenosegeräusche an den Halsarterien, Differenz der Karotispulse (kann bei Stenose der Carotis interna fehlen), farbkodierte Doppler-Sonographie, MR-Angiographie.
1.5.7.2.4 Transitorische ischämische Attacke n (TIA)
1.5.7.2.4.1 Definition
Flüchtige zerebrale Durchblutungsstörungen mit fokalen neurologischen Ausfallserscheinungen. Dauer einige Sekunden bis 10 Minuten, maximal 24 h.
Hinweis auf drohenden Hirninfarkt (kumulatives 5-Jahresrisiko 35–50 %). Gleichbleibendes Anfallsmuster spricht für Atherothrombose, unterschiedliches für rezidivierende Mikroembolien (aus Atheromen oder aus dem Herzen).
1.5.7.2.4.2 Karotis-System
Ipsilaterale Sehstörungen (Amaurosis fugax, homonyme Hemianopsie), kontralaterale sensomotorische Störungen (Schwäche und/oder Taubheitsgefühl im Arm und Gesicht, flüchtige Aphasien).
1.5.7.2.4.3 Vertebralis-Basilaris-System
Lage- oder Drehschwindel mit Übelkeit und Erbrechen ohne Ohrgeräusche, Gang- und Standunsicherheit, Doppelbilder, alternierende Hemiparesen mit und ohne Sensibilitätsbeteiligung, vorübergehende beidäugige Erblindung, Dysarthrie, Dysphagie, Blitzsynkopen (Tonusverlust der Beine mit nur kurzem Bewusstseinsverlust).
1.5.7.2.5 Aortenbogensyndrom
Gleichzeitige Behinderung der Blutzufuhr zum Gehirn und zu den Armen durch obliterierende Prozesse an den Ostien des Aortenbogens (Truncus brachiocephalicus, linke A. carotis communis, linke A. subclavia). Nachweis durch FKDS und Angiographie.
1.5.7.2.5.1 Umgehungskreisläufe
Zur distalen A. sublavia aus der Aorta thoracica (via Interkostalarterien und A. thoracica interna) und aus der retrograd durchströmten ipsilateralen Vertebralis unter Blutentzug aus dem Gehirn (Steal-Effekt).
1.5.7.2.5.2 Symptome
Transitorische ischämische Attacken (überwiegend vom Vertebralis-basilaris-Typ) und Schmerzen in den Armen bei manueller Tätigkeit auf der kollateral durchbluteten Seite, Puls- und Blutdruckdifferenzen an den Armen, supraklavikuläre Gefäßgeräusche, beim Steal-Syndrom inverser Fluss im Vertebralis-Sonogramm.
1.5.7.2.6 Takayasu-Arteriitis
Riesenzellarteriitis unklarer Ursache bei jungen Frauen, fast nur in Asien vorkommend. Befällt vorwiegend die vom Aortenbogen abgehenden Arterien. In der Frühphase Fieber und hohe BKS. Neben den transitorischen ischämischen Attacken Augenveränderungen (Blutstase, Irisatrophie, Katarakt, Optikusatrophie) und trophische Störungen an Fingernägeln, Mundschleimhaut (Ulzerationen), Nasenseptum (Perforation), Ohrmuscheln und Zähnen (Zahnausfall), dazu Claudicatio masticatoria.
1.5.7.2.6.1 Konservative Therapie
-
Allgemeine Maßnahmen: Kontrolle der atherogenen Risikofaktoren (Hypertonie, Hyperlipämie, Rauchen, Diabetes mellitus).
-
Glukokortikoide: Nur bei Takayasu-Krankheit.
-
Antikoagulation: Zur Prophylaxe transitorischer ischämischer Attacken und Hirninfarkte, auch bei asymptomatischen Stenosen. Indiziert sind Plättchenaggregationshemmer (ASS, ASS + Dypyridamol oder Clopidogrel), alternativ Phenprocoumon.
1.5.7.2.6.2 Chirurgische Therapie
Beschränkt sich im Wesentlichen auf die offene Karotisthrombendarteriektomie mit Patchplastik aus Kunststoff. Das bei der Indikationsstellung zu berücksichtigende Komplikations- plus Mortalitätsrisiko beträgt auch bei geübten Operateuren bis zu 6 %.
-
Asymptomatische Karotisstenosen: Bei Stenosen von >70 % keine Indikation zur TEA. Selbst bei schweren Stenosen von 80–99 % ist ein Erfolg fraglich. Zunehmend an Bedeutung gewinnt das Karotisstenting mit Protektionssystemen, die Embolien aus Atherommaterial verhindern (Komplikationsrate 2–10 %).
-
Symptomatische Karotisstenosen: Bei Stenosen von >70 % ist die TEA indiziert. Bei symptomatischen Stenosen von 30–69 % ist der Nutzen der TEA fraglich.
-
Symptomatische Vertebralisstenosen: Bei Abgangsstenosen Transposition der zentralen gesunden A. vertebralis auf die A. carotis communis (Letalität nahe 0 %, neurologiche Komplikationen in 1 % der Fälle). Bei Stenosen im V2-Segment Anlage eines Bypass vom V1- zum V3-Segment (Komplikationsrate um 3 %).
1.6 Krankheiten der Venen
1.6.1 Pathophysiologie
1.6.1.1 Normale Hämodynamik
1.6.1.1.1 Zentraler und peripherer Venendruck
Referenzebene für den Venendruck ist der rechte Vorhof. Dort beträgt der normale Druck unabhängig von der Körperlage etwa 0 mmHg (zentraler Venendruck), weil sich die Pumpleistung des Herzens dem venösen Rückfluss genau anpasst. Der periphere Venendruck wird vom hydrostatischen Druck bzw. Sog der Blutsäule zwischen rechtem Vorhof und Messpunkt bestimmt. Beim völlig stillstehenden Menschen liegt auf den Fußvenen ein hydrostatischer Druck von etwa 90 mmHg. Der Venendruck in anderen Körperebenen beträgt je nach Entfernung vom Herzen 0–90 mmHg, in den Halsvenen 0 mmHg, in den Sinus der Schädelkalotte –10 mmHg (aufrechte Körperhaltung).
1.6.1.1.2 Muskelpumpe
Das System der Beinvenen besteht aus tiefen, kommunizierenden (Vv. perforantes) und oberflächlichen Venen (◘ Abb. 1.28). Venenklappen öffnen sich in zentripetaler und schließen sich in zentrifugaler Richtung. Bei Muskelkontraktion, besonders beim Gehen, werden die tiefen Venen komprimiert. Es erfolgt ein zentripetaler Blutfluss mit Absaugung der oberflächlichen Venen. Die Pumpwirkung senkt den Venendruck in den Füßen von 90 auf 25 mmHg und verbessert den venösen Rückfluss zum Herzen. In ◘ Abb. 1.29 ist das Venensystem der Beine dargestellt.


Blutströmung in den Beinvenen
1.6.1.1.3 Kreislaufwiderstände und Blutfluss im Venensystem
Physiologische Strömungswiderstände entstehen beim Eintritt der großen Venen in den Brustraum: Abknicken der Armvenen an der ersten Rippe, Kompression der Halsvenen durch den atmosphärischen Druck, Kompression der Bauchvenen durch intraabdominalen Druck. Bedeutsam ist der Einfluss der Atmung. In der oberen Körperhälfte nimmt der venöse Rückfluss inspiratorisch zu (Sogwirkung auf die V. cava superior), exspiratorisch ab. In der unteren Körperhälfte nimmt der venöse Rückfluss inspiratorisch ab (Kompression der infradiaphragmalen Venen durch Tiefertreten des Zwerchfells), exspiratorisch zu (Aufhebung der Kompressionswirkung). Bei rein thorakaler Atmung (Frauen, Jugendliche, Astheniker) keine wesentliche Atemabhängigkeit der Flussraten in der unteren Körperhälfte.
1.6.1.2 Pathologische Hämodynamik
1.6.1.2.1 Klappeninsuffizienz der oberflächlichen Venen (V. saphena)
Mangelhafter Abfall von Venenvolumen und -druck im Gehen durch Rückstrom in die insuffiziente V. saphena magna an der Einmündung in die V. femoralis. Aus der ektasierten V. saphena fließt das Blut über die Vv. perforantes in die tiefen Venen zurück („Privatkreislauf“). Im Verlauf kann es zur Klappeninsuffizienz der Perforansvenen und der tiefen Venen mit weiterer Verschlechterung des venösen Rückflusses kommen. Mit der Druck- und Volumenbelastung nimmt auch die Varikose zu.
1.6.1.2.2 Klappeninsuffizienz der Vv. perforantes
Während der Muskelkontraktion Reflux des Blutes aus den tiefen Venen an die Oberfläche und während der Muskelerschlaffung von der Oberfläche in die Tiefe. Der abnorme Fluss nach außen dominiert und verstärkt die Ektasie der oberflächlichen Venen. Die Muskelpumpe arbeitet weniger effizient.
1.6.1.2.3 Klappeninsuffizienz der tiefen Venen
Weitgehende Ineffizienz der Muskelpumpe: Bei Muskelkontraktion regurgitiert Blut in die Peripherie, bei Muskelerschlaffung strömt herzwärts gepumptes Blut wieder zurück. In der Regel besteht gleichzeitig eine Klappeninsuffizienz der Vv. perforantes.
1.6.1.2.4 Venenverschluss
Nur ausgedehnte bzw. Hauptvenenverschlüsse bilden ein Strömungshindernis, das zur Stauung und Ischämie führt. Chronische Verschlüsse sind in Ruhe durch Kollateralvenen meistens kompensiert. Erst bei Belastung kommt es wegen herabgesetzter venöser Kapazität und Drainagefähigkeit distal des Verschlusses zu Stauungen. Die Kollateralvenen können sich unter der Druck- und Volumenbelastung varikös erweitern.
1.6.2 Untersuchung smethoden
1.6.2.1 Inspektion und Palpation
Untersuchung im Liegen und Stehen zur Erkennung von Varizen, Venektasien, Hautverfärbungen (Zyanose, Pigment) und trophischen Störungen. Durch Palpation werden Venenstränge, Druckschmerz, Faszienlücken im Bereich insuffizienter Vv. communicantes und Ödeme erfasst. Messung von Umfangsdifferenzen.
1.6.2.2 Einfache Funktionsprüfungen
1.6.2.2.1 Perthes-Versuch
Zur Prüfung der Durchgängigkeit der tiefen Beinvenen und der Suffizienz der Vv. perforantes bei Unterschenkelvarizen. Nach Anlegen einer Staubinde oberhalb der Varizen müssen sich diese beim Gehen entleeren. Andernfalls liegt eine Abflussbehinderung in den tiefen Beinvenen und/oder eine Klappeninsuffizienz der Vv. perforantes vor.
1.6.2.2.2 Trendelenburg-Versuch
Zur Prüfung der Klappenfunktion von V. saphena magna und Vv. perforantes bei Oberschenkelvarizen. Nach Ausstreichen der Varizen im Liegen mit angehobenem Bein wird die V. saphena magna am oberen Ende mit einem Stauschlauch abgeklemmt. Danach lässt man den Patienten aufstehen. Tritt innerhalb 30 Sekunden keine Wiederauffüllung der Varizen ein, sind die kommunizierenden Venen intakt, andernfalls, insuffizient. Rasche massive Varizenauffüllung im Stehen nach Lösung der Stauung zeigt Klappeninsuffizienz der V. saphena an.


Abb. 1.29a.b Oberflächliche Stammvenen (Vv. Saphena magna et parva) mit den klinisch wichtigen Perforansvenen. a Ansicht von vorn-medial, b Ansicht von hinten
1.6.2.3 Ultraschalluntersuchungen
Mit den zur Verfügung stehenden Methoden lassen sich Stenosen, Verschlüsse und abnorme Strömungsverhältnisse in den Venen der Extremitäten vollständig abklären.
1.6.2.3.1 Doppler-Stiftsonde
Misst Strömungsrichtung und Flussgeschwindigkeit (◘ Abb. 1.30). Geeignet zur Erfassung der Klappeninsuffizienz bei Varikose und zum Thrombosenachweis im Beckenbereich und in der Leistenregion.


Unauffälliger Dopplerbefund einer V. poplitea mit zentral gerichtetem Blutfluss bei Exspiration (E) und fehlendem Blutfluss bei Inspiration (I), Valsalvae-Manöver (V) und Kompression des Oberschenkels (OK). Nach OK beschleunigter Fluss nach zentral, der durch Kompression der Wade (WK) weiter zunimmt
1.6.2.3.2 B-Bild
Zugänglich sind sämtliche Abschnitte des tiefen Beinvenensystems und die meisten Abschnitte des tiefen Armvenensystems. Mit dem Sondenkompressionstest im Querschnitt lassen sich komplette Thrombosen (Venenlumen erweitert und nicht komprimierbar) sicher nachweisen und lokalisieren. Inkomplette Thrombosen sind weniger gut zu erfassen.
1.6.2.3.3 Konventioneller Duplex
Kombiniert morphologische B-Bild-Darstellung und Strömungsmessung mittels PW-Doppler. Bei inkompletten Thrombosen kann mit dem PW-Doppler die Strömung im Restlumen nachgewiesen werden. Bei Varikose kann der Klappenbesatz des oberflächlichen und tiefen Venensystems geprüft werden, außerdem die Klappenschlussfähigkeit der Perforansvenen.
1.6.2.3.4 Farbduplex
Erleichterte Beurteilung von Gefäßabschnitten, die einem Sondenkompressionstest schwer zugänglich sind. Verbessert wird insbesondere die Diagnostik im Bereich der V. iliaca sowie im distalen Abschnitt der V. femoralis superficialis. Bei Klappeninsuffizienz wird der Rückstrom durch Farbumschlag sichtbar. Das Auffinden der Perforansvenen wird erheblich erleichtert.
1.6.2.4 Phlebographie
1.6.2.4.1 Untersuchungstechnik
In eine Fußrückenvene wird jodhaltiges Kontrastmittel injiziert. Eine Staubinde oberhalb des Knöchels lässt das Kontrastmittel in das tiefe Venensystem abfließen. Der Röntgenkipptisch ist dabei auf 45° geneigt. Zur Darstellung der Beckenetage wird er zum Schluss horizontal gestellt. Thrombophlebitisrisiko 0,7 %.
1.6.2.4.2 Befunde
Darstellung des tiefen Venensystems, Erfassung von frischen und rekanalisierten Thromben, sekundärer Klappeninsuffizienz und Rethrombosen. Das oberflächliche Venensystem füllt sich nur bei Insuffizienz der Perforansklappen und bei Mündungsklappeninsuffizienz der V. saphena magna oder V. saphena parva.
1.6.2.4.3 Indikationen
Zweifelhafte Befunde der Farbduplexsonographie und zum sicheren Ausschluss tiefer Venenverschlüsse vor der Operation einer primären Varikose.
Übersicht Venenerkrankungen
-
Varikosen
-
Primäre
-
Sekundäre
-
-
Venenthrombosen
-
Oberflächliche Thrombophlebitis
-
Becken- und tiefe Beinvenenthrombosen
-
Postthrombotisches Syndrom
-
Tiefe Armvenenthrombosen
-
1.6.3 Varikose n
1.6.3.1 Definition
Varizen sind sackförmig oder zylindrisch erweiterte, häufig gewundene oberflächliche Venen, deren Klappen nach Überschreitung eines kritischen Durchmessers insuffizient werden. Häufigster Sitz ist das oberflächliche Venensystem der Beine (V. saphena magna, V. saphena parva).
1.6.3.2 Primäre Varikose
1.6.3.2.1 Ätiologie
Genetisch bedingte Anlage- bzw. Entwicklungsanomalie des oberflächlichen Venensystems. Erbliche Disposition in 75 % der Fälle nachweisbar. Starke Zunahme der Erkrankungshäufigkeit mit dem Lebensalter. Begünstigende Faktoren: Stehen im Beruf, ungenügende körperliche Aktivität, Adipositas, Gravidität, Kontrazeptiva. Frauen sind häufiger betroffen als Männer.
1.6.3.2.2 Einteilung
Nach Lokalisation und Ausdehnung lassen sich folgende Typen der Varikose unterscheiden (◘ Abb. 1.31):


Varizenztypen. a Besenreiser, b retikuläre Varikose, c Varikose der V. saphena magna ohne Insuffizienz der Mündungsklappen, d kombinierte Varikose der V. saphena magna und der V. saphena parva
-
Besenreiser: kutane Mikrovarizen (<1 mm im Durchmesser), angeordnet um eine „Nährvene“ (nur kosmetisch relevant)
-
retikuläre Varizen: entwickeln sich in dünnen Hautvenen, die im Ober- und Unterschenkel ein Venennetz bilden, klinisch ohne Bedeutung
-
Stamm - und Seitenastvarikose:
-
Varikose der V. saphena magna und parva, klinisch am bedeutsamsten
-
Varikose der V. saphena accessoria lateralis; analog kann die V. saphena accessoria medialis betroffen sein
-
-
Perforansvarikose: Insuffizienz der Vv. perforantes.
Bei fortgeschrittener Stammvarikose kommt es, wie erwähnt, zur Erweiterung und Klappeninsuffizienz der Perforansvenen, die das oberflächliche (epifasziale) mit dem tiefen (subfaszialen) Venensystem verbinden. Durch den Blutrückfluss nach außen entstehen an den Durchtrittsstellen der Perforansvenen durch die Faszie rundliche Vorwölbungen (blow outs) der Oberflächenvenen, sog. Perforansvarizen. Sie zeigen den Übergang in eine venöse Insuffizienz an. Ihre Prädilektionsstellen sind in ◘ Abb. 1.29 markiert.
1.6.3.2.3 Schweregrade
Die Schweregrade der Stammvarikose (◘ Tab. 1.4) zeigen an, wie weit die dopplersonographisch zu erfassende Insuffizienz der Venenklappen von proximal nach distal fortgeschritten ist.
1.6.3.2.4 Klinik
Verschiedene Varizenformen auch von größerer Ausdehnung können jahre- und jahrzehntelang beschwerdefrei bleiben. Mit der Kaliberzunahme und der Ausbreitung der Klappeninsuffizienz nach distal treten jedoch immer häufiger Beschwerden und Komplikationen auf. Unter klinischen Aspekten lassen sich folgende Stadien der primären Varikose unterscheiden:
-
Stadium I: Varikose meist geringeren Grades ohne Beschwerden und Komplikationen.
-
Stadium II: Varizen mit Beschwerden (Dysästhesien, Juckreiz, Schweregefühl, Spannungsgefühl, geringer Schwellungsneigung und Wadenkrämpfen). Keine Komplikationen.
-
Stadium III: Deutliche Varikosis mit Beschwerden wie im Stadium II. Dazu trophische Hautstörungen (Hyper- und Depigmentierung, Atrophie, Induration, Rötung, Ekzeme), Ödeme erst bei Insuffizienz der tiefen Venen (◘ Abb. 1.32).
Abb. 1.32 
Ausgeprägte Unterschenkelvarikosis mit trophischen Veränderungen der Haut und der Nägel
-
Stadium IV: Ausgedehnte Varikose mit Klappeninsuffizienz an verschiedenen Perforansvenen. Beschwerden und Komplikationen wie im Stadium III. Zusätzlich florides Ulkus cruris (häufig auf einem Krampfaderpolster, meistens trocken, manchmal in ekzematöser Umgebung).


1.6.3.3 Sekundäre Varikose
1.6.3.3.1 Ätiologie
Hauptsächlich Thrombosen der Becken- und tiefen Beinvenen. Selten venöse Abflussstörungen durch Venenkompression im Becken oder Schwangerschaft.
1.6.3.3.2 Pathogenese
Die Zirkulationssperre führt distal des Thrombus zur Dilatation der tiefen Venen und zur Erhöhung des Venendrucks, die sich über die Perforansvenen in den venösen Hautplexus fortpflanzt, der ebenfalls dilatiert wird. Es entstehen umschriebene, kissen- oder knäuelartige Venektasien und dermale Mikrovarizen. Der Kollateralkreislauf in das oberflächliche Venensystem durch die erweiterten und klappeninsuffizienten Perforansvenen kann im Verlauf zur Stammvarikose führen. Auch nach Rekanalisation eines Thrombus geht der Rückfluss nach außen weiter, wenn die tiefen Venenklappen zerstört wurden.
1.6.3.3.3 Differenzialdiagnosen
Die klinische Unterscheidung von primären und sekundären Varizen ist nicht mit letzter Sicherheit möglich, zumal beide Formen auch nebeneinander bestehen können. Vor der Entfernung oder Verödung anscheinend primärer Varizen muss in jedem Fall ausgeschlossen werden, dass sie zum Umgehungskreislauf eines tiefen Venenverschlusses gehören. Methodisch ist das mit den beschriebenen Ultraschall-Doppler-Verfahren möglich, die sowohl die Strömungsverhältnisse als auch anatomische Veränderungen in den oberflächlichen, den tiefen und den Perforanzvenen erfassen. In allen Zweifelsfällen bringt die Phlebographie Klarheit.
1.6.3.3.4 Therapie
1.6.3.3.5 Konservative Maßnahmen
Kompressionsverbände (bei frischen Ödemen mit Rückbildungstendenz), in leichten Fällen Stützstrümpfe, sonst Kompressionsstrümpfe mit adäquatem Kompressionsdruck (13–60 mmHg), die während der Bettruhe abzulegen sind. Aktive Bewegungstherapie (Schwimmen, Radfahren) zur Aktivierung der Muskelpumpe. Kalte Güsse, keine warmen Thermalbäder. Diuretika gegen Ödeme. Der Nutzen der Venenpharmaka (Rosskastanienextrakt etc.) ist umstritten.
1.6.3.3.6 Sklerosierung
Verschluss der varikös erweiterten Venen mit Verödungsmitteln (Varigloban, Aethoxysklerol), die eine künstliche Thrombose erzeugen. Indiziert bei Seitenast-, Besenreiser- und retikulären Varizen. Auch bei Stammvarikose der V. saphena parva möglich. Je distaler die Lokalisation der Varizen und je dystrophischer die Haut, desto eher ist die Sklerotherapie angezeigt. Besenreiser und Teleangiektasien können auch mit einem Laser verschlossen werden.
1.6.3.3.7 Operativ
Indiziert bei Stammvarikose, insuffizienten Perforansvenen und Seitenastvarikose. Ziel ist, nur den klappeninkompetenten Abschnitt der Vene zwischen proximalem (meist Mündungsklappe) und distalem Insuffizienzpunkt zu beseitigen. Unter dem Aspekt der Venenerhaltung als Gefäßersatzmaterial für Bypässe kann die V. saphena magna am Unterschenkel fast immer belassen werden. Unterschenkelvarizen gehören ebenso wie die Einmündungen der unteren Perforansvenen meist zur hinteren Bogenvene, die sich gesondert entfernen lässt. Methode: Abtragung des Mündungssegmentes der Stammvene von der V. femoralis bzw. V. poplitea und Ligatur aller hier einstrahlenden Venenäste. Einführung der Babcocksonde von distal in das Gefäßlumen, Befestigung des Sondenkopfes an der Stammvenenmündung, anschließend Invaginationsextraktion (Stripping) der Stammvene von proximal nach distal. Die Perforansvenen müssen unterbunden werden (gezielte Freilegung und subfasziale Ligatur oder endoskopische Dissektion). Seitenastvarizen werden über Stichinzisionen mit feinen Klemmen extrahiert.
1.6.4 Venenthrombose n
1.6.4.1 Thrombogenese
Die Kausalfaktoren lassen sich am besten nach der Virchow-Trias gliedern:
-
Gefäßwandschaden
-
Verlangsamung der Blutströmung bzw. Stase
-
gesteigerte Gerinnbarkeit des Blutes.
Das nicht seltene Zusammentreffen dieser Faktoren steigert das Thromboserisiko erheblich.
1.6.4.1.1 Gefäßwandschaden
Traumatisch, entzündlich oder durch Kompression. Insgesamt selten.
1.6.4.1.2 Verlangsamung der Blutströmung
Bettlägerigkeit, vor allem im höheren Lebensalter, Wochenbett, langes Sitzen (Flugzeug), chronische venöse Insuffizienz, Herzinsuffizienz, Abflussbehinderung (Schwangerschaft, abdominale Tumoren).
1.6.4.1.3 Gesteigerte Gerinnbarkeit des Blutes
Ausführliche Darstellung im Hämatologiekapitel (▶ Kap. 7).
-
Erworbene Thrombophilie: Einschwemmung von Gewebsthrombokinase in die Blutbahn (bei Operationen, Traumen, Entbindungen, aus Tumoren und Leukämiezellen). Phospholipidantikörper, Antikörper gegen Cardiolipin, Polyglobulie, Thrombozytose, Kontrazeptiva. Nephrotische Syndrome.
-
Hereditäre Thrombophilie: Mangel an Protein C, Protein S, Heparin-Kofaktor II oder Antithrombin III. Protein-C-Resistenz des Faktor V. Hereditäre Fibinolysedefekte.
1.6.4.2 Oberflächliche Thrombophlebitis
1.6.4.2.1 Klinik
Meistens am Ober- oder Unterschenkel lokalisierter schmerzhafter, druckempfindlicher, derber Venenstrang mit Erwärmung, Rötung und leichter ödematöser Schwellung der Haut in diesem Bereich. Schmerzen bei Muskelanspannung. Fieber und Leukozytose möglich. Sehr geringe Emboliegefahr, kein postthrombotisches Syndrom.
1.6.4.2.2 Sonderformen
-
Varikophlebitis: Thrombose in einer Krampfader, oft einschmelzend.
-
Septische Thrombophlebitis: Hauptsächlich durch liegende Verweilkatheter.
-
Phlebitis migrans: Schubweiser Verlauf und sprunghaftes Übergreifen auf andere Körperpartien. Subfebrile Temperaturen, Leukozytose, erhöhte Blutsenkung. Vorkommen bei Infekten, Lupus erythematodes, Allergie, Malignomen, Initialphase der Thrombangiitis obliterans.
1.6.4.2.3 Therapie
Nichtsteroidale Antiphlogistika für einige Tage, heparinhaltige Externa, Kälteapplikation, Kompressionsverbände, rasche Mobilisierung. Bei Bettruhe Gefahr des Übergreifens auf das tiefe Venensystem. Antikoagulation nur bei größerer Ausdehnung und Gefährdung der tiefen Venen. Bei septischer Thrombophlebitis Kühlung, Heparingel, Antibiotika, Antiphlogistika, keinen Druckverband.
1.6.4.3 Becken - und tiefe Beinvenenthrombosen
Am häufigsten sind Zweietagenthrombosen: Thrombose der tiefen Unterschenkelvenen und der V. poplitea oder Thrombose der V. femoralis superficialis und der V. iliaca. Einetagenthrombosen der Becken-, Oberschenkel- oder Unterschenkelvenen kommen relativ selten vor.
1.6.4.3.1 Klinik
1.6.4.3.2 Subjektive Symptome
Schweregefühl, Spannungsgefühl und Krampfneigung im Bein, Schmerzen beim Aufsetzen des Fußes. Schmerzen entlang den Venenbahnen oder in der Leiste.
1.6.4.3.3 Objektive Symptome
Beinschwellung, eindrückbare Ödeme, subkutane Kollateralvenen, Zyanose der Extremität und folgende Zeichen:
-
Payr: Druckschmerz der Plantarmuskulatur.
-
Homans: Wadenschmerz bei Dorsalflexion des Fußes.
-
Meyer: Druckschmerz bei Wadenmuskelkompression von dorsal gegen die Tibia.
-
Lowenberg: Schmerz bei Blutdruckmanschettendruck an der Wade unter 150 mmHg.
-
Bisgaard: Palpationsschmerz der Regio calcaneomalleolaris am gestreckten Bein.
Oft besteht leichtes Fieber. Bei stationär unter Immobilisation auftretenden Thrombosen sind die klinischen Symptome oft unvollständig und gering ausgeprägt. In etwa 14 % der Fälle ist eine Lungenembolie das Initialsymptom.
1.6.4.3.4 Besondere Formen
-
Phlegmasia alba dolens: Hochgradiges, blasses, schmerzhaftes Ödem eines Beines bei Oberschenkel- bzw. Beckenvenenthrombose.
-
Phlegmasia rubra dolens: Plötzliche schmerzhafte Schwellung einer Extremität mit Rotfärbung der Haut bei ausgedehnten Venenthrombosen mit Periarteriitis.
-
Phlegmasia coerulea dolens: Ausgedehnte, den Rückfluss weitgehend blockierende tiefe Phlebothrombose, die von arteriellen Spasmen begleitet sein kann. Pralle Anschwellung des Beines mit zyanotischer Verfärbung. Im Verlauf distale Gangrän.
1.6.4.3.5 Diagnostik
Venenthrombosen im Oberschenkel lassen sich mit der Doppler-Stiftsonde nachweisen (◘ Abb. 1.33). Lokalisation und Ausdehnung der Thromben sind in allen Etagen mit der konventionellen und noch genauer mit der farbkodierten Duplexsonographie festzustellen. Die Phlebographie ist in den meisten Fällen entbehrlich geworden. Ein normaler duplexsonographischer Befund schließt eine Bein- oder Beckenvenenthrombose aus.


Pathologischer Dopplerbefund bei einem 54-jährigen Patienten mit tiefer Oberschenkelvenenthrombose
1.6.4.3.6 Primäre Thromboseprophylaxe
Indiziert bei chirurgischen Patienten peri- und postoperativ und bei allen immobilisierten Patienten, insbesondere solchen mit fieberhaften und konsumierenden Krankheiten. Ferner bei Benommenheit, Lähmungen und Herzinsuffizienz.
1.6.4.3.7 Physikalische Maßnahmen
Postoperative Frühmobilisation, Bettgymnastik (aktive Bewegungsübungen, passives Durchbewegen, Kompression der Ober- und Unterschenkel), gut sitzende Antiemboliestrümpfe, elastische Bandage der Beine.
1.6.4.3.8 Medikamentöse Prophylaxe
Niedermolekulares Heparin (z. B. Enoxaparin 1-mal 40 mg s.c. pro Tag). Alternativ unfraktioniertes Heparin 2-mal 5000 IE s.c. (Low-Dose-Heparin). Bei Heparinunverträglichkeit (HIT II) ist Lepirudin eine gleichwertige Alternative, kann aber bei wiederholter Anwendung zur Allergie führen.
1.6.4.3.9 Therapie
Ziel ist die Vermeidung von Lungenembolien und einer postthrombotischen venösen Insuffizienz.
1.6.4.3.10 Allgemeine Maßnahmen
Hochlagerung und Wickeln des Beines (zur Verbesserung der venösen Zirkulation, gegen Ödeme und Ablösung von Thromben). Bettruhe bei schmerzhaften Schwellungen für wenige Tage.
1.6.4.3.11 Antikoagulation
Sofort nach Erkennung der Thrombose mit unfraktioniertem oder fraktioniertem Heparin einleiten. Gerinnungshemmend wirken beide Heparine im Komplex mit Antithrombin III im Plasma. Unfraktioniertes Heparin inhibiert Thrombin und Faktor Xa, fraktioniertes niedermolekulares Heparin dagegen überwiegend Faktor Xa (auch an Plättchen gebundenen) und damit hauptsächlich die Thrombinbildung (▶ Kap. 7).
-
Fraktionierte niedermolekulare Heparine (NMH): Mittel der Wahl wegen einfacher Handhabung bei gleich guter Wirkung wie Standard-Heparin. Bei Tumorpatienten mit tiefer Becken- und Beinvenenthrombose wird sogar ein Überlebensvorteil erzielt. Die NMH werden bei der Thrombosetherapie nach Körpergewicht dosiert und ausschließlich subkutan injiziert. Kontrollen der Blutgerinnung während der Behandlung sind nicht erforderlich. Im Handel sind: Enoxaparin (Clexane), Nadroparin (Fraxiparin), Tinzaparin (Innohep) und Certoparin (Mono-Embolex). Die Wirkungsstärke wird in mg oder Anti-Xa-Einheiten angegeben. Dosierungsbeispiele: Enoxaparin 2-mal 1 mg (= 100 IE)/kg/Tag; Tinzaparin 1-mal 175 Anti-Xa IE/kg/Tag.
-
Unfraktioniertes Heparin: Initial Bolus von 5000–10000 IE (oder 80 IE/kg) i. v., anschließend etwa 1000 IE (oder 18 IE/kg) pro Stunde per Perfusor. Die aPPT sollte auf den zweifachen Wert steigen (Kontrollen alle 4–6 h).
Komplikationen der Heparintherapie sind Blutungen (Antidot Protaminsulfat) und eine heparininduzierte Thrombopenie Typ II (mit Thrombosen und disseminierter intravaskulärer Gerinnung).
-
Übergang auf orale Antikoagulation mit Phenprocoumon (Marcumar) oder Warfarin (Coumadin): Die Umstellung wird nach 1–7 Tagen begonnen bis zu einem INR >2,0. Solange wird die Heparintherapie überlappend fortgesetzt.
-
Dauer der Antikoagulation: Bei postoperativer und idiopathischer TVT 3–6 Monate, bei rezidivierender idiopathischer TVT minimal 12 Monate, nach kleiner Lungenembolie minimal 6 Monate, nach massiver Lungenembolie und bei hereditären Thrombophilien unbefristet.
1.6.4.3.12 Thrombolyse
Mit Urokinase oder t-PA (rekombinanter Plasminogen Human-Aktivator). Indiziert bei Thrombusalter <7 Tagen vor allem bei Mehretagenthrombosen. Wegen des Blutungsrisikos nur bei ileofemoraler tiefer Venenthrombosen bei jüngeren Patienten zu erwägen. Verhindert bei Erfolg ein postthrombotisches Syndrom (▶ Abschn. 1.16.1).
1.6.4.3.13 Thrombektomie
Indiziert bei Phlegmasia coerulea dolens, rezidivierender Lungenembolie, flottierenden Thromben und Kontraindikation gegen eine Lysetherapie. Das Thrombusalter sollte 3–4 Tage, bei Beckenvenenthrombose 10 Tage nicht überschreiten. Technik: Venotomie der V. femoralis communis. Einführung eines zentralen Blockadeballons in die V. cava inferior zum Schutz gegen Lungenembolien. Extraktion des Thrombus aus der V. iliaca mittels Fogarty-Katheter. Manuelles Auspressen der Thromben aus dem Ober- und Unterschenkel.
1.6.4.3.14 Cava-Schirm
Indiziert nur bei rezidivierenden Lungenembolien unter adäquater Antikoagulanzienbehandlung. Trotzdem können die Filter thrombosieren. Der Schirmfilter wird über die V. jugularis eingeführt und unterhalb der Nierenveneneinmündung in der V. cava passager oder permanent verankert.
1.6.4.4 Postthrombotisches Syndrom
Chronische venöse und lymphatische Insuffizienz, die sich einige Jahre nach nicht optimaler Behandlung einer tiefen Becken- oder Beinvenenthrombose entwickelt.
1.6.4.4.1 Pathogenese
Nach unvollständiger Rekanalisation und/oder Zerstörung der tiefen Venenklappen bleibt der Venendruck im Bein erhöht. Die venöse Hypertonie führt zur Ödembildung, die durch Blockierung des subfaszialen Lymphtransportes noch verstärkt wird. Die Folge ist eine Ernährungsstörungen der Haut, die zu Läsionen führen kann.
1.6.4.4.2 Klinik
Ziehende dumpfe Schmerzen in den Beinen, die beim Stehen und langen Gehen zunehmen.
-
Rein ödematöse Form: Meist peripher beginnendes anfänglich weiches Ödem, das später härter wird, die ganze Extremität befallen und in ein Lymphödem übergehen kann. Letzteres kann durch Erysipelschübe verstärkt werden.
-
Variköse Form: Ausbildung sekundärer Varizen, meist regellos angeordnet, selten unter dem Bild einer Stammvarikose. Zahlreiche insuffiziente Vv. perforantes (besonders der Cockett-Gruppe). Manchmal dominieren in indurierten Hautpartien kleinkalibrige Venektasien („Canyon“-Varizen).
-
Trophisch-ulzeröse Form: Hautveränderungen durch venöse Stase: Stauungsekzem, Dermoepidermitis, Parakeratose, Hämosiderose, schmerzhafte Indurationen. Mikroulzera, längliche Ulzera über insuffizienten Cockett-Venen und größere Erosivgeschwüre im Unterschenkelbereich.
1.6.4.4.3 Diagnostik
Methode der Wahl zur Erkennung des Rekanalisationsgrades, der Klappenzerstörung und des Umgehungskreislaufs über die Perforansvenen ist die Farbduplextechnik. Zur morphologischen Beurteilung des Venensystems dient die Phlebographie.
1.6.4.4.4 Therapie
Konsequente Kompressionsbehandlung (Strumpfklasse 2 oder 3). Bei floridem Ulkus Anwendung von Pelotten und Kompressionsverband. Bei rekanalisierten Venen Ligatur der Perforansvenen und Stripping der Sekundärvarizen möglich.
1.6.4.5 Tiefe Armvenenthrombosen
1.6.4.5.1 Lokalisation
Überwiegend handelt es sich um eine isolierte Thrombose der V. subclavia oder der V. axillaris (Paget-v.-Schrötter-Syndrom).
1.6.4.5.2 Ätiologie
Abflussbehinderung an der Thoraxapertur (Halsrippe), abrupte Schulterzerrung, zentraler Verweilkatheter, Thrombophilie, Venenkompression durch Hämatom oder Tumor (Lymphom).
1.6.4.5.3 Klinik
Ödematöse Schwellung im Schulterbereich livid-rötliche Verfärbung, subkutane Kollateralvenen über dem M. pectoralis, Druck und Bewegungsschmerz in der Schulter.
1.6.4.5.4 Diagnostik
Farbduplex und Phlebographie.
1.6.4.5.5 Therapie
Antikoagulation mit Heparin mit Übergang auf Cumarine. Hochlagerung mit Kompressionsverband, später Kompressionsärmel. Bei frischen Thromben auch systemische oder Katheterlyse. Keine Embolektomie. Resektion der 1. Rippe, falls diese komprimiert. Zu einem postthrombotischen Syndrom kommt es an den Armen nicht.
1.7 Krankheiten der Lymphgefäße
1.7.1 Funktionen des Lymphgefäßsystems
1.7.1.1 Bildung und Zusammensetzung der Lymphe
Lymphe ist interstitielle Flüssigkeit, die in den Lymphbahnen fließt. Vom Ultrafiltrat des arteriellen Kapillarschenkels werden 90 % durch Rückresorption in die Venen und 10 % auf dem Lymphweg in den Blutkreislauf zurücktransportiert. Von größter Bedeutung ist der Lymphweg für den Rücktransport der ultrafiltrierten Proteine. Sie können vom venösen Kapillarschenkel nicht resorbiert werden und erreichen in der interstitiellen Flüssigkeit Konzentrationen von 2–5 g/100 ml. Proteinansammlungen im Interstitium würden den kolloidosmotischen Druck des Gewebes erhöhen und die Wasserrückresorption im venösen Kapillarschenkel stark beeinträchtigen. Auf dem Lymphweg werden auch Zellen und Bakterien transportiert. Darmlymphe enthält in Form von Chylomikronen die Masse des resorbierten Nahrungsfettes. Durch den Abtransport der interstitiellen Flüssigkeit mit ihrem hohen Proteingehalt über die Lymphbahnen sinkt der interstitielle Flüssigkeitsdruck auf -6 bis -7 mmHg. Das ist ein Sicherheitsfaktor gegen Ödeme, die sich erst bei Drücken über 0 mmHg ansammeln können.
1.7.1.2 Lymphbahnen und Lymphtransport
Der Lymphfluss beginnt in den Lymphkapillaren, deren Endothelzellen an den Rändern nicht verschmelzen, sondern sich so überlappen, dass der Einstrom frei, der Rückstrom aber blockiert ist. Die ableitenden Lymphgefäße besitzen glatte Muskulatur und dicht aufeinander folgende Klappen, die sich nur in zentripetaler Richtung öffnen. Werden die Segmente zwischen den Klappen durch zufließende Lymphe gedehnt, kontrahieren sich die Wandmuskeln reflektorisch und befördern die Lymphe weiter. Diese Lymphpumpe wird durch Kontraktionen der Skelettmuskulatur, durch Körperbewegungen, Arterienpulsationen und Kompression von außen verstärkt. Sie hat auf das Interstitium eine ansaugende Wirkung. Die Lymphe aus der unteren und linken oberen Körperregion fließt über den Ductus thoracicus in den linken Venenwinkel zwischen V. jugularis und V. subclavia, die aus der rechten oberen Körperregion über den Ductus lymphaticus dexter in den rechten Venenwinkel. Gesamtflussrate in Ruhelage 120 ml/Std. Bei offenen Lymphgefäßen wird der Lymphfluss durch Muskelarbeit und alle Faktoren gesteigert, die den interstitiellen Flüssigkeitsdruck erhöhen: Anstieg des Kapillardrucks, Zunahme der Kapillarpermeabilität und der Eiweißkonzentration der interstitiellen Flüssigkeit und einen herabgesetzten kolloidosmotischen Druck des Plasmas. Bis zu einem Anstieg des interstitiellen Flüssigkeitsdrucks auf –2 mmHg, was einer mehr als 10-fachen Steigerung des Lymphflusses entspricht, bleibt das Interstitium „trocken“. Höhere Drücke lassen seinen Flüssigkeitsgehalt steigen und führen zum Ödem , das nach einem Volumenzuwachs von 30 % in Erscheinung tritt. Die maximale Transportkapazität des Lymphgefäßsystems wird bei einem interstitiellen Flüssigkeitsdruck von 2 mmHg erreicht, weil das Ödem die Lymphkapillaren und Sammelgefäße erweitert und damit die Lymphpumpe schwächt. Außerdem werden große Lymphgefäße durch den Gewebedruck komprimiert.
1.7.2 Pathogenese der Ödem e
Die zum Ödem führende Steigerung des interstitiellen Flüssigkeitsdrucks kann durch verschiedene Störungen der kapillaren Flüssigkeitsdynamik zustande kommen. Erst auf dem erhöhten interstitiellen Druckniveau wird die im arteriellen Kapillarschenkel filtrierte Flüssigkeitsmenge im venösen Schenkel rückresorbiert. Bei gegebenem interstitiellen Flüssigkeitsdruck hängt der Schweregrad des Ödems von der Dehnbarkeit (Compliance) der Gewebespalten ab, die bei chronischen Ödemen zunimmt.
Ödeme können durch folgende Mechanismen entstehen:
-
Erhöhter Kapillardruck: Der Kapillardruck kann durch venöse Stauung (Venenthrombose, Herzinsuffizienz) oder durch Erweiterung der Arteriolen (durch Histamin bei Urticaria und angioneurotischem Ödem) ansteigen. Dadurch wird die Ultrafiltration solange überschüssig, bis der Anstieg des interstitiellen Flüssigkeitsdrucks die venöse Rückresorption kompensierend erhöht.
-
Hypoproteinämie: Die Verminderung des kolloidosmotischen Drucks bewirkt ein Übergewicht der Ultrafiltration im arteriellen Kapillarschenkel gegenüber der Rückresorption im venösen, das durch den Anstieg des interstitiellen Flüssigkeitsdrucks ausgeglichen wird. Hypoproteinämie kann durch Verbrennungen, proteinverlierende Enteropathie, Proteinurie und Eiweißmangelernährung entstehen.
-
Obstruktion der Lymphbahnen: Kein Abtransport ultrafiltrierter Plasmaproteine. Eiweißkonzentration und Kolloidosmotischer Druck der interstitiellen Flüssigkeit werden genauso hoch wie im Plasma. Verminderung der venösen Rückresorption, die durch einen erheblichen Anstieg des interstitiellen Drucks mit der Ultrafiltration ins Gleichgewicht gebracht wird. Besonders starke Ödeme haben einen hohen Eiweißgehalt der Ödemflüssigkeit, die gerinnen kann. Vorkommen der Lymphbahnverlegung bei Filariasis, nach Lymphangitiden und Lymphknotenentfernung, ferner bei Fehlbildungen (▶ unten).
-
Steigerung der Kapillarpermeabilität: Durch Eiweißaustritt ins Interstitium nimmt dessen kolloidosmotischer Druck zu, während der des Plasmas sinkt. Zum Ausgleich der eingeschränkten Rückresorption muss der interstitielle Flüssigkeitsdruck erheblich steigen. Vorkommen bei allergischen Reaktionen und Verbrennungen.
1.7.3 Untersuchungsmethoden
1.7.3.1 Lymphographie
1.7.3.1.1 Farbstoff-Lymphangiographie
Markierung der epifaszialen Lymphgefäße mit Patentblauviolett (Triphenylmethan), das interdigital injiziert wird. Keine Darstellung bei Aplasie. Abflussstörungen nach entzündlichen Prozessen in der Subkutis.
1.7.3.1.2 Lymphsequenzszintigraphie mit Radioisotopen (198Au)
Bei Transportstörungen keine Darstellung der Lymphknotenstationen. Injektion zwischen erste und zweite Zehe, lateral der Achillessehne oder in die Wadenmuskulatur.
1.7.3.1.3 Lymphographie mit Röntgenkontrastmittel (Lipiodol, ultrafluid)
Injektion in ein peripheres Lymphgefäß nach Farbstoffmarkierung und Freilegung (5–8 ml in 120 min). Füllungsbilder nach der Injektion, Speicherbilder zur Beurteilung der Lymphknotenstruktur 24 h später. Pathologische Strukturen bei Lymphomen, Füllungsausfall der Lymphknoten bei Metastasen.
1.7.3.2 Biopsie
Gewebeentnahme aus Lymphgefäßen und Lymphknoten zur histologischen Untersuchung.
1.7.4 Akute Lymphangitis
1.7.4.1 Ätiologie und Pathogenese
Lymphogene zentripetale Ausbreitung eines bakteriellen Erregers in den peripheren Lymphgefäßen verursacht eine Lymphbahnentzündung, die auch auf den regionalen Lymphknoten übergreift. Die Eintrittspforte in der Haut kann diskret sein. Häufigste Erreger Streptokokken und Staphylokokken.
1.7.4.2 Klinik
Feinkalibrige gerötete subkutane Stränge manchmal druckempfindlich, meistens von einem erkennbaren Infektionsherd (Furunkel, Abszess, Wunde etc.) ausgehend. Schwellung der regionalen Lymphknoten. Je nach Schwere auch Fieber und Leukozytose.
1.7.4.3 Differenzialdiagnosen
Abgrenzung gegen Phlebitis (derber erhabener Strang, keine Lymphknotenschwellung).
1.7.4.4 Therapie
Ruhigstellung, kühlende Umschläge, Wundversorgung, Antibiotika (Penicillin, Cephalosporine).
1.7.5 Lymphödem
1.7.5.1 Ätiologie und Pathogenese
Es handelt sich um Ödeme durch Störungen des Lymphabflusses, denen eine Aplasie oder Hypoplasie der Extremitätenlymphbahnen, eine Lymphangiektasie (Klappeninsuffizienz) oder eine Obstruktion der Lymphbahnen zugrunde liegen kann.
1.7.5.2 Primäres Lymphödem
Familiäre und sporadische Formen (Nonne-Milroy, de Meige) mit Hypoplasie oder Aplasie der Lymphgefäße an einem oder beiden Beinen, teils bei der Geburt vorhanden, teils im Kindesalter, nach der Pubertät oder später manifest werdend. Im Verlauf zusätzliche Lymphangitiden durch Infektion des ödematösen Gewebes, vor allem Erysipelschübe. Bei der kongenitalen intestinalen Lymphangiektasie besteht ein Lymphödem des Darmes mit Eiweiß- und Fettausscheidung in das Darmlumen und Beinödemen durch sekundäre Hypoproteinämie.
1.7.5.3 Sekundäres Lymphödem
Verlegung oder Zerstörung des Lymphgefäßsystems aus verschiedenen Ursachen:
-
Entzündliches Lymphödem: Chronische oder rezidivierende Lymphangitis und Lymphadenitis bei Erysipel, Filariasis, Granuloma venerum, sekundär infizierte Fußmykosen und Ekzeme, postthrombotische Beinulzera. Auch allergische Ödeme haben eine entzündliche Note.
-
Lymphödem bei regionalen Lymphknotenerkrankungen: Tuberkulose, Lymphogranulomatose, Metastasen von Sarkomen und Karzinomen.
-
Postoperatives oder traumatisches Lymphödem: Nach Lymphknotenausräumung, Bestrahlung, Verletzungen, Verbrennungen.
-
Lymphödem durch Blockierung des Abflusses: Übergreifen von krankhaften Prozessen auf die Lymphgefäße, retroperitoneale Fibrose, Unterleibstumoren.
1.7.5.4 Klinik
Spannungsgefühl, auch Brennen in der betroffenen Extremität, Einschränkung der Beweglichkeit. Die Schwellung kann das ganze Bein oder nur Ober- oder Unterschenkel befallen. Zunächst teigig-weich, wird sie zunehmend derb und nicht eindrückbar. Groteske Ausmaße werden als Elephantiasis bezeichnet. Frühzeichen: Einbeziehung der Zehen und Fußrücken mit indurierter Schwellung, die bei Ödemen anderer Ursache fehlt. Weitere Abklärung durch Lymphographie.
1.7.5.5 Therapie
Intermittierende Hochlagerung der Beine, Kompressionsstrümpfe oder Kompressionshosen, Lymphdrainage (ohne sofort anschließende Kompression unwirksam) und pneumatische Massage. Saluretika nach Bedarf. Behandlung und Prophylaxe von Streptokokken- und Pilzinfektionen. Bei den sekundären Formen Behandlung der Grundkrankheit. Unterbrochene Lymphbahnen (nach Trauma oder Lymphknotenausräumung) können durch mikrochirurgische Lymphgefäßtransplantation überbrückt werden (Spendergefäße aus dem Oberschenkel). Alternativ kommt eine lymphovenöse Anastomosierung in Betracht. Bei Elephantiasis lassen sich die ödematösen Gewebemassen durch Exzision des Subkutangewebes verkleinern.
1.8 Grundlagen und Methoden der kardiologischen Diagnostik
1.8.1 Kardialer Zyklus
Der kardiale Zyklus besteht aus Systole und Diastole (◘ Abb. 1.34).


Herzzyklus (Ziffern I bis IV = Herztöne)
1.8.1.1 Systole
1.8.1.1.1 Isovolumetrische Kontraktion
Auch Anspannungszeit genannt. Mit Beginn der Ventrikelkontraktion überschreitet der Kammerdruck den niedrigen Vorhofdruck. Durch den Druckgradienten werden die AV-Klappen geschlossen und angespannt; dabei entsteht der 1. Herzton. Sein Hauptanteil stammt von der Mitralklappe, die mit größerer Wucht zugeschlagen wird. Die Klappenschwingungen erzeugen in den Vorhöfen die c-Welle. Ohne Volumenverschiebung, bei geschlossenen Semilunar- und AV-Klappen steigt der intraventrikuläre Druck schnell bis zum diastolischen Aorten- bzw. Pulmonalisdruck an. Dauer der isovolumetrischen Kontraktion etwa 0,5 ms. Positiv inotrop wirkende Pharmaka (Digitalis, Metaproterenol) verkürzen sie.
1.8.1.1.2 Austreibungsphase
Beginnt mit Öffnung der Semilunarklappen, die mit dem Beginn des Steilanstieges der Aortendruckkurve zusammenfällt. Nur unter abnormen Bedingungen entsteht ein Öffnungston (Ejektionsklick). Etwa die Hälfte des Kammervolumens wird im ersten Viertel der Systole ausgeworfen, dann sinkt die Austreibungsgeschwindigkeit und erreicht schließlich den Wert Null. Die Ejektionsfraktion (Schlagvolumen/enddiastolisches Ventrikelvolumen) beträgt durchschnittlich 70 %. Das Ende der Austreibungsphase markiert der Semilunarklappenschluss, der den 2. Herzton erzeugt und eine Inzisur der Aortendruckkurve bewirkt. Dauer der Austreibungsphase etwa 0,22 s. Ablesung an der Karotispulskurve möglich (Distanz vom Beginn des Steilanstieges bis zur Inzisur). Zu Beginn der Austreibung sinkt der Vorhofdruck infolge Tiefertretens der Ventilebene (x-Tal) und steigt dann mit zunehmender Vorhoffüllung zur v-Welle an, die kurz vor der AV-Klappenöffnung ihr Maximum erreicht.
1.8.1.2 Diastole
1.8.1.2.1 Isovolumetrische Relaxation
Beginnt mit dem Schluss der Semilunarklappen. Durch die Erschlaffung des Kammermyokards sinkt der intraventrikuläre Druck schnell ab. Die Phase der isovolumetrischen Relaxation endet, sobald der Kammerdruck den Vorhofdruck unterschreitet und dadurch die AV-Klappen aufgestoßen werden. Dauer der isovolumetrischen Relaxation 0,08 s.
1.8.1.2.2 Phase der schnellen (frühdiastolischen) Füllung
Unmittelbar nach Öffnung der AV-Klappen ist der Druckgradient zwischen Vorhof und Kammer am größten. Die Klappenöffnung erzeugt normalerweise keinen Ton. Das ruckartig in die Kammer einschießende Blut spannt die Ventrikelmuskulatur an. Dies führt bei Kindern und Jugendlichen zum 3. Herzton oder Füllungston. Die Phase der frühdiastolischen Füllung umfasst das erste Drittel der Diastole. Unter raschem Druckausgleich zwischen Vorhof und Kammer wird bereits der größte Teil des Füllungsvolumens vom Ventrikel aufgenommen. Der Vorhofdruck sinkt.
1.8.1.2.3 Phase der langsamen Füllung (Diastase )
Umfasst das mittlere Drittel der Diastole mit nur noch geringer Blutaufnahme der Kammer. Bei Tachykardien ist diese Phase verkürzt, ohne dass die Kammerfüllung leidet. Bei behinderter schneller Füllungsphase (z. B. Mitralstenose) ist der Blutzufluss während der Diastase kompensatorisch erhöht. In dieser Situation beeinträchtigen Tachykardien Kammerfüllung und Herzleistung.
1.8.1.2.4 Vorhofsystole (spätdiastolische Füllungsphase)
Die Vorhofkontraktion (a-Welle der Vorhofdruckkurve) am Ende der Diastole erzeugt einen Druckgradienten mit Gefälle zum Ventrikel. Die Folge ist ein ruckartiger Bluteinstrom in die Kammern, der zur Anspannung der Kammerwand führt. Dabei entsteht der 4. Herzton oder Vorhofton. Er ist phonokardiographisch als tieffrequente Schwingung etwa 0,12 s nach Beginn der P-Zacke nachzuweisen, wegen geringer Intensität aber nur beim Kind und bei pathologischer Steigerung der Vorhofaktivität zu hören. Am Ende der Vorhofsystole besteht Druckausgleich zwischen Vorhöfen und Kammern, der Bluteinstrom ist minimal, in den Kammern herrscht der sog. enddiastolische Druck (rechte Kammer 0–8 mmHg, linke Kammer 4–12 mmHg).
1.8.2 Auskultation
1.8.2.1 Auskultationsbereiche
Sie liegen nicht direkt über den Klappen, nach denen sie benannt sind. Aorten- und Pulmonalareal sind stromabwärts der jeweiligen Klappe lokalisiert, das Trikuspidalareal über dem rechten Ventrikel, das Mitralareal über der Spitze. Es handelt sich um Regionen, die dem optimalen Wahrnehmungsbereich der verschiedenen Schallquellen entsprechen.
-
Mitralareal: Region der Herzspitze. Punctum maximum der von der Mitralklappe fortgeleiteten Schallphänomene. Zugleich Punctum maximum harmloser Strömungsgeräusche, die an der Aortenklappe entstehen. Sehr laut ist hier auch das Systolikum der Aortenstenose.
-
Trikuspidalareal: 4. bis 5. ICR am linken Sternalrand. Hierhin projizieren sich die Geräusche von der Trikuspidalklappe, deren zusätzliches wichtiges Kriterium eine inspiratorische Intensitätszunahme ist. Durch inspiratorische Sogwirkung auf die extrathorakalen Venen nehmen der venöse Zustrom und das rechtsventrikuläre Schlagvolumen zu, in der Exspiration geschieht das Gegenteil.
-
Pulmonalareal: 2. ICR links parasternal. Auskultationspunkt der von der Pulmonalklappe ausgehenden Schallerscheinungen. Nur hier ist der Pulmonalklappenschlusston und damit die Spaltung des 2. Tones zu hören. Auch der Aortenklappenschlusston wird hier deutlich gehört. Bei Überlagerung beider Töne kann der 2. Herzton hier sein Punctum maximum haben.
-
Aortenareal (1. und 2. ICR rechts parasternal): Projektionsbereich der Aortenklappe. Punctum maximum des Systolikums der Aortenstenose. Auch akzidentelle systolische Geräusche der Aortenklappe sind hier deutlich zu hören.
-
Erb-Punkt (3. ICR am linken Sternalrand): Projektionspunkt aller Klappen, wenngleich nicht deren optimaler Wahrnehmungspunkt. Punctum maximum des diastolischen Geräusches der Aorteninsuffizienz und der Pulmonalinsuffizienz. Systolikum der Pulmonalstenose (Fortleitung nach oben), der Subaortenstenose (keine wesentliche Fortleitung zur Aorta) und des Ventrikelseptumdefektes (Punctum maximum im 4. ICR).
1.8.2.2 Zur Auskultationstechnik
1.8.2.2.1 Stethoskop
Empfehlenswert sind Doppelkopfstethoskope. Membranteil: Optimal für die Wahrnehmung hochfrequenter Töne und Geräusche, da die tiefen Frequenzen weggefiltert werden (2. Herzton, Mitralöffnungston, Diastolikum der Aorteninsuffizienz). Glockenteil: Optimal für die Wahrnehmung mittel- und tiefrequenter Schallphänomene, da die hohen Frequenzen weggefiltert werden (3. Herzton, Vorhofton, Diastolikum der Mitralstenose).
1.8.2.2.2 Untersuchungsgang
Systematisches Aufsuchen aller 5 Auskultationsareale. Den Patienten normal atmen lassen, um die physiologische inspiratorische Spaltung des 2. Tones (Pulmonalareal) und atemabhängige Schwankungen der Geräuschintensität (Trikuspidalklappenphänomene) zu erfassen. Zunächst Identifizierung des 1. und 2. Herztones zur Bestimmung von Systole (Intervall zwischen 1. und 2. Ton) und Diastole (Intervall zwischen 2. und 1. Ton). Hilfsmittel: simultanes Tasten des Karotispulses, dessen Pulswelle mit der Systole zusammenfällt. Herztöne und Geräusche gesondert analysieren, Punctum maximum bestimmen.
1.8.2.2.3 Phonokardiographie
Herzschallschreibung mittels eines Phonokardiographen (Mikrophon-Verstärker-Registriergerät). Vorteile: Exakte zeitliche Bestimmung der Herzschallphänomene (durch simultane Aufzeichnung von EKG und Druckkurven), Erfassung nicht hörbarer tiefer Frequenzen, Analyse der Schallfrequenzen (durch Schallfilter für verschiedene Frequenzbereiche), Analyse der Geräuschformen, Dokumentation des Herzschallbefundes.
1.8.2.3 Auskultationsbefunde
1.8.2.3.1 1. Herzton
Punctum maximum Herzspitze, manchmal 4. ICR links parasternal. Lautstärke abhängig von der Steilheit des Druckanstieges im linken Ventrikel und von der Stellung der Mitralklappensegel zum Zeitpunkt des Klappenschlusses (normalerweise sind die Klappensegel nach der Vorhofkontraktionsphase einander weitgehend genähert, sie sind „gestellt“.
-
Lauter 1. Herzton bei:
-
Mitralstenose (mit beweglichen Klappensegeln) und verkürzter PQ-Zeit, weil die Klappen in beiden Fällen aus geöffneter Stellung zugeschlagen werden.
-
Hyperzirkulation (Fieber, Anämie, Thyreotoxikose), Erregung und körperlicher Belastung, weil sich der linke Ventrikel forciert kontrahiert. Bei diesen Zuständen gesteigerter Herzaktivität ist auch der 2. Herzton akzentuiert.
-
-
Leiser 1. Herzton bei Mitralinsuffizienz (fehlender Klappenschluss) und verlängerter PQ-Zeit (Klappe bei Systolenbeginn weitgehend geschlossen).
-
Wechselnde Lautstärke beim totalen AV-Block. Abschwächung des 1. Herztones (und des 2. Herztones) bei Emphysem und Perikarditis exsudativa (großer Abstand zum Ohr), akuter Herzinsuffizienz durch Infarkt (verlangsamter Druckanstieg) und im Schock.
1.8.2.3.2 2. Herzton
Besteht aus 2 Komponenten: Aortenton (Schluss der Aortenklappe: II A) und Pulmonalton (Schluss der Pulmonalklappe: II P), abgekürzt. Entsprechend dem höheren Druckgradienten zwischen Gefäß und Ventrikel in der frühen Diastole ist der Aortenton wesentlich lauter. Der Pulmonalton ist nur im Pulmonalareal hörbar, der Aortenton überall (Punctum maximum über der Herzbasis).
-
Physiolgische Spaltung des 2. Tones: II A und II P fallen in expiratorischer Atemruhe zusammen, während der Inspiration verschiebt sich II P hinter II A (wegen inspiratorischer Zunahme des rechtsventrikulären Schlagvolumens und Verlängerung der rechtsventrikulären Systole): dum-ta→dum-tra → dum-ta-ta. Dieses Phänomen ist nur über dem Pulmonalareal wahrnehmbar, an den übrigen Auskultationsstellen ist der 2. Herzton mit dem Aortenton identisch. Bei älteren Menschen fehlt die physiologische Spaltung des 2. Tones.
-
Spaltung des 2. Tones in normaler Reihenfolge: II A vor II P mit breitem Spaltungsintervall (in Inspiration und Exspiration): Rechtsschenkelblock (verspäteter Pulmonalklappenschluss), Pulmonalstenose (verlängerte Austreibungszeit), Mitralinsuffizienz (verkürzte Austreibungszeit des linken Ventrikels, Vorverlegung von II A). In allen Fällen expiratorische Verkleinerung des Spaltungsintervalls. Atemunabhängige (fixierte) Spaltung des 2. Herztones nur bei Vorhofseptumdefekt (konstante Volumenbelastung des rechten Ventrikels durch Shunt-Blut).
-
Spaltung des 2. Tones in umgekehrter Reihenfolge: (II P vor II A (paradoxe Spaltung) bei Verspätung des Aortenklappenschlusses (Linksschenkelblock, offener Ductus Botalli, Aortenstenose). Charakteristische inspiratorische Verkleinerung und exspiratorische Vergrößerung des Spaltungsintervalls (wegen inspiratorischer Verzögerung des Pulmonalklappenschlusses rückt II P zu II A auf). Die Spaltung kann inspiratorisch verschwinden.
-
Lauter Aortenton: Bei Hypertonie (Punctum maximum im Aortenareal).
-
Leiser Aortenton: Aortenstenose, Aorteninsuffizienz, Hypotonie.
-
Lauter Pulmonalton: Bei Drucksteigerung im kleinen Kreislauf (pulmonale Hypertonie. Cor pulmonale, Mitralvitien etc.).
-
Leiser Pulmonalton: Pulmonalstenose.
1.8.2.3.3 3. Herzton
Frühdiastolischer Füllungston, beim Erwachsenen stets pathologisch, wenn hörbar. Ursache: ruckartige Dehnung und Anspannung der Ventrikelwand bei erhöhtem Füllungsdruck und/oder Füllungsvolumen (Mitralinsuffizienz, Herzinsuffizienz mit Vorhofstauung und vergrößerter Restblutmenge, die schon eine Vordehnung bewirkt, Hyperzirkulation mit Tachykardie).
1.8.2.3.4 4. Herzton
Vorhofton, beim Erwachsenen vor allem hörbar, wenn der Vorhof erhöhte Kontraktionsleistung vollbringt: Füllung eines hypertrophierten Ventrikels erfordert die Erzeugung eines höheren enddiastolischen Ventrikeldruckes durch den Vorhof (Hypertonie, Aortenstenose, Pulmonalstenose). Noch kein Insuffizienzzeichen. Auftreten beim Infarkt allerdings Vorzeichen der drohenden Linksinsuffizienz.
1.8.2.3.5 Systolische Extratöne
Frühsystolisch kann es bei starker Dilatation der großen Gefäße zum Gefäßdehnungston oder Ejektionsklick kommen.
-
Aortaler Ejektionsklick: Aortenaneurysma, mittelgradige Aortenstenose (valvulär), Aorteninsuffizienz, Aortenlues, Fallot-Tetralogie. Punctum maximum an der Herzspitze.
-
Pulmonaler Ejektionsklick: Pulmonale Hypertonie, Pulmonalstenose (valvulär), Vorhofseptumdefekt, idiopathische Pulmonalisektasie. Punctum maximum im 3. ICR links parasternal.
-
Mesosystolischer Klick: Mittel- bis spätsystolischer Extraton, pathognomonisch für einen Mitralklappenprolaps in den linken Vorhof.
1.8.2.3.6 Galopprhythmus
Dreier-Rhythmus (1. Herzton, 2. Herzton, Zusatzton), der nur bei höherer Herzschlagfrequenz wie ein Pferdegalopp imponiert. Klinische Bedeutung nicht von der Frequenz, sondern von der Art des Dreier-Rhythmus abhängig (◘ Tab. 1.5).
1.8.2.3.7 Diastolische Extratöne
Öffnungstöne stenosierter AV-Klappen.
-
Mitralöffnungston (MÖT): Frühdiastolischer Extraton bei Mitralstenose. Intervall zwischen Aortenton und MÖT (II-MÖT-Intervall) atemunabhängig. Punctum maximum 4. und 5. ICR von der Herzspitze zum Sternalrand.
-
Trikuspidalöffnungston (TÖT): Bei Trikuspidalstenose. Wird während der Inspiration lauter. Das II-TÖT-Intervall nimmt inspiratorisch zu.
1.8.2.3.8 Systolische Geräusche
1.8.2.3.8.1 Austreibungsgeräusche
Entstehen durch Wirbelbildung an den Semilunarklappen bei:
-
organischer Aorten- und Pulmonalstenose (Druckaustreibungsgeräusch: spindelförmiges Pressstrahlgeräusch).
-
relativer Aorten- und Pulmonalstenose (Volumenaustreibungsgeräusch: leiser und weicher als organische Stenosegeräusche). Vorkommen: Begleitsystolikum bei Aorteninsuffizienz, Systolikum bei Vorhofseptumdefekt und Hyperzirkulation. Ursache: Das normal weite Ostium ist für ein vergrößertes Schlagvolumen relativ zu eng, daher Wirbel erzeugende Zunahme der Strömungsgeschwindigkeit des Blutes.
Die Austreibungsgeräusche beginnen stets nach dem 1. Herzton, weil sie erst nach der Semilunarklappenöffnung einsetzen können.
1.8.2.3.8.2 Refluxgeräusche
Entstehen bei systolischem Reflux durch insuffiziente AV-Klappen und Ventrikelseptumdefekte. Charakteristika: sofort mit dem 1. Herzton einsetzend, bandförmig, holosystolisch (da der Druckgradient für den Reflux in der frühesten Systole entsteht und erst nach dem 2. Herzton verschwindet). Vorkommen: Mitral- und Trikuspidalinsuffizienz, Ventrikelseptumdefekt.
1.8.2.3.9 Diastolische Geräusche
1.8.2.3.9.1 Einströmgeräusche
Entstehen bei organischer oder relativer AV-Klappenstenose durch Wirbelbildung während der Kammerfüllung.
-
Organische Mitral- und Trikuspidalstenose: Protodiastolisches, nach dem MÖT bzw. TÖT einsetzendes Decrescendogeräusch (Druckgradient nimmt schnell ab) und präsystolisches Crescendogeräusch (spätsystolischer Anstieg des Druckgradienten durch Vorhofkontraktion).
-
Relative Klappenstenose: Protodiastolisches Trikuspidalströmungsgeräusch (Vorhofseptumdefekt, Hyperzirkulation) und protodiastolisches Mitralströmungsgeräusch (Mitralinsuffizienz). In beiden Fällen brüske Kammerfüllung aus volumenüberlasteten Vorhöfen durch die relativ zu enge AV-Klappe. Die protodiastolischen Einströmgeräusche sind vom 2. Herzton deutlich abgesetzt (Intervallgeräusche), mittel- bis tieffrequent, rau.
1.8.2.3.9.2 Refluxgeräusche
Entstehen bei Aorten- und Pulmonalinsuffizienz durch den diastolischen Reflux aus den großen Gefäßen in die Ventrikel. Charakteristika: frühdiastolisches oder Sofortgeräusch (da der Reflux beim 2. Herzton beginnt) von Decrescendocharakter (da der Druckgradient schnell absinkt), hochfrequent, oft leise.
1.8.2.3.10 Anastomosengeräusche
Entstehen an der Kommunikationsstelle zweier Gefäße mit unterschiedlichem Blutdruck. Die Geräuschintensität ist dem Druckgradienten und damit der Strömungsgeschwindigkeit des Blutes an der Verbindungsstelle proportional. Vorkommen: Offener Ductus Botalli. Charakteristika: Kontinuierliche systolisch-diastolische Geräusche, die bis zum Ende der Systole anschwellen und in der Diastole abschwellen. Ein Druckgradient bleibt in der Diastole bestehen, daher die Bezeichnung als „Maschinengeräusch“.
1.8.2.3.11 Lautstärken-Skala der Herzgeräusche
-
Grad I: Sehr leise, nur bei großer Konzentration zu hören, kann beim ersten Aufsetzen des Stethoskops überhört werden.
-
Grad II: Leise, wird sofort gehört.
-
Grad III: Laut, jedoch ohne tastbares Schwirren.
-
Grad IV: Noch lauter, mit tastbarem Schwirren.
-
Grad V: Sehr laut, schon beim Aufsetzen der Stethoskopkante zu hören.
-
Grad VI: Sehr laut, auch nach Abheben des Stethoskops von der Brustwand zu hören (Distanzgeräusch).
1.8.3 Perkussion
-
Absolute Herzdämpfung: Kleiner Bezirk in der Mitte des Herzfeldes, der nicht von Lungengewebe überlagert ist. Dämpfung schon bei leiser Perkussion. Verschwindet bei ausgeprägtem Emphysem, das an Rechtsherzbelastung denken lässt.
-
Relative Herzdämpfung: Entspricht annähernd der Herzform und Herzgröße. Die Perkussionsfigur reicht aber nur zur groben Orientierung aus.
1.8.4 Palpation
-
Aortenareal: Schwirren bei Aortenstenose, Pulsationen bei Dilatation der Aorta ascendens.
-
Pulmonalareal: Schwirren bei Pulmonalstenose, Schnappen bei pulmonaler Hypertonie (durch Klappenschluss), verstärkte Pulsationen bei hoher Durchflussrate (Vorhofseptumdefekt).
-
Linke Parasternalregion: Abnorme Pulsationen bei Vergrößerung bzw. Hypertrophie des rechten Ventrikels (bei Druckbelastung „hebend“, bei Volumenbelastung „schleudernd“). Spätsystolisches Schleudern bei Mitralinsuffizienz, Schwirren bei Ventrikelseptumdefekt (4. ICR).
-
Epigastrium: Pulsationen bei Dilatation und Hypertrophie des rechten Ventrikels, besonders in Kombination mit Zwerchfelltiefstand infolge Emphysems.
-
Spitzenregion: Systolisches Schwirren bei Mitralinsuffizienz. Diastolisches Schwirren bei Mitralstenose. Hebender Spitzenstoß bei Aortenstenose und Hypertonie, schleudernder Spitzenstoß von größerer Flächenausdehnung bei Aorteninsuffizienz und Mitralinsuffizienz.
1.8.5 Röntgenuntersuchung
Thoraxaufnahme in 2 Ebenen, Herzfernaufnahme (2 m) zur Größenbestimmung. Mit dieser Standarduntersuchung werden Form- und Größenänderungen erfasst. Zur Analyse des Röntgenbildes muss man die Anatomie der Herzsilhouette kennen (◘ Abb. 1.35a, b).


Herzsilhouette und Größenbestimmung des Herzens: a Herzprojektion im Sagitalbild (posterior-anteriorer Strahlengang), b Herzprojektion im Linksseitenbild (dextro-sinistraler Strahlengang), c Schema zur Größenbestimmung des Herzens (VC = V. cava superior, RA = rechtes Atrium, RV = rechter Ventrikel, PU = Pulmonalarterie, LA = linkes Atrium, LV = linker Ventrikel, AO = Aorta, MR = Medianabstand rechts, ML = Medianabstand links, TDC = Transversaldurchmesser des Thorax)
1.8.5.1 Größenbestimmung
Der Transversaldurchmesser des Herzens setzt sich aus dem größten Medianabstand links (ML) und dem größten Medianabstand rechts (MR) zusammen. Er wird zum Transversaldurchmesser des Thorax (TDC) in Beziehung gesetzt. Normalerweise ist der Transversaldurchmesser des Herzens nicht größer als der halbe Transversaldurchmesser des Thorax (◘ Abb. 1.35c).
1.8.6 Elektrokardiographie
1.8.6.1 Entstehung des EKG
1.8.6.1.1 Ruhepotenzial
Im Ruhezustand sind die Herzmuskelfasern außen positiv, innen negativ elektrisch geladen (◘ Abb. 1.36). Das Ruhepotenzial der Fasermembran entsteht durch die Auswärtsdiffusion von Kaliumionen, deren Konzentration innen 30-mal höher ist als außen. Dieser Konzentrationsgradient bleibt bestehen, da die Membran für die Anionen des Faserinnern (organisch gebundene Phosphat- und Sulfatgruppen, Proteinanionen) undurchlässig ist. Die zurückgehaltenen Anionen erzeugen an der Innenseite ein negatives elektrisches Potenzial, das so lange steigt, bis im Gleichgewichtszustand ebenso viele Kaliumionen nach außen diffundieren wie durch die Anziehungskraft der Anionen (Elektrodiffusion) nach innen zurückgeholt werden. Das ist bei dem Wert des Ruhepotenzials von –90 mV der Fall. Die Konzentration der Natriumionen ist außen 10-fach höher als innen. Dieser Konzentrationsgradient kann sich nicht ausgleichen, weil die ruhende Faser für frei diffundierende Natriumionen praktisch undurchlässig ist. Erzeugt und aufrecht erhalten werden die in entgegengesetzter Richtung verlaufenden Konzentrationgradienten von Natrium- und Kaliumionen durch die „Natrium-Kalium-Pumpe“ der Zellmembran (Mg++-abhängige Na+/K+-ATPase). Sie befördert unter Energieverbrauch für 3 Natriumionen nach außen 2 Kaliumionen nach innen, entfernt also positive Ladung aus dem Zellinnern. Die Ionenbewegungen durch die Membran der Herzmuskelfasern erfolgen in spezifischen, vom Membranpotenzial gesteuerten Ionenkanälen, die aus Proteinkomplexen mit einer Pore bestehen. Heute sind deren Gene weitgehend bekannt.


Ruhepotenzial der Herzmuskelfaser
1.8.6.1.2 Aktionspotenzial
Es entsteht durch die Ionenströme an der Herzmuskelfasermembran während der Erregungsausbreitung und Erregungsrückbildung.
1.8.6.1.2.1 Depolarisation
Wenn das RuhepotenzZial über den Schwellenwert ansteigt (weniger negativ wird), öffnen sich für sehr kurze Zeit die Natriumkanäle und lassen Na+-Ionen ins Zellinnere strömen. In dieser Phase sind die K+-Kanäle gschlossen. Die Folge ist eine Ladungsumkehr an der Zellmembran (◘ Abb. 1.37). Die Na+-Kanäle werden nach der Ladungsumkehr sofort inaktiviert und lassen nur wenig Natriumionen einwärts fließen, bis sie definitiv geschlossen werden. Das entstandene Potenzial breitet sich schnell auf die Nachbarschaft aus und lässt die Depolarisation fortschreiten. Eine Ausnahme machen die Schrittmacherzellen des Sinusknotens und AV-Knotens. Bei ihnen erfolgt die Depolarisation durch den Einstrom von Calciumionen.


Ionenströme während des Aktionspotenzials (Depolarisation und Repolarisation) der Herzmuskelfaser
1.8.6.1.2.2 Repolarisation
In dieser Phase wird das Ruhepotenzial wiederhergestellt. Gleich nach der Ladungsumkehr öffnen sich K+-Kanäle, zuerst langsame, danach schnelle, durch die Kaliumionen auswärts diffundieren. Verzögert wird die Repolarisation durch die Öffnung von Ca++-Kanälen des L-Typs, durch die Calciumionen ins Zellinnere fließen. Sie bewirken die Freisetzung weiterer Calciumionen aus dem endoplasmatischen Retikulum und setzen zusammen mit diesen die Muskelkontraktion in Gang. Später wird das Calcium teils in das endoplasmatische Retikulum zurückgepumpt, teils durch einen Na+/Ca++-Austauscher nach außen befördert (▶ Abschn. 1.9.1).
1.8.6.1.3 Erregungsausbreitung
In ◘ Abb. 1.38 ist die Aufzeichnung des Aktionspotenzials der Einzelfaser dargestellt. Während der Erregungsausbreitung wird jede Einzelfaser an ihrer Außenfläche zu einem elektrischen Dipol: Negativer Pol auf der schon erregten, positiver Pol auf der noch unerregten Seite. Dieser Dipol stellt eine elektrische Spannungsquelle dar, die über 2 außen anliegende Elektroden mit einem Voltmeter gemessen werden kann. Ein positiver Ausschlag resultiert, wenn die Polung des Faserdipols mit der des Messgerätes übereinstimmt, ein negativer Ausschlag bei entgegengesetzter Polungsrichtung. Am Ende der Depolarisationsphase ist die gesamte Außenfläche negativ geladen und der Dipol verschwunden. Das Messgerät zeigt auf Null. In der Repolarisationsphase entsteht ein Faserdipol in umgekehrter Richtung mit negativem Ausschlag am Messgerät. (Im Experiment an der Einzelfaser beginnt die Repolarisation an der zuerst depolarisierten Stelle).


Aktionspotenzial der Einzelfaser (nach Guyton): A = Depolarisationsphase, B = Zustand der Vollerregung (totale Depolarisation), C = Repolarisationsphase, D = Ruhepotenzial (totale Repolarisation)
1.8.6.1.4 Aktionspotenzial des Herzens
Wie die Einzelfaser wird das ganze Herz während des Erregungsablaufes zu einem Dipol, der allerdings aus zahlreichen Einzeldipolen zusammengesetzt ist. Seine Richtung und Größe ändern sich fortlaufend. In der EIektrokardiographie wird der Dipol als Vektor – in der Form eines Pfeiles – dargestellt. Es entsprechen: die Pfeilspitze dem positiven, das Pfeilende dem negativen Pol des Dipols, die Pfeilrichtung der Dipolachse und die Pfeillänge der Größe der Potenzialdifferenz des Dipols (◘ Abb. 1.39).


Vektorielle Darstellung des Erregungsablaufes am Herzen (schraffierte Zonen elektronegativ): a–d Momentanvektoren der Depolarisation, e Stadium der Vollerregung (totale Depolarisation), f Momentanvektor der Repolarisation
Die Erregung der Kammern beginnt auf der linken Seite des Septums, erfasst dann die subendokardialen Kammerabschnitte und dringt von dort zur Außenseite des Herzens vor. Die ◘ Abb. 1.39a–d zeigt einige der nacheinander entstehenden Summationsvektoren, auch Momentanvektoren genannt, weil sie nur für einen Augenblick das elektrische Spannungsfeld des Herzens repräsentieren. Der größte Momentanvektor ist erreicht, wenn etwa die Hälfte des Kammermyokards erregt ist und negative und positive Ladungen einander die Waage halten. Er wird als elektrische Herzachse bezeichnet und zeigt die Hauptausbreitungsrichtung der Erregung an. Im Stadium der Vollerregung fehlen Potenzialdifferenz und Vektor. Es folgt die Erregungsrückbildung (Repolarisation), die von der Spitze zur Basis verläuft, so dass die Hauptvektoren von Depolarisation und Repolarisation die gleiche Richtung haben.
1.8.6.2 EKG-Ableitungen
1.8.6.2.1 Frontalebene
Die Ableitungselektroden werden am linken Arm, am rechten Arm und am linken Bein angelegt. Die Ableitungsachsen entsprechen den Seiten des gleichseitigen Einthoven-Dreiecks mit dem Herzen im Mittelpunkt (◘ Abb. 1.40). An den Ecken dieses Dreiecks kommen die genannten Extremitäten mit dem elektrischen Spannungsfeld des Herzens in Berührung. Die Polung der Ableitungen ist aus der Abbildung ersichtlich:


EKG-Ableitung in der Frontalebene: a Einthoven-Dreieck, b Axiales System der Standardableitungen
-
Ableitung I: rechter Arm → linker Arm
-
Ableitung II: rechter Arm → linkes Bein
-
Ableitung III: linker Arm → linkes Bein
Jede Ableitung erfasst von einem Momentanvektor des Herzens nur den Teil, der sich senkrecht auf die Ableitungsachse projiziert (◘ Abb. 1.40a). Für die Projektion ist allein der Winkel zwischen Momentanvektor und Ableitungsachse maßgebend. Er ändert sich nicht, wenn man die Ableitungsachsen ohne Richtungsänderung bis zum Mittelpunkt des Dreiecks verschiebt (◘ Abb. 1.40b). Durch diese Umzeichnung erhält man eine axiale Anordnung der Ableitungen, in der jede Ableitung durch ihren Winkel mit der Horizontalachse gekennzeichnet ist.
Aus der ◘ Abb. 1.40b geht hervor, dass die Winkel oberhalb der Horizontalen im Gegenuhrzeigersinn von 0–180° gemessen werden und ein negatives Vorzeichen erhalten. Unterhalb der Horizontalen erfolgt die Winkelmessung im Uhrzeigersinn von 0–180° mit positivem Vorzeichen. Zeigt der projizierte Vektor auf den positiven Pol der Ableitungsachse gibt es im EKG einen nach oben gerichteten (positiven) Ausschlag, zeigt er zum negativen Pol, ist der Ausschlag im EKG nach unten gerichtet (negativ).
Die bipolaren Einthoven-Ableitungen der Extremitäten werden durch die unipolaren Extremitätenableitungen nach Goldberger zu einem hexaxialen System erweitert. Bei den Goldberger-Ableitungen wird jeweils eine Extremität mit dem positiven Pol des Gerätes verbunden, während man die beiden anderen an den negativen Pol anschließt. Daraus ergeben sich Ableitungsachsen, die durch die Ecken und den Mittelpunkt des Einthoven-Dreiecks verlaufen und im hexaxialen System den Winkel zwischen 2 Standardableitungen teilen. Die Goldberger-Ableitungen haben folgende Bezeichnungen:
-
aVL: unipolare positive Elektrode am linken Arm
-
aVR: unipolare positive Elektrode am rechten Arm
-
aVF: unipolare positive Elektrode am linken Fuß
Das EKG-Bild der Goldbergerableitungen fügt sich hinsichtlich Höhe und Richtung der Ausschläge zwischen die Bilder der benachbarten Standardableitungen ein, mit Ausnahme von aVR, die in umgekehrter Richtung gepolt ist. Nach Umpolung zu -aVR, die in vielen EKG-Geräten möglich ist, ergibt sich ein Übergangsbild zwischen dem Bild von Ableitung I und II.
1.8.6.2.2 Horizontalebene
Der Erregungsablauf in der Horizontalebene wird durch die Brustwandableitungen (nach Wilson) erfasst. Die präkordiale Tastelektrode (positiver Pol) wird an der Brustwand in den Positionen V1, V2, V3, V4, V5 und V6 angelegt (◘ Abb. 1.41). Die Gegenelektrode (Wilson-Zentral- oder Sammelelektrode) liegt im Schnittpunkt der Ableitungsachsen und wird durch Zusammenschluss der 3 Extremitätenableitungen über hochohmige Widerstände erhalten. Die EKG-Kurve zeigt einen positiven Ausschlag, wenn der auf die Ableitungsachse projizierte Vektor in Richtung auf die Tastelektrode verläuft, einen negativen Ausschlag bei umgekehrter Verlaufsrichtung.


Ableitungsachsen des Brustwand-EKG
1.8.6.3 Grundform des EKG
Das Kurvenbild des normalen Erregungsablaufes ist in ◘ Abb. 1.42 dargestellt.


Grundform des EKG
1.8.6.3.1 Vorhofteil
-
P-Zacke: Depolarisationsphase beider Vorhöfe (Erregungsausbreitung). Der vordere Anteil ist dem rechten, der hintere dem linken zuzuordnen. Doppelgipfligkeit kommt vor. Die Repolarisation der Vorhöfe ist im EKG nicht sichtbar. Dauer bis 0,1 s.
-
PQ-Strecke: Überleitungszeit vom Vorhof zur Kammer. Genauer: Zeit vom Beginn der Vorhoferregung bis zum Beginn der Kammererregung. Umfasst Überleitung vom Sinusknoten zum AV-Knoten, vom AV-Knoten zum His-Bündel und von dort über beide Schenkel zu den Aufzweigungen der Purkinje-Fasern im Myokard. Dauer 0,12‒0,20 s.
1.8.6.3.2 Kammerteil
-
QRS-Komplex: Depolarisationsphase der Kammern (Erregungsausbreitung). Dauert vom Beginn der Kammererregung bis zur Vollerregung beider Kammern. Normalwert bis 0,10.
-
ST-Strecke: Stadium der Vollerregung (genauer: Stadium der langsamen Repolarisation mit negativer Ladung an der Oberfläche der Muskelfasern). Verläuft normalerweise in der isoelektrischen Linie.
-
T-Zacke: Phase der schnellen Repolarisation (Erregungsrückbildung), Nachschwankung.
-
Kammerendteil: ST-T Abschnitt, gesamte Repolarisationsphase.
-
QT-Strecke: Entspricht der Gesamtdauer der Kammererregung (Depolarisation + Repolarisation). Normalwert stark frequenzabhängig (Normbereich für jede Frequenz aus Tabellen und Nomogrammen abzulesen).
-
U-Zacke: Nachpotenzial (flach, häufig nicht erkennbar), das sich an die vollständige Repolarisation der Kammern anschließt und bereits in die Diastole fällt. Entstehung nicht sicher geklärt.
1.8.6.4 Lagetypen des EKG
Das Kurvenbild in den Extremitätenableitungen des EKG (Frontalebene) hängt von dem Winkel (α) ab, den die elektrische Herzachse mit der Horizontalen bildet. Die elektrische Herzachse liegt in der Hauptausbreitungsrichtung der Kammererregung (größter in die Frontalebene projizierter Momentanvektor der Erregungsausbreitung) und entspricht (mit Ausnahme der überdrehten Lagetypen) etwa der Frontalprojektion der anatomischen Herzachse. Die Konstruktion der mittleren elektrischen Herzachse ist aus ◘ Abb. 1.43 ersichtlich. In 2 Extremitätenableitungen werden vom Hauptausschlag der QRS-Gruppe die entgegengesetzten Ausschläge abgezogen. Dann trägt man die erhaltenen Partialvektoren auf der entsprechenden Ableitungsachse ab und errichtet an der Vektorspitze die Senkrechte. Die Verbindung vom Schnittpunkt der Senkrechten zum Nullpunkt ergibt die elektrische Herzachse.


Konstruktion der elektrischen Herzachse (aus Ableitung I und III: Winkel α 59°)
Die verschiedenen Lagetypen sind in ◘ Abb. 1.44 zusammengestellt. Im Kindes- und Jugendalter überwiegt der Steiltyp, in den mittleren Jahren der Mittellagetyp, jenseits des 50. Lebensjahres der Linkstyp. Die T-Zacke ist im allgemeinen der Kammeranfangsschwankung gleichgerichtet (konkordant). Ausnahmen: In Ableitung I beim Rechtstyp, in Ableitung III beim Mitteltyp gelegentlich negativ.


Lagetypen des EKG (Extremitätenableitungen)
1.8.6.5 Normales Brustwand-EKG
Das normale Brustwand-EKG hat bei allen Lagetypen des Extremitäten-EKG annähernd das gleiche Kurvenbild (◘ Abb. 1.45). Die Höhe der R-Zacken ist der unter den Ableitungsstellen vorhandenen Muskelmasse des Herzens proportional. Die Größe der R-Zacke nimmt von V1 bis V5 (manchmal nur bis V4) zu, in V6 ab. Die Ableitungspunkte V1 und V2 liegen über dem relativ muskelschwachen rechten Ventrikel, V3 im Grenzbereich, V4 an der Herzspitze, V5 und V6 an der Seitenwand des linken Ventrikels. Mit den hohen R-Zacken in den linkspräkordialen Ableitungen (V4–V6) korrespondieren tiefe S-Zacken in den rechtspräkordialen Ableitungen (V1 und V2), da der auf die seitlichen Ableitungen gerichtete positive Momentanvektor in den Ableitungen V1 und V2 in negativer Richtung (von der Brustwand weg) verläuft. Als Übergangszone bezeichnet man den Bereich des Überganges von dominierender S-Zacke zur dominierender R-Zacke (in ◘ Abb. 1.45 zwischen V1 und V2). Bei Hypertrophie der rechten Kammer verschiebt sich die Übergangszone nach links (→ V4), bei Hypertrophie der linken Kammer nach rechts (→ V1, V2).


Normales Brustwand-EKG
1.8.6.6 EKG bei Hypertrophie der Vorhöfe
Das EKG bei Vorhofhypertrophie ist in ◘ Abb. 1.46 dargestellt:


Vorhof-EKG: a normal, b P-destrokardiale, c P-sinsitrokardiale, d P-kardiale
-
P-dextrokardiale oder -pulmonale: Zeichen der Überlastung des rechten Vorhofes (Cor-pulmonale, Trikuspidalvitien, kongenitale Vitien mit Rechtsherzüberlastung). Es überwiegt der stärker nach rechts gedrehte Hauptvektor des rechten Vorhofes. Der Summationsvektor (elektrische Achse) der Vorhoferregung wird dadurch steiltypisch: PI flach, PII und PIII abnorm hoch, aber nicht über 0,10 s verbreitert, spitz und eingipfelig; in VI Anfangsteil deutlich positiv und zugespitzt (◘ Abb. 1.46b).
-
P-sinistrokardiale oder mitrale: Zeichen der Überlastung des linken Vorhofes (Mitralvitien, auch schwere Hypertonie und Aortenvitien). Es überwiegt der nach links gerichtete Vektor des linken Vorhofes. Der Summationsvektor dreht nach links, das Vorhof-EKG wird in seinem hinteren Abschnitt linkstypisch; wegen Verlängerung der Erregungsausbreitung wird die P-Zacke breiter als 0,10 s. PI und PII sind gekerbt oder doppelgipfelig, PIII ist biphasisch. P in V1 deutlich biphasisch mit breitem negativem hinteren Abschnitt (◘ Abb. 1.46c).
-
P-kardiale: Zeichen der Überlastung beider Vorhöfe (Vorhofseptumdefekt, rechts dekompensierte Mitral- und Aortenvitien). P-Zacke im vorderen Abschnitt rechtstypisch (in II und III und V1 deutlich positiv) im hinteren Abschnitt linkstypisch (in III und V1 negativ). In allen Ableitungen breiter als 0,10 s (◘ Abb. 1.46d).
1.8.6.6.1 Anmerkung
Die P-Zacke unterliegt starken Einflüssen des vegetativen Nervensystems:
-
Vagus-P: linkstypisch, flaches P in I und II, negatives P in III
-
Sympathikus-P: rechtstypisch, großes spitzes P in II und III, flaches P in I; tritt unter Belastung auf.
Vagus- und Sympathikus-P sind reversibel und nicht pathologisch.
1.8.6.7 EKG bei Hypertrophie der Kammern
Man unterscheidet:
-
konzentrische Widerstandshypertrophie durch überwiegende Faserverdickung (z. B. Aortenstenose, Hypertonie)
-
exzentrische Volumenhypertrophie durch überwiegende Faserverlängerung (z. B. Aorteninsuffizienz, Vorhofseptumdefekt).
Die Diagnose der Hypertrophie wird aus dem Brustwand-EKG gestellt: Größenzunahme der R-Zacke über dem hypertrophierten Ventrikel, Vertiefung der korrespondierenden S-Zacke auf der Gegenseite.
1.8.6.7.1 Besonderheiten
-
Widerstandshypertrophie: Depolarisation normal (QRS unter 0,10 s), Repolarisation wegen systolischer Kompression der Kammerwand durch den erhöhten Innendruck verlangsamt (mit dem Schweregrad zunehmende Diskordanz des Kammerendteils zur Kammeranfangsschwankung).
-
Volumenhypertrophie: Depolarisation wegen verzögerter Erregungsausbreitung im dilatierten Ventrikel verlangsamt (QRS-Verbreitung auf 0,11–0,12 s), Repolarisation normal (Konkordanz von Kammeranfangsschwankung und Kammerendteil). Erst in fortgeschrittenen Stadien Diskordanz des Kammerendteils.
1.8.6.7.2 Rechtshypertrophie
Steil- oder Rechtstyp im Extremitäten-EKG. Überhöhtes R in V1, tiefes S in V5 (◘ Abb. 1.47). Sokolow-Index: R(V1)+S(V5) >1,05 mV.


Kammer-EKG bei Rechtshypertrophie: a Widerstandshypertrophie, b Volumenhypertrophie
1.8.6.7.2.1 Zusätzliche Besonderheiten
-
Widerstandhypertrophie: QRS normal. In V1–V3 konvexe ST-Senkung und biphasisches oder negatives T; das gleiche in III (◘ Abb. 1.47a).
-
Volumenhypertrophie: QRS gering verbreitert. In V1 zweite R-Zacke (R’-Zacke). Isoelektrisches oder flach positives T in III und V1. Bei ausgeprägten Kammerendteilveränderungen spricht man von Rechtshypertrophie mit Rechtsschädigungszeichen (◘ Abb. 1.47b).
1.8.6.7.3 Linkshypertrophie
Linkstyp im Extremitäten-EKG (nicht obligatorisch) mit großer QRS-Amplitude; Überhöhtes R in V5 und V6, tiefes S in V1 und V2 (◘ Abb. 1.48). Sokolow-Index: S(V1)+(V5) >3,5 mV.


Kammer-EKG bei Linkshypertrophie: a Widerstandshypertrophie, b Volumenhypertrophie
1.8.6.7.3.1 Zusätzliche Besonderheiten
-
Widerstandshypertrophie: QRS normal. In V4–V6 konvexe ST-Senkung und biphasisches oder negatives T, das gleiche in I. ST-Hebung und positives T in III (◘ Abb. 1.48a).
-
Volumenhypertrophie: QR-Zeit in V5 und V6 verlängert (0,055 s). T in V5 und V6 positiv. Bei ausgeprägten Kammerendteilveränderungen spricht man von Linkshypertrophie mit Linksschädigungszeichen (◘ Abb. 1.48b).
1.8.6.7.3.2 Anmerkung
Bei gleichzeitiger Hypertrophie beider Ventrikel ist das EKG häufig stumm, da sich die Veränderungen gegenseitig kompensieren.
1.8.6.8 EKG bei Innenschichtschaden
Innenschichtschäden wirken sich nur auf den Kammerendteil des EKG (langsame und schnelle Repolarisationsphase) aus: Muldenförmige ST-Senkung, T-Abflachung oder negatives T. Ursache ist eine Erregungsabschwächung der Innenschicht, die im Stadium der Vollerregung (ST-Strecke) zu einer Potenzialdifferenz zwischen innen (geringe Elektronegativität) und außen (größere Elektronegativität) führt. Der ST-Vektor ist dem QRS-Vektor entgegengerichtet. Genese: Hypoxisch, toxisch, entzündlich.
-
Diffuser Innenschichtschaden (beide Ventrikel): Muldenförmige ST-Senkung und T-Abflachung in allen Ableitungen (◘ Abb. 1.49a).
Abb. 1.49 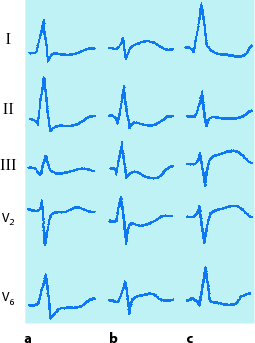
EKG bei Innenschichtschaden: a diffus, b rechtsventrikulär, c linksventrikulär
-
Rechtsventrikulärer Innenschichtschaden: Muldenförmige ST-Senkung und T-Abflachung in II, III, V2–V4. Entgegengesetzte Veränderungen in I und V6 (◘ Abb. 1.49b).
-
Linksventrikulärer Innenschichtschaden: Muldenförmige ST-Senkung und T-Abflachung in I, II, V5 und V6. Entgegengesetzte Veränderungen in III und V2 (◘ Abb. 1.49c).


1.8.6.9 EKG bei Außenschichtschaden
Außenschichtschäden entstehen in der Regel diffus (Perikarditis).
-
Frisches Stadium: ST-T Hebung in allen Ableitungen. Erregungsabschwächung außen. ST-Vektor hat die gleiche Richtung wie der QRS-Vektor. Erregungsausbreitung (QRS-Komplex) normal (◘ Abb. 1.50a).
Abb. 1.50 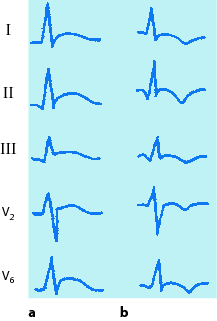
EKG bei Außenschichtschäden: a frisches Stadium, b Folgestadium
-
Folgestadium: Bei isoelektrischer ST-Strecke terminal negatives T. Erregungsverlängerung der Außenschicht. Beginn der schnellen Repolarisationsphase innen, daher verläuft der T-Vektor zum QRS-Vektor entgegengesetzt (◘ Abb. 1.50b).


1.8.6.10 EKG bei Elektrolytstörungen
1.8.6.10.1 Hypokaliämie
Bewirkt Erhöhung des Ruhepotenzials (Quotient Ka : Ki steigt). Schnelle Depolarisation aber verlangsamte Repolarisation.
EKG (◘ Abb. 1.51a): ST-Senkung, T-Abflachung unter zunehmender Vergrößerung der U-Welle, schließlich TU-Verschmelzungswelle. Deutlichste Ausprägung in V5 und V6. QRS normal. Ventrikuläre Extrasystolen.


EKG bei Kaliumveränderungen (obere Kurve normal, darunter zunehmende Schweregrade): a Hypokaliämie, b Hyperkaliämie
1.8.6.10.2 Hyperkaliämie
Bewirkt Verkleinerung des Ruhepotenzials, Verlangsamung der Depolarisation, Beschleunigung der Repolarisation.
EKG (◘ Abb. 1.51b): Zuerst Vergrößerung der T-Zacke (Zeltform), Schwund der U-Welle, dann erhöhter ST-Abgang, P-Abflachung, QRS-Verbreiterung, T-Negativität, PQ-Verlängerung (AV-Block).
1.8.6.10.3 Hypokalzämie
Verlängert das Stadium der Vollerregung (ST-Strecke) durch Verzögerung der langsamen Phase der Repolarisation.
EKG (◘ Abb. 1.52) Verlängerung der QT-Dauer durch lange ST-Strecke mit normaler T-Zacke. Die mechanische Systole (Ende beim 2. Herzton) kann hinter der elektrischen zurückbleiben (Hegglin-Syndrom).


EKG bei Hypo- und Hyperkalzämie
1.8.6.10.4 Hyperkalzämie
Verkürzt das Stadium der Vollerregung durch Beschleunigung der langsamen Phase der Repolarisation.
EKG (◘ Abb. 1.52): ST und folglich auch QT verkürzt; die T-Zacke geht unmittelbar aus dem abfallenden Schenkel der R-Zacke hervor.
1.8.6.11 Belastungs-EKG (Ergometrie )
1.8.6.11.1 Methode
Registrierung der Extremitäten- und Brustwandableitungen des EKG unter dosierter, stufenweise gesteigerter körperlicher Belastung am Fahrradergometer (im Sitzen oder Liegen). Durchführung in Gegenwart des Arztes.
1.8.6.11.2 Diagnostische Bedeutung
Nachweis von myokardialen Ischämiereaktionen, belastungsinduzierten Rhythmusstörungen, inadäquaten Frequenzsteigerungen (Störungen der Sinusknotenfunktion) und einer Belastungshypertonie. Zugleich wird die kardiopulmonale Belastbarkeit (Trainingszustand) erfasst. Indizien der Koronarinsuffizienz (▶ dort).
1.8.6.12 Langzeit-EKG
1.8.6.12.1 Methode
EKG-Registrierung auf Band oder elektronischem Speicher über 24 h, gewöhnlich unter Alltagsbelastungen mit Anfertigung eines Protokolls durch den Patienten.
1.8.6.12.2 Diagnostische Bedeutung
Nachweis intermittierender, insbesondere tachykarder Rhythmusstörungen als Erklärung für Schwindelanfälle und Herzsensationen. Erfassung von bedrohlichen ventrikulären Rhythmusstörungen. Effektivitätskontrolle jeder antiarrhythmischen Therapie. Erfassung intermittierender Bradyarrhythmien und Pausen zur Dokumentation vor Schrittmacherimplantation.
1.8.7 Invasive elektrophysiologische Untersuchungen
1.8.7.1 Methode
Einführung multipolarer Katheterelektroden in die Herzhöhlen auf venösem oder arteriellem Weg zur Registrierung der elektrischen Aktivität der Vorhöfe und Kammern.
1.8.7.2 Anwendung
Differenzierung atrioventrikulärer Leitungsstörungen (His-Bündel-EKG). Lokalisierung von Reentrykreisen und ektopischen Reizbildungsherden bei supraventrikulären und ventrikulären Tachyarrhythmien zur Vorbereitung der Ablationstherapie. Therapiekontrolle mittels Elektrostimulation.
1.8.8 Echokardiographie
1.8.8.1 Prinzip
Die Echokardiographie ist ein Verfahren zur Abbildung des Herzens und seiner Innenstrukturen durch reflektierten Ultraschall, mit dem auch Bewegungsabläufe an Herzklappen und Herzwand studiert werden können. Der zusätzliche Einsatz von Doppler- und Farbduplexfunktionen ermöglicht es, den Blutfluss im Herzen darzustellen und zu quantifizieren.
1.8.8.2 M-Mode-Echokardiographie
1.8.8.2.1 Methode
Der Schallkopf wird im 3. oder 4. Interkostalraum links parasternal aufgesetzt. Er sendet einen dünnen Ultraschallstrahl aus, mit dem sich die Längsschnittebene des Herzens in verschiedenen Richtungen ausloten lässt (◘ Abb. 1.53a). Da sich das Herz ständig bewegt, tun es auch die in Lichtpunkte transformierten Echos, die in einem schmalen Bündel von seinen Grenzflächen zurückgeworfen werden. Am Bildschirm des Oszilloskops werden die Echos fortlaufend gegen die Zeit aufgezeichnet, so dass sie als Linien erscheinen, deren Abstände denen der Grenzfächen des Herzens entsprechen. Das so gewonnene M-Mode-Echokardiogramm (von motion) wird auf Papier ausgedruckt und erlaubt es, die Abstände der Grenzflächen während eines Herzzyklus mit hoher zeitlicher Auflösung zu messen. Zur Illustration sind in ◘ Abb. 1.53b die M-Mode-Bilder von den 4 in ◘ Abb. 1.53a eingezeichneten Schallstrahlpositionen wiedergegeben. Das mitlaufende EKG markiert die Herzzyklen (Ende der Diastole am Beginn von QRS).


M-Mode-Echokardiogramm. a Schallstrahlrichtungen, b M-Mode-Bild. In der spitzennahen Position (1) folgen aufeinander: Echos der Brustwand, Echos der Vorderwand des rechten Ventrikels (ARV), echofreie rechte Herzhöhle (RV), Echos der rechten (RS) und der linken Seite (LS) des interventrikulären Septum, echofreie linke Herzhöhle (LV), Echos des hinteren Papillarmuskels (PPM), der Hinterwand des linken Ventrikels (PLV), des Perikards (PER) und die intensiven Echos der Lunge. In der nächst höheren Position (2) kommen das Endokard (EN), die Sehnenfäden und das vordere (AMV) und hintere (PMV) Segel der Mitralklappe zur Darstellung. Die linke Herzhöhle hat hier ihren größten enddiastolischen Durchmesser. Weiter aufwärts, in Position (3), verschwindet das hintere Mitralsegel, während das vordere noch gut abgebildet ist. Hinten erscheint die Hinterwand des linken Vorhofes (PLA). In der höchsten Position (4) passiert der Schallstrahl die Aortenwurzel mit den Aortenklappen (AV) und der Vorder- und Hinterwand der Aorta (Ao). Die Aortenhinterwand fällt mit der Vorderwand des linken Vorhofes (LA) zusammen


2-D-Ebenen (AO = Aorta, PA = Pulmonalarterie, RA/LA = rechtes/linkes Atrium, RV/LV = rechter/linker Ventrikel)


Schallkopfpositionen bei der 2-D-Echokardiographie


Schnittbilder: a Längsschnittebene, b Aortenklappenebene, c 4-Kammerblick (LV/RV = linker/rechter Ventrikel, LA/RA = linker/rechter Vorhof, PA = Pulmonalarterie, r = rechtskoronare Aortenklappentasche. l = linkskoronare Aortenklappentasche, a = akoronare Aortenklappentasche
1.8.8.2.2 Auswertung
-
Gemessen werden folgende Standardparameter:
-
enddiastolischer und endsystolischer Durchmesser des linken Ventrikels
-
enddiastolischer Durchmesser des rechten Ventrikels
-
endsystolische und enddiastolische Wanddicke der linksventrikulären Hinterwand und des interventrikulären Septums
-
enddsystolischer Durchmesser des linken Vorhofs
-
enddiastolischer Durchmesser der Aortenwurzel
-
-
Berechnet werden:
-
Fractional shorting (FS): Prozentuale Verkürzung des links-ventrikulären enddiastolischen Durchmessers in der Systole
-
linksventrikuläre (enddiastolische und endsystolische) Volumina zur Bestimmung der Ejektionsfraktion
-
-
Qualitativ beurteilt werden:
-
Mitralklappe
-
Aortenklappe
-
Kontraktilität von Septum und linksventrikulärer Hinterwand
-
1.8.8.3 Zweidimensionale (2-D-)Echokardiographie
1.8.8.3.1 Methode
Mit dem B-Mode werden Schnittbilder des Herzens gewonnen.
1.8.8.3.2 Standardebenen (◘ Abb. 1.54)
-
Längsachsenebene (in der Längsachse des Herzens, senkrecht zur Körperoberfläche)
-
Kurzachsenebene (senkrecht zur Längsachsenebene und zur Körperoberfläche)
-
Vierkammerebene (parallel zur Körperoberfläche)
1.8.8.3.3 Schallkopfpositionen (◘ Abb. 1.55) und Schnittbilder (◘ Abb. 1.56)
-
links parasternal in der Längsachse zur Abbildung der Längsschnittebene (◘ Abb. 1.56a)
-
links parasternal um 90° im Uhrzeigersinn in die kurze Achse gedreht für die Abbildung der Aortenklappenebene (◘ Abb. 1.56b)
-
apikal oder subkostal für den 4-Kammerblick (◘ Abb. 1.56c).
Einen Zweikammerblick mit der linksventrikulären Ausflussbahn (sog. Dreikammerblick) erhält man von apikal durch Drehung des Schallkopfs um 90° im Gegenuhrzeigersinn. Durch Schwenken des Schallkopfs in der Kurzachsenebene können Mitralklappenebene, Papillarmuskelebene und Apexebene dargestellt werden, mit einem suprasternal aufgesetzten Schallkopf Aorta und A. pulmonalis in 2 Ebenen.
Die ◘ Abb. 1.57 zeigt 4 Originalbilder von der 2-D-Echokardiographie eines Herzgesunden.


Transthorakale Echokardiographie eines Herzgesunden. Oben: links: parasternaler Längsachsenschnitt; rechts: parasternaler Querachsenschnitt in Höhe der Papillarmuskeln. Unten: links: apikaler 4-Kammerblick; rechts: apikaler 4-Kammerblick mit Farbdoppler (rot: Blutfluss zum Schallkopf hin, blau: Blutfluss vom Schallkopf weg).(LV/RV = linker/rechter Ventrikel, LA/RA = linker/rechter Vorhof, IVS = interventrikuläres Septum, IAS = interatriales Septum, PW = Posterolateralwand, AO = Aorta, AK = Aortenklappe, MK = Mitralklappe, PM = Papillarmuskel
1.8.8.3.4 Auswertung
Zu messen bzw. zu beurteilen sind:
-
Dimensionen: Form, Größe und Volumina der Herzhöhlen (Ejektionsfraktion), Wanddicke der Kammern, Echogenität des Endokards
-
Funktionen: Globale und regionale Kontraktilität, Akinesie, Dyskinesie
-
Strukturveränderungen: Intrakardiale Thromben und Tumoren, Perikardergüsse, Zysten, Veränderungen der Morphologie und Beweglichkeit der Herzklappen
1.8.8.4 Dreidimensionale (3-D-)Echokardiographie
1.8.8.4.1 Methode
Die Technik befindet sich noch in der Entwicklung. Nach zwei Prinzipien werden dreidimensionale Daten gesammelt. Bei einem Vorgehen wird zunächst eine Serie von zweidimensionalen Bildern gesammelt, deren räumliche Position bekannt ist. Danach erfolgt eine dreidimensionale Rekonstruktion. Das neuere Verfahren verwendet Schallköpfe mit rechtwinkliger Anordnung der Kristalle, die direkt einen dreidimensionalen Scan liefern.
1.8.8.4.2 Anwendung
Aus dem dreidimensionalen Datenset können gewünschte zweidimensionale Ebenen abgebildet werden. An Strukturen, z. B. Herzklappen, ergeben die summierten zweidimensionalen Ebenen pseudodimensionale Bilder, an denen endokarditische Läsionen genauer zu identifizieren sind.
1.8.8.5 Transösophageale Echokardiographie (TEE)
1.8.8.5.1 Methode
Eine schallkopftragende Sonde (2-D-Tranducer, auch mit Dopplerfunktionen) wird nach Lokalanästhesie der Rachenwand (mit Spray) in den Ösophagus eingeführt. Die Platzierung des Schallkopfes erfolgt im oberen und mittleren Ösophagus sowie am Mageneingang (transgastrische Bilder). Es lassen sich horizontale und longitudinale Schnittbilder in verschiedenen Ebenen gewinnen. Die Nähe zu den Herzstrukturen erlaubt die Verwendung hochfrequenter Schallköpfe (5–10 MHz) mit besserer Ortsauflösung als bei der transthorakalen Echokardiographie. Die ◘ Abb. 1.58 zeigt das transösophageale Echo eines Gesunden. Man blickt auf die Herzhöhlen von hinten.


Transösophageale Echokardiographie eines Herzgesunden (LA = linkes Atrium, LV/RV = linker/rechter Ventrikel, MK = Mitralklappe, AK = Aortenklappe)
1.8.8.5.2 Anwendung
Die Methode liefert optimale Echoaufnahmen der Vorhöfe, Vorhofohren und der Herzklappen bzw. Herzklappenprothesen. Man erkennt Vorhofthromben, Vorhoftumoren, Vorhofseptumdefekte und Vegetationen auf den Herzklappen bei Endokarditis. Klappenprothesen können beurteilt und Aortendissektionen erkannt werden. Die tieferen Horizontal- und Längsschnitte erlauben eine Beurteilung der Herzkammern, wenn die transthorakale Echokardiographie nicht gelingt. Die transösophageale Echokardiographie wird auch zur Überwachung der Ventrikel- und Herzklappenfunktion bei Herzoperationen und beim katheterinterventionellen Verschluss eines persistierenden Foramen ovale (PFO) und eines Atriumseptumdefektes (ASD) eingesetzt.
1.8.8.6 Kontrastechokardiographie
1.8.8.6.1 Rechtes Herz
2-D-Echokardiographie nach intravenöser Injektion eines Kontrastmittels (Echovist), das nicht die Lunge passiert. Indikationen: ASD, Ventrikelseptumdefekt (VSD), komplexe angeborene Vitien, Trikuspidal- und Pulonalklappeninsuffizienz (bessere Darstellung durch Signalverstärkung).
1.8.8.6.2 Linkes Herz
2-D-Echokardiographie nach intravenöser Injektion eines lungengängigen Kontrastmittels (Levovist, Optison, Sonovue). Indikationen: Verstärkung von Dopplersignalen im linken Ventrikel, Verbesserung der LV-Konturerkennung.
1.8.8.7 Doppler-Echokardiographie
1.8.8.7.1 Methode
Kombination von zweidimensionaler Echokardiographie und Dopplersonographie, die ein nichtinvasives Studium der Hämodynamik des Herzens ermöglicht:
-
Farbdoppler: Bei apikaler Position des Schallkopfes stellt sich in der Diastole das durch die AV-Klappen strömende Blut als rote Farbwolke dar, im linken Ventrikel wegen hoher Flussgeschwindigkeit mit zentralem Aliasing (Farbumschlag in orangegelb bis türkisblau). Mit der Vorhofkontraktion folgt ein zweiter Schub der Ventrikelfüllung. In der Systole zeigt sich im linksventrikulären Ausflusstrakt (LVOT) ein blau kodierter Fluss in Richtung Aortenklappe und im linken Vorhof eine rote Farbwolke durch den Bluteinstrom aus den Lungenvenen (◘ Abb. 1.57).
-
PW-Doppler: Dient zur Messung der Flussgeschwindigkeit des Blutes (nicht des Volumenflusses) in wählbaren Tiefenbereichen. Das Sample-Volumen wird im 2-D-Echokardiogramm an gewünschter Stelle positioniert. Eine Limitierung besteht bezüglich der Eindringtiefe und der Erfassung hoher Flussgeschwindigkeiten, da diese ein Alaising hervorrufen.
-
CW-Doppler: Dient zur Messung unbegrenzter und damit maximaler Flussgeschwindigkeiten entlang des Schallstrahles, der im B-Bild ausgerichtet wird. Tiefenselektive Messungen der Flussgeschwindigkeit sind nicht möglich.
-
Tissue-Doppler (Gewebedoppler): Durch Veränderung der Amplituden- und Geschwindigkeitsfilter der Dopplergeräte wurde es möglich, Richtung und Geschwindigkeiten der Wandbewegungen des Herzens, insbesondere des linken Ventrikels zu registrieren.
1.8.8.7.2 Anwendung
Der Farbdoppler gibt eine schnelle und relativ umfassende Information über Blutströmungsrichtung, ungefähre Blutflussgeschwindigkeit sowie Lokalisation und Ausdehnung von Turbulenzen, die am Alaising-Phänomen zu erkennen sind. Halbquantitativ zu erfassen sind z. B. der systolische Reflux bei der Mitralinsuffizienz und der diastolische Reflux bei der Aorteninsuffizienz. Bei der Mitralstenose und der Aortenstenose stellen sich die Stenosejets als Turbulenzen dar. Zur quantitativen Messung der Flussgeschwindigkeiten ist die gezielte Zuschaltung des PW- bzw. CW-Dopplers unverzichtbar. Der PW-Doppler erfasst die Richtung und Geschwindigkeit des Blutflusses an den Herzklappen und an umschriebenen Positionen in Vorhöfen und Kammern. Er dient zur Bestimmung der diastolischen Funktion der Ventrikel (◘ Abb. 1.8). Mit großer Empfindlichkeit weist er den retrograden Fluss an insuffizienten und die Flussbeschleunigung an stenosierten Klappen nach. Durch ein Mapping mit vielen Messpunkten im linken Vorhof lässt sich die Ausdehnung des Regurgitationsjets bei der Mitralinsuffizienz bestimmen. Analoges gilt für die Aorteninsuffizienz. Mit der gepulsten Dopplertechnik können aber nicht die maximalen Flussgeschwindigkeiten gemessen werden. Mit dem CW-Doppler gelingt die Messung der maximalen Flussgeschindigkeit, wobei Fluss- und Dopplerrichtung weitgehend übereinstimmen müssen. Aus der Vmax lassen sich die Druckgradienten an den Herzklappen berechnen und damit z. B. die Schweregrade der Aortenstenose und der Mitralstenose bestimmen. Bei der Aortenstenose kann man aus der Vmax vor der Stenose (mit dem PW-Doppler gemessen) und der Vmax in der Stenose (mit dem CW-Doppler) die Öffnungsfläche errechnen. Geschwindigkeitsmessung und Bestimmung der Querschnittsfläche an den Herzklappen ermöglichen die Berechnung des Herzminutenvolumens. Sehr hilfreich ist die Dopplertechnik für den nichtinvasiven Shuntnachweis auf Vorhof- und Kammerebene. Mit dem Tissue-Doppler lassen sich Störungen der diastolischen Funktion des linken Ventrikels erfassen und regionale Störungen der Wandbeweglichkeit (◘ Abb. 1.59).


Gewebedoppleruntersuchung eines Herzgesunden mit synchroner systolischer (S) und diastolischer (E, A) Bewegung der basalen septalen (gelb) u. der basalen lateralen (rot) linksventrikulären Wand mit normalen Wandbewegungsgeschwindigkeiten
1.8.8.8 Stressechokardiographie
1.8.8.8.1 Methode
Echokardiographische Untersuchung der Wandbewegung der Herzkammern unter Belastung zur Erfassung regionaler ischämischer Kontraktionsstörungen.
1.8.8.8.2 Belastungsarten
Aktive Belastung am Fahrradergometer im Liegen. Passive Belastung durch die Pharmaka Dobutamin (Sympathikomimetikum) oder Dipyridamol (Vasodilatator, der über einen Steal-Effekt Ischämien provoziert). Am Fahrradergometer und unter pharmakologischem Stress wird während der Belastung geschallt. Letztere Methode wird angewandt, wenn eine Maximalbelastung am Fahrrad wegen Muskelschwäche oder Gliedmaßenproblemen nicht möglich erscheint.
1.8.8.8.3 Durchführung
In Ruhe, während stufenweise gesteigerter Belastung und am Belastungsende wird in 3–5 Schnittebenen (Vierkammerblick, Zweikammerblick, apikaler Dreikammerblick, parasternale Längsachse, parasternale kurze Achse) je ein Herzzyklus im Echokardiographen festgehalten und auf dem Auswertungscomputer zur Speicherung übertragen.
1.8.8.8.4 Auswertung
Von jeder Schnittebene gibt der Computer bei der sog. Quad-Screen-Darstellung 4 synchronisierte Bewegungsbilder der gespeicherten Herzzyklen nebeneinander wieder (Ruhe, 2 Belastungsstufen und Belastungsende). Verglichen werden visuell die Wandbewegungen in den basalen, mittleren und apikalen Segmenten der Ventrikelwand und folgenden Kategorien zugewiesen: Normokinesie, Hypokinesie, Akinesie und Dyskinesie. Beurteilt wird auch die Wanddickenzunahme.
1.8.8.8.5 Beurteilung
Der Nachweis einer Belastungsischämie gelingt früher als mit dem Belastungs-EKG. Aus der Lokalisation der Wandbewegungsstörung lässt sich zuverlässig auf das betroffene Koronargefäß schließen. Narben bleiben akinetisch, Aneurysmen dyskinetisch, noch vitale Myokardsegemente, die sich in Ruhe schlecht kontrahieren zeigen unter niedrig dosiertem Dobutaminstress noch eine kontraktile Reserve.
1.8.9 Nuklearmedizinische Methoden
1.8.9.1 Myokardszintigraphie
Myokardszintigraphie
Abbildung des Myokards mit Radioisotopen, die nach intravenöser Applikation im Herzmuskel reversibel gespeichert werden. Die Bildgewinnung erfolgt meistens mit SPECT-Technik (single-photon emission computed tomographie)
1.8.9.1.1 Thalliumszintigraphie
201Thallium wird als ein kaliumanaloges Kation über das Natrium-Kalium-ATPase-System aktiv von den Herzmuskelzellen aufgenommen (Extraktionsrate 85 % bei einem Transit durch den Koronarkreislauf). Seine Anreicherung im Herzmuskel ist (a) vom koronaren Blutfluss, (b) von der ATPase-Aktivität und damit von der metabolischen Integrität der Herzmuskelzellen abhängig. Etwa 30 Minuten nach der Lokalisation im Herzmuskel beginnt die Rückverteilung des 201Thallium in den Körper (physikalische Halbwertszeit 74 h). Herzinfarkte führen zu persistierenden regionalen Speicherdefekten im Ruheszintigramm, Koronarstenosen zu Speicherdefekten im Belastungsszintigramm, die 2‒4 h nach der Belastung wieder verschwinden, da sich in dieser Zeit auch im ischämischen Myokard 201Thallium anreichert. Man injiziert das Isotop auf dem Höhepunkt der Belastung. Die Aktivität über dem linken Ventrikel wird innerhalb von 5 Minuten nach der Injektion und 3–4 h nach der Belastung gemessen. Die Treffsicherheit der Thalliumbelastungsszintigraphie bei Patienten mit Koronarinsuffizienz beträgt 87 % (71–93 %). Infarktnachweis (Gold-Spot-Scanning) innerhalb 24 h möglich.
Die Myokardszintigraphie wird auch mit verschiedenen 99mTc-markierten Substanzen durchgeführt. Gebräuchlich ist 99mTc-MIBI (Methoxy-Isobutyl-Isonitril). Es lagert sich als lipophile Substanz durch Diffusion in Muskelzellen ein. Die regionale Verteilung im Myokard entspricht immer der aktuellen Perfusion während der Injektion. Der Radiotracer wird nur in der Ischämiediagnostik eingesetzt. Für die Bildgebung besitzt er günstigere physikalische Eigenschaften.
1.8.9.1.2 Infarktszintigraphie mit 99mTc-Pyrophosphat (Hot Spot Scanning)
Selektive Ansammlung des Tracers im infarzierten Myokard 60–90 Minuten nach intravenöser Injektion, allerdings erst 12 h nach Infarkteintritt, wahrscheinlich durch Bildung von Calciumpräzipitaten in geschädigten Mitochondrien. Optimale Abbildung bei einem Infarktalter von 24–96 h. Treffsicherheit 86 %.
1.8.9.2 Radionuklid-Ventrikulographie
1.8.9.2.1 Methode
Die Binnenräume des Herzens werden nach Markierung der Erythrozyten mit 99mTc mit einer Szintillationskamera (Ein- oder Vielkristallsystem) in schneller Bildfolge aufgenommen. Die Datenerfassung und -verarbeitung erfolgt durch einen Computer. Es wurden 2 Verfahren entwickelt, die im Wesentlichen gleiche Informationen liefern. Die Markierung der Erythrozyten erfolgt in vivo. Zunächst wird eine reduzierende Substanz (Zinn-Pyrophosphat) injiziert, 15–30 Minuten später 99mTc-Pertechnetat.
-
First-Transit-Ventrikulographie: Gewinnung von Sequenzbildern während der ersten Herzpassage der als Bolus injizierten markierten Erythrozyten. Dabei zeitlich getrennte Darstellung des rechten und linken Ventrikels ohne Überlappung mit der Möglichkeit, für jeden Ventrikel die Ejektionsfraktion (EF) zu bestimmen und die regionalen Wandbewegungen beider Ventrikel zu messen. Nachweis eines Links-Rechts-Shunts: Die Transitkurve eines ausgeblendeten Lungenfeldes fällt im absteigenden Schenkel unvollständig ab, da der Tracer rezirkuliert. Nachweis eines Rechts-Links-Shunts: Verfrühtes Erscheinen der Aktivität in der Aorta. Die Zeit-Aktivitäts-Kurve der linken Kammer gibt Aufschluss über die systolischen und diastolischen Zeitintervalle, über Füllung und Entleerung sowie Volumen und Ejektionsfraktion der linken Kammer.
-
Ventrikulographie nach intravasaler Gleichverteilung des Indikators (equilibrium blood pool imaging): Beginn der Messungen nachdem sich die markierten Erythroyten gleichmäßig in der Blutbahn verteilt haben. Dadurch schwächere Aktivität über dem Herzen als bei der First-Transit-Methode (1 : 200) und gleichzeitige Markierung beider Vorhöfe und Kammern, die besondere Projektionen zur getrennten Untersuchung des rechten und linken Ventrikel erfordern. Die Tracerverdünnung wird ausgeglichen, indem man bis zu 400 Einzelzyklen im Computer speichert, der sie zu einem repräsentativen Zyklus addiert. Man gewinnt Zeit-Aktivitäts-Kurven, aus denen die Ejektionsfraktion genau berechnet werden kann, ferner Bilder der Herzkammern in allen Phasen des Zyklus, die eine globale und regionale Messung der Wandbewegungen erlauben. Die Messungen lassen sich ohne erneute Tracergabe innerhalb der physikalischen Halbwertszeit des 99mTc (6 h) beliebig oft wiederholen, so dass Untersuchungen unter körperlicher Belastung möglich sind. Eine First-Transit-Ventrikulographie kann vorgeschaltet werden. Sie erfasst den rechten Ventrikel besser.
1.8.9.2.2 Diagnostische Bedeutung
Genaueste Methode zur Bestimmung der Ejektionsfraktion und der Herzvolumina. Letztere sind durch Bezug auf die Strahlung einer entnommenen Blutprobe zu ermitteln. Die Ejektionsfraktion lässt sich anhand der Zeitaktivitätskurve über der untersuchten Kammer bestimmen. Die Ejektionsfraktion gibt Aufschluss über den Schweregrad einer muskulären Herzinsuffizienz. Bei koronarer Herzkrankheit sind Abnahme der Ejektionsfraktion während ergometrischer Belastung und Störungen der regionalen Wandbewegung (Dyskinesie, Akinesie, Aneurysmen) zu erfassen. Beim Cor pulmonale ist die Ejektionsfraktion des rechten Ventrikels herabgesetzt, in manchen Fällen auch des linken. Klappeninsuffizienzen sind anhand der Differenz der Schlagvolumina beider Ventrikel zu quantifizieren (deutliches Überwiegen des linksventrikulären Schlagvolumens bei Aorten- und Mitralinsuffizienz). Bei Herzklappenfehlern ist die Bestimmung der effektiven LV-Ejektionsfraktion wichtig. Sie ergibt sich aus der Differenz zwischen der EF in Ruhe und der EF bei submaximaler ergometrischer Belastung. Normalwerte: Anstieg der EF um 5–10 %. Darunter liegende Werte zeigen eine beginnende Erschöpfung der myokardialen Adaptation an Druck- und Volumenbelastung an.
1.8.9.3 Positronen-Emissions-Tomographie (PET)
1.8.9.3.1 Methode
Der Herzmuskel wird mit natürlichen Substanzen des Stoffwechels perfundiert, die mit Positronen-emittierenden Isotopen, z. B. 11 C, 15O, 18 F markiert wurden. Das Verfahren dient dazu, den Stoffwechsel und die regionale Durchblutung des Herzmukels zu bestimmen und vitales von abgestorbenem Gewebe zu unterscheiden. Die meisten Tracer werden im Zyklotron durch Beschuss der Atome mit Protonen erzeugt. Das vom Atomkern aufgenommene Proton strahlt mit kurzer Halbwertszeit ein Positron ab und geht dabei in ein Neutron über. Im Gewebe stößt das Positron nach wenigen Millimetern auf ein freies Elektron, das die gleiche Masse, aber eine negative Ladung besitzt. Beide vereinigen sich zum Positronium, dessen Masse sich sofort in Energie verwandelt und zwar in zwei Photonen (Gammastrahlung), die in genau entgegengesetzter Richtung abgestrahlt werden. Die Detektoren zu ihrer Messung sind im Messgerät kreisförmig dicht nebeneinander angeordnet, so dass jedes Protonenpaar von einem Detektorenpaar aufgefangen wird, das sich genau gegenüberliegt. Aus dem Zeitabstand mit dem die Photonen eintreffen, lässt sich ihr Ausgangspunkt bestimmen. Alle aus einer Gewebsschicht eintreffenden Strahlenimpulse werden zu einer computertomographischen Darstellung verarbeitet.
1.8.9.3.2 Diagnostische Anwendung
In der Kardiologie wird mit der PET am häufigsten der Glukosestoffwechsel des Myokards untersucht. Dazu injiziert man Desoxyglukose, die mit 18 F markiert ist. Fluordesoxyglukose wird von stoffwechselaktiven Zellen als FDG-6-Phosphat aufgenommen, aber nicht weiter metabolisiert und deshalb angreichert. Ein positiver Speicherungstest zeigt lebensfähiges Myokard an, auch wenn es sich nicht mehr kontrahiert (hibernating und stunned myocardium). Die für die Indikationsstellung zur Revaskularisierung nach Infarkt wichtige Vitalitätsprüfung gelingt mittels PET am zuverlässigsten. Wegen hoher Kosten leider selten verfügbar.
1.8.10 Herzkatherisierung und Angiokardiographie
1.8.10.1 Diagnostische Indikationen
-
Angeborene und erworbene Vitien: Sicherung der Diagnose und Bestimmung des für die Operationsindikation maßgeblichen Schweregrades. Gelingt nicht selten allein durch die Echokardiographie.
-
Koronare Herzkrankheit: Feststellung der genauen anatomischen Ausdehnung, Indikationsstellung zur Bypass-Operation. Ausschluss einer koronaren Herzkrankheit vor Herzklappenoperationen und bei unklaren präkordialen Beschwerden. Nachweis von Restenosen nach Revaskularisierung.
-
Kongestive Kardiomyopathie: Ausschluss einer koronaren Herzkrankheit, evtl. Klärung der Ursache durch Myokardbiopsie.
-
Pulmonale Hypertonie: Nachweis und Bestimmung des Schweregrades, Klärung der Ursache (primäre Form, rezidivierende Lungenembolien, Mitralstenose, Links-Rechts-Shunt).
-
Intensivmedizin: Kontrolle der Herzfunktion durch laufende intrakardiale oder intrapulmonale Druckmessung bei Schockzuständen und Herzinsuffizienz.
-
Herzrhythmusstörungen: His-Bündel-EKG, diagnostische Elektrostimulation (▶ Abschn. 1.10).
1.8.10.2 Therapeutische Indikationen
Darstellung der Eingriffe und Methoden bei den einzelnen Erkrankungen.
1.8.10.2.1 Messgrößen
Diagnostische Informationen erhält man durch die Bestimmung nachstehender Parameter.
1.8.10.2.1.1 Druckmessung
Gemessen wird intrakardial und intravaskulär. Erfasst werden Druckgradienten über stenosierten Klappen, typische Vorhofdruckkurven bei Klappeninsuffizienz sowie intraventrikuläre Drucksteigerungen bei Myokardinsuffizienz, Hypertrophie mit herabgesetzter Compliance, Myokardischämie und konstriktiver Perikarditis. Die ◘ Tab. 1.6 zeigt die Normalwerte im Bereich des Herzens.
1.8.10.2.1.2 Bestimmung des Herzminutenvolumens (HMV)
Erfasst werden die Förderleistung des Herzens in Ruhe und unter Belastung, das Vorwärts- und das Regurgitationsvolumen bei Klappeninsuffizienzen sowie die durch Shunts fließenden Blutmengen. Das Herzminutenvolumen lässt sich nach dem Fick-Prinzip aus der O2-Aufnahme der Lunge und der arteriovenösen Differenz des O2-Gehaltes des Blutes berechnen. Wenn das venöse Blut mit einem 02-Gehalt von 160 ml/l in die Lunge fließt und das arterielle Blut 200 ml O2/l enthält, nimmt jeder Liter Blut in der Lunge 40 ml/O2 auf. Das ergibt bei einer O2-Absorption der Lunge von 200 ml/min einen Durchfluss von 5 l Blut/min, was einem normalen Herzminutenvolumen entspricht. Das HMV kann auch mit der Farbstoffverdünnungsmethode (Cardiogreen) oder mit der Thermodilutionsmethode (Injektion kalter Kochsalzlösung) bestimmt werden. Bei diesen Verfahren erfolgt die Berechnung des Blutflusses aus der Transportzeit für eine bestimmte Indikatormenge und der mittleren Indikatorkonzentration am distalen Messpunkt.
Aus den Minutenvolumina und Drücken können die Gesamtwiderstände im großen und kleinen Kreislauf sowie die Offnungsflächen stenosierter Herzklappen berechnet werden.
1.8.10.2.2 Angiographie
Darstellung der Herzinnenräume, herznahen Gefäße und Koronararterien mit Röntgenkontrastmittel. Die Kontrastmittelinjektion erfolgt mit einem Druckinjektor oder manuell. Dabei wird das Durchleuchtungsbild des Bildverstärkers durch eine Filmkamera aufgenommen (Cineangiographie).
1.8.10.2.3 Katheterisierung des rechten Herzens
Am einfachsten mit dem Swan-Ganz-Balloneinschwemmkatheter . Dieser wird perkutan in eine Vene der Ellenbeuge eingeführt, nach dem Eintritt in das Herz an seinem Ende aufgeblasen und unter fortlaufender Druck- und EKG-Kontrolle bis in die Lungenarterienperipherie vorgeschoben. Dort verschließt der Ballon am Katheterende einen peripheren Lungenarterienast, so dass es zwischen Katheteröffnung und Lungenkapillaren zum Druckausgleich kommt. In dieser Position wird der Kapillarverschlussdruck (PCP) gemessen, der dem Lungenkapillardruck gleichzusetzen ist. Während der Sondierung werden die Drücke im rechten Vorhof, in der rechten Kammer, der Pulmonalarterie und in der PCP-Stellung (pulmonary wedge pressure) gemessen. Aus der Pulmonalarterie wird gemischtvenöses Blut zur Bestimmung des O2-Gehaltes (für die Messung des Minutenvolumens nach dem Fick-Prinzip) entnommen. Falls keine Mitralstenose vorliegt, entspricht der Mitteldruck im Pulmonalkapillarsystem (PCPm) dem Mitteldruck im linken Vorhof (LAPm) und dem enddiastolischen Druck im linken Ventrikel (LVEDP). Wenn der pulmonale Gefäßwiderstand nicht erhöht ist, hat der diastolische Pulmonalarteriendruck (PAPd) die gleiche Höhe. Die Rechtsherzeinschwemmkatheteruntersuchung gibt deshalb auch über die Funktion des linken Ventrikels Aufschluss. Die Messungen werden in Ruhe und unter dosierter Belastung mit dem Fahrradergometer durchgeführt. Je nach Schweregrad sind bei der Herzinsuffizienz die PCP-Drücke schon in Ruhe oder erst bei Belastung erhöht. Auch bei der Koronarinsuffizienz kommt es unter der Belastung zum Anstieg des PCPm. Als Insuffizienzsymptom kann das Herzminutenvolumen schon in Ruhe herabgesetzt sein oder erst unter Ergometerbelastung inadäquat werden.
Zur Angiographie wird ein halbsteifer Rechtsherzkatheter durch die V. femoralis (perkutan mit Seldinger-Technik) unter Röntgenkontrolle in das rechte Herz und die A. pulmonalis vorgeschoben. Auf der rechten Herzseite lassen sich kongenitale Vitien (Ebstein-Syndrom, Pulmonalstenose, Fallot-Tetralogie) und Trikuspidalklappenfehler nachweisen, mittels Angiographie der A. pulmonalis Lungenembolien, abnorme Pulmonalvenenmündungen sowie Thromben und Tumoren im linken Vorhof.
1.8.10.2.4 Katheterisierung des linken Herzens
Bevorzugt auf retrogradem Weg mit einem perkutan (Seldinger-Technik) in die A. femoralis eingeführten Katheter, der durch die Aorta und das Aortenostium in den linken Ventrikel vorgeschoben wird, die Mitralklappe jedoch nicht passieren kann (◘ Abb. 1.60). Die Kathetereinführung ist auch über die A. brachialis oder A. radialis möglich. Linker Vorhof und linker Ventrikel können ggf. auf transseptalem Weg katheterisiert werden. Dabei wird der Katheter vom rechten Vorhof mittels einer Punktionsnadel durch das Vorhofseptum in den linken Vorhof eingeführt und bis in den linken Ventrikel vorgeschoben.


Normale Lävokardiographie
Die Angiographie der linken Herzseite ergibt Füllungsbilder vom linken Ventrikel, die auch zur Berechnung des endsystolischen und enddiastolischen Volumens dienen. Sie erlauben den Nachweis regionaler Störungen der Wandbewegung bzw. Ventrikelfunktion. Durch Kontrastmittelinjektion in die Aorta gelingt der halbquantitative Nachweis einer Aorteninsuffizienz. Auch Aortenektasie und Aortendissektion werden erfasst. Am systolischen Reflux in den linken Vorhof ist die Mitralinsufizienz zu erkennen und ihr Schweregrad zu beurteilen.
1.8.10.2.5 Koronarangiographie
Selektive Darstellung beider Koronararterien (◘ Abb. 1.61) mittels spezieller Katheter, die über A. femoralis, A. brachialis oder A. radialis retrograd am Abgang der Arterien aus der Aorta platziert werden (▶ Abschnitt koronare Herzkrankheit).


Unauffällige Koronarangiographie der linken (RIVA, RCX) und rechten Koronararterie (RCA) bei einer 51-jährigen Patientin mit atypischen pektanginösen Beschwerden
1.8.11 Computertomographie
Das kontrastverstärkte Kardio-CT mit modernen Scannern ermöglicht die Darstellung struktureller Veränderungen des Herzens (Perikardprozesse, Aneurysmen, Narben des Kammermyokards, Klappenverkalkungen, Lungenembolie). Ein normaler Längsschnitt wird in ◘ Abb. 1.62a gezeigt. Auch die Koronargefäße lassen sich bis auf die Peripherie sichtbar machen (◘ Abb. 1.62b).


Kardio-CT (nach KM-Gabe). a Längsschnitt durch das Herz eines 58-jährigen Herzgesunden. b Normale linke Koronararterie (RIVA/RCX) eines 60-jährigen Mannes mit unklaren retrosternalen Beschwerden (LA = linker Vorhof, LV/RV = linker/rechter Ventrikel, AO = Aorta, PA = Pulmonalarterie, AK = Aortenklappe, MK = Mitralklappe) (Sammlung Dr. Langer, Bad Oeynhausen)
1.8.12 Magnetresonanztomographie (MRT)
1.8.12.1 Methode
Sie basiert auf dem als Spin bezeichneten Drehimpuls der Atomkerne mit ungerader Protonen- oder Neutronenzahl und wird deshalb auch als Kernspintomographie bezeichnet. Zur Bildgebung bei der MRT (MRI: magnetic resonance imaging) werden nur Wasserstoffatome genutzt. Mit den Protonen rotiert ihre elektrische Ladung und erzeugt ein eigenes kleines Magnetfeld, durch das sich die Protonen in einem sehr starken äußeren Magnetfeld parallel und antiparallel ausrichten. Ihr Drehimpuls wird durch das äußere Magnetfeld entsprechend der magnetischen Feldstärke um viele Zehnerpotenzen beschleunigt. Legt man ein solches Magnetfeld an, lassen sich die Protonen durch einen Radiofrequenzimpuls (der die gleiche Frequenz haben muss wie der Drehimpuls der Protonen) zur Seite ablenken. Nach Abschalten des Impulses kehren sie in die Ausgangslage zurück (Relaxation) und strahlen dabei als Kernspinresonanz Radiofrequenzen ab, die von Empfängerspulen aufgefangen und zum Bildaufbau auf den Computer übertragen werden.
Die Intensität des Signals hängt erstens von der Geschwindigkeit ab, mit der sich der abgelenkte Magnetvektor wieder im äußeren Magnetfeld ausrichtet. Diese Zeitkonstante T1 (Longitudinalrelaxationszeit) beträgt 200‒3000 ms. Zweitens wird die Signalintensität von der Geschwindigkeit bestimmt, mit der sich der transversale Magnetvektor zurückbildet. Das geschieht in der Zeitkonstante T2 (Transversalrelaxationszeit), die zwischen 20 und 400 ms variiert. Da sich die Zeitkonstanten der Gewebe deutlich unterscheiden, kommen Schnittbilder mit guten Kontrasten zustande. Da es möglich ist, den Signalempfang für eine bestimmte Periode nach dem Radiofrequenzimpuls zu verzögern, gelingt es, entweder die T1- oder die T2-Differenzen der Gewebe hervorzuheben. Man spricht von T1- oder T2-gewichteten Bildern.
Für die Bildgebung gibt es zwei technische Versionen: Spin-Echo-Imaging (S-E) und Gradient-Echo-Imaging (G-E). Beim S-E-Imaging wird der Magnetvektor der Protonen um 90° abgelenkt; hier erscheint das strömende Blut schwarz. Beim G-E-Imaging beträgt die Ablenkung 20–60°, wodurch eine schnellere Bildfolge möglich wird; hier erscheint das Blut hell. Cine-MRT ist eine dynamische G-E-Imaging Technik mit der in einer Ebene 10–20 Bilder pro Herzzyklus gewonnen werden können. Eine noch schnellere Bilderzeugung ist mit der Ultrafast-Technik (in 100–300 ms) und dem Echoplanar-Imaging (in 40–50 ms) möglich.
1.8.12.2 Diagnostische Bedeutung
Als tomographisches bildgebendes Verfahren mit sehr guter Kontrastierung hat die MRT in der kardiologischen Diagnostik viele Anwendungsmöglichkeiten.
1.8.12.2.1 Herzanatomie
Sehr genau können Größe der Herzkammern, Myokardmasse und intrakardiale Massen bestimmt und Klappenanomalien und Perikarderkrankungen erfasst werden. Überlegen gelingt die Darstellung von angeborenen Herzfehlern, Aortenaneurysmen und der Rechtshypertrophie bei pulmonaler Hypertonie. Infarktnarben sind gut zu erkennen. Bis auf die peripheren Aufzweigungen lassen sich auch die Koronararterien darstellen.
1.8.12.2.2 Herzfunktion
Ausgezeichnet ist die systolische Ventrikelfunktion zu bestimmen, weniger gut die diastolische. Man erkennt auch Stenosen und Insuffizienzen der Herzklappen und intrakardiale Shunts.
Gegenüber dem Kardio-CT, das ähnliche Anwendungsmöglichkeiten bietet, hat die MRT den Vorteil der fehlenden Strahlenbelastung. In der kardiologischen Praxis kommen beide Verfahren noch selten zum Einsatz, weil sie sehr teuer, nicht überall verfügbar und weitgehend durch konventionelle Methoden (Röntgen, Echokardiographie, Angiographie, Isotopenszintigraphie) zu ersetzen sind.
1.9 Herzinsuffizienz
1.9.1 Normalwerte und Determinanten der Herzleistung
1.9.2 Herzminutenvolumen (Herzzeitvolumen, cardiac output)
Unter Ruhebedingungen 5‒6 l/min. Abhängig von Alter, Größe und Geschlecht. Mit dem Alter abnehmend, bei Frauen 10‒20 % niedriger als bei Männern. Bei körperlicher Belastung mit der Sauerstoffaufnahme zunächst linear (bis 70 % des Maximum), dann flacher ansteigend. Sportler erreichen 35‒38 l/min.
1.9.3 Herzindex (cardiac index)
Auf die Körperoberfläche bezogenes Herzminutenvolumen, Normalbereich liegt bei 3,0–3,5 l/min.
1.9.4 Vorlast (preload)
Enddiastolische Kammerwanddehnung gegenüber der Ausgangslänge. Genauer die für Kontraktionsstärke maßgebende Vordehnung der Sarkomere.
Nach dem Starling-Gesetz nimmt die bei der Muskelkontraktion freigesetzte mechanische Energie bis zur optimalen Überlappung von Aktin- und Myosinfilamenten zu, bei weiterer Dehnung wieder ab. Bei der Überdehnung des Herzmuskels wird nicht die optimale Sarkomerenlänge überschritten, sondern die Muskelfasern verschieben sich gegeneinander (slippage). Dabei kommt es gleichfalls zur Verminderung der Kontraktionsleistung.
Die Vorlast wird im Wesentlichen vom venösen Rückfluss bestimmt, an den sich die Förderleistung des Herzens innerhalb physiologischer Grenzen automatisch anpasst. Ist die optimale Sarkomerenlänge erreicht, kann die Pumpleistung des Herzens nur noch durch Zunahme der Schlagfrequenz und der Kontraktilität sowie durch eine Verminderung der Nachlast (▶ unten) gesteigert werden. Einen wichtigen Beitrag zur Vorlast leistet die Vorhofkontraktion. Ihr Wegfall (Vorhofflimmern, elektrischer Kammerschrittmacher) vermindert die maximale Pumpleistung, was sich vor allem bei einer Herzinsuffizienz nachteilig auswirkt.
Für die Vorlast gibt es nur indirekte Maße, den enddiastolischen Füllungsdruck und die diastolische Zunahme des Kammervolumens. Bei gegebener Vorlast ist der enddiastolische Füllungsdruck umso höher, je schwerer sich der Herzmuskel dehnen lässt. Häufige Ursachen einer verminderten Dehnbarkeit (Compliance) und damit erschwerten Füllung der Kammern sind Hypertrophie, Ischämie, Myokardfibrose und obstruktive Perikarderkrankungen.
1.9.5 Kontraktilität (inotroper Zustand)
Die nicht vom Preload, sondern von inneren Faktoren, hauptsächlich vom Anstieg der zytoplasmatischen Ca++-Konzentration bestimmte Kontraktionskraft des Herzmuskels. Eine Kontraktilitätssteigerung (positiv inotroper Effekt) bewirkt bei jeder Vor- und Nachlast eine Zunahme des Schlagvolumens, der Druckanstiegs- und der Kontraktionsgeschwindigkeit. Eine Kontraktilitätsherabsetzung (negativ inotroper Effekt) hat das Gegenteil zur Folge.
Positiv inotrop wirken Catecholamine, Sympathikusstimulation, Digitalis, Xanthine, Calciumionen, Steigerungen der Herzfrequenz und postextrasystolische Intervalle. Negativ inotrop wirken β-Rezeptorenblocker, Chinidin, Calciumantagonisten, Barbiturate und die Azidose.
Der für die Kontraktilität maßgebende Calciumeinstrom in das Zytoplasma der Muskelfasern wird durch das Aktionspotenzial induziert. Zum einen öffnet es Membrankanäle für einen langsamen Ca++-Einstrom von außen. Zum anderen setzt es aus dem sarkoplasmatischen Retikulum große Ca++-Mengen frei, so dass die intrazelluläre Ca++-Konzentration auf das Hundertfache ansteigt.
Oberhalb einer Schwellenkonzentration werden Calciumionen an das Troponin C im Troponin-Tropomyosin-Komplex gebunden, der dadurch seinen blockierenden Effekt auf die aktiven Stellen der Aktinfilamente verliert. Myosinquerbrücken und Aktin können sich nun verbinden, worauf es unter Bindung und Spaltung von ATP am Myosinkopf zur Kraftentwicklung und Kontraktion kommt. Die Zahl der Aktin-Myosin-Brücken und damit die systolische Kraftentfaltung nimmt mit der intrazellulären Ca++-Konzentration zu.
Mit der Repolarisation endet der Ca++-Einstrom. Calciumionen werden unter Energieverbrauch überwiegend von der sarkoplasmatischen Retikulum Calcium-ATPase (SERCA2a) in das sarkoplasmatische Retikulum zurückgepumpt. Sie lösen sich vom Troponin C, so dass die aktiven Stellen des Aktins wieder blockiert werden und die Relaxation eintritt. Der „Ca++-Puls“ dauert nur 0,2–0,3 s. Nach neuesten Erkenntnissen wird die Aktivität der Calciumpumpe SERCA2a von einem zytoplasmatischen Protein moduliert, dem SUMO1 (small ubiquitin-like modifier type 1), das sich mit der Calciumpumpe an der der Membran des endoplasmatischen Retikulum verbindet. Bei Herzinsuffizienz sind beide weniger aktiv mit der Folge, dass die Calciumkonzentration im Zytoplasma steigt und die Kontraktilität abnimmt. Im Tierversuch konnte durch Hochregulation von SUMO1 per Virustransfektion eine deutliche Verbesserung der Herzleistung erreicht werden.
Viele Interventionen, die zur Steigerung oder Herabsetzung der Kontraktilität führen, greifen in die Calcium-Kinetik der Herzmuskelfasern ein:
-
Sympathikomimetika: Steigern den Ca++-Einstrom in die Zellen durch Öffnung zusätzlicher (vom Aktionspotenzial unabhängiger) Calciumkanäle. Sie vergrößern damit den intrazellulären Calciumpool und die bei der Kontraktion freigesetzte Ca++-Menge. Auch der Rücktransport in das sarkoplasmatische Retikulum wird beschleunigt und damit die Relaxation.
-
β-Rezeptorenblocker: Setzen die beschriebene Wirkung endogener und exogener Catecholamine und damit Kontraktilität und Relaxationsgeschwindigkeit herab.
-
Digitalis: Steigert die Bindungskapazität für Ca++ an der Innenfläche der Zellmembran, wodurch die vom Aktionspotenzial freigesetzte Ca++-Menge und die Kontraktilität zunehmen. Erst bei überhöhter Dosis wird der Ca++-Transport aus der Zelle gehemmt.
-
Frequenz-Kraft-Beziehung: Kontraktilitätssteigerung bei Frequenzanstieg und nach Extrasystolen, vermutlich infolge stärkerer Auffüllung der Ca++-Speicher durch die höhere Zahl von Aktionspotenzialen/min.
-
Calciumantagonisten: Hemmen den erregungsbedingten Ca++-Einstrom in die Myokardzellen und in die glatten Muskelzellen der Gefäße. Der im isolierten Herzmuskel deutliche negativ inotrope Effekt tritt am suffizienten Herzen klinisch nicht in Erscheinung (Kompensation durch Catecholamine), sondern erst bei Herzinsuffizienz und gleichzeitiger Gabe von β-Rezeptorenblockern. Bei den Caliciumantagonisten überwiegt die vasodilatatorische Wirkung mit ihren Konsequenzen für die Nachlast (▶ unten). Hinzu kommen antiarrhythmische Eigenschaften dieser Substanzen.
Als Messgrößen für die Kontraktilität dienen experimentell die Rate der Druckänderung im linken Ventrikel in Beziehung zurzeit (dP/dt) und zwar bei einem diastolischen Ventrikeldruck von 40 mmHg oder die maximale dP/dt während der Systole. In der Praxis Austreibungsgrößen wie Ejektionsfraktion (EF), prozentuale systolische Verkürzung des linksventrikulären Durchmessers (FS) und die maximale Beschleunigung des aortalen Blutflusses. Für alle Parameter der Kontraktilität des im Organismus schlagenden Herzens gilt einschränkend, dass sie auch von Vor- und Nachlast beeinflusst werden können.
1.9.6 Nachlast (afterload)
Darunter versteht man die aktive Spannungs- oder Kraftentwicklung, auch Stress genannt, pro Querschnittseinheit der Ventrikelwand während der Austreibungsphase. Je mehr von der mechanischen Energie der Kontraktion für die Spannungsentwicklung verbraucht wird, desto weniger bleibt für die Faserverkürzung, also für das Schlagvolumen übrig.
Vorlast und Kontraktilität determinieren zusammen die Kontraktionsenergie, die Nachlast beeinflusst die Förderleistung der Herzkammer.
Vom gesunden linken Ventrikel wird eine Zunahme der Nachlast durch eine Zunahme von Vorlast und Kontraktilität kompensiert. Wenn Vorlast- und Kontraktilitätsreserve erschöpft sind, kann die Förderleistung nur durch eine Senkung der Nachlast gesteigert werden. Die Determinanten der Nachlast sind der Aortendruck während der Systole und das enddiastolische Ventrikelvolumen. Denn nach dem Laplace-Gesetz nimmt die Wandspannung pro Querschnittseinheit für eine bestimmte Drucksteigerung im Ventrikel mit dem Ventrikelvolumen zu. Durch eine Wandhypertrophie kann die Nachlast normalisiert werden, da die einzelne Muskelfaser jetzt weniger Spannung erzeugen muss.
1.9.7 Herzfrequenz
Die Bedeutung der Herzfrequenz für die kardiale Förderleistung ergibt sich aus der Beziehung:
Herzminutenvolumen = Schlagvolumen × Schlagfrequenz
Da das Schlagvolumen nur begrenzt steigerungsfähig ist (Zunahme von Vorlast und Kontraktilität), kann die Anpassung des Minutenvolumens an größere körperliche Belastungen nur durch Erhöhung der Herzfrequenz erfolgen. Erst bei sehr hohen Frequenzen nimmt die Förderleistung wieder ab, weil die Diastolendauer für eine ausreichende Kammerfüllung zu kurz wird. Mit der Erhöhung der Schlagfrequenz ist auch ein positiv inotroper Effekt verbunden (s. oben).
Voraussetzung für die Zunahme des Minutenvolumens ist, dass die Frequenzsteigerung von einem Anstieg des venösen Rückflusses begleitet wird, wie es bei körperlicher Belastung der Fall ist. Stimuliert wird der Sinusknoten bei der Belastung durch Dehnung des Vorhofes, Sympathikusaktivierung und Vagushemmung. Steigert man die Herzfrequenz unter Ruhebedingungen, so kommt es nicht zur Vergrößerung des Minutenvolumens, weil enddiastolisches Ventrikelvolumen und Schlagvolumen kleiner werden. Eine Ausnahme bildet die Ruhetachykardie bei Herzinsuffizienz, die durch eine reflektorische Sympathikusstimulation entsteht und die Förderleistung verbessert.
Durch Bradykardien wird die Herzleistung auch bei intakter Ventrikelfunktion herabgesetzt. Extreme Bradykardien (AV-Block, Sinusknotenschädigung) führen zur Ruheinsuffizienz.
1.9.8 Definition
Herzinsuffizienz bedeutet Einschränkung der normalen Herzfunktion mit herabgesetzter Förderleistung und/oder Blutrückstau. Man unterscheidet 2 Schweregrade:
-
Kompensierte Herzinsuffizienz: Ungenügendes Herzminutenvolumen und/oder manifeste Lungenstauung erst unter körperlicher Belastung unterschiedlichen Grades (Belastungsinsuffizienz, latente Insuffizienz).
-
Dekompensierte Herzinsuffizienz: Ungenügendes Herzminutenvolumen und/oder manifeste Lungenstauung bereits unter Ruhebedingungen (Ruheinsuffizienz).
1.9.9 Pathogenese
1.9.10 Seitenlokalisation der Herzinsuffizienz
-
Linksherzinsuffizienz: Mit 80 % am häufigsten. Vorkommen: Koronare Herzkrankheit, Hypertonie, Aortenklappenfehler etc. Bei suffizientem rechten Ventrikel Begünstigung der Lungenstauung. Oft in globale Insuffizienz übergehend.
-
Rechtsherzinsuffizienz: Bei obstruktiven Lungenerkrankungen, Mitralstenose, pulmonaler Hypertonie, Pulmonalklappenvitien, Trikuspidalinsuffizienz, Septumdefekten mit Links-Rechts-Shunt etc. Förderleistung ebenso herabgesetzt wie bei Linksinsuffizienz. Blutrückstau nur im großen Kreislauf (Ausnahme: Mitralstenose). Im Allgemeinen kein Übergang in Globalinsuffizienz.
-
Doppel- oder Globalinsuffizienz: Primär bei Myokarditis, Anämie, Beri-Beri und extremer Bradykardie. Am häufigsten sekundär aus einer Linksinsuffizienz entstehend. Führt zu Stauungszeichen im großen und kleinen Kreislauf.
1.9.11 Pathogenetische Mechanismen
-
Mechanische Überbeanspruchung: Dafür gibt es 2 in ihren Ursachen und Auswirkungen unterschiedliche Möglichkeiten:
-
Volumenbelastung: Durch ein abnorm gesteigertes Minutenvolumen mit entsprechend vergrößertem venösen Rückfluss (high output failure), Shunts, Klappeninsuffizienzen etc. Sie erweitert den betroffenen Ventrikel und zwingt ihn, schon unter Ruhebedingungen ein erhöhtes Schlag- und Minutenvolumen auszuwerfen.
-
Druckbelastung: Durch erhöhten Austreibungswiderstand, der eine entsprechende Steigerung des systolischen Ventrikeldrucks erfordert.
-
-
Kontraktilitätsverlust: Durch anatomische oder funktionelle Veränderungen des Myokards (Myokardinsuffizienz): Bei Myokarditis, Koronarinsuffizienz, Myokardinfarkt, Kardiomyopathie, Anämie, Hypoxie, Intoxikationen, Azidose etc. Sekundär kommt es auch bei mechanischer Belastung mit Hypertrophie zur Kontraktilitätsminderung.
-
Störungen der diastolischen Kammerfüllung: Bei Stenosen der AV-Klappen, extremer Tachykardie, herabgesetzter Dehnbarkeit der Ventrikel (Endo- und Myokardfibrose, Ventrikelhypertrophie, ischämische Störung der Relaxation, Amyloidablagerungen, Pericarditis constriktiva, Perikardergüsse) und bei dilatiertem Ventrikel, da die Dehnbarkeit (Compliance) mit zunehmender Dilatation abnimmt. Folge der erschwerten Füllung des linken Ventrikels ist ein Druckanstieg auf der venösen Seite des kleinen Kreislaufs, der zur Lungenstauung und zum Lungenödem führen kann, bevor das Minutenvolumen unter die Bedarfsgröße sinkt.
1.9.12 Kompensationsmechanismen
-
Sympathikusstimulation und Vagushemmung: Steigern in Sekundenschnelle Kontraktilität und Herzfrequenz wirken daher schnell einem Herzversagen entgegen.
-
Zunahme der Vorlast durch renale Flüssigkeitsretention: Akutes Nachlassen der Förderleistung erhöht das Restvolumen im Ventrikel und führt durch den zunächst nicht verminderten venösen Rückfluss zur Erhöhung des enddiastolischen Volumens und damit nach dem Starling-Gesetz zur Steigerung der Kontraktionskraft.
Langfristig wird die Vorlast bei kardialer Pumpschwäche durch eine kompensatorische Hypervolämie erhöht, die auf folgende Weise entsteht: Drosselung der Nierendurchblutung durch sympathikusinduzierte Vasokonstriktion → Herabsetzung der glomerulären Filtration → Aktivierung des Renin-Angiotensin-Aldosteron-Systems → Verminderung der renalen Wasser- und Salzausscheidung → Hypervolämie → Zunahme des venösen Rückflusses und der Ventrikelfüllung.
Wenn die Vorlast durch die Hypervolämie genügend steigt, um das Minutenvolumen zu normalisieren, lässt die Sympathikusstimulation nach, andernfalls bleibt sie bestehen. Herzglykoside können den Sympathikus entlasten bzw. seine kontraktilitätssteigernde Wirkung unterstützen. Die Flüssigkeitsretention geht auch nach Ausschöpfung der Vorlastreserve weiter, wenn sich das Minutenvolumen nicht normalisiert. Die progrediente Hypervolämie führt dann zu Ödemen und zur Überdehnung des linken Ventrikels (slippage), dessen Kontraktilität dadurch noch weiter abnimmt. Diuretika können durch Beseitigung der Überdehnung eine deutliche Besserung der Kontraktionskraft des linken Ventrikels bewirken. Es sollte aber nicht zu stark entwässert werden, weil ein gewisser Grad von Hypervolämie durch Ausschöpfung der Vorlastreserve kompensierend wirkt.
Eine Lungenstauung entsteht durch Rückstau in den Lungenkreislauf bei Insuffizienz des linken Ventrikels. Steigt der hydrostatische Druck in den Lungenkapillaren über den kolloidosmotischen Druck des Blutplasmas, kommt es zum Lungenödem. Die Bedingungen dafür sind Blutrückstau und ein Ungleichgewicht der Starling-Kräfte beider Ventrikel. Bei einer Insuffizienz beider Ventrikel ist die Gefahr eines Lungenödems wesentlich geringer.
-
Hypertrophie: Erhöhte Dauerbelastung beantwortet das Herz mit einer Zunahme der Muskelmasse im betroffenen Abschnitt und vergrößert dadurch seine Kontraktionskraft. Die Form der Hypertrophie hängt vom Belastungstyp ab:
-
Exzentrische Hypertrophie: Entsteht bei Volumenüberlastung durch den stimulierenden Effekt des erhöhten enddiastolischen Drucks (diastolischer Wandstress). Es kommt zur Vermehrung der Sarkomeren überwiegend in der Längsrichtung und damit zur Vergrößerung des Ventrikels ohne wesentliche Zunahme der Wanddicke.
-
Konzentrische Hypertrophie: Entsteht bei Drucküberlastung, stimuliert durch den systolischen Wandstress. Die Sarkomere vermehren sich parallel zueinander, so dass die Wanddicke zunimmt und das Ventrikelvolumen unverändert bleibt bzw. leicht abnimmt.
-
Spätfolgen der Hypertrophie: Missverhältnis von Muskelmasse zur Vaskularisierung bei hohem O2-Verbrauch, Ischämie, Fibrosierung des subendokardialen Myokards, das infolge schlechter Blutversorgung degeneriert, Kontraktilitätsverlust bis zum finalen Myokardversagen.
-
Sportherz: Physiologische Hypertrophie leichteren Grades, durch Sportarten mit Volumenbelastung vom exzentrischen Typ (Langstreckenlauf, Schwimmen etc.), bei solchen mit Druckbelastungen vom konzentrischen Typ (Gewichtheben, Ringen, Rudern).
-
1.9.13 Klinische Diagnostik
1.9.14 Anamnese
Die Bedeutung der Anamnese ergibt sich nicht zuletzt daraus, daß die New York Heart Association (NYHA) die Schweregrade der Herzinsuffizienz nach der subjektiven Einschätzung der Belastungstoleranz klassifiziert hat:
-
Grad I: Belastungstoleranz nicht herabgesetzt.
-
Grad II: Belastungstoleranz gering herabgesetzt.
-
Grad III: Belastungstoleranz deutlich herabgesetzt. Weniger als die normale Belastung erzeugt Beschwerden.
-
Grad IV: Belastungstoleranz hochgradig herabgesetzt. Beschwerden können schon in Ruhe vorhanden sein, treten bei jeder geringsten Belastung auf.
Als limitierende Faktoren für die körperliche Belastung gelten in dieser Klassifikation Schwäche, Herzklopfen, Atemnot und anginöse Herzbeschwerden.
1.9.15 Symptome des reduzierten Herzminutenvolumens
-
Verminderung der körperlichen Leistungsfähigkeit: Rasche Erschöpfung mit Schweregefühl in den Gliedern infolge mangelhafter Muskeldurchblutung.
-
Zerebrale Symptome: Durch Absinken des Blutdrucks Schwindelgefühl bei körperlicher Belastung, Kopfschmerzen, Verwirrtheit, Gedächtnisstörungen, Angstzustände, Schlafstörungen, bei alten Menschen mit zerebraler Gefäßsklerose auch Delirien und Psychosen.
-
Renale Symptome: Nykturie durch Aufhebung der kompensatorischen renalen Vasokonstriktion in Ruhelage. Oligurie und Azotämie durch intensive permanente renale Vasokonstriktion bei schwerer dekompensierter Herzinsuffizienz.
-
Intestinale Symptome: Leibschmerzen infolge intestinaler Ischämie.
1.9.16 Symptome der venösen Stauung
-
Linksherzinsuffizienz: Stauungserscheinungen im Lungenkreislauf:
-
Dyspnoe: Kurzatmigkeit bei immer geringeren Anstrengungen (Belastungsdyspnoe), zuletzt auch in Ruhe (Ruhedyspnoe). Inspiratorische feinblasige basale Rasselgeräusche können hörbar werden. Abgrenzung gegen pulmonale Dyspnoe (Emphysem, chronische Obstruktion mit Husten und Auswurf, Besserung durch Bronchodilatoren) in Grenzfällen schwierig, erst durch Nachweis des erhöhten Lungenkapillardrucks möglich.
-
Orthopnoe: Atemnot bei Flachlagerung, die nach Aufsetzen oder Aufstehen verschwindet, da der venöse Rückfluss abnimmt. Auch bei schwerer obstruktiver Lungenerkrankung zum besseren Einsatz der Atemhilfsmuskulatur vorkommend.
-
Paroxysmale nächtliche Dyspnoe: Nächtliches Erwachen aus REM-Phasen des Schlafes mit schwerer Atemnot, die zu raschem Aufstehen zwingt und nur allmählich abklingt. Verursacht durch plötzliche Volumenverschiebung in den kleinen Kreislauf. Manchmal Bronchospastik (Asthma cardiale). Bedrohlich Hinweis auf Linksherzversagen.
-
Stauungsbronchitis: Chronischer, meist trockener Husten, der auf antibronchitische Behandlung nicht anspricht. Nachts oft verstärkt. Auch Stauungsblutungen kommen vor. Im Sputum Herzfehlerzellen.
-
Lungenödem: Nach trockenem Husten einsetzende schwerste Dyspnoe mit beschleunigter, vertiefter Atmung unter Einsatz der Hilfsmuskulatur, begleitet von Erregung, Unruhe, Angst, Tachykardie, kaltem Schweiß, zentraler und peripherer Zyanose. Mittelblasige inspiratorische Rasselgeräusche bis in die oberen Lungenabschnitte, auch Spastik. Schaumiges, manchmal blutig tingiertes Sputum.
-
-
Rechtsherzinsuffizienz: Stauungszeichen im großen Kreislauf:
-
Erhöhung des Venendrucks: Durch Abflussbehinderung in den rechten Ventrikel. Erkennbar an fortgeleiteten Pulsationen der V. jugularis interna (die im Gegensatz zur V. jugularis externa ohne Zwischenschaltung von Venenklappen mit dem rechten Vorhof kommuniziert).
-
Hepatojugulärer Reflux: Anstieg des Jugularvenendrucks bei Kompression des rechten oberen Abdomens mit der Hand, der normalerweise keinen Druckanstieg bewirkt. Ursachen des abnormen Druckanstiegs: Erhöhter Venentonus, und Verschiebung eines relativ großen Volumens aus dem blutüberfüllten Abdomen zum Herzen.
-
Halsvenenfüllung: In aufrechter Haltung indirektes Zeichen eines erhöhten Vorhofdrucks, sofern keine Abflussbehinderung vor dem Herzen besteht (z. B. durch eine Struma).
-
Stauungsleber: Lebervergrößerung, oft vor der Ausbildung peripherer Ödeme nachweisbar. Bei akuter Leberstauung Schmerzen durch Kapseldehnung. Abgrenzung gegen andere Ursachen der Lebervergrößerung durch positiven hepatojugulären Reflux, bei Trikuspidalinsuffizienz durch fortgeleiteten Jugularvenenpuls und sonographischen Nachweis erweiterter Lebervenen.
-
Aszites: Durch langdauernde Druckerhöhung in den Lebervenen und den Venen, die das Peritoneum drainieren (Trikuspidalinsuffizienz, Pericarditis constrictiva).
-
Intestinale Symptome: Völlegefühl, Anorexie, Übelkeit, Meteorismus, Obstipation.
-
Renale Symptome: Stauungsalbuminurie und -hämaturie.
-
Periphere Ödeme: Durch Erhöhung des Venendrucks infolge mechanischer Abflussbehinderung und durch die kompensatorische renale Flüssigkeitsretention bedingt. In aufrechter Haltung an Knöcheln und Unterschenkeln, im Liegen über dem Kreuzbein, in schweren Fällen generalisiert (Anasarka).
-
-
Globalinsuffizienz: Summation der Stauungszeichen von Links- und Rechtsherzinsuffizienz. Als zusätzliches Symptom treten Pleuraergüsse auf. Die Pleurahöhlen werden von den Venen und Lymphgefäßen der viszeralen und der parietalen Pleura drainiert. Bei Linksherzinsuffizienz ausreichende Drainage durch die parietale Pleura. Bei Rechtsherzinsuffizienz kann eine starke Venendruckerhöhung (durch Erschwerung des venösen Abflusses und des Lymphabflusses aus dem Ductus thoracicus in die obere Hohlvene) zum Pleuraerguss und zur Dyspnoe führen. Meistens besteht bei Pleuraergüssen eine Globalinsuffizienz. Aus ungeklärten Gründen entwickeln sich Stauungsergüsse oft auf der rechten Seite zuerst bzw. stärker.
1.9.17 Kardiovaskuläre Symptome
-
Herzvergrößerung: Im dekompensierten Stadium in der Regel vorhanden, bei druckhypertrophierten Herzen (Aortenstenose, Hypertonie, HOCM, Cor pulmonale) später als bei volumenbelasteten (Aorteninsuffizienz, Trikuspidalinsuffizienz). Keine Vergrößerung bei ganz akuter Insuffizienz (Lungenembolie, Herzinfarkt) und bei Concretio pericardii. Linksverbreiterung perkutorisch und durch Palpation des Spitzenstoßes festzustellen. Bei Rechtshypertrophie parasternale und epigastrische Pulsationen. Genaue Analyse durch Röntgenbild und Echokardiogramm.
-
3. Herzton (protodiastolischer Galopprhythmus): Frühdiastolischer Füllungston bei überdehntem Ventrikel. Linksventrikulärer 3. Herzton niederfrequent, oft leise, am besten in Linksseitenlage über der Herzspitze und rechtsventrikulärer 3. Herzton links parasternal zu hören (Trikuspidalinsuffizienz).
-
Relative AV-Klappeninsuffizienz: Bei starker Kammerdilatation mit Erweiterung des Klappenringes Mitral- bzw. Trikuspidalinsuffizienz, bei letzterer atemabhängige Intensitätsschwankung des holosystolischen Refluxgeräusches.
-
Pulsus alternans: Abwechselnd folgen ein schwacher und ein kräftiger Herzschlag aufeinander mit entsprechendem Blutdruckunterschied. Nachweis durch Zunahme der Korotkow-Töne beim Ablassen des Manschettendruckes während der Blutdruckmessung. Differenzen von 10–20 mmHg. Zeichen einer schweren Kontraktilitätsstörung des linken Ventrikels.
-
Inadäquate Herzfrequenz: Ruhetachykardie und inadäquater Frequenzanstieg unter körperlicher Belastung als Zeichen der kompensatorischen Sympathikusstimulation und Vagushemmung.
-
Kutane Vasokonstriktion: Im Rahmen der Umverteilung des Minutenvolumens zugunsten der lebenswichtigen Organe. Die damit verbundene Behinderung der Wärmeabgabe kann subfebrile Temperaturen hervorrufen.
1.9.18 Apparative Diagnostik
1.9.19 Röntgenaufnahmen des Thorax in zwei Ebenen
Herzbefunde: Erkennung des Ausmaßes der Herzdilatation und abnormer Konfigurationen der Herzsilhouette. Nachweis von Verkalkungen des Perikards, der Pleura, Lungenstauung und Pleuraergüssen.
1.9.20 Elektrokardiogramm (EKG)
Erfasst tachykarde und bradykarde zur Insuffizienz führende Rhythmusstörungen, frische Infarkte und alte Infarktnarben. Mittels Ergometrie sind Belastungsgrenzen zu objektivieren.
1.9.21 Echokardiographische Untersuchung
Ermöglicht eine umfassende Analyse der Anatomie und Hämodynamik des Herzens. Auf den Funktionsgrad ist aus dem Durchmesser des linken Ventrikels, der Ejektionsfraktion und der relativen systolischen Durchmesserverkürzung (FS) zu schließen. Die ◘ Abb. 1.63 zeigt im Echokardiogramm eines Patienten mit dekompensierter dilatativer Kardiomyopathie einen stark erweiterten dünnwandigen linken Ventrikel und eine Dilatation des linken Vorhofs. Mit der Dopplermessung des transmitralen Einflusses werden diastolische Funktionsstörungen erfasst.


Echokardiographischer 4-Kammerblick eines 50-jährigen Patienten mit dilatativer Kardiomyopathie (RA/LA = rechter/linker Vorhof, RV/LV = rechter/linker Ventrikel)
1.9.22 Rechtsherzeinschwemmkatheter
Die Durchführung in Ruhe und bei dosierter ergometrischer Belastung gibt Aufschluss über das Minutenvolumen, das bei insuffizienten Herzen nicht adäquat ansteigt. Bei Herzinsuffizienz werden Drucksteigerungen bzw. Stauungszeichen im kleinen Kreislauf und ein erhöhter enddiastolischer Füllungsdruck des linken Ventrikels erfasst. Der periphere Widerstand lässt sich bestimmen. In der Intensivmedizin kann der pulmonale Kapillardrucks (PCP) fortlaufend kontrolliert werden.
1.9.23 Labordiagnostik
Frühindikator der Herzinsuffizienz ist ein erhöhter BNP-Spiegel. Bestimmt wird häufig der aminoterminale Abschnitt des Prohormons (NT-proBNT), der neben BNP mit höherer Halbwertszeit zirkuliert. Die ◘ Abb. 1.64 zeigt die Korrelation zwischen NT-pro-BNP-Konzentrationen und NYHA-Klassen der Herzinsuffizienz. In der NYHA-Klasse IV können die NT-pro-BNP-Konzentrationen auf >5.000 pg/ml steigen.


NT-proBNP-Werte korrelieren mit dem Schweregrad der Herzinsuffizienz. Je höher der NT-proBNP-Wert, desto schwerer die Erkrankung und desto schlechter die Prognose (Roche Diagnostics)
Die natriuretischen Peptide können auch bei ventrikulärer Hypertrophie, Herzklappenerkrankungen, Vorhofflimmern, akuter und chronischer Myokardischämie, Lungenembolie, Anämie und Nereninsuffizienz erhöht sein.
1.9.24 Therapie
Die Herzinsuffizienz ist ein progredientes Leiden. Es schreitet mit zunehmender Geschwindigkeit fort. In einigen Fällen kann es durch rechtzeitige kausale Therapie der zugrunde liegenden Herzkrankheit abgewendet, behoben oder stark verlangsamt werden. Dazu gehören z. B. operable angeborene und erworbene Vitien, ischämische Kardiomyopathien, Myokarditiden und Hypertonien.
In den nicht reparablen Fällen ist es am wichtigsten, das Herz dauerhaft zu entlasten. Das beginnt mit der Beschränkung körperlicher Aktivitäten im häuslichen und beruflichen Alltag und kann einen Berufswechsel oder die vorzeitige Berentung notwendig machen. Auf jeden Fall sind erschöpfende körperliche Anstrengungen und übermäßiger psychischer Stress zu vermeiden. Zu achten ist auf ausgiebige Nachtruhe und Ruhepausen am Tage. Die Ernährung sollte salzarm und nicht überkalorisch sein. Große Mahlzeiten sind zu vermeiden, weil sie den Kreislauf belasten. Übergewicht ist längerfristig zu reduzieren. Wenn trotz optimaler medikamentöser Therapie die NYHA-Klasse IV erreicht ist, kann manchmal eine strikte wochenlange Bettruhe noch zu erstaunlicher Besserung führen, wie man es bei hospitalisierten Kandidaten für eine Herztransplantation beobachtet hat.
1.9.25 Medikamentöse Therapie
Die Mehrzahl der Patienten in den NYHA-Klassen I und II der Herzinsuffizienz wird mit einem Programm körperlicher Entlastung und diätetischer Vorsorge auskommen. Viele Patienten werden sogar keine ärztliche Hilfe in Anspruch nehmen. Es sollte aber schon bald mit einer protektiven medikamentösen Therapie begonnen werden.
-
Inhibitoren des Renin-Angiotensin-Systems: Mit der Ausschaltung des Angiotensin II senken sie den peripheren Widerstand und damit das Afterload. Mit der Drosselung der Aldosteronsekretion wirken sie einer Hypervolämie entgegen und vermindern das Präload des Herzens. Als wichtiger Effekt kommt hinzu, dass sie durch Hemmung des lokalen Renin-Angiotensin-Systems Hypertrophie und Fibrose des Herzmuskels (nachteiliges Remodeling nach Infarkt) hemmen. Kürzlich wurde gezeigt, dass sie auch die Aktivierung von Blutmonozyten (zur Adhärenz am Endothel und wahrscheinlich zur Freisetzung von Zytokinen) unterdrücken. Wegen dieser Eigenschaften sind die Inhibitoren des RAAS in allen Stadien der Herzinsuffizienz indiziert und bei vorgeschädigten Herzen und Hypertonie schon zur Prophylaxe. Bei zu hoher Dosierung drohen Blutdruckabfall, Hyperkaliämie und Einschränkung der Nierenfunktion. Das Serumkreatinin sollte nicht über 2,5 mg/dl steigen.
Die beiden Substanzklassen ACE-Blocker und Angiotensin-II-Rezeptor-Antagonisten sind in ihrer Wirkung gleichwertig. Letztere, die AT1-Rezeptor-Antagonisten haben jedoch den Vorteil, keinen chronischen Hustenreiz zu verursachen, den die ACE-Blocker in bis zu 20 % der Fälle auslösen.
-
ACE-Blocker :
-
Captopril: Initialdosis 6,25 mg, Maximaldosis bis 4-mal 50–100 mg/Tag
-
Enalapril-Maleat: Initialdosis 2-mal 2,5 mg/Tag, Maximaldosis 2-mal 10–20 mg/Tag
-
Lisinopril: Initialdosis 1-mal 2,5–5 mg/Tag, Maximaldosis 1-mal 10–20 mg/Tag
-
-
Angiotensin-Rezeptor-Antagonisten:
-
Lorsartan: Initialdosis 1-mal 12,5 mg/Tag, Maximaldosis 1-mal 50 mg/Tag
-
Irbesartan: Initialdosis 1-mal 75 mg/Tag, Maximaldosis 300 mg/Tag
-
Valsartan: Initialdosis 1-mal 80 mg/Tag, Maximaldosis 1-mal 16 mg/Tag
-
Candesartan: Initialdosis 1-mal 4 mg/Tag, Maximaldosis 1-mal 16 m/Tag
-
-
Diuretika: Ihr Einsatz erfolgt zur Verhinderung oder Beseitigung von Ödemen. Sie wirken der renalen Wasser- und Salzretention bei der Herzinsuffizienz entgegen, die umso stärker wird, je weiter das Herzminutenvolumen absinkt. Patienten mit kardialen Ödemen trinken viel, weil die Verkleinerung des arteriellen Blutvolumens das Durstzentrum im Zwischenhirn stimuliert. Auf diese Weise wird die Ödembildung im Sinne eines Circulus vitiosus gefördert. Die Auswahl des Diuretikum richtet sich nach dem Schweregrad der Ödeme bzw. Stauungszeichen und nach der Nierenfunktion.
-
Thiazide: Genügen in leichten bis mittelschweren Fällen.
-
Hydrochlorothiazid: Intialdosis 1-mal 25 mg/Tag, Maximaldosis 1-mal 100 mg/Tag
-
Chlorthalidon: Initialdosis 1-mal 50 mg/Tag, Maximaldosis 100 mg/Tag
-
Am besten werden Thiazide mit einem kaliumsparenden Diuretikum kombiniert
-
Kombinationspräparate: Dytide H (Hydrochlorothiazid 25 mg + Triampteren 50 mg) 1-mal 1–2 Tabl./Tag oder Moduretik: (Hydrochlorothiazid 25 mg + Amilorid 2,5 mg) 1-mal 1–2 Tabl./Tag
-
-
Schleifendiuretika: Indiziert in schweren Fällen und bei eingeschränkter Nierenfunktion.
-
Furosemid: Initialdosis 1–2-mal 20–40 mg/Tag p.o. oder 20 mg i. v., Maximaldosis 400 mg/Tag p.o. oder 400 mg i. v./Tag mit Perfusor (nicht bei dekompensierter Aortenstenose!)
-
Torasemid: Initialdosis 1–2-mal 10 mg/Tag p.o. oder 0,5 mg i. v., Maximaldosis 200 mg/Tag p.o. oder i. v. mit Perfusor/24 h
-
-
-
Aldosteronantagonisten: In einer Studie an 1663 Patienten mit Herzinsuffizienz der NYHA-Klassen II und IV (RALES-Studie) wurde unter Standardmedikation (ACE-Blocker plus Schleifendiuretikum) die zusätzliche Gabe von Spironolacton (12,5–25 mg/Tag) gegen Plazebo getestet. Ergebnis: In der Spironolacton-Gruppe besserten sich die Symptome signifikant, und das Mortalitätsrisiko ging gegenüber der Plazebo-Gruppe um 30 % zurück. Zu erklären ist dieser Effekt zum einen damit, dass erhöhte Aldosteronspiegel den Herzmuskel schädigen und fibrosefördernd wirken. Zum anderen ist anzuführen, dass Aldosteron erst durch spezifische Antagonisten ausgeschaltet wird, weil die Plasmakonzentrationen unter langfristiger Behandlung mit ACE-Blockern nicht zurückgehen. Neuerdings steht mit Eplerenon ein Aldosteronantagonist zur Verfügung, der keine hormonalen Nebenwirkungen hat und deshalb vor allem für Frauen zu empfehlen ist. Unter Beachtung des Kaliumspiegels und der Nierenfunktion (Serumkreatinin) sollten niedrig dosierte Aldosteronantagonisten jetzt in den NYHA-Stadien III und IV prinzipiell eingesetzt werden.
-
β-Adrenorezeptor-Antagonisten (β-Blocker )
-
Wirkungsmechanismus: Die Steigerung des Sympathikustonus mit Frequenzanstieg und positiv inotropem Effekt ist ein wichtiger Kompensationsmechanismus bei akuter Herzinsuffizienz. Deshalb kann eine Volldosis von β-Rezeptorenblockern bei Herzinsuffizienz leicht zur akuten Dekompensation führen. Bei chronischer Herzinsuffizienz wirkt sich dagegen ein permanent erhöhter Tonus des adrenergen Nervensystems schädlich aus. Die stärksten nachteiligen Effekte werden von Noradrenalin über die β1-Rezeptoren vermittelt (Myotoxizität, Apotose, Myozytenwachstum), während positive Inotropie und Chronotropie über β1- und β2-Rezeptoren mit gleicher Intensität induziert werden. Mit fortschreitender Herzinsuffizienz nimmt der Anteil der β1-Rezeptoren ab, der bei Gesunden >70 % beträgt. Das Verhältnis β1: β2: α1 liegt zuletzt bei 2 : 1 : 1.
Die protektive Wirkung von β-Blockern zeigte sich, als bei Patienten mit chronischer Herzinsuffizienz (NYHA II und III), die durch Diuretika, ACE-Blocker und Digitalis stabilisiert waren, langsam steigende Dosen von β1-Blockern gegeben wurden. Zu verzeichnen war eine Besserung der klinischen Symptome und ein Rückgang der Gesamttodesfälle sowie des akuten Todes. Auch die Häufigkeit der Rehospitalisierung wegen Dekompensation ging zurück. Ein positiv-inotroper Effekt über β2-Rezeptoren bleibt erhalten.
In einer Meta-Analyse von mehreren großen randomisierten kontrollierten Studien an Patienten mit Herzinsuffizienz wurde kürzlich gezeigt, dass die Effekt der β-Blockertherapie mit der Reduzierung der Herzfrequenz korreliert. Eine weitere große Meta-Analyse an 12.209 Patienten aus mehreren Studien ergab, dass der Überlebensvorteil vom Grad der Frequenzreduktion des Herzens und nicht von der Dosis des β-Blockerdosis abhängig war. Sehr wirksam lässt sich die Herzfrequenz mit dem neuen Sf-Kanalblocker Ivabradin reduzieren. Man hat deshalb begonnen, diese nicht negativ inotrope Substanz bei der Herzinsuffizienz zu testen.
-
Präparate und Dosierungen: Als klinisch geeignet erwiesen sich in zahlreichen Studien selektive β1-Rezeptor-Antagonisten (Metoprolol-Succinat, Bisoprolol) und β-Blocker-Vasodilatatoren (Carvedilol, Bucindolol). Erstere werden bei Herzinsuffizienz toleriert, weil β2- und α1-Rezeptoren nicht blockiert werden und weiter positiv-inotrope adrenerge Stimulation vermitteln. Letztere, zwei nichtselektive β-Blocker, weil der relativ mäßige negativ-inotrope Effekt durch die Vasodilation kompensiert wird, die das Afterload senkt.
Für alle β-Blocker gilt, dass ihre Anwendung bei der Herzinsuffizienz mit extrem niedrigen Dosierungen (1/8 bis 1/16 der Zieldosis) begonnen werden muss mit vorsichtiger Steigerung in Abständen von 1–2 Wochen. Die hohe Zieldosis ist erforderlich, um die β1-Rezeptoren weitgehend auszuschalten. Die Zieldosis muss auf Dauer beibehalten werden. Der therapeutische Effekt stellt sich erst im Laufe einiger Monate ein.
-
Metoprolol-Succinat (Beloc-Zok): Initialdosis 1-mal 12,5 mg/Tag, Zieldosis 1-mal 190 mg/Tag.
-
Bisoprolol (Concor): Initialdosis 1-mal 1,25 mg/Tag, Zieldosis 1-mal 5 mg/Tag (<75 bis 80 kg), 10 m/Tag (75 bis >85 kg).
-
Carvedilol (Dilatrend): Initialdosis 2-mal 3,125 mg/Tag, Zieldosis 2-mal 25 mg/Tag (<75 bis 85 kg), 2-mal 50 mg/Tag (75 bis >85 kg). Das Mittel scheint bei NYHA III etwas vorteilhafter zu sein, führt aber leichter zur Hypotonie.
-
-
Indikationen: Bei Herzinsuffizienz der NYHA-Klassen II und III, bei euvolämischen Patienten der Klasse IV. Bei Dekompensation unter β-Blockade sind Phosphodiesterase-Inhibitoren angezeigt (▶ unten), da sie nicht antagonisiert werden.
-
-
Kontraindikationen: Bradykardie, AV-Leitungsstörungen, hydropische Herzinsuffizienz.
-
Herzglykoside (Digitalis): Bei der Herzinsuffizienz werden sie zur Steigerung der Inotropie des Myokards und zur Verlangsamung der Kammerfrequenz bei Vorhofflimmern und Vorhofflattern angewendet.
-
Wirkungsmechanismus: Die positive Inotropie beruht auf einer Vergrößerung des intrazellulären Calciumpools, die während der Systole zur gesteigerten Freisetzung von Ca++ aus dem zytoplasmatischen Retikulum führt. Induziert wird die Calciumanreicherung durch die selektive Hemmung der Na+/K+-ATPase in der Zellmembran, die einen Anstieg der intrazellulären Na+-Konzentration, zur Folge hat. Der erhöhte Na+-Spiegel steigert die Aktivität der ebenfalls in der Zellmembran lokalisierten Na+/Ca++-ATPase, so dass es im Austausch gegen Na+ zu einem verstärkten Ca++-Einstrom in die Zelle kommt.
Der inhibierende Effekt auf den Sinus- und den AV-Knoten kommt durch eine Steigerung der efferenten Vagusimpulse mit entsprechend vermehrter Freisetzung von Acetylcholin zustande. Acetylcholin erhöht die K+-Permeabilität der Zellmembran und bewirkt dadurch eine Hyperpolarisation, die zur Verlangsamung der Depolarisation und der Erregungsleitung führt.
-
Therapeutischer Nutzen: Digitalis ist ein wichtiges Mittel zur Normalisierung der Herzfrequenz bei tachykardem Vorhofflimmern und Vorhofflattern. Beide können eine Herzinsuffizienz herbeiführen oder verschlimmern. Bei einer Herzinsuffizienz mit Sinusrhythmus reduziert Digitalis die Symptome und die Häufigkeit der Krankenhausbehandlungen. Die Überlebensdauer der Patienten wird kontrollierten Studien zufolge jedoch nicht verlängert. Die Anwendung erfolgt in den NYHA-Klassen III und IV. Die gebräuchlichen Vertreter der Digitalisglykoside sind Digoxin und Digitoxin.
-
Pharmakokinetik und Dosierung: Digitalisglykoside kumulieren im Körper und werden erst nach Speicherung bestimmter Mengen therapeutisch wirksam. Nach der Aufsättigung muss die tägliche Abklingquote durch die Erhaltungsdosis ersetzt werden. Anhand der Plasmaspiegel kann die Dosierung kontrolliert werden.
-
Digoxin und Derivate (Acetyldigoxin, β-Methyldigoxin): Applikation per os oder intravenös. Enterale Resorption 50–80 %. Renale Ausscheidung 70 %. Tägliche Abklingquote 30 %. Aufsättigung mit Digoxin: 2-mal 0,25 mg/Tag p.o. für 3 Tage oder 2-mal 0,25 mg/Tag i. v. für 2 Tage. Erhaltungsdosis 1-mal 0,25–0,375 mg/Tag. Dosisanpassung bei eingeschränkter Nierenfunktion. Therapeutische Serumspiegel 0,9‒2,0 ng/ml. Nach neueren Studien sollte ein Spiegel von 1,2 ng/ml nicht überschritten werden. Vom Acetyldigoxin (Novodigal) gibt es Tabletten zu 0,1 und 0,2 mg und Ampullen zu 0,4 mg.
-
Digitoxin: Applikation per os, selten intravenös. Enterale Resorption 90–100 %. Renale Ausscheidung 25 %. Tägliche Abklingquote 10 %. Aufsättigung: 3-mal 0,05–0,1 mg/Tag p.o. für 3–5 Tage. Therapeutische Plasmakonzentrationen 10–30 ng/ml (wegen starker Bindung an Plasmaproteine höher als für Digoxin). Erhaltungsdosis durchschnittlich 0,07 mg. Keine Dosisanpassung bei eingeschränkter Nierenfunktion. Wegen niedriger Abklingquote schlechter steuerbar als Digoxin.
-
-
Digitalisintoxikation: Entsteht bei Überdosierung oder verminderter Digitalistoleranz (organische Herzerkrankungen, hohes Alter, Hypokaliämie, Hypothyreose).
-
Kardiale Symptome: Sinusbradykardie, SA-Block, AV-Block unterschiedlichen Grades, Vorhofextrasystolen, Vorhof- und Knotentachykardien, Vorhofflimmern, Kammerextrasystolen, Kammertachykardien, auch Kammerflimmern. Diese Symptome sind unspezifisch, verschwinden aber nach Dosiskorrektur oder Absetzen des Digitalispräparates.
-
Extrakardiale Symptome: Anorexie, Übelkeit, Erbrechen durch Reizung medullärer Zentren. Gelbsehen, Kopfschmerzen, Müdigkeit, depressive Verstimmung.
-
Therapie: Digitalis absetzen oder niedriger dosieren. Kaliummangel ausgleichen. Gegen Bradykardie und Erregungsleitungsstörungen Atropin. Bei Tachykardien Lidocain oder Phenytoin. In den schwersten Intoxikationsfällen Neutralisierung des Digoxin mit spezifischen Antikörpern.
-
-
-
Katecholamine: Diese Sympathikomimetika von stark positiv inotroper Wirksamkeit werden nur bei akuter Herzinsuffizienz, kardiogenem Schock und im Endstadium der chronischen Herzinsuffizienz eingesetzt, wenn alle anderen Möglichkeiten der medikamentösen Therapie ausgeschöpft sind. Sie können nur intravenös appliziert werden.
-
Dobutamin: Verbessert die Ventrikelfunktion durch direkte Stimulation der β1- und β2-Rezeptoren des Myokards. Die β1-Rezeptoren des Sinusknotens werden kaum stimuliert, so dass die Herzfrequenz nur wenig zunimmt. Der periphere Widerstand ändert sich kaum, weil neben vasokonstriktorischen α1-Rezeptoren auch vasodilatatorische β2-Rezeptoren stimuliert werden. Die Applikation erfolgt durch kontinuierliche Infusion (2,5–10 µg/kg/min). Anwendung bei akuter und bei schwer dekompensierter chronischer Herzinsuffizienz. Nach mehrtägiger Infusion lässt die Wirkung nach. Intermittierende Kurzzeitinfusionen bleiben dagegen effektiv.
-
Dopamin: Die kardiovaskulären Effekte des Dopamin sind dosisabhängig:
-
Niedrige Dosis (1,5–3,5 µg/kg/min): Durch Stimulation vaskulärer D1-dopaminerger Rezeptoren Vasodilatation im renalen, koronaren und mesenterialen Flussbett. Steigerung der Diurese und Na+-Ausscheidung.
-
Mittlere Dosis (4–10 µg/kg/min): Positiv inotrope Wirkung durch direkte Stimulation beider β-Rezeptoren am Myokard und Noradrenalinfreisetzung an den sympathischen Nervenenden, die auch den Blutdruck und die Herzfrequenz erhöht.
-
Hohe Dosis (10,5–21,5 µg/kg/min): Bewirkt durch direkte Stimulation der vaskulären α1-Rezeptoren eine periphere Vasokonstriktion mit Blutdruckanstieg und Drosselung der Nierendurchblutung. Der positiv inotrope Effekt des Dobutamin ist stärker als der des Dopamin. Letztes wird in mittlerer bis hoher Dosierung eingesetzt, wenn der Blutdruck abgefallen ist. Oft werden beide Katecholamine kombiniert.
-
-
-
Phosphodiesterase-Inhibitoren: Sie verzögern den Abbau des intrazellulären cAMP, das der second messenger der adrenalen Stimulation ist. Durch Steigerung der Kontraktilität des Herzens bei gleichzeitiger Absenkung des peripheren Widerstands wird das Herzzeitvolumen vergrößert. Verfügbar sind Milrinom und Enoximon. Die Applikation erfolgt durch Infusion. Anwendung beim Versagen der Katecholamine, mit denen sie kombiniert werden können.
-
Vasodilatatoren: Bei schwerer Herzinsuffizienz kann es zu einer massiven, gegen ACE-Blocker resistenten Vasokonstriktion kommen. Dabei spielt vielleicht die Freisetzung von Endothelin aus geschädigten Endothelzellen eine Rolle. In diesen Fällen werden starke Vasodilatatoren eingesetzt, von denen zwei genannt seien:
-
Natrium-Nitroprussid: Infusion mit 0,1–3,0 µg/kg/min. Die Wirkung setzt sehr schnell ein ist kurzdauernd.
-
Nitroglycerin: Infusion zunächst mit 20 µg/min, dann schrittweise erhöhen auf maximal 400 µg/kg/min.
-
1.9.26 Biventrikuläre Elektrostimulation (Resynchronisation )
Bei intraventrikulären Leitungsstörungen, speziell beim Linksschenkelblock erfolgt die Kontraktion der Ventrikel nicht mehr völlig synchron. Das trägt im Falle einer Herzinsuffizienz deutlich zur Verminderung der Herzleistung bei. Die Resynchronisation gelingt durch biventrikuläre Elektrostimulation. Dazu werden drei Schrittmacherelektroden gelegt, eine in den rechten Vorhof, eine in den rechten Ventrikel und die dritte in den Koronarsinus, die von dort den linken Ventrikel stimuliert (◘ Abb. 1.65). Erzielt werden eine Steigerung der Ejektionsfraktion und eine Verbesserung um eine oder zwei NYHA-Klassen. Häufig bildet sich auch eine relative Mitralinsuffizienz um ein bis zwei Schweregrade zurück.


Röntgenthorax eines 45-jährigen Patienten mit schwerer dilatativer Kardiomyopathie links vor und rechts 6 Monate nach kardialer Resynchronisationstherapie (CRT) mittels eines biventrikulären Schrittmachersystems (1 Pfeil = Vorhofelektrode, 2 Pfeile = Ventrikelelektrode, 3 Pfeile = Koronarsinuselektrode)
1.9.27 Assistierte Zirkulation und Kunstherz
-
Intraaortale Ballongegenpulsation (IABP): Die schon 1953 eingeführte Technik ist leicht anzuwenden und hat wenig Komplikationen.
-
Indikationen: Kardiogener Schock nach Herzinfarkt, Infarktkomplikationen (Septumperforation, akute Mitralinsuffizienz durch Papillarmuskelabriss), low-output-Syndrom nach Herzoperationen, Das Herzminutenvolumen kann um 10–20 % gesteigert werden. Anwendungsdauer 24–48 h.
-
Kontraindikationen: Aorteninsuffizienz und Aortenaneurysma.
-
-
Ventrikuläre Assistenzsysteme: Es handelt sich um Blutpumpen, die das Herz durch komplette Aufrechterhaltung des großen, kleinen oder gesamten Kreislaufs entlasten. Sie können nur am freigelegten Herzen mit Hilfe der Herz-Lungen-Maschine installiert werden.
-
Linksventikuläre Assistenzsysteme (Linksherzbypass): Das Blut wird nach Kanülierung des linken Vorhofes oder der Spitzenregion des linken Ventrikels in die Aorta ascendens gepumpt.
-
Rechtsventrikuläre Assistenzsysteme (Rechtsherzbypass): Das Blut wird nach Kanülierung des rechten Vorhofes in den Hauptstamm der Pulmonalarterie gepumpt.
-
Biventrikuläre Assistenzsysteme: Kombination der beiden Assistenzsysteme.
-
-
Primär erfolgte der Einsatz der ventrikulären Assistenzsysteme beim kardialen Postkardiotomieschock, wenn die Patienten nicht von der Herz-Lungen-Maschine abgehängt werden konnten. Zur passageren Assistenz, für mehrere Stunden bis zu wenigen Tagen werden kontinuierliche Zentrifugalpumpen und pulsatile pneumatische Membranpumpen benutzt.
Seit sich die Herztransplantation als definitive Therapie der Herzinsuffizienz etabliert hat, werden die ventrikulären Assistenzsysteme bei bedrohten Patienten der Warteliste als Bridge-to-Transplantation installiert. Langfristig, über mehrere Monate, einsetzbare Systeme sind die extrakorporale pneumatische Thoratec-Pumpe (für linksventrikuläre, rechtsventrikuläre und biventrikuläre Assistenz) und voll implantierbare elektrische linksventrikuläre Assistenzsysteme. Bei letzteren erfolgt die Energiezufuhr transkutan mittels einer Magnetspule oder über transkutane Kabel, die mit einer tragbaren Steuer- und Batterieeinheit verbunden sind.
-
Kunstherz: Die Implantation eines künstlichen Herzens als Langzeitherzersatz erfolgte erstmals 1983. Damit wurden bei 5 Patienten Überlebenszeiten bis zu 622 Tagen erzielt. Inzwischen gibt es verschiedene pneumatische und elektrische Kunstherzen, die anstelle des Herzens an die großen Gefäße angeschlossen werden. Man wendet sie aber nur noch als Bridge-to-Transplantation an. Die Ergebnisse scheinen dabei weniger gut zu sein als mit den implantierbaren linksventrikulären Assistenzsystemen.
1.9.28 Linksventrikuläre Aneurysmaresektion
Große linksventrikuläre Aneurysmen sind gewöhnlich anterolateral lokalisiert. Sie entstehen nach Herzinfarkt und gehen in 50 % der Fälle mit einer Herzinsuffizienz einher, da sich die vernarbte Aneurysmawand nicht an der Kontraktion beteiligt. Weitere Komplikationen: Angina pectoris, relative Mitralinsuffizienz, tachykarde Rhythmusstörungen, Wandthromben. In diesen Fällen ist die Resektion des Aneurysmas indiziert (Dor-Plastik), meistens in Verbindung mit einer kompletten Revaskularisation. Die Rekonstruktion des linken Ventrikels führt in 80 % der Fälle zu einer deutlichen Besserung der Symptome und der kardialen Funktion. Die Überlebensrate nach 5 Jahren liegt bei 84 %.
1.9.29 Herztransplantation
Dank verbesserter Verfahren der Immunsuppression hat sich die Herztranplantation zu einer etablierten Therapie der terminalen Herzinsuffizienz entwickelt. Anders als bei der Nierentransplantation genügt es, wenn Spender und Empfänger in den ABO-Blutgruppen übereinstimmen. In Deutschland werden jährlich über 500 Herztransplantationen durchgeführt. Die Überlebensrate beträgt nach einem Jahr 80–85 %, nach 5 Jahren etwa 70 %, nach 10 Jahren etwa 50 %. Leider bedingt der Mangel an Spenderherzen Wartezeiten von 12–18 Monaten, die viele Patienten nicht überleben.
-
Indikationen: Irreversible terminale Herzerkrankungen, die weder medikamentös noch durch konventionelle operative Maßnahmen gebessert werden können. Etwa 50 % entfallen auf die dilatative Kardiomyopathie, 40 % auf koronare Herzerkrankungen im Endstadium, 10 % auf kongenitale und erworbene Vitien.
-
Kontraindikationen: Erhöhter Lungengefäßwiderstand (würde eine Herz-Lungen-Transplantation erfordern), akute und chronische Infektionen, Malignome oder Systemerkrankungen mit schlechter Prognose, ausgedehnte Lungenparenchymerkrankungen, Leberinsuffizienz, akute peptische Ulzera, bestehende Drogen- oder Alkoholabhängigkeit.
-
Chirurgische Technik: Exzision des Empfängerherzens auf Vorhofebene, Durchtrennung der Aorta ascendens und der A. pulmonalis. Anastomosierung des Spenderherzens, zuerst am linken, danach am rechten Vorhof. Zuletzt Verbindung von A. pulmonalis und Aorta ascendens. Variante: Bikavale Anastomose, also Exzision des rechten Vorhofes und des linken Vorhofes bis auf das Einmündungsgebiet der Lungenvenen.
-
Immunsuppression: Standard ist die Kombination von Cyclosporin A, Azathioprin und Prednisolon. Gegen akute Abstoßungsreaktionen werden auch Antikörper gegen T-Lymphozyten eingesetzt. Neuere Immunsuppressiva sind Tacrolimus (für Cyclosporin A) und Mofetil (für Azathioprin).
-
Komplikationen nach Herztransplantation:
-
Abstoßungsreaktionen: Am häufigsten in den ersten 4 Wochen, später zunehmend seltener. Sicherster Nachweis durch endomykardiale Biopsie über die rechte V. jugularis interna aus dem rechten Ventrikel. Behandlung mit Glukokortikoidstoß.
-
Infektionen: Am häufigsten durch Zytomegalievirus und Pneumocystis carinii.
-
Transplantationsvaskulopathie: Arteriosklerose der Kranzgefäße auf dem Boden einer immunologischen Intimaschädigung. Inzidenz 5‒10 % pro Jahr. Nach 5 Jahren weisen 40–50 % der Patienten angiographische Veränderungen auf. Statine wirken hier besonders günstig.
-
1.10 Rhythmusstörungen des Herzens
1.10.1 Normale Reizbildung und Erregungsleitung
1.10.1.1 Spezifisches Muskelsystem des Herzens
Das spezifische Muskelsystem dient der Reizbildung und Erregungsleitung (Reizleitungssystem) und gliedert sich in die folgend beschriebenen Bereiche (◘ Abb. 1.66).


Spezifisches Muskelsystem des Herzens: a Reizleitungssystem mit akzessorischen Strukturen, b His-Bündel- und Oberflächen-EKG
1.10.1.1.1 Sinusknoten (Sinuatrialknoten)
Physiologischer Schrittmacher des Herzens, der im Winkel zwischen V. cava superior und dem Dach des rechten Vorhofes lokalisiert ist (◘ Abb. 1.66).
1.10.1.1.2 Atriale Leitungsbahnen
Spezifische, die Erregung beschleunigt leitende Fasern zwischen Sinusknoten und AV-Knoten sowie zwischen beiden Vorhöfen (◘ Abb. 1.66).
1.10.1.1.3 AV-Knoten
Lokalisiert am Boden des rechten Vorhofes neben dem Vorhofseptum. Enthält Verbindungsfasern zum Vorhof und eigentliche Knotenfasern und Übergangsfasern zum His-Bündel, die das Trigonum dextrum durchsetzen. An allen übrigen Stellen sind die Vorhöfe durch das Herzskelett gegen die Kammern elektrisch isoliert (◘ Abb. 1.66).
1.10.1.1.4 His-Purkinje-System
Es besteht aus dicken, schnell leitenden Purkinje-Fasern und erstreckt sich vom His-Bündel über die beiden Tawara-Schenkel zum subendokardialen Netz der Purkinje-Fasern und von dort mit seinen Endaufzweigungen bis in das innere Wanddrittel der Kammern. Der rechte Tawara-Schenkel ist einsträngig, der linke teilt sich in einen breiten hinteren und schmalen vorderen Ast. Die Fasern des spezifischen Muskelsystems stehen mit denen des Arbeitsmyokards der Vorhöfe und Kammern in synzytialer Verbindung (◘ Abb. 1.66).
1.10.1.2 Schrittmacherfunktion (Automatie )
1.10.1.2.1 Sinusknoten
Zur Impulsbildung ist der Sinusknoten durch spontane Depolarisation befähigt. Das elektrische Ruhepotenzial der Sinusfasern, die keine kontraktilen Fibrillen enthalten, ist mit –55 mVolt relativ niedrig. Wie die transmembrane Potenzialmessung in der ◘ Abb. 1.67 zeigt, bleibt es in der Diastole nicht konstant, sondern nimmt kontinuierlich ab. Wenn das Schwellenpotenzial (–40 mVolt) erreicht ist, kommt es zum nächsten Aktionspotenzial. Neuerdings konnte gezeigt werden, dass der diastolische Depolarisationsstrom bis durch einen besonderen Kationenkanal der Zellmembran erfolgt, den sog. If-Kanal (f für „funny“). Durch ihn fließem hauptsächlich Na+-Ionen in das Zellinnere. Das nach Erreichen des Schwellenpotenzials beginnde Aktionspotenzials wird anders als im Arbeitsmyokard durch den schnellen Einstrom von Ca++-Ionen ausgelöst, für den sich spannungsabhängige Ca++-Kanäle öffnen. Die Repolarisation erfolgt durch den Auswärtsstrom von K+-Ionen (▶ Abschn. 1.8.6).


Schrittmacherfunktion des Sinusknotens: spontane diastolische Depolarisation und Aktionspotenzial
Somit ist die Entladungsfrequenz des Sinusknotens und damit die Herzfrequenz i von der Geschwindigkeit des diastolischen Depolarisationsstromes durch die If-Kanäle abhängig. Diese Kanäle werden durch Sympathikus und Vagus in der Weise moduliert, dass die Herzfrequenz vom Sympathikus beschleunigt und vom Vagus verlangsamt wird. Für den If-Kanal steht jetzt mit der Substanz Ivabradin ein spezifischer Inhibitor zur Verfügung, mit dem die Herzfrequenz zu therapeutischen Zwecken gesenkt werden kann.
1.10.1.2.2 Nachgeordnete Schrittmacher
Auch der proximale Abschnitt des His-Bündels (sekundäres Reizbildungszentrum) und die His-Purkinje-Fasern (tertiäres Reizbildungszentrum) sind zur Automatie (Selbsterregung) befähigt. Ihre Impulsfrequenz beträgt aber nur 40–60 bzw. 15–40/min, da sie ein höheres Ruhepotenzial haben und in der Diastole langsamer spontan depolarisieren. Sie werden normalerweise vom Aktionspotenzial des Sinusknotens entladen, bevor sie einen Impuls bilden können. Erst wenn der Sinusknoten ausfällt oder blockiert wird, übernehmen nachgeordnete Schrittmacher die Steuerung der Herzfrequenz. Dem Arbeitsmyokard fehlt normalerweise die Automatieeigenschaft. Es wird durch das fortgeleitete Aktionspotenzial depolarisiert.
1.10.1.3 Ruhe- und Aktionspotenzial des Arbeitsmyokards
Das Ruhepotenzial einer Zelle des Arbeitsmyokard liegt mit ‒90 mV nahe am K+-Gleichgewichtspotenzial.
Der Ablauf des Aktionspotenzials ist in ◘ Abb. 1.68 dargestellt:


Aktionspotenzial der Arbeitsmyokardfaser
-
der schnellen Depolarisation (Phase 1) folgt
-
die langsame (Phase 2) und die schnelle (Phase 3) Repolarisation
-
das Ruhepotenzial (Phase 4) entspricht dem diastolischen Potenzial und bleibt im Gegensatz zu den Schrittmacherzellen konstant. Es kann zu Anfang eine kurze negative Nachschwankung (Hyperpolarisation) aufweisen, die im EKG einer U-Welle entspricht.
1.10.1.4 Erregungsleitung
Nach der Depolarisation in den Sinusknotenfasern fließt ein positiver Ionenstrom (Ca++) durch die Ionenkanäle (Nexus) der Glanzstreifen, die an der Kontaktzone zwischen den Herzmuskelfasern liegen. Er hebt das Ruhepotenzial in den angrenzenden Fasern zum Schwellenpotenzial an und bringt damit auch diese zur Depolarisation. Auf diese Weise breitet sich die Erregung über das ganze Herz aus.
Die vom Sinusknoten ausgehende Erregung erreicht den AV-Knoten über schnell leitende spezifische Bahnen, bevor die Erregungsausbreitung durch das langsam leitende Arbeitsmyokard der Vorhöfe beendet ist. Im AV-Knoten wird die Erregungsleitung stark verzögert, damit die Kammern nicht vor Ablauf der Vorhofkontraktion erregt werden. Nach Austritt aus dem AV-Knoten wird die Erregung mit hoher Geschwindigkeit (2,5 m/s) über das His-Bündel und die Tawara-Schenkel in beide Kammern fortgeleitet.
1.10.1.5 Erregbarkeit
Erregbarkeit ist die Fähigkeit der Herzmuskelfasern, auf einen Impuls (der Schrittmacherzellen des Herzens oder exogener Reizquellen wie elektrische Schrittmacher) mit einem Aktionspotenzial zu antworten.
1.10.1.5.1 Refraktärphasen
Während das Aktionspotenzial abläuft, ändert sich die Ansprechbarkeit der betroffenen Herzmuskelfaser gegenüber neuen Reizimpulsen. Zu unterscheiden ist zwischen:
-
Absoluter Refraktärperiode (Plateauphase): Völlige Unerregbarkeit der Muskelfasern vom Beginn der Repolarisation bis zum Ende der Plateauphase (Phase 2 und 3 in ◘ Abb. 1.68). Die Dauer der absoluten Refraktärperiode nimmt mit der Aktionspotenzialdauer zu.
-
Relativer Refraktärperiode (Repolarisation): Erregbarkeit durch Reize erhöhter Intensität, die nach der Plateauphase der Repolarisation fortlaufend zunimmt (Phase 3 in ◘ Abb. 1.68). Die Aktionspotenziale unvollständig repolarsierter Zellen haben jedoch eine verminderte Anstiegsgeschwindigkeit und werden verlangsamt fortgeleitet.
Die effektive Refraktärperiode bezeichnet den Zeitraum bis zur ersten fortgeleiteten Erregung.
1.10.2 Elektrophysiologische Mechanismen der Arrhythmien
1.10.2.1 Potenzialverlust der ruhenden Fasern
Die meisten Rhythmusstörungen resultieren aus einer regionalen Verminderung des Ruhepotenzials im Leitungssystem und Arbeitsmyokard des Herzens, die vielerlei Ursachen haben kann, z. B.:
-
erhöhte K+-Konzentration außerhalb oder verminderte K+-Konzentration innerhalb der Zellen
-
Steigerung der Membranpermeabilität für Na+ und
-
Verminderung der Permeabilität für K+.
Eine akute Ischämie beispielsweise führt durch Hemmung der Na+/K+-Pumpe (ATP-Mangel) zum Anstieg der äußeren und Absinken der inneren K+-Konzentration, ferner zum Anstieg der inneren Ca2+-Konzentration, durch den das maximale diastolische Membranpotenzial ebenfalls weniger negativ wird.
1.10.2.2 Heterotope Automatie
Nach Herabsetzung des Ruhepotenzials (auf etwa –60 mV) durch unvollständige Repolarisation oder partielle Depolarisation können Myokardfasern der Vorhöfe und Kammern die Fähigkeit zur Automatie erlangen, die ihnen beim normalen Ruhepotenzial (–90 mV) fehlt.
Purkinje-Fasern, auf –50 mV gebracht, depolarisieren schneller und durch einen anderen ionalen Mechanismus als bei ihrem normalen Ruhepotenzial von –90 mV. Die spontane Entladungsfrequenz von Purkinje- und Myokardfasern mit herabgesetztem Ruhepotenzial kann in Gegenwart von Katecholaminen 200/min erreichen. Die Aktionspotenziale ähneln denen des normalen Sinus- und AV-Knotens (Depolarisation durch langsame Kanäle).
Die Automatiefrequenz wird durch Katecholamine und Hyperkalzämie gesteigert. Durch Verapamil wird sie stärker gehemmt als durch Inhibitoren der schnellen Kanäle.
Ein durch Depolarisation induzierte heterotope Automatie wurde bei Ischämie und Infarkt nachgewiesen.
1.10.2.3 Getriggerte Aktivität
Impulsbildung durch Nachpotenziale, die einem Aktionspotenzial vor oder nach Beendigung der Repolarisation folgen und das Schwellenpotenzial für eine neue Erregung erreichen.
Getriggerte Aktionspotenziale können einzeln, in Salven oder als Dauerrhythmus auftreten, je nachdem, in welchem Maße sie durch eigene Nachpotenziale triggernd wirken. Die ionalen Mechanismen sind nicht genau bekannt und wahrscheinlich uneinheitlich.
1.10.2.4 Erregungsleitungsstörungen
Ein herabgesetztes Ruhepotenzial vermindert die Anstiegsgeschwindigkeit des Aktionspotenzials und damit seine Fortleitung. Es kann zur progredienten Leitungsverzögerung (decremental conduction) und schließlich zum lokalen Block kommen. Natürlich wird die Erregungsleitung auch durch Gewebeläsionen und Narben blockiert.
1.10.2.5 Reentry
1.10.2.5.1 Definition
Wiedererregung des Herzens durch Eintritt eines an umschriebener Stelle verzögert fortgeleiteten Impulses in ein nicht mehr refraktäres Areal.
Der Reentry-Mechanismus ist eine häufige Ursache von Rhythmusstörungen. Zu unterscheiden sind zwei Formen.
1.10.2.5.2 Kreisende Erregung
Der ankommende Impuls trifft auf ein in antegrader Richtung unerregbares Gewebe (unidirektionaler Block), umgeht es mit langsamer Geschwindigkeit, erregt es auf retrogradem Weg und gelangt dann zum Ausgangspunkt der Kreisbahn zurück (◘ Abb. 1.69).


Kreisende Erregung: unidirektionaler Block (links) und Reentry (rechts)
Wenn das Gewebe proximal der blockierten Stelle inzwischen wieder erregbar geworden ist, breitet sich der Impuls von hier aus erneut über das Herz aus. Eine andere Variante ist ein Impuls, der sich kreisförmig um ein unerregtes Zentrum bewegt und in einem Abschnitt der Kreisbahn stark verzögert fortgeleitet wird. Wenn er diesen Abschnitt verlässt, trifft er auf Gewebe, dessen Refraktärperiode inzwischen beendet ist. Ein solcher kreisförmiger Reentry-Mechanismus liegt dem Vorhofflattern zugrunde. Die Kreisbahn kann auch von Fasern gebildet werden, die aneinander grenzen (Längsdissoziation der Erregungsausbreitung).
Das Kreisen des Impulses wird unterbrochen, wenn er bei seiner Rückkehr auf refraktäres Gewebe trifft. Um das zu erreichen, kann man die Erregungsleitung auf der Kreisbahn beschleunigen oder die langsam leitende Strecke ganz blockieren.
1.10.2.5.3 Reflektion
Der Impuls durchläuft eine Leitungsfaser, die zwischen ihrem proximalen und distalen Segment eine unerregbare Zone aufweist. Diese Zone leitet den Depolarisationsstrom nur passiv, d. h. ohne zusätzliches Aktionspotenzial, und deshalb stark verlangsamt fort. Wenn seine Intensität ausreicht, löst der Depolarisationsstrom im diastalen Segment wieder ein Aktionspotenzial aus, das mit normaler Geschwindigkeit weitergeleitet wird. Gleichzeitig läuft es durch die unerregbare Zone langsam in das proximale Segment zurück und bewirkt hier eine erneute Erregung, falls die Refraktärzeit inzwischen abgelaufen ist.
1.10.3 Diagnostik von Herzrhythmusstörungen
Eine Zusammenfassung der Untersuchungen zur Diagnose von Rhythmusstörungen des Herzen enthält ◘ Tab. 1.7.
Bei der Anamnese stehen folgende Fragen im Vordergrund:
-
Art der Rhythmusstörung: langsam oder schnell, regelmäßig oder unregelmäßig
-
Dauer der Störung
-
zusätzliche Symptome (z. B. Schwäche, Synkope, Schwindel, Dyspnoe).
Von Bedeutung sind auch kardiale Grunderkrankungen des Patienten und die Einnahme von Medikamenten.
Zur körperlichen Untersuchung gehört ein kompletter internistischer Status mit besonderem Augenmerk auf Zeichen von Herzinsuffizienz, arteriellem Hypertonus sowie auf Erkrankungen der Lunge und Schilddrüse.
1.10.4 Therapeutische Maßnahmen bei Herzrhythmusstörungen
Therapie der Herzrhythmusstörungen
-
Medikamentöse Therapie
-
Antiarrhythmika Klasse I A–C
-
Antiarrhythmika Klasse II
-
Antiarrhythmika Klasse III
-
Antiarrhythmika Klasse IV
-
-
Apparative Therapie
-
transthorakale Elektrokonversion
-
Herzschrittmacher
-
implantierbarer Kardioverter-Defibrillator (ICD)
-
automatische externe Defibrillatoren (AED)
-
1.10.4.1 Antiarrhythmika
Die Klassifizierung der Antiarrhytmika nach Vaughan Williams (◘ Tab. 1.8) berücksichtigt nicht alle Wirkungen und lässt einige Überschneidungen außer Betracht. Auch variiert die Wirkung der Antiarrhythmika mit dem Gewebetyp, dem Grad akuter und chronischer Gewebeschäden, der Herzfrequenz, dem Membranpotenzial und weiteren Faktoren. Sie wird ferner durch Effekte auf Vagus oder Sympathikus bestimmt. In Deutschland sind Chinidin duriles® und Disopyramid nicht mehr im Handel.
1.10.4.1.1 Anwendungsbeispiele
1.10.4.1.1.1 Lidocain (Xylocain®)
Ein Lokalanästhetikum, das sich bei der akuten intravenösen Therapie ventrikulärer Arrhythmien bewährt hat. Gegen Vorhofarrhythmien ist es unwirksam. Lidocain blockiert offene und inaktivierte Na+-Kanäle und setzt die Automatie herab. Es verstärkt aber Herzblock und Herzinsuffizienz und wird deshalb nach Herzinfarkt nicht routinemäßig angewendet. Dosierung: Infusion von 3–4 mg/kg in 2 Minuren, danach 3-mal je 50 mg über 8 Minuten. Erhaltungsdosis 1–4 mg/min.
1.10.4.1.1.2 Sotalol
Ein nicht selektiver β-Blocker, der durch die Hemmung von K+-Kanälen auch das Aktionspotenzial verlängert. Die AV-Überleitung wird verzögert, die Automatie gebremst. Anwendung bei ventrikulären Taxhyarrhythmien und Vorhofflimmern. Überdosierung kann torsades de pointes induzieren. Als Infusion 20 mg während 5 Minuten, danach 1 mg/min über 20 Minuten. In Tablettenform 2–3×80 mg/Tag.
1.10.4.1.1.3 Calciumantagonisten (Verapamil, Diltiazem)
Hauptwirkung ist die Verzögerung der AV-Überleitung, durch die beide Mittel alle AV-Knotentachykardien beenden. Bei Vorhofflimmern wird die Kammerfrequenz herabgesetzt. Der Sinusrhythmus wird kaum beeinflusst, unter gleichzeitiger Einnahme von β-Blockern aber bedrohlich absinken. Die Kombination mit β-Blockern ist daher zu vermeiden. Dosierung: 5 mg Verapamil langsam i. v. während 2 Minuten; nach 5–10 Minuten wiederholen. Bei Bedarf Infusion mit 5–10 mg/h.
1.10.4.1.1.4 Flecainid (Tambocor®)
Bewirkt eine länger anhaltende Blockade des Natriumkanals. Indiziert bei supraventrikulären und bedrohlichen Kammertachykardien. Kontraindiziert nach Herzinfarkt. Dosierung: Infusion mit 2 mg/kg i. v. alle 12 h. Erhaltungstherapie: 2×50 mg/p.o./Tag; maximale Tagesdosis 300 mg. Sorgfältige Überwachung der Patienten!
1.10.4.1.1.5 Propafenon (Rytmonorm®)
Verlängert PR- und QRS-Dauer. Anwendung zur chronischen Tachykardien incl. Vorhofflimmern. Dosierung: 2×150–300 mg/Tag p.o.
1.10.4.1.1.6 β-Rezeptorenblocker (Metoprolol, Bisoprolol, Propranolol)
Durch Hemmung des Calciumeinstroms bremsen sie den Sinusthythmus bei Thyreotoxikose und Phäochromozytom. Bei letzterem müssen sie mit α-Blockern kombiniert werden. Vorhofflimmern und -flattern lassen sich nicht beheben, doch wird die Kammerfrequenz durch Blockade der AV-Überleitung herabgesetzt. Das kurzwirksame Elmolol (Breviblock®) verlangsamt supraventrikuläre Tachykardien schnell (500 µg/kg i. v. über 2–3 Minuten). Kammertachykardien bei verlängerter QT-Dauer sprechen an, nicht aber nach Infarkt.
1.10.4.1.1.7 Amiodaron (Cordarex®)
Anwendung notfallmäßig bei ventrikulären Tachykardien und ventrikulären Tachyarrhythmien. Die lipophile Substanz geht in viele Gewebe und wird sehr langsam eliminiert. Sie hemmt inaktivierte Na+-Kanäle mit langer Erholungszeit, vermindert den Calciumstrom und K+-Kanäle. Amiodaron ist ein potenter Inhibitor der Automatie und verlängert das Aktionspotenzial. Die Summation der Effekte ergibt eine starke antiarrhythmische Wirksamkeit. Vorhofflimmern lässt sich beseitigen und langfristig unterdrücken. Auch Kammertachykardien werden beherrscht. Dosierung: 15 mg/min für 10 Minuten, 1 mg/min für 3 h, danach 0,5 mg/min; p.o. 3×200 mg/Tag für 5 Tage, danach 1×200 mg/Tag (TSH-Kontrollen). Gravierend sind bei längerer Anwendung die Nebenwirkungen: Thyreotoxikose wegen des Jodgehalts, therapierefraktäre Lungenfibrose, periphere Neuropathie mit Muskelschwäche.
1.10.4.1.1.8 Dromedaron (Multaq®)
Ein nicht-jodiertes Derivat des Amiodaron. Zugelassen zur Erhaltung des Sinustrhythmus nach Vorhofflimmern. Wirksamkeit, aber auch Nebenwirkungen schwächer als bei Amiodaron. Kontraindiziert bei fortgeschrittener Herzinsuffizienz.
1.10.4.1.1.9 Vernakalant (Brinavess®)
Es verlängert selektiv die artriale Refraktärperiode und wirkt so antiarrhythmisch. Indiziert zur Konversion des kurze Zeit bestehenden Vorhofflimmerns (<7 Tage). Infusion über 10 Minuten mit 3 mg/kg. Zweiter Versuch nach 15 Minuten. Unwirksam bei länger bestehendem Flimmern.
1.10.4.1.1.10 Verapamil (Isoptin®)
Anwendung bei supraventrikulären Tachykardien (nicht mit β-Blockern kombinieren!) und zur Frequenzregulierung bei Vorhofflimmern. Dosierung: akut 5–10 mg i. v. über 1–2 min. Erhaltungsdosis: 3×40–120 mg p.o.
1.10.4.1.1.11 Adenosin
Endogenes Nukleosid, das K+-Kanäle aktiviert. Mittel der ersten Wahl zur Beendigung von AV-Knotentachykardien. Dosierung: 8–18 mg schnell i. v.
1.10.4.1.2 Allgemeine Nebenwirkungen
1.10.4.1.2.1 Verlangsamung der Sinusknotenfrequenz
Bei intaktem Sinusknoten sind im Allgemeinen die Nebenwirkungen gering und tolerabel. Am stärksten wirken β-Rezeptorenblocker, die vor allem den Frequenzanstieg unter Belastung hemmen.
Bei vorgeschädigtem Sinusknoten können alle Antiarrhythmika erhebliche Bradykardien hervorrufen.
1.10.4.1.2.2 Erregungsleitungsstörungen
Es können ein sinuaurikulärer sowie AV-Block 1. bis 3. Grades, eine QRS-Verbreiterung sowie Schenkelblöcke auftreten. Erregungsleitungsstörungen drohen vor allem bei vorgeschädigtem Reizleitungssystem. Am wenigsten bei den Substanzen der Klasse III.
1.10.4.1.2.3 Verlängerung der Erregungsdauer (QT-Zeit)
Normaler therapeutischer Effekt bei Substanzen der Klasse III, ansonsten bei Überdosierungen und Hypokaliämie.
Die Verlängerung der QT-Zeit ist bei Überdosierungen und bei Hypokaliämie ein Hinweis auf drohende Kammertachykardien (Torsades de pointes).
1.10.4.1.2.4 Arrhythmogene Wirkung
Bei keinem Antiarrhythmikum auszuschließen. Wahrscheinlich durch Reentry, wenn z. B. durch Hemmung der Erregungsleitung ein unidirektionaler Block induziert wird, ohne dass gleichzeitig die effektive Refraktärzeit zunimmt oder durch die Umwandlung eines bidirektionalen in einen unidirektionalen Block. Möglicherweise auch durch Nachpotenziale.
1.10.4.1.2.5 Negativ inotrope Wirkung
Durch Hemmung des langsamen Calciumeinstroms und der intrazellulären Calciumkinetik. Ausgeprägt durch β-Rezeptorenblocker und Disopyramid, die zur Herzinsuffizienz führen können. Bei den übrigen Substanzen der Klasse I und den Calciumantagonisten durch Überdosierung möglich und im Allgemeinen nur bei vorbestehender Herzinsuffizienz relevant. Keine Herabsetzung der Kontraktilität durch Amiodaron.
1.10.4.2 Transthorakale Elektrokonversion
Die elektrische Kardioversion beseitigt mit hoher Erfolgsquote tachykarde Rhythmusstörungen, die auf einem Reentry-Mechanismus beruhen:
-
Vorhofflattern und viele Fälle von Vorhofflimmern
-
AV-Reentry-Tachykardien
-
Tachykardien bei WPW-Syndrom
-
die meisten Kammertachykardien
-
Kammerflattern und Kammerflimmern
Unwirksam ist die Defibrillation bei Tachykardien, die von einem Automatiezentrum ausgehen, weil dieses sofort wieder aktiv wird. Dazu gehören Parasystolie, einige Fälle von Vorhofflimmern, manche Vorhoftachykardien, AV-junktionale Tachykardien und seltene Fälle von Kammertachykardie.
1.10.4.2.1 Technik und Wirkungsmechanismus
Ein Gleichstromimpuls von hoher elektrischer Energie (50–200 Joule) wird durch den Brustkorb in das Herz geleitet. Er depolarisiert fast alle nichtrefraktären Herzmuskelzellen und schaltet damit die Reentrykreise ab. Neue Defibrillatoren geben nicht mehr einen monophasischen, sondern einen biphasischen Gleichstromimpuls ab. Dabei wird die Polarität des Schocks nach Abfall der Ausgangsspannung um 65 % umgeschaltet. Die biphasische Defibrillation ist deutlich effektiver. Sie benötigt weniger Energie und ist auch bei adipösen Patienten effektiv. Die Applikation erfolgt in Kurznarkose über zwei großflächige Elektroden, von denen eine im rechten 2. Interkostalraum aufgesetzt wird, die andere im Bereich der Herzspitze (beide mit Elektrodengel). Gelingt die Kardioversion damit nicht, kann eine Elektrode ventral und die andere dorsal aufgesetzt werden, bevor man mit einem geeigneten Katheter intrakardial kardiovertiert. Mit Ausnahme sehr schneller Tachykardien wird der Elektroschock synchron zum QRS-Komplex abgegeben, damit er nicht in die vulnerable Phase der Ventrikel fällt.
Zur biphasischen Defibrillation werden für die Vorhöfe 25–50 J, für die Kammern 100–200 J benötigt. Bei monophasischem Schock betragen die entsprechenden Werte <100 J und 200–360 J.
1.10.4.2.2 Antikoagulation
Wenn die Tachyarrhythmie länger als 48 h bestanden hat, ist vor der Kardioversion wegen Thromboemboliegefahr eine Antikoagulation mit Phenprocoumon (Marcumar) erforderlich. Oft gelingt der Nachweis eines Vorhofthrombus in transösophagealen Echo (◘ Abb. 1.70). Die Patienten sollen mindestens 4 Wochen auf INR-Werten von 2–3 gewesen sein. Nach der Konversion muss die Antikoagulation 3–4 Wochen fortgesetzt werden, weil die mechanische Systole vorübergehend ausbleiben kann, während die elektrische Systole schon wieder vorhanden ist. Eine dringende Kardioversion kann unter Heparinschutz vorgenommen werden, wenn das transösophageale Echokardiogramm im linken Vorhof keinen Thrombus erkennen lässt.


Transösophageale Echokardiographie einer 56-jährigen Patientin mit Vorhofflimmern und Nachweis eines Thrombus im linken Vorhofohr (LAA)
1.10.4.3 Herzschrittmacher
1.10.4.3.1 Technik und Funktion
Herzschrittmacher sind implantierbare Impulsgeber, die den Herzmuskel durch elektrische Stimulation depolarisieren und zur Kontraktion bringen. Sie bestehen aus Energiequelle, Schaltkreis und Elektroden. Als Energiequelle dienen Lithium-Jod-Batterien mit 5- bis 15-jähriger Betriebsdauer. Die Mikrochip-Schaltkreise moderner Schrittmacher gestatten die Programmierung komplexer Schrittmacherfunktionen mittels Radiowellen auf transkutanem Wege. Die dazu verwendeten Programmiergeräte können das eingespeicherte Programm für Kontrollzwecke vom Schrittmacher abfragen. Die Elektroden bestehen aus isolierten Drahtsonden mit einem Kopf, der Verankerungshilfen (Anker, Spiralen, Schrauben) trägt. Im Regelfall verwendet man transvenös (V. cephalica, V. subclavia) eingeführte Elektrodensonden und verlegt den Impulsgeber in die Pektoralisregion unter die Muskelfaszie (◘ Abb. 1.71). Die typische transvenöse Schrittmacherimplantation ist ein kleiner, in Lokalanästhesie durchführbarer Eingriff, der auch sehr alten Patienten zugemutet werden kann. Zur Überbrückung von Notfallsituationen oder passageren Blockierungen (frischer Infarkt, postoperative Komplikationen) führt man eine temporäre Elektrostimulation mit transvenösen Sonden (V. jugularis, V. subclavia) und externem Impulsgeber durch.


Röntgenthorax (p. a. und lateral) einer 72-jährigen Patientin mit DDD-Schrittmacher (1 Pfeil: Vorhofelektrode, 2 Pfeile: Ventrikelelektrode) mit Sick-Sinus-Syndrom und Synkope
Herzschrittmacher haben 2 Basisfunktionen:
-
Herzstimulation (Pacing)
-
Wahrnehmung spontaner elektrischer Impulse des Herzens (detection sensing), die ihnen die Information für das richtige Timing der Impulsabgabe liefern.
Durch das Sensing wird erreicht, dass der Schrittmacher das Herz nur bei Bedarf (demand) stimuliert (paced), d. h. wenn die Herzfrequenz unter die Basisfrequenz des Schrittmachers sinkt. Spontane QRS-Komplexe können die Schrittmacheraktion entweder durch Hemmung der Impulsabgabe unterdrücken (Inhibition) oder indem sie einen Schrittmacherimpuls triggern, der noch in die absolute Refraktärphase der Kammern fällt (Trigger-Mechanismus). Die meisten Schrittmacher sind frequenzadaptiert (rate response), d. h., sie können ihre Frequenz einer körperlichen Belastung anpassen. Dazu dienen Sensoren die u. a. Muskelaktivität (Vibrationen), Atemfrequenz, Q-T-Intervall und Sauerstoffsättigung registrieren (◘ Tab. 1.9).
Aus der Möglichkeit, Schrittmachersonden mit Sensing- und Pacing-Funktion im Vorhof und im Ventrikel zu platzieren, resultiert eine Reihe von Schrittmachersystemen, zu deren Kennzeichnung ein internationaler Fünf-Buchstaben-Code eingeführt wurde, meist werden nur 3‒4 Buchstaben verwendet. Die Buchstaben haben folgende Bedeutung:
-
1. Buchstabe: stimulierte Kammer (A = Atrium, V = Ventrikel, D = A und V)
-
2. Buchstabe: Ort des Sensing (A, V, D oder 0)
-
3. Buchstabe: Betriebsart zur Unterdrückung spontaner Impulse (I = Inhibition, T = triggern, D = beide Mechanismen)
-
4. Buchstabe: Programmierbarkeit (R = frequenzadaptiert, M = multiprogrammierbar, 0 = keine)
-
5. Buchstabe: Tachyarrhythmiefunktion: (B = Aktivität, N = normale Frequenkonkurrenz, S = Scanning, E = extern).
1.10.4.3.1.1 Indikationen
Herzschrittmacher werden eingesetzt bei:
-
Dysfunktion des Sinusknotens
-
atrioventrikulärem Block
-
hypersensitivem Karotissinus
1.10.4.3.1.2 Komplikationen
-
Postoperative Komplikationen: Drucknekrosen mit Hautperforation, Infektionen (Schrittmachertasche, Elektrodensonden oder beide), Thrombose und Embolie (Oberarm- und Schultervenen, V. cava superior, rechter Vorhof, Insertionsstelle in der rechten Kammer, Pneumothorax, Perikarderguss, Perikardtamponade).
-
Hämodynamische Konsequenzen: Minderung der Herzleistung bei Wegfall der Vorhof-Kammer-Koordination, die bei sequentieller Vorhof-Kammerstimulation vermieden wird, Vorhofpfropfungswellen bei retrograder Überleitung der Kammerimpulse auf die Vorhöfe;
-
Elektrodenkomplikationen: Sondenbruch, Sondendislokation, Sondenperforation. Exit-Block (ineffektiver Schrittmacherimpuls außerhalb der Refraktärzeit vorausgegangener Systolen) und Entrance-Block (Wegfall des Sensing) sind die Folgen.
-
Störungen des Schrittmachersystems: vorzeitige Batterieerschöpfung, elektromagnetische Interferenzen (Diathermie, Elektrokauter, Magnetresonanztomographie).
1.10.4.4 Implantierbarer Kardioverter-Defibrillator (ICD)
1.10.4.4.1 Technik
Der Elektroschock (bis 40 J) zur Defibrillierung (bei Kammerflimmern) oder Konversion (bei Kammertachykardien) wird vom subpektoral implantierten Impulsgeber (mit Lithiumjodid-Batterie) über einen Katheter im rechten Ventrikel appliziert (◘ Abb. 1.72). Dabei dient das Defibrillatorgehäuse als Gegenpol. Der rechtsventrikuläre Katheter besorgt auch das Sensing. Durch einen Vorhofkatheter kann das System zusätzlich als vorhofgesteuerter Herzschrittmacher arbeiten. Ausgelöste Schocks werden vom Gerät gespeichert.


Röntgenthorax p. a. eines 49-jährigen Patienten 6 h nach Implantation eines Defibrillators mit linksseitigem Pneumothorax
1.10.4.4.2 Indikationen
Überstandenes Kammerflimmern und lebensbedrohliche ventrikuläre Tachykardien, die medikamentös nicht beherrscht werden können. Die Implantation ist dringend erforderlich, wenn bei der elektrophysiologischen Untersuchung, Kammertachykardien und Kammerflimmern induziert werden können.
1.10.4.5 Automatischer externer Defibrillator (AED)
Der AED kann bei plötzlich auftretendem Kammerflimmern vom Ersthelfer im Notfall eingesetzt werden bevor der Notarzt eintrifft. Sie sind schon an vielen öffentlichen Orten zu finden. Diese Geräte können automatisch defibrillationswürdige Rhythmen erkennen und die Defibrillation empfehlen. Die Überlebenschancen bei Herzstillstand durch Kammerflimmern steigen, je früher eine Defibrillation durchgeführt wird.
1.10.5 Einteilung von Herzrhythmusstörungen
Herzrhythmusstörungen werden nach Mechanismus und Ort der Störung eingeteilt (◘ Tab. 1.10).
1.10.6 Supraventrikuläre Reizbildungsstörungen
Supraventrikuläre Reizbildungsstörungen
-
Sinustachykardie
-
Sinusbradykardie
-
Sinusarrhythmie
-
supraventrikuläre Extrasystolen
-
Vorhoftachykardien
-
multifokale Vorhoftachykardie
-
nichtparoxysmale AV-junktionale Tachykardie
-
AV-Knoten-Reentrytachykardie
-
Vorhofflattern
-
Vorhofflimmern
1.10.6.1 Sinustachykardie
1.10.6.1.1 Definition
Herzfrequenz 100–200/min bei normalem Erregungsursprung im Sinusknoten.
1.10.6.1.2 Ätiologie
Steigerung des Sympathikustonus bei körperlicher Anstrengung, seelischer Erregung, Psychosen, Fieber, Hyperthyreose, Anämie, kompensatorisch bei Herzinsuffizienz. Analog wirken Sympathikomimetika und Anticholinergika. Nicht selten ist die unangemessene Tachykardie (inappropriate tachycardy) bei organisch gesundem Herzen, die im Deutschen als hyperkinetisches Herzsyndrom bezeichnet wird. Bei meist noch normalem Ruhepuls führt jede körpeliche Anstrengung zu einer inadäquaten erheblichen Tachykardie, die als störend empfunden wird und wegen der abnorm gesteigerten Herzarbeit relativ schnell zur körperlichen Erschöpfung führt. Die Patienten leiden dadurch auch psychisch. Die Ursache ist ungeklärt. Psychotherapeutische Therapie hilft nicht. Erfolge verspricht die Blockade der If-Kanäle am Sinusknoten (▶ Abschn. 1.10.1).
1.10.6.1.3 EKG
Zeichen der Sympathikotonie: Steiltypische P-Zacken, tiefer Abgang der ST-Strecke mit ansteigendem Verlauf, Abflachung der T-Zacke. T- und P-Zacke können sich überlagern.
1.10.6.1.4 Therapie
Elimination der kausalen Faktoren. Optimale Dämpfung der Tachykardie mit Ivabradin, dem spezifischen Blocker des If-Kanals im Sinusknoten. Tranquilizer bei Erregungstachykardien, die mit Hyperventilation verbunden sein können.
1.10.6.2 Sinusbradykardie
1.10.6.2.1 Definition
Herzfrequenz unter 60/min bei normalem Erregungsursprung im Sinusknoten.
1.10.6.2.2 Ätiologie
-
Vagotonie: Chronisch bei trainierten Sportlern. Paroxysmal bei Karotisdruck, hypersensitivem Karotissinus, vasovagalen Synkopen (▶ Abschn. 1.3.4) und Hirndrucksteigerung.
-
Kranker Sinusknoten: Altersveränderungen, organische Herzkrankheiten, Infektionen, Kollagenosen, Hypothyreose, Verschlussikterus, Traumen.
-
Pharmaka: β-Rezeptorenblocker, Digitalis (cholinergischer Effekt), Antiarrhythmika (meistens cholinergischer Effekt), Antihypertensiva (Reserpin, Clonidin, α-Methyldopa).
1.10.6.2.3 Klinik
Bei jungen Erwachsenen kann die Herzfrequenz im Schlaf auf 35–40/min sinken. Extreme Bradykardien kommen bei Langstreckenläufern vor (Schlaf: 31/min, Ruhelage: 37/min). Im Alter kann die Ruhefrequenz unter 50/min betragen, ohne dass Kreislaufstörungen auftreten. Symptome verursachen inadäquate persistierende Bradykardien durch Absinken des Herzminutenvolumens (Schwächeanfälle, Verwirrtheit, Schwindelgefühl, Gleichgewichtsstörungen, Synkopen) oder durch Stauung (Atemnot, Ödeme).
Organisch bedingte Sinusbradykardien gehen oft mit paroxysmalen atrialen Tachyarrhythmien (Vorhofflimmern, Vorhofflattern, Vorhoftachykardie) einher, die zu anginösen Beschwerden und Herzklopfen führen können.
Infolge der verlängerten Sinusknotenerholungszeit treten beim Umschlagen der Tachyarrhythmien in den Sinusrhythmus bei manchen Patienten Synkopen auf. Man spricht vom Bradykardie-Tachykardie-Syndrom. Offenbar ist in diesen Fällen auch die Impulsbildung der Ersatzschrittmacherzellen in den Vorhöfen und im AV-Knoten gestört. Schlägt der bradykarde Sinusrhythmus in ein bradykardes Vorhofflimmern um, muss eine zusätzliche AV-Leitungsstörung angenommen werden. Da es sich jeweils um Vorhofstrukturen handelt, ist es verständlich, dass Sinusknoten, Vorhofmyokard und AV-Knoten bei Vorhoferkrankungen gleichzeitig betroffen sein können bzw. nacheinander in Mitleidenschaft gezogen werden.
1.10.6.2.4 Diagnostik
1.10.6.2.5 Atropin-Test
Frequenzanstieg nach 1 mg Atropin i. v. normalerweise auf über 50 % des Ausgangswertes im Liegen. Bei gestörter Generatorfunktion des Sinusknotens Anstieg unter 25 % des Ausgangswertes bzw. auf weniger als 90/min.
1.10.6.2.6 EKG
Bei erhöhtem Vagustonus flache P-Zacken, relativ lange PQ-Zeit (die sich unter Belastung verkürzt) und hohe T-Zacken in V2–V6. Klinisch relevante Bradykardien erfordern Aufzeichnung im 24-h-Langzeit-EKG. Bei Sinusknotenaffektionen und Sympathikusblockade kein adäquater Frequenzanstieg im Belastungs-EKG. Die Sinusknotenerholungszeit nach schneller Schrittmacherstimulation des rechten Vorhofs (Zeitintervall zwischen der letzten stimulierten und der ersten spontanen Vorhofaktion) ist bei Dysfunktion des Sinusknotens häufig verlängert (>1600 ms). SA(sinuatriale)-Blockierungen ▶ Abschn. 1.10.9.
1.10.6.2.7 Therapie
In passageren Fällen Atropin oder Sympathikomimetika (Orciprenalin sublingual). Alle potentiell frequenzsenkenden Pharmaka absetzen. In symptomatischen Fällen Schrittmacherimplantation.
1.10.6.3 Sinusarrhythmie
1.10.6.3.1 Definition
Phasische Variationen der Länge des Sinuszyklus, bei der die Differenz zwischen maximaler und minimaler Zykluslänge 120 ms übersteigt.
1.10.6.3.2 Ätiologie
Respiratorische Arrhythmie durch reflektorische Hemmung des Vagustonus während der Inspiration; ein physiologisches Phänomen, bei jüngeren Individuen ausgeprägter. Nichtrespiratorische Form bei Sinusknotenschädigung und Digitalisintoxikation.
1.10.6.3.3 Klinik
Meistens symptomlos. Bei langen Sinuspausen Schwindelgefühl, Palpitationen, selten Synkopen. Körperliche Belastung steigert die Frequenz.
1.10.6.3.4 EKG
Bei der respiratorischen Form zyklische Verkürzung der P-P-Intervalle während der Inspiration und Verlängerung während der Exspiration, Konstanz beim Atemanhalten. Bei der nichtrespiratorischen Form keine Relation der Frequenzschwankungen zur Atmung.
1.10.6.3.5 Therapie
Die Therapie entspricht dem Vorgehen bei Bradykardie.
1.10.6.4 Supraventrikuläre Extrasystolen
1.10.6.4.1 Definition
Vorzeitige Herzschläge, die den Sinusrhythmus unterbrechen. Der Erregungsursprung liegt oberhalb der Teilungsstelle des His-Bündels. Nach dem Ursprungsort können Sinusextrasystolen, Vorhofextrasystolen und AV-Extrasystolen unterschieden werden. Letztere gehen nicht von Knotenfasern aus, sondern vom knotennahen Abschnitt des His-Bündels.
1.10.6.4.2 Pathogenese
Der gewöhnlich konstante zeitliche Abstand vom vorhergehenden Normalschlag (feste Kuppelung) zeigt, dass die Extrasystole von der Normalerregung induziert wird. Meistens liegt ein Reentry-Mechanismus vor, der durch die inhomogene Refraktärzeit bedingt ist.
1.10.6.4.3 Ätiologie
Kommt bei Patienten mit organisch gesunden Herzen (seltener als Kammerextrasystolen) vor sowie bei Myokarditis, koronarer Herzkrankheit, Infektionen (auch fokal), Hyperthyreose. Aber auch eine Hypokaliämie, Genussmittel (Tabak, Alkohol, Kaffee) und Pharmaka (Digitalis, Narkotika, Antiarrhythmika, Sympathikomimetika) können supraventrikuläre Extrasystolen auslösen. In jedem Fall ist nach Kausalfaktoren zu suchen.
1.10.6.4.4 Klinik
Hämodynamisch sind einzelne Extrasystolen bedeutungslos. Herzklopfen resultiert aus der größeren Kontraktionskraft des postextrasystolischen Herzschlages. Als Herzstolpern wird der vorzeitige Einfall der Extrasystolen, als Aussetzen des Herzens die postextrasystolische Pause empfunden.
Supraventrikuläre Extrasystolen können Vorboten eines Vorhofflimmerns oder -flatterns sein.
1.10.6.4.5 EKG
-
Extrasystolische P-Zacke: Bei einer Sinusarrhythmie mit der des Sinusrhythmus identisch. Bei Vorhofextrasystolen deformiert, bisweilen in der T-Zacke der vorausgegangenen Normalerregung verborgen (◘ Abb. 1.73). PQ-Zeit verkürzt oder verlängert. Bei AV-Extrasystolen (◘ Abb. 1.74) retrograde Erregung der Vorhöfe (P in Abl. II und III negativ); P kann vor, im oder hinter dem QRS-Komplex liegen. Keine P-Zacke bei retrograder Blockierung.
Abb. 1.73 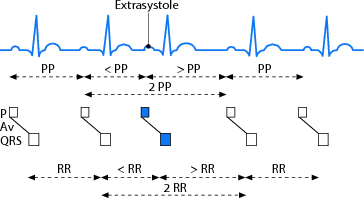
Extrasystole des Vorhofes
Abb. 1.74 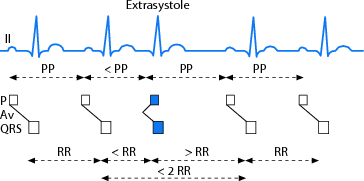
Extrasystole des AV-Knotens
-
QRS: Nicht verbreitert, bei früh einfallenden Extrasystolen manchmal verbreitert und deformiert (wegen unvollständiger Repolarisation im Leitungssystem der Kammern: aberrierende Leitung).
-
Postextrasystolische Pause: Länger als ein normaler R-R-Abstand, meistens nicht voll kompensierend, d. h. das Intervall von der vorausgehenden zur nachfolgenden R-Zacke ist kürzer als 2 normale RR-Intervalle.
-
Periodizität: Extrasystolen (ES) können sich in fixierten Abständen wiederholen: Bigeminie (nach jedem Normalschlag 1 ES), Trigeminie (nach jedem Normalschlag 2 ES), 2:1-, 3:1-Extrasystolie (nach jedem zweiten bzw. dritten Normalschlag eine ES).




1.10.6.4.6 Therapie
Eine medikamentöse antiarrhythmische Therapie ist nur bei relevanten Symptomen notwendig.
1.10.6.5 Vorhoftachykardien
1.10.6.5.1 Definition
Tachykardien mit Erregungsursprung im Vorhof.
1.10.6.5.2 Klassifikation
Nach dem Entstehungsmechanismus sind folgende 3 Typen zu unterscheiden:
-
Intraatriale Reentrytachykardie: Die Lokalisation des Reentry-Kreises ist nicht genau untersucht. Sie kann durch programmierte elektrische Stimulation ausgelöst und bei der elektrophysiologischen Untersuchung verifiziert werden. Hat paroxymalen Charakter und folgt stets auf eine Vorhofextrasystole. Patienten mit dieser Form haben meistens organische Herzkrankheiten, sind symptomatisch, aber hämodynamisch kompensiert. Sehr häufig besteht eine 2:1-Blockierung mit Kammerfrequenzen von 90–120/min.
-
Vorhoftachykardie durch ektopische Automatie: Lokalisation des ektopischen automatischen Fokus an der Crista terminalis des rechten Vorhofes oder an der Basis der Pulmonalvenen im linken Vorhof. Die Automatie kann durch Vorhofstimulation weder ausgelöst noch beendet werden. Diese häufigste Form der Vorhoftachykardie kommt bei gesunden jungen Erwachsenen und Kindern vor. Gewöhnlich tritt sie nach körperlichen Belastungen auf, steigert die Frequenz und bleibt lang dauernd bestehen. In chronischen Fällen kann es zu einer sekundären Kardiomyopathie kommen.
-
Getriggerte Vorhoftachykardie: Verursacht durch Nachpotenziale. Kann durch hochfrequente Vorhofstimulation induziert und elektrophysiologisch von der Reentrytachykardie abgegrenzt werden. Hat stets paroxysmalen Charakter. Seltenste Form mit einem Durchschnittsalter der Patienten von 60 Jahren. Kommt bei Gesunden und Herzkranken vor, typischerweise bei Digitalisintoxikation.
1.10.6.5.3 Klinik
Gesunde haben oft keine oder nur geringe Beschwerden. Sehr hohe Frequenzen können zu Schwindelgefühl und Synkopen führen. Bei Herzkranken sind anginöse Beschwerden und Dyspnoe möglich. In jedem Fall wird der Kreislauf nachteilig beeinflusst.
1.10.6.5.4 EKG
Vorhoffrequenz 140–240/min. Bei hohen Frequenzen AV-Überleitung im Verhältnis 1:2 oder höher. P-Zacken ähnlich wie in Phasen mit Sinusrhythmus. P-P-Abstände konstant (bei ektopischer Automatie leicht variierend). Im Gegensatz zum Vorhofflattern liegt zwischen den P-Zacken eine isoelektrische Linie. QRS ist nicht verbreitert, ohne aberrierende Leitung (◘ Abb. 1.75).


Tachykarde Herzrhythmusstörungen. a supraventrikuläre paroxysmale Tachykardie, b Vorhofflattern, Vorhofflimmern, c Vorhofflimmern, d ventrikuläre Tachykardie, e Kammerflattern, f Kammerflimmern
1.10.6.5.5 Therapie
Karotisdruck und Adenosin sind im Gegensatz zu Knotentachykardien unwirksam die Therapiemöglichkeiten bei Vorhoftachykardien sind in ◘ Tab. 1.11 aufgeführt.
1.10.6.6 Multifokale Vorhoftachykardie
1.10.6.6.1 Definition
Seltene supraventrikuläre Tachykardie aus mindestens 3 atrialen Foki. Sie feuern unabhängig von einander, so dass eine Arrhythmie resultiert.
1.10.6.6.2 Klinik
Kommt fast nur bei schweren Lungenkrankheiten mit Hypoxie vor.
1.10.6.6.3 EKG
Mindestens 3 unterschiedliche Formen von P-Zacken und/oder PR-Intervallen. Vorhoffrequenz durchschnittlich 100/min.
1.10.6.6.4 Therapie
Im Vordergrund steht die Behandlung der Grundkrankheit. Außerdem vorsichtige Gabe von β-Blockern oder Calciumantagonisten möglich.
Bei einer multifokalen Vorhoftachykardie keine Kardioversion einsetzen.
1.10.6.7 Nichtparoxysmale AV-junktionale Tachykardie
1.10.6.7.1 Definition
Supraventrikuläre Tachykardie durch gesteigerte junktionale Automatie (ektopischer Fokus im oberen His-Bündel).
1.10.6.7.2 Ätiologie
Posteroinferiorer Herzinfarkt, Myokarditis, Herzoperationen, am häufigsten Digitalisintoxikation. Selten bei sonst gesunden Individuen.
1.10.6.7.3 Klinik
An diese Form ist zu denken, wenn die Kammerfrequenz bei digitalisbehandeltem Vorhofflimmern plötzlich regelmäßig wird, während das Vorhofflimmern bestehen bleibt.
1.10.6.7.4 EKG
Konstante Kammerfrequenz zwischen 70 und 130/min mit schmalem QRS-Komplex. Steigerung des Vagustonus verlangsamt die Frequenz, Herabsetzung des Vagustonus erhöht sie. Die retrograde Vorhoferregung ist meistens blockiert. Die Vorhöfe werden vom Sinusknoten, einem atrialen oder einem zweiten junctionalen Fokus mit unabhängiger Frequenz kontrolliert (AV-Dissoziation).
1.10.6.7.5 Therapie
Bei Digitalis-Intoxikation Glykosid absetzen, zusätzlich Lidocain. Eine Kardioversion ist meist erfolglos. Katheterablation ist möglich. Spontane Rückbildung unter Behandlung des Grundleidens.
1.10.6.8 AV-Knoten-Reentrytachykardie
1.10.6.8.1 Definition
Paroxysmale Tachykardie durch Reentry-Erregung innerhalb des AV-Knotens. Häufigste Form der paroxysmalen supraventrikulären Tachykardien (PSVT) mit einem Anteil von 50 % (Vorhofflimmern und -flattern nicht mitgerechnet).
1.10.6.8.2 Ätiologie
Nicht durch organische Herzkrankheiten verursacht, oft in früher Jugend beginnend. Späteres Zusammentreffen mit organischen Herzkrankheiten wahrscheinlich zufällig.
1.10.6.8.3 Pathogenese und EKG
Der AV-Knoten hat in diesen Fällen einen langsamen Leitungsweg (l) mit kurzer und einen schnellen Leitungsweg (s) mit langer Refraktärzeit (◘ Abb. 1.76a). Beim Sinusrhythmus und bei langsamer Vorhofstimulation leitet der schnelle Weg mit normaler PQ-Zeit. Eine frühe Vorhofextrasystole trifft den schnellen Weg refraktär an, wird antegrad auf dem langsamen Weg durch den AV-Knoten zum His-Bündel geleitet (◘ Abb. 1.76a). Gleichzeitig wird retrograd der schnelle Weg erregt. Die auf dem schnellen Weg zurückkehrende Erregung durchläuft und aktiviert retrograd den Vorhof und geht innerhalb des Knotens auf die wieder erregbar gewordene langsame Bahn über, womit sich ein Reentry-Kreis schließt (◘ Abb. 1.76b). Neben dieser gewöhnlichen gibt es eine ungewöhnliche Variante mit umgekehrter Kreisbewegung: Die antegrade Erregung geht über den schnellen, die retrograde über den langsamen intranodalen Leitungsweg. Bei der gewöhnlichen Form liegt die stets negative P-Zacke wegen der schnellen retrograden Leitung im oder dicht hinter dem QRS-Komplex, bei der ungewöhnlichen Form wegen langsamer retrograder Leitung dicht vor dem nächsten QRS-Komplex. Die Kammerfrequenz beträgt 150–250/min. Der QRS-Komplex ist schmal, das P/QRS-Verhältnis 1:1. Spontaner, durch Karotisdruck oder Pharmaka induzierter Block in retrograder Richtung beendet die Tachykardie.


AV-Knoten-Reentrytachykardie
1.10.6.8.4 Klinik
Häufigste Form der paroxysmalen supraventrikulären Tachykardie. Das Manifestationsalter liegt zwischen dem 30. und 40. Lebensjahr, der Anteil des weiblichen Geschlechts beträgt 70 %. Im Anfall Herzklopfen, Erregung, Angst und Unruhe sowie Schwindelgefühl bis zu Synkopen infolge Abnahme des Herzminutenvolumens und der zerebralen Durchblutung. Viele Anfälle klingen im Liegen evtl. unter Sedierung bald ab.
Abgrenzung der AV-Knoten-Reentrytachykardie gegen atriale Tachykardien durch Karotisdruck- oder Valsalva-Manöver.
1.10.6.8.5 Therapie
Der Anfall endet nicht selten spontan (◘ Abb. 1.77). Zur Anfallbeendigung wird zuerst die einseitige Karotismassage (5–10 s) eingesetzt, die einen Vagusimpuls mit Freisetzung von Acetylcholin auslöst. Bei Fehlschlag Adenosin (6 mg als Bolus i. v.) oder Verapamil (5–10 mg über 2 min), nötigenfalls elektrische Konversion. Anfallsprophylaxe mit Verapamil, Diltiazem oder β-Blocker (nicht mit Calciumantagonisten kombinieren). Kurativ mit hoher Erfolgsquote ist die RF-Katheterablation, möglichst der langsam leitenden Bahn.


Spontan terminierte AV-Knoten-Reentrytachykardie
1.10.6.8.6 Prognose
Bei organisch Herzgesunden ist die Prognose günstig.
1.10.6.9 Vorhofflattern
1.10.6.9.1 Definition
Atriale Reentrytachykardie mit regelmäßiger Frequenz, gleichförmiger Morphologie und einer Frequenz über 240/min. Die Kammerfrequenz ist meist durch einen fixierten 2:1 AV-Block gebremst.
1.10.6.9.2 Klassifikation
Das Vorhofflattern wird in 2 Formen eingeteilt:
-
Typ I: Häufigste Variante ist die isthmusabhängige Variante mit einem Makro-Reentry-Kreis im rechten Vorhof. Er verläuft dorsal der Trikuspidalklappe unter Einbeziehung von Septum, vorderem Vorhofdach, Seitenwand und einem Muskelstrang, dem sog. Isthmus, der vor der Einmündung der V. cava inferior beginnt und zwischen dem Ostium des Sinus-coronarius- und dem Trikuspidalklappenring zum Septum zieht. Die langsame Erregungsleitung im Isthmus ermöglicht den Wiedereintritt der Erregung nach einer Kreisbahn. Diese kann im Uhrzeigersinn oder im Gegenuhrzeigersinn durchlaufen werden, wobei letztere Richtung stark überwiegt. Die Flatterfrequenz beträgt 250–320/min.
Die nichtisthmusabhängige Variante hat einen fixierten Reentry-Kreis, der nicht den Isthmus einbezieht. Er muss durch elektrophysiologische Untersuchung lokalisiert werden. Die Flatterfrequenz liegt zwischen 250–320/min. Charakteristisch für beide Varianten des Typs I ist, dass sich das Flattern durch schnelle Vorhofstimulation unterbrechen und durch RF-Katheterablation beseitigen lässt.
-
Typ II: Atypisches Vorhofflattern, das vom Typ I durch folgende Kriterien zu unterscheiden ist:
-
Flatterfrequenz 350–450/min
-
keine Unterbrechung durch Vorhofstimulation
-
schwie RF-Katheterablation möglich
-
der Reentrykreis ist nicht fixiert, die P-Zacke variabel
Der Entstehungsmechanismus entspricht anscheinend dem des groben Vorhofflimmerns.
-
1.10.6.9.3 Ätiologie
Rheumatische Vitien, hauptsächlich Mitralstenose, Vorhofseptumdefekt, Perikarderkrankungen, chronische obstruktive Lungenkrankheiten, offene Herzoperationen, Hyperthyreose. Selten kommt paroxysmales Vorhofflattern auch bei klinisch Gesunden vor.
1.10.6.9.4 Klinik
Neben der paroxysmalen gibt es eine chronische Form, die bei normalen Kammerfrequenzen gut toleriert wird. Beschwerden und hämodynamische Auswirkungen hängen von der Pulsfrequenz ab. Wechselseitiger Übergang in ein Vorhofflimmern ist häufig. Da sich eine Seite der Vorhöfe kontrahiert, während die andere relaxiert ist, wird von den Vorhöfen wenig Blut gefördert, was die gesamte Herzleistung beeinträchtigt.
1.10.6.9.5 EKG
Sägezahnfömige P-Zacken ohne horizontale Zwischenstrecken (◘ Abb. 1.75b). Bei der gewöhnlichen Form mit Erregungskreisen im Gegenuhrsinn sind die P-Zacken biphasisch, beim seltenen Kreisen im Uhrzeigersinn aufrecht. AV-Blockierungen 2:1 seltener 4:1. Beim atypischen Flattern ungleichmäßige P-Zacken mit sehr hoher Frequenz (◘ Abb. 1.78).


Typisches Vorhofflattern mit negativen sägezahnartigen P-Zacken in den Ableitungen II, III, aVF und positiver P-Zacke in V1
1.10.6.9.6 Therapie
Zur Beendigung der Attacke zuerst Digoxin i. v. plus β-Blocker oder Verapamil zwecks Reduzierung der Kammerfrequenz. Danach ist bei typischem Vorhofflattern die kurative RF-Katheterablation indiziert.
Beim Versagen der Medikamente oder in bedrohlicher Kreislaufsituation elektrische transthorakale Kardioversion (25–50 Wattsekunden) oder hochfrequente Vorhofstimulation per Katheter im Ösophagus oder rechten Vorhof.
Bei atypischen Vorhofflattern Behandlung wie bei Vorhofflimmern.
Eine Antikoagulation ist umstritten, bei intermittierendem Vorhofflimmern jedoch dringend indiziert.
1.10.6.10 Vorhofflimmern
1.10.6.10.1 Definition
Völlig ungeordnete, unkoordinierte Vorhoferregung ohne effektive Vorhofkontraktion, die mit unregelmäßiger meist beschleunigter Kammertätigkeit verbunden ist.
1.10.6.10.2 Klassifikation
Es werden folgende Formen des Vorhofflimmern unterschieden:
-
paroxysmales VF: spontane Terminierung innerhalb 7 Tagen
-
kurz persistierendes VF: <1 Jahr anhaltend
-
lang persistierendes VF: >1 Jahr anhaltend
Die Erfahrung zeigt, dass paroxysmales Vorhofflimmern im Laufe von Jahren unter Verkürzung der anfallsfreien Intervalle in langdauernd persistierendes übergeht.
1.10.6.10.3 Pathogenese
Beim Vorhoflimmern haben sich multiple kleine Reentrykreise ausgebildet, denen Inhomogenität der Leitungsgeschwindigkeit und Refraktärität im Vorhofmyokard zugrunde liegt. Durch eine Verkürzung der Aktionspotenzialdauer und damit der Refraktärzeit in Verbindung mit einer Reduktion der Leitungsgeschwindigkeit wird die Entstehung kleiner Reentrykreise begünstigt. Elektrophysiologisch werden die schnellen Na+-Ströme bei der Depolarisaion und die Ca++-Einwärtsstöme in der Plateauphase der Repolarisation herabgesetzt. Das paroxysmale Vorhofflimmern wird durch schnelle Triggerimpulse induziert, die zu 97 % von Foci in den Muskelbrücken ausgehen, die sich vom linken Vorhof ein kurzes Stück weit in die Pulmonalvenen erstrecken. Nach Beseitigung der fokalen Triggerung durch RF-Ablation hört das Vorhofflimmern auf. Permanentes Vorhofflimmern erhält sich nur teilweise durch Triggeraktivität aus den Pulmonalvenen. Hier tragen strukturelle Veränderungen (Fibrosierung mit verlängerten Leitingszeiten, Vorhofdilatation) und elektrisches Remodeling (Verkürzung von Aktionspotenzials und Refraktärzeit) zur Entstehung und Aufrechtserhaltung des Vorhofflimmerns bei.
1.10.6.10.4 Ätiologie
Mitralstenose und andere Vitien mit Überdehnung des linken Vorhofes, koronare Herzkrankheit, akuter Infarkt mit Linksinsuffizienz, Hypertonie, Kardiomyopathien, Vorhofmyxome, Hyperthyreose, Alkoholismus. Nach Bypass-Operationen tritt oft mehrtägiges Vorhofflimmern auf. Nicht selten kommt Vorhofflimmern ohne erkennbare Ursache und ohne Hinweis auf eine strukturelle Herzerkrankung vor.
1.10.6.10.5 Klinik
Unbehandelte Patienten haben meistens eine Tachyarrhythmia absoluta mit Pulsdefizit infolge schlechter Kammerfüllung. Die Kammerfrequenz kann in unbehandelten Fällen mit intaktem AV-Knoten 150–180/min erreichen. Die Flimmerfrequenz der Vorhöfe schwankt zwischen 350 und 600/min. Häufige Beschwerden während der Tachykardie sind Schwäche, Atemnot, Beklemmungsgefühl in der Brust und innere Unruhe. Durch die unökonomische Herztätigkeit können selbst kräftige Herzen nach einiger Zeit hämodynamisch dekompensieren. Auch Synkopen kommen vor. Die Normalisierung der Kammerfrequenz führt dann schnell zur Rekompensation. Bei insuffizienten Herzen droht auch im Falle normaler Kammerfrequenzen die Dekompensation, da mit dem Wegfall der Vorhofkontraktion die Inotropie schwächer wird. Durch Thrombenbildung in den stillstehenden Vorhöfen kann es zu Thromboembolien kommen, besonders bei Mitralstenosen und insuffizienten vergrößerten Herzen, auch bei Hyperthyreose. Bei idiopathischem Vorhofflimmern nimmt das Thromboembolierisiko jenseits des 70. Lebensjahres deutlich zu.
1.10.6.10.6 Diagnostik
Die Diagnose ergibt sich aus dem EKG (◘ Abb. 1.79): Keine P-Zacken, stattdessen unregelmäßige kleine Flimmerwellen etwa 400–700/min von ungleicher Form und Größe, oft nur in Abl. V1 zu erkennen (◘ Abb. 1.75c). Absolute Arrhythmie der Kammern mit schmalem QRS-Komplex. Kammerfrequenz 60–140/min, abhängig von der AV-Überleitung, die durch verborgene Leitung (concealed conduction) gehemmt ist.


Tachykardes Vorhofflimmern
Zur Klärung der Ursache ist eine vollständige kardiologische Untersuchung einschließlich Echokardiographie und Ischämiediagnostik. In jedem Fall ist die Schilddrüsenfunktion zu überprüfen.
1.10.6.10.7 Therapie
Nach Möglichkeit Beseitigung der Ursache durch Behandlung des Grundleidens (Mitralkommissurotomie, Thyreostatika etc.). Die Therapiemaßnahmen zum Vorhofflimmern sind in ◘ Tab. 1.12 übersichtlich zusammengestellt.
Vorhofflimmern durch ein Automatiezentrum kann bei 70–85 % der Patienten durch Ablation des Fokus an der Einmündung der Pulmonalvenen in den linken Vorhof geheilt werden.
1.10.7 Präexzitationssyndrom e
Präexzitationssyndrome
-
Wolff-Parkinson-White-Syndrom
-
Präexzitation durch Mahaim-Fasern
-
verkürztes AV-Intervall
1.10.7.1 Wolff-Parkinson-White-Syndrom
1.10.7.1.1 Definition
Das Wolff-Parkinson-White-(WPW-)Syndrom ist eine Reentrytachykardie, die aufgrund einer akzessorischen Muskelbrücke (Kent-Bündel) zwischen Vorhof und Kammer mit antegrader und retrograder Leitung entsteht (◘ Abb. 1.66a, ◘ Abb. 1.80b).


Präexzitation: Darstellung exzitatorischer Leitungsbahnen, des EKG und His-Bündel-Elektrogramm. a normaler Befund, b atrioventrikulärer Bypass (WPW-Syndrom), c nodoventrikuläres Bündel (Mahaim-Fasern), d AV-Knoten-Bypass (H = His-Bündel, K = Kent-Bündel, A = Atrium, V = Ventrikel, d = Delta-Welle) (nach J. Gallacker)
1.10.7.1.2 Ätiologie
Angeborener Defekt, der sich meist im frühen Erwachsenenalter manifestiert.
1.10.7.1.3 Varianten
Die akzessorische Bahn kann an verschiedenen Stellen der Vorhof-Kammer-Grenze lokalisiert sein. Ihre Auswirkungen hängen von ihrer Leitungsgeschwindigkeit ab, die von Fall zu Fall und auch beim einzelnen Patienten variiert:
-
Sinusrhythmus mit Früherregung der Kammern: Die Kammern werden antegrad über den AV-Knoten und das akzessorische Muskelbündel erregt. Je höher die Leitungsgeschwindigkeit des Bündels, desto größer der Bereich des von ihm erregten Kammermyokards.
EKG: Die vorzeitige Erregung eines Kammerabschnitts durch die akzessorische Leitung bewirkt eine Verkürzung des AV-Intervalls (<0,12 s) und eine Verbreiterung des QRS-Komplexes (>0,12 s) durch eine träge ansteigende R-Zacke bzw. träge abfallende Q-Zacke (◘ Abb. 1.81). Das flache Anfangsstück des QRS-Komplexes nennt man Delta-Welle. Sie repräsentiert den vorzeitig erregten Kammerbezirk. Der QRS-Komplex ist genau um den Betrag verbreitert, um den das AV-Intervall verkürzt ist. Aus der Polarität der Deltawellen in den 12 Ableitungen des EKG kann auf die Lokalisation des Bündels geschlossen werden. Die T-Zacke ist meist diskordant zur Hauptausschlagrichtung von QRS.
Abb. 1.81 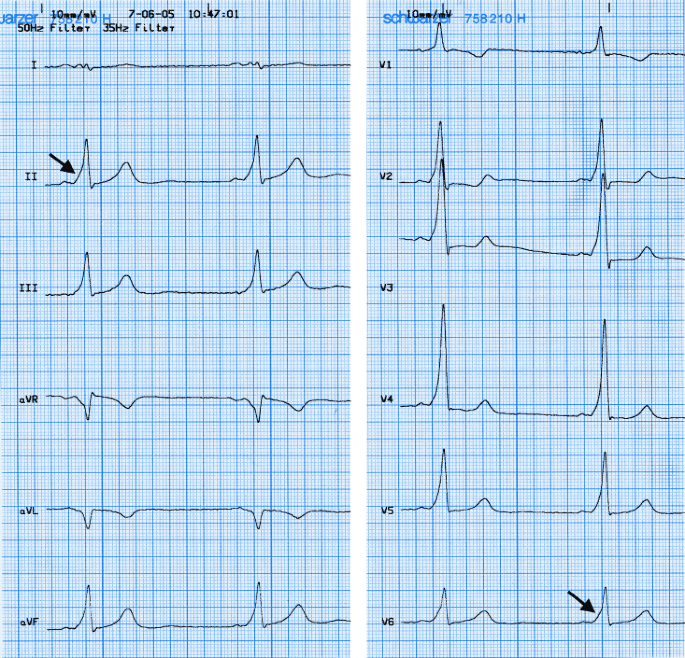
EKG einer 28-jährigen Patientin mit WPW-Syndrom und intermittierenden Tachykardien: positive Delta-Welle in II, III, aVF, V2–V6
-
Orthodrome AV-Reentrytachykardie: Eine supraventrikuläre oder ventrikuläre Extrasystole, die das akzessorische Bündel noch refraktär antrifft, durchläuft dann folgende Kreisbahn: AV-Knoten und His-Bündel → Tawara-Schenkel → Kammern → retrograd die wieder leitungsfähige akzessorische Bahn zum Vorhof zurück, der inzwischen erneut erregbar geworden ist. Anschließend kreist die Erregung im Gegenuhrzeigersinn weiter. Bei dieser orthodromen Tachykardie bleibt der QRS-Komplex schmal, da die Kammererregung auf normalem Weg erfolgt. Die Deltawelle verschwindet. Die negative P-Zacke liegt im frühen ST-Segment. Kammerfrequenz 150–250/min. Bei sehr langsamer retrograder Leitung des Kent-Bündels kann sich eine orthodrome Reentrytachykardie auch ohne Extrasystole entwickeln.
-
Antidrome AV-Reentrytachykardie: Bei dieser seltenen Variante kreist die Erregung im Uhrzeigersinn. Eine Extraystole trifft den AV-Knoten refraktär an, wird antegrad durch das Kent-Bündel zum Ventrikel geleitet und retrograd über den AV-Knoten zum Vorhof zurück.
EKG: QRS verbreitert mit Delta-Welle. Negatives P im ST-Segment.
-
AV-Reentrytachykardie bei verborgenem WPW-Syndrom: In diesen seltenen Fällen leitet das Kent-Bündel nur retrograd. Es kann zu orthodromen Reentrytachykardien kommen, die schwer zu diagnostizieren sind, weil das EKG im Sinusrhythmus keine Delta-Welle aufweist. Die negative P-Zacke liegt im Bereich des ST-Intervalls. Klärung erlaubt die elektrophysiologische Untersuchung.


1.10.7.1.4 Klinik
Viele Patienten mit Delta-Welle im EKG bleiben asymptomatisch. Bei sonst Gesunden werden auch die paroxysmalen Tachykardien relativ gut toleriert. Sehr hohe Frequenzen können zu Schwindelgefühl führen, bei Koronarkranken zu Angina pectoris.
Lebensbedrohlich ist hinzutretendes Vorhofflimmern. Da das akzessorische Bündel eine kurze Refraktärzeit hat, droht Kammerflimmern.
1.10.7.1.5 Therapie
Asymptomatische Patienten mit Früherregung werden nicht behandelt. Anfallbeendigung mit Karotisdruck, Adenosin oder Verapamil i. v. Mittel der Wahl bei symptomatischen Patienten ist die definitive Behandlung mit RF-Katheterablation des akzessorischen Bündels.
Bei Vorhofflimmern darf Verapamil nicht i. v. gegeben werden, weil es Kammerflimmern auslösen kann. Im Notfall elektrische Kardioversion.
1.10.7.2 Präexzitation durch Mahaim-Fasern
Nach der Lokalisation der Mahaim-Fasern (◘ Abb. 1.66) unterscheidet man 2 Formen.
1.10.7.2.1 Nodoventrikuläres Bündel
Verbindung zwischen AV-Knoten und rechtem Ventrikel (◘ Abb. 1.80c). Präexzitation mit verkürzter PQ-Zeit und Delta-Welle kann bei Sinusrhythmus fehlen und erst nach Vorhofstimulation wegen Verzögerung der AV-Überleitung sichtbar werden. Reentrytachykardie antegrad über das akzessorische Bündel, retrograd von den Kammern über die Tawara-Schenkel und das His-Bündel zum AV-Knoten zurück. QRS hat dabei die Form wie beim Linksschenkelblock.
1.10.7.2.1.1 Klinik
Symptome und Therapie der Reentrytachykardie bei nodoventrikulärer Verbindung entsprechen denen beim WPW-Syndrom. Vorhofflattern und -flimmern führen nicht zu bedrohlicher Kammertachykardie, weil die Impulse einen Teil des AV-Knotens passieren müssen.
1.10.7.2.1.2 Therapie
Entsprechend wie beim WPW-Syndrom.
1.10.7.2.2 Faszikuloventrikuläres Bündel
Bei normaler AV-Leitung ist die PQ-Zeit hier normal. Da das Bündel langsamer leitet als die Purkinje-Fasern ergibt sich keine Deltawelle, sondern nur eine leichte Deformierung der R-Zacke. Im His-Bündel-EKG ist die AH-Zeit normal, die HV-Zeit verkürzt. Spezifische Arrhythmien für dieses Bündel sind nicht bekannt.
1.10.7.3 Verkürztes AV-Intervall
1.10.7.3.1 Definition
Vorzeitige Kammererregung durch ein atriofaszikuläres Bündel.
1.10.7.3.2 Klinik
Häufiges Phänomen, wahrscheinlich ohne pathologische Bedeutung.
1.10.7.3.3 EKG
PQ 0,12 s oder kürzer, keine Deltawelle, QRS-Komplex normal (◘ Abb. 1.80d). In der Mehrzahl der Fälle nimmt die PQ-Zeit bei rascher Vorhofstimulation zu, nur bei etwa 10 % der Fälle bleibt sie verkürzt. Demnach scheint die verkürzte PQ-Zeit überwiegend auf einer beschleunigten AV-Knotenleitung und nicht auf einer Umgehung des AV-Knotens zu beruhen.
1.10.8 Ventrikuläre Reizbildungsstörungen
Ventrikuläre Reizbildungsstörungen
-
ventrikuläre Extrasystolen
-
Kammertachykardien
-
Kammerflattern und Kammerflimmern
1.10.8.1 Ventrikuläre Extrasystolen (VES)
1.10.8.1.1 Definition
Vorzeitige Extraschläge mit Erregungsursprung in den Ventrikeln (Purkinje-Fasern oder Myokard).
Einteilung der Extrasystolen
-
Monomorphe Extrasystolen: mehrere Extrasystolen mit identischer Konfiguration im EKG
-
Monotope Extrasystolen: Extrasystolen mit identischer Konfiguration und gleichem Ursprung
-
Couplet: 2 aufeinander folgende VES
-
Bigeminus: regelmäßige Abfolge eines Normalschlages und einer Extrasystole
-
Trigeminus: jeder VES folgen 2 Normalschläge
-
Triplet: 3 VES
-
3 oder mehr sukzessive VES: ventrikuläre Tachykardie
Gelegentliche VES kommen häufig bei Gesunden vor, mit dem Alter zunehmend.
1.10.8.1.2 Pathogenese
In Betracht kommen Reentry-Mechanismus, fokale ektopische Impulsbildung und triggernde Nachpotenziale.
1.10.8.1.3 Ätiologie
-
organisch: Myokarditis, Infektionen, koronare Herzkrankheit und Infarkt
-
toxisch: Digitalis, Anästhesie, Antiarrhythmika
-
metabolisch: Elektrolytstörungen (Hypo- und Hyperkaliämie), Hypoxie, Azidose, Urämie
-
mechanisch: Katheter, Traumen
1.10.8.1.4 Klinik
Symptome wie bei supraventrikulären Extrasystolen (▶ Abschn. 1.1.5). VES sollten immer Anlass zu einer gründlichen Herzuntersuchung und zur Fahndung nach versteckten Entzündungsherden (Tonsillen, Zähnen, Nebenhöhlen) sein. Die Behandlung des Grundleidens ist vorrangig. Auf Tabak, Alkohol und Kaffee sollte versuchsweise verzichtet werden. Eine Belastungsextrasystolie kann auf koronaren Durchblutungsstörungen oder Drucksteigerung in den Ventrikeln beruhen. Eine Ruheextrasystolie, die unter Belastung verschwindet ist meistens harmlos, muss es aber nicht sein.
Die Bedeutung ventrikulärer Extrasystolen hängt von der klinischen Situation ab und wird nicht mehr nach der Lown-Klassifikation bemessen.
In Abwesenheit einer organischen Herzerkrankung mindern VES die Lebenserwartung nicht und erfordern auch keine Einschränkung der körperlichen Aktivität. Dagegen bedeuten ventrikuläre Arrythmien bei Patienten mit koronarer Herzkrankheit eine Verdoppelung des Risikos, am Herzinfarkt und an allen anderen Mortalitätsursachen zu sterben. Dabei ist die Arrhythmie eher ein Marker für die Herzkrankheit als die unmittelbare Todesursache. Patienten mit VES, die bei elektrophysiologischer Untersuchung keine Kammertachykardie bekommen, haben ein geringes Risiko, einen akuten Herztod zu erleiden. Bei Patienten nach frischem Herzinfarkt mit 1–10 VES pro Stunde ist das Risiko eines Kammerflimmerns erhöht. Ein Kammerflimmern wird allerdings in der Hälfte der Fälle ohne vorausgehende ventrikuläre Rhythmustörungen (VES, R-auf-T-Phänomen, Bigeminus, multiforme QRS-Komplexe, Salven von 2, 3 und mehr Komplexen) beobachtet. Umgekehrt bleibt bei der Hälfte der Patienten mit solchen Rhythmusstörungen ein Kammerflimmern aus. Demnach ist die prognostische Bedeutung ventrikulärer Arrhythmien unsicher.
1.10.8.1.5 EKG
Vorzeitig einfallende, verbreiterte (QRS >0,11 s), schenkelblockartig deformierte Kammerkomplexe ohne Korrelation zur P-Zacke. Bei rechtsventrikulärem Ursprung Linksschenkelblockbild, bei linksventrikulärem Ursprung Rechtsschenkelblockbild. Die QRS-Verbreiterung und Schenkelblockbilder beruhen auf der langsamen myokardialen Erregungsleitung. Sie hat auch zur Folge, dass die Erregungsrückbildung an der zuerst erregten Stelle beginnt, woraus sich eine zur Hauptausschlagsrichtung von QRS diskordante T-Zacke ergibt (◘ Abb. 1.82).


Kammerextrasystole mit kompensatorischer Pause
Die postextrasystolische Pause ist meist kompensatorisch (R – R* + R*– R = 2 R – R), da der Sinusknoten nur selten durch eine retrograde (ventrikuloatriale) Leitung gelöscht wird. Andernfalls ist die postextrasystolische Pause nicht kompensierend. Die Sinuserregung trifft auf einen durch die VES refraktär gewordenen Ventrikel. Erst der nächste Sinusimpuls wird übergeleitet. Sehr frühe diastolische VES beginnen in Höhe der T-Zacke (R- auf T-Phänomen) und disponieren zu Kammertachykardien und Kammerflimmern. Interponierte VES fügen sich zwischen zwei R-Zacken ein, ohne deren Abstand wesentlich zu verändern. Monomorphe VES sind als monotop (unifokal) aufzufassen. Auch polymorphe VES können monotop sein, sie haben dann ein konstantes Kopplungsintervall und weichen infolge variabler Erregungsausbreitung in der Form voneinander ab. Polytope VES sind polymorph und zeigen ein variables Kopplungsintervall.
1.10.8.1.6 Therapie
Elimination der auslösenden Faktoren, falls erkennbar. Korrektur der Elektrolytwerte. Auslassversuch mit Digitalis. Bei Patienten mit unklarer rhythmologischer Situation (z. B. Synkopen fraglicher Ursache) ist eine elektrophysiologische Untersuchung angezeigt. Symptomatische Patienten sind vorübergehend antiarrhythmisch zu behandeln, zunächst mit β-Blockern, wenn nötig mit Amiodaron. Patienten mit einer Ejektionsfraktion <30 % nach Myokardinfarkt oder infolge DCM erfüllen nach großen Studien (MADIT I, MADIT II) die Kriterien für die Implantation eines automatischen Defibrillators (ICD).
1.10.8.2 Kammertachykardien
1.10.8.2.1 Definition
Drei oder mehr aufeinander folgende ektopische Kammerkomplexe, deren Frequenz 100/min überschreitet.
Zu unterscheiden sind:
-
ventrikuläre Dauertachykardien (>30 s: sustained type), die hämodynamische Symptome (Hypotonie, Synkopen) verursachen und dringend therapiebedürftig sind
-
intermittierende ventrikuläre Tachykardien (<30 s: non-sustained type): verlaufen oft asymptomatisch.
1.10.8.2.2 Mechanismen
Überwiegend Reentry, seltener Triggerung durch Nachpotenziale und fokale Automatie.
1.10.8.2.3 EKG
Frequenz bis 250/min, meist 130–170/min und ganz regelmäßig. AV-Dissoziation (Kammern schneller als Vorhöfe, am besten in V1 sichtbar) für VT beweisend. QRS Verbreiterung (>120 ms, meist >140 ms), mit Achsenabweichung gegenüber dem QRS im Sinusrhythmus nach links oder rechts.
-
Konkordanz der QRS-Komplexe in den Brustwandableitungen. Bei positiver Konkordanz (QRS in V1–V6 überwiegend positiv) Erregungsursprung im posterobasalen linken Ventrikel. Bei negativer Konkordanz (QRS in V1–V6 überwiegend negativ) Erregungsursprung im anterolateralen linken Ventrikel.
-
QRS-Morphologie. Bei Rechtsschenkelblockbild sprechen biphasisches QRS in V1 und RS (R:S-Verhältnis <1) in V6 für VT und gegen aberrierende Leitung einer SVT, die in V1 zum QRS-Komplex führt. Beim Linksschenkelblockbild sprechen ein breites initiales r (>30 ms) und eine breite (>70 ms) sowie geknotete S-Zacken für VT. Die ◘ Abb. 1.83 zeigt das Original-EKG einer nichtanhaltenden (non sustained) Kammertachykardie.
Abb. 1.83 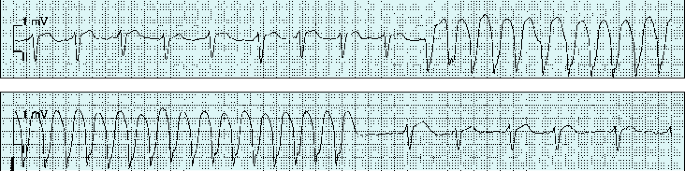
Nichtanhaltende ventrikuläre Tachykardie


Alle Kammertachykardien sind sehr ernst zu nehmen, da sie das Risiko des akuten Herztodes durch Übergang in Kammerflimmern beinhalten.
1.10.8.2.4 Allgemeine Therapie
Medikamentös sind manchmal β-Blocker oder Calciumantagonisten und β-Blocker wirksam. Am zuverlässigsten ist die Wirkung von Amiodaron. In manchen Fällen ist die RF-Katheterablation möglich. Sicheren Schutz gewährt ein implantierter Defibrillator.
1.10.8.2.5 Kammertachykardie bei koronarer Herzkrankheit
Hochgradige Gefährdung durch Übergang in Kammerflimmern. Mechanismen: Hauptsächlich Reentry im Narbengewebe, im Frühstadium eines Infarkts auch Automatie und Nachpotenziale. Risikofaktoren: Herabgesetzte linksventrikuläre Ejektionsfraktion, intermittierende VT, Nachpotenziale, Alternans der T-Welle (von Schlag zu Schlag), starre Sinusfrequenz (Wegfall des Vaguseinflusses), durch programmierte elektrische Stimulation induzierbare VT.
1.10.8.2.6 Ischämische ventrikuläre Tachykardie ohne koronare Arteriosklerose
Bei jungen Patienten mit abnormem Abgang der Koronararterien, Embolie der Koronararterien durch Thromben oder Vegetationen. Relative Ischämie bei Hypertrophie, schwerer Hypertonie und Aortenstenose.
1.10.8.2.7 Kammertachykardie bei Myokarditis
Vorkommen bei Myokardsarkoidose, akute Myokarditiden und Trypanosomiasis (Chagas-Krankheit). Behandlung des Grundleidens. Vorübergehend β-Blocker.
1.10.8.2.8 Kammertachykardien mit/ohne Kardiomyopathie
Es werden 3 Formen unterschieden, die in ◘ Tab. 1.13 dargestellt sind.
1.10.8.2.9 Langes QT-Syndrom
-
Definition: Arrhythmogene Anomalie mit Anfälligkeit für lebensbedrohliche Kammerarrhythmien, die angeboren oder erworben sein kann.
-
Genetisch bedingtes langes QT-Syndrom: Nach dem Erbgang ist das autosomal-dominante Romano-Ward-Syndrom vom seltenen autosomal-rezessiven Jervell- und Lange-Nielsen-Syndrom zu unterscheiden, das mit Taubheit assoziiert ist. Molekularbiologisch nachgewiesen sind 6 unterschiedliche genetische Defekte, von denen 5 Ionenkanäle der Myozyten modifizieren und einer das Membranmolekül Ankyrin betrifft. Gemeinsam ist allen eine Verlängerung der QT-Zeit mit genspezifischen Unterschieden des ST-T-Abschnitts (◘ Abb. 1.84).
Abb. 1.84 
Long-QT-Syndrom bei einer 23-jährigen Patientin (QT-Zeit 480 ms bei einer Herzfrequenz von 60/min). Genetisch: LQT5, KCNE1 (2 % aller LQT). Vor 5 Jahren ICD-Implantation nach Kammerflimmern und Reanimation mit 3-maliger Defibrillation
-
Symptombeginn ist häufig schon in den ersten beiden Lebensdekaden einschließlich der Neugeborenenperiode. Der Schweregrad variiert von häufigen Synkopen, die zum akuten Herztod führen können, bis zu subklinischen Formen ohne Arrhythmien. Das Risiko von Ereignissen ist bei einem QT-Intervall >500 ms um über 50 % höher als bei einem Intervall <500 ms.
-
Varianten: Die häufigsten Varianten sind:
-
LQT1: Häufigste Variante mit einem Gendefekt am Kaliumkanal Iks, der die Repolarisation verzögert. Erhöhtes Risiko bei physischem und psychischem Stress, besonders beim Schwimmen. Auf solche Situationen entfallen 90 % der Todesfälle.
-
LQT2: Gendefekt am Kaliumkanal Ikr mit Verzögerung der Repolarisation. Erhöhtes Risiko beim Erwachen und bei emotionaler Belastung.
-
LQT3: Der genetische Defekt betrifft einen Natriumkanal und verursacht eine Steigerung des späten Na+-Einwärtsstroms. Die meisten Ereignisse passieren in Ruhe und im Schlaf (64 %) und nur 4 % bei Anstrengungen.
-
-
Die Synkopen werden häufig durch Torsades de Pointes verursacht. Anhaltende Kammertachykardien können in Kammerflimmern übergehen. Patienten, von denen Angehörige in jungen Jahren plötzlich verstarben, haben ein hohes Mortalitätsrisiko.
-
Therapie: Asymptomatische Patienten der Typen LQT1 und LQT2 mit komplexen Arrhythmien oder positiver Familienanamnese erhalten hochdosierte β-Blocker. Wenn Synkopen auftreten ist ein ICD indiziert. Bei dem seltenen LQT3-Typ sind β-Blocker nicht indiziert. Sie scheinen auf Mexiletin anzusprechen.
-
-
Erworbenes langes QT-Syndrom:
-
Ätiologie: Verursacht werden kann es durch verschiedene Pharmaka: Chinidin, Procainamid, Sotalol, Amiodaron, Disopyramid, Phenothiazine oder trizyklische Antidepressiva, Terfenadin und Erythromycin. Weitere Risikofaktoren sind Hypokaliämie, Hypomagnesiämie, flüssige Proteindiät, Hunger, bradykardes Vorhofflimmern und Mitralklappenprolaps.
-
Klinisch manifestiert es sich mit Synkopen durch Torsades de Pointes, Kammertachykardien und Kammerflimmern.
-
Therapie: Gegen Torsades de Pointes 1–2 g Magnesiumsulfat i. v., bei Bradykardie Schrittmacherstimulation der Vorhöfe oder Ventrikel.
-


Auslösende Pharmaka absetzen und künftig strikt meiden. Hypokaliämie ausgleichen.
1.10.8.2.10 Brugada-Syndrom
1.10.8.2.10.1 Ätiopathogenese
Genetischer Defekt der Na+-Kanäle mit Repolarisationsstörung im Epikard des rechten Ventrikels, der in der Repolarisationsphase zu einem Spannungsgradienten zwischen Epi- und Endokard führt und zur Reentry-VT führen kann. Vorkommen bei jungen Erwachsenen hauptsächlich in Asien.
1.10.8.2.10.2 EKG
EKG im Sinusrhythmus: Inkompletter Rechtsschenkelblock mit starker ST-Hebung in V1–V3. Hohe Rate an plötzlichem Herztod.
1.10.8.2.10.3 Therapie
Primärtherapie mit implantierbarem Defibrillator.
1.10.8.2.11 Katecholaminerge polymorphe ventrikuläre Tachykardie (CPVT)
1.10.8.2.11.1 Ätiopathogenese
Autosomal-dominant erbliche Krankheit, deren Gendefekte Proteine betrifft, die den intrazellulären Calciumtransport regulieren oder Calcium im sarkoplasmatischen Retikulum binden. Neben eindeutig familiärem gibt es sporadisches Vorkommen.
1.10.8.2.11.2 Klinik
Bei organisch gesunden Herzen treten unter körperlicher und psychischer Belastung schon im Kindesalter Synkopen und Krämpfe auf, auch akute Todesfälle.
1.10.8.2.11.3 EKG
Kennzeichnend ist die Auslösung durch adrenale Stimulation mit Rechtsschenkelblockbild während der Tachykardie bei unauffälligem Ruhe-EKG.
1.10.8.2.11.4 Therapie
Gutes Ansprechen auf β-Blocker.
1.10.8.2.12 Arrhythmogene rechtsventrikuläre Dysplasie
Genetisch bedingte progrediente Kardiomyopathie, die bei jungen Menschen im rechten Ventrikel mit fleckförmiger Fettinfiltration und Myokardzellendegeneration beginnt und zur Dilatation und Kontraktionsschwäche führt. Beginn der klinischen Manifestation mit ventrikulärer Extrasystolie und schnellen ventrikulären Reentrytachykardien, teils mit Synkopen.
1.10.8.2.12.1 EKG
-
VT-EKG: Linksschenkelblockbild (wegen rechtsventrikulären Ursprungs der Tachykardie).
-
EKG im Sinusrhythmus: inkompletter Rechtsschenkelblock mit gekerbter S-Zacke und negativer T-Welle in V1 und V2.
1.10.8.2.12.2 Therapie
Eine RF-Katheterablation kann versucht werden. Wegen der Progredienz der Rhythmusstörungen ist in der Regel ein ICD indiziert.
1.10.8.3 Kammerflattern und Kammerflimmern
1.10.8.3.1 Definition
Hochfrequente Erregung der Kammern mit sofortigem Sistieren der Blutförderung (Herz- und Kreislaufstillstand). Häufigste Ursache des plötzlichen Herztodes.
1.10.8.3.2 Pathogenese
Multiple Reentrykreise infolge inhomogener Erregungsleitungsgeschwindigkeit und Refraktärität in den Herzkammern.
1.10.8.3.3 Ätiologie
Koronare Herzkrankheit (nur in etwa 25 % der Fälle ein frischer Myokardinfarkt), Kardiomyopathien, Präexzitationssyndrom mit Vorhofflimmern, QT-Verlängerungssyndrome, Cor pulmonale, Elektrounfall, toxische Herzschädigung durch Glykoside, Narkotika, Katecholamine etc.
1.10.8.3.4 Klinik
Bewusstseinsverlust, Apnoe, Blutdruckabfall auf Null.
1.10.8.3.5 EKG
-
Kammerflattern: einwellige, große, regelmäßige Oszillationen mit einer Frequenz von 150–300/min. Von einer schnellen ventrikulären Tachykardie manchmal schwer zu unterscheiden (◘ Abb. 1.77e).
-
Kammerflimmern: hochfrequente, unregelmäßige biphasische Undulationen von unterschiedlicher Amplitude und Form. QRS, ST und T fehlen. Auslösung durch eine früh- oder spätsystolische VES oder eine VT (◘ Abb. 1.77f).
1.10.8.3.6 Therapie
Reanimation mit elektrischer Defibrillation. Der überlebte Herzstillstand ist heute eine eindeutige Indikation zur Implantation eines Defibrillators.
1.10.9 Erregungsleitungsstörungen
Erregungsleitungsstörungen
-
Sinuatriale Blockierungen
-
Atrioventrikuläre Blockierungen
-
Intraventrikuläre Blockierungen
1.10.9.1 Sinuatriale Blockierungen
1.10.9.1.1 Definition
Blockierungen der Erregungsleitung vom Sinusknoten zum Vorhofmyokard.
1.10.9.1.2 EKG
-
SA-Block 1. Grades: Verlängerte sinuatriale Leitungszeit ohne Leitungsausfall. Im Oberflächen-EKG nicht erkennbar. Direkter Nachweis im intrakardialen Sinusknoten-Elektrogramm. Indirekter Nachweis durch vorzeitige atriale Einzelstimulation möglich.
-
SA-Block 2. Grades Typ I (Wenckebach-Periodizität): Ausfall der sinuatrialen Leitung nach progressiver Verlängerung der Leitungszeit. Da die Zunahme des Überleitungsintervalls zwischen dem ersten und zweiten Schlag der Periodizität am größten ist und dann progressiv kleiner wird, resultiert im EKG ein Herzschlagausfall nach zunehmender P-P-Verkürzung (◘ Abb. 1.85a).
Abb. 1.85a–c 
Sinuaurikuläre Leitungsstörungen. a SA-Block 2. Grades (Typ I), b SA-Block 2. Grades (Typ II), c SA-Block 3. Grades
-
SA-Block 2. Grades Typ II: Intermittierender Leitungsausfall ohne vorangehende Verlängerung der Leitungszeit. Ausfall eines Herzschlages, so dass ein Abstand von einem doppelten (oder vielfachen) P-P-Intervall entsteht (◘ Abb. 1.85b).
-
SA-Block 3. Grades: Totale Blockierung der Sinusimpulse. Nach präautomatischer Phase tritt ein Knotenersatzrhythmus auf (mit normalem QRS-Komplex ohne vorausgehende P-Zacke) (◘ Abb. 1.85c).


1.10.9.1.3 Ätiologie
Extreme Vagotonie, organische Sinusknotenschädigung (Ischämie, Entzündung, Degeneration), Pharmaka (Digitalis, Chinidin und andere Antiarrhythmika), besonders bei vorbestehender Sinusknotendysfunktion.
1.10.9.1.4 Klinik
Langsamer Puls bzw. pausenartiger Pulsausfall. Bei höhergradiger Bradykardie Absinken des Herzminutenvolumens mit Schwindelgefühl, Atemnot, evtl. auch Synkopen (Adam-Stokes-Anfälle). Im Rahmen eines Sinusknotensyndroms oft mit Störungen der Sinusknotenautomatie, atrialen Tachyarrhythmien und AV-Leitungsstörungen kombiniert.
1.10.9.1.5 Therapie
Pharmaka mit leitungsverzögernder Wirkung absetzen. In akuten Situationen können Orciprenalin (Alupent) und Atropin versucht werden. Bei rezidivierenden SA-Blockierungen Indikation zur Schrittmacherimplantation.
1.10.9.2 Atrioventrikuläre Blockierungen
1.10.9.2.1 Definition
Blockierungen der Erregungsleitung von den Vorhöfen zu den Kammern.
1.10.9.2.2 Ätiologie
Verlängerungen des PQ-Intervalls sind häufig ein stabiler Restzustand nach Myokarditiden (Diphtherie, rheumatisches Fieber u. a.). Ausgelöst durch Medikamente (Digitalis, Antiarrhythmika) oder kongenitale Defekte (ASD, VSD). AV-Blöcke 2. und 3. Grades kommen nach Herzinfarkt, bei Kardiomyopathien und Vitien aller Art, nach Traumen und bei primärer Degeneration des Leitungssystems (Lev-Krankheit) vor.
1.10.9.2.3 Diagnostik
1.10.9.2.4 Standard-EKG
-
AV-Block 1. Grades: Gleichmäßige Verlängerung des AV-Intervalls ohne Leitungsausfall. PQ-Zeit > 0,20 s. Bei normalem QRS-Komplex ist die Leitungsstörung überwiegend im AV-Knoten (87 %), seltener im His-Bündel (13 %) lokalisiert, bei einem Schenkelblock zu 50–80 % distal des His-Bündels in den noch leitenden Faszikeln. Auch Kombinationen kommen vor (◘ Abb. 1.86a).
Abb. 1.86 
AV-Leitungsstörungen. a AV-Block 1. Grades (PQ konstant verlängert), b AV-Block 2. Grades Typ I (Wenkebach-Periodik), c AV-Block 2. Grades Typ II, d AV Block 3. Grades (totaler Block)
-
AV-Block 2. Grades Typ I (Mobitz-I-Block, Wenckebach-Typ): Partieller AV-Block mit progressiver Verlängerung des AV-Intervalls bis zum Leitungsausfall (P-Zacke ohne QRS), der sich periodisch wiederholt (Wenckebach-Periodik). Die Leitungsstörung liegt im AV-Knoten (◘ Abb. 1.86b).
-
AV-Block 2. Grades Typ II (Mobitz-II-Block): Partieller AV-Block mit konstanten AV-Intervallen. Bei normaler oder konstant verlängerter PQ-Zeit folgt auf einzelne zeitgerechte P-Zacken kein QRS-Komplex. Lokalisation der Leitungsunterbrechung im AV-Knoten oder His-Purkinje-System. Bei vorbestehendem bifaszikulären Block erfolgt die Leitungsunterbrechung im noch leitenden Faszikel (trifaszikulärer Block). Das Verhältnis der übergeleiteten zu den blockierten Impulsen kann 2:1, 3:1 oder 4:1 betragen (◘ Abb. 1.86c).
-
AV-Block 3. Grades (kompletter oder totaler AV-Block): Vollständige Blockierung der Vorhof-Kammer-Überleitung im AV-Knoten, im His-Bündel oder in beiden Tawara-Schenkeln zugleich (trifaszikulärer Block) (◘ Abb. 1.86d). Vorhöfe und Kammern schlagen unabhängig voneinander. Vorhoferregung meistens durch den Sinusknoten (80 %), selten durch Vorhofflimmern oder -flattern (20 %). Kammererregung durch ein AV-junktionales (sekundäres) oder ein ventrikuläres (tertiäres) Automatiezentrum, je nach Lokalisation des Blockes:
-
AV-Knotenblock: AV-junktionaler Ersatzrhythmus (Frequenz 40–60/min, bei Belastung zunehmend, QRS-Komplexe normal).
-
Block im His-Bündel: Kammerersatzrhythmus mit Ersatzzentrum im unteren Teil des His-Bündels oder in den Faszikeln (Frequenz unter 40/min), oft instabil. QRS beim tiefen His-Bündel-Ersatzrhythmus meistens deformiert, bei faszikulärem Ersatzrhythmus verbreitert);
-
Infra-His-Block: ventrikulärer Kammerersatzrhythmus (Frequenz 30–40/min), instabil, QRS-Komplexe verbreitert.
-


1.10.9.2.5 His-Bündel-Elektrogramm
Erlaubt die abschnittsweise Messung der AV-Überleitung (◘ Abb. 1.87 und ◘ Tab. 1.14).


His-Bündel-Elektrogramm (A-H-Intervall 110 ms, H-V-Intervall 35 ms)


Rechtsschenkelblock: QRS 0,15 s, hohe, gesplitterte R-Zacken in V1–3, tiefe, breite S-Zacken in V5–6


Inkompletter Rechtsschenkelblock: QRS 0,11 s, Rechtsverspätung in V1–2


Linksschenkelblock: QRS 0,18 s, breite, plumpe R-Zacken in V5–6


Inkompletter Linksschenkelblock QRS 110 ms, keine initiale R-Zacke in V1, kein septales Q in V6


Linksanteriorer Hemiblock, überdrehter Linkslagetyp


Linksposteriorer Hemiblock, überdrehter Rechtslagetyp


Rechtsschenkel- plus linksanteriorer Hemiblock (bifaszikulärer Block)
1.10.9.2.6 Methode
Über die V. femoralis wird unter Röntgenkontrolle ein bipolarer Elektrodenkatheter in den rechten Ventrikel vorgeführt und dann so weit zurückgezogen, dass die Spitze mit dem Elektrodenpaar dem septalen Segel der Trikuspidalklappe und dem oberen Septum anliegt. An dieser Stelle werden die Aktionspotenziale A (vom AV-Knoten-nahen Anteil des rechten Vorhofes), H (vom His-Bündel) und V (vom Kammermyokard, im Septum beginnend) registriert. Der AV-Knoten selbst erzeugt kein messbares Aktionspotenzial. Simultan wird eine Ableitung des Oberflächen-EKG aufgezeichnet. Zur exakten Bestimmung von V kann ein zweiter Elektrodenkatheter zur Spitze des rechten Ventrikels vorgeführt werden.
1.10.9.2.7 Messwerte
-
A-H-Zeit: intranodale Leitungszeit (normal: 70–100 ms)
-
H-V-Zeit: Leitungszeit vom His-Bündel zur Kammer (normal: 35–55 ms)
1.10.9.2.8 Klinik
AV-Blockierungen können transitorisch (reversibel) oder permanent (irreversibel) sein. Der kongenitale komplette AV-Block , meistens im AV-Knoten lokalisiert, wird wegen eines frequenten junktionalen Ersatzrhythmus oft gut toleriert. Der erworbene komplette AV-Block entsteht selten akut (Infarkt, Herzoperationen, Traumen). Meistens geht ihm eine langsam progrediente Degeneration des unteren Reizleitungssystems voraus (analog der chronisch progredienten Affektion des oberen Reizleitungssystems beim Sick-Sinus-Syndrom). Dabei können die drei Grade der AV-Blockierung durchlaufen werden, oder die Faszikel fallen nacheinander aus. Symptomatisch werden AV-Blockierungen 2. und 3. Grades durch zerebrale Ischämie und bradykarde Herzinsuffizienz. Der komplette AV-Block ist durch eine AV-Dissoziation mit Wegfall des inotropen Effektes der Vorhofkontraktion gekennzeichnet.
1.10.9.2.9 Zerebrale Ischämie
Beim Übergang zum totalen Block in der Schaltpause zum Ersatzrhythmus bzw. bei extremer Bradykardie, besonders unter körperlicher Belastung, abhängig von der Dauer der Asystolie:
-
Dauer 3–6 s: Schwindel, Schwarzwerden vor den Augen, Bewusstseinstrübung
-
Dauer 10–20 s: Synkope (Adams-Stokes-Syndrom)
-
Dauer 25–40 s: Krämpfe
-
Dauer 60 s: Atemstillstand.
1.10.9.2.10 Bradykarde Herzinsuffizienz
Folgen sind:
-
Größenzunahme des Herzens aufgrund eines Anstiegs des enddiastolischen Volumens
-
kompensatorische Vergrößerung des Schlagvolumens
-
Schlagvolumenhochdruck (Anstieg des systolischen, Abfall des diastolischen Drucks)
-
Lungenstauung mit Ruhe- bzw. Belastungsdyspnoe, Ödeme
-
Absinken des Herzminutenvolumens mit Leitungsabfall und Schwindelgefühl, auch Präkordialschmerz
-
durch Systolenausfall kommt es beim AV-Block 2. Grades zur Arrhythmie
-
AV-Dissoziation: Wechselnde Lautstärke des 1. Herztones (leise bei großem, laut bei kurzem P-R-Intervall) und Kanonenwellen im Jugularvenenpuls (hohe A-Welle bei Kontraktion des rechten Vorhofes gegen geschlossene Trikuspidalklappe)
1.10.9.2.11 Therapie
-
AV-Block 1. Grades: Behandlung der Grundkrankheit, Verlaufsbeobachtung.
-
AV-Block 2. Grades Typ I: Bei Blockierung im AV-Knoten oft vorübergehend (Infarkt, Myokarditis, Digitalis). Leitungsverbesserung durch Atropin oder Ipratropiumbromid.
-
AV-Block 2. Grades Typ II: Akuttherapie mit Orciprenalin (Alupent) zur Verbesserung der AV-Überleitung und Verkürzung der präautomatischen Pause ventrikulärer Ersatzzentren. Passagere oder permanente Schrittmachertherapie.
-
AV-Block 3. Grades: Kongenitaler Block oft nicht behandlungsbedürftig. Temporäre Elektrostimulation bei reversiblem AV-Knotenblock durch Digitalisintoxikation oder Hinterwandinfarkt. Notfalltherapie mit Orciprenalin. In den meisten Fällen Dauertherapie mit Schrittmacherimplantation.
1.10.9.3 Intraventrikuläre Blockierungen
1.10.9.3.1 Definition
Verlangsamung oder Unterbrechung der Erregungsleitung in den Tawara-Schenkeln bzw. in den drei Faszikeln.
1.10.9.3.2 EKG
Nach dem Grad der Leitungsstörung unterscheidet man zwischen komplettem und inkomplettem Schenkelblock . Die Merkmale der Schenkelblöcke im Kammer-EKG ergeben sich aus den veränderten Vektoren der Erregungsausbreitung. Der ST/T-Abschnitt verhält sich zur Hauptausschlagrichtung von QRS stets diskordant, da die Erregungsrückbildung anders als bei normaler Erregungsausbreitung im zuerst erregten Abschnitt beginnt. Die Seitenlokalisation geht nur aus den Brustwandableitungen eindeutig hervor. Die normale Kammererregung erfasst zuerst die linke Seite des Septums mit einem initialen Vektor nach rechts. Danach werden beide Kammern erregt mit dem Hauptvektor nach links. Folglich ist der initiale Ausschlag in V1 positiv, in V6 negativ. Der Hauptausschlag ist V1 negativ und in V6 positiv.
Die Formen von kompletten und inkompletten Schenkelblöcken sind in ◘ Tab. 1.15 übersichtlich zusammengefasst.
1.10.9.3.3 Ätiologie
Inkomplette Schenkelblockbilder können ohne organische Herzerkrankung vorkommen. Meistens liegt bei den intraventrikulären Leitungsstörungen jedoch ein organischer Befund am Herzen vor. Herzinfarkt, koronare Herzkrankheit, Hypertonien mit Linksherzschädigung, Vitien und Kardiomyopathien sind die dominierenden Ursachen.
1.10.9.3.4 Klinik
Intraventrikuläre Leitungsstörungen sind bei voll kompensierten Herzen asymptomatisch. Ein ischämisch bedingter Linksschenkelblock kann anfangs erst bei Belastung auftreten und sich in der Ruhe zurückbilden. Die unkoordinierte Kontraktion, besonders beim Linksschenkelblock, verstärkt eine bestehende Herzinsuffizienz.
1.10.9.3.5 Therapie
Behandlung der vorliegenden Grundkrankheit. Bei kardialer Insuffizienz linksventrikuläre oder biventrikuläre Schrittmacherstimulation.
1.11 Entzündliche Herzkrankheit en
Entzündliche Herzkrankheiten
-
rheumatische Karditis
-
infektiöse Endokarditis
-
Myokarditis
-
Perikarditis
-
akute Perikarditis
-
Perikarderguss und Herztamponade
-
chronische konstriktive Perikarditis
-
1.11.1 Rheumatische Karditis
1.11.1.1 Ätiologie und Pathogenese
Akute nichteitrige Pankarditis als immunologische Zweitkrankheit nach einem Racheninfekt mit β-hämolytischen Streptokokken der Gruppe A. Es handelt sich um eine Manifestation des rheumatischen Fiebers, das im Abschnitt über die entzündlichen Gelenkkrankheiten ausführlich abgehandelt ist (▶ Abschn. 9.1.5). Die meisten erworbenen Herzklappenfehler gingen vor der Antibiotika-Ära auf das rheumatische Fieber zurück. Heute ist die Mehrzahl durch degenerative Veränderungen bedingt.
1.11.1.2 Klinik
Bei der ersten von Fieber und Arthritis dominierten Attacke bleibt die Herzbeteiligung in etwa der Hälfte der Fälle unbemerkt und wird erst viel später bei der Manifestation eines Klappenfehlers erkannt. Frühe Hinweise sind perikarditischer Brustschmerz und Zeichen der Herzschwäche (Dyspnoe, Stauungszeichen im großen und kleinen Kreislauf). Die überwiegend jugendlichen Patienten sind tachykard mit akzidentellem systolischem Geräusch. In schweren Fällen holosystolisches Spitzengeräusch infolge relativer Mitralinsuffizienz. Die initialen verrukösen Klappenauflagerungen verursachen noch keinen vitiumtypischen Auskultationsbefund. Im Vordergrund steht anfangs die Myokarditis, der histologisch Aschoff-Knötchen zugrunde liegen. Eine Begleitperikarditis manifestiert sich mit typischem systolisch-diastolischem Maschinengeräusch.
1.11.1.3 Diagnostik
Vorausgegangener Streptokokkeninfekt. Im EKG verlängertes AV-Intervall, Abflachung oder Negativierung der T-Wellen, selten Arrhythmien. Im Echokardiogramm sind Linksherzdilatation und Perikardergüsse zu erkennen. Die Röntgenaufnahme des Thorax deckt eine Größenzunahme des Herzschattens auf.
1.11.1.4 Laborbefunde
-
spezifisch: α-hämolytische Streptokokken der Gruppe A im Rachen- oder Tonsillenabstrich, hoher Antistreptolysin-Titer
-
unspezifisch: starke Blutsenkungsbeschleunigung, Erhöhung von CRP und α2-Globulinen im Serum.
1.11.1.5 Therapie
Antipyretisch (Aspirin oder nichtsteroidale Antirheumatika), in schweren Fällen Glukokortikoide. Dazu Penicillin zur Eradikation der Streptokokken (▶ unten).
1.11.1.6 Verlauf und Prognose
Schwere Myokarditiden können vor allem bei Kindern zum Tode führen. Mittelschwere und leichtere Formen klingen im Verlauf einiger Monate ab und hinterlassen selten bleibende Myokardschäden. Die Valvulitis schreitet aber unter Verdickung und Vernarbung der Klappen oft fort und führt nach Jahren zum manifesten Klappenfehler. Entscheidend für die Progredienz sind rezidivierende Streptokokkeninfekte, die am Herzen in 50 % der Fälle wieder rheumatische Schübe induzieren, während die Gelenke oft nur beim ersten Ausbruch der Krankheit betroffen sind. Daraus ergibt sich die Notwendigkeit zur Langzeitrezidivprophylaxe mit Penicillin (1,2 Mio. IE Penicillin i.m. monatlich), die bei Kindern mindesten bis zum 18. Lebensjahr durchgeführt werden sollte.
1.11.2 Infektiöse Endokarditis
1.11.2.1 Definition
Mikrobielle Infektion des Endokards, überwiegend durch Bakterien, vor allem an den Herzklappen, die unbehandelt eine 100%ige Mortalität hat. Klinisch ist eine Verlaufsform mit rascher Klappendestruktion von einer schleichenden Verlaufsform mit Anämie und Milzschwellung zu unterscheiden. Beide haben ein unterschiedliches Erregerspektrum.
1.11.2.2 Ätiologie und Pathogenese
Voraussetzungen für die Entstehung einer Endokarditis sind:
-
die Anwesenheit der Erreger im Blut (Bakteriämie) und
-
Endokardanomalien mit Verlust der Thromboresistenz.
1.11.2.3 Bakteriämie
Ausgangspunkt sind häufig Läsionen der dicht besiedelten Mundschleimhaut, besonders am Zahnfleisch. Ferner kommen Hautdefekte, Entzündungsprozesse am Urogenitalsystem und am Darmtrakt als Erregerquelle in Betracht. Große Bedeutung kommt der durch diagnostische oder therapeutische Interventionen verursachten Bakteriämie zu (▶ Endokarditisprophylaxe).
1.11.2.4 Endokardanomalien
-
Klappenveränderungen nach rheumatischer Karditis
-
mechanische Läsionen:
-
auf der atrialen Seite der Klappensegel bei Mitral- und Trikuspidalinsuffizienz
-
auf der ventrikulären Seite bei Aorten- und Pulmonalinsuffizienz
-
auf der Niederdruckseite intrakardialer Kurzschlussverbindungen
-
auf der ventrikulären Seite bei Mitral- und Trikuspidalstenosen
-
auf der aortalen Seite bei Stenosen im linksventrikulären Ausflusstrakt
-
-
Mitralklappenprolaps mit ausgeprägter myxomatöser Degeneration
-
kalzifizierende Klappendegeneration vorzugsweise im Alter.
Zunehmende Bedeutung erlangen intrakardiale Transplantate (Herzklappenprothesen, Elektroden) aus Polymermaterial.
1.11.2.5 Bildung von Vegetationen
Nach Infektion nichtbakterieller Mikrothromben entsteht durch Zelldetritus, Thrombozyten und Fibrin eine in den ersten 3 Wochen besonders embolieträchtige „Vegetation“ mit Tendenz zur Größenzunahme. Die bakterielle Entzündung kann das Klappengewebe zerstören und sich unter Abszedierung über die Klappenränder hinaus ausdehnen. Aus den Vegetationen lösen sich regelhaft mehr oder weniger große Fragmente als Emboli ab.
1.11.2.6 Häufige ätiologische Mikroorganismen
-
Staphylokokken: S. aureus, S. epidermidis
-
Streptokokken: Streptokokken der Viridans-Gruppe, Streptococcus bovis, Streptococcus pneumoniae
-
Enterokokken: E. faecalis, E, faecium
-
gramnegative Erreger: HACEK-Gruppe (Haemophilus, Actinobazillus, Cardiobacterium hominis, Eikenella corrodens, Kingella kingae), Pseudomonas ssp., Neisseria gonorrhoeae
-
andere Erreger: Listeria monocytogenes, Rickettsia, Chlamydien, Bartonella quintana, Bartonella henselae, Anaerobier (insbesondere Peptostreptokokken)
-
Pilze: Candida albicans, Candida-Spezies, Histoplasma, Aspergillus-Spezies
1.11.2.7 Klinik
1.11.2.8 Inkubationszeit
Bei etwa 30 % der Patienten lässt sich kein Ereignis eruieren, das vor Ausbruch der Endokarditis zur Bakteriämie geführt haben könnte. Das Intervall wechselt mit dem Erreger. Bei Streptokokken-Endokarditis wurden Inkubationszeiten von 2 bis mehreren Wochen beobachtet. Bei der Staphylokokken-Endokarditis ist das Intervall wegen der Aggressivität der Erreger meistens kurz. Nach vorausgegangener Zahnbehandlung wurden Intervalle zwischen 4 Tagen und 8 Wochen beschrieben.
1.11.2.9 Symptome
Remittierendes Fieber (80–90 %), bei akuten Verlaufsformen bis 40 °C, auch mit Schüttelfrost. Bei subakuten Verläufen sind die Temperaturen oft subfebril mit Schweißausbrüchen. Häufig Unwohlsein, Appetitmangel, Gewichtsverlust. Außerdem Dyspnoe (20–40 %), Myalgien und Arthralgien (15–30 %), Kopfschmerzen (15–40 %), Verwirrtheit (10–20 %), zerebrale Insulte und Hirnembolie (13–20 %), abdominale Schmerzen (5–15 %), Brustschmerzen (8–35 %), Symptome durch embolische Ereignisse (40 %).
1.11.2.10 Diagnostik
1.11.2.11 Körperliche Untersuchungsbefunde
Klappeninsuffizienzgeräusche (80–85 %), die diagnostisch wegweisend sind, wenn sie während der Infektion neu auftreten, neurologische Auffälligkeiten (20–40 %), Milzschwellung (15–50 %), Immunkomplex-Glomerulonephritis (<15 %). Lungenembolien mit fleckigen, oft wechselnden Infiltraten sind für die Endokarditis des rechten Herzens typisch. In 10 % der Fälle periphere Manifestationen: Petechien, subunguale Hämorrhagien, Osler-Knötchen (erbsgroß, hellrot, druckschmerzhaft, hauptsächlich an Fingerbeeren oder Zehen), nicht schmerzende Janeway-Läsionen an Handflächen und Fußsohlen durch Mikroembolien (◘ Abb. 1.95), Roth-Flecken (ovale Retinablutung mit blassem Zentrum).


Ausgeprägte periphere Embolisationen bei einem 46-jährigen Patienten mit fulminanter Staphylokokken-Endokarditis
1.11.2.12 Laborbefunde
Normochrome Anämie mit herabgesetztem Serumeisen. In akuten Fällen Leukozytose mit Linksverschiebung im Differenzialblutbild. Blutsenkung stark beschleunigt, CRP deutlich erhöht, Albuminurie und Mikrohämaturie (50 %).
1.11.2.13 Echokardiographie
Mit der transthorakalen Echokardiographie werden nur 65 % der Vegetationen erkannt und keine mit einem Durchmesser <2 mm (◘ Abb. 1.96). Empfindlicher ist die transösophageale Echokardiographie, die >95 % aller Vegetationen und ihre Größe erfasst (◘ Abb. 1.97). Entdeckt werden auch Abszesse und Fisteln.


Akute Staphylokokken-Mitralklappenendokarditis mit großer flottierender Vegetation am vorderen Mitralklappensegel (AML) und Begleitperikarderguss (PE), PML = hinteres Mitralklappensegel


Transösophageale Echokardiographie eines 71-jährigen Patienten mit einer akuten Staphylokokken-Aortenklappenprothesenendokarditis (AKE), großen flottierenden Vegetationen und paraprothetischen Abszessen (MK = Mitralklappe)
1.11.2.14 Erregernachweis in der Blutkultur
Gelingt in 95 % der Fälle, unter antibiotischer Therapie sind es nur 72 %. Nach Absetzen der Antibiotika kann es eine Woche oder länger dauern, bis Kulturen positiv werden. Innerhalb von 24 h sind mindestens dreimal aus verschiedenen venösen Punktionsstellen 2 Testflaschen mit je 10 ml Blut zu beschicken, eine zur aeroben, die andere zur anaeroben Kultur. Die Körpertemperatur ist für den Zeitpunkt der Blutentnahme unerheblich. Die Erreger werden identifiziert und auf ihre Antibiotikaempfindlichkeit getestet. Dabei wird die minimale Hemmkonzentration (MHK) bestimmt.
Diagnostische Haupt- und Nebenkriterien der infektiösen Endokarditis sind in ◘ Tab. 1.16 zusammengestellt. Wenn 2 Hauptkriterien oder 1 Hauptkriterium und 3 Nebenkriterien erfüllt sind, ist die klinische Diagnose Endokarditis begründet.
1.11.2.15 Antibiotische Therapie
1.11.2.16 Prinzip
Zur Überwindung des Expositionsschutzes der Erreger in den Vegetationen muss ein hoher Diffusionsgradient erzielt werden. Hohe bakterizide Serumspiegel (durch parenterale Applikation) sowie eine Behandlungsdauer von in der Regel 4–6 Wochen sind unverzichtbar. Enterokokken werden von Penicillin und Vancomycin nicht abgetötet. Durch die Kombination mit einem Aminoglykosid wird ein synergistischer Effekt erreicht. Er resultiert aus unterschiedlichen Wirkungsmechanismen. Penicilline und Vancomycin greifen an der Zellmembran an, Aminoglykoside sehr intensiv im Zellinnern.
1.11.2.17 Praktische Durchführung
Für die häufigsten Erreger sind die therapeutischen Alternativen in ◘ Tab. 1.17 zusammengestellt.
1.11.2.18 Anmerkungen
Die intravenösen Applikationen erfolgen als Kurzinfusionen von 30 Minuten, zuerst immer mit den β-Laktamantibiotika. Unter Therapie mit Gentamicin und/oder Vancomycin sind die Serumspiegel zu kontrollieren. Bei Beteiligung intrakardial implantierten Polymermaterials, Staphylokokken-, Enterokokken- und Pilzendokarditiden besteht unabhängig von bereits eingetretenen Komplikationen (▶ unten) primär die Indikation zur Operation. Polymer-assoziierte Endokarditiden erfordern zudem die höchstdosierte Kombinationstherapie mit synergistisch wirksamen Antibiotika.
1.11.2.19 Chirurgische Therapie intrakardialer Komplikationen
1.11.2.20 Indikationen
-
Klappeninsuffizienz: Hohe Mortalität, da das Myokard an die akute Volumenbelastung nicht adaptiert ist. Lässt sich die konsekutive Lungenstauung medikamentös nicht rasch kompensieren, besteht eine dringliche Operationsindikation.
-
Instabile Prothesen: Dehiszenzen durch perivalvuläre Infektionen erfordern in der Regel eine Reoperation.
-
Unkontrollierte Infektion: Wenn die antibiotische Therapie versagt, besonders bei Endokarditiden durch gramnegative Erreger, Staphylokokken, Enterokokken und Pilze. Bei Endokarditiden durch S. aureus oder Enterokokken, falls die Sepsis nach 48-stündiger Antibiotikatherapie fortbesteht und der Entzündungsprozess sich lokal ausbreitet (Erregungsleitungsstörungen, Abklatschvegetationen, Destruktion supportiver Strukturen).
-
Große Vegetationen: Bei großen flottierenden Gebilden kommt wegen drohender oder erfolgter Embolien die Operation in Betracht.
-
Akutes Nierenversagen: Entstanden durch die Infektion oder Medikamente, hat eine schlechte Prognose, die durch die Operation gebessert werden kann.
1.11.2.21 Operative Eingriffe
-
Meistens Klappenersatz oder Prothesenwechsel unter Antibiotikaschutz.
-
Entfernung von Vegetationen und entzündlich verändertem Gewebe mit Klappen-erhaltender Rekonstruktion.
Die antibiotische Therapie ist postoperativ für 6 Wochen fortzusetzen.
1.11.2.22 Endokarditisprophylaxe
1.11.2.23 Indikationen
Die American Heart Association hat dazu 2007 neue Empfehlungen herausgegeben, die von den früheren Leitlinien erheblich abweichen. Man geht davon aus, dass nur eine extrem kleine Fallzahl von infektiöser Endokarditis durch eine Antibiotikaprophylaxe vor zahnärztlichen Eingriffen verhindert werden kann, auch wenn sie hundertprozentig effektiv ist. Denn transitorische Bakteriämien kommen weitaus häufiger bei der täglichen Zahnpflege vor. Nach den neuen Richtlinien ist eine Antibiotikaprophylaxe vor dentalen Prozeduren nur noch bei Patienten indiziert, die wegen bestimmter kardialer Anomalien durch eine infektiöse Endokarditis im höchsten Grade gefährdet wären. Es handelt sich um nachstehende Patientengruppen:
-
Patienten mit Klappenersatz (mechanische und biologische Prothesen)
-
Patienten mit überstandener Endokarditis
-
Patienten mit angeborenen Herzfehlern
-
Patienten mit zyanotischen Herzfehlern, die nicht oder palliativ mit systemisch-pulmonalem Shunt operiert sind
-
Patienten mit operierten Herzfehlern mit Implantation von Conduits (mit oder ohne Klappe) oder residuellen Defekten, d. h. turbulenter Blutströmung im Bereich des prothetischen Materials
-
alle Patienten mit operativ oder interventionell unter Verwendung von prothetischem Material behandelten Herzfehlern in den ersten 6 Monaten nach Operation
-
herztransplantierte Patienten, die eine kardiale Valvulopathie entwickeln
Risikoprozeduren sind alle Zahneingriffe, die zur Bakteriämie führen können: Manupulationen an der Gingiva, der periapikalen Zahnregion oder mit Perforationen der oralen Mukosa. Dazu können auch die Entnahme von Biopsien und die Plazierung kieferorthopädischer Bänder gezählt werden. Vor herzchirurgischen Eingriffen ist auf orale Hygiene und Zahngesundheit zuachten.
Bei Risikopatienten sind kardiovaskuläre und Blasenkatheter auf ein Minmum zu beschränken und Infektionen mit Bakteriämie prompt und vor Gewebsinzisionen zu behandeln. Keine Endokarditisprophylaxe wird bei Eingriffen am Urogenital- und Gastrointestinaltrakt empfohlen.
In ◘ Tab. 1.18 sind die medikamentösen Maßnahmen der Endokarditisprophylaxe zusammengestellt.
1.11.3 Myokarditis
1.11.3.1 Ätiologie
1.11.3.2 Infektiöse Myokarditis
Entzündliche Myokardläsionen durch pathogene Organismen, von denen hier nur die wichtigsten aufgeführt werden.
-
Virusarten: Coxsackie-Virus A und B, Enteroviren, Echovirus, Adenovirus, Influenzavirus, Hepatitisvirus B, Zytomegalievirus, EBV (infektiöse Mononukleose), Parvo-Virus B19, Herpesviren.
-
Bakterien: Streptokokken (rheumatisches Fieber, Scharlach), Korynebakterien (Diphtherie), Salmonellen, Meningokokken, Clostridien.
-
Spirochäten: Treponema pallidum (Syphilis), Leptospiren (Morbus Weil, Borrelia recurrentis (Rückfallfieber) und burgdorferi (Lyme-Krankheit).
-
Pilze: Disseminierte Moniliasis.
-
Protozoen: Trypanosoma cruzi (Trypanosomiasis), Toxoplasma gondii (Toxoplasmose).
-
Metazoen: Trichinen( Trichinose).
1.11.3.3 Nichtinfektiöse Myokarditis
Entzündliche Läsionen immunologischer Genese bei Medikamentenallergie (Penicillin, Methyldopa, Sulfonamide, Streptomycin u. a.), Kollagenkrankheiten und Riesenzellmyokarditis. Entzündliche Reaktionen auf toxische Einwirkungen: Katecholamine (Phäochromozytom), Kohlenmonoxid, Lithium, Arsen, Blei, Röntgenstrahlen.
1.11.3.4 Pathogenese und Pathologie
1.11.3.5 Mechanismen der Myokardschädigung
Invasion der Myokardzellen (z. B. durch Echovirus), Toxinbildung (z. B. Diphtherie) oder sekundäre Immunreaktionen (z. B. rheumatisches Fieber).
1.11.3.6 Myokardläsionen
Interstitielle entzündliche Infiltrate, Myokardnekrosen, Desintegration der Myokardfasern, Granulome. Fokaler oder diffuser Befall, je nach Schwere der Erkrankung. Sekundäre interstitielle Fibrose.
1.11.3.7 Klinik
1.11.3.8 Verlaufsformen
Nach dem klinischen Bild lassen sich 3 Varianten der Myokarditis unterscheiden:
-
Subklinische Myokarditis bei akuten Infektionskrankheiten: Nachweis der Myokardbeteiligung nur in autoptisch untersuchten Fällen, klinisch nicht relevant.
-
Klinisch manifeste Myokarditis bei akuten Infektionskrankheiten: Neben den Zeichen der Infektionskrankheit wird die Herzbeteiligung evident.
-
Myokarditis als isolierte Erkrankung: Die Zeichen der Herzaffektion stehen im Vordergrund. Begleitendes Fieber weist auf entzündliches Geschehen hin. Endokarditis, Perikarditis und Herzinfarkt sind abzugrenzen.
1.11.3.9 Symptome
Schwäche, Herzklopfen, präkordiale Sensationen, Herzschmerz bei Perikardbeteiligung, Dyspnoe bei Herzinsuffizienz.
1.11.3.10 Körperliche Befunde
In leichten Fällen nur Tachykardie (über den fieberbedingten Grad hinausgehend). Bei Herzinsuffizienz Abschwächung des 1. Herztones, protodiastolischer Galopp, oft weitere Rhythmusstörungen (atriale und ventrikuläre). Stauungszeichen im großen und kleinen Kreislauf mit Zyanose.
1.11.3.11 Diagnostik
Virologische, bakteriologische, immunologische und serologische Untersuchungen bzw. der Nachweis nichtinfektiöser Noxen sind zur ätiologischen Klärung erforderlich, da die Symptome der Myokarditis keine Spezifität besitzen. Bei Virusmyokarditiden gelingt der Nachweis meistens nur bioptisch und nur selten serologisch. Wichtige Hinweise ergeben sich auch aus extrakardialen Manifestationen der zugrundeliegenden infektiösen Erkrankungen (z. B. Arthritis). Bei generalisierten Infektionskrankheiten stets auf Myokardbeteiligung achten (inadäquate Sinustachykardie, EKG-Veränderungen), da auch latente Myokardschäden die Rekonvaleszenz verzögern oder beeinträchtigen können.
1.11.3.12 EKG
Veränderungen des ST/T-Abschnittes, in klinisch latenten Fällen oft der einzige Hinweis auf die Myokarditis. Nicht selten atrioventrikuläre und intraventrikuläre Reizleitungsstörungen, bei Nekrosen auch Q-Zacken.
1.11.3.13 Echokardiographie
Erfasst Herzdilatation und Herabsetzung der Verkürzungs- und Ejektionsfraktion.
1.11.3.14 Röntgenuntersuchung des Thorax
Nachweis von Herzverbreiterung und Lungenstauung.
1.11.3.15 Therapie
Spezifische antibiotische Therapie bei bekanntem Erreger. Penicillin bei rheumatischem Fieber. Kortikoide bei rheumatischem Fieber, Kollagenosen und versuchsweise bei entzündlichen Kardiomyopathien (nach Endomyokardbiopsie), nicht bei Virusmyokarditis. Im fieberhaften Stadium Bettruhe. Symptomatische Behandlung der Rhythmusstörungen. Behandlung der Herzinsuffizienz mit ACE-Blockern, Diuretika, β-Blockern und Aldosteronantagonisten.
Bei einer Myokarditis besteht gegenüber Digitalispräparaten eine erhöhte Empfindlichkeit.
Parainfektiöse Myokarditiden heilen meistens nach mehreren Wochen aus.
1.11.4 Perikarditis
1.11.4.1 Ätiologische Klassifikation
1.11.4.2 Idiopathische Perikarditis
Ursache ungeklärt (nicht identifiziertes Virus, Autoimmunerkrankung?). Von der Virusperikarditis klinisch nicht zu unterscheiden, wie diese meistens gutartig mit Ausheilung in 3–6 Wochen.
1.11.4.3 Virusperikarditis
Am häufigsten durch Coxsackie-Virus B und Echovirus Typ 8, seltener durch die Viren von Mumps, Influenza, infektiöser Mononukleose, Hepatitis B und Varizellen. In der Regel gutartig verlaufend. Das Myokard kann beteiligt sein. Tuberkulöse Perikarditis exsudativa: Früher die häufigste Form, Anteil jetzt unter 10 %. Entstehung per continuitatem aus befallenen mediastinalen Lymphknoten oder hämatogen. Weniger akut als die idiopathische Perikarditis und die Virusperikarditis. Übergänge von akuter fibrinöser zu adhäsiver und konstriktiver Perikarditis mit Verkalkung (Panzerherz).
1.11.4.4 Eitrige Perikarditis
Durch verschiedene Erreger (Pneumokokken, Staphylokokken, Streptokokken u. a.), hämatogen, lymphogen oder per continuitatem (Pneumonie, Mediastinitis, subdiaphragmaler Abszess). Meistens Komplikation einer septischen Allgemeininfektion.
1.11.4.5 Perikarditis nach Herzinfarkt
Zu unterscheiden ist zwischen der durch Katheterintervention selten gewordenen Begleitperikarditis durch Nekrose und Ischämie, die nach 12 h bis 10 Tagen auftritt und der immunologisch bedingten diffusen Perikarditis des Dressler-Syndroms, das 2‒3 Wochen (eine Woche bis wenige Monate) nach dem Infarkt beginnt. Beide Formen sind gegen Postinfarktangina und Infarktrezidiv abzugrenzen. Urämische Perikarditis: Früher erkrankten daran bis zu 50 % der urämischen Patienten. Unter Dialysebehandlung haben 15‒20 % hämodynamisch nicht signifikante Perikardergüsse und nur 1,3–5 % Ergüsse von hämodynamischer Signifikanz, die sich nach Steigerung der Dialysefrequenz häufig bessern. Die Pathogenese ist ungeklärt (toxische stickstoffhaltige Metaboliten, Infektionen, immunologische Mechanismen nach urämischen oder heparininduzierten Blutungen in den Perikardspalt?).
1.11.4.6 Immunoreaktive Perikarditis
Bei rheumatischem Fieber, Kollagenosen und Postperikardiotomie-Syndrom. Promptes Ansprechen auf Glukokortikoide.
1.11.4.7 Neoplastische Perikarditis
Sekundärer Befall des Perikards vor allem bei Bronchialkarzinom, Mammakarzinom, Morbus Hodgkin und Leukämie, seltener bei gastrointestinalen Tumoren, Sarkomen und Melanom. Primärtumoren des Perikards (Mesotheliom, Fibrosarkom) sind äußerst selten.
1.11.4.8 Strahlen-Perikarditis
Relativ häufige Komplikation der Megavolt-Therapie bei Mammakarzinom und Morbus Hodgkin, innerhalb von 12 Monaten nach Beendigung der Bestrahlung auftretend.
1.11.4.9 Akute Perikarditis
1.11.4.9.1 Klinik
Anhaltender scharfer, stechender Präkordialschmerz mit Intensitätszunahme beim Atmen und Husten, im Liegen stärker als im Sitzen mit vorgebeugtem Oberkörper. Der Schmerz hält Tage bis Wochen an, reagiert nicht auf Nitroglycerin, aber auf Analgetika. In schweren Fällen Symptome der Herztamponade durch Perikarderguss (▶ unten). Allgemeinerscheinungen: Müdigkeit, Abgeschlagenheit, Appetitmangel, Fieber.
1.11.4.9.2 Diagnostik
1.11.4.9.3 Herzauskultaltion: Perikardreiben
An umschriebener Stelle über dem Herzen charakteristisches systolisch-diastolisches Reibegeräusch (Lokomotivgeräusch). Manchmal ist das Reiben zu tasten.
1.11.4.9.4 EKG
Veränderungen entstehen durch Schädigung des subepikardialen Myokards mit Verletzungsstrom, dessen Vektor bei diffusem Befall des Subepikards in Richtung des QRS-Vektors verläuft. Das typische Perikarditis-EKG durchläuft folgende 4 Stadien:
-
Stadium I: ST-Hebung in allen Ableitungen des Extremitäten- und Brustwand-EKG, außer in aVR und V1, wo die ST-Strecke gesenkt sein kann. Die ST-Strecken sind nach oben konkav, nicht konvex wie beim Infarkt. Positives T in allen Ableitungen mit ST-Erhöhung.
-
Stadium II: Nach einigen Tagen Rückkehr der ST-Strecke zur Grundlinie unter Abflachung der T-Zacke.
-
Stadium III: Inversion der T-Zacken bei isoelektrischem ST. Im Gegensatz zum Infarkt werden die T-Zacken erst nach Rückbildung der ST-Hebung negativ; außerdem entstehen weder Q-Zacken noch Potenzialverluste der R-Zacken, da Nekrosen ausbleiben.
-
Stadium IV: Rückbildung der T-Negativität zum Ausgangsbefund innerhalb von Wochen bis Monaten.
In knapp 50 % der Fälle werden verschiedene Abweichungen von diesem Verlauf beobachtet: Isolierte Senkung der ST-Strecke, direkter Übergang von Stadium I nach IV, Beschränkung der ST-Hebung auf einige Ableitungen, Negativierung von T bei noch gehobener ST-Strecke, persistierende T-Negativität. Die Abgrenzung gegen das bei jungen Männern nicht seltene EKG der normalen frühen Repolarisation mit ST-Hebungen ergibt sich aus dem Ausbleiben von ST/T-Veränderungen. Die bei etwa 25 % der Patienten auftretenden Rhythmusstörungen betreffen hauptsächlich Sinusknoten und Vorhöfe (Nachbarschaft von Sinusknoten und persinodalem Gewebe mit dem entzündeten Perikard): Sinustachykardie, Vorhofflattern, Vorhofflimmern, paroxysmale supraventrikuläre Tachykardien.
1.11.4.9.5 Echokardiographie
Nachweis und Lokalisation eines begleitenden Perikardergusses.
1.11.4.9.6 Röntgenuntersuchung des Thorax
Oft ohne auffälligen Befund. Nur bei größerem Perikarderguss Herzverbreiterung. Keine pulmonalen Stauungszeichen.
1.11.4.9.7 Laborbefunde
Unspezifische Entzündungszeichen (hohe BKS, Leukozytose), CK und CK-MB meistens normal, doch kann die CK-MB erhöht sein. Troponin ist meistens negativ.
1.11.4.9.8 Zur ätiologischen Klärung
Tuberkulinreaktion, virologische Untersuchungen, Blutkulturen, Harnstoff- und Kreatininbestimmung, serologische Untersuchungen (Antistreptolysinreaktion, antinukleäre Faktoren, Rheumafaktor, heterophile Mononukleoseantikörper).
1.11.4.9.9 Therapie
Schmerzstillung bevorzugt mit Antiphlogistika, wenn nötig auch mit starken Analgetika, evtl. mit Morphium. Bei infektiöser Ursache Antibiotika. Bei idiopathischer und immunologischer Form Glukokortikoide, ggf. Colchicum.
1.11.4.10 Perikarderguss und Herztamponade
1.11.4.10.1 Definition
Komplikation der akuten oder chronischen Perikarditis jeglicher Ätiologie. Zur akuten Tamponade führt außerdem das Hämoperikard bei Ruptur des Herzens oder einer aneurysmatischen Aorta.
1.11.4.10.2 Pathogenese
Die Perikardflüssigkeit (normal 15–50 ml) wird vom Mesothel der parietalen Perikardoberfläche sezerniert und auf dem Lymphweg abtransportiert. Ein Perikarderguss resultiert, wenn die Sekretion die Transportkapazität der Lymphbahnen überschreitet. Das ist bei vielen Perikarditiden der Fall. Für die hämodynamischen Auswirkungen ist nicht die Menge des Perikardergusses, sondern der zur Herzkompression führende Anstieg des intraperikardialen Drucks maßgebend. Bei normalem Herzbeutel steigt der intraperikardiale Druck oberhalb eines Ergussvolumens von 150–200 ml steil an, während der langsam und chronisch gedehnte Herzbeutel mehr als einen Liter Erguss ohne wesentliche Druckerhöhung aufnehmen kann. Die Kompression durch den Perikarderguss erschwert die diastolische Füllung des Herzens. Folglich muss der Füllungsdrucks steigen. Aus der Kompression wird eine Tamponade des Herzens, wenn der intraperikardiale Druck den diastolischen Druck des rechten Vorhofes und der rechten Kammer erreicht. Es resultiert eine Einflussstauung vor dem rechten Herzen mit veränderter Dynamik des venösen Rückflusses. Ansaugeffekt nur während der Systole durch Druckabfall im rechten Vorhof, nicht mehr in der frühen Diastole nach Öffnung der Trikuspidalklappe. Da die Kompression während des ganzen Herzzyklus bestehen bleibt, fallen diastolische Kammerfüllung, Schlagvolumen, Herzminutenvolumen und Blutdruck bis zum kardiogenen Schock ab.
1.11.4.10.3 Klinik
Keine oder geringe präkordiale Beschwerden bei Ergüssen ohne Drucksteigerung. Bei großen Ergüssen mechanische Kompression der Trachea mit Hustenreiz, der Lunge mit Atemnot und des Ösophagus mit Dysphagie.
Eine akute Herztamponade führt rasch zum Kreislaufschock.
1.11.4.10.4 Diagnostik
1.11.4.10.5 Körperlicher Untersuchungsbefund
Verschwinden des perikardialen Reibegeräusches, Abschwächung des 1. Herztones, Verschwinden des Spitzenstoßes, perkutorische Herzverbreiterung, Stauung der Halsvenen. Bei beginnender Tamponade inspiratorischer Blutdruckabfall um mindestens 10 mmHg (verstärkte Füllung des rechten Ventrikels verdrängt das Septum nach links und verkleinert das linksventrikuläre Schlagvolumen).
1.11.4.10.6 EKG
Niedervoltage von QRS und T bei stärkerer Ergussbildung (◘ Abb. 1.98), daneben evtl. Perikarditiszeichen (▶ oben). Bei Herztamponade und massivem Erguss elektrischer Alternans.


Akute Perikarditis mit Herztamponade. Low-voltage und P-pulmonale (durch Drucksteigerung im rechten Vorhof)
1.11.4.10.7 Echokardiographie
Ergüsse stellen sich als relativ echofreie Zonen dar. Der Ergusssaum erlaubt Rückschlüsse auf das Ergussvolumen. Bei großen Ergüssen bzw. großer Tamponade „schwingendes Herz“ mit schnellen unzureichenden Kontraktionen. Kompression des rechten Vorhofs und rechten Ventrikels. Inspiratorische Zunahme der Flussgeschwindigkeit in der A. pulmonalis, verbunden mit abnehmender Flussgeschwindigkeit in der Aorta.
1.11.4.10.8 Röntgenuntersuchung des Thorax
Verbreiterung der Herzsilhouette bei Ergüssen ab 250 ml (Boxbeutelform).
1.11.4.10.9 Untersuchung des Herzbeutelpunktats
Bakteriologisch und zytologisch.
1.11.4.10.10 Therapie
Subxiphoidale Perikardpunktion, wenn nötig Drainage über einige Tage. Nicht ungefährlicher Eingriff, der nach Lokalanästhesie möglichst unter Durchleuchtungskontrolle, alternativ unter echokardiographischer Führung streng aseptisch durchgeführt werden sollte. Bei rezidivierendem Perikarderguss kommt eine operative Perikardfensterung in Betracht.
1.11.4.11 Chronische konstriktive Perikarditis
1.11.4.11.1 Ätiologie
Früher dominierte die Tuberkulose als Krankheitsursache. Heute bleiben die meisten Fälle ungeklärt. Eine initiale akute Perikarditis ist oft nicht nachzuweisen. Akute Perikarditiden jeder Ursache können aber vorausgehen.
1.11.4.11.2 Pathogenese
Behinderung der diastolischen Füllung des Herzens durch narbige Verdickung und Schrumpfung des Perikards, in 50 % der Fälle mit ausgedehnter Verkalkung. Die progrediente Zunahme des Füllungswiderstandes bewirkt einen venösen Rückstau im großen Kreislauf mit Ödembildung und einen Anstieg des enddiastolischen Füllungsdrucks unter Angleichung des Druckniveaus im rechten und linken Herzen. Bei hochgradiger Schrumpfung des Perikards werden die Herzkammern nur in der frühen Diastole gefüllt. Die Förderleitung des Herzens nimmt ab, die Stauung im großen Kreislauf zu.
1.11.4.11.3 Klinik
Im fortgeschrittenen Stadium präsentieren sich die Patienten mit hydropischer Herzinsuffizienz, Leber- und Milzvergrößerung, Aszites und Beinödemen. Auf den ersten Blick ähnelt das Krankheitsbild einer Leberzirrhose, zumal das Herz nicht verbreitert erscheint. Die ausgeprägte Halsvenenstauung und ein frühdiastolischer Extraton weisen auf die Herzerkrankung hin.
1.11.4.11.4 Diagnostik
1.11.4.11.5 EKG
Unspezifische Abflachung oder Negativierung der T-Wellen in den Brustwandableitungen. Infolge des erhöhten Vorhofdrucks manchmal Vorhofflimmern.
1.11.4.11.6 Echokardiographie
Typisch sind Perikardverdickung, Dilatation der Vorhöfe und der V. cava inferior, abrupter diastolischer Füllungsstop in den Kammern bei guter Kontraktilität, atemabhängige Änderungen der Flussgeschwindigkeit durch die Mitralklappe. Abgrenzung gegen restriktive Kardiomyopathie mit dem Tissue-Doppler: Bei Perikarditis normale, bei restriktiver Kardiomyopathie herabgesetzte Gewebegeschwindigkeit des linksventrikulären Myokards.
1.11.4.11.7 Röntgenuntersuchung des Thorax
Normale Herzgröße. Manchmal sind Kalkeinlagerungen im Perikard nachzuweisen.
1.11.4.11.8 MRT und CT
Lassen die Perikardverdickung eindeutig erkennen.
1.11.4.11.9 Herzkatheter
Typischer enddiastolischer Druckausgleich zwischen rechtem und linkem Ventrikel. Präoperativ Koronarangiographie zum Ausschluss einer Mitbeteiligung der Koronararterien.
1.11.4.11.10 Therapie
Die konservativen Möglichkeiten erschöpfen sich mit Entwässerung durch salzarme Kost und Diuretika. Eine Tuberkulose ist gezielt zu behandeln. Definitve Besserung ist nur durch die Perikardektomie zu erzielen (◘ Abb. 1.99). Dabei werden nach Längssternotomie beide Ventrikel und die A. pulmonalis freipräpariert (Dekortikation). Die hintere Schale verbleibt, da ihr Ablösen zu gefährlich ist. Letalität 3‒10 %. Postoperativ tritt im Laufe einiger Monate eine weitgehende Normalisierung der Herzfunktion ein. Mit dem Eingriff sollte nicht lange gewartet werden, weil die Verschwielung auf das Myokard übergreifen kann.


Perikardektomie. Röntgenbilder und Computertomogramm eines 58-jährigen Patienten mit Perikarditis calcarea vor und nach operativer Perikardektomie (Sammlung Dr. Langer, Bad Oeynhausen)
1.12 Erworbene Herzklappenfehler
Erworbene Herzklappenfehler
-
Mitralstenose und -insuffizienz
-
Mitralklappenprolapssyndrom
-
Aortenstenose und -insuffizienz
-
Trikuspidalstenose und -insuffizienz
-
Erworbene Pulmonalstenose und -insuffizienz
1.12.1 Mitralstenose
1.12.1.1 Ätiologie und Pathologie
Die Stenose der Mitralklappe entsteht fast immer als Komplikation des rheumatischen Fiebers, das manchmal auch ohne Gelenkmanifestationen verläuft. Anamnestisch geben etwa 60 % der Patienten eine Gelenkerkrankung an, 12 % eitrige Anginen und 3 % Scharlach. Die Stenosierung der Mitralklappe kommt durch Fusion der Kommissuren zustande der sich Verdickung und Verkalkung der Klappen und eine Verkürzung der Sehnenfäden anschließen kann (◘ Abb. 1.100). Der Stenosierungsprozess ist langsam progredient, teils durch rheumatische Rezidive, teils durch mechanische Läsionen nach initialer entzündlicher Schädigung. Intervall zwischen erstem rheumatischem Schub und signifikanter Mitralstenose 2‒10 Jahre.


Hochgradig stenosierte, stark kalzifizierte Mitralklappe
1.12.1.2 Pathophysiologie
Die Mitralstenose behindert die Füllung des linken Ventrikels. Am verengten Mitralostium besteht ein diastolischer Druckgradient zwischen linkem Vorhof und linkem Ventrikel. Der Druckgradient steigt mit dem Schweregrad der Stenose und mit dem Herzminutenvolumen, also bei körperlicher Anstrengung. Mitralöffnungsfläche normal 4–6 cm2, bei leichter Stenose >2,5 cm2, bei mittelschwerer 1,1–2,5 cm2, bei schwerer 1,0 cm2 und weniger.
-
Auswirkungen nach rückwärts: Blutstau und Druckanstieg im linken Vorhof (Hypertrophie → Dilatation → Vorhofflimmern → Zunahme der Dilatation, Tendenz zur Thrombenbildung). Passiver Rückstau in die Lungenvenen und -kapillaren (Stauungslunge mit Hämosiderose, auch Hämoptoe → Lungenödem), Adaptative Drucksteigerung in der A. pulmonalis und im rechten Ventrikel (passive pulmonale Hypertonie). In fortgeschrittenen Fällen kommt es durch intensive Konstriktion der Lungenarteriolen zu einer schweren reaktiven pulmonalen Hypertonie (Rechtshypertrophie → Dilatation des rechten Herzens → Rechtsinsuffizienz → sekundäre Trikuspidalinsuffizienz). Die pulmonale Vasokonstriktion (zweite Stenose) wirkt der Lungenstauung bei körperlicher Belastung entgegen, verhindert aber einen adäquaten Anstieg des Herzminutenvolumens. Trotz Hyalinbildung in den Arteriolen bildet sich die reaktive pumonale Hypertonie nach der Mitralkommissurotomie weitgehend zurück.
-
Auswirkungen nach vorwärts: Kompensation der Stenose durch verlängerten diastolischen Bluteinstrom in den linken Ventrikel. Bei Tachykardie ist dieser Kompensationsmechanismus gestört, die Lungenstauung entsprechend größer. Das Herzminutenvolumen nimmt sukzessive ab. Die Verkleinerung bewirkt Abnahme der linksventrikulären Muskelmasse, Schwindelgefühl bei Belastung und Hautblässe mit zyanotischem Einschlag wegen O2-Ausschöpfung des Kapillarblutes.
1.12.1.3 Klinik
Leichtere Grade der Mitralstenose sind asymptomatisch und können es über 20 Jahre und länger bleiben. Die Verschlimmerung erfolgt hauptsächlich durch rezidivierende entzündliche Schübe. Frühsymptom ist eine Belastungsdyspnoe aufgrund des Rückstaus in die Lunge mit Herabsetzung der Vitalkapazität. Allmählich kommt es zum Leistungsabfall und zur chronischen Stauungsbronchitis mit viel Husten und gelegentlich blutigem Sputum. Die Ruptur einer Bronchialvene kann zur Hämoptyse führen. Im Gesicht entwickelt sich auf blassem Grundton eine lividrote Verfärbung der Wangen mit Teleangiektasien („Mitralgesicht“). Die progrediente Dilatation des linken Vorhofs verursacht schließlich Vorhofflimmern. Halsvenenstauung, Leberschwellung und Ödeme zeigen im Spätstadium eine dekompensierte pulmonale Hypertonie an. Der Übergang von der NYHA-Klasse II in die Klassen III und IV vollzieht sich von Fall zu Fall mit unterschiedlicher Geschwindigkeit.
1.12.1.4 Diagnostik
1.12.1.5 Auskultationsbefund
Paukender 1. Herzton (Mitralklappe wird aus geöffneter Stellung zugeschlagen). Apikaler hochfrequenter Mitralöffnungston, 0,04–0,11 s nach der aortalen Komponente des 2. Herztones (durch Anspannung der an den Rändern verklebten Klappensegel). Präsystolisches Crescendogeräusch (Einstromgeräusch) während der Vorhofsystole, ein Frühsymptom, das bei Vorhofflimmern verschwindet. In schweren Fällen von pulmonaler Hypertonie Dilatation des Pulmonalklappenringes mit frühdiastolischem Pulmonalinsuffizienzgeräusch im 2.–3. ICR links parasternal (Graham-Steell-Geräusch) und relative Trikuspidalinsuffizienz mit holosystolischem Geräusch links parasternal, dessen Intensität (im Gegensatz zur Mitralinsuffizienz) inspiratorisch zunimmt.
1.12.1.6 EKG
Steil- bis Rechtslagetyp, P-mitrale, Vorhofextrasystolen, im Verlauf häufig Vorhofflimmern. Bei pulmonaler Hypertonie Zeichen der Rechtshypertrophie und Rechtsschädigung.
1.12.1.7 Röntgenaufnahme des Thorax
Steiler Abfall des linken Herzrandes, Herztaille durch linken Vorhof und prominenten Pulmonalbogen verstrichen (Mitralkonfiguration), Kernschatten durch vergrößerten linken Vorhof im Frontalbild. Im Seitenbild Einengung des Retrokardialraumes im oberen Anteil durch den linken Vorhof, Vorwölbung des rechten Ventrikels in den Retrosternalraum bei Rechtsinsuffizienz.
1.12.1.8 Echokardiographie mit Doppler
Erlaubt die sichere Diagnose der Mitralstenose, die Bestimmung des Schweregrades und die Beurteilung der Klappenmorphologie (Valvotomiefähigkeit).
-
M-Mode: Linker Vorhof vergrößert bzw. dilatiert. Klappensegel verdickt, eingeschränkt beweglich. DE-Amplitude verkleinert. EF-Slope abgeflacht. Multiple parallele Echos im Bereich der Klappensegel. Frühdiastolische Vorwärtsbewegung des hinteren Segels.
-
2-D-Echo und transösophageales Echo: Differenzierung zwischen Klappenverklebung und Klappenverkalkung, deren Ausmaß und Verteilung zu erfassen sind. Nachweis des diastolischen Vorwölbens (doming) und der fehlenden Separation der Segel. Bestimmung der Mitralöffnungsfläche (MÖF) in der kurzen Achse durch Umfahren der Öffnung im diastolischen Standbild (◘ Abb. 1.101). Beurteilung des subvalvulären Klappenapparates (Sehnenfäden, Papillarmuskel).
Abb. 1.101 
Echokardiographischer parasternaler Querschnitt bei einer 48-jährigen Patientin mit höhergradiger Mitralstenose. Links: verplumpte, sich wenig öffnende Mitralklappe (Öffnungsfläche 0,8 cm2). Rechts: nach katheterinterventioneller Valvotomie Zunahme der Öffnungsfläche auf 1,8 cm2 durch Wiedereröffnung der verklebten Suturen
-
Doppler:
-
Farbdoppler: Unterhalb der Mitralklappenebene hochturbulenter Fluss mit mosaikartigem Farbmuster
-
PW-Doppler: A-Welle bei Sinusrhythmus erhöht, hohe Flussgeschwindigkeit
-
CW-Doppler: Maximale und mittlere Flussgeschwindigkeit erhöht. Daraus errechnet sich der mittlere diastolische Druckgradient: gering <5 mmHg, mäßig 5–10 mmHg, beträchtlich 10–15 mmHg, schwer >15 mmHg. Aus der Druckhalbierungszeit (PHT) kann die Mitralöffnungsfläche berechnet werden.
-
-
Mitralklappen-Score (Wilkens): Bewertet 4 Parameter der Mitralklappe mit den Graden 1‒4:
-
Grad 1: Mobilität
-
Grad 2: subvalvuläre Dicke
-
Grad 3: Dicke der Klappensegel
-
Grad 4: Kalzifizierung
-


Der Mitralklappen-Score ist ein wichtiges Kriterium bei der Indikationsstellung zur mitralen Ballonvalvotomie.
1.12.1.9 Herzkatheter
Da die moderne Echokardiographie umfassende Informationen für die Beurteilung einer Mitralstenose liefert, wird der Herzkatheter nur bei besonderen Indikationen eingesetzt: Diskrepanz zwischen klinischen und echokardiographischen Befunden, chronische obstruktive Lungenerkrankung (welchen Anteil hat die Mitralstenose?), Verdacht auf Vorhofmyxom.
1.12.1.10 Koronarangiographie
Indikationen sind Angina pectoris, Risikofaktoren einer koronaren Herzkrankheit bei Männern >40 und Frauen >50 Jahre.
1.12.1.11 Therapie
1.12.1.12 Konservative Maßnahmen
In leichten asymptomatischen Fällen ggf. β-Blocker zur Senkung der Herzfrequenz. Bei Dyspnoe Diuretika, bei Vorhofflimmern Digitalis und Dauerantikoagulation (Marcumar). Im Frühstadium kann die elektrische Kardioversion nach Antikoagulation noch erfolgreich sein.
1.12.1.13 Perkutane Ballon-Valvotomie (BMV)
Schematische Darstellung der BMV in ◘ Abb. 1.102. Indiziert bei unkomplizierter Mitralstenose (an den Kommissuren verklebte Klappen ohne ausgedehnte Verkalkung und ohne starke Schrumpfung der Sehnenfäden). Der Wilkens-Score sollte ≤8 betragen, die Öffnungsfläche <1,6 cm2. Bei Vorhofflimmern genügen <2 cm2. Mortalität des Eingriffs 1–2 %.


Schema einer Mitralvalvotomie mittels Ballonkatheter (Inoue-Technik)
1.12.1.14 Operative Valvotomie /Mitralklappenersatz
Die geschlossene Valvotomie ist durch die BMV abgelöst worden. Die offene Valvotomie wird mit kardiopulmonalem Bypass durchgeführt (kaum noch). In Betracht kommt sie bei Patienten, deren Mitralklappe für eine BMV zu stark verzogen und zu sehr verkalkt ist. Korrigieren lassen sich damit nur leichte und mittelschwere Mitralstenosen. Ein Mitralklappenersatz ist indiziert bei Klappenöffnungsflächen <1,5 cm2 in den NYHA-Klassen III und IV und bei Klappenöffnungsflächen <1 cm2 in NYHA-Klasse II und bei schwerer pulmonaler Hypertonie (Operationsmortalität 2–10 %).
1.12.2 Mitralinsuffizienz
1.12.2.1 Definition
Schlussunfähigkeit der Mitralklappe mit Reflux in den linken Vorhof durch Veränderungen der Klappensegel, des Klappenringes und des Halteapparates mit Chordae und Papillarmuskeln.
1.12.2.2 Ätiologie und Pathologie
1.12.2.3 Rheumatische Mitralklappenerkrankung
Verursacht weniger als die Hälfte aller Fälle von reiner Mitralinsuffizienz. Entsteht durch Schrumpfung der Klappensegel, auch der Sehnenfäden.
1.12.2.4 Bakterielle Endokarditis
Entzündliche Klappenläsionen, auch Sehnenfadenabriss, der zum Flottieren führt, und Zerstörung des Anulus fibrosus durch Klappenringabszesse.
1.12.2.5 Myxomatöse Degeneration
In den USA dominierende Ursache. Abnorme Ansammlung saurer Mucopolysaccharide in der mittleren Schicht der Klappensegel und im Zentrum der Sehnenfäden, auch im Anulus fibrosus, als Folge eines gestörten Kollagenstoffwechsels. Dadurch ballonförmige Deformierung der Klappen, Verlängerung der Sehnenfäden und Erweiterung des Klappenringes. Zunächst nur systolischer Mitralklappenprolaps in den linken Vorhof (s. unten), dann auch Reflux, also Mitralinsuffizienz.
1.12.2.6 Koronare Herzkrankheit und Infarkt
Dysfunktion oder Ruptur der Papillarmuskeln (eines der sechs Köpfe mit Lösung des zugehörigen Sehnenfadens oder Abriss des ganzen Muskels) durch Ischämie, Fibrose oder Nekrose.
1.12.2.7 Linksventrikuläre Dysfunktion
Schlussunfähigkeit der Mitralklappe infolge Erweiterung des Klappenringes bei Dilatation des linken Ventrikels (z. B. kongestive Kardiomyopathie), durch Verlagerung der Papillarmuskeln bei hypertrophischer Kardiomyopathie.
1.12.2.8 Mitralklappenringverkalkung
Beeinträchtigt die systolische Verkleinerung des Mitralklappenringes durch zirkuläre Muskelkontraktion. Vorkommen: idiopathisch, Diabetes mellitus, Hypertonie, Marfan-Syndrom, Hyperparathyreoidismus. Insuffizienzgrad der Mitralklappe meistens gering.
1.12.2.9 Kongenitale Defekte
Spaltbildungen, vor allem im vorderen Segel, oft mit defektem AV-Kanal kombiniert.
1.12.2.10 Traumen
Segel- oder Papillarmuskelruptur bei stumpfen Brustkorbtraumen.
1.12.2.11 Vorhofmyxom
Schlussunfähigkeit der Klappe durch Tumorprolaps in den linken Ventrikel. Meistens zugleich Einstromhindernis.
1.12.2.12 Pathophysiologie
Der linke Ventrikel muss ein vergrößertes Schlagvolumen auswerfen, weil ein Teil davon durch die insuffiziente Mitralklappe zurückfließt. Nach der Größe der Regurgitationsfraktion unterscheidet man:
-
Grad I: geringe Insuffizienz (<15 %)
-
Grad II: mäßige Insuffizienz (15–30 %)
-
Grad III: mittlere Insuffizienz (30–50 %)
-
Grad IV: schwere Insuffizienz (>50 %)
Die Mehrförderung erreicht der linke Ventrikel bei nicht zu starkem Reflux allein durch eine Vergrößerung der Ejektionsfraktion. Auch wenn eine mäßige Dilatation erfolgt, hält sich die Mehrarbeit des linken Ventrikels in Grenzen, da der Reflux in ein Niederdruckgebiet erfolgt. Der linke Ventrikel ist unabhängig von der Refluxgröße als voll suffizient anzsehen, so lange sein endsystolischer Durchmesser den Wert von 40 mm nicht überschreitet und die Ejektionsfraktion >0,6 ist. Ein Anstieg über diesen Wert bedeutet, dass eine ernste myokardiale Schädigung eingetreten ist, die auch nach einem Klappenersatz fortbesteht. Der systolische Rückfluss führt zur Hypertrophie und Dilatation des linken Vorhofes, im Verlauf auch zum Vorhofflimmern. Der Rückstau kann sich in den Lungenkreislauf und den rechten Ventrikel fortpflanzen und eine Rechtsinsuffizienz mit relativer Trikuspidalinsuffizienz zur Folge haben.
1.12.2.13 Klinik
In den meisten Fällen schreitet die Mitralinsuffizienz langsam fort. Die ersten Symptome treten gewöhnlich erst nach vielen Jahren auf. Zu diesem Zeitpunkt ist der linke Ventrikel oft noch voll suffizient. Geklagt wird über Müdigkeit, Erschöpfung, Belastungsdyspnoe, später über Rhythmusstörungen, Stauungsbronchitis und Ödeme. Vorhofflimmern bewirkt wegen Wegfall der Vorhofkontraktion eine plötzliche Verschlimmerung.
1.12.2.14 Diagnostik
1.12.2.15 Auskultationsbefund
Leiser 1. Herzton (Schlussunfähigkeit verhindert deutlichen Klappenschlusston). Holosystolisches Refluxgeräusch an der Herzspitze mit Fortleitung in die Axilla. 3. Herzton (diastolischer Füllungston, infolge abrupten frühdiastolischen Bluteinstroms aus dem endsystolisch gestauten linken Vorhof).
1.12.2.16 EKG
P-mitrale (sinistroatriale) in V1, mit breitem negativem hinteren Abschnitt, häufig Vorhofflimmern. Linkslagetyp und Linkshypertrophie (vom Volumentyp), in 15 % der Fälle Rechtshypertrophie bzw. biventrikuläre Hypertrophie. Normalbefund in leichten Fällen.
1.12.2.17 Echokardiographie mit Doppler
Eindeutiger Refluxnachweis mit dem Farbdoppler im 4-Kammerblick. Die Größe der Jetfläche in Prozent der Vorhoffläche erlaubt eine Semiquantifizierung der Mitralinsuffizienz. Auch die Breite des Jets an seinem Eintritt in den Vorhof erlaubt eine Abschätzung des Schweregrades (<0,3 cm geringe Mitralinsuffizienz, >0,5 cm schwere Mitralinsuffizienz). Im M-Mode erhöhte Verkürzungsfraktion, linker Vorhof gering bis deutlich vergrößert. Im 2-D-Echo Erkennung der Ursache (Prolaps, Verkalkung, Stenosekomponente). CW-Doppler: Semiquantifizierung der Mitralinsuffizienz nach Signalintensität. Am wichtigsten ist die Bestimmung des endsystolischen Durchmessers des linken Ventrikels und seiner Ejektionsfraktion (im 2-D-Echo mit M-Mode-Zuschaltung) zur Beurteilung der systolischen Funktion.
1.12.2.18 Röntgenaufnahme des Thorax
Bei geringer Mitralinsuffizienz normal. Bei stärkerer Regurgitation im Frontalbild Linksverbreiterung mit verstrichener Herztaille; im Seitenbild Vergrößerung des linken Vorhofes und des linken Ventrikels mit Einengung des gesamten Retrokardialraumes. Zeichen der Lungenstauung bei Linksinsuffizienz.
1.12.2.19 Radionuklid-Ventrikulographie
Zur Beurteilung des Schweregrades der Mitralinsuffizienz wird die Änderung der Ejektionsfraktion unter ergometrischer Belastung bestimmt. Ein ausbleibende Anstieg (normal 5–10 %) zeigt eine beginnende Erschöpfung der myokardialen Adaptation an.
1.12.2.20 Linksventrikuläre Angiokardiographie
Initiale Messung des enddiastolischen Ventrikeldrucks. Darstellung der Mitralklappe nach intraventrikulärer Kontrastmittelinjektion, Abschätzung des Regurgitationsvolumens nach der Anfärbung des linken Vorhofes. Bestimmung der Regurgitationsfraktion aus der Messung des Gesamtschlagvolumens und dem mit anderer Methode (z. B. Fick-Prinzip) ermittelten Vorwärtsschlagvolumens.
1.12.2.21 Magnetresonanztomographie
Ermöglicht akkurate Messungen des Rückflusses in den linken Vorhof sowie des enddiastolischen und endsystolischen Ventrikelvolumens.
1.12.2.22 Konservative Therapie
1.12.2.23 Indikationen
Schwere chronische Mitralinsuffizienz, NYHA I, ESVI <40 ml/m2, ESD <40 mm, EF >70 %. Bei einer Mitralinsuffizienz ab Grad II Nachlastsenkung und Einschränkung der körperlichen Belastung (keinen Leistungssport). Bei asymptomatischer schwerer chronischer Mitralinsuffizienz mit noch normaler Ventrikelfunktion Nachlastsenkung mit ACE-Blockern oder Angiotensin-II-Rezeptor-Antagonisten. Wenn in diesen Fällen Vorhofflimmern besteht oder die Grenzwerte der normalen Ventrikelfunktion unterschritten sind (Ejektionsfraktion ≤0,60, endsystolischer Durchmesser ≥45 mm), besteht Operationsindikation. Diese ist auch bei allen symptomatischen Patienten gegeben, sofern die Ejektionsfraktion nicht unter 0,30 liegt. Solche schwersten Fälle werden durch die Operation nicht gebessert. Bei dilatativer Kardiomyopathie kann eine relative Mitralinsuffizienz durch die medikamentöse Behandlung, biventrikuläre Schrittmacherstimulation und bei Grad III auch durch Operation gebessert werden. Bei Mitralinsuffizienz durch Myokardischämie ist durch Revaskularisierung eine Besserung erreichbar.
1.12.2.24 Operative Therapie
1.12.2.25 Indikationen
Schwere chronische Mitralinsuffizienz, NYHA II, endsystolisches Volumen >50 ml/m2, endsystolischer Durchmesser >45 mm, pathologischer Belastungstest (ΔEF<10 %), ggf. neu aufgetretenes Vorhofflimmern.
1.12.2.26 Mitralklappenplastik
Eine erfolgreiche Reparatur der Mitralklappe ist hämodynamisch und für die Ventrikelfunktion vorteilhafter als ein Klappenersatz und macht eine Dauerantikoagulation überflüssig. Wenn technisch möglich, sollte sie durch Exzision pathologischer Anteile, Anheften abgerissener Chordae oder Annulus-Raffen (evtl. mit Metallring nach Carpentier) versucht werden. Operationsletalität 2–4 %.
1.12.2.27 Klappenersatz
Mit Kunstoff- oder Bioprothese (Schweineklappen mit oder ohne Bügel). Operationsletalität in Deutschland 4,5 %. Mechanische Prothesen sind lebenslang haltbar, biologische Klappen nur etwa 15 Jahre. Erstere erfordern eine Dauerantikoagulation mit Phenprocoumon, letztere nur eine Antikoagulation für die ersten 3 Monate.
1.12.2.28 Prognose
Nach erfolgreicher Operation ist die Prognose sehr gut, wenn rechtzeitig operiert wurde. Wird bei gegebener Indikation nicht operiert, beträgt die Mortalität pro Jahr 5 %. Todesursachen sind hauptsächlich Herzinsuffizienz und plötzliches Kammerflimmern.
1.12.3 Mitralklappenprolapssyndrom
1.12.3.1 Definition
Systolisches Vorwölben eines oder beider Mitralklappensegel, das zu Schmerzen und Rhythmusstörungen führen kann.
1.12.3.2 Ätiologie und Pathologie
1.12.3.3 Primärer Mitralklappenprolaps
Idiopathisch mit hereditärer Komponente, bei sonst normalem Herzen. In den meisten Fällen symptomlos. Abgrenzung gegen Normvarianten schwierig. Häufigkeitsangaben (bei jungen Frauen bis 10 %) daher noch unsicher. Meistens myxomatöse Degeneration bzw. Proliferation mit Vergrößerung der Klappensegel und Verlängerung der Sehnenfäden. Übergänge in ausgeprägte Mitralklappeninsuffizienz.
1.12.3.4 Sekundärer Mitralklappenprolaps
Am häufigsten bei koronarer Herzkrankheit nach Infarkt durch Papillarmuskelschädigung, selten bei rheumatischer Klappenaffektion, bei Marfan-Syndrom in 90 % der Fälle.
1.12.3.5 Pathophysiologie
Vergrößerung der Klappenfläche, Verlängerung der Sehnenfäden oder Papillarmuskelschwäche führen in der zweiten Hälfte der Systole zur ballonförmigen Vorwölbung der Segel in den linken Vorhof, häufig auch zum Auseinanderweichen der Klappenränder mit spätsystolischem, hämodynamisch unbedeutendem Reflux in den linken Vorhof. Verkleinerung des Ventrikelvolumens (Stehen, Inspiration, Valsalva-Manöver) verstärkt den Prolaps. Durch Abriss von Sehnenfäden und Fortschreiten der Klappendegeneration kann sich eine schwere Mitralinsuffizienz entwickeln. Die Entstehung der Herzschmerzen (reflektorische Koronarspasmen?) und die Tendenz zu Rhythmusstörungen (Zerrung der Papillarmuskeln bzw. der Muskelfasern an den Segeln?) beim Mitralklappenprolaps sind nicht geklärt.
1.12.3.6 Klinik
Charakteristisch sind belastungsunabhängige stechende Schmerzen im Bereich der Herzspitze, die bei jungen Patienten oft bagatellisiert werden und in vielen Fällen ausbleiben. Die Diagnose ist daher oft ein Zufallsbefund. Häufige Begleiterscheinung sind ventrikuläre und supraventrikuläre Extrasystolen. Seltener kommen suprventrikuläre Tachykardien sowie Vorhofflattern und -flimmern vor, ganz selten treten Kammertachykardien und -flimmern auf.
1.12.3.7 Diagnostik
1.12.3.8 Auskultationsbefund
Mesosystolischer Klick, 0,14 s nach dem 1. Herzton. Bei systolischem Reflux spätsystolisches Crescendogeräusch über die Spitze mit Fortleitung in die Axilla. Beide Phänomene werden im Stehen lauter.
1.12.3.9 Echokardiographie und Doppler
2-D-Echo: Prolapsnachweis in parasternaler und apikaler langer Achse. Mitralsegel evtl. verdickt (◘ Abb. 1.103). Bei signifikanter Mitralinsuffizienz Vergrößerung des linken Vorhofes. M-Mode: In unterschiedlichen Phasen der Systole muldenförmige dorsale Auslenkung einer oder beider Segel. Farbdoppler: Darstellung des systolischen Refluxes. Refluxsignal auch im CW-Doppler. Verlaufkontrolle.


Echokardiographischer 4-Kammerblick einer 42-jährigen Patientin mit Prolaps des hinteren Mitralsegels (links) und assoziierter mittelgradiger Mitralinsuffizienz (rechts sind im Farbdoppler 2 Regurgitationsjets zu erkennen)
1.12.3.10 EKG
Bei asymptomatischen Patienten gewöhnlich normal. Einige asymptomatische und viele symptomatische Patienten haben biphasische T-Zacken und unspezifische Veränderungen der ST-Strecken in den Ableitungen II, aVF und III, mitunter auch in den anterolateralen Brustwandableitungen.
1.12.3.11 Therapie
Bei systolischem Reflux Endokarditisprophylaxe. In symptomatischen Fällen bessern β-Rezeptorenblocker Schmerzen und Rhythmusstörungen. Bei starkem Reflux Behandlungsstrategie wie bei Mitralinsuffizienz.
1.12.4 Aortenstenose
1.12.4.1 Definition
Valvuläre, subvalvuläre oder supravalvuläre Stenose des Aortenostiums.
1.12.4.2 Ätiologie und Pathologie
1.12.4.3 Rheumatische Aortenklappenstenose
Verkleinerung der Klappenöffnung durch Verschmelzung der Klappensegel an den Kommissuren nach rezidivierender rheumatischer Valvulitis. Dazu Verdickung der Klappen durch Fibrosierung und Verkalkung an beiden Oberflächen. Durch narbige Schrumpfung oft Insuffizienzkomponente. Indizien für die rheumatische Ursache: Rheumatisches Fieber, oder gehäufte Anginen in der Anamnese und Mitbefall der Mitralklappe.
1.12.4.4 Degenerative (senile) kalzifizierende Aortenklappenstenose
Behinderung der Klappenöffnung durch massive Kalkablagerungen, beginnend an der Basis der Klappensegel ohne Verschmelzung der Kommissuren. Bei kongenitalen bikuspidalen Klappen infolge abnormer mechanischer Beanspruchung schon im mittleren, bei trikuspidalen Klappen erst im höheren Lebensalter. Häufigste Form der isolierten Aortenstenose (◘ Abb. 1.104).


Kalzifizierte hochgradig stenosierte Aortenklappe
1.12.4.5 Kongenitale Aortenklappenstenose
-
Valvuläre Stenose: Meistens Fusion und Verdickung zweier Taschenklappen. Bikuspidale Klappen als Anlagedefekt.
-
Supravalvuläre Stenose: Uhrglasförmige Einengung oberhalb der intakten Aortenklappe mit Verdickung aller Wandschichten.
-
Subvalvuläre Stenose: Fibröse Ringe 5–10 mm unterhalb der intakten Klappenebene. In ungünstigen Fällen bleistiftdünne Verengung und Fibrosierung der ganzen linksventrikulären Ausflussbahn.
Einige dieser Defekte manifestieren sich klinisch erst im Erwachsenenalter.
1.12.4.6 Pathophysiologie
Ein signifikanter Austreibungswiderstand für den linken Ventrikel entsteht erst bei Verkleinerung der normal ca. 3,0 cm2 großen Klappenöffnungsfläche auf die Hälfte. Er wird durch eine Drucksteigerung im linken Ventrikel kompensiert, die zu einem Druckgradienten an der Klappe führt. Infolge der Klappenstenose geht die laminare in eine turbulente Blutströmung über und erzeugt dadurch ein systolisches Austreibungsgeräusch. Der Druckanstieg in der Aorta verzögert sich. Bei körperlicher Belastung nimmt der Druckgradient an der stenosierten Klappe mit dem Quadrat des Blutflusses zu. Im Stadium der Linksinsuffizienz fällt mit dem systolischen Ventrikeldruck auch der Druckgradient ab und täuscht dann einen geringeren als den tatsächlichen Stenosierungsgrad vor. Daher wird für den Schweregrad der Aortenstenose nicht der Druckgradient an der Klappe, sondern die Klappenöffnungsfläche herangezogen. Eine Aortenöffnungsfläche (AÖF) von <1 cm2 bzw, AÖF <0,6 cm2/m2 zeigt eine schwere Aortenstenose an.
Die chronische Druckbelastung induziert eine konzentrische Hypertrophie des linken Ventrikels. Sie bewirkt, dass sich die erhöhte Nachlast auf eine entsprechend erhöhte Zahl vom Muskelfasern verteilt. Auch der linke Vorhof hypertrophiert, da die Compliance des linken Ventrikels mit zunehmender Muskelmasse abnimmt. Trotzdem kommt es infolge der erschwerten Kammerfüllung zu einer diastolischen Funktionsstörung mit Rückstau in den kleinen Kreislauf. Man darf sie nicht mit einer Linksherzinsuffizienz verwechseln, denn in diesem Stadium ist die Kontraktilität des linken Ventrikels meistens noch gut.
Dank der Linkshypertrophie bleibt die Aortenstenose lange Zeit kompensiert. Zur Dekompensation führt eine Abnahme der Kontraktilität des hypertrophierten Myokards durch Veränderungen der Ultrastruktur und Vermehrung des interstitiellen Bindegewebes. Hinzu kommt eine ischämische Myokardschädigung, weil der erhöhte Sauerstoffbedarf (Hypertrophie, Druckarbeit) infolge ungenügender koronarer Perfusion (niedriger Blutdruck, Kompression der Innenschicht) zuletzt nicht mehr gedeckt wird. Diastolische und systolische Funktion verschlechtern sich abrupt, wenn durch das Hinzutreten eines Vorhofflimmerns, die Vorhofkontraktion wegfällt.
1.12.4.7 Klinik
Schwindelgefühl bis zu Synkopen während oder unmittelbar nach körperlicher Belastung, wenn die Förderleistung des Herzens durch die stenosierte Aortenklappe dem erhöhten Bedarf nicht mehr genügt und der Blutduck plötzlich abfällt. Angina pectoris bei Anstrengungen durch Ischämie in der komprimierten Innenschicht des linken Ventrikels. Belastungsdyspnoe durch die diastolische Funktionsstörung des linken Ventrikels, verstärkt im Stadium der Dekompensation, die relativ schnell auch zur Insuffizienz des nicht angepaßten rechten Ventrikels führt.
1.12.4.8 Diagnostik
1.12.4.9 Auskultationsbefund
-
Lautes, raues, spindelförmiges systolisches Austreibungsgeräusch mit Punctum maximum am Erb-Punkt und im 2. ICR rechts mit Ausstrahlung in beide Karotiden und zur Herzspitze. Je schwerer die Stenose, desto später in der Systole liegt das Maximum des Geräusches. Bei schwerer Linksinsuffizienz wird es leise.
-
Aortenöffnungston (Ejektionklick 0,06 s nach dem 1. Herzton), so lange die Klappe noch beweglich ist.
-
2. Herzton im 2. ICR rechts abgeschwächt.
-
Vorhofton (4. Herzton) über der Spitze infolge ruckartiger Dehnung des linken Ventrikels bei der Kontraktion des hypertrophierten linken Vorhofs.
1.12.4.10 Palpationsbefund
Hebender und verbreiterter Herzspitzenstoß. Pulsus parvus und tardus bei niedrigem Blutdruck. Sicherung der Diagnose durch Karotispulsschreibung: Hahnenkammkurve infolge trägen systolischen Druckanstiegs in der Aorta.
1.12.4.11 EKG
Zeichen der Linkshypertrophie und Linksschädigung: Sokolow-Index über 3,5 mV, ST-Senkung und biphasisches T in den Ableitungen V3 bis V6. Zeichen der Hypertrophie und Dilatation des linken Vorhofes. P in V1 mit später Negativität. Durch übergreifende Verkalkung auf das Reizleitungssystem relativ häufig atrioventrikuläre und intraventrikuläre Blockierungen. Linksanteriorer Hemiblock dominiert.
1.12.4.12 Echokardiographie mit Doppler
M-Mode und 2-D-Echo (◘ Abb. 1.105): Verdickte, echodichte Klappensegel mit eingeschränkter bis aufgehobener Separation. Hypertrophie des linken Ventrikels. Poststenotische Dilatation der Aortenwurzel, durch Planimetrie der Öffnungsfläche quantitativ zu erfassen. Linker Vorhof evtl. vergrößert. Farbdoppler: Turbulenz in der Klappenebene. PW-Doppler Geschwindigkeitsmessung im linksventrikulären Ausflusstrakt. CW-Doppler: Erhöhte Flussgeschwindigkeit im Klappenlumen. Berechnung des Druckgradienten und der Aortenöffnungsfläche möglich.


Echokardiographie einer 72-jährigen Patientin mit schwerer kalzifizierter Aortenstenose. Links: parasternaler Längsschnitt mit geringer Separation der Aortenklappe (AK) u. konzentrisch hypertrophiertem linken Ventikel (IVS, PW). Mitte: parasternaler Querschnitt mit planimetrisch ermittelter Öffnungsfläche von 0,7 cm2. Rechts: CW-Doppler mit einem mittleren transaortalen Druckgradienten von 68 u. einem maximalen von 95 mmHg
1.12.4.13 Röntgenaufnahme des Thorax
Im asymptomatischen Stadium kaum verändert. Später Rundung des linken Ventrikelbogens, im Seitenbild Vorwölbung der linken Kammer in den Retrokardialraum. Im fortgeschrittenen Stadium „Aortenkonfiguration“ des Herzens (Holzschuhform).
1.12.4.14 Herzkatheter
Messung des transvalvulären maximalen und mittleren Druckgradienten, Ausschluss einer Aorteninsuffizienz und einer sekundären Mitralinsuffizienz. Rechtsherzeinschwemmkatheter zur Bestimmung der Druckwerte in kleinen Kreislauf und des Herzminutenvolumens. Koronarangiographie präoperativ zur Erfassung von Kranzgefäßstenosen.
1.12.4.15 Konservative Therapie
1.12.4.16 Indikationen
Solange auch bei schwerer chronischer AS NYHA I, normale linksventrikuläre Funktion und unauffälliger Belastungstest (ΔEF >5 %) bestehen.
1.12.4.17 Maßnahmen
Kein Therapiebedarf, Einschränkung der körperlichen Aktivität (kein Leistungssport). Endokarditisprohylaxe vor Eingriffen mit dem Risiko einer Bakteriämie. Bei Vorhofflimmern Frequenznormalisierung, Dauerantikoagulation, Operationsindikation überprüfen.
1.12.4.18 Kathetergeführter Aortenklappenersatz
Indiziert bei Patienten, die wegen hohen Alters oder schwerer Komorbiditäten eine Operationsletalität von >10 % hätten. Voraussetzung ist eine symptomatische degenerative Aortenklappenstenose mit Verkalkungen und eine Klappenringgröße zwischen 18 und 27 mm. Vor der Implantation wird die Stenose mit einem Ballonkatheter aufgedehnt. Gebräuchlich sind die selbstexpandierende Core Valve (mit Schweineperikardklappe) und das ballonexpandierende Edwards-SAPIEN-System (mit Rinderperikardklappe). Die Insertion der Klappe kann transfemoral oder transapikal erfolgen. Der letztere Weg erfordert eine anteriore Mikrothorakotomie und die Punktion der Herzspitze durch den Chirurgen mit anschließendem Nahtverschluss der Punktionsstelle. Die 30-Tage-Mortalität liegt durchschnittlich bei 10 %.
1.12.4.19 Operative Therapie
1.12.4.20 Indikationen
Schwere chronische AS, NYHA II, bei NYHA I mit folgenden Zusatzbefunden: pathologischem Belastungstest (IIa), + LV-Dysfunktion (IIa), + AÖF <0,6 cm2 (IIb), + ventrikuläre Tachykardien (IIb), + LV-Hypertrophie ≥15 mm (IIb).
1.12.4.21 Eingriff
Klappenersatz mit Kunststoff- oder Bioprothese (◘ Abb. 1.106), letztere bei alten Patienten. In 90 % der Fälle gute Langzeitergebnisse. Operationsletalität etwa 3 %.


Verschiedene Herzklappenprothesen. a Monokippscheibenprothese, b Doppelflügelprothese(mechanische Prothesen); c Bioprothese, d Stentless-Bioprothese
1.12.5 Aorteninsuffizienz
1.12.5.1 Definition
Schlussunfähigkeit der Aortenklappe mit diastolischem Rückfluss in den linken Ventrikel.
1.12.5.2 Ätiologie und Pathologie
1.12.5.3 Erkrankungen der Aortenklappe
-
Rheumatische Aorteninsuffizienz: Schlussunfähigkeit der Klappe infolge narbiger Schrumpfung nach Valvulitis bei rheumatischem Fieber.
-
Bakterielle Endokarditis: Schlussunfähigkeit durch Zerstörung oder Perforation der Segel, auch durch Vegetationen, die eine vollständige Apposition der Segel verhindern.
-
Seltene entzündliche Läsionen: Ausgelöst durch rheumatoide Arthritis, generalisierter Lupus erythematodes, Morbus Bechterew, Whipple-Krankheit.
-
Myxomatöse Degeneration: Schlussunfähigkeit der Klappe durch Segelprolaps (Marfan-Syndrom) und im höheren Alter. Bei bikuspidaler Klappe durch Überbeanspruchung schon in mittleren Lebensjahren.
-
Traumen: Segelabriss bei stumpfen Brustkorbtraumen.
1.12.5.4 Erkrankungen der Aortenwurzel
Schlussunfähigkeit der Aortenklappe infolge Dilatation der Aorta ascendens mit Erweiterung des Klappenringes bei:
-
Aortitiden: Syphilis, Morbus Bechterew, Reiter-Syndrom, Psoriasis, Colitis ulcerosa.
-
Aortendissektion: Deformierung des Aortenringes und Verschiebung einzelner oder aller Semilunarklappen ventrikelwärts (◘ Abb. 1.107)
Abb. 1.107a,b 
Echokardiographie eines 58-jährigen Patienten mit schwerer Aorteninsuffizienz auf der Basis eines nichtdisseziierten Aortenaneurysmas. a Parasternaler Längsschnitt mit Ektasie der Aorta ascendens (79 mm) und dilatiertem linken Ventrikel, b parasternaler Querschnitt in Höhe der Aortenklappe mit großer enddiastolischer Regurgitationsfläche, c FD-Doppler mit breitem transaortalem Rückstromsignal in den linken Ventrikel (AO = Aorta, LV = linker Ventrikel, LA = linker Vorhof, AML = vorderes Mitralsegel, PML = hinteres Mitralsegel, AK = Aortenklappe, RVOT = rechtsventrikulärer Ausflusstrakt, RCC = rechtskoronare Semilunarklappe, ACC = akoronare Semilunarklappe, LCC = linkskoronare Semilunarklappe)
Abb. 1.108 
Echokardiographischer 4-Kammerblick einer 67-jährigen Patienten mit höhergradiger Trikuspidalinsuffizienz. a Vergrößerter rechter Vorhof (RA) und Schlussunfähigkeit der Trikuspidalklappe (TK), b FD-Doppler mit deutlicher transtrikuspidaler Regurgitation in den rechten Vorhof, c CW-Doppler mit Bestimmung der maximalen Regurgitationsgeschwindigkeit und Abschätzung des systolischen Pulmonalarteriendrucks (ca. 35 mmHg)
-
Zystische Medianekrose: Idiopathisch, Marfan-Syndrom




1.12.5.5 Pathophysiologie
1.12.5.6 Akute Klappeninsuffizienz
Vorkommen bei Aortendissektion, traumatischem Klappenabriss und infektiöser Endokarditis. Diastolischer Rückfluss in den nicht angepassten linken Ventrikel (Regurgitationsfraktion 50 %) → geringer Anstieg des enddiastolischen Volumens, aber starker Anstieg des enddiastolischen Drucks im linken Ventrikel mit Rückstau in den Lungenkreislauf → Anstieg des gesamtem, aber Abfall des Vorwärts-Schlagvolumens des linken Ventrikels → geringer Anstieg des systolischen und deutlicher Abfall des diastolischen Blutdrucks → drohender Kreislaufschock.
1.12.5.7 Chronische Klappeninsuffizienz
Vorkommen bei rheumatischer Valvulits und myxomatöser Klappendegeneration. Allmählich zunehmender diastolischer Rückfluss → exzentrische Hypertrophie des linken Ventrikels (durch Verlängerung der Muskelfasern ohne Zunahme der Wanddicke) → kompensierende Zunahme des enddiastolischen Volumens bei nur minimalem Anstieg des enddiastolischen Drucks → erhebliche Zunahme des gesamten bei normalem Vorwärtsschlagvolumen und normalem endsystolischem Ventrikelvolumen → starker Anstieg des systolischen und Abfall des diastolischen Blutdrucks (Pulsus celer et altus). Das sind die Kennzeichen des Kompensationsstadiums. Im Verlauf kommt es durch Druck- plus Volumenbelastung zum Kontraktilitätsverlust des hypertrophierten Myokards (Degeneration, Fibrose, Ischämie) und damit zur Dekompensation: Verkleinerung der Ejektionsfraktion → Anstieg des endsystolischen und enddiastolischen Ventrikelvolumens mit Linksdilatation → Lungenstauung → Insuffizienz des nicht angepassten rechten Ventrikels mit peripheren Ödemen.
1.12.5.8 Klinik
Im chronischen kompensierten Stadium, das viele Jahre dauern kann, sind die Patienten beschwerdefrei und genügend leistungsfähig. Bei beginnender Dekompensation treten Belastungsdyspnoe, Stenokardien und Erschöpfung auf, schließlich paroxysmale nächtliche Dyspnoe und Ödeme. Diesen Symptomen kann schnell das terminale Herzversagen folgen.
1.12.5.9 Diagnostik
1.12.5.10 Auskultationsbefund
-
Protodiastolisches Decrescendogeräusch: Unmittelbar nach dem 2. Herzton, hochfrequent, mit Punktum maximum im 3. ICR links. In leichten Fällen kurz, in schweren während der ganzen Diastole, in schwersten nur bis zur Mitte der Diastole hörbar. Lautstärke nicht mit dem Schweregrad korrelierend.
-
Systolisches Volumenaustreibungsgeräusch: An gleicher Stelle infolge relativer Aortenstenose bei dem vergrößerten Schlagvolumen. Leiser oder fehlender Aortenklappenschlusston.
-
Austin-Flint-Geräusch: Raues, vom Aortenton abgesetztes diastolisches Decrescendogeräusch über der Spitze (ähnlich wie bei Mitralstenose), nur bei schwerer Aorteninsuffizienz durch vorzeitigen partiellen Mitralklappenschluss, den der Rückstrom aus der Aorta verursacht.
1.12.5.11 Palpationsbefund
Herzspitzenstoß verbreitert und nach links verlagert. Sichtbare Pulsation des Brustkorbs und der Halsschlagadern, Kapillarpuls im Nagelbett. Pulsus celer et altus. Vergrößerung der Blutdruckamplitude bei normalem Mitteldruck.
1.12.5.12 EKG
Linkslagetyp und Zeichen der Linkshypertrophie (erhöhter Sokolow-Index), zunächst ohne Linksschädigungszeichen (ST-Senkung, biphasisches T in V3 bis V6), die auch im Verlauf weniger ausgeprägt sind als bei der Aortenstenose. Im Spätstadium Schenkelblockbilder, nicht selten auch Vorhofflimmern.
1.12.5.13 Echokardiographie und Doppler (◘ Abb. 1.107)
Nachweis der Aorteninsuffizienz mit dem Farbdoppler, der den diastolischen Reflux als mosaikfarbenen Jet im Klappenbereich und unterhalb der Aortenklappe erkennen lässt. Je breiter der Farbjet in der Klappenebene, desto höher der Insuffizienzgrad. Eine semiquantitative Bestimmung des Insuffizienzgrades gelingt mit dem CW-Doppler: Durch Messung der maximalen Rückflussgeschwindigkeit, des Geschwindigkeitsslopes und der Druckhalbierungszeit über der Aortenklappe während der Diastole. Je steiler der Slope und je kürzer die Druckhalbierungszeit desto schwerer die Aorteninsuffizienz. Wichtig sind die Insuffizienzzeichen des linken Ventrikels im M-Mode und 2-D-Echo: endsystolischer Diameter >55 mm, enddiastolischer Diameter >75 mm, eine Verkürzungsfraktion (FS) <25 % und eine Ejektionsfraktion <0,5.
1.12.5.14 Herzkatheter
Präoperativ indiziert, wenn nach echokardiographischer Untersuchung am Schweregrad noch Zweifel bestehen. Man appliziert einen Kontrastmittelbolus in die Aorta ascendens und schätzt das in den Ventrikelbereich abfließende Kontrastmittelvolumen ab. Bei Älteren ist präoperativ die Koronarangiographie angezeigt.
1.12.5.15 Röntgenaufnahme des Thorax
Linksverbreiterung des Herzens, Einengung des Retrokardialraumes, Elongation der Aorta mit betontem Aortenknopf.
1.12.5.16 Konservative Therapie
1.12.5.17 Indikationen
Bis zu schwerer chronischer AI mit NYHA I, ESDI <25 mm/m2, EF ≥55 %, unauffälligem Belastungstest.
1.12.5.18 Maßnahmen
Nachlastsenkung, bevorzugt mit ACE-Blockern oder AT1-Antagonisten. Keine bradykardisierenden Mittel, um die Diastolendauer nicht zu verlängern.
1.12.5.19 Operative Therapie
1.12.5.20 Indikationen
Schwere chronische Aorteninsuffizienz, NYHA II, ESDI >25 mm/m2, ESD >50 mm, EDD >70 mm, pathologischer Belastungstest (ΔEF <5 %), Ektasie der Aorta aszendens ≥55 mm.
1.12.5.21 Klappenersatz
Durch eine Kunststoff- oder Bioprothese ist bei einem endsystolischen Durchmesser von 50 mm und einer FS <25 % indiziert. Schon bei einem endostolischen Durchmesser von 55 mm bleibt die Ventrikeldysfunktion postoperativ vermindert. Die Operationsindikation ist unabhängig davon, ob Beschwerden bestehen oder nicht. Häufig sind die Patienten in der NYHA-Klasse II. Operationsletalität um 3 %. Nichtoperierte Patienten haben nach erstmaliger Linksinsuffizienz eine Lebenserwartung von 1–2 Jahren. Postoperativ Dauerantikoagulation, bei Bio-Klappen nur antithrombotische Therapie (100 mg ASS).
1.12.6 Trikuspidalstenose
1.12.6.1 Definition
Verengung der Trikuspidalklappe mit Rückstau vor dem rechten Ventrikel.
1.12.6.2 Ätiologie und Pathologie
In den meisten Fällen durch rheumatisches Fieber verursacht, häufig mit einer Mitralstenose kombiniert. Seltene Ursachen sind: Vorhofmyxom, Karzinoidsyndrom, eosinophile Endomyokarditis, Traumen durch Schrittmacherelektroden. Es kommt zur Klappenfibrosierung und Fusion von zwei oder drei Kommissuren.
1.12.6.3 Pathophysiologie
Kriterium für den Schweregrad der Stenose ist der Druckgradient an der Trikuspidalklappe: <3 mmHg (gering), 3–5 mmHg (mäßig), >5 mmHg (schwer). Die Kontraktionswelle des rechten Vorhofes pflanzt sich in die V. jugularis interna fort und kann die Höhe der Ventrikeldruckkurve erreichen. Der Anstieg des mittleren Vorhofdrucks führt zur Erhöhung des systemischen Venendrucks mit Dilatation der V. jugularis, Leberstauung, Aszites und Beinödemen. Inspiratorisch nimmt der Druckgradient zu, exspiratorisch ab. Das Herzminutenvolumen ist schon in Ruhe herabgesetzt und steigt bei Belastung nicht adäquat an. Bei begleitender Mitralstenose bleiben pulmonale Stauungszeichen aus.
1.12.6.4 Klinik
In leichten Fällen keine Beschwerden. Bei mittleren und höheren Stenosegraden Müdigkeit, Leistungsabfall, Zeichen der isolierten Rechtsinsuffizienz ohne Lungenstauung.
1.12.6.5 Diagnostik
1.12.6.6 Auskultationsbefund
Oft durch den Befund einer gleichzeitig vorliegenden Mitralstenose überlagert. Öffnungston und diastolisches Einstromgeräusch am unteren linken Sternalrand, letzteres bei Inspiration zunehmend. Punctum maximum der analogen Phänomene der Mitralstenose dagegen über der Spitze und ohne atemabhängige Intensitätsschwankungen.
1.12.6.7 EKG
Meistens Sinusrhythmus. Hohe P-Zacke mit nach rechts gerichtetem Vektor bei fehlenden Hypertrophiezeichen des rechten Ventrikels.
1.12.6.8 Echokardiographie mit Doppler
Anlotung im 4- und rechten 2-Kammerblick. Klappensegel verdickt, verkalkt, bewegungseingeschränkt. Rechter Vorhof erweitert. Mit dem CW-Doppler ist der Druckgradient zu bestimmen.
1.12.6.9 Röntgenaufnahme des Thorax
Vergrößerung des Herzschattens durch Vorwölbung des rechten Herzrandes.
1.12.6.10 Rechtsherzkatheter
Direkte Bestimmung des Druckgradienten über der Trikuspidalklappe.
1.12.6.11 Therapie
1.12.6.12 Konservative Therapie
Diuretika zur Beseitigung der Ödeme, auch präoperativ.
1.12.6.13 Operative Therapie
Indiziert bei einem Druckgradienten über 5 mmHg und einer Öffnungsfläche unter 2 cm2. Primär ist eine Ballonvalvuloplastie indiziert. Bei ungenügendem Effekt kommt die offene Valvuloplastie mit Umwandlung der trikuspidalen in eine funktionell bikuspidale Klappe in Betracht. Das letzte Mittel ist der Klappenersatz, bevorzugt mit einer Bioprothese.
1.12.7 Trikuspidalinsuffizienz
1.12.7.1 Definition
Schlussunfähigkeit der Trikuspidalklappe mit Rückfluss in den rechten Vorhof.
1.12.7.2 Ätiologie und Pathologie
1.12.7.3 Relative Trikuspidalinsuffizienz
Weitaus häufigste Form. Entsteht durch Erweiterung des Klappenringes bei Dilatation des rechten Ventrikels aus folgenden Ursachen: Mitralklappenfehler, pulmonale Hypertonie und Cor pulmonale, Pulmonalstenose, Infarkt des rechten Ventrikels, Myokarditis, Kardiomyopathie, Thyreotoxikose. Der Klappenapparat bleibt intakt. Reversibilität bei Rückbildung der Rechtsherzdilatation.
1.12.7.4 Organische Trikuspidalinsuffizienz
Durch rheumatisches Fieber (meistens mit Trikuspidalstenose kombiniert), infektiöse Endokarditis (bevorzugt bei Drogenabhängigen), myxomatöse Degeneration, Karzinoidsyndrom, Endomyokardfibrose, Traumen. Entzündungsprozesse führen zur Verdickung, Verkürzung und Versteifung der Segel ohne signifikante Fusion der Kommissuren.
1.12.7.5 Pathophysiologie
Systolischer Rückfluss durch die schlussunfähige Trikuspidalklappe in den rechten Vorhof. Zunahme der Pendelblutmenge mit dem Schweregrad der Klappeninsuffizienz und der Höhe des systolischen Drucks im rechten Ventrikel. Bei drucküberlastetem rechten Ventrikel (pulmonale Hypertonie, Pulmonalstenose) deshalb schlechte, bei normalem rechtsventrikulären Druck relativ gute Toleranz der Trikuspidalinsuffizienz. Anstieg des zentralen Venendrucks, Stauung der Halsvenen, Leberstauung, Aszites, Beinödeme.
1.12.7.6 Klinik
In leichten Fällen kaum Beschwerden. Stauungserscheinungen im großen Kreislauf entsprechend dem Schweregrad: Pulsationen am Hals, Druckgefühl im Oberbauch durch Leberschwellung, Rhythmusstörungen, vor allem Vorhofflimmern. Bei vorbestehender Lungenstauung führt eine relative Trikuspidalinsuffizienz zum Nachlassen der Dyspnoe und der pulmonalen Stauungszeichen, während das Herzminutenvolumen weiter absinkt.
1.12.7.7 Diagnostik
1.12.7.8 Auskultationsbefund
Holosystolisches Regurgitationsgeräusch und 3. Herzton am unteren linken Sternalrand. Intensitätszunahme des Geräusches während der Inspiration, die erst bei starker Dilatation des rechten Ventrikels fehlt (Schlagvolumen nicht mehr steigerungsfähig). Bei leichten Trikuspidalinsuffizienzen nur inspiratorisches Systolikum, das auf die erste Hälfte der Systole beschränkt bleibt.
1.12.7.9 Jugularvenenpuls
Ausgeprägte V-Welle (während der Ventrikelkontraktion) unter Verschwinden der systolischen Abwärtsbewegung (x‘).
1.12.7.10 EKG
R‘-Zacke in V1 (inkompletter Rechtsschenkelblock), bei pulmonaler Hypertonie P-pulmonale und Rechtshypertrophie. Häufig Vorhofflimmern.
1.12.7.11 Echokardiographie mit Doppler (◘ Abb. 1.108)
Anlotung im 4- und rechten 2-Kammerblick. Vergrößerung des rechten Vorhofes evtl. auch des rechten Ventrikels. Klappensegel unverändert oder verdickt, evtl. mit Auflagerungen (Endokarditis). Der Farbdoppler zeigt den systolischen Reflux an, dessen Größe aus der Ausdehnung der Jetfläche geschätzt werden kann. Mit dem CW-Doppler lässt sich der maximale systolische Pulmonlarteriendruck abschätzen. Im M-Mode paradoxe Speptumbewegung nach links, Dilatation des rechten Ventrikels und Rechtshypertrophie.
1.12.7.12 Röntgenaufnahme des Thorax
Im Frontalbild Verbreiterung des rechten Herzrandes durch Vergrößerung des rechten Vorhofes und Erweiterung der Pulmonalbogens. Im Seitenbild Einengung des Retrosternalraumes durch Vergrößerung des rechten Ventrikels.
1.12.7.13 Herzkatheter
Typische Druckkurve im rechten Vorhof. Systolischer Ventrikeldruck >60 mmHg spricht für relative Klappeninsuffizienz, ein Druck <40 mmHg für eine primäre Klappenläsion. Die rechtsventrikuläre Angiokardiographie lässt den Reflux erkennen. Etwas Kontrastmittel fließt aber am Katheter entlang zurück.
1.12.7.14 Therapie
1.12.7.15 Konservative Therapie
Diuretika gegen Ödeme.
1.12.7.16 Operative Therapie
Bei relativer Insuffizienz entbehrlich, wenn keine pulmonale Hypertonie vorliegt. Andernfalls Annuloplastie mit Carpentier-Ring. Bei organischer Insuffizienz Rekonstruktion oder Klappenersatz mit Bioprothese. Bei bakterieller Endokarditis gelegentlich Exzision der Klappe (wird gut toleriert), Klappenersatz erst nach Beseitigung der Infektion einige Monaten später.
1.12.8 Erworbene Pulmonalstenose
1.12.8.1 Ätiologie und Pathologie
Sehr seltene Manifestation des rheumatischen Fiebers, nur in großen Höhen wegen des dort erhöhten Pulmonalarteriendrucks häufiger vorkommend. Viele Fälle wurden bei malignem Karzinoidsyndrom mit Einengung des Klappenringes und Klappenfusion beobachtet. Manchmal werden rechtsventrikulärer Ausflusstrakt und Hauptstamm der A. pulmonalis durch mediastinale Tumoren komprimiert. Die große Mehrzahl der Pulmonalstenosen ist angeboren.
1.12.8.2 Pathophysiologie
Austreibungswiderstand für den rechten Ventrikel, der zur Hypertrophie und Rechtsherzinsuffizienz führen kann. Die erworbenen Fälle von Pulmonalstenose sind gewöhnlich weniger schwer als die angeborenen.
1.12.8.3 Klinik
Häufig nur Herzklopfen und Missempfindungen im Brustkorb, da der Druckgradient meistens niedrig ist. Bei Rechtshypertrophie Pulsation im Epigastrium.
1.12.8.4 Diagnostik
1.12.8.5 Auskultationsbefund
Lautes Austreibungsgeräusch im 2. ICR am linken Sternalrand.
1.12.8.6 EKG
P-pulmonale und Rechtshypertrophiezeichen (rSR‘ in V1).
1.12.8.7 Echokardiographie mit Doppler
M-Mode und 2-D-Echo: Rechtshypertrophie, auch Dilatation mit paradoxer Septumbewegung. In der parasternalen kurzen Achse verdickte, echodichte Klappensegel. Poststenotische Dilatation der A. pulmonalis. Farbdoppler: Turbulenter Stenosefluss. CW-Doppler: Erhöhte maximale Flussgeschwindigkeit, Berechnung des Druckgradienten.
1.12.8.8 Röntgenaufnahme des Thorax
Im Frontalbild Vorspringen des Pulmonalbogens, bei Dekompensation Vorwölbung des rechten Herzrandes infolge Vorhofdilatation. Im Seitenbild Einengung des Retrosternalraumes durch den hypertrophierten rechten Ventrikel.
1.12.8.9 Herzkatheter
Genaue Messung des Druckgradienten. Darstellung der Ausflussbahn und der Klappe mit Kontrastmittel.
1.12.8.10 Therapie
Primär Ballonvalvuloplastie (bei Gradient >50 mm Hg). Nur in schweren Fällen Klappenplastik oder Prothesenimplantation. Die Prognose ist bei Kindern besser als bei Erwachsenen.
1.12.9 Erworbene Pulmonalinsuffizienz
1.12.9.1 Ätiologie und Pathologie
1.12.9.2 Sekundäre Insuffizienz
Bei allen Formen der erworbenen pulmonalen Hypertonie durch Erweiterung des Klappen-rings.
1.12.9.3 Primäre Insuffizienz
Durch Klappenläsionen beim rheumatischen Fieber, infektiöser Endokarditis, rheumatoider Arthritis, Karzinoid, Traumen und Marfan-Syndrom.
1.12.9.4 Pathophysiologie
Bei primärer Klappenläsion Volumenüberlastung des rechten Ventrikels mit Dilatation und exzentrischer Hypertrophie.
Bei primärer pulmonaler Hypertonie besteht eine konzentrische Rechtshypertrophie. Das Regurgitationsvolumen ist gewöhnlich gering. Die Rechtsinsuffizienz resultiert überwiegend aus der erhöhten Druckbelastung.
1.12.9.5 Klinik
Bei pulmonaler Hypertonie Dyspnoe, Hämoptoe und Zeichen der Rechtsinsuffizienz. Bei primärer Klappeninsuffizienz wenig Beschwerden, in schweren Fällen Brustschmerz und Leistungsschwäche. Verstärkte epigastrische Pulsationen.
1.12.9.6 Diagnostik
1.12.9.7 Auskultationsbefund
Am oberen linken Sternalrand systolisches Volumenaustreibungsgeräusch mit frühem Maximum und kurzes diastolisches Regurgitationsgeräusch.
1.12.9.8 EKG
Bei vorbestehender pulmonaler Hypertonie Zeichen der Rechtshypertrophie. Bei primärer Klappeninsuffizienz Normalbefund oder inkompletter Rechtsschenkelblock.
1.12.9.9 Echokardiographie mit Doppler
-
2-D-Echo und M-Mode: Rechter Ventrikel dilatiert, evtl. hypertrophiert. Klappenring evtl. dilatiert.
-
Farbdoppler: Zeigt den Reflux an, die Jetgröße das Ausmaß der Insuffizienz.
-
CW-Doppler: Darstellung des Insuffizienzjets und semiquantitative Bestimmung des Schweregrades.
1.12.9.10 Röntgenaufnahme des Thorax
Prominenz des Pulmonalbogens, im Seitenbild Dilatation des rechten Ventrikels.
1.12.9.11 Herzkatheter
Druckkurve in der A. pulmonalis und im rechten Ventrikel gleichen sich an. Die enddiastolischen Drucke werden identisch.
1.12.9.12 Therapie
1.12.9.13 Konservative Therapie
Diuretika bei Ödemen. Behandlung der Grundkrankheit.
1.12.9.14 Operative Therapie
Nur in schweren Fällen von primären Klappenläsionen Klappenersatz.
1.13 Angeborene Herzfehler
Klassifizierung der angeborenen Herzfehler im Erwachsenenalter
-
Azyanotische Vitien mit Links-Rechts-Shunt
-
Vorhofseptumdefekt (ASD) und fehlmündende Lungenvenen
-
Atrioventrikularkanal
-
Ventrikelseptumdefekt (VSD)
-
Ductus Botalli apertus
-
-
Azyanotische Vitien ohne Shunt
-
Aortenklappenstenose
-
Aortenisthmusstenose
-
isolierte Pulmonalstenose
-
-
Zyanotische Vitien mit Rechts-Links-Shunt
-
Fallot-Tetralogie
-
Ebstein-Anomalie
-
Trikuspidalatresie
-
gemeinsamer Ventrikel
-
Truncus arteriosus communis
-
Transposition der großen Arterien
-
1.13.1 Allgemeines
1.13.1.1 Definition
Bei der Geburt vorhandene kardiovaskuläre Fehlbildungen, die sich sofort oder später klinisch manifestieren. Vorkommen bei 0,8 % aller Lebendgeborenen.
1.13.1.2 Ätiologie und Pathogenese
Die anatomischen Abnormitäten resultieren aus Störungen der embryonalen Herzentwicklung in den ersten 8 Wochen der Fetalzeit. Gleiche Fehlbildungen können unterschiedliche Ursachen haben. Durch chromosomale Aberrationen und Einzelgendefekte entstehen weniger als 10 % der Fälle. Ein-Gen-Mutationen liegen z. B. dem familiären Vorhofseptumdefekt und Ventrikelseptumdefekt zugrunde. Chromosomenanomalien mit multiplen Herzfehlbildungen sind z. B. die Trisomie und das Turner-Syndrom.
Die Mehrzahl der angeborenen Herzfehler ist auf multifaktorielle, vielfach ungeklärte Umwelteinflüsse zurückzuführen, zum Teil im Zusammenwirken mit disponierenden Genen. Gesicherte teratogene Faktoren von seiten der Mutter sind Rötelinfektion, Lupus erythematodes, Alkoholismus und teratogene Medikamente (Lithium, Retinol). Das Risiko für Eltern, nach einem Kind mit angeborenem Herzfehler ein zweites herzkrankes Kind zu bekommen, ist nur 2–10 %, so dass von einer neuen Schwangerschaft nicht abgeraten werden muss.
1.13.1.3 Häufigkeit
Auf 1000 Lebendgeburten ist durchschnittlich mit 8 angeborenen Herzfehlern zu rechen. In 25 % der Fälle liegen zusätzlich extrakardiale Anomalien vor. In diese Angaben sind bikuspidale Aortenklappe und Mitralklappenprolaps nicht einbezogen.
In ◘ Tab. 1.19 ist die Häufigkeitsverteilung aus einer größeren Sammelstatistik wiedergegeben.
1.13.1.4 Pathophysiologie
1.13.1.5 Vitien mit Links-Rechts-Shunt
Querverbindung zwischen großem und kleinem Kreislauf, durch die infolge des höheren Drucks auf der linken und des geringeren Füllungs- bzw. Strömungswiderstandes auf der rechten Seite arterielles Blut im Kurzschluss (Shunt) in den Lungenkreislauf zurückgepumpt wird. Besteht seit der Geburt systemarterieller Druck im Lungenkreislauf, kommt es nicht zur physiologischen Reduktion der pulmonalen arteriellen Widerstände in den ersten Lebensjahren. Die pulmonalen Widerstände bleiben auf der Höhe der systemarteriellen Widerstände stehen. Fällt der pulmonale Widerstand zunächst unter den systemarteriellen Widerstand, dann kann der gesteigerte Lungendurchfluss früher oder später durch Intimaveränderungen an den Lungenarteriolen zur irreversiblen Widerstandserhöhung im kleinen Kreislauf mit pulmonaler Hypertonie und schließlich zur Shuntumkehr mit Zyanose führen (Eisenmenger-Reaktion).
1.13.1.6 Vitien ohne Shunt
▶ Abschn. 1.12.
1.13.1.7 Vitien mit Rechts-Links-Shunt
Bei diesen Fehlbildungen gelangt venöses Shuntblut direkt in den großen Kreislauf. Es setzt die arterielle Sauerstoffsättigung herab.
1.13.1.8 Folgen
-
zentrale Zyanose bei arterieller O2-Sättigung <85 % bzw. einer Konzentration des reduzierten Hb >3 g/dl
-
reaktive kompensatorische Polyzythämie mit Thromboembolie- und Blutungsgefahr sowie Einschränkung der Nierenfunktion infolge Herabsetzung des zirkulierenden Plasmavolumens
-
Uhrglasnägel- und Trommelschlegelfinger infolge hypoxisch induzierter Zunahme der Kapillaren, des Bindegewebes an den Endphalangen und Ablagerung von Megakaryozyten.
-
hypoxisch bedingte Verminderung der Infektresistenz (Akne, von der Haut ausgehende Sepsis, Hirnabszesse).
1.13.2 Vorhofseptumdefekt (ASD)
1.13.2.1 Definition
1.13.2.2 Sinus-venosus-Defekt
Hochsitzend, an der Einmündung der V. cava superior, die über dem Defekt reitet. Meistens kombiniert mit Lungenvenentransposition (Venen aus der rechten Lunge münden in den rechten Vorhof).
1.13.2.3 Sekundumdefekt
Defekt in Septummitte. Der physiologische Defekt im Septum primum wird nicht durch das Septum secundum geschlossen. Häufigste Form des ASD.
1.13.2.4 Primumdefekt
Tiefsitzend, AV-klappennah. Oft zusätzliche Spaltbildung am vorderen Mitralklappensegel (partieller AV-Kanal).
1.13.2.5 Kompletter AV-Kanal
Primumdefekt, der in einen hochsitzenden Ventrikelseptumdefekt übergeht. Dazu Fehlbildung der AV-Klappen (keine Vereinigung von vorderem und hinterem Endokardkissen).
1.13.2.6 Pathophysiologie
Links-Rechts-Shunt auf Vorhofebene. Volumenbelastung des rechten Herzens und des Lungenkreislaufs. Minutenvolumen des kleinen Kreislaufs bei mittleren Defekten 2- bis 3-fach, bei schweren bis 6-fach größer als das des großen Kreislaufs. Blutfluss durch den ASD von links nach rechts wegen der größeren Compliance des rechten gegenüber der des linken Ventrikels. Mit zunehmender Rechtshypertrophie Abnahme der rechtsventrikulären Compliance und des Shuntvolumens bis zur Shuntumkehr. Shuntfolgen: Dilatation und Volumenhypertrophie des rechten Vorhofes und Ventrikels, Dilatation des Truncus pulmonalis, der zentralen und peripheren Lungenarterien und der Lungenvenen, Weitstellung der pulmonalen Widerstandsgefäße. Die pulmonale Vasodilatation senkt den Austreibungswiderstand des rechten Ventrikels. Nur geringer Druckanstieg in der Pulmonalarterie und im rechten Ventrikel bei Lungendurchflusserhöhung auf das 3- bis 4-fache des Herzminutenvolumens. Doch wird der Spielraum für eine pulmonale Widerstandssenkung bei körperlicher Belastung enger. Vom 3. bis 4. Lebensjahrzehnt an fortschreitende Verminderung der Compliance der Lungengefäße und Rhythmusstörungen (Vorhofflimmem). Bei 15 % der Patienten obstruktive Veränderungen der kleinen Lungenarterien und Arteriolen mit Widerstandserhöhung im kleinen Kreislauf, pulmonaler Hypertonie, zusätzlicher Druckhypertrophie des rechten Ventrikels, Shuntumkehr und finaler Rechtsinsuffizienz.
1.13.2.7 Klinik
Sekundumdefekte im Säuglings- und Kindesalter meist asymptomatisch. Im Jugend- und frühen Erwachsenenalter allenfalls Klagen über Müdigkeit und verminderte körperliche Leistungsfähigkeit. Beschwerden können bis ins 6. Lebensjahrzehnt und später ausbleiben. Die meisten Patienten über 40 Jahre sind symptomatisch: Belastungsdyspnoe, Druckgefühl in der Herzgegend, Rhythmusstörungen. Bei sekundärer pulmonaler Hypertonie progrediente Rechtsinsuffizienz mit zentraler Zyanose bei Shuntumkehr. Primumdefekt oft früher manifest und schwerer verlaufend. Teils wegen begleitender Mitralinsuffizienz, die den Links-Rechts-Shunt verstärkt, teils durch Reizleitungsstörungen (Läsionen am AV-Knoten und His-Bündel) und durch turbulenten Blutstrom. Die obstruktive Lungengefäßerkrankung setzt früher ein. Beim kompletten AV-Kanal Wachstumsstörungen, respiratorische Infekte und Herzinsuffizienz bereits im Säuglingsalter. Ohne Operation erreichen nur 5 % das Erwachsenenalter.
1.13.2.8 Diagnostik
1.13.2.9 Auskultationsbefund
Spindelförmiges systolisches Austreibungsgeräusch am linken Sternalrand mit Punctum maximum im 2. ICR links und fixierte (atemunabhängige) breite Spaltung des 2. Herztones im Pulmonalareal (wegen Verspätung des Pulmonalklappenschlusses). Bei großem Shuntvolumen diastolisches trikuspidales Strömungsgeräusch am linken unteren Sternalrand. Bei Primumdefekt mit Mitralinsuffizienz zusätzlich holosystolisches Spitzengeräusch durch die Mitralinsuffizienz.
1.13.2.10 Palpationsbefund
Hebende Pulsation des rechten Ventrikels in der mittleren und unteren linken Parasternalregion und im Epigastrium.
1.13.2.11 EKG
Beim Sekundumdefekt Steil- oder Rechtslagetyp mit inkomplettem Rechtsschenkelblock (rSR’ in V3R) und Zeichen der Rechtshypertrophie. Beim Primumdefekt häufig AV-Block 1. Grades, fast immer überdrehter Linkslagetyp (linksanteriorer Hemiblock), dazu inkompletter Rechtsschenkelblock und Rechtshypertrophiezeichen, letztere bei Eisenmenger-Syndrom am ausgeprägtesten. Bei beiden Formen oft Vorhofflattern und Vorhofflimmern.
1.13.2.12 Echokardiographie und Doppler
M-Mode: Vergrößerung des rechten Ventrikels mit paradoxer systolischer Septumbewegung (nach anterior). 2-D-Echo: Dilatation des rechten Ventrikels und der A. pulmonalis. Direkte Erkennung des ASD von subkostal. Auswascheffekt im Kontrastecho. Sehr gute Erfassung des ASD mit der transösophagealen Farbdoppler-Echokardiographie (◘ Abb. 1.109).


Transösophageale Echokardiographie vor und nach katheterinterventionellem Verschluss eines offenen Foramen ovale. a Links-Rechts-Shunt in Ruhe und Shuntumkehr unter Valsava Manöver. b Amplatz-Occluder verschließt das Foramen ovale (IAS = Vorhofseptum, Pfeile: Occluder)
1.13.2.13 Röntgenaufnahme des Thorax
Nach beiden Seiten verbreitertes Herz mit verstrichener Herztaille, Prominenz des Pulmonalbogens und Erweiterung der zentralen und peripheren Lungenarterien ohne Vergrößerung des linken Vorhofs und linken Ventrikels. Im Seitenbild kann der rechte Ventrikel links randbildend werden und engt den Retrosternalraum ein. Bei sekundärer Obstruktion der Lungengefäße verschwindet die verstärkte Lungengefäßzeichnung bei zunehmender Erweiterung der Pulmonalarterie und ihrer Hauptstämme.
1.13.2.14 Rechtsherzkatheter
Weitgehend ersetzt durch die Echokardiographie. Noch indiziert bei Assoziation mit pulmonaler Gefäßerkrankung.
1.13.2.15 Therapie
Bei nicht zu großen Sekundumdefekten und offenem Foramen ovale gelingt der Verschluss mit beidseitigen Schirmen oder Schalen, die mittels transkutanem Katheter appliziert werden (◘ Abb. 1.110).


Devices für den katheterinterventionellen Verschluss eines offenen Foramen ovale/ASD
Der operative Verschluss des Defektes erfolgt durch Naht oder Patch aus Perikard oder prothetischem Material unter extrakorporaler Zirkulation. Indikation bei jedem ASD mit signifikantem Links-Rechts-Shunt (Relation der Flussraten über 1,5:1,0). Operationszeitpunkt optimal zwischen 3. und 6. Lebensjahr, im Allgemeinen bei Stellung der Diagnose, auch jenseits des 45. Lebensjahres. Die Operation ist nicht mehr indiziert bei schwerer pulmonaler Hypertonie mit nur noch geringem Shuntvolumen.
1.13.2.16 Lutembacher-Syndrom
Seltene Kombination eines Vorhofseptumdefektes mit einer Mitralstenose, die fast immer rheumatischen Ursprungs ist. Häufiger bei Erwachsenen als bei Kindern. Die Mitralstenose verstärkt den Links-Rechts-Shunt, so dass die Patienten frühzeitig symptomatisch werden (Dyspnoe, Schwäche, Herzklopfen). Andererseits bewirkt der ASD eine Druckentlastung des linken Vorhofs und der Lungenvenen (geringere Lungenstauung, kein Lungenödem). Zusätzlich hört man einen lauten 1. Herzton und einen Mitralöffnungston über der Spitze: hoher Druck. Diagnose der Mitralstenose mittels Echokardiographie.
1.13.3 Ventrikelseptumdefekt (VSD)
Lokalisation häufiger im membranösen als im muskulären Teil des Septums. Defektgröße nach Durchmessern:
-
klein: bis 0,5 cm, vollständig drucktrennend
-
mittelgroß: 0,5–1,5 cm, unvollständig drucktrennend
-
groß: 1,5–3,0 cm, mit vollständigem Druckangleich
Spontanverschlussrate bis zum Alter von 3 Jahren etwa 40 % (überwiegend der kleinen und mittelgroßen Defekte, von den großen nur 7 %). Einige Verschlüsse erst nach 10 Jahren.
1.13.3.1 Pathophysiologie
1.13.3.2 Allgemeine Hämodynamik
Links-Rechts-Shunt auf Kammerebene mit Volumenbelastung von rechtem Ventrikel, Lungenarterien, Lungenvenen, linkem Vorhof und linkem Ventrikel. Bei sekundärer Widerstandserhöhung im Lungenkreislauf Druckbelastung des rechten Ventrikels und des rechten Vorhofes.
1.13.3.3 Drucktrennender VSD (Typ Roger)
Hämodynamisch unbedeutender Links-Rechts-Shunt (maximal 3 l/min). Drücke im Lungenkreislauf bei gleichzeitiger Abnahme des Lungengefäßwiderstandes normal.
1.13.3.4 Unvollständig drucktrennender VSD
Shunt-Volumen von hämodynamischer Relevanz ab einer Relation der Minutenvolumina des kleinen und großen Kreislaufs von 2:1. Erreicht werden Relationen bis 4:1. Mäßige Drucksteigerung im rechten Ventrikel und in der Pulmonalarterie. In einigen Fällen sekundäre pulmonale Widerstandszunahme mit entsprechender Verkleinerung des Links-Rechts-Shunts und Druckhypertrophie des rechten Ventrikels. Selten Shuntumkehr.
1.13.3.5 Großer VSD mit Druckangleich
Beide Ventrikel arbeiten wie eine einzelne Pumpe mit zwei Ausgängen. Das Shuntvolumen hängt von der Relation der peripheren Widerstände im kleinen und großen Kreislauf ab. Postnatal ist der pulmonale Widerstand noch relativ groß, der Shunt relativ klein. Mit der physiologischen Abnahme des pulmonalen Widerstands in den ersten Lebensmonaten nimmt der Links-Rechts-Shunt zu. Der Druck im rechten Ventrikel und in der A. pulmonalis steigt im ersten Lebensjahr durch adaptative Kontraktion der Lungenarteriolen, die von langsam progredienten obstruktiven Gefäßveränderungen abgelöst wird. Säuglinge mit ungenügender pulmonaler Vasokonstriktion werden im ersten Lebensjahr zunehmend rechtsinsuffizient. Bei den anderen nimmt der Shunt unter klinischer Besserung nach dem ersten Lebensjahr ab. Im Verlauf schreitet die Obstruktion der Lungengefäße unter Verkleinerung des Links-Rechts-Shunts fort. Aus einer zunächst überwiegend hämodynamischen wird eine pulmonale Widerstandshypertonie, wobei der rechtsventrikuläre Druck auf der Höhe des linksventrikulären bleibt. Bis zum Shuntausgleich im späten Kindes- oder Jugendalter wird der große VSD zunehmend besser toleriert. Danach kommt es durch Shuntumkehr zur Verschlimmerung mit zentraler Zyanose und Polyglobulie (Eisenmenger Komplex).
1.13.3.6 Komplikationen
Infektiöse Endokarditis (4 % der Erwachsenen mit VSD aller Größen), Aorteninsuffizienz durch Prolaps eines Klappensegels in den VSD (ab 5. Lebensjahr, 6 % der Fälle), infundibuläre Pulmonalstenose (im Säuglings- und frühen Kindesalter bei großem Shunt in 8–15 % der Fälle).
1.13.3.7 Klinik
Normale Entwicklung und Leistungsfähigkeit bei drucktrennendem VSD. Bei unvollständig drucktrennendem VSD mit größerem Shunt kann die Entwicklung gestört sein, die Leistungsbreite eingeschränkt. Bei großem VSD mit Druckangleich gehemmte körperliche Entwicklung, Leistungsminderung, Dyspnoe, Lungeninfekte, in manchen Fällen letale Herzinsuffizienz im ersten Lebensjahr. Danach zunehmende Stabilisierung. Auch ohne Operation trotz Shuntumkehr erreichen viele Patienten mit großem VSD ein mittleres Lebensalter. Finale Komplikationen: infektiöse Endokarditis, Rechtsinsuffizienz, Ruptur der Pulmonalarterie, Thromboembolie bei Polyglobulie.
1.13.3.8 Diagnostik
1.13.3.9 Auskultationsbefund
Lautes, bandförmiges holosystolisches Geräusch mit tastbarem Schwirren am linken unteren Sternalrand (3.–5. ICR). Bei Defekten im muskulären Septum oft holosystolisches Decrescendogeräusch oder Spindelform des Geräusches. Spindelform auch bei sehr kleinem VSD. Abnahme der Lautstärke mit Verminderung des Links-Rechts-Shunts. Kein Geräusch mehr nach Shuntumkehr. Bei großem Links-Rechts-Shunt apikales tieffrequentes diastolisches Mitralströmunsgeräusch.
1.13.3.10 Palpationsbefund
Herzspitzenstoß und präkordiale Pulsation bei großen Shunts verstärkt.
1.13.3.11 EKG
Unauffällig bei VSD mit hämodynamisch nicht signifikantem Links-Rechts-Shunt und normalem Pulmonalarteriendruck. Bei unvollständig drucktrennendem VSD mit signifikantem Shunt linksventrikuläre Volumenhypertrophie (hohes R in aVL, I, V5 und V6 mit deutlichem Q in diesen Ableitungen). Bei großem VSD mit Druckausgleich Zeichen der Rechtshypertrophie oder biventrikulären Hypertrophie. Ausgeprägte Rechtshypertrophie bei Eisenmenger-Komplex.
1.13.3.12 Echokardiographie und Doppler
2-D-Echo: Nur große Defekte direkt sichtbar, die anderen im Kontrastecho. Bei Links-Rechts-Shunt Auswaschphänomen, bei Rechts-Links-Shunt Kontrastmittelübertritt in den linken Ventrikel. Shuntnachweis mit dem Farbdoppler und PW-Doppler, Druckgradientmessung mit dem CW-Doppler. Beide Vorhöfe und Kammern sind bei kleinen und großen Defekten zunächst normal groß. Später dilatieren linker Vorhof und linker Ventrikel.
1.13.3.13 Röntgenaufnahme des Thorax
Bei kleinen Defekten normale Form und Größe des Herzens. Bei mittleren Defekten Dilatation und Pulsation des Pulmonalbogens und der zentralen Lungenarterien, vermehrte Lungengefäßzeichnung. Bei Defekten mit großem Links-Rechts-Shunt schmale Aorta, Vergrößerung des linken Ventrikels und linken Vorhofes, Dilatation und Pulsation des Pulmonalstammes, der zentralen und peripheren Lungenarterien. Bei großem VSD mit Shuntumkehr starke Erweiterung der zentralen Lungenarterien unter Reduzierung der peripheren Lungengefäßzeichnung.
1.13.3.14 Herzkatheter und Angiokardiographie
Shuntnachweis durch Erfassung eines O2-Sättigungssprunges zwischen rechtem Vorhof und rechtem Ventrikel. Dazu muss das Shuntvolumen mindestens 20 % des Minutenvolumens im großen Kreislauf betragen. Quantitative Bestimmung des Shunts nach dem Fick’schen Prinzip (wie beim ASD, ▶ oben). Bestimmung des systolischen Druckgradienten zwischen linkem und rechtem Ventrikel zur Erfassung von Drucktrennung und Druckangleich. Dazu werden systolischer Pulmonalarteriendruck und arterieller Blutdruck simultan gemessen. Erfassung der pulmonalen Hypertonie. Bestimmung des pulmonalen Gefäßwiderstandes. Direkter Nachweis auch der kleinen Defekte mit Kontrastmittelinjektion in den linken bzw. rechten Ventrikel.
1.13.3.15 Therapie
1.13.3.16 Operativer Defektverschluss
Keine Indikation bei Kindern und Erwachsenen mit normalem Pulmonalarteriendruck und einer Minutenvolumenrelation zwischen kleinem und großem Kreislauf von 1,5:1. Größere Defekte müssen verschlossen werden, bevor der pulmonale Gefäßwiderstand irreversibel steigt. Er sollte höchstens ein Drittel des Widerstandes im großen Kreislauf betragen. Große Defekte mit Druckangleich werden schon im ersten Lebensjahr operiert, die anderen kurz vor dem Schulalter. Ein Eisenmenger-Komplex bei Erwachsenen ist nicht operabel, allenfalls durch Herz-Lungen-Transplantation oder Defektverschluss und einseitiger Lungentransplantation.
1.13.3.17 Komplikationen der Operation
Totaler AV-Block bei 1 %, bifaszikulärer Block bei 10–25 %, vermindertes Minutenvolumen unter Belastung als Folge der Ventrikulotomie in manchen Fällen. Operationsmortalität 1–9 %.
1.13.3.18 Konservative Therapie
Endokarditisprophylaxe in allen nicht operierten Fällen. Behandlung der Herzinsuffizienz in inoperablen Fällen.
1.13.4 Ductus Botalli apertus
1.13.4.1 Pathoanatomie
Persistenz der fetalen Gefäßverbindung zwischen Pulmonalarterienstamm und Aorta, dicht unterhalb vom Abgang der linken A. subclavia. Länge des Duktus 2–4 cm. Weite 1–1,5 cm. Verschließt sich normalerweise sofort nach der Geburt. Je nach dem Obliterationsgrad kann ein enger, mittelgroßer und großer Duktus bestehen bleiben.
1.13.4.2 Pathophysiologie
Links-Rechts-Shunt von der Aorta in die Pulmonalarterie und damit in den Lungenkreislauf. Enger Duktus hämodynamisch unbedeutend. Mittelgroßer Duktus bedeutet Volumenbelastung von Lungenarterien, Lungenvenen, linkem Vorhof, linkem Ventrikel und Aortenbogen, mit der Tendenz zur sekundären pulmonalen Widerstandshypertonie. Großer Duktus verursacht großen Shunt mit Druckangleich und eine pulmonale Hypertonie, der früher oder später eine irreversible pulmonale Widerstandserhöhung mit Shuntumkehr und Eisenmenger-Reaktion folgt.
Durch den Abfluss aus der Aorta in das Niederdruckgebiet des Lungenkreislaufs wird die Windkesselfunktion der Aorta gestört (wie bei der Aorteninsuffizienz). Es resultiert eine große Blutdruckamplitude mit herabgesetztem diastolischem Druck. In allen Fällen besteht die Gefahr der bakteriellen Endarteriitis an der Einmündung des Duktus in die Pulmonalarterie.
1.13.4.3 Klinik
Bei kleinem Duktus keine kardiovaskulären Funktionsstörungen, aber Risiko der Endarteriitis bzw. Endokarditis. Bei mittelgroßem Duktus je nach Volumenbelastung Einschränkung der Leistungsfähigkeit, Belastungsdyspnoe, Herzklopfen, Rhythmusstörungen, auch Übergang in sekundäre pulmonale Widerstandshypertonie ist möglich. Beim seltenen großen Duktus manchmal schon postnatal Linksinsuffizienz. Im weiteren Verlauf hängt die Shuntgröße von der Entwicklung des pulmonalen Gefäßwiderstandes ab. Durch entsprechende Shuntverkleinerung können die Patienten vorübergehend asymptomatisch werden, ehe es nach Shuntumkehr zur typischen Zyanose in der unteren Körperhälfte und zur Rechtsinsuffizienz kommt, manchmal schon im Kindesalter.
1.13.4.4 Diagnostik
1.13.4.5 Auskultationsbefund
Lautes, kontinuierliches Geräusch (systolisch-diastolisch), das nach dem 1. Herzton beginnt, Spindelform hat und in Höhe des 2. Herztones seine maximale Lautstärke („Maschinengeräusch“) erreicht. Lokalisiert im 2. ICR links parasternal oder unterhalb der linken Klavikula. Bei Shuntumkehr fehlt dieses Geräusch. Bei großem Shuntvolumen Mitralströmungsgeräusch über der Spitze.
1.13.4.6 Palpationsbefund
Verbreiterter Spitzenstoß, pulsierende Halsgefäße, Pulsus celer et altus und große Blutdruckamplitude.
1.13.4.7 EKG
Linkshypertrophie bei entsprechender Volumenbelastung. Mit zunehmendem pulmonalen Widerstand Zeichen der Rechtshypertrophie.
1.13.4.8 Echokardiographie und Doppler
Keine direkte Darstellung im 2-D-Echo. Im suprasternalen Fenster Nachweis eines turbulenten Flusses zwischen Aorta descendens und linker Pulmonalarterie.
1.13.4.9 Röntgenaufnahme des Thorax
Nur bei größerem Shunt entsprechende Vergrößerung des linken Vorhofes und Ventrikels sowie Prominenz des Aorten- und Pulmonalbogens. Lungengefäßzeichnung verstärkt, bei pulmonaler Hypertonie vermindert. MRT (mit Kontrastmittel): Schonende sichere Nachweismöglichkeit (◘ Abb. 1.111).


MRT (nach KM-Gabe) eines offenen Ductus arteriosus Botalli (AO = Aorta, PA = Pulmonalarterie)
1.13.4.10 Herzkatheteruntersuchung
Indirekter Shuntnachweis durch sprunghafte Zunahme der O2-Sättigung in der Pulmonalarterie (nicht bei geringem Shunt). Direkter Shuntnachweis durch Kontrastmittelinjektion in die Aorta bzw. in die Pulmonalarterie.
1.13.4.11 Therapie
1.13.4.12 Katheterverfahren
Als primäre Behandlungsmethode hat sich der schonende Verschluss per Katheter mit Schirmen oder Spiralen etabliert (◘ Abb. 1.112).


40-jährige Patientin mit bekanntem persistierenden Ductus arteriosus Botalli. Interventioneller Verschluss mit einem Occluder bei zunehmender Dilatation des linken Ventrikels
1.13.4.13 Operation
Der klassische Eingriff ist die Durchtrennung des Duktus zwischen zwei Klemmen. Die Stümpfe werden vernäht. Bei minimal invasivem Vorgehen wird der Duktus mit einem Metallclip verschlossen. Zweck des Duktusverschlusses: Verbesserung der Hämodynamik, Verhütung der irreversiblen Widerstandserhöhung im Lungenkreislauf und Endokarditisprophylaxe. Indikation im Erwachsenenalter in allen Fällen mit hörbarem Shuntgeräusch. Patienten mit kleinem stummen offenen Duktus und solche mit Eisenmenger-Reaktion werden nicht operiert. In den meisten Fällen mit mittelgroßem und großem Duktus erfolgt die Operation schon im Säuglings- bzw. Kindesalter. Bei Frühgeburten schließt man den häufig offenen Ductus Botalli mit intravenös appliziertem Prostaglandin.
1.13.5 Kongenitale Aortenstenose
Darstellung bei den erworbenen Herzklappenfehlern (▶ Abschn. 1.12).
1.13.6 Aortenisthmusstenose (Koarktation der Aorta )
1.13.6.1 Pathoanatomie
1.13.6.2 Juxtaduktale Koarktation
Stenose der Aorta im Einmündungsbereich des Ductus Botalli bzw. in Höhe des Lig. arteriosum durch eine Verdickung und Einstülpung der posterolateralen Aortenwand. In der Fetalzeit verhält sich diese Vorwölbung in das Lumen wie eine Bifurkation, die den kleineren Teil des Blutes aus dem Duktus in die aszendierende, den größeren Teil in die deszendierende Aorta leitet. Die Stenose wird erst wirksam, wenn das aortale Ende des Duktus postnatal obliteriert. Das geschieht um so früher, je größer die Einstülpung ist und je schneller der Duktus obliteriert. Nach der Geburt bilden sich folgende Kollateralkreisläufe aus:
-
A. subclavia → A. thoracica interna → Aa. intercostales → Aorta unterhalb der Stenose.
-
A. subclavia → A. thoracica interna → A. epigastrica superficialis → A. iliaca.
1.13.6.3 Hypoplasie des Aortenisthmus
Stenosierung durch einen unterentwickelten Isthmus (Aortensegment zwischen linker A. subclavia und Ductus Botalli) mit meistens offen bleibendem Duktus (infantile Form der Koarktation). In fast allen Fällen weitere Fehlbildungen: großer VSD, Endokardkissendefekte, komplette oder inkomplette Transposition der großen Arterien. Die untere Körperhälfte erhält Blut aus dem Ductus Botalli (Rechts-Links-Shunt).
1.13.6.4 Pathophysiologie
An der Stenose entsteht ein Druckgradient mit Hypertonie in der oberen und Hypotonie in der unteren Körperhälfte. In der Pathogenese des Hochdrucks spielt das Renin-Angiotensin-System die wichtigste Rolle. Hochdruckfolgen: Linkshypertrophie, Hirnblutungen, sekundäre Arteriosklerose der Hirn- und Koronargefäße.
1.13.6.5 Klinik
Wenn die Stenose erst im Laufe von 6–9 Monaten einen höheren Grad erreicht, können sich ein ausreichender Kollateralkreislauf und eine kompensatorische Linkshypertrophie entwickeln. Der Kreislauf kann dann bis ins Erwachsenenalter kompensiert bleiben. Zahlreiche Fälle bleiben mangels Symptomen Jahre und Jahrzehnte unentdeckt. Häufigste Fehldiagnose juvenile Hypertonie.
Das Fehlen der Fußpulse führt zur Diagnose.
Im fortgeschrittenen Stadium: Kopfschmerzen, Schwindelgefühl, Nasenbluten, Schulterschmerzen, kalte Füße, Parästhesien in den Beinen.
1.13.6.6 Komplikationen
Aortenaneurysma mit Ruptur, bakterielle Endokarditis (distal der Stenose und an der Aortenklappe).
1.13.6.7 Diagnostik
1.13.6.8 Auskultationsbefund
Spätsystolisches, oft über den 2. Herzton hinausreichendes Geräusch zwischen linkem Schulterblatt und Wirbelsäule, sowie im 4.‒5. ICR links parasternal. Dazu kontinuierliches Geräusch über den hinteren Interkostalräumen (Kollateralgefäße).
1.13.6.9 Palpationsbefund
Fehlender oder abgeschwächter Femoralispuls bei kräftigem Radialispuls. Verbreiterter Spitzenstoß. Tastbare und sichtbare Pulsation erweiterter Interkostalarterien.
1.13.6.10 EKG
Alle Übergänge vom Normalbefund zur Linkshypertrophie.
1.13.6.11 Echokardiographie und Doppler
Im suprasternalen Fenster Stenosenachweis mit dem Farbdoppler. Bestimmung des Druckgradienten mit dem CW-Doppler. Im M-Mode Linkshypertrophie.
1.13.6.12 Röntgenaufnahme des Thorax
Fehlen oder atypische Form des Aortenknopfes, Rippenusuren durch Kollateralgefäße. Normale Herzgröße oder Linksverbreiterung. Im Seitenbild ist die Einschnürung der Aorta im Stenosebereich manchmal sichtbar.
1.13.6.13 MRT (nach KM-Gabe)
Zur präoperativen Lokalisation der Stenose und zur Bestimmung ihrer Längenausdehnung. Bevorzugt wird heute die Darstellung mittels MR-Angiographie (◘ Abb. 1.10).
1.13.6.14 Therapie
1.13.6.15 Ballondilatation
Bei geeigneter Morphologie als primärer Eingriff möglich. In der Regel wird auch ein Stent appliziert (◘ Abb. 1.11). An Spätkomplikation können sich allerdings Restenosen und Aneurysmen entwickeln.
1.13.6.16 Operative Therapie
Klassische Methode ist die Resektion mit anschließender End-zu-End-Anastomose, evtl. unter Zuhilfenahme einer Gefäßprothese. Operationszeitpunkt: Bei asymptomatischen Kindern im 4.–6. Lebensjahr, sonst nach Stellung der Diagnose. Mittlere Lebenserwartung ohne Operation 43 Jahre. Operationsmortalität unter 5 %. Häufige paradoxe hypertensive Reaktion in den ersten postoperativen Tagen durch starke Angiotensinfreisetzung. Zu beherrschen mit Captopril oder Enalapril.
1.13.7 Kongenitale Pulmonalstenose
1.13.7.1 Pathoanatomie
Pulmonalstenosen sind von seltenen Ausnahmen abgesehen kongenitalen Ursprungs. Nach dem Sitz unterscheidet man folgende Formen:
-
Valvuläre Pulmonalstenose: Häufigste Ursache der isolierten Obstruktion der rechtsventrikulären Ausflussbahn. Entsteht durch Verlötung der Pulmonaltaschenklappen in der Mitte oder gegen Ende der embryonalen Entwicklung. Alle Schweregrade bis zur Pulmonalatresie.
-
Infundibuläre Pulmonalstenose: Als isolierte Anomalie sehr selten, entsteht durch abnormes ringförmiges Muskelbündel in der Ausflussbahn.
-
Supravalvuläre Pulmonalarterienstenose: Lokalisiert im Truncus, im Bereich der Bifurkation und an multiplen Stellen der Peripherie. Auch kombinierte zentrale und periphere Stenosen kommen vor, häufig bei Rötelembryopathie mit anderen kardiovaskulären Fehlbildungen.
1.13.7.2 Pathophysiologie
Austreibungswiderstand für den rechten Ventrikel mit Druckbelastung, die zur Hypertrophie führt. Den Schweregrad bestimmt der Druckgradient über der Pulmonalklappe: leicht: bis 40 mmHg, mittelgradig: 50–60 mmHg, schwer: >60 mmHg.
1.13.7.3 Klinik
Leichte Stenosen sind asymptomatisch, bedingen aber ein erhöhtes Endokarditisrisiko. Mittelgradige Stenosen schränken die Leistungsfähigkeit ein. Bei schweren Stenosen Belastungsdyspnoe, anginaähnliche Brustschmerzen, Synkopen, Zeichen der Rechtsinsuffizienz.
1.13.7.4 Diagnostik
1.13.7.5 Auskultationsbefund
Lautes systolisches Austreibungsgeräusch im 2. ICR links parasternal, inspiratorisch lauter werdend. Pulmonaler Ejektionsklick nur bei valvulärer Stenose. Spaltung des 2. Herztones mit leisem P2, 4. Herzton am unteren linken Sternalrand.
1.13.7.6 Palpationsbefund
Pulsation am linken unteren Sternalrand und im Epigastrium infolge Rechtshypertrophie. Tastbares Schwirren am mittleren oder oberen linken Sternalrand.
1.13.7.7 EKG
Bei schwerer Stenose Zeichen der Rechtshypertrophie und Rechtsschädigung. Deutliches P-dextrokardiale.
1.13.7.8 Echokardiographie und Doppler
-
2-D-Echo: Hypertrophierter, oft dilatierter rechter Ventrikel, Darstellung der Stenose im subkostalen Fenster
-
Farb-Doppler: Mosaikbild über der Pulmonalklappe
-
CW-Doppler: Stenosejet, Berechnung des Druckgradienten
1.13.7.9 Röntgenaufnahme des Thorax
Normale Herzgröße. Poststenotische Dilatation des Pulmonalbogens bei herabgesetzter Lungengefäßzeichnung (helle Lungenfelder).
1.13.7.10 Rechtsherzkatheter
Zur Diagnose des Schweregrades indiziert. Druckmessung im rechten Ventrikel und in der Pulmonalarterie. Erfassung einer Rechtsinsuffizienz, die zur Erhöhung des enddiastolischen Ventrikeldrucks führt.
1.13.7.11 Angiokardiographie
Präoperative Darstellung. Ausschluss zusätzlicher Fehlbildungen. Darstellung infundibulärer Stenosen.
1.13.7.12 Therapie
Bei mittelgradigen und schweren Stenosen wird der perkutanen transluminalen Ballondilatation in jedem Lebensalter der Vorzug gegeben. Alterativ kommt die offene Kommissurotomie ohne Herz-Lungen-Maschine in Betracht (sehr niedriges Risiko). Infundibulumstenosen werden mit Hilfe der Herz-Lungen-Maschine reseziert.
1.13.8 Morbus Fallot
1.13.8.1 Pathoanatomie
Als Fallot-Tetralogie bezeichnet, hat diese Fehlbildung 4 anatomische Merkmale.
Die 4 anatomischen Merkmale der Fallot-Tetralogie
-
Pulmonalstenose: Meistens infundibulär, bei 25 % zusätzlich valvulär. Die schwach durchströmte Pulmonalarterie kann sekundär unterentwickelt sein. In seltenen Fällen besteht eine Atresie.
-
Großer Ventrikelseptumdefekt mit Druckangleich
-
Reitende Aorta: liegt über dem Septumdefekt
-
Hypertrophie des rechten Ventrikels
1.13.8.2 Pathophysiologie
Funktionell liegt die Kombination eines großen, druckangleichenden VSD mit einer Obstruktion der rechtsventrikulären Ausflussbahn vor. Wenn der rechtsventrikuläre Austreibungswiderstand größer ist als der periphere Widerstand im großen Kreislauf, resultiert ein Rechts-Links-Shunt (zyanotische Form), im umgekehrten Fall ein Links-Rechts-Shunt (azyanotische Form).
1.13.8.3 Zyanotische Form (klassischer Morbus Fallot)
Verminderte Lungenperfusion und Rechts-Links-Shunt bewirken eine schwere Hypoxämie (arterielle-O2-Sättigung kann auf 50 % sinken) mit lebensbedrohlichen hypoxischen Zuständen (durch Spasmen des Infundibulums), Zyanose, Trommelschlegelfingern und sekundärer Polyglobulie. Eine Erhöhung des peripheren Widerstandes (Hockerstellung) und eine Verminderung der Kontraktilität des Infundibulums (Sedativa, β-Rezeptorenblocker) leiten vermehrt Blut in den Lungenkreislauf und vermindern Hypoxämie und Zyanose.
1.13.8.4 Azyanotische Form (Links-Rechts-Shunt)
Entspricht der Situation beim großen VSD mit Druckangleich, doch bleibt wegen der Pulmonalstenose ein wesentlicher Anstieg des Pulmonalarteriendrucks aus. Dennoch kann es im Verlauf durch Zunahme der infundibulären Pulmonalstenose zur Shuntumkehr kommen.
1.13.8.5 Klinik
Zyanose bei Geburt oder im Verlauf des ersten Lebensjahres überwiegt. Anfälle von intensiver Verstärkung der Zyanose zwischen dem 2. und 9. Monat, später seltener. Im Anfall Hyperpnoe, Bewusstseinstrübung, auch Synkopen und Konvulsionen, bisweilen Exitus. Je nach Schweregrad der Pulmonalstenose alle Übergänge von starker bis geringer Leistungsbeschränkung.
1.13.8.6 Komplikationen
Infektiöse Endokarditis, paradoxe Embolien (aus Venenthrombosen in den großen Kreislauf), Hirnabszesse, Thrombosen infolge Polyglobulie. Selten Rechtsherzinsuffizienz, da der rechte Ventrikel durch den VSD drainiert wird.
Im Erwachsenenalter präsentieren sich fast nur noch Patienten mit operierter Tetralogie. Bis zu 25 % der Patienten haben Restenosen mit erheblich erhöhten rechtsventrikulären Drücken, einige auch einen residualen VSD. Fast immer besteht ein Rechtsschenkelblock und oft eine ventrikuläre Extrasystolie. In symptomatischen Fällen ist eine Reoperation indiziert.
1.13.8.7 Diagnostik
1.13.8.8 Auskultationsbefund
Lautes, spindelförmiges systolisches Austreibungsgeräusch durch die infundibuläre Pulmonalstenose in der Mitte des linken Sternalrandes, tiefer lokalisiert und um ein bis zwei Grade leiser als bei valvulärer Pulmonalstenose. Bei azyanotischer Form mit Links-Rechts-Shunt zusätzliches Holosystolikum des VSD links parasternal.
1.13.8.9 EKG
Ausgeprägter Rechtslagetyp und Zeichen der Rechtshypertrophie.
1.13.8.10 Echokardiographie
2-D-Echo: Fehlende Kontinuität der anteponierten Aortenvorderwand mit dem interventrikulären Septum. Nachweis der infundibulären und valvulären Pulmonalstenose.
1.13.8.11 Röntgenaufnahme des Thorax
Wenig vergrößertes Herz mit ausgeprägter Taille wegen des kleinen Pulmonalkonus. Angedeutete Schuhform, da sich die vergrößerte rechte Kammer an der Bildung der Herzspitze beteiligt und die linke Kammer nach dorsal drängt. Bei der azyanotischen Form Prominenz des Pulmonalbogens und Hypervaskularisation der Lunge.
1.13.8.12 Herzkatheter
Nachweis des druckangleichenden VSD. Bestimmung des Druckgradienten an der Pulmonalklappe und des pulmonalen Blutflusses.
1.13.8.13 Angiokardiographie
Ermöglicht die Sicherung der Diagnose durch Darstellung der infundibulären und valvulären Pulmonalstenose, des VSD und einer evtl. vorliegenden Hypoplasie der Pulmonalarterie.
1.13.8.14 Magnetresonanztomographie
Liefert eine umfassende Diagnose und hat die Angiokardiographie weitgehend verdrängt.
1.13.8.15 Therapie
1.13.8.16 Totale operative Korrektur
Verschluss des VSD mit Kunststoffpatch, Ausräumung der hypertrophierten Infundibulummuskulatur, ggf. Valvulotomie der Pulmonalklappe. Bei engem Infundibulum Erweiterungsplastik mit Kunstoffpatch. Bei Hypoplasie des Ausflusstraktes und der Pulmonalarterie Implantation eines Conduits (Gefäßprothese mit Klappe) zwischen rechtem Ventrikel und distaler Pulmonalarterie. Operationszeitpunkt: Schon im Säuglingsalter bzw. bei Stellung der Diagnose, möglichst vor dem 5. Lebensjahr.
1.13.8.17 Palliative Operation
Bei zu kleinen Pulmonalarterienästen droht nach VSD-Verschluss eine akute Rechtsinsuffizienz. Deshalb zunächst Blalock-Taussig-Anastomose zwischen A. subclavia und gleichseitigem Pulmonalarterienast mit einer Gefäßprothese. Totalkorrektur nach ausreichendem Wachstum der Pulmonalarterienäste.
1.13.8.18 Prognose
Unter etwa 1000 publizierten Fällen war das funktionelle Ergebnis 10 Jahre nach der Totalkorrektur in 90–95 % der Fälle gut. Nach 30 Jahren überlebten in einem kleineren Patientenkollektiv 86 %.
1.13.9 Ebstein-Anomalie
1.13.9.1 Pathoanatomie
Ventrikelwärts verlagerte, fehlgebildete und undichte Trikuspidalklappe. Ein variabler Teil der Einflussbahn des rechten Ventrikels wird atrialisiert. In mehr als 50 % der Fälle zusätzlich ASD oder offen bleibendes Foramen ovale.
1.13.9.2 Pathophysiologie
Verkleinerte Füllungkapazität und geringe Compliance des rechten Ventrikels bewirken eine verminderte Lungendurchströmung und gewöhnlich einen Rechts-Links-Shunt auf Vorhofebene. Der Schweregrad der anatomischen und funktionellen Abweichungen variiert beträchtlich.
1.13.9.3 Klinik
Hypoxie, Zyanose, Zeichen der Rechtsinsuffizienz im fortgeschrittenen Stadium. Von bedrohlichem Zustandsbild im Säuglingsalter bis zu leichtem Verlauf mit fast normaler Lebenserwartung alle Übergänge.
1.13.9.4 Diagnostik
1.13.9.5 Auskultationsbefund
Holosystolikum am linken unteren Sternalrand, S3- und S4- Galopprhythmus.
1.13.9.6 Röntgenaufnahme des Thorax
Kugelförmiges, beiderseits verbreitertes Herz (Rotation nach links durch massive Dilatation des rechten Vorhofes einschließlich seines Kammeranteiles).
1.13.9.7 EKG
Rechtshypertrophie, Rechtsschenkelblock, auch WPW-Syndrom. Neigung zu Vorhoftachykardien.
1.13.9.8 Echokardiographie und Doppler
Sichern die Diagnose. Im apikalen 4-Kammerblick beträgt der Abstand zwischen MK- und TK-Ebene mehr als 8 mm/m2 Körperoberfläche. Farb-, PW- und CW-Doppler weisen den Reflux nach.
1.13.9.9 Magnetresonanztomographie
Gibt Einblick in die Größe und Funktion des rechten Ventrikels.
1.13.9.10 Herzkatheter
Normale Drücke im rechten Vorhof und Ventrikel, keine pulmonale Hypertonie, Nachweis des Rechts-Links-Shunts bei ASD. Lokalisation der Trikuspidalklappe im Angiokardiogramm.
1.13.9.11 Therapie
Operation bei entsprechendem Schweregrad. Raffung des verlagerten Trikuspidalklappenringes oder Klappenersatz. Bei ausgeprägter Hypoplasie des rechten Ventrikels hohe Operationsletalität. Eventuell Herztransplantation.
1.13.10 Trikuspidalatresie
1.13.10.1 Pathoanatomie und Pathophysiologie
Keine Verbindung zwischen rechtem Vorhof und rechter Kammer, die bei fehlender Einflussbahn hypoplastisch ist. Rechts-Links-Shunt auf Vorhofebene, meistens durch großen Sekundumdefekt, seltener durch weit offenes Foramen ovale. Vollständige arteriovenöse Blutmischung im linken Vorhof. Aus dem linken Ventrikel fließt das Blut in die Aorta, durch einen VSD unterschiedlicher Größe in den hypoplastischen rechten Ventrikel und von dort in die meistens hypoplastische Pulmonalarterie, die zusätzlich in der valvulären Region stenosiert sein kann. Bei der häufigen Transposition der großen Arterien Hyperperfusion der Lunge und etwas behinderter Zufluss zur Aorta. Die Zyanose richtet sich nach der meistens eingeschränkten Lungenperfusion. Ähnlicher Verlauf wie bei schwerer Fallot-Tetralogie mit zyanotischen Attacken und ausgeprägter Polyglobulie.
1.13.10.2 Diagnostik
Durch Echokardiographie, MRT und Angiokardiographie.
1.13.10.3 Therapie
Bei Neugeborenen Palliativeingriffe zum Überleben (Blalock-Taussig-Shunts oder End-zu-Seit-Verbindung zwischen V. cava superior und Pulmonalarterie). Kranke Kinder ohne Pulmonalstenose erhalten ein Pulmonalisbändchen zur Drosselung der Lungenperfusion. Definitive Korrektur ab dem 4. Lebensjahr mit einer modifizierten Fontan-Operation. Dabei verbindet man den rechten Vorhof mit der Bifurkation der Pulmonalarterie. Zuvor wird der Pulmonalarterienstamm durchtrennt und übernäht. Der ASD wird verschlossen. Die Lungenperfusion erfolgt dann nur durch den zentralen Venendruck.
1.13.11 Gemeinsamer (singulärer) Ventrikel
1.13.11.1 Pathoanatomie und Pathophysiologie
Fehlen der Kammerscheidewand oder Reduzierung auf eine niedrige posteroinferiore Leiste. Meist ist eine ‒ die rechte oder linke Kammer ‒ hypoplastisch. Es liegt eine komplette arteriovenöse Mischung vor.
1.13.11.2 Therapie
1.13.11.3 Palliativmaßnahme
Besteht keine Pulmonalstenose, muss die Lungenperfusion mit einem Pulmonalisbändchen gedrosselt werden. Im Falle einer Pulmonalstenose kann eine Blalock-Taussig-Anastomose notwendig werden.
1.13.11.4 Korrekturoperation
Nach dem 4. Lebensjahr Fontan-Operation wie bei Trikuspidalatresie, ergänzt durch den Verschluss der Trikuspidalklappe mit einem Patch.
1.13.12 Transposition der großen Gefäße
1.13.12.1 Pathoanatomie
Häufigste Form der Arterientransposition. Die Aorta entspringt aus dem anatomisch rechten Ventrikel, die Pulmonalarterie aus dem anatomisch linken Ventrikel. Folglich zirkuliert das venöse Blut im großen und das arterialisierte Blut im kleinen Kreislauf. Die Lebensfähigkeit basiert auf Kommunikationen zwischen beiden geschlossenen Kreisläufen: offenes Foramen ovale, ASD, VSD, persistierender Ductus Botalli. Eine oder mehrere dieser Verbindungen werden realisiert. Die Shunts sind bidirektional, mit gleichgroßen Flussraten in beiden Richtungen, da sich sonst einer der Kreisläufe leeren würde. Die arterielle Sauerstoffsättigung im großen Kreislauf hängt von der ausgetauschten Blutmenge ab. Sie ist bei offenem Foramen ovale ungenügend, relativ groß bei der Kombination mit ASD.
1.13.12.2 Klinik
Dyspnoe und Zyanose bei der Geburt, progrediente Hypoxämie, globale Herzinsuffizienz.
1.13.12.3 Diagnostik
Diagnose mit Echokardiographie, MRT und Angiokardiographie.
1.13.12.4 Therapie
1.13.12.5 Palliative Ballonseptotomie
Einreißen des Vorhofseptums mittels Ballonkatheter kurz nach der Geburt.
1.13.12.6 Operative Korrektur
Neuerdings in den ersten beiden Wochen durch den sog. arteriellen Switch. Man verbindet dabei den proximalen Abschnitt der Aorta mit der Bifurkation der Pulmonalis und den proximalen Anteil der Pulmonalis mit dem distalen Teil der Aorta ascendens. Außerdem werden die Kranzgefäße transponiert.
1.13.13 Anatomisch korrigierte Transposition der großen Arterien
1.13.13.1 Pathoanatomie
Der anatomisch rechte Ventrikel mit Trikuspidalklappe liegt auf der linken Seite und erhält oxygeniertes Blut aus den Lungenvenen, das er in die an seiner Vorderseite entspringende Aorta pumpt. Der anatomisch linke Ventrikel mit Mitralklappe liegt auf der rechten Seite und verbindet den rechten Vorhof mit der an seiner Hinterseite abgehenden Pulmonalarterie. Bei dieser anatomischen Konstellation ist die Zirkulation normal.
1.13.13.2 Klinik
Klinisch relevant erscheint der Herzfehler erst bei den häufigen zusätzlichen Fehlbildungen: VSD, ASD, AV-Block 3. Grades. Im Verlauf kommt es häufig zur Insuffizienz der Trikuspidalklappe.
1.13.13.3 Diagnostik
EKG, Echokardiographie und Herzkatheter mit Angiokardiographie.
1.13.13.4 Therapie
Patienten ohne zusätzliche Fehlbildungen am Herzen können bis zum sechsten Dezenium beschwerdefrei bleiben, andere erkranken früher. Chirurgische Maßnahmen beschränken sich auf die Beseitigung von Scheidewanddefekten und AV-Klappeninsuffizienzen sowie auf Schrittmacherimplantation. Bei Versagen des rechten Ventrikels, der den großen Kreislauf versorgt, bleibt nur die Herztransplantation.
1.13.14 Truncus arteriosus communis
1.13.14.1 Anatomie und Pathophysiologie
Über einem großen VSD entspringt ein großes median gelegenes Gefäß mit 3 oder 4 Taschenklappen, das der Aorta entspricht. Etwa 3 cm oberhalb der Klappenebene gehen die Pulmonalarterien ab, entweder getrennt oder aus einem Truncus. Es resultiert ein Links-Rechts-Shunt mit Hyperperfusion der Lunge, die schnell zur Eisenmenger-Reaktion mit Shuntumkehr und Zyanose führt.
1.13.14.2 Klinik
In den ersten 3 Lebensmonaten werden 91 % der Kinder symptomatisch mit Trinkschwierigkeiten, starkem Schwitzen und Gedeihstörungen. Bei vermehrter Lungendurchblutung Dyspnoe und Tachypnoe, bei verminderter Lungendurchblutung ausgeprägte Zyanose, die nach Shuntumkehr eintritt.
1.13.14.3 Diagnostik
Echokardiographie, MRT, Herzkatheter und Angiokardiographie.
1.13.14.4 Therapie
1.13.14.5 Palliativmaßnahme
Bei Herzinsuffizienz und pulmonaler Hypertonie Banding der Pulmonalarterien.
1.13.14.6 Operative Korrektur
Innerhalb des ersten Lebensjahres VSD-Verschluss mit einem Patch und Verlegen eines klappentragenden prosthetischen Conduits zwischen dem rechten Ventrikel und den Pulmonalarterien.
1.14 Kardiomyopathien
1.14.1 Definition
In der Klassifikation der WHO von 1980 wurden Kardiomyopathien als Herzmuskelerkrankungen unbekannter Genese definiert und in dilatative, hypertrophe und restriktive Kardiomyopathien unterteilt. Abgetrennt davon wurde die Kategorie der spezifischen Herzmuskelerkrankungen mit bekannter Ursache.
1.14.2 Klassifikation
In der Klassifikation der WHO und der International Society and Federation of Cardiology von 1995 werden alle Herzmuskelerkrankungen unter dem Begriff der Kardiomyopathie zusammengefasst und in 2 Hauptkategorien gegliedert.
Die erste Hauptkategorie umfasst als pathophysiologische Einheiten dilatative, hypertrophische und restriktive Kardiomyopathien, sowie die arrhythmogene rechtsventrikuläre Kardiomyopathie und die unklassifizierte Kardiomyopathie. Soweit bekannt, werden die Kausalfaktoren genannt.
Die zweite Hauptkategorie umfasst alle spezifischen Herzmuskelerkrankungen mit definierter Ätiologie:
-
ischämische Kardiomyopathie
-
valvuläre Kardiomyopathie
-
hypertensive Kardiomyopathie
-
inflammatorische Kardiomyopathie (Myokarditiden)
-
metabolische Kardiomyopathie (Glykogenspeicherkrankheit, endokrine Störungen u. a.)
-
generalisierte Systemkrankheiten (Sarkoidose, Leukämien)
-
Muskeldystrophien (Duchenne, Becker-Typ, myotonische Dystrophie)
-
neuromuskuläre Erkrankungen (Friedreich-Ataxie, Noonan-Syndrom)
-
Überempfindlichkeit und Toxine (Alkohol, Strahlen, Pharmaka u. a.)
-
peripartale Kardiomyopathie
Im folgenden Kapitel werden nur die Kardiomyopathien der ersten Kategorie abgehandelt.
Kardiomyopathien der ersten Kategorie
-
Dilatative Kardiomyopathie (DCM)
-
Hypertrophische Kardiomyopathie (HCM)
-
Restriktive Kardiomyopathie (RCM)
1.14.3 Dilatative Kardiomyopathie (DCM)
1.14.3.1 Vorkommen
In den USA beträgt die Inzidenz 5–8/100.000 mit steigender Tendenz. Anscheinend werden leichte und asymptomatische Fälle unvollständig erfasst.
1.14.3.2 Ätiologie
Etwa 30 % der Fälle sind genetisch bedingt und zwar mit heterogenem Erbgang. Die meisten familiären Fälle werden autosomal-dominant übertragen. Daneben kommen rezessiv-erbliche und X-chromosomal übertragene Fälle vor. Die genetischen Defekte sind vielfältig. Sie betreffen Gene die für das Zytoskelett der Myozyten, die kontraktilen Proteine (Myosin, Troponin) und Moleküle der Kernmembran kodieren. Insgesamt hat man Mutationen an 14 verschiedenen Genen identifiziert. Welche funktionellen Defizite sie im einzelnen bewirken, ist noch ungenügend geklärt.
Über weitere Kausalfaktoren gibt es keine gesicherten Erkenntnisse. Diskutiert werden subklinische Virusmyokarditiden, da man gelegentlich diskrete entzündliche Infiltrate findet. Auch an eine autoimmunologische Genese wird gedacht mit humoralen und zellulären Immunreaktionen gegen Strukturen, die vielleicht durch Viren antigen geworden sind. Bei der DCM werden Zytokine exprimiert, darunter TNF-α und von ihm abhängige Zytokine, ferner das vasokonstriktorische Endothelin. Sie scheinen von pathogenetischer Bedeutung zu sein, da bei DCM kürzlich gezeigt werden konnte, dass Pentoxifyllin eine symptomatische und funktionelle Besserung bewirkt.
Ungeklärt ist auch, ob Toxine, endokrine Faktoren oder eine mikrovaskuläre Hyperreaktivität mit Spasmen eine Rolle spielen.
Eine dilatative Kardiomyopathie als Endstadium bekannter Herzmuskelaffektionen kommt in erster Linie bei chronischem Alkoholismus, Kokainabusus und Aids vor, auch unter der Chemotherapie von Malignomen, speziell durch Doxorubicin.
1.14.3.3 Pathogenese
Ventrikeldilatation mit Volumenhypertrophie ohne Zunahme der Wanddicke. Herzklappen intakt, relative Mitralinsuffizienz häufig. Intrakavitäre Thromben, bevorzugt im Bereich der Herzspitze. Kranzgefäße nicht stenosiert. Histologisch unspezifische interstitielle und perivaskuläre Fibrose.
1.14.3.4 Pathophysiologie
Progrediente Linksinsuffizienz, oft globale Herzinsuffizienz durch zunehmenden Kontraktilitätsverlust des Myokards. Kompensatorische Dilatation des linken Ventrikels mit meist inadäquater Wandverdickung, Zunahme des enddiastolischen und endsystolischen Volumens. Abfall der Ejektionsfraktion unter 50 %, diastolischer Druckanstieg im linken Ventrikel, wegen Zunahme der Compliance geringer als nach dem Ausmaß der Dilatation zu erwarten. Erst bei manifester Linksinsuffizienz steigen die Drücke im linken Ventrikel, linken Vorhof, im Lungenkreislauf und im rechten Herzen an, entsprechend auch der zentrale Venendruck. Herzminutenvolumen zunächst normal dann absinkend mit sekundärer Azotämie.
1.14.3.5 Klinik
Gewöhnlich mehrjähriges asymptomatisches Vorstadium. Symptombeginn mit allmählich zunehmender Belastungdyspnoe, Rhythmusstörungen, Herzsensationen und nächtlichen Dyspnoeattacken. Tachykardes Vorhofflimmern beschleunigt die Linksinsuffzienz durch den Wegfall der Vorhofkontraktion. Allmähliches Fortschreiten der Erkrankung zur therapierefraktären kardialen Insuffizienz mit Ruhedyspnoe, Lungenstauung und peripheren Ödemen. Gefürchtet sind systemische und pulmonale Embolien.
1.14.3.6 Diagnostik
1.14.3.7 Auskultationsbefund
Oft 3. Herzton (protodiastolischer Galopprhythmus) und Ruhetachykardie, holosystolische Geräusche bei relativer Mitral- und Trikuspidalinsuffizienz.
1.14.3.8 Palpationsbefund
Herzspitzenstoß nach links verlagert und verbreitert, aber nicht hebend. Halsvenen oft gestaut. Leberschwellung und Ödeme (können bei isolierter Linksinsuffizienz fehlen).
1.14.3.9 EKG
Sinustachykardie (bei Insuffizienz). Linksventrikuläre Repolarisationsstörungen, häufig Linksschenkelblock. Atriale und ventrikuläre Arrhythmien im 24-h-EKG.
1.14.3.10 Echokardiogramm
Im M-Mode- und 2-D-Echo Dilatation des linken Ventrikels ohne Zunahme der Wanddicke mit herabgesetzter FS und Ejektionsfraktion. Dilatation des linken Vorhofes, in fortgeschrittenen Fällen auch des rechten Herzens. Fehlen der systolischen Dickenzunahme des Septums. Oft relative Mitralinsuffizienz.
1.14.3.11 Röntgenaufnahme des Thorax
Vergrößerung des linken Ventrikels oder des ganzen Herzens. Oft Zeichen der Lungenstauung.
1.14.3.12 Kardio-CT (nach KM-Gabe)
Optimale Darstellung des vergrößerten linken Ventrikels (◘ Abb. 1.113, ◘ Abb. 1.64).


Cardio-CT (nach KM-Gabe) eines 58-jährigen Patienten mit dilatativer Kardiomyopathie (AO = Aorta, AK = Aortenklappe, LA = linker Vorhof, MK = Mitralklappe, LV = linker Ventrikel)
1.14.3.13 Herzkatheteruntersuchung
-
Rechtsherzeinschwemmkatheter: Erhöhte Druckwerte bei manifester Linksinsuffizienz, Herabsetzung des Minutenvolumens, zunächst unter Belastung, dann auch in Ruhe.
-
Koronarangiographie: Ausschluss einer koronaren Herzkrankheit.
1.14.3.14 Therapie
1.14.3.15 Allgemeinmaßnahmen
Weitgehende körperliche Schonung, bei Dekompensation längere Bettruhe. Beseitigung von Übergewicht, Kochsalzbeschränkung, Alkoholverzicht.
1.14.3.16 Medikamentös Therapie
Siehe Ausführungen über die Therapie der Herzinsuffizienz (▶ Abschn. 1.9). Zusätzlich Dauerantikoagulation zwingend bei Vorhofflimmern.
1.14.3.17 Herzschrittmacher
Evtl. biventrikuläre Schrittmacherstimulation, ggf. mit Defibrillator oder alleiniger Defibrillator.
1.14.3.18 Herztransplantation
Einzige längerfristige Überlebenschance. Ohne Transplantation sterben 25‒40 % der Patienten innerhalb von 5 Jahren nach Stellung der Diagnose, die meisten davon im ersten Jahr.
1.14.4 Hypertrophische Kardiomyopathie (HCM)
1.14.4.1 Häufigkeit
Die Prävalenz in der allgemeinen Bevölkerung wird mit 1:500 angegeben.
1.14.4.2 Ätiologie
Die HCM ist ein Erbleiden mit langsam progredienter, symmetrischer oder asymmetrischer Hypertrophie des linken Ventrikels, von der meistens das Septum am stärksten betroffen ist. Es tritt in mindestens 50 % der Fälle familiär auf, mit autosomal-dominantem Erbgang. Die sporadischen Fälle sind nach molekularbiologischen Untersuchungen auf Neumutationen zurückzuführen. Mit HCM sind mindestens 10 unterschiedliche Gene assoziiert. Bisher hat man 150 differente Mutationen identifiziert. Sie betreffen die Gene für die schwere β-Myosinkette (40 %), für das Troponin (15 %), das Myosin-bindende Protein C (20 %) und das α-Tropomyosin (5 %). Die meisten Fälle von familiärer HCM werden von 1–3 mutanten Genen verursacht. Wie sich die Mutationen im einzelnen auf Struktur und Funktion des Herzmuskels auswirken, ist noch nicht geklärt. Die Expression des Phänotyps zeigt eine beachtliche Variationsbreite hinsichtlich des Schweregrades der Hypertrophie, der klinischen Symptome, des Manifestationsalters und des Verlaufes. Morphologische Krankheitszeichen werden bei 25 % der Verwandten ersten Grades von Patienten mit HCM gefunden. In vielen Fällen ist die Ausprägung der Hypertrophie jedoch milder und mehr lokalisiert.
1.14.4.3 Pathogenese
Ausmaß und Verteilungsmuster der Hypertrophie variieren von Patient zu Patient. Am stärksten ist der linke Ventrikel betroffen und hier meistens asymmetrisch im Bereich der anterolateralen Wand und im vorderen Septumabschnitt. Relativ selten sind hinterer Septumabschnitt und posterobasale Wand am stärksten hypertrophiert. In Japan kommt eine apikale Hypertrophie häufig vor. Histologisch zeigen die Herzmuskelfasern häufiger eine wirbelförmige Anordnung als bei Hypertrophien aus anderen Ursachen.
1.14.4.4 Pathophysiologie
Für die Funktion des Herzens ergeben sich aus der Hypertrophie folgende Konsequenzen:
1.14.4.5 Hyperkontraktilität des linken Ventrikels
Beschleunigte Austreibung des Blutes (90 % in der ersten Hälfte der Systole) und Erhöhung der Ejektionsfraktion. Verkleinerung des endsystolischen Volumens.
1.14.4.6 Dynamische Obstruktion der linksventrikulären Ausflussbahn
Entsteht in Fällen von asymmetrischer Septumhypertrophie wegen Verlagerung des vorderen Mitralsegels in die Ausflussbahn (SAM). Während der Systole kommt es zu einer intraventrikulären Stenose mit oft erheblichem Druckgradienten, besonders postextrasystolisch und bei anderen Bedingungen gesteigerter Kontraktilität. Das Obstruktionsphänomen tritt aber nur bei etwa 25 % der Patienten auf und ist bei ihnen nicht konstant. Die obstruktive Variante hat die Bezeichnung hypertrophe obstruktive Kardiomyopathie (HOCM) erhalten.
1.14.4.7 Diastolische Dysfunktion
Beeinträchtigung der diastolischen Füllung des linken Ventrikels durch Steifheit (verminderte Compliance) der verdickten Wand. Verkleinerung des enddiastolischen Volumens und der Füllungskapazität, mangelhafte Steigerungsfähigkeit des Minutenvolumens unter Belastung, Anstieg des enddiastolischen Ventrikeldrucks mit Drucksteigerung im linken Vorhof, in den Lungenvenen und Lungenkapillaren, Dyspnoe durch Lungenstauung.
1.14.4.8 Rhythmusstörungen
Die Disposition zu Arrhythmien resultiert aus der Überlastung der sekundär dilatierenden Vorhöfe und der Hypertrophie des Kammermyokards mit atypischem Faserverlauf. Häufig sind Vorhofflimmern und Kammerextrasystolie. Die größte Gefahr geht von Kammertachykardien aus, die in Kammerflimmern übergehen können und hauptsächlich unter körperlicher Anstrengung auftreten. Kammerflimmern ist die häufigste Todesursache bei Patienten mit HCM.
1.14.4.9 Klinik
Symptombeginn am häufigsten im 3. Lebensjahrzehnt, aber auch schon im Kindesalter und erst nach dem 60. Lebensjahr. Bereits im Stadium der klinischen Latenz ist die Septumhypertrophie echokardiographisch nachzuweisen. Frühsymptome sind Belastungsdyspnoe, Herzklopfen und Schwindel. Die Leistungsfähigkeit nimmt ab. Es kann zu anginösen Herzschmerzen und Synkopen kommen. Eine muskuläre Herzinsuffizienz tritt nicht auf. Die meisten Symptome sind Ausdruck der diastolischen Dysfunktion. Oft stehen Rhythmustörungen klinisch im Vordergrund.
1.14.4.10 Diagnostik
1.14.4.11 Auskultationsbefund
Bei HOCM spindelförmiges systolisches Geräusch zwischen Spitze und linkem Sternalrand mit Lautstärkenzunahme unter Valsalva-Manöver. Bei sekundärer Mitralinsuffizienz Holosystolikum über der Spitze. Meistens 4., manchmal auch 3. Herzton.
1.14.4.12 EKG
Ausgeprägte Zeichen der Linkshypertrophie mit deszendierenden ST-Strecken und negativem T in den Brustwandableitungen (bei Fehlen einer Hypertonie oder Aortenstenose). Extreme T-Negativitäten im Brustwand-EKG deuten auf eine apikale Hypertrophie hin, die eine relativ gute Prognose hat. In 20–50 % der Fälle Pseudoinfarkt-Q in Abl. II, aVF und III, auch in V4‒V6.
1.14.4.13 Echokardiographie und Doppler
Asymmetrische Hypertrophie des linken Ventrikels, meistens des Septums (Wanddicke 1,3-mal größer als die der Hinterwand). Verkleinerte enddiastolische und endsystolische Volumina. Bei Obstruktion SAM-Phänomen (systolische Vorwärtsbewegung des vorderen Mitralsegels). Der Farbdoppler erfasst die systolische Obstruktion der linksventrikulären Ausflussbahn in Ruhe und unter Valsalva-Manöver, der CW-Doppler die schwere Obstruktion. Septumdicke im M-Mode >15 mm.
1.14.4.14 Röntgenaufnahme des Thorax
Linker Ventrikel normal groß oder durch Zunahme der Muskelmasse vergrößert. Erweiterung des linken Vorhofes.
1.14.4.15 Herzkatheter
Für die Diagnose entbehrlich. Bei retrograder Sondierung des linken Ventrikels ergeben sich erhöhte enddiastolische Drücke. Auch ein systolischer Druckgradient im linken Ventrikel ist nachzuweisen.
1.14.4.16 Therapie
1.14.4.17 Medikamentöse Therapie
β-Rezeptorenblocker gegen Angina pectoris und Synkopen. Calciumantagonisten (Verapamil, Diltiazem) verbessern die Relaxation und senken die Inotropie.
Calciumantagonisten dürfen nicht mit β-Blockern kombiniert werden, weil es zur extremen Bradykardie führen kann.
Digitalis, Diuretika, Vasodilatatoren und Nitrate möglichst vermeiden. Gegen Rhythmusstörungen am besten Amiodaron. Bei Vorhofflimmern Dauerantikoagulation.
1.14.4.18 Schrittmacher
DDD-Schrittmachertherapie kann erforderlich werden, auch biventrikuläre Stimulation. Bei Kammertachykardien Implantation eines Defibrillators.
1.14.4.19 Interventionelle Eingriffe
Bei ausgeprägter Obstruktion Kontrastechokardiographie-gesteuerte Septumablation durch Alkoholinjektion in die Septumarterie. Chirurgische Myektomie von Septumteilen.
1.14.5 Restriktive Kardiomyopathie (RCM)
1.14.5.1 Häufigkeit
Zahlen zur Inzidenz liegen nicht vor. Die RCM ist jedoch die seltenste unter den drei Formen der Kardiomyopathie.
1.14.5.2 Ätiologie
Die ätiologisch ungeklärten Fälle werden als idiopathische RCM bezeichnet. Gelegentlich wurde familiäres Vorkommen beobachtet mit Mutationen am Gen für Troponin I. Kennzeichen der RCM ist eine diastolische Funktionsstörung beider Ventrikel infolge herabgesetzter Dehnbarkeit (Compliance) des Myokards bei normaler oder nur geringer Störung der systolischen Funktion.
Folgende spezifische Erkrankungen führen zur sekundären RCM:
-
Myokardiale Krankheiten
-
nichtinfiltrativ: hypertrophische Kardiomyopathie, diabetische Kardiomyopathie, Pseudoxanthoma elasticum
-
infiltrativ: Amyloidose, Sarkoidose, Morbus Gaucher, Morbus Hurler, Fettinfiltration
-
Speicherkrankheiten: Hämochromatose, Morbus Fabry, Glykogenspeicherkrankheiten
-
-
Endomyokardiale Krankheiten
-
endomyokardiale Fibrose, hypereosinophiles Syndrom, Karzinoid-Syndrom, Karzinommetastasen
-
ionisierende Strahlen
-
Antrazykline
-
Pharmaka (Serotonin, Methysergid, Ergotamin, Quecksilberverbindungen, Busulfan)
-
1.14.5.3 Pathogenese
Bei der idiopathischen RCM besteht meistens eine interstitielle Fibrose. In manchen Fällen ergibt die Biopsie leicht hypertrophierte Myozyten ohne sonstige Auffälligkeiten.
1.14.5.4 Pathophysiologie
Die diastolische Funktionsstörung besteht darin, daß die diastolische Füllung infolge einer abnormen Rigidität der Ventrikel erschwert ist. Der Ventrikeldruck fällt in der frühen Diastole steil ab (dip), steigt rasch wieder an und erreicht gleich ein Plateau. Dip und Plateau ergeben ein „Quadratwurzelzeichen“. Bei normalem oder etwas erniedrigtem Füllungsvolumen bleiben die enddiastolischen Drücke erhöht. Anders als bei der konstriktiven Perikarditis ist der linksventrikuläre Füllungsdruck mindestens 5 mmHg höher als der rechtsventrikuläre.
1.14.5.5 Isolierter Befall des rechten Ventrikels
Stauung vor dem rechten Herzen (Leberschwellung, Aszites, Ödeme) mit Druckerhöhung in den Jugularvenen und im rechten Vorhof. Keine Drucksteigerung im Lungenkreislauf. Vorkommen bei Endomyokardfibrose und Hypereosinophilie-Syndrom (Löffler).
1.14.5.6 Isolierter Befall des linken Ventrikels
Stauung vor dem linken Ventrikel mit Lungenstauung, pulmonaler Hypertonie und Rechtshypertrophie. Vorkommen bei schwerer Linkshypertrophie, Amyloidose, Sarkoidose, Endomyokardfibrose und Löffler-Syndrom.
1.14.5.7 Biventrikulärer Befall
Anstieg des links- und rechtsventrikulären Füllungsdrucks. Zentraler und pulmonaler Venendruck erhöht. Systolischer Druckanstieg im rechten Ventrikel und in der A. pulmonalis. Wegen der eingeschränkten Funktion des rechten Ventrikels keine stärkere Lungenstauung, so dass klinisch die Zeichen der Rechtsinsuffizienz dominieren. Häufigste Manifestation der restriktiven Kardiomyopathie. Bei Endomyokardfibrose und Löffler-Syndrom kommen oft die hämodynamischen Auswirkungen einer Trikuspidal- und/oder Mitralinsuffizienz hinzu.
1.14.5.8 Klinik
Durch Steigerung der Herzfrequenz kann das Minutenvolumen nur begrenzt erhöht werden, weil die Ventrikelfüllung bei Verkürzung der Diastole abnimmt. Daher Belastungsintoleranz, Dyspnoe und Schwäche. Atriale und ventrikuläre Arrhythmien sind häufig. Plötzlicher Herztod durch Kammerflimmern kommt vor. Erregungsleitungsstörungen (AV-Block) bevorzugt bei Sarkoidose und Amyloidose. Murale Thromben rufen Embolien hervor. Halsvenen meistens gestaut. Der Herzspitzenstoß ist im Gegensatz zur Pericarditis constrictiva tastbar.
1.14.5.9 Diagnostik
1.14.5.10 Auskultationsbefund
3. Herzton (protodiastolischer Galopp), bei AV-Klappenbeteiligung holosystolisches Geräusch der Mitral- oder Trikuspidalinsuffizienz.
1.14.5.11 EKG
Linkshypertrophiezeichen bei primärer Linkshypertrophie, Niedervoltage, unspezifische Veränderungen des ST/T-Abschnittes.
1.14.5.12 Echokardiographie und Doppler
M-Mode und 2-D-Echo: Verdicktes (bzw. infiltriertes) Myokard mit verminderter Kontraktionsamplitude. Doppler: Im transmitralen Flussprofil (◘ Abb. 1.8) hohe frühdiastolische Flussgeschwindigkeit (E-Welle) und niedrige spätdiastolische (A-Welle). Im Tissue-Doppler niedrige Gewebegeschwindigkeit als wichtiges Unterscheidungsmerkmal gegenüber der Pericarditis constrictiva.
1.14.5.13 Röntgenaufnahme des Thorax
Geringe oder fehlende Herzvergrößerung. Bei fortgeschrittener Amyloidose und Hämochromatose Kardiomegalie und Lungenstauung, auch Pleuraergüsse. Nachweis von pulmonalen Sarkoidosezeichen.
1.14.5.14 Herzkatheter
Erhöhte enddiastolische Ventrikeldrucke und typische Ventrikeldruckkurven mit enddiastolischer Dip-Plateau-Konfiguration.
1.14.5.15 Endomyokardbiopsie
Nachweis von Infiltration und Fibrose.
1.14.5.16 Kernspintomographie
Ausschluss einer konstriktiven Perikarditis.
1.14.5.17 Spezielle Laboruntersuchungen
Nachweis der Amyloidose evtl. durch Rektumbiopsie, der Sarkoidose durch Lymphknotenbiopsie, der Hämochromatose durch Leberbiopsie und Bestimmung von Serumeisen, Serumferritin und Hämochromatosegen, des Löffler-Syndroms durch das Differenzialblutbild.
1.14.5.18 Therapie
Behandlung des Grundleidens. Antikoagulation. Bei Endokardfibrose operative Ausschälung möglich.
1.15 Koronare Herzkrankheit (KHK)
1.15.1 Allgemeines
1.15.1.1 Definition
Ischämische Erkrankung des Myokards durch eine obliterierende Atherosklerose der Koronararterien mit den klinischen Manifestationen Angina pectoris, stumme Ischämie, plötzlicher Herztod, Herzinfarkt und ischämische Kardiomyopathie.
1.15.1.2 Epidemiologie
Verbreitung weltweit. Nach den Tumorerkrankungen zweithäufigste Todesursache in den zivilisierten Ländern. Häufigkeitsanstieg nach dem 50. Lebensjahr. Frauen sind bis zur Menopause relativ geschützt.
1.15.1.3 Risikofaktoren
Was für die Atherosklerose der mittleren und großen Arterien allgemein gilt, trifft in besonderem Maße für die koronare Herzkrankheit zu. Ihre Ursache ist multifaktoriell, wobei man nicht von Kausalfaktoren spricht, sondern von Risikofaktoren, weil sie nicht obligatorisch zur Erkrankung führen. Das mit bestimmten exogenen und endogenen Faktoren verbundene Risiko wurde durch große prospektive Langzeitstudien ermittelt, letztlich also auf epidemiologischem Wege.
Es zeigte sich, dass Risikofaktoren bei ihrem Zusammentreffen das Risiko nicht additiv, sondern potenzierend erhöhen.
-
Lebensalter: Vor dem 50. Lebensjahr ist die KHK beim Fehlen von Risikofaktoren sehr selten. Im höheren Alter kommt sie jedoch auf rein degenerativer Basis vor, durch eine genetische Disposition begünstigt. Natürlich nimmt mit dem Alter die Einwirkungsdauer endogener und exogener Risikofaktoren zu.
-
Zigarettenrauchen: Das Risiko ist der Zahl der täglich gerauchten Zigaretten proportional. Zwei Jahre nach Aufhören des Rauchens liegt das Risiko nur noch wenig über dem des Nichtrauchers. Infarktpatienten unter 40 Jahren sind ganz überwiegend starke Raucher (>20 Stück/Tag).
-
Hypertonie: Das KHK-Risiko steigt kontinuierlich mit dem Blutdruckniveau. Systolische und diastolische Hypertonie sind von gleicher Relevanz. Wahrscheinlich ist für die mechanische Belastung der Gefäßwand der arterielle Mitteldruck maßgebend.
-
Hypercholesterinämie: Maßgebend ist das LDL-Cholesterin, dessen Nüchternwert im Serum <150 mg betragen sollte. Das HDL-Cholesterin hat dagegen einen protektiven Effekt.
-
Diabetes mellitus Typ 2 und gestörte Glukosetoleranz: Die Kriterien sind Nüchternblutzuckerwerte von 125 mg/dl, bzw. 110 mg/dl. Der atherogene Effekt beruht wahrscheinlich auf der begleitenden Hyperlipidämie. Das LDL-Cholesterin sollte deshalb auf 100 mg/dl gesenkt werden.
-
Adipositas: Gewichtsreduktion erhöht das HDL-Cholesterin, verbessert außerdem die Glukosetoleranz.
-
Hyperhomozysteinämie: Bewirkt eine Endothelschädigung. Wurde bei Infarktpatienten abnorm häufig festgestellt. Mit einer Kombination von Vitamin B6, Vitamin B12 und Folsäure zu beheben.
-
Chlamydia-pneumoniae-Infektion: Infarktpatienten waren gehäuft seropositiv. Erreger-Material wurde in vielen atherosklerotischen Plaques gefunden.
1.15.1.4 Pathogenetische Faktoren
1.15.1.5 Fatty Streaks
Schon bei Kindern und Jugendlichen nachweisbare oberflächliche Fettablagerungen in der Intima. Sie werden als Initialläsion der Atheromatose aufgefasst, können sich aber zurückbilden. Histologisch handelt es sich um eine Schicht aus fettspeichernden Makrophagen mit eingestreuten T-Lymphozyten und glatten Muskelzellen. Das Fett wurde als LDL-Lipoprotein von den Endothelzellen eingeschleust, die LDL-Rezeptoren tragen und LDL-Moleküle durch schwache Oxidation modifizieren können. Zur Phagozytose der oxidierten LDL dringen Monozyten durch Endothelspalten in die Intima ein, wo sie zu Makrophagen werden. Teils transportieren die Makrophagen das gespeicherte Fett (freies und verestertes Cholesterin) wieder in die Blutbahn zurück, teils bleiben sie sesshaft, werden aktiviert und stimulieren durch Freisetzung von Interleukinen ihre eigene und die Proliferation von glatten Muskelzellen. Außerdem bewirken sie die Einwanderung von T-Lymphozyten.
1.15.1.6 Diffuse Intimaverdickungen
Bilden sich an Stellen mit erhöhtem Wandstress. Bestehen aus glatten Muskelzellen mit einer bindegewebigen Umhüllung und können T-Lymphozyten und fettspeichernde Makrophagen enthalten. Auch extrazellulär werden Lipide abgelagert. Anscheinend findet aber keine Weiterentwicklung zu ausgeprägten Plaques statt.
1.15.1.7 Fibröser Plaque
Fortgeschrittene atherosklerotische Läsion in Form einer weißlichen Vorwölbung in das Gefäßlumen. Unter der endothelbesetzten bindegewebigen Kappe liegen proliferierte glatte Muskelzellen zusammen mit zahlreichen Makrophagen und T-Zellen. Glatte Muskelzellen und Makrophagen haben Lipide gespeichert. Unterhalb der zellreichen Zone liegt eine nekrotische lipidhaltige Masse.
1.15.1.8 Sekundäre Thrombose und Plaqueruptur
Risse und Fissuren der Plaques führen zu Auflagerung von Plättchenthromben, durch deren Inkorporation die Plaques weiter wachsen. Schließlich kann es unabhängig von der Plaquegröße zur Ruptur mit frischer Thrombose an der Oberfläche kommen, die das Lumen vollständig verschließt. Die Mehrzahl der Myokardinfarkte kommt durch akute Thrombosierung zustande.
1.15.1.9 Pathophysiologische Faktoren
1.15.1.10 Koronarperfusion
Die Blutversorgung des Herzens erfolgt durch die rechte (RCA) und linke Kranzarterie (LCA). Letztere verzweigt sich nach kurzem Hauptstamm in zwei große Äste, den R. interventrikularis anterior (RIVA) und den R. circumflexus (RCX). Zusammen sind es 3 epikardiale Arterienstämme, die selektiv stenosieren können. Man spricht folglich von Ein-, Zwei- und Dreigefäßerkrankung und beim Befall der proximalen LCA von einer Hauptstammstenose. Die epikardialen Arterien sind nur Leitungsgefäße. Die Regulation des Blutflusses obliegt den Widerstandsgefäßen im Myokard (Arteriolen, präkapilläre Sphinkter).
Durch metabolische Autoregulation wird der koronare Blutfluss automatisch dem Sauerstoffverbrauch bzw. Stoffwechsel des Myokards angepasst. Bei Anstieg des O2-Bedarfs erweitern sich die Widerstandsgefäße, bei Absinken des O2-Bedarfs nimmt ihr Lumen ab. Bei 80%iger Stenose einer epikardialen Arterie kann der Ruhebedarf an O2 durch maximale Vasodilation noch gedeckt werden.
Das autonome Nervensystem wirkt auf die Koronarperfusion hauptsächlich indirekt über seinen Effekt auf den Myokardstoffwechsel. Sympathikusstimulation steigert den myokardialen Sauerstoffverbrauch erheblich und auf diesem Wege die koronare Vasodilation. Vagusstimulation bewirkt über Drosselung der Herzfrequenz das Gegenteil.
1.15.1.11 Determinanten des myokardialen Sauerstoffverbrauchs
-
Gesamtverbrauch: Pro Minute und 100 g Gewebe unter Stillstandsbedingungen 1,5 ml, bei körperliche Ruhe über 8 ml, bei maximaler Belastung bis 80 ml O2.
-
Systolische Wandspannung: (Druckarbeit): Fast linearer Anstieg des O2-Verbrauchs mit der systolischen Spannungsentwicklung in den Muskelfasern des linken Ventrikels. Somit erhöhter O2-Verbrauch bei Hypertonie und bei Dilatation des insuffizienten Herzens.
-
Systolische Faserverkürzung (Volumenarbeit): Der Auswurf des Schlagvolumens erfordert wesentlich weniger Sauerstoff als die Spannungsentwicklung.
-
Herzschlagfrequenz: Fast linearer Anstieg des O2-Verbrauchs mit Zunahme der Schlagfrequenz, weil Arbeitsleistung und Kontraktilität größer werden.
-
Kontraktilitätssteigerung durch positiv inotrope Substanzen: Erheblicher Anstieg des O2-Verbrauchs bei erregungsbedingter Ausschüttung von Noradrenalin und Adrenalin und durch Katecholamine wie Dobutamin, auch durch Digitalis.
-
Basalstoffwechsel: Relativ geringer O2-Verbrauch für alle nicht mit der Kontraktion zusammenhängenden Prozesse (unter Ruhebedingungen 19 % des Gesamtbedarfs).
1.15.1.12 Myokardiale Ischämie
Ungenügende Sauerstoffzufuhr infolge inadäquater Koronarperfusion.
-
Ursachen: Obstruktion der epikardialen Koronararterien durch atheromatöse Plaques, mikrovaskuläre Dysfunktion mit inadäquater Dilatationsreserve, Stenosierung der Koronarostien bei Aortitis luica, Arteriitis der Kranzgefäße, Embolie, inadäquater Perfusionsdruck bei Aortenstenose, arterieller Hypotonie und im Kreislaufschock.
-
Folgen der Myokardischämie:
-
Lokale Stoffwechselstörung: Gewebehypoxie, Laktatazidose, Freisetzung schmerzauslösender Substanzen.
-
Kontraktilitätsverlust: Bis zur vollständigen Akinesie im ischämischen Bezirk, wahrscheinlich infolge einer Störung der ATP-abhängigen Ca++-Freisetzung aus dem Sarkolemm und dem sarkoplasmatischen Retikulum. Ferner durch die Azidose. Wenn 20 % von der Wand des linken Ventrikels akut akinetisch werden, kommt es zur Linksinsuffizienz, bei 40 % zum kardiogenen Schock.
-
Störung der Relaxation: Verzögerte und unvollständige Erschlaffung des Ventrikelmyokards in der Diastole. Wahrscheinlich durch eine Störung der energieabhängigen Ca++-Aufnahme in das sarkoplasmatische Retikulum nach der Kontraktion. Die Abnahme der Compliance erschwert die diastolische Kammerfüllung. Der enddiastolische Druck steigt.
-
Elektrophysiologische Störungen: Beruhen überwiegend auf lokaler Herabsetzung des Membranpotenzials bzw. Störungen der Repolarisation. Manifestationen: Ischämische EKG-Zeichen, Erregungsleitungsstörungen, Rhythmusstörungen, im Extremfall Kammerflimmern.
-
Morphologische Veränderungen: Trübe Schwellung und feintropfige Verfettung (reversible Hypoxie- und Ischämiefolgen). Nach Gefäßverschluss innerhalb 20 Minuten reversible Mitochondrienschädigung (Schwellung, Granulabildung), danach irreversible Membranschäden mit Enzymaustritt und Nekrose (Infarkt).
-
Koronare Herzkrankheit
-
Angina pectoris
-
Stumme Ischämie
-
Mikrovaskuläre Ischämie („Syndrom X“)
-
Akuter Myokardinfarkt
1.15.2 Angina pectoris
1.15.2.1 Definition
Klinische Manifestation der koronaren Herzkrankheit mit anfallsartigen reversiblen Brustschmerzen infolge myokardialer Ischämie.
1.15.2.2 Klassifikation
1.15.2.3 Stabile Angina pectoris
Schmerzattacken bei einer bestimmten, den Sauerstoffbedarf des Myokards steigernden körperlichen oder emotionalen Belastung. Die Schmerzschwelle kann nach den Mahlzeiten, bei Kälte und Wind herabgesetzt sein. Am Kranzgefäßsystem bestehen in der Regel fixierte obstruktive Läsionen.
1.15.2.4 Instabile Angina pectoris (Präinfarktangina )
Schmerzattacken, die auf eine rasch progrediente, bedrohliche Ischämie hinweisen:
-
Verschlimmerung einer stabilen belastungsinduzierten Angina pectoris durch Zunahme der Schwere, Dauer und Häufigkeit der Anfälle.
-
Angina pectoris in Ruhe und bei geringster Belastung.
-
Innerhalb eines Monats neu aufgetretene Angina pectoris bei geringer Belastung. Reversible EKG-Veränderungen im Anfall (ST-Senkung oder -Hebung, T-Inversion). Angiographisch meistens schwere Mehrgefäßerkrankung. Plötzliche Zunahme der Lumeneinengung durch Spasmen oder Thromben.
Infarktausschluss nur durch EKG und fehlenden Enzymanstieg möglich.
1.15.2.5 Variant-Angina (Prinzmetal )
Schmerzattacken, die in Ruhe, meistens in der zweiten Nachthälfte auftreten, mit transmuraler Ischämie (ST-Hebung) und Rhythmusstörungen. Sie werden nicht durch körperlichen oder emotionalen Stress ausgelöst. Nur wenige Patienten haben Schmerzattacken bei körperlicher Belastung. Die Ischämie wird durch Spasmen der epikardialen Koronararterien ausgelöst, die sich fixierten Stenosierungen überlagern können (Kombination von stabiler und Variant-Angina). Meistens ist das Koronarangiogramm bis auf minimale Läsionen unauffällig.
1.15.2.6 Klinik
1.15.2.7 Anfallauslösende Faktoren
Körperliche Anstrengung, Arbeit mit den Händen oberhalb des Kopfes, kalte Witterung, Spaziergang nach dem Essen oder gegen den Wind, emotionaler Stress während körperlicher Belastung (Wettkampfsport), seelische Erregung (Angst, Ärger, Freude).
1.15.2.8 Schmerzqualität und Begleiterscheinungen
Dumpfer, quälender Druck, ein Schweregefühl oder Brennen in der Brust, oft mit Enge- und Beklemmungsgefühl verbunden, durch Atmung und Lageänderung unbeeinflusst. Innerhalb weniger Minuten zur maximalen Intensität anschwellend, dann allmählich nachlassend. Manchmal nur leichtes unbestimmtes Druckgefühl. Bei schweren Attacken Angst, Erregung, Schweißausbruch und Tachykardie mit Blutdruckanstieg (Sympathikusstimulation). Trotz ischämischer EKG-Zeichen können Schmerzen ausbleiben (stille Ischämie), besonders bei Diabetikern (autonome Denervation im Bereich der sensorischen Endplatten der intrakardialen sympathischen Nerven), manchmal nach Herzinfarkt, wenn sympathische Endplatten zerstört sind.
1.15.2.9 Schmerzlokalisation
Ganz überwiegend retrosternal. Schmerzen über der Herzspitze sind meistens nicht koronaren Ursprungs. Gelegentlich Schmerzen nur in der linken Schulter und im linken Arm, selten im rechten Arm, im Unterkiefer, in der unteren Halswirbelsäule oder in der linken Interskapularregion. Ausstrahlung zur Innenseite des linken Armes, zur linken Schulter, gelegentlich zum rechten Arm oder in beide Arme zugleich.
1.15.2.10 Schmerzdauer
Eine halbe Minute bis zu 30 Minuten. Bei längerer Dauer besteht Infarktverdacht.
1.15.2.11 Nitroglycerineffekt
Nachlassen der Schmerzen 45 Sekunden bis 5 Minuten nach sublingualer Applikation von Nitroglycerin als Kapsel oder Spray. Ausbleibender Effekt bei beginnender Myokardnekrose oder nichtkoronarer Schmerzursache.
Differenzialdiagnose des Brustschmerzes: Generell fehlt bei den nichtkoronaren Brustschmerzen die typische Belastungsabhängigkeit der Angina pectoris. Die Ruhe-Angina ist durch den positiven Nitroglycerineffekt abzugrenzen, der sonst nur noch beim Ösophagusspasmus vorkommt. Angina-ähnliche Schmerzzustände können bei folgenden Erkrankungen auftreten: Aortendissektion (Schmerz sofort maximal), Mitralklappenprolaps (oberflächlicher, mehr über der Spitze lokalisiert), Perikarditis (lässt im Sitzen nach), pulmonale Hypertonie (Lungenembolie), Pleuritis (atemabhängiger Schmerz), Spontanpneumothorax (Dyspnoe), Tietze-Syndrom (Druckschmerz der Kostosternalgelenke bzw. des Rippenknorpels), zervikale Radikulitis (Röntgenbefund an der HWS), Refluxösophagitis mit Spasmen (Hiatushernie).
1.15.2.12 Diagnostik
1.15.2.13 Anamnese
Feststellung des persönlichen koronaren Risikoprofils (familiäre Infarktbelastung, Hypertonie, Diabetes mellitus, Hyperlipidämie, Zigarettenkonsum). Wichtig sind auch Krankheitszustände mit Hypoxie (schwere Anämie, Asthma bronchiale, chronische obstruktive Lungenkrankheit, zyanotische Vitien). Nicht zuletzt sollten beruflicher und privater Stress eruiert werden.
1.15.2.14 Ruhe-EKG
In etwa 50 % der Fälle normal. Häufig, aber unspezifisch und deshalb diagnostisch nicht verwertbar sind Veränderungen des ST-T-Abschnittes, linksanteriorer Hemiblock, Schenkelblocks und diverse Rhythmusstörungen, insbesondere ventrikuläre Extrasystolen. Nur reversible Veränderungen im Angina-pectoris-Anfall (vor allem ST-Senkung oder -Hebung) zeigen eine Ischämie an. Alte Infarktnarben (pathologische Q-Zacken) bestätigen das Vorliegen einer koronaren Herzkrankheit.
1.15.2.15 Belastungs-EKG
Ergometerbelastung (Fahrrad im Liegen oder Sitzen, Laufband) unter fortlaufender EKG-Kontrolle (12 Ableitungen) und intermittierender Blutdruckmessung, wobei die Belastung stufenweise, z. B. alle 2 min um 25‒50 Watt, gesteigert wird. Ausbelastung bei 85 % der maximalen altersabhängigen Herzfrequenz (220 minus Alter). Belastungsabbruch bei Auftreten anginöser Beschwerden und einer horizontalen ST-Senkung um ≥0,1 mV, ferner bei ST-Hebung, polytopen VES, supraventrikulären Arrhythmien, Blutdruckanstieg auf 250/115 mmHg Blutdruckabfall und Erschöpfung. Bei Patienten mit typischer Angina-pectoris-Anamnese sollte keine Belastung, sondern gleich eine Koronarangiographie durchgeführt werden. Nach Ende der Belastung 6 Minuten Erholung unter EKG-Kontrolle. Ein Defibrillator muss sicherheitshalber bereitstehen.
Ischämiekriterium ist eine horizontale ST-Senkung ab 0,1 mV in den Extremitätenableitungen und ab 0,2 mV in den Brustwandableitungen, eine isolierte T-Negativierung jedoch nicht (◘ Abb. 1.114). Je früher und stärker sie auftritt und je länger sie anhält, desto höhergradig die Koronarstenosen. Die ST-Senkung erscheint in den Ableitungen mit dominanter R-Zacke und nimmt mit der Höhe der R-Zacke zu. Als Null-Linie für die Messung der ST-Senkung dient die TP-Linie, bei Tachykardie die PQ-Linie. Ursache der ST-Senkung ist eine Ischämie (der am schlechtesten durchbluteten) Innenschicht, die früher repolarisiert oder in der Diastole depolarisiert bleibt. Eine ST-Senkung mit steilem Anstieg der ST-Strecke ist nicht pathologisch. Wenn die ST-Strecke schon im Ruhe-EKG verändert ist, sind ST-Senkungen unter Belastung nicht eindeutig zu bewerten (Digitalismedikation, Hypokaliämie, schwere Linkshypertrophie, Schenkelblocks, WPW-Syndrom).


Ischämiereaktion im Belastungs-EKG


Zur echokardiographischen Beurteilung und Lokalisation einer koronaren Herzkrankheit werden die Wandbewegungen des linken Ventrikels in 16 Segmenten hinsichtlich ihrer Kontraktilität beurteilt. Das Schema zeigt die Zuordnung der Segmente zu den Koronargefäßen
Es kommen falsch positive und falsch negative Befunde vor. Die Sensitivität des Belastungs-EKG variiert bei Eingefäßerkrankung von 25–71 % und erreicht bei 3-Gefäßerkrankung etwa 86 %. Von anginösen Beschwerden begleitete ST-Senkungen sind fast immer ein Ischämiesymptom. ST-Hebungen sind Ausdruck einer transmuralen Ischämie und als bedrohliches Zeichen anzusehen.
1.15.2.16 Myokardszintigraphie
Ermöglicht den direkten Nachweis belastungsinduzierter Ischämien und übertrifft damit das Belastungs-EKG an diagnostischer Treffsicherheit (74–96 %). Unmittelbar vor Beendigung der Ergometerbelastung wird 201Thallium injiziert. Im sofort nach der Belastung aufgenommenen Szintigramm stellen sich ischämische Bezirke als totale oder partielle Speicherdefekte dar, die im zweiten Szintigramm nach 2‒4 h verschwunden sind, während Infarktnarben permanente Defekte entstehen lassen. Strahlenbelastung geringer als bei Thoraxdurchleuchtung. Indikationen:
-
typische oder atypische Angina pectoris mit normalem oder grenzwertigem Belastungs-EKG
-
pathologisches Belastungs-EKG ohne Angina pectoris
-
Verdacht auf koronare Herzkrankheit bei Interpretationsproblemen des Belastungs-EKG (Schenkelblockbilder, ST-T-Veränderungen im Ruhe-EKG, Linkshypertrophie, Digitalisierung)
-
mittelgradige Koronarstenosen zur Prüfung der hämodynamischen Relevanz
1.15.2.17 Stressechokardiographie (◘ Abb. 1.115)
Ein methodisch etwas aufwendiges, aber sehr sicheres Verfahren ohne Strahlenbelastung zur Aufdeckung einer koronaren Herzkrankheit (Sensitivität 74–96 %). Indikationen:
-
Angina pectoris bei negativem Belastungs-EKG
-
atypische Angina pectoris
-
Risikopatienten
-
Prüfung der hämodynamischen Relevanz einer Stenose
-
Kontrolle des Revaskularisierungserfolges
1.15.2.18 Rechtsherzeinschwemmkatheter
Erfasst die Auswirkungen der koronaren Herzkrankheit auf die diastolische und systolische Ventrikelfunktion. In Ruhe und unter Ergometerbelastung werden der Lungenkapillardruck (PCP) als Referenzwert für den enddiastolischen Füllungsdruck des linken Ventrikels und das Minutenvolumen bestimmt. Sobald es unter der Belastung zur Ischämie kommt, steigt der PCP deutlich an, weil die Compliance des linken Ventrikels abnimmt. Bei stärkeren Graden der Ischämie nimmt gleichzeitig das Minutenvolumen nicht mehr belastungsadäquat zu. Als indirekte Methode nur noch wenig gebräuchlich.
1.15.2.19 Koronarangiographie und Ventrikulographie
Die kombinierte Darstellung der Koronararterien (◘ Abb. 1.116) und des linken Ventrikels mit Kontrastmitteln ermöglicht die definitive Diagnose bzw. den Ausschluss einer koronaren Herzkrankheit. Sie erfasst den Schweregrad und die Lokalisation der Arterienobstruktion und der durch Infarkte eingetretenen Wandschädigung. Damit bildet sie die Grundlage für Eingriffe zur Revaskularisierung.


61-jähriger Patient mit koronarer 2-Gefäßerkrankung. Computertomographische (oben) und koronarangiographische (unten) Darstellung einer 70%igen Stenose der rechten Koronararterie (RCA) und einer 70 %igen Stenose des Ramus marginalis I (RCX = R. circumflexus)


Primäres Stenting einer 90%igen Stenose der rechten Koronararterie bei einem 68-jährigen Patienten mit progredienter belastungsabhängiger Angina-pectoris-Symptomatik
-
Indikationen: Im Hinblick auf das sehr geringe Risiko der Untersuchung einerseits und der Möglichkeit der gleichzeitigen Ballondilatation signifikanter Stenosen andererseits, sollte die Koronarangiographie bei jeder typischen Angina pectoris, bei klinisch dringendem Verdacht auf KHK und bei objektivierter Ischämiereaktion angestrebt werden. Diese Empfehlung wird durch die Beobachtung gestützt, dass bei relativ milder Angina pectoris nicht selten hochgradige Koronarstenosen gefunden werden.
-
Methoden: Retrograde Sondierung der Koronararterien und des linken Ventrikels nach dem Verfahren von Judkins: Mit der Seldinger-Technik werden über die A. femoralis transkutan verschiedene Katheter zur Ventrikulographie und zur Sondierung der rechten und linken Koronararterie eingeführt. Die Kontrastmittelinjektion wird auf dem Fernsehschirm beobachtet und mit der Kinobildaufnahmetechnik aufgezeichnet. Bei Stenosierung der Beckenarterien erfolgt die Sondierung nach der Methode von Jones über die A. brachialis dextra oder die A. radialis.
1.15.2.20 Allgemeine Maßnahmen
1.15.2.21 Instruktionen
Erklärung der koronaren Herzkrankheit, auch den nächsten Angehörigen. Anginaschmerz als Warnsignal darstellen. Die anfallsauslösenden Faktoren benennen: Kälte, große Höhe, Schwüle, feuchte Luft, körperliche Belastung und Aufregungen aller Art. Entsprechende Anpassung der Lebensweise und leichtes Training unterhalb der Anginaschwelle empfehlen (Koronarsportgruppe).
1.15.2.22 Maßnahmen gegen die Risikofaktoren
Absolutes Rauchverbot, optimale Blutdruckeinstellung (nach Langzeitprotokoll), optimale Diabeteskontrolle inklusive Gewichtsreduktion, Senkung der LDL-Werte, unter den Normalbereich.
1.15.2.23 Medikamentöse Therapie
1.15.2.24 Organische Nitrate
-
Substanzen: Nitroglycerin (Glyceroltrinitrat), Isosorbitdinitrat (ISDN), Isosorbit-5-Mononitrat (IS-5-MN), Pentaerythrityltetranitrat (PETN).
-
Wirkungsmechanismus: Relaxation der glatten Muskelzellen in der Gefäßwand von Venen und Arterien nach Metabolisierung zu Nitroxid (NO), das auch endogen als Vasodilatator gebildet wird (▶ Abschn. 1.1.1). Zur Vasodilation führt folgende Reaktionskette: NO → Aktivierung der Guanylatcyclase (nach Umwandlung in S-Nitrosothiol) → Bildung von cGMP aus GTP → Absinken der intrazellulären Ca++-Konzentration → Relaxation der Muskelzellen.
-
Kreislaufeffekte:
-
Bei der üblichen (relativ niedrigen) Dosierung überwiegt die Relaxation der Venen: → Blutpooling im Venensystem → Abnahme des venösen Rückflusses → Herabsetzung des enddiastolischen Volumens → Abnahme von Prä- und Afterload → Reduzierung des myokardialen O2-Verbrauchs.
-
Relaxation der epikardialen Koronararterien und Beseitigung von Koronarspasmen: → Zunahme des koronaren Blutflusses, bevorzugt in den ischämischen Bezirken, weil hier die Widerstandsgefäße durch die Autoregulation weitgestellt sind.
-
Abnahme des Arteriolenwiderstandes: Schon bei niedrigen Dosen im Bereich des Gesichts (Flush) und der Meningen (Kopfschmerz). Erst bei höheren Dosen auch systemisch: → Blutdruckabfall, reflektorische Tachykardie.
-
-
Nitrattoleranz: Nachlassen des therapeutischen Effektes bei Applikation in kurzen Intervallen, unter Dauerinfusion schon nach einem Tag. Mögliche Ursachen: Mangel an Sulfhydrilgruppen, Volumenexpansion durch venöses Pooling, gegenregulatorische Sympathikusaktivierung. Vermeidbar durch Therapieunterbrechung für 8–12 h pro Tag, am besten in der Nacht. Vielleicht können ACE-Blocker der Nitrattoleranz entgegenwirken.
-
Therapeutische Anwendung: Nitrolingual wird von der Mundschleimhaut resorbiert und wirkt nach 1–2 Minuten. Zerbeißkapseln (0,8 mg) oder Sprays (0,4 mg je Sprühstoß) sind die Mittel der Wahl für den Anfall. Die Dosis kann nach 5 Minuten wiederholt werden. Bei der instabilen Angina pectoris kann eine Infusion nötig werden. Diese Patienten gehören auf eine Intensivstation. Auch ISDN wird sublingual resorbiert beginnt aber erst nach 3‒6 Minuten zu wirken (Tabl. zu 5–10 mg, Sprühstoß 1,25 mg). Für die perorale Langzeittherapie zur Anfallsprophylaxe dienen ISDN (2–3×20–60 mg/Tag), IS-5-MN (2–3×20–40 mg/Tag) und PETN (2×80 mg/Tag). Von nitratähnlicher Wirkung und ein Plättchenaggregationshemmer ist Molsidomin (2×8 mg/Tag).
-
Nebenwirkungen: Flush, Kopfschmerzen, oft nach einigen Tagen nachlassend. Bei höherer Dosis Schwindelgefühl und Hypotonie bis zum Kreislaufkollaps. Die Kreislaufwirkung ist im Stehen größer als im Liegen. Bei Nitratunverträglichkeit kann alternativ Molsidomin eingesetzt werden.
1.15.2.25 β-Rezeptorenblocker
-
Substanzen: Nichtselektive (Propanolol) und kardioselektive (Metoprolol, Atenolol) Blocker.
-
Wirkungsmechanismus: Alle β-Blocker setzen die Schlagfrequenz und Kontraktilität des Herzens sowie den Blutdruck herab. Damit reduzieren sie den myokardialen Sauerstoffverbrauch. Therapeutisch sind die kardioselektiven β1-Blocker vorzuziehen, da von ihnen die vasodilatatorischen β2-Rezeptoren der koronaren und peripheren Arterien nicht gehemmt werden.
-
Therapeutische Anwendung: Mittel der Wahl für die Langzeittherapie der Angina pectoris, da die Arbeitskapazität erhöht und die Anfallshäufigkeit gesenkt wird. Mittlere Tagesdosen: Metoprolol 2×50 mg, Atenolol 1×50–100 mg. Die Kombination mit Nitraten ist sinnvoll. Auf Intensivstationen werden β-Blocker zusätzlich zu Nitraten eingesetzt, um die Patienten zu stabilisieren. Nebenwirkungen: Bronchialspasmen, periphere zirkulatorische Vasokonstriktion, selten Koronarspasmen, herabgesetzte Gegenregulation bei Hypoglykämie, Verstärkung einer Bradykardie.
1.15.2.26 If-Kanalblocker
Der If-Kanalblocker Ivabradin hemmt den Frequenzanstieg unter Belastung und verbessert bei Koronarkranken die Gehstrecke. Substanzen: Bisher nur Ivabradin, das die If-Kanäle des Sinusknotens hemmt.
-
Wirkungsmechanismus: Durch Hemmung der diastolischen Depolarisation des Sinusknotens wird die Herzfrequenz herabgesetzt und damit auch die Angina-pektoris-Schwelle.
-
Therapeutische Anwendung: Zugelassen für Patienten mit stabiler Angina pektoris, bei denen eine Kontraindikation oder Unverträglichkeit für Betablocker besteht. Die Wirksamkeit ist vergleichbar oder besser. Kombination mit β-Bockern zulässig.
-
Nebenwirkungen: In 15 % der Fälle Phosphene (Helligkeiten im Gesichtsfeld), die sich unter der Therapie meistens zurückbilden.
-
Dosierung: 1–2×5 mg p.o./Tag.
1.15.2.27 Calciumantagonisten
-
Substanzen: Dihydropyridine (Nifedipin, Nicardipin, Amlodipin, Felodipin, Isrodipin), Phenylalkylamine (Verapamil), Benzodiazepine (Diltiazem).
-
Wirkungsmechanismus: Hemmung des Ca++-Einstroms in die glatten Muskelzellen der Arterien, Arteriolen und der Herzmuskelzellen. Kein Effekt auf die Venen, daher in Gegensatz zu den Nitraten kein venöses Pooling.
-
Kreislaufeffekte: Blutdrucksenkung durch periphere Vasodilation, Lösung von Koronarspasmen, Herabsetzung der myokardialen Kontraktilität (negative Inotropie).
-
Therapeutische Anwendung: Nifedipin (3×10–20 mg/Tag) ist wegen seiner spasmolytischen Wirkung das Mittel der Wahl bei nachgewiesener Prinzmetal-Angina, im Anfall (zusammen mit Nitrolingual) und langfristig zur Anfallsprophylaxe. Bei der stabilen Angina pectoris werden Calciumantagonisten erst eingesetzt, wenn β-Rezeptorenblocker nicht toleriert werden oder kontraindiziert sind. Sie reduzieren die Anfallshäufigkeit und verbessern die Arbeitstoleranz. Dihydroperidine dürfen nur in Retardform und nur in Kombination mit β-Rezeptorenblockern gegeben werden, weil ihre starke periphere Vasodilation eine Sympathikusaktivierung induziert, die den myokardialen O2-Verbrauch erhöht. Die eigene negativ-inotrope Wirkung ist zu schwach. Verapamil und Diltiazem sind schwächer vasodilatatorisch und stärker negativ inotrop wirksam und können einzeln gegeben werden, nicht aber in Kombination mit β-Blockern. Bei instabiler Angina pectoris sind Calciumantagonisten kontraindiziert.
1.15.2.28 Ranolazin
Ein neues zusätzliches Mittel zur Behandlung der stabilen Angina pectoris, die auf andere Mittel nicht ausreichend angesprochen hat. In vielen Fällen nimmt die körperliche Belastbarkeit zu. Das Piperazidinderivat hat eine einzigartige Wirkung. Es hemmt den späten Natriumeinstrom in die Myokardzellen und vermindert dadurch die zytoplamatische Calciumkonzentration, die der Natriumeinstrom ansteigen lässt. Zugleich wird einer Verlängerung des Aktionspotenzials und damit der QT-ZeitIntervalls entgegengewirkt. Die Relaxationszeit wird länger, womit der diastolische Koronarfluss zunimmt.
-
Dosierung: Initial 2×375 mg/Tag, nach 2–4 Wochen 2×500 mg/Tag, maximal 2×750 mg/Tag.
1.15.2.29 Antikoagulation
-
Inhibitoren der Thrombozytenaggregation:
-
Acetylsalicylsäure (ASS): Inaktiviert die Cyclooxygenase der Thrombozyten und setzt dadurch ihre Aggregationsneigung herab. Ausgedehnte Studien haben gezeigt, dass ASS das Fortschreiten der koronaren Herzkrankheit verzögern und das Infarktrisiko verkleinern kann. Je schwerer die Angina pectoris, desto dringender ist ASS (100–200 mg/Tag) indiziert.
-
Clopidogrel und das analoge Ticlopidin: Hemmen die Bildung von Fibrinbrücken zwischen den Plättchen und damit die Thrombenbildung. Gegen Ticlopidin entwickelt sich häufig eine Allergie.
-
Prasugel : Bioaktivierung zuverlässiger als bei Clopidogrel.
-
Ticagrelor : Reversibler Inhibitor der ADP-induzierten Plättchenaggregation, schneller und stärker wirksam als Clopidogrel.
-
-
Inhibitoren der Blutgerinnung:
-
Phenprocoumon, Warfarin: Angezeigt, wenn Aggregationshemmer nicht vertragen werden.
-
Heparine: Für die Soforttherapie der instabilen Angina pectoris, bei der sich oft eine Plaquethrombose entwickelt.
-
1.15.2.30 Revaskularisation
Im Prinzip bei jeder symptomatischen koronaren Herzkrankheit indizeirt, wenn die Koronarangiographie eine bedrohliche Stenosierung aufdeckt. Das gilt auch für Fälle mit stummer Ischämie. Die zur Verfügung stehenden Methoden haben sehr hohe Erfolgsquoten bei niedrigem Risiko und sind auch im höheren Alter anzuwenden.
1.15.2.31 Perkutane koronare Revaskularisation (PCR)
Erstmals 1977 von Grüntzig durchgeführt, kommt sie dank vieler technischer Verbesserungen jetzt häufiger zum Einsatz als die Bypass-Operation. Letztere wird in den meisten Fällen bei Hauptstammstenose der LCA und multiplen Stenosen angewandt, ferner bei vitalem Myokard distal von Totalverschlüssen und verkalkten ungünstig gelegenen Stenosen. Bei Stenosen, die beiden Methoden zugänglich sind, ist die Mortalität vergleichbar, die Restenoserate aber für die PCR (mit unbeschichteten Stents) deutlich höher (20–50 % versus 7–10 %). Restenosen lassen sich aber meistens wieder eröffnen. Als Sofortmaßnahme ist der PCR bei instabiler Angina pectoris angezeigt.
1.15.2.32 Ballonkatheter
Die zuerst eingeführte, als perkutane transluminale koronare Angioplastie (PTCA) bezeichnete Methode. Ein Katheter mit aufblasbarem länglichem Ballon an der Spitze wird über die Fermoralarterie mit einem Führungsdraht in die Koronararterie vorgeschoben. Der im stenosierten Lumen platzierte Ballon wird dann für 1–3 Minuten mit 2–20 atm aufgeblasen. Der Dilatationserfolg ist fast immer mit einer lokalen Dissektion der Gefäßwand verbunden, wie sich durch intrakoronare Ultraschalluntersuchung nachweisen lässt. Die Wandschädigung führt in 3 % der Fälle zum akuten thrombotischen Gefäßverschluss und einer hohen Restenoserate (bis 40 % in 6 Monaten).
1.15.2.33 Stentimplantation (◘ Abb. 1.117)
Ein kollabiertes, auf einen Ballon montiertes Drahtgeflecht wird mit einem Katheter in den stenosierten Gefäßabschnitt eingeführt und durch Aufblasen des Ballons zur Entfaltung gebracht. Wandläsionen entstehen dabei kaum. Die Restenoserate sinkt um die Hälfte. Dennoch entstehende Restenosen basieren auf neointimaler Hyperplasie, die neuerdings durch intrakoronare β- oder γ-Bestrahlung gehemmt werden kann. Der Dilatation folgt eine 4-wöchige Nachbehandlung mit ASS (100 mg/Tag) und Clopidogrel (75 mg/Tag). ASS wird langfristig weiter verordnet. Inzwischen werden 70–80 % aller dilatierenden Eingriffe mit der Stenttechnik durchgeführt.
1.15.2.34 Medikamente freisetzende Stents (drug eluting stents )
Es handelt sich um Stents, die mit einem Polymer beschichtet sind, aus dem über einige Wochen zytostatische und immunsuppressive bzw. antiproliferative Substanzen freigesetzt werden. Sie können, wie sich gezeigt hat, die zur Restenose führende Proliferation der Neointima in dilatierten Stenosen supprimieren, wenn auch nicht ganz verhindern. Der CYPHER-Stent eluiert Sirolimus, das die durch Wachstumsfaktoren und Zytokine induzierte Zellproliferation hemmt. In der SIRUS-Studie an 1058 Patienten betrug nach 8 Monaten das Zielgefäßversagen (Revaskularisierungsbedarf, Herzinfarkt, Tod) mit dem CYPHER-Stent 8,6 %, mit unbeschichtetem Stent dagegen 21 %. Der TAXUS-Stent setzt aus einem Polymer den Mitoseinhibitor Paclitaxel frei und erzielt ähnlich günstige Ergebnisse. Die Entwicklung auf diesem Gebiet geht weiter. Nach DE-Stentimplantation ASS + Clopidogrel für 6 Monate.
1.15.2.35 Brachytherapie bei In-Stent-Stenose n
Zur Verhütung von Restenosen hat man eine katheterapplizierte lokale Radiotherapie angewandt. In den dilatierten Stenosebereich im Stent wurden bei liegendem Katheter für kurze Zeit Isotope mit Gamma- oder Betastrahlung eingebracht. Mit dieser Methode senkte sich die Restenoserate um bis zu 50 %. Der günstige Effekt hielt häufig jahrelang an. Da ein aufwendiger Strahlenschutz erforderlich ist, benutzt man meistens die Drug-eluting-Stents auch zur Therapie der In-Stent-Stenosen.
1.15.2.36 Chirurgische Revaskularisierung
Indiziert vor allem bei mindestens 50%iger Hauptstammstenose und bei komplizierten multiplen Stenosen:
-
Aortokoronarer Venenbypass mit V. saphena magna (ACVB): In der Regel sind mehrere Stenosen zu überbrücken. Das kann mit Einzeltransplantaten (single grafts) mit zweifachem Anschluss eines y-förmigen Transplantats (jump-graft) oder mit einem sequenziellen Bypass (sequential graft) geschehen, bei dem mit einer Vene mehrere Arterien distal der Stenose anastomosiert werden.
-
A.-mammaria-interna-Bypass (IMA-Bypass): Anastomosierung der linken IMA (LIMA) hauptsächlich mit dem R. descendens anterior (RIVA) der LCA, aber auch mit dem linken R. circumflexus. Für Stenosen der RCA kann die rechte IMA (RIMA) verwendet werden. Bei gut zugänglichem RIVA lässt sich der Eingriff auch mit minimal invasiver Technik durchführen, also ohne Herz-Lungen-Maschine.
-
Minimal-invasive Methoden:
-
Off pump coronary artery bypass (OPCAP): Operation am schlagenden Herzen ohne Einsatz der Herz-Lungen-Maschine, aber mit Spaltung des Sternum. Mit dem Octopus-Stabilisator wird der Anastomosenbereich ruhiggestellt. Als Bypass dient die A. mammaria interna.
-
Minimally invasive direct coronary artery bypass (MIDCAB): Zugang durch einen interkostalen Schnitt unter der linken Brust. Immobilisierung der Anastomosenstelle und Anlegung eines Bypass mit der A. mammaria interna. Nur zur Revaskularisierung an der Vorderwand geeignet.
-
-
Ergebnisse: Die Operationsletalität beträgt mit beiden Methoden in unkomplizierten Fällen 2 %, bei perioperativen Infarkten 4 %. Sie ist höher, wenn im kardiogenen Schock operiert werden muss. Nach 8–10 Jahren sind 80 % der Mammaria-Implantate offen, aber nur 40–50 % der V.-saphena-Segmente.
1.15.3 Stumme Ischämie
Die schmerzlose myokardiale Ischämie ist ein häufiges Phänomen. Es kann dazu führen, dass eine koronare Herzkrankheit ganz übersehen oder in ihrem Schweregrad unterschätzt wird. Man hat die stumme Ischämie in 2 Typen unterteilt:
-
Typ I: Patienten mit obstruktiver koronarer Herzkrankheit, die zu keiner Zeit anginöse Schmerzen haben. Bei ihnen ist das erste Symptom der Myokardinfarkt und selbst dieser kann stumm verlaufen. Die Ischämie lässt sich aber bei einer Vorsorgeuntersuchung im Belastungs-EKG nachweisen.
-
Typ II: Patienten mit symptomatischer koronarer Herzkrankheit, bei denen auch stumme Ischämien beobachtet werden. Schon beim Belastungs-EKG treten oft ischämische ST-Senkungen auf, ohne dass Schmerzen angegeben werden. Besonders eindrucksvoll haben Untersuchungen mit dem ambulatorischen oder Langzeit-EKG gezeigt, dass manche ST-Senkungen von anginösen Schmerzen begleitet werden, andere jedoch nicht. Beschwerdefreiheit ist demnach kein Indiz für eine optimale antiischämische Therapie. Medikamentös lassen sich auch die stummen Ischämien zum Verschwinden bringen.
1.15.3.1 Mechanismen der stummen Ischämie
Folgende Möglichkeiten werden diskutiert:
-
Bei sehr langsamer Progredienz einer Eingefäßstenose können durch simultane Ausbildung von Kollateralen Beschwerden ausbleiben.
-
Bei Diabetikern lässt sich Schmerzfreiheit mit autonomer Neuropathie erklären.
-
Patienten mit stummer Ischämie haben auch für andere Schmerzformen eine hohe Schwelle.
-
Solche Patienten produzieren reichlich endogene Opioide (Endorphine).
-
Patienten mit stillen Ischämien vom Typ II nehmen erst höhergradige Ischämien als Schmerzen wahr.
1.15.4 Mikrovaskuläre Ischämie („Syndrom X “)
1.15.4.1 Definition
Angina pectoris und objektivierbare myokardiale Ischämie bei normalem Koronarangiogramm, also ohne Stenose der epikardialen Kranzarterien.
1.15.4.2 Ätiologie und Pathogenese
Inadäquate vasodilatatorische Reserve der myokardialen Mikrozirkulation, wahrscheinlich durch eine Dysfunktion des Endothels. Die Ursache ist ungeklärt. Als sekundäres Phänomen kommt diese Störung bei Hypertonie mit Linkshypertrophie vor.
1.15.4.3 Klinik
Das Syndrom wird bei 25 % der Patienten beobachtet, die wegen stabiler Angina pectoris eine Koronarangiographie erhalten. Meistens handelt es sich um Frauen in der Prämenopause. Symptome sind typische Anfälle von Angina pectoris bei körperlicher Belastung und Erregungzuständen (Adrenalinausschüttung), die zu erhöhtem myokardialen O2-Verbrauch führen. Die Patienten sind oft psychisch labil und verängstigt.
1.15.4.4 Diagnostik
Ruhe-EKG meistens normal. Im Belastungs-EKG typische ischämische ST-Senkung. Auch ohne ST-Senkung kann eine Ischämie vorliegen, wie Stressechokardiographie und Thallium-Szintigraphie gezeigt haben. Gesichert wird die Diagnose durch das negative Koronarangiogramm.
1.15.4.5 Therapie und Prognose
Im Anfall hilft Nitroglycerin sublingual. Nitrate steigern aber die Belastungstoleranz nicht. Vorbeugend sind β-Blocker oder Calciumantagonisten indiziert, aber auch ACE-Blocker und Statine. Vielversprechend ist der neu entwickelte Blocker des späten Na+-Einstroms Vernakalant. Er hemmt die Natrium- und Calciumüberlastung des Myokards und verbessert den diastolischen Koronarfluss. Die Prognose ist sehr gut. Myokardinfarkte treten nicht auf. Im Verlauf kann es aber zu fokalen Fibrosierungen kommen.
1.15.5 Akuter Myokardinfarkt
1.15.5.1 Definition
Regionale ischämische Nekrose des Herzmuskels meistens auf dem Boden einer koronaren Atherosklerose. Infarkte ohne koronare Atherosklerose sind sehr selten.
1.15.5.2 Nomenklatur
Die praktischen Richtlinien des American College of Cardiology und der American Heart Association gehen beim infarktverdächtigen Brustschmerz (▶ unten) in Übereinstimmung mit der Europäischen und der Deutschen Gesellschaft für Kardiologie vom akuten koronaren Syndrom (ACS) aus, das dann mit EKG (12 Ableitungen) und Biomarkern (Troponin, CK) wie folgt zu differenzieren ist:
-
ACS mit ST-Hebung: Diagnose Myokardinfarkt (STEMI ), mit Q-Zacke (QwMI) oder ohne Q-Zacke (NQMI). Die frühere Unterscheidung von transmuralem und nichttransmuralem Infarkt aufgrund einer Q-Zacke im ersten Fall ist unsicher und für die Therapie nicht relevant.
-
ACS ohne ST-Hebung: Es kann sich um einen Myokardinfarkt handeln (NSTEMI) oder um eine instabile Angina pectoris. Die Entscheidung fällt nach dem Troponinspiegel im Serum. Selten treten beim NSTEMI Q-Zacken auf, die einen Infarkt beweisen. Patienten mit ACS ohne ST-Hebung sind keine Kandidaten für eine Fibrinolyse. Sie werden antiischämisch behandelt und bei hohem Risiko (Troponin-Erhöhung, ST-Senkung >0,1 mV, hämodynamische Instabilität, Rhythmusinstabilität, Diabetes) umgehend der Koronarographie, ggf. einer PTCA zugeführt.
1.15.5.3 Differenzialdiagnosen
-
Kardiale: instabile Angina pectoris (Troponin steigt nicht an), Perikarditis, Myokarditis (Troponin steigt nicht an)
-
Extrakardiale: Aortendisektion, Lungenembolie
Abklärung durch Echokardiographie, CT oder MR-Angiographie.
1.15.5.4 Pathogenese und Pathologie
Der akute Infarkt mit ST-Hebung entsteht in über 90 % der Fälle durch eine frische Koronarthrombose, auf dem Boden eines rupturierten atherosklerotischen Plaques. Bei langsam verlaufendem Verschlussprozess kann durch Kollateralenentwicklung ein Infarkt ausbleiben. Andere Ursachen des akuten Koronararterienverschlusses: persistierender Gefäßspasmus, Koronararterienembolie (z. B. bei Endokarditis), Arteriitis, Traumen mit lokaler Thrombose. Der Infarkt ohne ST-Hebung entsteht durch anhaltende Ischämie der subendokardialen Region bei fortgeschrittener koronarer Atherosklerose und geht nicht immer mit einem akuten koronaren Syndrom einher. Ischämische Läsionen werden nach 20–120 Minuten irreversibel. Etwa am 4. Tag nach Infarkteintritt beginnt der Abtransport der nekrotischen Muskelfasern. Nach der 4. bis 6. Woche hat sich in der Infarktzone eine stabile Bindegewebenarbe mit eingestreuten Muskelfasern gebildet. Komplikationen: Herzwandruptur, Septumruptur, Papillarmuskelabriss, ventrikuläres Aneurysma, wandständige Thromben mit Embolisierung.
Topographie und Terminologie. Myokardinfarkte sind in der Regel auf das Versorgungsgebiet der verschlossenen bzw. stenosierten Koronararterien und auf den linken Ventrikel beschränkt. Nur in 10 % der Fälle ist der rechte Ventrikel mitbetroffen (bei 30 % der Fälle von posteroinferiorem Infarkt). Die Bezeichnungen Vorder-, Seiten- und Hinterwandinfarkt beziehen sich deshalb nur auf den linken Ventrikel, nicht auf das Herz als Ganzes (◘ Abb. 1.118):


Topographie des Herzinfarktes
-
Vorderwandinfarkte: Betreffen das Versorgungsgebiet des R. interventricularis anterior der linken Koronararterie (RIVA): Vorderwand, vordere Seitenwand, und Spitze des linken Ventrikels, dazu die vorderen 2/3 des Septums. Zu unterscheiden sind:
-
Vorderwandspitzeninfarkt ( ◘ Abb. 1.118a ): Ausgedehnter Vorderwandinfarkt bei proximalem RIVA-Verschluss.
-
Anteroseptaler Infarkt (◘ Abb. 1.118b ): Rudimentärer supraapikaler Vorderwandinfarkt durch Verschluss septaler Äste des RIVA. Spitze und Seitenwand sind nicht betroffen.
-
Apikaler Infarkt (◘ Abb. 1.118c ): Kleiner auf die Ventrikelspitze beschränkter Infarkt durch Verschluss im distalen Segment des RIVA.
-
-
Seitenwandinfarkte: Betreffen Teile des Versorgungsgebietes des R. circumflexus der linken Koronararterie (RCX) oder den R. diagonalis des RIVA. Zu unterscheiden sind:
-
Hinterwandinfarkte (◘ Abb. 1.118f): Betreffen das Versorgungsgebiet der rechten Koronararterie, beim Linksversorgungstyp das des RCX. Zu unterscheiden sind:
-
Posteroinferiorer Infarkt: Diaphragmaler Infarkt, lokalisiert in der dem Zwerchfell anliegenden inferioren Hinterwand. Verschluss des R. posterolateralis der RCA, seltener aus dem RCX.
-
Posteriorer Infarkt: strikter posteriorer oder posterobasaler Infarkt, lokalisiert im freien, superioren Teil der Hinterwand. Verschluss der RCA bzw. des R. interventricularis posterior. Der rechte Ventrikel kann mitbetroffen sein.
-
1.15.5.5 Pathophysiologie
1.15.5.6 Störungen der Ventrikelfunktion
Frisch infarzierte Myokardareale werden akinetisch oder dyskinetisch, d. h. in der Systole nach außen gedrängt. Die Auswirkungen auf die Pumpfunktion des linken Ventrikels hängen vom prozentualen Verlust an kontraktiler Substanz ab: Herabsetzung der Ejektionsfraktion (ab 10 %), Zunahme von enddiastolischem Druck und Volumen durch Störung der systolischen Funktion (ab 15 %), klinische Symptome der Herzinsuffizienz (ab 25 %), kardiogener Schock (ab 40 %). Bei großen Infarkten sinkt mit dem Schlagvolumen der Blutdruck und damit der koronare Perfusionsdruck, wodurch die Ischämie zunehmen kann. Zugleich vergrößert die Ventrikeldilatation das Afterload und damit den myokardialen O2-Verbrauch, der zusätzlich durch kompensatorische Sympathikusstimulation ansteigt. So kann ein Circulus vitiosus mit progredienter Linksinsuffizienz zustande kommen. Zusätzliche mechanische Belastungen entstehen bei Mitralinsuffizienz durch Papillarmuskelabriss und bei Septumperforation.
1.15.5.7 Störungen des Herzrhythmus
Kammerflimmern ist die häufigste Todesursache im Frühstadium des Myokardinfarktes und auch im Folgestadium zu fürchten. Ihm gehen oft ventrikuläre Extrasystolen und Kammertachykardien voraus. Auch supraventrikuläre Arrhythmien kommen vor. Pathogenetische Faktoren für die elektrische Instabilität sind ischämisch bedingte Potenzialverluste der ruhenden Fasern des Arbeitsmyokards bzw. der Leitungsbahnen. Es entstehen dadurch Verletzungsströme und Reentrykreise. An der ischämischen Schädigung sind Effekte der Hypoxie (Azidose, zelluläre Kaliumverluste) beteiligt. Hinzu kommen lokale Katecholaminfreisetzung und zirkulierende freie Fettsäuren in erhöhter Konzentration. Funktionsstörungen des Sinusknotens und Leitungsstörungen im AV-Knoten sind überwiegend beim Hinterwandinfarkt anzutreffen, weil diese Strukturen meistens von der rechten Koronararterie perfundiert werden. Blockierungen der Tawara-Schenkel findet man in der Regel beim Vorderwandinfarkt, da der größte Teil des Septums im Versorgungsbereich des RIVA liegt.
1.15.5.8 Klinik
1.15.5.9 Anamnese
Bei etwa 50 % der Patienten beginnen die Infarktsymptome in Ruhe, bei 8 % aus dem Schlaf heraus, woran zu erkennen ist, dass Plaqueruptur und Thrombose belastungsunabhängig vorkommen. Nur bei 13 % der Patienten tritt der Infarkt im Anschluss an schwere körperliche Anstrengung auf. Eine vorbestehende stabile Angina pectoris wird von 20–60 % der Patienten angegeben. Etwa ein Viertel der nicht tödlichen Infarkte verläuft stumm oder führt die Patienten nicht zum Arzt.
1.15.5.10 Symptome
Der Infarktschmerz ist meistens von höchster Intensität und in der Tiefe hinterm Sternum lokalisiert. Ausstrahlungen können in den linken Arm, ins Epigastrium, in die Nackenregion und in den rechten Arm erfolgen. Der Schmerz ist anhaltend und spricht nicht auf Nitrate an, denn aus dem ischämischen ist ein Nekroseschmerz geworden. Er kann von Schwäche und Atemnot begleitet sein. Beim Hinterwandinfarkt kommen neben epigastrischer Schmerzlokalisation Übelkeit, Erbrechen und Stuhldrang als vagale Reflexe vor. Selten ist das Initialsymptom eine vagovasale Synkope mit extremer Bradykardie.
1.15.5.11 Körperliche Untersuchung
Die Patienten erscheinen gewöhnlich schwerkrank, sind durch Schmerz und Angst stark erregt und motorisch unruhig. Eine Linksinsuffizienz gibt sich durch kompensatorische Sympathikusstimulation mit Hautblässe, kaltem Schweiß und Tachykardie zu erkennen. Es kann eine Lungenstauung mit Dyspnoe, Stauungshusten und Katarrh über den basalen Lungenabschnitten vorliegen. Blutdruck und Pulsfrequenz sind erregungsbedingt erhöht, manchmal normal oder herabgesetzt. Patienten mit verengten Hirngefäßen können durch die Hypotonie einen zerebralen Insult erleiden. Ein Absinken des systolischen Drucks unter 90 mmHg ist schockverdächtig. Für einige Tage kann sich leicht- bis mittelgradiges Fieber einstellen.
Über der Herzspitze kann eine abnorme Pulsation zu tasten sein. Oft ist der 1. Herzton abgeschwächt und ein 3. Herzton zu hören. Am 2. oder 3. Tag tritt in etwa 20 % der Fälle ein Perikardreiben auf.
1.15.5.12 EKG
Das EKG beweist nicht immer sofort, aber meistens innerhalb von Stunden das Vorliegen eines Myokardinfarktes und erlaubt seine Lokalisation.
1.15.5.13 Allgemeine EKG-Veränderungen beim akuten Infarkt
Jede Phase des EKG ist auf bestimmte Weise betroffen:
-
Erregungsausbreitung (QRS): Das infarzierte Gebiet ist elektrisch inert. Da die Verbindungen zur gesunden Umgebung unterbrochen sind, leitet es keinen Strom mehr und kann mangels intakter Zellen auch nicht mehr elektrisch erregt werden. Damit entfallen an dieser Stelle bei der Erregungsausbreitung die positiven Partialvektoren, und es kommt ein entgegengerichteter negativer Summationsvektor zustande, der sich in den Extremitätenableitungen als Q-Zacke, in den Brustwandableitungen als R-Verlust dokumentiert. Wenn sich der usprünglich positive Initialvektor im Infarktgebiet nur verkleinert, aber nicht negativ wird, resultiert eine R-Reduktion. Q-Zacken und R-Verlust sind sichere Nekrosezeichen. Da sie in einigen Fällen ausbleiben, unterscheidet man zwischen Q-Zacken-Infarkt und Nicht-Q-Zacken-Infarkt. Beide können transmural oder nichttransmural sein. Deshalb wurde die frühere Einteilung in transmuralen und nichttransmuralen Infarkt aufgegeben.
-
Stadium der Vollerregung (ST-Strecke): Das EKG des frischen Infarktes zeigt eine vom absteigenden R-Schenkel hochabgehende starke ST-Hebung, die eine Kuppel- oder Plateauform aufweist und die T-Zacke einbezieht (monophasische Deformierung). Dieses Phänomen geht nicht vom nekrotisierten Infarktareal, sondern von der angrenzenden ischämisch geschädigten Verletzungszone aus. Es basiert auf einem Verletzungsstrom während der Vollerregung oder in der Diastole. Die ST-Hebung ist ein reversibles Ischämiezeichen, kein Indiz für eine Nekrose.
-
Erregungsrückbildung (T-Abschnitt): Im ischämischen Frühstadium, das meistens nicht erfasst wird, überhöhtes T wegen Verspätung der Repolarisation in der ischämischen Innenschicht. Sobald die Ischämie auch die Außenschicht erfasst, dreht sich der T-Vektor und die T-Zacke wird negativ. Im Verlauf nimmt die T-Negativität allmählich ab. Eine flache T-Negativität kann langfristig bestehen bleiben.
1.15.5.14 Stadien des Infarktablaufes
Repräsentativ sind für den Vorderwandinfarkt die Ableitungen V1‒V4, für den Hinterwandinfarkt die Ableitungen II, aVF und III (◘ Abb. 1.119).


Infarktstadien im EKG
-
Akutes Stadium: Monophasische Erhöhung des ST-T-Abschnittes, gefolgt von der Negativierung des initialen QRS-Vektors (Q-Zacke oder R-Verlust). Letztere fehlt beim Nicht-Q-Zacken-Infarkt, der durch weitere Tests zu sichern ist (▶ unten). Dauer: wenige Stunden bis einige Tage.
-
Zwischen- oder Folgestadium: Rückbildung der ST-Hebung, die T-Zacke wird spitz-negativ und erscheint gleichschenklig (koronares T). Q-Zacke bzw. R-Verlust unverändert. Dauer: mehrere Tage bis einige Wochen.
-
Endstadium: Weitgehende, nicht selten aber unvollständige Normalisierung des ST-T-Abschnittes. Tiefes Q und R-Verlust bleiben von seltenen Ausnahmen abgesehen bestehen.
1.15.5.15 Elektrokardiographische Infarktlokalisation
Die Infarktlokalisationen sind in ◘ Abb. 1.20 dargestellt:


Infarktlokalisation im EKG
-
Vorderwandspitzeninfarkt: Extr. Abl.: Q-Zacke und monophasische ST-T-Deformierung in I und II. BWA: R-Verlust und monophasische ST-T-Deformierung in V1–V5.
-
Anteroseptaler Infarkt: Extr. Abl.: unauffällig. BWA: R-Verlust und monophasische ST-T-Deformierung in V1–V3.
-
Apikaler Infarkt: Extr. Abl.: Q-Zacke und monophasische ST-T-Deformierung in I. BWA: R-Verlust und monophasische ST-T-Deformierung in V3 und/oder V4.
-
Vorderseitenwandinfarkt: Extr. Abl.: Q-Zacke und monophasische ST-T-Deformierung in I. BWA: R-Verlust und ST-T-Deformierung in V4–V6.
-
Hinterseitenwandinfarkt: Extr. Abl.: Q-Zacke und monophasische ST-T-Deformierung in II und III. BWA: Q-Zacken und monophasische ST-T-Deformierung in V4–V6.
-
Hinterwandinfarkt: Extr. Abl.: Q-Zacke und monophasische ST-T-Deformierung in II und III. BWA: ST-Senkung in V1–V3.
-
Innenschichtinfarkt: Oft in fast allen Ableitungen tiefe muldenförmige ST-Senkungen bei erhaltenen R-Zacken. Die Diagnose kann nur im Zusammenhang mit positiven Biomarkern gestellt werden. Es ist ein NSTEMI (▶ oben).
1.15.5.16 Labordiagnostik
1.15.5.17 Kreatininkinase (CK)
Sie kommt im menschlichen Organismus in Form von Dimeren der Untereinheiten CK-M (Muskel) und CK-B (Brain) vor, die sich zu 3 Isoenzymen kombinieren können: CK-MM (Skelettmuskeltyp), CK-BB(Gehirntyp) und CK-MB (Herzmuskeltyp). Die CK-Aktivität besteht im Skelettmuskel zu 96 % aus dem Isoenzym CK-MM und zu 4 % aus dem Isoenzym CK-MB. Dagegen beträgt das Verhältnis CK-MM zu CK-MB im Herzmuskel 60 % zu 40 %. In der Regel treten CK-BB-Aktivitäten im Serum nicht auf.
Die Gesamt-CK ist in der Diagnostik das Leitenzym. Bei erhöhter Gesamt-CK-Aktivität kann durch Bestimmung des Anteils der CK-MB zwischen Herz- und Skelettmuskelschäden differenziert werden.
Beim Infarkt liegt der CK-MB-Anteil über 6 %. Der Anstieg der CK- und CK-MB-Werte ist 6 h nach Schmerzbeginn signifikant und nach 18–20 h maximal. Zur Norm sinken die Werte nach 48 h ab. Die CK- und CK-MB-Aktivität sollte in Abständen von je 6 h zweimal wiederholt werden, weil dem Anstieg der Aktivitäten höchste Beweiskraft für einen Infarkt zukommt. Fehlerquellen bei alleiniger Bestimmung der Gesamt-CK: Muskeltraumen, i.m. Injektionen, körperliche Anstrengung, Alkoholintoxikation, Lungenembolie. Die Bestimmung der CK-MB-Masse ist zuverlässiger als die der CK-MB-Aktivität.
Ein Quotient CK-MB-Masse/CK-Aktivität >2,5 zeigt einen Infarkt an.
1.15.5.18 Herzspezifische Troponine
Der Troponin-Komplex besteht aus 3 Untereinheiten: Troponin C (bindet Ca++), Troponin I (bindet an Aktin und hemmt die Aktin-Myosin-Interaktion), Troponin T (bindet an Tropomyosin und damit den Komplex an Aktin). Vom herzspezifischen Troponinkomplex (cTn) sind 6 % der Untereinheit cTnT und 3 % der Untereinheit cTnI im Zytoplasma gelöst. Beide steigen im Serum beim Myokardinfarkt auf das 20-fache an, wobei der Normalwert dicht an der Nachweisgrenze liegt. Eine signifikante Erhöhung ist schon nach 3 h nachweisbar und am Krankenbett mit einem einfachen qualitativen Schnelltest zu eruieren. Beim STEMI ist das Testergebnis nicht abzuwarten. Der Test ist hochempfindlich und manchmal schon bei instabiler Angina pectoris deutlich positiv, als Beweis für Mikronekrosen in diesen Fällen. Erhöhte Werte bleiben beim cTnI 7–10 Tage, beim cTnT 10–14 Tage bestehen, so dass auch länger zurückliegende Schmerzereignisse als Infarkte identifiziert werden können. Durch den Troponin-Test ist die CK-Bestimmung für die Infarktdiagnose überflüssig geworden. „Falsch“ positive Werte kommen vereinzelt bei Patienten mit Niereninsuffizienz (Kreatinin >2,5 mg(dl) vor, auch bei anderen Krankheiten mit Myokardzellschädigung (z. B. Myokarditis, Lungenembolie, dekompensierte Herzinsuffizienz, Contusio cordis, Transplantatabstoßung). Die Einführung der Troponinbestimmung im Blut zur Erfassung des Myokardinfarkts auch bei negativem EKJ war eine Pionierleistung des Heidelberger Kardiologen Hugo A. Katus, durch die das Leben vieler Infarktpatienten gerettet werden konnte.
1.15.5.19 Unspezifische Parameter
Anstieg der Enzyme GOT und LDH und der Blutsenkungsgeschwindigkeit. Leukozytose (bis 15.000/mm3). Das Gesamtcholesterin kann absinken, der Nüchternblutzucker steigen.
1.15.5.20 Diagnostik mit bildgebenden Verfahren
1.15.5.21 Echokardiographie
Im 2-D-Echo sind stets regionale Kontraktionsanomalien (Hypokinesie, Akinesie oder Dyskinesie) zu erkennen. Schnell lässt sich die linksventrikuläre Funktion beurteilen (Ventrikeldurchmesser, Ejektionsfraktion), deren Absinken prognostisch ungünstig ist. Erkannt werden wandständige Thromben, Aneurysmen, Perikardergüsse und Perforationen (◘ Abb. 1.21). Mit dem Farbdoppler lassen sich Septumperforation und Mitralinsuffizienz durch Papillarmuskelabriss diagnostizieren. Erschwert wird die Beurteilung, wenn alte Infarktnarben vorliegen.


Echokardiographischer 4-Kammerblick bei einem 59-jährigen Patienten mit überlebter gedeckter Ventrikelperforation nach protrahiertem Hinterwandinfarkt, die erst 4 Monate später diagnostiziert und operativ mittels Resektion u. Patchübernähung therapiert wurde
1.15.5.22 Ventrikulographie
Erfasst regionale Hypokinesien des linken Ventrikel und Aneurysmen (◘ Abb. 1.122).


Lävokardiographie eines 49-jährigen Patienten mit implantiertem Defibrillator (LV-EF: 25 %), der nach Vorderwandinfarkt ein großes Vorderwandspitzenseptumaneurysma ausgebildet hat
1.15.5.23 MRT (nach KM-Gabe)
Nachweis von Infarktnarben und wandständigen Thromben (◘ Abb. 1.123).


MRT (nach KM-Gabe) eines 51-jährigen Patienten mit erlittenem Vorderwandinfarkt, Ausbildung einer Vorderwandnarbe u. eines apikalen Ventrikelthrombus
1.15.5.24 Röntgenaufnahne des Thorax
Bei der Erstuntersuchung häufig normal, auch wenn das enddiastolische Volumen des linken Ventrikels erhöht ist. Kardiomegalie und Lungenstauung können sich mit einer Latenz von 12–24 h entwickeln und sind als Zeichen der Linksinsuffizienz prognostisch ungünstig.
1.15.5.25 Szintigraphische Untersuchungen
Infarktszintigraphie mit 99mTc-Pyriphosphat zur selektiven Darstellung des Infarktes bei einem Infarktalter von 24–96 h. Geeignet zur genauen Lokalisation und Größenbestimmung.
1.15.5.26 Therapie in der Prähospitalphase
Ein Herzinfarkt ist zu vermuten, wenn ein Anfall von schwerer Angina pectoris nicht auf sublinguales Nitroglycerin anspricht.
Am besten ist es, dem Patienten einen venösen Zugang zu legen, zur Schmerzstillung Morphium zu geben und 500 mg ASS und falls zur Hand 5000 IE Heparin (oder 30 mg Enoxaparin i. v. + 1 mg/kg s. c.) zu injizieren. Schnellstmöglich ist der Transport in die Klinik zu veranlassen, der dann vom Arzt zu begleiten ist. Die Mortalität des akuten Myokardinfarktes beträgt rund 30 %, wobei die Hälfte der Todesfälle vor der Ankunft im Krankenhaus eintritt, gewöhnlich durch Kammerflimmern. Der Rettungswagen sollte daher mit EKG und Defibrillator ausgestattet sein.
1.15.5.27 Therapie in der Hospitalphase
Um keine Zeit zu verlieren, werden viele Patienten mit dem Rettungswagen in eine Klinik eingeliefert, ohne dass sie ein Arzt gesehen hat. Die Erstversorgung hat dann auf der Intensivstation stattzufinden, wo alle Möglichkeiten zur Kreislaufüberwachung gegeben sind. Die Ziele sind: Schmerzbefreiung, hämodynamische Stabilisierung, Begrenzung der Infarktgröße und Beherrschung der Komplikationen, besonders der Rhythmusstörungen.
Bei außerhalb reanimierten, noch bewusstlosen Patienten mit stabilem Kreislauf hat sich Vorbeugung von Hirnschäden eine vorübergehende Unterkühlung (Cool-Guard) bewährt. Angewendet wird meistens die invasive Kühlung mit Kühldevices. Das Blut wird aus der Femoralvene in das Kühlgerät geleitet und wieder zurückgepumpt. Die Temperatur sollte für 24 h bei 32–34° C gehalten wetrden.
1.15.5.28 Schmerzbekämpfung
Mittel der Wahl ist Morphium: 2–4 mg i. v. alle 5 Minuten, bis zum Wirkungseintritt. Mit dem Schmerz geht auch der erhöhte Sympathikustonus zurück, der den O2-Verbrauch des Myokards steigert. Bei Auftreten von Übelkeit ist Dimenhydrinat (Supp.), bei vagotoner Bradykardie Atropin (0,5 mg i. v.) indiziert.
Die Schmerzbekämpfung ist die erste und wichtigste Maßnahme.
1.15.5.29 Plättchenaggregationshemmer
Acetylsalicylsäure (ASS): Sofort 500 mg i. v. oder eine Kautablette (160–325 mg) zur bukkalen Resorption verabreichen und diese Dosis täglich wiederholen. Nach großen Studien senkt ASS die Infarktmortalität in den ersten 35 Tagen signifikant, wahrscheinlich durch Herabsetzung des Thromboxan-A2-Spiegels im Blut. Zusätzlich ist ein zweiter Aggregationshemmer zu geben. Alternativen: Ticagrelor 75 mg, Prasugel 60 mg, Clopidogrel 600 mg.
1.15.5.30 Nitroglycerin
Auch wenn es den Infarktschmerz nicht beseitigt, hat es Vorteile. Es senkt den Blutdruck und durch venöses Pooling das Präload. Damit wird der myokardiale O2-Verbrauch reduziert. Außerdem erweitert es infarktnahe Gefäße. Man gibt 3 Dosen von 0,4 mg sublingual im Abstand von je 5 Minuten. Bei günstigem Ansprechen kann im Falle erneuter Verschlimmerung eine Infusion (0,5–1,0 mg/Std.) angeschlossen werden. Kontraindikationen: systolischer Blutdruck <100 mmHg, rechtsventrikulärer Infarkt mit Halsvenenstauung.
1.15.5.31 β-Rezeptorenblocker
Indiziert bei Patienten mit Tachykardie und Hypertonie ohne akute Dekompensationszeichen. Sie reduzieren den O2-Verbrauch, Infarktgröße und die Häufigkeit ventrikulärer Arrhythmien. Dosis: Metoprolol in Einzeldosen von 5 mg alle 2–5 Minuten bis zu 3 Dosen, danach orale Weiterbehandlung mit 1–2×50–100 mg/Tag. Der Blutdruck soll nicht unter 100 mmHg sinken, die Pulsfrequenz nicht unter 60/min.
1.15.5.32 Thrombolyse
1.15.5.33 Indikationen
Zur Reperfusion nur indiziert, wenn kein Katheterlabor zur Verfügung steht bzw. bei zu langem Transportweg (>1 h). Ferner nur beim Infarkt mit ST-Hebung. Infarktpatienten mit ST-Senkung und erhöhten Markern erhalten antiischämische Mittel (s. oben) und Heparine (5000 IE unfraktioniertes Heparin i. v. oder Enoxaparin 30 mg i. v. plus 1 mg/kg s.c.). Bei fehlendem Lyseerfolg, erneuter oder progredienter Angina pectoris umgehende PTCA.
1.15.5.34 Thrombolytische Substanzen
Ihr Wirkungsmechanismus wird im ▶ Kap. 7, Abschnitt Thrombophilie beschrieben. Für die Thrombolyse beim Infarkt wird mit Vorteil der humane nichtantigene Plasminogenaktivator vom Gewebetyp verwendet, der gentechnologisch hergestellt als Alteplase oder t-PA verfügbar ist. Weniger gebräuchlich sind Streptokinase und Urokinase. Alteplase-Dosierung: Bolus von 15 mg, danach 0,75 mg/kg in 30 Minuten (nicht >50 mg), anschließend in der folgenden Stunde 0,5 mg/kg i. v. (Gesamtdosis ≤100 mg). Gleichzeitig wird heparinisiert bis zu einer PTT vom 1,5- bis 2-fachen des Ausgangswertes.
1.15.5.35 Der thrombolytische Effekt
Gemessen am Rückgang der Mortalität ist ein Thrombolysebeginn innerhalb der ersten Stunde optimal. Doch lässt sich ein so früher Behandlungsbeginn selten realisieren. Deutlich positiv sind die Ergebnisse noch bis zu 6 h nach Symptombeginn. Lohnend können sie noch nach 7–12 h sein, wenn bis dahin eine ST-Hebung bestanden hat. In etwa 30 % der Fälle erfolgt innerhalb von 24 h eine Wiederöffnung des Gefäßes durch spontane Thrombolyse. Die erzielte Reperfusion begrenzt die Infarktgröße, verbessert die Ejektionsfraktion reduziert das vergrößerte enddiastolische Ventrikelvolumen und beseitigt die ST-Hebung. Jenseits des 75. Lebensjahres sind die Erfolge der Thrombolyse bescheiden (10 gerettete Leben auf 1000 Behandlungen). Es besteht vor allem ein erhöhtes zerebrales Blutungsrisiko.
1.15.5.36 Komplikationen
Blutungen, intrakraniale Hämatome, allergische Reaktionen.
1.15.5.37 Absolute Kontraindikationen
Hirnblutungen irgendwann, zerebrale Insulte im letzen Jahr, systolischer Blutdruck >180 mmHg, diastolischer Blutdruck >110 mmHg, Verdacht auf Aortendissektion, innere Blutungen.
1.15.5.38 Relative Kontraindikationen
Antikoagulanzienbehandlung mit Coumarinderivaten (INR >2), vorausgegangene Operation oder Reanimation, hämorrhagische Diathese, Schwangerschaft, hämorrhagische diabetische Retinopathie.
1.15.5.39 Primäre perkutane koronare Intervention
Die primäre perkutane koronare Intervention (◘ Abb. 1.124) ist eine optimale Sofortmaßnahme bei Infarktpatienten mit ST-Erhöhung. Innerhalb der ersten 5 h mit Ballondilatation und Stent durchgeführt, wird die Infarktgröße auf 10–11 % des linken Ventrikels begrenzt. Die Thrombolyse ist weniger effektiv. Verbessert werden Früh- und Spätergebnisse der Infarkttherapie. Der Eingriff wird unter Heparinschutz und Infusion eines GP-IIb/IIIa-Inhibitors (Abciximab) durchgeführt, der sehr effektiv die Thrombozytenaggregation hemmt und die Mikrozirkulation in der Randzone des Infarktes steigert. Nach vorausgegangener Thrombolyse wird eine PTCA nur bei persistierender ST-Hebung und fortdauernder Angina pectoris durchgeführt.


61-jährige Patientin mit akutem ST-Hebungsinfarkt über der Vorderwand. Ventrikulographisch ist eine Akinesie der Vorderwand und der Herzspitze zu erkennen. Koronarangiographisch zeigt sich, dass der R. interventricularis anterior (RIVA) proximal verschlossen ist. Mittels Draht, Ballon und Stent kann der RIVA erfolgreich rekanalisiert werden (RCX = R. cirmumflexus)


Echokardiographischer 4-Kammerblick einer 38-jährigen Patientin mit rezidivierenden Lungenembolien: Oben: akutes Cor pulmonale mit Vergrößerung des rechten Ventrikels und des rechten Vorhofes sowie Linksverschiebung des Septums. Unten: Normalisierung des Echobefundes 48 h nach Beginn einer Vollheparinisierung
1.15.5.40 Komplikationen und ihre Therapie
1.15.5.41 Linksherzinsuffizienz und kardiogener Schock
Infarktareale von 20–25 % des linken Ventrikels haben eine deutliche Funktionseinschränkung zur Folge, Areale ab 40 % führen zum kardiogenen Schock (Blutdruck <90 mmHg, linksventrikulärer Füllungsdruck >20 mmHg, Herzindex <1,8 l/min/m2). Durch Thrombolyse und PTCA sind diese Komplikationen erheblich seltener geworden. Die Messung des pulmonalen Kapillardrucks, des Minutenvolumens und des peripheren Widerstandes mit dem Rechtsherzeinschwemmkatheter ermöglicht die rechtzeitige Diagnose und die Steuerung der Therapie, die ‒ wie bei anderen Ursachen der Herzinsuffizienz und des Kreislaufschocks ‒ erfolgt (▶ Abschn. 1.9).
Digitalisglykoside sind beim Herzinfarkt unwirksam.
Eine notfallmäßige PTCA unter intraaortaler Gegenpulsation kann die Mortalität senken.
1.15.5.42 Herzwandruptur , Septumdefekt , Papillarmuskelabriss
Schwerste, meist tödliche Komplikationen, bei denen als Ultima ratio eine chirurgische Korrektur zu versuchen ist, wenn die Zeit dazu bleibt (◘ Abb. 1.121).
1.15.5.43 Arrhythmien
Im Akutstadium droht Kammerflimmern, das auf der Intensivstation schnell durch transthorakale elektrische Defibrillation zu beseitigen ist. Rezidiven kann mit Amiodaron ggf. mit β-Blockern vorgebeugt werden, sofern die Schilddrüsenfunktion normal ist. Sicheren Schutz gibt ein transvenös implantierbarer Kardioverter-Defibrillator (ICD). Bei ventrikulären Tachykardien >150/min ebenfalls Defibrillation, bei Frequenzen <150/min Amiodaron i. v. (15 mg/min für 10 min, dann 1 mg/min für 6 h, danach 0,5 mg/h für 18 h). Supraventrikuläre Tachykardien sind wie üblich zu behandeln, bedrohliche Bradykardien mit Atropin (0,3–0,6 mg i. v. alle 3–10 min), nötigenfalls mit passagerer oder permanenter Schrittmacherimplantation.
1.15.5.44 Perikarditis
Eine relativ häufige, am Perikardreiben und im Echokardiogramm leicht zu erkennende Komplikation, die nicht mit einem Ischämieschmerz zu verwechseln ist. In solchen Fällen ist mit Antikoagulanzien große Vorsicht geboten, da ein Hämoperikard droht.
1.15.5.45 Dressler-Syndrom (Postmyokardinfarktsyndrom )
Akutes Krankheitsbild mit stark beeinträchtigtem Allgemeinbefinden, hohem Fieber, Brustschmerz, Perikarditis und Pleuritis, das bei 1–3 % der Patienten in einem Abstand von 3 Wochen bis wenigen Monaten nach dem Herzinfarkt auftritt, in seltenen Fällen schon nach einer Woche.
Die Diagnose ergibt sich aus den Symptomen von Perikarditis, Pleuritis, dem Fieber und einer maximalen Blutsenkungbeschleunigung.
Es handelt sich wahrscheinlich um eine Autoimmunreaktion gegen perikardiale oder myokardiale Antigene, die durch den Infarkt freigesetzt wurden. Therapie der Wahl: Corticosteroide, die prompt zur Besserung führen, manchmal aber wegen Rezidivneigung über längere Zeit in niedriger Dosis weiter verordnet werden müssen. Im Zeitalter der Herzkatheter ist diese Komplikation selten geworden.
1.15.5.46 Linksventrikuläres Aneurysma
Umschriebene paradoxe Wandbewegung nach außen während der Systole, der eine anatomische Aussackung folgen kann. Die Wand besteht überwiegend aus festem Narbengewebe. Hämodynamisch wird das effektive Schlagvolumem vermindert, weil Blut in das Aneurysma fließt. Die globale Pumpfunktion wird herabgesetzt. Außerdem besteht die Gefahr der Thrombosierung und Embolisierung. Das Aneurysma entwickelt sich innerhalb von Wochen oder Monaten nach dem Infarkt, stellt also eine Spätkomplikation da. Bei stärkerer funktioneller Beeinträchtigung des linken Ventrikels muss es chirurgisch reseziert werden.
1.15.5.47 Nachbehandlung und sekundäre Prävention
1.15.5.48 Antikoagulation
Nach den neuesten europäischen Leitlinien sind nach akutem Koronarsyndrom für 12 Monate ASS und Clopidogrel indiziert. Der bewährte Standard ist die unbefristete Gabe von ASS (100 mg/Tag per os). Nicht wenige Patienten reagieren aber auf ASS mit Gastritiden, intestinalen Blutungen und Ulzera. In diesen Fällen ist ASS durch einen anderen Plättchenaggregationshemmer zu ersetzen (▶ oben).
1.15.5.49 β-Rezeptorenblocker
Seit der routinemäßigen Anwendung von Thrombolyse und primärer Angioplastie haben sie auf die Infarktmortalität einen geringeren Einfluss als zuvor. Weiterhin sind sie jedoch zur Langzeittherapie nach Infarkt indiziert, sofern keine Kontraindikationen bestehen (Bradykardie, Herzblock, Asthma). Bei Herzinsuffizienz sind β1-Blocker in langsam steigender Dosierung indiziert (▶ Abschn. 1.9).
1.15.5.50 ACE-Inhibitoren oder Angiotensin-II-Rezeptor-Antagonisten
Sie reduzieren die Mortalität nach Myokardinfarkt deutlich und sollten schon nach 24 h gegeben werden. Den größten Nutzen haben Patienten mit herabgesetzter linksventrikulärer Funktion. Der systolische Blutdruck sollte aber nicht unter 110 mmHg gesenkt werden.
1.15.5.51 Aldosteronantagonisten
Sie verbessern nach einem Infarkt die Ventrikelfunktion und wirken antifibrotisch. Männer erhalten Spironolacton, Frauen (wegen Heiserkeit) Eplerenon.
1.15.5.52 Statine
Als Inhibitoren der HMG-CoA-Reduktase blockieren sie die Cholesterinsynthese in der Leber und bewirken eine starke Senkung des LDL-Cholesterins im Serum. Mehrere große kontrollierte Studien an Patienten mit koronarer Herzkrankheit haben gezeigt, dass Statine unter Herabsetzung des LDL-Cholesterins das Risiko von kardiovaskulären Ereignissen und des Todes erheblich senken. In der Heart-Protection Study (2002) führt die Reduktion des LDL-Cholesterin um 40 mg/dl zu einer Abnahme der koronaren Ereignisse um 25 %. Später wurde nachgewiesen, dass der Effekt auf die ultrasonographische Progredienz der Läsionen und auf klinische Ereignisse bei Absenkung des LDL-Cholesterins auf <70 mg/dl am stärksten ist. Sie wurde mit 80 mg Atorvastatin pro Tag erreicht. Hinzu kommt bei den Statinen, vor allem bei hoch dosiertem Atorvastatin, ein entzündungshemmender Effekt, der die Atherombildung additiv hemmt. Als Indikator der Entzündungshemmung erwies sich das C-reaktive Protein (CRP), dessen im Normbereich liegende Serumkonzentration durch Atorvastatin supprimiert wurde. CRP gehört zu den „Akute-Phase-Proteinen“, die von den Leberzellen unter dem Einfluss verschiedener Zytokine vermehrt gebildet werden und die Funktion von Opsoninen haben. Wichtig sind direkte Hinweise auf Angriffspunkte der Statine im Immunsystem. Zum einen hemmen sie die durch Interferon-γ gesteigerte Bildung von MHC-Molekülen der Klasse II, die zur Antigenpräsentation an CD4-T-Lymphozyten und deren Aktivierung dienen (▶ Kap. 8, adaptive Immunität). Zum anderen kann Atorvastatin T-Zellrezeptoren in vitro inaktivieren, indem es der Zellmembran Cholesterin entzieht, das der Verankerung von Co-Rezeptoren dient. Diese Beobachtungen weisen eher auf eine immunsuppressive als auf eine primäre entzündungshemmende Wirkung der Statine hin. Es bleibt zu fragen, ob der Entzug von Membrancholesterin auch für andere Nebenwirkungen der Statine verantwortlich ist. Kürzlich wurde über mehrere Fälle von reversiblem, demenzähnlichem Gedächtnisverlust durch Statine berichtet.
1.15.5.53 Korrektur der Risikofaktoren
Die Mortalität nach Myokardinfarkt sinkt deutlich, wenn das Rauchen ganz eingestellt und ein erhöhtes LDL-Cholesterin dietätisch und medikamentös unter 100 mg/dl gesenkt wird. Sehr wichtig ist eine optimale Diabeteseinstellung (HbA1c <6,5 %). Eine Hyperhomozysteinämie sollte durch Vitamin B12 und Folsäure beseitigt werden.
1.15.5.54 Kardiopulmonale Reanimation
Kardiopulmonale Reanimation
Wiederbelebungsmaßnahmen bei Kreislaufstillstand (Kammerflimmern, pulslose Kammertachykardie, pulslose elektrische Aktivität).
1.15.5.54.1 Neue Empfehlungen
Im Advanced Cardiovascular Life Support (ACLS) der American Heart Association (AHA) von 2005 wurden die bisherigen Empfehlungen in mehreren Punkten geändert. Jetzt gilt, dass der ersten Defibrillation 150 Thoraxkompressionen mit Beatmung vorzuschalten sind, wenn der Kreislaufstillstand unbeobachtet eintrat oder länger als 4–5 Minuten zurückliegt. Nach jeder Defibrillation soll der Puls nicht mehr sofort, sondern erst nach weiteren 5 Kompressionszyklen geprüft werden. Außerdem beginnt man die Reanimation stets mit Thoraxkompressionen und nicht mit Atemspenden.
1.15.5.54.2 Initiale Diagnostik
Prüfung der Bewusstlosigkeit durch lautes Ansprechen und Schütteln. Überstrecken des Kopfes und Vorziehen des Kinns zur Öffnung der Atemwege. Hören auf Atemgeräusche, achten auf Thoraxbewegungen. Bei Reaktionslosigkeit des Patienten möglichst 2. Person herbeirufen. Sauerstoffgabe, Monitor anschließen.
1.15.5.54.3 BLS-(Basic Life Support -)Phasen
Jede Phase dauert 2 Minuten und umfasst 5 Zyklen von je 30 Thoraxkompressionen und 2 Ventilationen. Dazu ist der Kranke auf eine feste Unterlage zu legen.
-
1. BLS-Phase: Sofortiger Beginn! Zur Herzdruckmassage seitlich vom Patienten knien und dessen Oberkörper freimachen. Handballen in der Mitte des Brustbeins des Patienten aufsetzen. Die zweite Hand unterstützend auf die erste legen und den Brustkorb senkrecht 4–5 cm tief bei gestreckten Armen mit einer Frequenz von 100/min komprimieren. Nach jeweils 30 Kompressionen den Mund auf den des Patienten legen, seine Nase mit zwei Fingern verschließen und 2-mal in die Atemwege des Patienten normal ausatmen. Falls die Beatmung scheitert, Thoraxkompressionen mit einer Frequenz von 100/min forstsetzen. Am Ende der 1. Phase Puls- und Rhythmuskontrolle.
Bei defibrillationswürdigem Rhythmus: Weiter wie folgt:
Defibrillator aufladen: In dieser Zeit Fortsetzung von Thoraxkompressionen und Beatmung im unveränderten Rhythmus. Anschließend Einmal-Defibrillation (mit monophasischen Defi 360 Joule, mit biphasischem Defi 200 Joule). Danach keine Rhythmus- bzw. Pulskontrolle, sondern weiter mit Thoraxkompressionen und Beatmung.
-
2. BLS-Phase: Fortsetzung mit Thoraxkompressionen und Beatmung über 5 Zyklen/2 Minuten. Im Laufe der Reanimation 1 mg Adrenalin i. v. oder intraossär (Malleolus medialis der Tibia); alternativ 40 IE Vasopressin i. v./intraossär. Am Ende der 2. Phase Puls- und Rhythmuskontrolle.
Bei defibrillationswürdigem Rhythmus: Weiter wie folgt:
Defibrillator aufladen: In dieser Zeit Thoraxkompressionen und Beatmung fortsetzen. Danach Einmal-Defibrillation wie bereits beschrieben.
-
3. BLS-Phase: 5 Zyklen in 2 Minuten wie bereits beschrieben. Antiarrhythmika erwägen: 300 mg Amiodaron i. v./intraossär oder 1–1,5 mg/kg Lidocain i. v./intraossär. Puls- und Rhythmuskontrolle.
Bei defibrillationswürdigem Rhythmus: Weiter wie folgt:
Defibrillator aufladen: In dieser Zeit Fortsetzung von Thoraxkompressionen und Beatmung im unveränderten Rhythmus. Anschließend Einmal-Defibrillation wie bereits beschrieben.
-
4. BLS-Phase: Thoraxkompressionen und Beatmung über 2 Minuten wie beschrieben. Während dieser Phase 1 mg Adrenalin bzw. 40 IE Vasopressin i. v./intraossär, Puls- und Rhythmuskontrolle.
Bei defibrillationswürdigem Rhythmus: Weiter wie folgt:
Defibrillator aufladen: In dieser Zeit Fortsetzung von Thoraxkompressionen und Beatmung im unveränderten Rhythmus. Anschließend Einmal-Defibrillation wie bereits beschrieben.
-
5. BLS-Phase: Durchführung wie in 4. BSL-Phase. Zusätzlich Antiarrhythmika erwägen: Amiodaron oder Lidocain wie bereits beschrieben. Am Ende der Phase Puls- und Rhythmuskontrolle.
Bei defibrillationswürdigem Rhythmus: Weiter wie folgt:
Defibrillator aufladen: In dieser Zeit Fortsetzung von Thoraxkompressionen und Beatmung im unveränderten Rhythmus. Anschließend Einmal-Defibrillation wie bereits beschrieben.
Bei nichtdefbrillationswürdigem Rhythmus nach der 1. BLS-Phase:
Wiederaufnahme von 5 BLS-Zyklen: Dazu 1 mg Adrenalin i. v. oder intraossär oder 40 IE Vasopressin i. v./i.o., 1 mg Atropin i. v./i.o., alle 3–5 min wiederholen (bis 3 Dosen).
Rhythmuskontrolle: Bei defibrillationswürdigem Rhythmus weiter wie in 3. BLS-Phase beschrieben. Andernfalls weitere BLS-Phasen anschließen. Ein Pacing wird bei Asystolie nicht mehr empfohlen.
1.15.5.54.4 Anmerkungen
-
Intubation: Frühe Durchführung nur durch gut geübte Person. Solange sich der Patient gut beatmen lässt, kann bis zur Rückkehr des spontanen Kreislaufs mit der Intubation gewartet werden.
-
Apparative Beatmung: Im Gegensatz zu früher können manuell getriggerte- und automatische Respiratoren verwendet werden.
-
Maßnahmen nach erfolgreicher Reanimation: Nach Kreislaufstillstand durch Kammerflimmern oder pulslose Kammertachykardie außerhalb des Krankenhauses sollte der Patient für 12–24 h auf 32–34 °C unterkühlt werden.
1.15.5.55 Rehabilitation
Sie umfasst alle Maßnahmen, die geeignet sind, die Folgen des Infarktes für die Lebensqualität des Kranken auf das erreichbare Minimum zu begrenzen und einem Infarktrezidiv vorzubeugen. Der zeitliche Ablauf der Rehabilitation lässt sich in folgende Phasen gliedern:
1.15.5.55.1 Akutphase
Im Akutkrankenhaus während der ersten 2‒4 Wochen nach Infarkteintritt.
1.15.5.55.1.1 Initiale Immobilisierung
Indiziert zur kontinuierlichen Überwachung und zur Begrenzung der Infarktgröße durch Minimierung des myokardialen Sauerstoffverbrauchs. Dauer einige Tage, mindesten bis zur Normalisierung der CK, damit jede Ausdehnung der Nekrose während der Mobilisierung sofort erkannt werden kann. Auch der Infarktschmerz und eine Fieberreaktion sollten vollständig abgeklungen sein. Verlängerte feste Bettruhe bei manifester Linksinsuffizienz, weiterbestehender instabiler Angina pectoris und bedrohlichen ventrikulären Rhythmusstörungen.
Das Herz muss sich „bei laufendem Motor“ regenerieren.
1.15.5.55.1.2 Frühmobilisierung
Vermeidet Verluste an körperlicher Arbeitskapazität, Hypovolämie, pulmonale Ventilationsstörungen, Muskelschwund, thromboembolische Komplikationen und orthostatische Intoleranz. Praktiziert werden selbständiges Aufstehen, Waschen, Rasieren, Essen am Tisch, Toilettengang, Spaziergänge und vorsichtige Belastung mit Übungen bis 50 Watt. Kontrolle durch Messung von Pulsfrequenz, Blutdruck und Langzeit-EKG. Vor der Entlassung Belastungs-EKG (mit Vorsicht), ggf. auch Stressechokardiographie.
1.15.5.55.2 Aufbauphase
Von der 3.–8. Woche. Zunächst wenn möglich als Anschlussheilbehandlung in einer kardiologischen Rehabilitationsklinik.
1.15.5.55.2.1 Körperliches Training
Dient der Wiederherstellung körperlicher Leistungsfähigkeit und der Stärkung des Selbstvertrauens.
Der Hauptakzent liegt dabei auf dynamischen Übungen (Gehen, Bergangehen, Gymnastik, Laufen, Radfahren, Ergometerübungen, Schwimmen), die durch rhythmische Kontraktion großer Muskelgruppen, isotonische Muskelaktivität (Faserverkürzung bei konstanter Spannung) und einen aeroben Muskelstoffwechsel gekennzeichnet sind. Dem gesteigerten venösen Rückfluss aus der beanspruchten Muskulatur passt sich das Herz mit einer Vergrößerung des Minutenvolumens an, wobei die Basisregulation durch Intrinsic-Mechanismen und bei höherer Belastung auch die nervale Regulation wirksam wird. Für das Herz entsteht in erster Linie eine Volumenbelastung. Der Druckanstieg, der viel Sauerstoff kostet, hält sich in Grenzen. Dynamisches Training steigert die körperliche Leistungsfähigkeit vor allem durch Vergrößerung und Vermehrung der Mitochondrien in der beanspruchten Skelettmuskulatur, deren Sauerstoffaufnahme und oxidative Kapazität zunimmt. Zugleich wird die Blutversorgung der Muskulatur durch Zunahme der Kapillarisierung verbessert. Beide Effekte zusammen steigern die periphere Sauerstoffextraktion bzw. -utilisation und entlasten dadurch bei submaximaler Belastung Herz und Kreislauf. Gegenüber dem untrainierten Zustand findet eine geringere Steigerung der Sympathikusaktivität statt. Kraftübungen (Muskelanspannung ohne Verkürzung) sind zurückhaltend einzusetzen, da sie mit anaerobem Muskelstoffwechsel und höherer Sympathikusaktivität verbunden sind. Das Training ist individuell zu dosieren und mit Puls-, Blutdruck- und EKG-Kontrollen zu überwachen.
Wettkampf aller Art ist zu vermeiden, weil er den Sympathikus in unkontrollierbarer Weise stimuliert.
1.15.5.55.2.2 Psychotherapie und Gesundheitsförderung
Psychotherapeutische Behandlung, Verhaltenstherapie, Sexualberatung. Gesundheitsförderung durch Autogenes Training. Ausschaltung der Risikofaktoren, insbesondere Raucherentwöhnung (in der gesamten Familie!).
1.15.5.55.2.3 Sozialtherapeutische Maßnahmen
Umsetzung, Umschulung, Berentung bei Berufs- oder Erwerbsunfähigkeit. Vermittlung in ambulante Koronargruppen.
1.15.5.55.3 Wiedereingliederung
Von der 12.–24. Woche. Ambulante Betreuung durch den Hausarzt und möglichst auch einer Koronargruppe. Zunehmende Belastung im Alltagsleben. Entscheidend für die Belastbarkeit ist letztlich die Größe des erlittenen Infarktes, die aus dem maximalen CK-Anstieg und dem echokardiographischen Befund abzuleiten ist. Nötigenfalls Eingliederung in eine leichtere Berufstätigkeit.
1.15.5.55.4 Erhaltungsphase
Ab 4.–6. Monat. Nach dieser Zeit kann meistens über die Arbeitsfähigkeit definitiv entschieden werden. Hausärztliche Überwachung, evtl. betriebsärztliche Kontrollen (Blutdruck etc). Weiterbetreuung in der Koronargruppe. Ausdauerbelastung beim Training bei 50–60 % der individuellen Maximalbelastung. Konsequente medikamentöse Langzeittherapie.
1.16 Pulmonale Herzkrankheiten
Pulmonale Herzkrankheiten
-
Lungenembolie
-
Thromboembolie
-
Nichtthrombogene Embolie
-
-
Pulmonale arterielle Hypertonie
-
Idiopathische und familiäre pulmonale Hypertonie
-
Krankheiten mit assoziierter pulmonaler arterieller Hypertonie
-
1.16.1 Lungenembolie
1.16.1.1 Thromboembolie
1.16.1.1.1 Ätiologie und Pathogenese
Der Verschluss von Pulmonalarterienästen durch verschleppte Thromben ist die häufigste Form der Lungenembolie. Inzidenzrate rund 1:1000 pro Jahr. In den USA werden jährlich über 250.000 Personen wegen Thromboembolie hospitalisiert. Ausgangspunkt ist meistens eine Thrombose der tiefen Beinvenen, die auf dem Boden einer hereditären oder erworbenen Thrombophilie entstehen kann (▶ Kap. 7). Die seltenen Armvenenthrombosen entstehen meistens traumatisch, auch auf dem Boden von Wandläsionen, die mit Kanülen und Venenkathetern gesetzt werden.
1.16.1.1.2 Pathophysiologie
1.16.1.1.3 Respiratorische Störungen
Durch Belüftung der nicht perfundierten Alveolen im embolisierten Lungenbezirk entsteht ein alveolärer Totraum. Die resultierende Störung des Ventilations-Perfusions-Verhältnisses führt zur Hypoxie und zur kompensatorischen Hyperventilation, die als Dyspnoe empfunden wird. Im Verlauf kann es durch Surfactant-Verlust und Kontraktion der Längsmuskulatur in den terminalen Luftwegen zu Atelektasen kommen. Die meisten emboliebedingten Lungenverschattungen im Röntgenbild stellen reversible Atelektasen dar. Lungeninfarkte sind selten, da die Lunge auf 2 Wegen mit Blut versorgt wird: durch den Bronchialkreislauf und die Pumonalarterie. Infarzierungen werden meistens durch periphere Embolien hervorgerufen, am ehesten bei gleichzeitiger Stauung im kleinen Kreislauf. Sie gehen mit Begleitpleuritis einher, manchmal mit blutigem Auswurf.
1.16.1.1.4 Hämodynamische Störungen
Die Verlegung von Lungenarterienästen erhöht den Widerstand im kleinen Kreislauf und damit die Nachlast für den rechten Ventrikel. Wegen der großen Compliance der Lungenarterien steigt der Pulmonalarteriendruck erst bei ausgedehnten Embolien. Von normalen Ausgangswerten kann er auf 80 mmHg steigen, bei vorbestehender Rechtshypertrophie noch höher. Die akute Druckbelastung des rechten Ventrikels führt durch Kompression der Ventrikelwand zur Ischämie und zur progredienten Rechtsherzinsuffizienz. Auch die Funktion des linken Herzens wird herabgesetzt, da sich das interventrikuläre Septum infolge Dilatation des rechten Ventrikels nach links vorwölbt und die Füllung des linken Ventrikels vermindert. Etwa bei 5 % der Thromboembolien kommt es zum kardiogenen Schock. Bei offenem Foramen ovale können Thromben in den großen Kreislauf eingeschwemmt werden und im Gehirn und anderen Organen ischämische Infarkte verursachen.
1.16.1.1.5 Klinik
Erstes und wichtigstes Zeichen ist eine plötzlich einsetzende unerklärliche Dyspnoe mit Tachypnoe, deren Stärke sich nach dem Schweregrad der Embolie richtet.
Tachykardie, Blutdruckabfall und Zyanose kommen bei massiven Embolien hinzu. Wenn sich eine Rechtsinsuffizienz entwickelt, treten Halsvenenstauung und Leberschwellung auf. Die relativ seltenen fulminanten Embolien führen abrupt zu Bewusstseinsverlust und kardiogenem Schock. Kleinere periphere Embolien mit Infarkt manifestieren sich nach mehrstündiger Latenzzeit mit Husten und atemabhängigem Pleuraschmerz ohne stärkere Dyspnoe. In der Hälfte dieser Fälle wird etwas Blut ausgehustet. Rezidivierende Mikroembolien können asymptomatisch verlaufen und schließlich zur irreversiblen chronischen pulmonalen Hypertonie führen.
1.16.1.1.6 Diagnostik
1.16.1.1.7 Anamnese
Familiäre Thrombosebelastung erfragen (genetische Thrombophilie) und erworbene Risikofaktoren für eine Thrombose eruieren.
1.16.1.1.8 Körperliche Untersuchung
Symptome einer tiefen Beinvenenthrombose? Auskultationsbefund der Lungen meistens normal. Akzentuierter Pulmonalton im 2. ICR links. Inspiratorisch verstärktes Systolikum links parasternal (Trikuspidalinsuffizienz)? Zeichen der Rechtsherzinsuffizienz?
1.16.1.1.9 Röntgenbild des Thorax
Normalbefund häufig! Ansonsten: Zwerchfellhochstand, keilförmige, mit der Basis an die Pleura grenzende Verschattung (Atelektase, seltener Infarkt), hypovaskularisierte Zonen (Westermark-Zeichen).
1.16.1.1.10 EKG
SIQIII-Typ (Rechtsdrehung des QRS-Vektors), inkompletter Rechtsschenkelblock, dazu ST-Hebung und T-Negativität in V1 und V2 bei rechtsventrikulärer Ischämie (◘ Abb. 1.120).
1.16.1.1.11 Echokardiographie (◘ Abb. 1.125)
Bei schwerer Lungenembolie großer rechter Ventrikel (durch Dilatation) bei kleinem linken Ventrikel. Paradoxe Septumbewegung nach links, erweiterte Pulmonalarterie. Manchmal noch Thromben im rechten Vorhof oder Ventrikel, bei offenem Foramen ovale auch im linken Vorhof.
Die Echokardiographie ist auf Intensivstationen bei kardiogenem Schock das wichtigste Instrument zur Unterscheidung von Lungenembolie, Aortendissektion und Perikardtamponade.
1.16.1.1.12 Duplexsonographie der tiefen Beinvenen
Nur in 65 % der Fälle ist ein Thrombus nachweisbar (fehlende Komprimierbarkeit).
1.16.1.1.13 Laborbefunde
Hypoxie und Hypokapnie, aber auch Normalbefunde bei der Blutgasanalyse. Nachweis von Fibrinspaltprodukten (D-Dimer) im Serum mit Latexagglutination, besser mit ELISA, aber nicht spezifisch. Ein negatives Resultat spricht aber gegen Embolie.
1.16.1.1.14 Lungenperfusionsszintigraphie
Wichtigster Screening-Test, da ein normales Szintigramm eine klinisch relevante Embolie ausschließt. Ein Speicherdefekt spricht für eine Embolie, wenn das konventionelle Röntgenbild des Thorax unauffällig ist. Im Ventilations-Perfusions-Scan wird der nicht perfundierte Lungenabschnitt noch belüftet (◘ Abb. 1.126).


54-jährige Patientin im Stadium NYHA III mit Verdacht auf rezidivierende Lungenembolien. a Das Ventilations- und Perfusionsszintigramm zeigt eine deutliche Minderperfusion der gesamten rechten Lunge (unten) bei nur geringer ventilatorischer Aktivitätsminderung im rechten Lungenoberlappen (oben), passend zu einer fast kompletten Thrombosierung der rechten Lungenstrombahn. b Operationspräparat nach rechtsseitiger Thrombendarterektomie


Thorax-CT im Spiralmodus nach Kontrastmittelgabe bei einer 67-jährigen Patientin mit schwerer Lungenembolie. Großer Embolus in der Bifurkation des Pulmonalarterienstammes, kleinere Thromben in der rechten Unterlappenarterie mit peripherer Parenchymverdichtung (PV) bei embolischem Lungeninfarkt und begleitendem Pleuraerguss (PLE)
1.16.1.1.15 Spiral-CT mit Kontrastmittelbolus (◘ Abb. 1.127)
Zunehmend diagnostischer Goldstandard. Erfasst Embolien in den Haupt-, Lappen- und Segmentästen mit einer Sensitivität von 73–97 % und einer Spezifität von 86–98 %.
1.16.1.1.16 Dreidimensionale Magnetresonanzangiographie mit Gadolinum
Annähernd von gleicher Aussagekraft wie die konventionelle pulmonale Angiographie mit Kontrastmittel. Ergänzend sind mit einer MRA der proximalen Beinvenen und der Beckenvenen Thromben in diesem Bereich sicher zu erfassen.
1.16.1.1.17 Konventionelle pulmonale Kontrastmittelangiographie
Bei unklaren Fällen, wenn ein Spiral-CT nicht verfügbar ist. Indiziert vor chirurgischer Therapie einer pulmonalen Hypertonie auf dem Boden rezidivierender Lungenembolien.
1.16.1.1.18 Differenzialdiagnosen
Pneumonie, Asthmaanfall, Pneumothorax, primäre pulmonale Hypertonie, Herzinfarkt, Lungenödem, Herztamponade, Aortendissektion, Rippenfraktur, Kostochondritis, psychogene Hyperventilation.
1.16.1.1.19 Therapie
1.16.1.1.20 Antikoagulation mit unfraktioniertem Heparin
Sofortmaßnahme bei Verdacht auf Lungenembolie noch vor Abschluss der Diagnostik. Dosis: 10.000 IE als Bolus i. v., anschließend Langzeitinfusion mit 1000 IE/h (18 IE/kg, maximal 1600 E/h). Die aPTT soll auf das 1,5- bis 2-fache des Ausgangswertes steigen. Heparin (als Heparin/Antithrombin-III-Komplex) stoppt die Thrombusbildung und ermöglicht eine zügige endogene Fibrinolyse. Alternativ hat sich Fondaparinux bewährt (5–7,5 mg/Tag s.c. für 5 Tage). Nach Erreichen des therapeutischen aPTT-Wertes kann mit oraler Antikoagulation (Phenprocoumon, Warfarin) begonnen werden. Die Heparininfusion ist aber mindestens 5 Tage fortzusetzen (bis alles Thrombin eliminiert ist). Der INR-Wert ist auf 2,0–3,0 einzustellen.
Die Antikoagulanzientherapie genügt für alle Patienten mit stabilem Kreislauf und normaler Kontraktilität des rechten Ventrikels im Echokardiogramm. Kontraindikationen: akute Blutungen, zerebrale Insulte, heparininduzierte Thrombopenie (mit intravaskulärer Gerinnung). Alternativ kann der reine Thrombininhibitor Hirudin gegeben werden. Bei Blutungskomplikationen lässt sich Heparin mit Protaminsulfat neutralisieren. Dauer der Antikoagulation mindestens 6 Monate, bei genetischen Thrombophilien mehrere Jahre, bei rezidivierenden Embolien evtl. lebenslang.
1.16.1.1.21 Vena-cava-Schirm (Greenfield-Filter )
Applikation über die V. jugularis in die V. cava inferior. Indikationen (sehr selten): Embolierezidive unter der Therapie, Kontraindikationen für Antikoagulation und Fibrinolyse. Komplikationen: Verschiebungen des Filters, Thrombose auf der proximalen Seite, Kavathrombose. In randomisierten Studien wurde die 2-Jahresmortalitat nicht gesenkt.
1.16.1.1.22 Thrombolyse
Als potenziell lebensrettend indiziert bei massiven Embolien mit kardiogenem Schock oder hämodynamischer Instabiltät, auch bei normalem Blutdruck, wenn der rechte Ventrikel im Echokardiogramm hypokinetisch ist. Die Lyse kann noch nach einem Intervall von bis zu 14 Tagen effektiv sein. Am kürzesten ist das Protokoll mit t-PA (100 mg während 2 h i. v.), wobei die Heparininfusion nicht unterbrochen wird. Alternativ kommen Urokinase und Streptokinase in Betracht. Der pulmonale Strömungswiderstand fällt schon nach mehreren Stunden ab. Kontraindikationen: aktive innere Blutungen, Hirninfarkt während der letzten 2 Monate, unter 10 Tagen Abstand nach großen Operationen, bakterielle Endokarditis.
1.16.1.1.23 Transvenöse Katheterembolektomie
Absaugung von Thromben oder Zertrümmerung mit speziellen Kathetern in Kombination mit der Thrombolyse. Indikation: bei schweren Embolien und im kardiogenen Schock.
1.16.1.1.24 Chirurgische Embolektomie
In schwersten Fällen mit Schocksymptomatik indiziert. Eingriff unter Einsatz der Herz-Lungen-Maschine oder nach Trendelenburg (Inzision des Pulmonalarterienhauptstammes). Letalität 25–40 %.
1.16.1.1.25 Prognose
Die Heparinbehandlung hat die Mortalität der Lungenembolie von 18–38 % auf unter 9 % gesenkt. Die Thrombolyse ist in schweren Fällen lebensrettend. Die spontane Thrombolyse dauert mehrere Wochen und kann unvollständig bleiben (16 % der Fälle). Patienten mit rezidivierenden Embolien und chronischer pulmonaler Hypertonie sind Kandidaten für eine pulmonale Thrombendarterektomie, die vielfach erfolgreich ist (Operationsmortalität 5–10 %; ◘ Abb. 1.126b).
1.16.1.1.26 Prävention
Sehr erfolgreich mit subkutan appliziertem Heparin in niedriger Dosis ohne Laborkontrollen durchzuführen. Statt unfraktioniertem Heparin (3×5000 IE/Tag) verwendet man meistens niedermolekulare Heparine (Enoxaparin, Dalteparin, Ardeparin, Reviparin, Tinzaparin, Nadroparin). Diese haben sind wegen besserer Bioverfügbarkeit niedriger zu dosieren und nur einmal oder zweimal täglich zu injizieren (z. B. Enoxaparin 2-mal 30 mg/Tag s.c., Dalteparin 1-mal 2500–5000 IE/Tag s.c.) durchgesetzt. Indikationen: Peri- und postoperative Prophylaxe tiefer Beinvenenthrombosen (Allgemeinchirurgie, Hüft- und Kniegelenkersatz, Neurochirurgie, Thorax- und Herzchirurgie). Prophylaxe bei bettlägerigen Schwerkranken und Schwerverletzten. Nach Hüft- und Kniegelenkersatz muss die Prophylaxe postoperativ mindestens einen Monat lang fortgesetzt werden, da Spätthrombosen drohen.
1.16.1.1.27 Physikalische Maßnahmen
Postoperative Frühmobilisation, Antiemboliestrümpfe bzw. elastische Bandagen der Beine. Bei Bewusstlosen intermittierende Kompression der Unter- und Oberschenkel.
1.16.1.2 Nichtthrombogene Embolien
1.16.1.2.1 Fettembolie
Fettembolisierung kommt bei 90 % der Patienten mit schweren Traumen vor und verläuft überwiegend asymptomatisch. Das klinisch manifeste Fettemboliesyndrom ist relativ selten. Es wird zu 90 % nach stumpfen Traumen mit Frakturen langer Röhrenknochen, Marknagelungen, sowie nach Hüft- und Kniegelenkersatz mit Zementeinspritzungen beobachtet. Seine Kennzeichen sind pulmonale Dysfunktion mit Tachypnoe und Hypoxie, in schweren Fälle auch pulmonale Hypertonie und rechtsventrikuläre Dysfunktion. In 70 % der Fälle folgen nach 2 Tagen multiple Petechien und mentale und neurologische Ausfallserscheinungen. Die Mortalität beträgt 10–20 %.
Bei der Marknagelung einer Femurfraktur konnte die Entstehung eines fulminanten Fettemboliesyndroms durch fortlaufende Registrierung des transösophagealen Echchokardiogramms genau verfolgt werden. Es kam zur massiven Einschwemmung echogener Fettmassen über den rechten Vorhof in den rechten Ventrikel und die A. pulmonalis. Außerdem trat embolisches Material durch ein offenes Formanen ovale in den linken Vorhof über und führte zu systemischen Fettembolien mit generalisierten Krämpfen, Hyperreflexie und Koma. Nach 32 h erfolgte der Exitus. Das Fettemboliesyndrom hat damit eine mechanische Erklärung gefunden. Bei einer Häufigkeit des offenen Formanen ovale von 20–34 % sind neurologische Ausfallserscheinungen durchaus plausibel. Eine spezifische Therapie gibt es nicht.
Das Fettemboliesyndrom wurde auch bei akuter Pankreatitis beobachtet. Das embolisierende Fett scheint hier durch Aggregation von Chylomikronen und VLDL unter dem Einfluss von C-reaktivem Protein und anderen Faktoren zu entstehen.
1.16.1.2.2 Tumorembolie
Entsteht durch Eindringen von Tumorgewebe in die großen Venen. Die Unterscheidung von einer Thrombembolie kann schwierig sein.
1.16.1.2.3 Embolien mit Amnionflüssigkeit
Vorkommen bei Blaseneinriss am Plazentarand. Führt zum Lungenödem durch Kapillardurchlässigkeit in den Alveolen.
1.16.2 Pulmonale arterielle Hypertonie
1.16.2.1 Definition
Drucksteigerung im kleinen Kreislauf mit erhöhter Rechtsherzbelastung, die zur Rechtshypertrophie und zur Rechtsherzinsuffizienz führen kann.
Klassifikation der pulmonalen arteriellen Hypertonie (Venedig 2003).
-
1. Pulmonale arterielle Hypertonie (PAH)
-
1.1. Idiopathisch (IPAH)
-
1.2. Familiär (FPAH)
-
1.3. Assoziiert mit (APAH)
-
1.3.1. Kollagengefäßkrankheiten
-
1.3.2. Kongenitale Links-rechts-Shunts
-
1.3.3. Portale Hypertension
-
1.3.4. HIV-Infektion
-
1.3.5. Pharmaka und Toxine
-
1.3.6. Andere (Schilddrüsenkrankheiten, Glykogenspeicherkrankheit, Morbus Gaucher, hereditäre hämorrhagische Teleangiektasie, Hämoglobinopathien, myeloproliferative Erkrankungen, Splenektomie)
-
1.4. Assoziiert mit signifikanter Venen- oder Kapillarbeteiligung
-
1.4.1. Pulmonale venookklusive Krankheit (PVOD)
-
1.4.2. Pulmonale kapillare Hämangiomatose (PCH)
-
1.5. Persistierende pulmonale Hypertonie der Neugeborenen
-
-
2. Pulmonale Hypertonie bei Linksherzerkrankungen
-
2.1. Linksseitige atriale oder ventrikuläre Krankheiten
-
2.2. Linksseitige Herzklappenerkrankungen
-
-
3. Pulmonale Hypertonie assoziiert mit Lungenkrankheiten und/oder Hypoxämie
-
3.1. Chronische obstruktive Lungenkrankheit
-
3.2. Interstitielle Lungenkrankheiten
-
3.3. Schlaf-Atem-Störungen
-
3.4. Krankheiten mit alveolärer Hypoventilation
-
3.5. Chronische Höhenluftexposition
-
3.6. Entwicklungsanomalien
-
-
4. Pulmonale Hypertonie durch chronische thrombotische und/oder embolische Krankheiten
-
4.1. Thromboembolische Obstruktion der proximalen pulmonalen Arterien
-
4.2. Thromboembolische Obstruktion der distalen pulmonalen Arterien
-
4.3. Nichtthrombotische pulmonale Embolien (Tumor, Parasiten, Fremdmaterial)
-
-
5. Verschiedenes (Sarkoidose, Histiozytose X, Lymphangiomatose, Kompression von Lungengefäßen, Adenopathie, Tumor, fibrosierende Mediastinitis)
1.16.2.2 Diagnostik
1.16.2.3 Anamnese
Hinweise auf Rechtsinsuffizienz (Belastungsdyspnoe, belastungsabhängige Synkopen, Beinödeme)? Sekundäre Angina pectoris? Hinweise auf diastolische Funktionsstörung des linken Ventrikels (nächtliche Dyspnoe)? Zeichen einer Lungenkrankheit (Husten, Auswurf, Asthma)? Hinweise auf Lungenembolie (Beinvenenthrombose, Pleuraschmerz mit Fieber)? Schlaf-Apnoe-Phasen? Leberkrankheit mit Aszites? Einnahme von Appetitzüglern? Angeborene Herzfehler? HIV-Infektion? Rheumatische Systemkrankheiten?
1.16.2.4 Körperlicher Untersuchungsbefund
Im 2. ICR links systolische Pulsation der dilatierten A. pulmonalis, Ejektionsklick, systolisches Strömungsgeräusch und enge Spaltung des 2. Herztones. Epigastrische Pulsationen infolge Rechtshypertrophie, Leberschwellung, Ödeme. Bei relativer Trikuspidalinsuffizienz holosystolisches Regurgitationsgeräusch links parasternal und prominente V-Welle im Jugularvenenpuls. Zyanose bei Lungenkrankheiten mit Hypoxämie oder im Finalstadium.
1.16.2.5 Röntgenuntersuchung des Thorax
Erweiterung der Pulmonalarterie und der Hauptäste mit Kalibersprung zur Peripherie. Bei obstruktiver Lungenkrankheit weist eine Erweiterung (>16 mm) des rechten deszendierenden Pulmonalarterienastes auf eine pulmonale Hypertonie hin. Einengung des Retrosternalraumes bei Dilatation des rechten Ventrikels.
1.16.2.6 Lungenfunktionsprüfung
Nachweis obstruktiver und restriktiver Ventilationsstörungen und Veränderungen der Blutgase. Bei Indikation Untersuchung im Schlaflabor.
1.16.2.7 Lungenszintigraphie
Das Perfusionsszintigramm dient zum Nachweis bzw. Ausschluss rezidivierender Lungenembolien als Ursache einer PAH.
1.16.2.8 Elektrokardiogramm
Steil- oder Rechtslagetyp; Zeichen der Rechtshypertrophie: hohe spitze P-Zacken in II, aVF, III und V1; zunehmende R-Zacke in V1, zuletzt rSR. Bei Emphysem periphere Niedervoltage. Spiroergometrie zur Feststellung der Belastungstoleranz.
1.16.2.9 Echokardiographie
Rechtskardiale Dilatation. Im 4-Kammerblick Verdickung der rechtsventrikulären Vorderwand (>5 mm). Häufig hyperkinetischer rechter Ventrikel. Bei relativer Trikuspidalinsuffizienz dopplersonographische Abschätzung der rechtskardialen Drücke.
1.16.2.10 Magnetresonanztomographie
Goldstandard zur Größenbestimmung des rechten Ventrikels und seiner Ejektionsfraktion.
1.16.2.11 Spiral-CT
Erlaubt die optimale Diagnostik des Emphysem und anderer Lungenveränderungen.
1.16.2.12 Rechtsherzeinschwemmkatheter
Ermöglicht durch Druckmessungen im kleinen Kreislauf (rechter Vorhof, rechter Ventrikel, Pulmonalarterie, Pulmonalkapillaren) und Bestimmung des Herzminutenvolumens in Ruhe und unter Ergometerbelastung den Schweregrad der pulmonalen Hypertonie genau zu bestimmen und eine Linksinsuffizienz als Ursache auszuschließen. Bei der Untersuchung kann der Effekt von Vasodilatoren getestet werden.
-
1. Stadium: Ruhedrücke normal. Anstieg des Pulmonalarteriendrucks unter leichter Belastung ohne Erhöhung des pulmonalen Kapillardrucks (Druckgradient im kleinen Kreislauf steigt). Vorhofmitteldruck und enddiastolischer rechtsventrikulärer Druck normal. Adäquate Zunahme des Herzminutenvolumens.
-
2. Stadium: Erhöhter Pulmonalarteriendruck in Ruhe ohne wesentlichen Anstieg des rechtsventrikulären enddiastolischen und des mittleren Vorhofdrucks. Starker Druckanstieg in der Pulmonalarterie und inadäquate Förderleistung unter Belastung.
-
3. Stadium: Erhöhter Pulmonalarteriendruck in Ruhe. Enddiastolischer Druck im rechten Ventrikel und mittlerer Vorhofdruck deutlich über 8 mmHg. Herzminutenvolumen an der unteren Grenze der Norm und nicht mehr steigerungsfähig (Dekompensierte Rechtsinsuffizienz).
1.16.2.13 Idiopathische und familiäre pulmonale Hypertonie (IPAH)
1.16.2.13.1 Vorkommen
Mit 2 Fällen pro Jahr auf eine Million Einwohner sehr selten. Frauen erkranken viermal häufiger als Männer, hauptsächlich im Alter zwischen 20 und 40 Jahren.
1.16.2.13.2 Ätiologie und Pathogenese
Die Ursache der idiopathischen PAH ist unbekannt. Die familiäre PAH wird autosomal-dominant vererbt. Pathogenetische Komponenten sind:
-
verminderte Produktion vasodilatatorischer Substanzen durch das Endothel
-
Proliferation der Intima und glatten Gefäßmuskelzellen
-
in situ Thrombose in den kleinen pulmonalen Arterien
1.16.2.13.3 Klinik
Treten meistens erst im fortgeschrittenen Stadium auf. Dyspnoe (60 %), Körperschwäche (19 %), Synkopen oder Präsynkopen (13 %) und Raynaud-Phänomen (10 %). Die Neigung zu Synkopen ist durch das fixierte Minutenvolumen bedingt. Es kann bei Anstrengungen nur begrenzt durch Steigerung der Schlagfrequenz erhöht werden. Im Verlauf kommt es zur Rechtsherzinsuffizienz. Haupttodesursachen sind Herzversagen und Kammerflimmern.
1.16.2.13.4 Symptomatische Therapie
Die hier beschriebenen Maßnahmen gelten auch für die anderen Formen der pulmonalen arteriellen Hypertonie.
1.16.2.13.5 Vasodilatatoren
-
Calciumantagonisten: Ein Teil der Patienten spricht auf Höchstdosen von Nifedipin (170 mg/Tag) oder Diltiazem (720 mg/Tag) an.
-
Iloprost-Inhalation: Die Substanz ist ein stabiles Analogon vom Prostacyclin mit vasodilatatorischer Wirkung auf die Pulmonalgefäße. Es wird als Aerosol mit 6–8 Inhalationen pro Tag appliziert (Tagesdosis 75–200 µg).
-
Sildenafil: Der Phosphodiesterase-5-Inhibitor wirkt vasodilatierend, indem er die intrazelluläre Konzentration von cGMP erhöht.
-
Endothelinrezeptor-Blocker: Verfügbar ist die Substanz Bosentan, das durch Vasodilation die Hämodynamik verbessert. Indiziert bei Rechtsinsuffizienz der Klassen III und IV (Initialdosis 2×62,5 mg/Tag, nach 4 Wochen verdoppeln, falls möglich).
-
NO-Inhalation: Bei akuter Dekompensation hilfreich, Langzeitanwendung schwierig.
1.16.2.13.6 Diuretika
Führen durch Verminderung des rechtsventrikulären Füllungsdruck zur Entlastung des rechten Herzens und schwemmen Stauungsödeme aus.
1.16.2.13.7 Digitalis
Steigert bei Rechtsinsuffizienz das Herzzeitvolumen und senkt den Noradrenalinspiegel. Indiziert bei Vorhofflimmern.
1.16.2.13.8 Orale Antikoagulation
Wirkt sekundären Thrombosierungen der kleinen Lungengefäße entgegen und verbessert nachweislich die Lebenserwartung (INR 2,0–3,0).
1.16.2.13.9 Lungentransplantation
Durch die moderne medikamentöse Therapie werden 2-Jahresüberlebensraten von 80 % erreicht. Im fortgeschrittenen Stadium besteht die Indikation zur einseitigen oder doppelseitigen Lungentransplantation. Danach kann die 5-Jahresüberlebensrate 60 % betragen. Selten kommt eine Herz-Lungen-Transplantation in Betracht.
1.16.2.14 Krankheiten mit assoziierter pulmonaler arterieller Hypertonie
1.16.2.14.1 Kollagenkrankheiten
Häufige Komplikation bei Sklerodermie (30 %) und dem CREST-Syndrom (50 %). Deutlich seltener bei Lupus erythematodes, Sjögren-Syndrom und Dermatomyositis.
1.16.2.14.1.1 Diagnostik
Durch immunserologische Untersuchungen und Biopsie (▶ Kap. Kollagenosen).
1.16.2.14.1.2 Klinik
Körperschwäche und Dyspnoe sind initiale Symptome der pulmonalen Hypertonie, die sich prognostisch ungünstig auswirkt.
1.16.2.14.1.3 Therapie
Wie bei IPAH, aber weniger erfolgversprechend.
1.16.2.14.2 Kongenitale Shunt-Vitien
Häufig bei großen posttrikuspidalen Shunts (VSD, offener Ductus Botalli). Vorkommen auch bei prätrikuspidalen Shunts (ASV, anomale Pulmonalvenendrainage). Durch den bestehenden Links-Rechts-Shunt kommt es zu einer chronischen Steigerung des pulmonalen Blutflusses, der das Endothel schädigt und über Mediahyperplasie und Intimaproliferation zur obliterierenden konzentrischen Fibrosierung mit starker pulmonaler Hypertonie führt. Rechtzeitige operative Korrektur kann eine Shuntumkehr verhindern. Allerdings tritt eine PAH manchmal Jahre oder Jahrzehnte nach erfolgreicher Operation auf. Sie ähnelt dann der IPAH. Bei gleicher symptomatischer Therapie ist die Langzeitprognose jedoch besser.
1.16.2.14.3 Pulmonale Hypertension
Eine pulmonale Hypertonie kommt bei portaler Hypertension mit und ohne Leberzirrhose vor. Die Pathogenese ist nicht geklärt. In beiden Fällen ist das Herzzeitvolumen und damit der pulmonale Blutfluss erhöht mit möglicher Wandschädigung. Zusätzlich könnten vasoaktive Mediatoren, Zytokine und Wachstumsfaktoren aus der überdehnten Pfortader eine Rolle spielen. Therapie wie bei IPAH. Leichte Grade der pulmonalen Hypertonie sind keine Kontraindikation für eine Leberttransplantation.
1.16.2.14.4 HIV-Infektion
Der Mechanismus durch den bei dieser Infektion eine pulmonale Hypertonie entstehen kann, ist unbekannt. Die spezifische Therapie hat auf die Komplikation keinen Einfluss. Symptomatische Behandlung wie bei IPAH.
1.16.2.14.5 Appetitzügler
Es ist gesichert, daß verschiedene Anorexiegene eine pulmonale Hypertonie verursachen können. Zu einer Epidemie kam es 1967 in Europa durch die Substanz Aminorex. Später führten Fenfluramin-haltige Präparate zu vielen Fällen. Klinisch ähnelt das Krankheitsbild der IPAH. Nach experimentellen Untersuchungen bewirkt Fenfluramin durch Hemmung spannungsabhängiger Kaliumkanäle eine pulmonale Vasokonstriktion. Betroffene Personen scheinen außerdem eine herabgesetzte basale NO-Produktion zu haben. Therapie wie bei IPAH, doch sprechen die Patienten gewöhnlich schlechter darauf an.
1.16.2.14.6 Pulmonale venookklusive Krankheit (PVOD)
Sehr seltene Ursache der pulmonalen Hypertonie. Es liegt eine Intimaproliferation und Fibrose der intrapulmonalen Venen und Venülen unklarer Ursache vor. Dadurch ist der pulmonale Kapillardruck erhöht. Eine wirksame Therapie gibt es nicht.
1.16.2.14.7 Pulmonale kapillare Hämangiomatose
Bei dieser sehr seltenen Krankheit sind in der Lunge Interstitium und Gefäßwände mit dünnwandigen Blutgefäßen infiltriert. Es kann zu Blutungen kommen. Eine Therapie ist nicht bekannt, die Prognose ist infaust.
1.16.2.14.8 Linksherzerkrankungen mit Lungenstauung
Für einen gestörten Abfluss aus den Lungenvenen in das linke Herz gibt es verschiedene Ursachen: linksventrikuläre Dysfunktion, Mitralvitien, Aortenvitien, Kardiomyopathien und Perikarderkrankungen. Wegen der großen Compliance der Lungenvenen wird ein Rückstau zunächst ohne Drucksteigerung aufgefangen. Höhere Grade des Rückstaus lassen den pulmonalen Venendruck und den Pulmonalarteriendruck in einem Verhältnis steigen, das den Druckgradient und damit auch den Blutfluss konstant hält. Wenn der pulmonale Venendruck 25 mmHg übersteigt, kommt es bei einem Drittel der Patienten zu einer Konstriktion der pulmonalen Widerstandsgefäße mit starkem Anstieg des Pulmonalarteriendrucks (bis 80 mmHg). Trotz dieses Druckanstiegs bleibt der Blutfluss konstant, weil auch der Strömungswiderstand zugenommen hat. Der Mechanismus der Vasokonstriktion ist nicht geklärt. Bei chronischer venöser Hypertension schwellen die pulmonalen Kapillarendothelien, ihre Basalmembran nimmt an Dicke zu und es entwickelt sich ein interstitielles Ödem, das allmählich in Fibrose übergeht. Auf der arteriellen Seite resultieren Mediahypertrophie der Arterien und Rechtsherzbelastung mit Hypertrophie und Insuffizienz. Die Therapie muss nach Möglichkeit die kardialen Ursachen der venösen Hypertension beseitigen. Wenn es gelingt, bildet sich die pulmonale Hypertonie meistens zurück.
1.16.2.14.9 Lungenkrankheiten und/oder Hypoxämie
In der Venedig-Klassifikation sind die in Betracht kommenden Krankheitszustände aufgeführt. Sie werden im Kapitel über die Erkrankungen der Atmungsorgane abgehandelt (▶ Kap. 2).
Hypertrophie und Dilatation des rechten Herzens infolge chronischer pulmonaler Hypertonie durch Erkrankungen des respiratorischen Systems (ohne Linksherzbeteiligung) werden als Cor pulmonale bezeichnet.
Der Widerstandserhöhung im kleinen Kreislauf können Verschluss oder Rarefizierung der Lungengefäße und Ventilationsstörungen mit hypoxisch bedingter Vasokonstriktion zugrunde liegen. Die Therapie richtet sich gegen das Grundleiden und mit den gleichen Mitteln wie bei der IPAH gegen die pulmonale Hypertonie.
1.16.2.14.10 Chronische Lungenembolie
Ganz überwiegend handelt es sich um Thromboembolien, selten um Fett- oder Tumorzellembolien. Neben symptomatischer Therapie wie bei IPAH ist eine Dauerantikoagulation mit Phenprocoumon oder täglichen subkutanen Injektionen von Enoxaparin erforderlich. Bei Verschlüssen größerer Äste der Pulmonalarterie kommt eine Embolektomie in Betracht.
Author information
Authors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Piper, W. (2013). Krankheiten des Herz-Kreislauf-Systems. In: Innere Medizin. Springer-Lehrbuch. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-33108-4_1
Download citation
DOI: https://doi.org/10.1007/978-3-642-33108-4_1
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-33107-7
Online ISBN: 978-3-642-33108-4
eBook Packages: Medicine (German Language)