
Zusammenfassung
Bereits vor über 150 Jahren konnte Charles Darwin durch zahlreiche wissenschaftliche Befunde und Überlegungen aufzeigen, dass sich die heutigen Organismen aus früheren Formen herleiten lassen und durch Evolution entstanden sein müssen. Darwins großes Verdienst bestand jedoch insbesondere darin, dass er einen Mechanismus, nämlich die natürliche Selektion, vorschlug, der die Entwicklung und Spezialisierung der Organismen im Verlauf der Evolution plausibel erklären kann. Obwohl Darwin damals noch kein Wissen über Zellbiologie, Biochemie, Genetik und Molekularbiologie besitzen und daher über die molekularen Mechanismen nichts sagen konnte, werden seine grundsätzlichen Vorstellungen immer noch als zutreffend angesehen. Heute sind wir jedoch in der Lage, Einzelheiten der Evolution und dazugehörige molekulare Mechanismen und Ursachen im Detail wesentlich besser verstehen und belegen zu können. Dazu dienen die Ausführungen in den Kap. 3 und 4 dieses Buches.
3.1 Einführung
Bereits vor über 150 Jahren konnte Charles Darwin durch zahlreiche wissenschaftliche Befunde und Überlegungen aufzeigen, dass sich die heutigen Organismen aus früheren Formen herleiten lassen und durch Evolution entstanden sein müssen. Darwins großes Verdienst bestand jedoch insbesondere darin, dass er einen Mechanismus, nämlich die natürliche Selektion, vorschlug, der die Entwicklung und Spezialisierung der Organismen im Verlauf der Evolution plausibel erklären kann. Obwohl Darwin damals noch kein Wissen über Zellbiologie, Biochemie, Genetik und Molekularbiologie besitzen und daher über die molekularen Mechanismen nichts sagen konnte, werden seine grundsätzlichen Vorstellungen immer noch als zutreffend angesehen. Heute sind wir jedoch in der Lage, Einzelheiten der Evolution und dazugehörige molekulare Mechanismen und Ursachen im Detail wesentlich besser verstehen und belegen zu können. Dazu dienen die Ausführungen in den Kap. 3 und 4 dieses Buches.
Die Evolution und Stammesgeschichte (Phylogenie) der Organismen lässt sich in Form von Stammbäumen anschaulich darstellen. Bereits Darwin dachte in Stammbäumen, wie man der einfachen Skizze in seinem „First Notebook on Transmutations of Species“, das er 1837 erstellte, entnehmen kann (◘ Abb. 3.1). Die Grundidee besteht darin, dass Organismen, die gleiche Merkmale tragen, auch nahe verwandt sein müssen und in den Bäumen benachbart positioniert sind, da sie diese Merkmale von einem gemeinsamen Vorfahren ererbt haben. Ein wichtiges Ziel der Biologie besteht deshalb darin, ein natürliches System der systematischen Beziehungen aller Lebewesen zu erstellen. Die Zuordnung nach dem Ähnlichkeitsprinzip wird jedoch dadurch erschwert, dass gleiche Merkmale nicht in jedem Falle homolog sind, sondern auch konvergent durch Anpassung an gemeinsame Lebensbedingungen entstanden sein können; d. h. sie sind analog (zur Diskussion der Homologie-Analogie-Problematik s. Abschn. 1.2.3). Bekannte Beispiele für analoge Merkmale sind die Flossen bei Fischen und Tintenfischen (Cephalopoda) und die Flügel bei Vögeln und pterygoten (geflügelten) Insekten. Für die Interpretation von Verwandtschaftsverhältnissen sind aber nur die homologen Merkmale hilfreich. Die Unterscheidung zwischen homologen und analogen Merkmalen ist in der Praxis häufig problematisch und kann zu widersprüchlichen Phylogeniehypothesen führen. Schon Charles Darwin beklagte diese Problematik:

a, b. Darstellung evolutionärer Beziehungen anhand eines Stammbaums. a Skizze eines hypothetischen Stammbaums von Charles Darwin (1837) im „First Notebook on Transmutation of Species“. b Schematische Darstellung eines Phylogramms: Taxa A und B, C und D sowie E und F sind jeweils Schwesterarten, die sich von einem gemeinsamen Vorfahren ableiten lassen. Taxa C, D, E und F bilden eine monophyletische Gruppe. Der blaue Pfeil weist auf den gemeinsamen Vorfahren hin, von dem sich die monophyletischen Gruppen ableiten lassen
a, b. Darstellung evolutionärer Beziehungen anhand eines Stammbaums. a Skizze eines hypothetischen Stammbaums von Charles Darwin (1837) im „First Notebook on Transmutation of Species“. b Schematische Darstellung eines Phylogramms: Taxa A und B, C und D sowie E und F sind jeweils Schwesterarten, die sich von einem gemeinsamen Vorfahren ableiten lassen. Taxa C, D, E und F bilden eine monophyletische Gruppe. Der blaue Pfeil weist auf den gemeinsamen Vorfahren hin, von dem sich die monophyletischen Gruppen ableiten lassen
Darwin schrieb 1857 an seinen Freund Thomas H. Huxley: „In regard to classification, & all the endless disputes about the ‚natural system‘ which no two authors define in same way, I believe it ought, in accordance with my heterodox notions, to be simply genealogical. – But as we have no written pedigrees, you will, perhaps, say this will not help much; but I think it ultimately will, whenever heterodoxy becomes orthodoxy, for it will clear away an immense amount of rubbish about the value of characters & – will make the difference between analogy & homology, clear. – The time will come I believe, though I shall not live to see it, when we shall have fairly true genealogical trees of each kingdom of nature …“ („In Hinblick auf die Klassifizierung der Organismen und die endlosen Dispute über das natürliche System, das von zwei Autoren niemals gleich definiert wird, habe ich die unorthodoxe Vorstellung, dass sie einfach genealogisch sein sollte. – Da wir aber keine dokumentierten Stammbäume besitzen, werden Sie vermutlich argumentieren, dass diese Aussage nicht weiterhilft. Aber letztendlich wird sie weiterführen, dann nämlich, wenn sich die unorthodoxe Sichtweise als richtig herausstellen wird. Denn genealogische Stammbäume werden die immensen Dummheiten, die über den Wert von Merkmalen geschrieben wurden, beseitigen und den Unterschied zwischen Analogie und Homologie klarstellen. – Ich glaube, dass einmal die Zeit kommen wird, obwohl ich es nicht erleben werde, dass wir ziemlich exakte genealogische Stammbäume für jedes Reich der Natur haben werden …“)
Die Evolutionsbiologie, die sich traditionell mit Fachgebieten wie vergleichender Anatomie und Morphologie, Entwicklungsbiologie, Biogeographie, Paläontologie und Verhaltensbiologie sowie Biochemie (Proteine, Sekundärstoffe), aber nicht mit der Erbsubstanz selbst beschäftigen konnte, hat in den letzten 40–50 Jahren durch die rasche Entwicklung der Molekularbiologie neue Werkzeuge erhalten, um Evolutionsvorgänge sowie deren Mechanismen und Ursachen molekular zu analysieren und besser zu verstehen.
Die vielen Millionen Basenpaare in den Genomen der heute lebenden Organismen stellen eine Blaupause der Evolutionsgeschichte und Phylogenie eines jeden Individuums dar. Punktmutationen haben das Genom im Verlauf der Zeit in kleinen Schritten verändert ( Abschn. 3.3). Durch Verdopplungen von DNA-Abschnitten oder ganzen Genomen, Insertionen und Inversionen wurden Genome umorganisiert oder vergrößert. Mobile DNA-Elemente, vor allem aber Viren, haben dazu beigetragen, dass DNA-Abschnitte horizontal, d. h. über Artgrenzen hinaus, verschoben wurden ( Abschn. 3.4 und EXKURS 3.4 Abschn. 3.4.3). Alle diese DNA-Veränderungen, die in der Vergangenheit auftraten, sind im Genom eines jeden Individuums und aller Arten gespeichert. Denn jede Zelle der heute lebenden Eukaryoten ist durch Fusion einer Ei- und Spermazelle oder durch Teilung aus einer Mutterzelle entstanden, und hat damit die DNA einer Vorgängerzelle erhalten. Wenn es gelingt, diese Informationen zu lesen und zu interpretieren, dann können wir die nahe, mittlere und ferne evolutionäre Vergangenheit aller Lebewesen rekonstruieren. Theoretisch lässt sich über die DNA-Analyse die Verwandtschaft der Lebewesen bis zur Entstehung des Lebens zurückverfolgen, ohne dass wir die Zwischenglieder oder Fossilien kennen. So wie der Archäologe aus alten Scherben auf frühere Kulturen oder der Paläontologe anhand von Fossilien auf die Phylogenie ausgestorbener Arten schließen kann, vermag die molekulare Evolutionsforschung anhand der DNA-Analyse die Entstehung des Lebens auf der Erde sowie die Phylogenie oder Phylogeographie einer Organismengruppe, Art oder Population zu rekonstruieren.
Die molekularen Methoden reichen von der bereits länger etablierten Allozymanalyse über neuere Verfahren, wie DNA-Fingerprinting, Mikrosatelliten-Analyse, bis hin zur DNA-Sequenzierung (inklusive des Next Generation Sequencing) und vergleichenden Genomanalyse ( Kap. 4). Genetische Unterschiede sind zwischen Individuen einer Population in der Regel klein, aber größer zwischen Arten oder Angehörigen verschiedener Gattungen oder Familien. Je länger zwei Organismen sich von einem gemeinsamen Vorfahren getrennt haben, desto größer sind die genetischen Unterschiede. Diese Tatsache ist die Grundlage für die molekulare Uhr, die in Abschn. 4.1.2 ausführlicher diskutiert wird.
Obwohl die Molekulare Evolutionsforschung eine noch junge Disziplin ist, kann man jetzt schon absehen, dass Darwins Vision vom natürlichen System bereits vielfach Realität geworden ist oder bald werden wird. Schon heute verfügen wir über verlässliche molekulare Stammbäume für ausgewählte Organismen (s. Kap. 4), und vermutlich werden die Evolutionsbiologen in nächsten Jahren derartige Stammbäume für alle wichtigen Tier-, Pflanzen- und Prokaryoten-Gruppen aufgestellt haben. Solche Projekte laufen häufig unter dem Überbegriff Assembling the Tree of Life (ATOL-Projekte). Neuerdings bestehen Bestrebungen, alle Organismen durch die Analyse von Markergenen zu identifizieren und zu typisieren. In Analogie zu den Strichcodes auf Industrieprodukten spricht man deshalb vom DNA-Barcoding.
3.2 Grundlagen der Molekularbiologie und Genetik
3.2.1 Aufbau der DNA
Desoxyribonucleinsäure (DNA) ist ein Makromolekül, das aus linear gekoppelten Nucleotiden aufgebaut ist (◘ Abb. 3.2). Jeder der vier Nucleotid-Bausteine besteht aus einer stickstoffhaltigen Base (◘ Tab. 3.1), d. h. einem heterozyklischen Kohlenstoffgerüst. Die Pyrimidinbasen Cytosin (C) und Thymin (T) weisen zwei N-Atome auf, die Purinbasen Adenin (A) und Guanin (G) jeweils vier N-Atome. Außerdem gehören Desoxyribose (eine Pentose) und eine Phosphatgruppe zu einem Nucleotidbaustein. Im Unterschied zur DNA findet man in der Ribonucleinsäure (RNA) Uracil (U) anstelle von Thymin und Ribose (der die Hydroxylgruppe in 2-Position fehlt) anstelle von Desoxyribose. DNA enthält also die Basen A, T, G und C, RNA die Basen A, U, G und C.

a, b. a Struktur der Bausteine der Nucleinsäuren und Aufbau von DNA und RNA. A: Adenin, G: Guanin, C: Cytosin, T: Thymin, U: Uracil; b Bei der Biosynthese der Nucleinsäuren wird die α-ständige Phosphatgruppe von Trinucleotiden (dNTPs) mit der freien 3‘-OH-Gruppe des bereits vorliegenden Stranges verknüpft
a, b. a Struktur der Bausteine der Nucleinsäuren und Aufbau von DNA und RNA. A: Adenin, G: Guanin, C: Cytosin, T: Thymin, U: Uracil; b Bei der Biosynthese der Nucleinsäuren wird die α-ständige Phosphatgruppe von Trinucleotiden (dNTPs) mit der freien 3‘-OH-Gruppe des bereits vorliegenden Stranges verknüpft
Die Basen sind N-glycosidisch mit der 1-Position der Pentose verknüpft. Ein solches Molekül wird als Nucleosid bezeichnet; trägt die 5-Position der Pentose einen Phosphatrest, so liegt ein Nucleotid vor (◘ Tab. 3.1).
Die Nucleotide stellen die Bausteine für DNA und RNA dar. Nucleotide sind über ein Phosphatrückgrat zu Polynucleotidketten verknüpft. Dabei wird jeweils die 5‘-Hydroxylgruppe (sprich „Fünf-Strich-Hydroxylgruppe“) einer Pentose über eine Phosphodiesterverbindung mit der 3‘-Hydroxylgruppe einer zweiten Pentose verknüpft (◘ Abb. 3.2). In einer Nucleinsäurekette haben die endständigen Nucleotide eine freie 5‘-Gruppe auf der einen Seite und eine freie 3‘-Gruppe auf der anderen Seite. Man hat sich darauf geeinigt, Nucleotidsequenzen in der 5‘→3'-Orientierung aufzuschreiben, wobei der 5'-Terminus links und das 3'-Ende rechts zu stehen kommt.
Zur Biosynthese der Nucleinsäuren (◘ Abb. 3.2) werden die jeweiligen Triphosphate benötigt, deren Phosphatesterbindungen besonders energiereich sind. In der fertigen Nucleinsäure liegen die jeweiligen Monophosphate vor. Nach Abspaltung eines Diphosphatrestes greift die α-Phosphatgruppe am freien 3‘-Ende des bereits bestehenden Nucleinsäurestranges an und bildet eine neue Phosphodiesterbindung.
Die DNA liegt als Doppelhelix vor, wobei die Basen A und T bzw. G und C sich jeweils komplementär gegenüberstehen (◘ Abb. 3.3). Die DNA-Doppelhelix weist einen Durchmesser von 2 nm auf. Die komplementäre Basenpaarung kommt durch Ausbildung von jeweils zwei bzw. drei Wasserstoffbrücken zwischen AT- bzw. GC-Paaren zustande (◘ Abb. 3.3). Die komplementäre Basenpaarung kann als Ergebnis einer molekularen Erkennungsreaktion angesehen werden.

Aufbau der DNA-Doppelhelix. Räumliche Orientierung der Basenpaare in der Doppelhelix (maßstabsgetreues Modell der DNA nach dem Moletomics™-Konzept, das die Bildung der großen und kleinen Furche zeigt; Herkunft: Quadbeck-Seeger) und Prinzip der komplementären Basenpaarung zwischen A und T bzw. G und C durch Ausbildung von Wasserstoffbrücken-Bindungen
Aufbau der DNA-Doppelhelix. Räumliche Orientierung der Basenpaare in der Doppelhelix (maßstabsgetreues Modell der DNA nach dem Moletomics™-Konzept, das die Bildung der großen und kleinen Furche zeigt; Herkunft: Quadbeck-Seeger) und Prinzip der komplementären Basenpaarung zwischen A und T bzw. G und C durch Ausbildung von Wasserstoffbrücken-Bindungen
Die beiden DNA-Stränge sind antiparallel angeordnet, d. h. wenn man auf die Helix blickt, läuft einer der Stränge in 5‘→3'-Richtung, während der Partnerstrang in 3'→5'-Richtung orientiert ist. Die nach außen durch Phosphatgruppen vielfach negativ geladene DNA-Doppelhelix wird bei Eukaryoten durch basische Histonproteine komplexiert (bilden die sogenannten Nucleosomen); bei Prokaryoten übernehmen Polyamine diese Rolle. Die Basen sind ins Helixinnere gerichtet und bilden planare Stapel aus (◘ Abb. 3.3). Das Innere der Helix ist wasserfrei, d. h. nur lipophile Substanzen, vor allem, wenn sie ebenfalls planar sind, können sich zwischen die Basenstapel einlagern („DNA-Interkalatoren“). Eine solche Interkalation führt meist zu Fehlern bei der Replikation, die zu Frame-shift-Mutationen, d. h. einem Verschieben des Leserasters, führen können (s. Abschn. 3.3.1).
Bedingt durch die Kooperativität vieler Wasserstoffbrücken und die lipophilen Wechselwirkungen zwischen den Basenstapeln ist die DNA-Doppelhelix sehr stabil und kann nur durch hohe Temperaturen in ihre beiden Einzelstränge getrennt werden. Dieser Vorgang wird auch als Schmelzen bezeichnet; T m kennzeichnet die Temperatur, bei der 50 % der DNA bereits einzelsträngig vorliegen. Tm ist abhängig vom GC-Gehalt der DNA, der zwischen den Organismengruppen deutlich schwankt (◘ Tab. 3.2). Je größer der GC-Gehalt, desto höher liegt die mittlere Schmelztemperatur (bedingt durch drei Wasserstoffbrücken in GC-Paaren gegenüber zwei Wasserstoffbrücken in AT-Paaren). Tm-Werte wurden in der Anfangszeit der molekularen Systematik als taxonomisches Merkmal, insbesondere für die Klassifizierung von Prokaryoten, herangezogen. Tm-Werte spielten ferner bei der heute kaum noch eingesetzten DNA-DNA-Hybridisierung in Phylogenieuntersuchungen eine wichtige Rolle (s. Abschn. 4.1.2).
3.2.2 Replikation
Schon Rudolf Virchow postulierte 1885, dass Zellen nicht de novo entstehen können, sondern immer nur aus der Teilung einer Mutterzelle hervorgehen. Sein Lehrsatz lautete „omnis cellula e cellula“. Jeder Zellteilung muss eine exakte Verdopplung des Genoms vorausgehen, d. h. aus jedem Chromosom entstehen zwei identische Chromatiden, die nach Trennung als identische Tochterchromosomen auf die Tochterzellen verteilt werden (s. Abschn. 3.3). Die Verdopplung der DNA, die als DNA-Replikation bezeichnet wird, verläuft semikonservativ. Dabei wird der DNA-Doppelstrang zunächst lokal in seine Einzelstränge getrennt, indem sich eine Replikationsgabel bildet. Die Einzelstränge dienen nun als Matrize für die Synthese der jeweils komplementären neuen Stränge (◘ Abb. 3.4). Die DNA-Replikation ist ein komplexer Vorgang, an dem mehrere Proteine und Enzyme beteiligt sind.

Schematische Darstellung der DNA-Replikation und Transkription. DNA-Polymerase-Komplex (Pol III: DNA-Polymerase III; SSB: single-strand binding protein; Transkription erfolgt mittels RNA-Polymerase
Schematische Darstellung der DNA-Replikation und Transkription. DNA-Polymerase-Komplex (Pol III: DNA-Polymerase III; SSB: single-strand binding protein; Transkription erfolgt mittels RNA-Polymerase
DNA-Polymerasen kopieren die ursprüngliche Basensequenz äußerst exakt (ihre Fehlerrate liegt während der eigentlichen Synthese bei 1 falsch eingebautem Nucleotid pro 10.000 Nucleotide). Spezielle Korrekturlese- und Reparaturfunktionen des Enzyms spielen eine große Rolle und sorgen dafür, dass die fertige Kopie fast fehlerfrei ist. Falsch gepaarte Nucleotide werden durch eine spezifische Exonuclease entfernt und dann durch DNA-Polymerase ersetzt; zuletzt wird die Phosphodiesterbindung mittels DNA-Ligase kovalent verknüpft ( Abschn. 3.3). Diese hohe, aber nicht absolute Genauigkeit war und ist für die Evolution von großer Wichtigkeit, denn das Erzeugen von Variabilität ist eine Grundvoraussetzung für evolutive Vorgänge. Zur Entfernung der in der DNA regelmäßig und häufig entstehenden Mutationen sind in der Evolution spezielle Reparaturenzyme selektiert worden, die in Abschn. 3.3.1 näher besprochen werden.
3.2.3 Vom Gen zum Protein
Ursprünglich, als man die DNA als Genträger noch nicht kannte, wurden Mutations- und Rekombinationseinheiten als ein Gen bezeichnet; in den 50er Jahren des 20. Jahrhunderts wurden Gene enger definiert und die „Ein-Gen-ein-Protein“-Hypothese aufgestellt („DNA makes RNA, which makes proteins“) (◘ Abb. 3.5). Heute wird das Gen allgemein als Transkriptionseinheit definiert, da inzwischen sowohl die Intron/Exonstruktur als auch die nicht-codierenden regulatorischen Sequenzen, die zu einem Gen gehören, sowie das alternative Spleißen erkannt wurden. Mit Exon bezeichnet man die DNA-Abschnitte innerhalb eines Gens, die eine Proteindomäne codieren, während Introns nicht-codierende Bereiche zwischen benachbarten Exons darstellen (◘ Abb. 3.6).

Vom Gen zum Protein. Im Zellkern der Eukaryoten finden Replikation und Transkription statt. Die mRNA wird aus dem Kern über Poren in der Kernmembran in das Cytoplasma transportiert. Im Cytoplasma erfolgt die Übersetzung (Translation) des mRNA-Codes in Aminosäuresequenzen mittels Ribosomen
Vom Gen zum Protein. Im Zellkern der Eukaryoten finden Replikation und Transkription statt. Die mRNA wird aus dem Kern über Poren in der Kernmembran in das Cytoplasma transportiert. Im Cytoplasma erfolgt die Übersetzung (Translation) des mRNA-Codes in Aminosäuresequenzen mittels Ribosomen

Schematische Darstellung der Struktur eines Eukaryotengens. Zu einem Gen zählen die regulatorischen Enhancer- und Promotorregionen ebenso wie die eigentliche Transkriptionseinheit, die aus Introns, Exons und nicht-codierenden Sequenzen (NCS) aufgebaut ist. Der DNA-Abschnitt, der oberhalb des Promotors liegt, wird 5‘-Upstream-Region genannt; entsprechend heißt der DNA-Abschnitt, der hinter einem Gen folgt, 3‘-Downstream-Region. Die CCAAT- und TATA-Box bezeichnet DNA-Sequenzen in Promotoren, an die Transkriptionsfaktoren binden
Schematische Darstellung der Struktur eines Eukaryotengens. Zu einem Gen zählen die regulatorischen Enhancer- und Promotorregionen ebenso wie die eigentliche Transkriptionseinheit, die aus Introns, Exons und nicht-codierenden Sequenzen (NCS) aufgebaut ist. Der DNA-Abschnitt, der oberhalb des Promotors liegt, wird 5‘-Upstream-Region genannt; entsprechend heißt der DNA-Abschnitt, der hinter einem Gen folgt, 3‘-Downstream-Region. Die CCAAT- und TATA-Box bezeichnet DNA-Sequenzen in Promotoren, an die Transkriptionsfaktoren binden
Der Fluss der Erbinformation verläuft bei allen Organismen vom Gen über die mRNA zum Protein (◘ Abb. 3.5). Nur Retroviren können RNA mittels reverser Transcriptase in DNA zurückübersetzen; aber in keinem Falle wurde ein Informationsfluss vom Protein zum Gen nachgewiesen. Durch Variation und Kombination der 20 Protein-bildenden Aminosäuren können Peptide und Proteine alle nur erdenklichen Raumstrukturen, aktive Zentren und Bindungsstellen bilden. Durch alternatives Spleißen können aus einem mRNA-Vorläufer, in dem noch Introns und Exons enthalten sind, fertige mRNAs gebildet werden, die unterschiedliche Kombinationen von Exons enthalten (◘ Abb. 3.29). Auf diese Weise können aus einem Gen diverse Proteine codiert werden, die sich in der Zusammenstellung der Exons unterscheiden. Durch das alternative Spleißen kann also die phänotypische Variabilität und Plastizität erhöht werden. Proteine sind aufgrund ihrer Strukturvariabilität in der Lage, ihre mannigfaltigen Aufgaben als Enzyme, Rezeptoren, Ionenkanäle, Transporter, Strukturproteine, Transkriptionsfaktoren, Wachstumsfaktoren und Hormone zu übernehmen. Proteine gehören somit zu den wichtigsten Werkzeugen der Zelle. Ihre Konformation
codierender Strang | 5‘-GGC | TCC | CTA | TTA | GCA | GTC | TGC | CTC | ATG-3‘ |
Templatestrang | 3‘-CCG | AGG | GAT | AAT | CGT | CAG | ACG | GAG | TAC-5‘ |
mRNA | 5‘-GGC | UCC | CUA | UUA | GCA | GUC | UGC | CUC | AUG-3‘ |
ist für molekulare Erkennungsreaktionen von entscheidender Bedeutung. Wird durch eine Genmutation eine Aminosäure in einem Protein ausgetauscht, so kann dies die Funktionsweise eines Proteins dann einschneidend verändern, wenn dadurch Raumstruktur oder Bindungsstellen verändert werden.
codierender Strang | 5‘-GGC | TCC | CTA | TTA | GCA | GTC | TGC | CTC | ATG-3‘ |
Templatestrang | 3‘-CCG | AGG | GAT | AAT | CGT | CAG | ACG | GAG | TAC-5‘ |
mRNA | 5‘-GGC | UCC | CUA | UUA | GCA | GUC | UGC | CUC | AUG-3‘ |
3.2.4 Transkription und Mosaikstruktur der Eukaryotengene
Bei Eukaryoten finden wir drei verschiedene RNA-Polymerasen, die DNA in mRNA (RNA-Polymerase II), in rRNA (RNA-Polymerase I) oder in andere funktionelle RNAs (z. B. tRNAs; RNA-Polymerase III) umschreiben. Dieser Prozess wird als Transkription bezeichnet.
Auch bei der Transkription wird die DNA-Doppelhelix lokal geöffnet, so dass die RNA-Polymerase die RNA (mRNA, rRNA oder tRNA) komplementär zum Template-DNA-Strang synthetisieren kann (◘ Abb. 3.4). Der Templatestrang dient somit als Matrize für die Synthese der mRNA. Der DNA-Strang, der dieselbe Basensequenz wie die mRNA aufweist (außer dass er T anstelle von U enthält), wird Nicht-Templatestrang oder (irreführenderweise) als codierender Strang bezeichnet. Üblicherweise wird die Sequenz des codierenden Stranges in 5'→3'-Orientierung abgebildet und auch so in Datenbanken hinterlegt.
Die Festlegung Template- oder Nicht-Templatestrang gilt nicht für ein komplettes Chromosom; innerhalb eines Chromosoms kann diese Funktion von Gen zu Gen wechseln, d. h. Gen A kann von einem der Stränge abgelesen werden, das benachbarte Gen B dagegen vom gegenüber liegenden komplementären Strang (s. ◘ Abb. 3.18).
Bei Eukaryoten sind die Protein-codierenden Gene meist aus Exons und Introns aufgebaut; wir sprechen deshalb auch von Mosaikgenen. Das bei der Transkription entstehende Primärtranskript wird anschließend noch im Zellkern so prozessiert („gespleißt“; abgeleitet von splicing), dass die jeweils nicht-codierenden Intronregionen, die durch GU- und AG-Sequenzen flankiert sind, entfernt werden. Am Spleißprozess sind snRNAs (small nuclear RNAs) beteiligt und wirken hier als Ribozyme, d. h. RNAs mit katalytischer Aktivität. Die wichtige Bedeutung der Intron-Exon-Struktur für die Evolution (Erhöhung der Variabilität; Generierung neuer Proteine) wird in Abschn. 3.4 ausführlich diskutiert.
Während bei Eukaryoten die Gene in der Regel streng linear hintereinander angeordnet sind, findet man bei Prokaryoten häufiger sich überlappende Gene, die entweder von demselben oder dem gegenüber liegenden komplementären DNA-Strang codiert werden. Dies ermöglicht eine erhöhte Informationsdichte, behindert aber die unabhängige Evolution der DNA-Sequenzen.
Die Transkription eines Gens wird durch benachbarte regulatorische DNA-Bereiche (Promotoren, enhancer) (◘ Abb. 3.6) mittels Transkriptionsfaktoren gesteuert, die darüber entscheiden, ob ein Gen angeschaltet und aktiv oder abgeschaltet und inaktiv ist. Von den rund 20.000 Protein-codierenden Genen des Menschen wird in einer einzelnen differenzierten Zelle nur immer ein kleiner Teil der Gene spezifisch angeschaltet, während die Mehrzahl der Gene inaktiv bleibt. Der Aufbau eines korrekten zellspezifischen Genexpressionssystems war ein wichtiges Ergebnis der frühen Evolution und Voraussetzung für die Entwicklung von höher differenzierten Metazoen. Genregulation, insbesondere das Abschalten von Genen, kann auch über RNA-Interferenz (RNAi) erfolgen. Sie beruht auf der Aktivität von microRNA-Molekülen, die komplementär zur Basensequenz von spezifischen Genen sind. Wenn microRNAi mit komplementären mRNAs hybridisieren, entstehen Doppelstränge, die enzymatisch von einem Enzymkomplex abgebaut werden. Auf diese Weise wird die Genexpression auf dem Weg vom Transkript zur Translation gestört. In der Molekularbiologie spielt die RNAi-Technik heute eine große Rolle, um gezielt Gene auszuschalten.
Bei der Genregulation und der Differenzierung spielt die Epigenetik eine entscheidende Rolle (s. EXKURS 5.9 Abschn. 5.7). Der Begriff Epigenetik wurde 1942 von Conrad H. Waddington geprägt als „the branch of biology which studies the causal interactions between genes and their products which bring the phenotype into being“ („der Zweig der Biologie, der die kausalen Wechselwirkungen zwischen Genen und ihren Produkten, die den Phänotyp hervorbringen, studiert“). Durch unterschiedliche Umweltbedingungen können ausgehend von einem singulären Genotyp diverse Phänotypen (Polyphänie) herausgebildet werden. Für die phänotypische Plastizität spielt die Epigenetik ebenfalls eine wichtige Rolle. Bei der Epigenetik geht es um die Methylierung von Cytosin (bei Eukaryoten) sowie eine enzymatische Modifikation der Histonproteine. In der Regel sind die Promotoren der Gene, die in einer Zelle exprimiert werden, nicht methyliert, während sie bei abgeschalteten Genen (silent) hoch methyliert sind. Nach jeder Replikation muss die Methylierung des frisch replizierten DNA-Stranges neu erfolgen; eine Störung der Methyltransferasen kann die Genexpression und Differenzierung von Zellen (und damit den Phänotyp) stark beeinflussen. Als kleinste Organisationseinheit des Chromatins ist die DNA in Nucleosomen verpackt. Unter dem mit basischen Kernfarbstoffen anfärbbaren Chromatin versteht man das Material (Komplex aus DNA und speziellen Proteinen, u. a. Histone), aus dem die Chromosomen aufgebaut sind. Wenn Gene transkribiert werden sollen, muss die Nucleosomorganisation kurzfristig aufgegeben werden. Dies wird durch eine lokale enzymatische Veränderung der Histonproteine erreicht.
Im Wesentlichen durch die Methylierung der DNA-Basen und durch enzymatische Modifizierung der Histonproteine wird die Differenzierung von Zellen gesteuert und festgelegt („Epigenese“, genetische Prägung oder imprinting). Die DNA ist in den Gameten noch nicht durch Methylierung modifiziert. Nach der Befruchtung ist die Zygote omnipotent; sukzessive erfolgt eine Programmierung des Erbmaterials in der nachfolgenden Embryonalentwicklung. Daher weisen differenzierte Zellen einen hohen Methylierungsgrad auf und sind daher nicht mehr in der Lage, sich in andere Zelltypen umzuwandeln.
DNA-Methylierung und Histonmodifizierungen werden bei der Zellteilung an die Tochterzellen vererbt; man spricht hier von einer epigenetischen Vererbung. Solche somatischen Veränderungen haben vermutlich aber keinen Einfluss auf die Nachkommen. Nur direkte Mutationen der Gameten können an die nachkommenden Generationen vererbt werden. Ausnahmen wurden bei Pflanzen nachgewiesen, was die Diskussion über die Hypothese der Vererbung erworbener Eigenschaften (Lamarck) wieder entfacht hat. Über die möglichen Einflüsse der Epigenetik auf die Evolution des Menschen geht der EXKURS 5.9 in Abschn. 5.7 ein.
3.2.5 Genetischer Code
Ein zentraler Fortschritt in der Anfangszeit der Molekularbiologie war die Entdeckung eines fast einheitlichen, kommalosen und nicht überlappenden genetischen Codes bei allen lebenden Organismen, der für Bakterien ebenso gilt wie für Pflanzen und Tiere. Er wird „kanonisch“ genannt und evolvierte offenbar in einer Epoche, bevor sich die Organismenreiche aufspalteten. Jeweils drei Nucleotide codieren für den Einbau einer spezifischen Aminosäure in das jeweilige Protein (◘ Abb. 3.7). Der weitgehend universelle Triplettcode beginnt an einem spezifischen Startsignal. Da Methionin (bei Eukaryoten) bzw. N-Formylmethionin (bei Bakterien und Chloroplasten) als erste Aminosäure in Polypeptide eingebaut wird, heißt das universelle Startcodon AUG (wesentlich seltener kommt GUG vor). Methionin bleibt jedoch nicht als die erste Aminosäure in den fertigen Proteinen erhalten, sondern wird nach der Translation in den meisten Fällen durch eine spezifische Protease wieder entfernt.

Überblick über den genetischen Code. Die meisten Aminosäuren werden von mehr als einem Triplett codiert. Unterschiede im Codon findet man meist in der dritten Triplettposition. Die Abkürzungen der Aminosäuren sind in Tab. 3.3 erklärt
Überblick über den genetischen Code. Die meisten Aminosäuren werden von mehr als einem Triplett codiert. Unterschiede im Codon findet man meist in der dritten Triplettposition. Die Abkürzungen der Aminosäuren sind in Tab. 3.3 erklärt
In den tierischen und pilzlichen (nicht aber pflanzlichen) Mitochondrien gibt es kleine Abweichungen vom universellen genetischen Code: z. B. wird auch AUA zur Initiation verwendet und codiert für Methionin, während dieses Codon in eukaryotischen Ribosomen für Isoleucin steht; AGG/AGA wird bei Vertebraten als Terminationscodon eingesetzt, während es sonst für Arginin codiert. UGA, das gewöhnlich als Stoppcodon eingesetzt wird, kann auch für Selenocystein („21. Aminosäure“) oder für Pyrrolysin (ein modifiziertes Lysinmolekül; „22. Aminosäure“) codieren.
Der Translationsstart beginnt bei AUG; dadurch ist die Sequenz der folgenden Codons festgelegt (der Leserahmen). Würde sich der Start der Translation auch nur um ein oder zwei Nucleotide verschieben, käme es zu einer Verschiebung des Leserahmens, einem frame shift. Dadurch würden sich die Codons verschieben und für gänzliche andere Aminosäuren codieren. Folglich würde ein gänzlich anderes neues Protein entstehen. Mutationen, die einen frame shift verursachen, führen meist zum Funktionsverlust in den veränderten Proteinen (s. Abschn. 3.3.1).
Bei einem Triplettcode mit vier Basen stehen theoretisch 43 = 64 Kombinationen zur Verfügung. Da aber nur 20 reguläre Aminosäuren in Proteinen (◘ Tab. 3.3) vorkommen, gibt es mehr Codons als eigentlich notwendig wären. In der frühen Evolution wurde dieses Problem so gelöst, dass einige Aminosäuren nicht von nur einem, sondern von zwei bis maximal sechs verschiedenen synonymen Codons (also Codons, die jeweils für dieselbe Aminosäure stehen) codiert werden (◘ Abb. 3.7). Häufig unterscheiden sich die Codons, die für dieselbe Aminosäure codieren, in der dritten Codonposition. Die zugehörigen tRNAs existieren grundsätzlich für jedes Codon; bei synonymen oder „degenerierten“ Codons, die alle dieselbe Aminosäure codieren, existiert häufig nur eine spezifische tRNA, die eine Fehlpaarung ( mismatching) in der dritten Codonposition toleriert. Insgesamt wurden im eukaryotischen System ca. 31 tRNAs, in Mitochondrien 22 tRNAs nachgewiesen. Eine Mutation in der dritten Codonposition beeinflusst das Ergebnis der Proteinbiosynthese meist nicht, da dieselbe Aminosäure eingebaut wird; die Konsequenz aus dieser für die molekulare Evolution so wichtigen Tatsache wird ausführlich in Abschn. 3.3.1 diskutiert.
3.2.6 Proteinbiosynthese (Translation)
Die Proteinbiosynthese erfolgt in den Ribosomen, die komplex aufgebaute Multienzymkomplexe (auch molekulare Maschinen genannt) darstellen. In den Ribosomen spielen verschiedene rRNAs eine wichtige Rolle (◘ Abb. 3.10, ◘ Abb. 3.11). Da die rRNAs durch interne Basenpaarung (Stammstrukturen) komplexe Raumstrukturen ausbilden, können sie als Gerüst für die richtige Anordnung der diversen Ribosomenproteine dienen. Sie sind zudem katalytisch aktiv, z. B. bei der Synthese von Peptidbindungen. Da die Proteinbiosynthese ein Prozess ist, der offenbar in der frühen Evolution entstand, ist es nicht verwunderlich, dass der Aufbau der Ribosomen in den verschiedenen Organismenbereichen grundsätzlich sehr ähnlich ist, wenn sich auch prokaryotische von eukaryotischen Ribosomen in Einzelheiten unterscheiden.
Ribosomale RNAs (rRNAs) gehören zu den häufigsten Makromolekülen in einer Zelle, alleine für E. coli schätzt man die Zahl der rRNA-Moleküle auf 38.000. Die zusammengehörigen rRNA-Gene liegen als Sequenzeinheiten (rDNA-Kassetten) vor, z. B. in der Abfolge 18S rDNA, 5,8S rDNA und 28S rDNA, die als komplette Einheit transkribiert werden (◘ Abb. 3.9). Nach der Transkription werden sie in die einzelnen rRNAs aufgespalten. Da diese rRNAs unterschiedlich groß sind, kann man sie mittels Ultrazentrifugation voneinander trennen. Die Größe der rRNA wird in Svedberg-Einheiten (S) angegeben; also 18S für 18 Svedberg-Einheiten. In den Genomen der Zellen kommen die rDNA-Kassetten in zahlreichen Kopien vor. Dies beruht sicher auf der Tatsache, dass diese Gene sehr häufig abgelesen werden müssen, um die große Zahl an rRNA-Molekülen zu produzieren, die jede Zelle zum Aufbau der zahlreichen Ribosomen benötigt.

Schematische Darstellung der ribosomalen Proteinbiosynthese von Pro- und Eukaryoten. Die große Ribosomenuntereinheit steht oben, die kleine Untereinheit, durch die die mRNA läuft, unten. In der A-Stelle hybridisiert jeweils die ankommende, mit einer Aminosäure (AS) beladene tRNA mit ihrem Anticodon an das entsprechende Triplett der mRNA. Dann kommt es zum Transfer des Peptidrestes, der auf der tRNA in der P-Stelle sitzt, auf die Aminosäure in der A-Stelle (Peptidyltransferase). Jetzt rückt das Ribosom drei Nucleotide weiter auf der mRNA und entlässt die freie tRNA aus der E-Stelle (Exit-Stelle); die tRNA mit dem verlängerten Peptidylrest rückt in die P-Stelle. Diese Schritte wiederholen sich, bis ein Stoppcodon erreicht wird. Die Knüpfung der Peptidbindung (roter Pfeil) wird durch die rRNA katalysiert.
Schematische Darstellung der ribosomalen Proteinbiosynthese von Pro- und Eukaryoten. Die große Ribosomenuntereinheit steht oben, die kleine Untereinheit, durch die die mRNA läuft, unten. In der A-Stelle hybridisiert jeweils die ankommende, mit einer Aminosäure (AS) beladene tRNA mit ihrem Anticodon an das entsprechende Triplett der mRNA. Dann kommt es zum Transfer des Peptidrestes, der auf der tRNA in der P-Stelle sitzt, auf die Aminosäure in der A-Stelle (Peptidyltransferase). Jetzt rückt das Ribosom drei Nucleotide weiter auf der mRNA und entlässt die freie tRNA aus der E-Stelle (Exit-Stelle); die tRNA mit dem verlängerten Peptidylrest rückt in die P-Stelle. Diese Schritte wiederholen sich, bis ein Stoppcodon erreicht wird. Die Knüpfung der Peptidbindung (roter Pfeil) wird durch die rRNA katalysiert.

rDNA-Kassetten. ITS: internal transcribed spacer; ETS: external transcribed spacer; IGS: intergenic spacer. Die Wiederholungseinheit umfasst ETS, 18S rDNA, ITS-1, 5,8S rDNA, ITS-2, 28S rDNA und IGS. Gene der 5S rRNA liegen außerhalb der RNA-Kassetten auf anderen Chromosomenabschnitten
rDNA-Kassetten. ITS: internal transcribed spacer; ETS: external transcribed spacer; IGS: intergenic spacer. Die Wiederholungseinheit umfasst ETS, 18S rDNA, ITS-1, 5,8S rDNA, ITS-2, 28S rDNA und IGS. Gene der 5S rRNA liegen außerhalb der RNA-Kassetten auf anderen Chromosomenabschnitten
Weil rRNAs und zugehörige Gene auch als Markergene für die molekulare Evolutionsforschung wichtig sind, wurden in ◘ Abb. 3.10 die Bausteine von prokaryotischen und eukaryotischen Ribosomen zusammengestellt. Da sich Mitochondrien und Chloroplasten aus Bakterien ableiten (s. EXKURS 3.1 Abschn. 3.2.7), finden wir in mtDNA und cpDNA erwartungsgemäß rRNAs, deren Aufbau und Sequenz denen der Bakterien weitgehend entsprechen (◘ Abb. 3.11, ◘ Abb. 3.17, ◘ Abb. 3.18) (man beachte, dass in Mitochondrien eine 12S rRNA anstelle der 23S rRNA der Prokaryoten vorkommt).

Vergleich des Aufbaus pro- und eukaryotischer Ribosomen, die Molekülmasse ist in Dalton angegeben. S bedeutet Svedberg-Sedimentationseinheit, über die man die Größe der Ribosomenuntereinheiten und die rRNA charakterisieren kann
Vergleich des Aufbaus pro- und eukaryotischer Ribosomen, die Molekülmasse ist in Dalton angegeben. S bedeutet Svedberg-Sedimentationseinheit, über die man die Größe der Ribosomenuntereinheiten und die rRNA charakterisieren kann

a–c. Struktur der 16S rRNA am Beispiel von a E. coli, b Saccharomyces cerevisiae und c Säugermitochondrien (Rind)
a–c. Struktur der 16S rRNA am Beispiel von a E. coli, b Saccharomyces cerevisiae und c Säugermitochondrien (Rind)
Insbesondere 16/18S rRNAs und 23/28S rRNAs weisen komplexe Raumstrukturen auf, die über große Bereiche der Organismen konserviert wurden (◘ Abb. 3.11). Obwohl RNAs als Einzelstränge vorliegen, bilden sie im wässrigen Milieu an vielen Stellen komplementäre Doppelstränge, sogenannte Stammstrukturen aus. Die Nucleotidsequenz dieser Bereiche der rRNAs wurde in der Evolution meist sehr stark konserviert. Anders sieht es bei den nicht basengepaarten Schleifen (loops) aus, in denen die Nucleotide zudem noch nachträglich durch Anhängen von weiteren chemischen Gruppen modifiziert werden. Dieses Phänomen beobachtet man insbesondere bei tRNAs, bei denen mehr als 50 modifizierte Nucleotide entdeckt wurden. Substituierte Basen sind Thiouracil, 5-Methylcytosin, Dihydrouracil, 2-Thiothymin, 2-Thiocytosin, N4-Acetylcytosin, 1-Methylhypoxanthin, 1-Methylguanin oder N6-Methyladenin. Insbesondere über diese ungepaarten Nucleotide können RNAs mit anderen Molekülen (meist Proteinen) wechselwirken. In den loops finden wir vergleichsweise viele Basensubstitutionen, Deletionen, Insertionen und Inversionen, die ein Alignment homologer Sequenzen erschweren (s. Abschn. 4.1.2). Unter Alignment versteht man die Anordnung von zwei oder mehreren Sequenzen zueinander, so dass identische Basenpositionen übereinander zu stehen kommen. Da die Nucleotidsequenzen der rDNA-Gene in der Evolution sehr stark konserviert wurden, sind sie für die molekulare Evolutionsforschung von großem Interesse, da man mit ihrer Hilfe Stammbäume über alle Organismengruppen hinweg erstellen kann. Der Tree of Life (Baum des Lebens) und die davon abgeleitete Einteilung der Organismen beruht u. a. auf der Analyse von konservierten rDNA-Genen (s. Abschn. 4.2).
3.2.7 Kern-, Mitochondrien- und Chloroplastengenom
Die Gene eines Organismus (in ihrer Gesamtheit auch als Genotyp zusammengefasst) sind die funktionellen Einheiten der Vererbung und enthalten die Bauanweisungen für RNAs, Struktur- und Membranproteine, Transkriptionsfaktoren sowie Enzyme (◘ Abb. 3.6), die für die Differenzierung und zum Aufbau der Zellen, Gewebe und des Gesamtorganismus und damit zur Ausbildung des Phänotyps notwendig sind. Ebenso steuern die Gene direkt oder indirekt alle zentralen Lebensvorgänge, sowohl Stoffwechsel und Organfunktion, Bewegung, Reizaufnahme und Erregungsleitung als auch Zellteilung, Fortpflanzung und diverse Verhaltensweisen. Da in einer Zelle oder einem Organismus nicht alle Gene gleichzeitig zur Expression kommen, bezeichnet man unter Phänotyp die jeweils in Erscheinung tretende Ausprägung der Gene. Am Phänotyp setzt die natürliche Selektion an, so dass die Variabilität des Phänotyps eine große Bedeutung hat.
Bei Prokaryoten liegt die DNA als ringförmiges Chromosom in einem Kernäquivalent vor, während sie in eukaryotischen Chromosomen, von denen der Mensch z. B. 46 und die Taufliege Drosophila 8 hat, linear aufgebaut ist (◘ Tab. 3.10). Eukaryotische Chromosomen sind im Zellkern vom Rest der Zelle abgegrenzt lokalisiert.
3.2.7.1 Genomgröße
Die Gesamtheit der DNA einer Zelle, eines Zellkerns oder eines Organells (Mitochondrien, Chloroplasten) wird als Genom bezeichnet. Betrachtet man das menschliche Kerngenom, so erkennt man schnell, welche gigantische Informationsmenge hier vorhanden ist. Würde man die DNA einer einzelnen Zelle des Menschen als Faden aufspannen, so wäre er 2 m lang. Bei etwa 1013 Zellen in unserem Körper beträgt die Gesamtlänge der DNA aller Zellen 2 ×1010 km. Man könnte damit einen DNA-Faden spannen, der mehrfach von der Erde bis zur Sonne und zurück reicht.
Von den 3 Mrd. Basen, die z. B. im haploiden Chromosomensatz des Menschen vorhanden sind, codieren jedoch nur ca. 1–3 % direkt für Peptide und Proteine (◘ Abb. 3.28). Die restliche DNA besteht aus RNA-Genen und nicht-codierenden Bereichen, die oft an Regulationsvorgängen beteiligt sind, keine Funktion besitzen oder deren Funktion man noch nicht kennt (s. Abschn. 3.4.3).
In den letzten Jahren hat sich die Genomik (genomics) als neues Teilgebiet der Genetik mit Riesenschritten etabliert, mit dem Ziel, komplette Genome molekular und funktionell zu charakterisieren. Im Rahmen des humanen Genomprojektes (HUGO; human genome organization) wurde die Nucleotidsequenz eines haploiden Chromosomensatzes des Menschen im Jahre 2001 fast komplett ermittelt. Bedingt durch die Entwicklung neuer leistungsfähiger DNA-Sequenziergeräte (insbesondere Next Generation Sequencer; s. Kap. 4) wächst die Zahl komplett sequenzierter Genome kontinuierlich. Über 2000 weitere Genome (davon 124 Archaea, 1845 Bacteria, 152 Eukaryota) sind bereits vollständig sequenziert (Stand Feb. 2012) (◘ Tab. 3.4). Diverse Großprojekte wurden initiiert, welche unser Wissen in den nächsten Jahren signifikant vergrößern werden; u. a. das Genome 10 K Project (G10 K), das 1000 Plant & Animal Reference Genomes Project (1000P&A), die 5000 Insect (i5 K) and other Arthropod Genome Initiative oder das Ten Thousand Microbial Genomes Project (10 K M). Über den Vergleich mit Nucleotidsequenzen, die aus umfangreichen organ- und gewebespezifischen cDNA- und EST-Banken (expressed sequence tags) gewonnen wurden, oder durch Konstruktion von Knock-out- oder Antisense-Mutanten (in denen einzelne Gene gezielt abgeschaltet wurden) sowie durch RNAi-Experimente wird im nächsten Schritt versucht, genomischen Sequenzen funktionelle Einheiten oder Gene zuzuordnen (Teilgebiet der Funktionellen Genomik). Die Funktionelle Genomik wird letztlich eine genaue Antwort auf die Frage liefern, welche Bereiche des Genoms eine Funktion haben (heute schätzt man die zum Überleben notwendige Information auf 85–90 % bei Bakterien und auf weniger als 10 % der Gesamt-DNA bei Vertebraten) und welche Teile lediglich als funktionsloses evolutionäres Erbe anzusehen sind (s. Abschn. 3.4). Da wir erst am Anfang dieser Forschung stehen, wird man sicher bald für viele der aus heutiger Sicht funktionslosen DNA-Abschnitte eine Funktion erkennen können.
Die Größe der Genome einiger Organismengruppen ist in ◘ Abb. 3.12 graphisch dargestellt. Betrachtet man die minimale Genomgröße in den Organismenreichen (d. h. nur die linke Seite der Balken in ◘ Abb. 3.12), so beobachtet man eine Zunahme, die im Wesentlichen parallel zur Organisationshöhe verläuft. Einfach aufgebaute Bakterien und Pilze haben kleinere Genome als komplex aufgebaute multizelluläre Organismen. Die maximale Genomgröße hat bei den Eukaryoten jedoch nur eine geringe Beziehung zur Entwicklungshöhe, denn viele Pflanzen und Amphibien haben Genome mit annähernd 1011 Basen, die damit das Genom des Menschen um ein bis zwei Größenordnungen übertreffen. Dieses Phänomen (sogenanntes C-Wert-Paradoxon) deutet schon darauf hin, dass die überdimensionierten Genome DNA-Bereiche aufweisen, die nicht mit dem Phänotyp im Zusammenhang stehen können (s. Abschn. 3.4). Offensichtlich kam es bei diesen Gruppen zu mehrfachen Verdopplungen der Genome (s. Abschn. 3.4.2).

Größe des haploiden Genoms bei einigen großen Organismengruppen. Die X-Achse hat eine logarithmische Skala
Größe des haploiden Genoms bei einigen großen Organismengruppen. Die X-Achse hat eine logarithmische Skala
3.2.7.2 Mitochondrien und Chloroplasten enthalten DNA
Eukaryotenzellen enthalten Erbinformation in den Chromosomen (Kerngenom) sowie in den Mitochondrien und Chloroplasten (s. EXKURS 3.1).
In den Mitochondrien ist die DNA ringförmig aufgebaut (◘ Abb. 3.17), wie bei den α-Proteobakterien, aus denen die Mitochondrien ursprünglich durch Endosymbiose entstanden sind (s. EXKURS 3.1). In Pflanzenzellen finden wir außerdem extranukleäre ringförmige DNA in den Chloroplasten (◘ Abb. 3.18), die sich ursprünglich aus Cyanobakterien entwickelt haben. Mitochondrien und Chloroplasten werden niemals de novo gebildet, sondern vermehren sich (ähnlich wie Bakterien) durch Teilung. Es liegt also eine klonale Vererbung vor. Bei jeder Zellteilung werden diese Organellen auf die Tochterzellen verteilt. Auch Replikation, Transkription und Proteinbiosynthese laufen heute noch in den Mitochondrien und Chloroplasten ab, jedoch sind diese Organellen nicht länger autonom. Sie importieren die meisten ihrer Proteine aus dem Cytoplasma. Die zugehörigen Gene waren ursprünglich einmal Bestandteil der Endosymbionten, wurden dann aber zunehmend in den Kern ausgelagert, so dass heute nur ein vergleichsweise kleiner Bausatz an Genen in Mitochondrien und Chloroplasten übrig geblieben ist (◘ Abb. 3.17 , ◘ Abb. 3.18). Im Gegensatz zu vielen Protein-codierenden Genen verblieben die tRNA- und rRNA-Gene in diesen Organellen. Mitochondrien sind strukturell sehr dynamische Organellen, die regelmäßig Fusionsprozesse mit anderen Mitochondrien einer Zelle durchlaufen und auf diese Weise ihre DNA offenbar durchmischen.

Doppel-, Drei- und Vierfachmembranen von Plastiden bei primärer bzw. sekundärer Endosymbiose. Oben: Cyanobakterien werden durch Phagocytose in eukaryotische Wirtszellen aufgenommen (1), wenn dabei die sogenannte Outer membrane der Cyanobakterien erhalten bleibt, können Plastiden mit drei Hüllmembranen entstehen (2). Unten: Entstehung komplexer Plastiden durch Aufnahme plastidenhaltiger Einzeller in phagotrophe Wirtszellen und nachfolgende Reduktion der Endocytobionten auf Plastide und Zellmembran (3). Geht eine der vier Hüllmembranen verloren (5), entstehen auch hier Plastiden mit drei Hüllmembranen. Durch unvollständige Reduktion des Endocytobionten (4) entsteht eine Situation, wie sie bei Cryptomonaden und Chlorarachnion beobachtet wird (vgl. ◘ Abb. 3.15)
Doppel-, Drei- und Vierfachmembranen von Plastiden bei primärer bzw. sekundärer Endosymbiose. Oben: Cyanobakterien werden durch Phagocytose in eukaryotische Wirtszellen aufgenommen (1), wenn dabei die sogenannte Outer membrane der Cyanobakterien erhalten bleibt, können Plastiden mit drei Hüllmembranen entstehen (2). Unten: Entstehung komplexer Plastiden durch Aufnahme plastidenhaltiger Einzeller in phagotrophe Wirtszellen und nachfolgende Reduktion der Endocytobionten auf Plastide und Zellmembran (3). Geht eine der vier Hüllmembranen verloren (5), entstehen auch hier Plastiden mit drei Hüllmembranen. Durch unvollständige Reduktion des Endocytobionten (4) entsteht eine Situation, wie sie bei Cryptomonaden und Chlorarachnion beobachtet wird (vgl. ◘ Abb. 3.15)

a, b. Beispiele für Endocytobiosen rezenter Einzeller. a das Grüne Paramecium, P. bursaria, ist durch intrazelluläre, einzellige Grünalgen (Chlorella lobophora) phototroph. b Glaucosphaera vacuolata, ein einzelliger Glaucocystophyt, mit Cyanellen, endocytischen Abkömmlingen von Cyanobakterien
a, b. Beispiele für Endocytobiosen rezenter Einzeller. a das Grüne Paramecium, P. bursaria, ist durch intrazelluläre, einzellige Grünalgen (Chlorella lobophora) phototroph. b Glaucosphaera vacuolata, ein einzelliger Glaucocystophyt, mit Cyanellen, endocytischen Abkömmlingen von Cyanobakterien

a, b. Nucleomorphen in den Zellen von a Pyrenomonas salina und b Chlorarachnion reptans. P: Plastiden mit Pyrenoiden, Py; S: Stärke oder entsprechende Speicherstoffe; N: Kern der Wirtszelle (in b außerhalb der Schnittebene); M: Mitochondrien. Pfeile: Nucleomorphen, in beiden Fällen in die Pyrenoide eingesenkt. Maßstab 1 µm. Elektronenmikroskop. Aufnahmen von H. Falk (a) und V. Speth (b)
a, b. Nucleomorphen in den Zellen von a Pyrenomonas salina und b Chlorarachnion reptans. P: Plastiden mit Pyrenoiden, Py; S: Stärke oder entsprechende Speicherstoffe; N: Kern der Wirtszelle (in b außerhalb der Schnittebene); M: Mitochondrien. Pfeile: Nucleomorphen, in beiden Fällen in die Pyrenoide eingesenkt. Maßstab 1 µm. Elektronenmikroskop. Aufnahmen von H. Falk (a) und V. Speth (b)

a, b. a Allgemeiner Stammbaum des Organismenreiches nach Sequenzvergleichen ribosomaler RNAs, Drei-Domänen-Konzept (verändert nach C. R. Woese). b Basis des allgemeinen Stammbaumes nach neueren Vorstellungen: Die Domäne der Eucarya ist sekundär durch eine Symbiogenese von Vertretern der beiden prokaryotischen Domänen entstanden (in Anlehnung an Martin u. Müller 1998, Rivera u. Lake 2004)
a, b. a Allgemeiner Stammbaum des Organismenreiches nach Sequenzvergleichen ribosomaler RNAs, Drei-Domänen-Konzept (verändert nach C. R. Woese). b Basis des allgemeinen Stammbaumes nach neueren Vorstellungen: Die Domäne der Eucarya ist sekundär durch eine Symbiogenese von Vertretern der beiden prokaryotischen Domänen entstanden (in Anlehnung an Martin u. Müller 1998, Rivera u. Lake 2004)

Schematische Übersicht über die Anordnung der Gene in Mitochondrien von Vögeln, Mensch und anderen Säugetieren (ohne Beuteltiere) sowie Amphibien (nach Mindell 1997). Im Vergleich zu anderen Vertebraten ist bei Vögeln ein Teil der mtDNA neu arrangiert, indem das ND6-Gen (links oben) zwischen Cytochrom b und der Kontrollregion inseriert wurde. Die Kontrollregion wird auch als D-Loop bezeichnet und enthält den Replikationsursprung (origin of replication). Der äußere DNA-Strang wird als H-Strang, der innere als L-Strang bezeichnet (H von heavy; L von light). ND: NADH-Dehydrogenase mit den Untereinheiten ND1 bis ND6; CO: Cytochrom-Oxidase mit den Untereinheiten COI bis COIII; ATP: ATPase mit den Untereinheiten ATPase 6 und 8; 12S: 12S rRNA-Gen; 23S: 23S rRNA-Gen. Die gelben Querstriche stellen tRNA-Gene dar; in vielen Fällen stehen sie zwischen Protein-codierenden Genen. Weiße Querstriche deuten die Grenzen von Genen an
Schematische Übersicht über die Anordnung der Gene in Mitochondrien von Vögeln, Mensch und anderen Säugetieren (ohne Beuteltiere) sowie Amphibien (nach Mindell 1997). Im Vergleich zu anderen Vertebraten ist bei Vögeln ein Teil der mtDNA neu arrangiert, indem das ND6-Gen (links oben) zwischen Cytochrom b und der Kontrollregion inseriert wurde. Die Kontrollregion wird auch als D-Loop bezeichnet und enthält den Replikationsursprung (origin of replication). Der äußere DNA-Strang wird als H-Strang, der innere als L-Strang bezeichnet (H von heavy; L von light). ND: NADH-Dehydrogenase mit den Untereinheiten ND1 bis ND6; CO: Cytochrom-Oxidase mit den Untereinheiten COI bis COIII; ATP: ATPase mit den Untereinheiten ATPase 6 und 8; 12S: 12S rRNA-Gen; 23S: 23S rRNA-Gen. Die gelben Querstriche stellen tRNA-Gene dar; in vielen Fällen stehen sie zwischen Protein-codierenden Genen. Weiße Querstriche deuten die Grenzen von Genen an

Übersicht über die Gene im Chloroplastengenom einer Alge (Nephroselmis olivacea) mit 200.799 Basenpaaren (nach Turmel et al. 1989). Das Genom enthält 200 Gene, davon 155 Protein-codierend und 45 RNA-Gene. Im Chloroplastengenom wurde ein DNA-Abschnitt verdoppelt und invers orientiert eingebaut; sogenannte inverted repeat, IRA und IRB. Einige Gene werden vom äußeren H-Strang, andere vom inneren L-Strang codiert. In vielen phylogenetischen Arbeiten wird das rbcL-Gen, das für die große Untereinheit der Rubisco codiert, als Markergen eingesetzt. Jedes Kästchen entlang der ringförmigen DNA entspricht einem Gen, das durch eine Abkürzung eindeutig gekennzeichnet wird, so dass man Chloroplastengenome untereinander vergleichen kann
Übersicht über die Gene im Chloroplastengenom einer Alge (Nephroselmis olivacea) mit 200.799 Basenpaaren (nach Turmel et al. 1989). Das Genom enthält 200 Gene, davon 155 Protein-codierend und 45 RNA-Gene. Im Chloroplastengenom wurde ein DNA-Abschnitt verdoppelt und invers orientiert eingebaut; sogenannte inverted repeat, IRA und IRB. Einige Gene werden vom äußeren H-Strang, andere vom inneren L-Strang codiert. In vielen phylogenetischen Arbeiten wird das rbcL-Gen, das für die große Untereinheit der Rubisco codiert, als Markergen eingesetzt. Jedes Kästchen entlang der ringförmigen DNA entspricht einem Gen, das durch eine Abkürzung eindeutig gekennzeichnet wird, so dass man Chloroplastengenome untereinander vergleichen kann
Das Chloroplastengenom (cpDNA) ist 120–200 kB groß und kommt 20- bis 40-mal in einem einzelnen Chloroplasten vor. Da eine Pflanzenzelle bis zu 40 Chloroplasten enthält, liegt die Gesamtzahl der cpDNA-Kopien zwischen 800 und 1600 pro Zelle. Auch in den Chloroplastengenomen ist die lineare Anordnung der Gene innerhalb der verschiedenen photosynthetisch aktiven Eukaryoten sehr ähnlich. Dies deutet auf einen gemeinsamen Ursprung hin. Auffällig ist eine inverse Verdopplung eines größeren Sequenzabschnitts (◘ Abb. 3.18).
Das Mitochondriengenom (mtDNA) ist bei Tieren mit ca. 14–19 kB deutlich kleiner als bei Pflanzen. Es enthält bei den meisten Tieren 13 Gene, die für Enzyme oder andere am Elektronentransport beteiligte Proteine codieren, und Gene für tRNAs sowie zwei für rRNAs. Die lineare Anordnung der mitochondrialen Gene ist bei den Eukaryoten weitgehend konstant, was auf einen gemeinsamen Ursprung der Mitochondrien hindeutet. Kleine Unterschiede, die auf einer Inversion von Genen beruhen, ergeben sich jedoch z. B. zwischen Säugetieren und Vögeln (◘ Abb. 3.17). Da jede tierische Zelle mehrere Hundert bis über 1000 Mitochondrien und jedes davon 5–10 mtDNA-Kopien enthält, liegt die Gesamtzahl der mtDNA-Kopien bei mehreren Tausend pro Zelle. Die mtDNA macht etwa 1 % der Gesamt-DNA-Menge einer Zelle aus.
Bislang ungeklärt ist, warum Pilze und vor allem Pflanzen Mitochondrien besitzen, deren DNA um ein Vielfaches größer ist als die der Tiere. Mitochondriengenome von Pilzen weisen große Längenunterschiede auf und enthalten zwischen 20 und 100 kB. Pflanzliche Mitochondrien haben dagegen sehr große Genome (über 140–2500 kB) und weisen z. T. Gene mit Intron-/Exonstruktur auf.
Bedingt durch die hohe Kopienzahl der mtDNA und cpDNA eignen sich diese Genome besonders gut für die molekulare Evolutionsforschung, da sie leichter zugänglich sind als single-copy-Gene der Kern-DNA, die nur in wenigen Kopien pro Zelle vorkommen ( Kap. 4). Für die Betrachtung der molekularen Systematik ist außerdem die Tatsache wichtig, dass Mitochondrien fast immer und Chloroplasten bei höheren Pflanzen bei ca. 70 % der Arten maternal (also nicht nach den Mendelschen Regeln; s. Abschn. 3.5.2) vererbt werden. Über die Analyse von mtDNA und cpDNA kann man streng genommen nur maternale Linien zurückverfolgen.
Experimentelle Befunde deuten jedoch daraufhin, dass bei der Befruchtung doch einige Mitochondrien aus den Spermien in die Eizelle (die sehr viele Mitochondrien aufweist) gelangen, so dass die Hypothese der rein maternalen Vererbung vermutlich nur begrenzt stimmt und im Einzelfall zu prüfen ist. Nach allgemeiner Ansicht unterliegen mtDNA und cpDNA außerdem keiner Rekombination (s. Abschn. 3.3.3). Überraschenderweise sind die DNA-Sequenzen der mtDNA in einem Individuum weitgehend identisch, was eigentlich auf Rekombinationsvorgänge oder Genkonversion hinweist. Vermutlich finden Rekombinations- und Genkonversionsvorgänge (s. Abschn. 3.3.3 u. 3.4.2) auch in der mtDNA bei Tier und Mensch statt.
Da sich die DNA-Polymerasen und Reparaturenzyme der Mitochondrien und Chloroplasten untereinander und von denen des Zellkerns unterscheiden, beobachtet man unabhängige und unterschiedlich schnelle Evolutionsraten der mtDNA und cpDNA. Bei Tieren ist die Rate der Nucleotidsubstitutionen in den Mitochondrien etwa zehnmal höher als die der DNA im Zellkern (ncDNA), da die mitochondriale Replikation eine höhere Fehlerrate aufweist. In Pflanzen dagegen ist die mtDNA am stärksten konserviert und die ncDNA am variabelsten; die cpDNA liegt in der Evolutionsgeschwindigkeit zwischen beiden Extremen.
Bakterien weisen zusätzlich zu einem ringförmigen Chromosom mehrere kleine ringförmige DNA-Moleküle auf, die Plasmide, welche häufig Gene enthalten, die für Antibiotikaresistenz codieren. Plasmidähnliche DNA-Elemente wurden auch in Mitochondrien und Chloroplasten nachgewiesen, in denen sie aber angeblich keine Funktion haben. Vielleicht helfen sie, über Rekombination die DNA-Sequenzen der Organell-DNA einheitlich zu halten; denn extrachromosomale Elemente der Bakterien ermöglichen auch bei den haploiden Prokaryoten Rekombinationsvorgänge.
3.2.7.2.1 EXKURS 3.1
3.2.7.2.1.1 Symbiogenese in der Zell- und Lebensevolution
Peter Sitte (Freiburg)
Das Periodensystem der Biologie: Zwei Zelltypen, drei Domänen
Zellen sind die kleinsten lebensfähigen Systeme. Sie sind enorm vielgestaltig – man denke nur an Bakterien oder die meist größeren, sehr verschiedenen Einzellerformen, schließlich an die ganz unterschiedlichen Zellen unseres Körpers. Dennoch lassen sie sich zwei Grundtypen zuordnen: Protocyten und Eucyten. Die meist sehr kleinen Protocyten der Bakterien und Archaeen (Archaebakterien) enthalten keinen von Membranen umhüllten Zellkern, ihre DNA-haltigen Nucleoide liegen ohne Membranumgrenzung in der Zelle. Dagegen verfügen die in der Regel viel größeren Eucyten aller übrigen Organismen über einen Zellkern, der von einer doppelten, von Porenkomplexen durchbrochenen Membranhülle umschlossen ist. Auch sonst gibt es eine Reihe fundamentaler Unterschiede (◘ Tab. 3.5). Nach ihrem Zellbau werden dementsprechend die beiden Großreiche der Prokaryota (der Monera im Sinne Ernst Haeckels; ihre Zellen entsprechen Protocyten) und der Eukaryota (mit Eucyten) unterschieden.
Inzwischen haben neben anderen Merkmalen vor allem Sequenzvergleiche ribosomaler RNAs bei den Prokaryoten eine tiefe Kluft zwischen Bakterien und Archaeen deutlich werden lassen (◘ Tab. 3.6). Daher wird heute das Gesamtreich aller zellulär gebauten Organismen in drei Domänen gegliedert: Bacteria, Archaea und Eukarya (◘ Abb. 3.16).
3.2.7.2.1.1.1 Frühe Lebensevolution als Zellevolution
Spuren von Lebewesen lassen sich bis in älteste Sedimente zurückverfolgen. Es hat demnach schon vor mehr als 3,5 (vermutlich sogar 4) Mrd. Jahren Leben auf unserem Planeten gegeben. Darüber, wie erste Zellen auf der unwirtlichen Ur-Erde in nur einer halben bis einer Jahrmilliarde entstehen konnten (die Erde ist 4,6 Mrd. Jahre alt), gibt es mangels konkreter Daten nur Hypothesen. Wahrscheinlich konnten sich unter einer weitgehend sauerstofffreien Atmosphäre und an unterseeischen heißen Quellen abiotisch verschiedene organische Moleküle bilden, darunter Aminosäuren, Zucker und organische Basen, schließlich auch Oligonucleotide und Peptide. In dieser präbiotischen, chemischen Evolution können sich letztlich auch selbstreplizierende Ribonucleinsäuren („RNA-Welt“) gebildet haben, eine Vorstellung, die durch den Nachweis enzymatisch aktiver RNAs (Ribozyme) gestützt wird. Somit war zwar noch nicht die Organisationsstufe von Zellen erreicht, doch gab es jetzt schon Vererbung. Damit waren alle wesentlichen Voraussetzungen für eine Evolution im Sinne Darwins erfüllt: Vermehrung, erbliche Variation durch Mutationen, Konkurrenz und Selektion. Dabei stand das Mutationsgeschehen wegen der zunächst noch geringen Präzision der Nucleinsäure-Vervielfältigung im Vordergrund. Die Evolution konnte vergleichsweise rasch zur Verbesserung der Replikation mit Hilfe von Proteinen führen, damit zu Hyperzyklen (zyklische Reaktionsfolgen zwischen RNAs und Proteinen) (Eigen u. Winkler 1975) und Translation, zu definierten Genen und über RNA-Genome schließlich zu DNA-Genomen, Transkription und Genregulation. Nach Ausbildung einer umgebenden Membran, über deren Entstehung unterschiedliche Hypothesen existieren, sollte es Zellen gegeben haben, die alle essenziellen biochemischen Komponenten und Vorgänge innerhalb einer Membran – der Zellmembran – vereinigten und in hohen Konzentrationen halten konnten. Mit dem Auftreten zellulärer Organismen war jene Evolutionsphase erreicht, die bis heute andauert. Dabei sind nach den freilich sehr lückenhaften Mikrofossilfunden Eucyten möglicherweise vor ca. 2 Mrd. Jahren aufgetreten. Reste von möglichen Vielzellern finden sich in Ablagerungen, die ca. eine Jahrmilliarde alt sind. Der dominante Teil der Lebensentwicklung war also Zellevolution.
Astronomen nehmen an, dass im Universum Abertausende erdähnliche Systeme existieren. Daher ist die Möglichkeit nicht auszuschließen, dass das Leben nicht auf unserer Erde entstand, sondern durch Meteoriteneinschlag aus anderen Welten importiert wurde.
3.2.7.2.1.1.2 Makroevolution und Großübergänge
In der Evolution der Organismen wurden und werden evolutive Fortschritte vorwiegend durch Rekombination im Gen-Pool der einzelnen Arten im Sinne immer besserer Anpassung an die jeweils gegebenen, veränderlichen Umwelten erzielt. Eine nicht unerhebliche Rolle spielen weiterhin Änderungen der Expressionsrate von Genprodukten, sowie die Integration und Expression von Genen, die von nicht verwandten Arten abstammen und via horizontalem Gentransfer in einen Organismus gelangen können. Nun muss es allerdings in der Phylogenese neben den zahlreichen kleineren Veränderungsschritten gelegentlich auch größere Sprünge gegeben haben. Neben graduellen Veränderungen, wie sie die Bildung von Unterarten und Arten beherrschen (Mikroevolution), wird für die Makroevolution also auch die mehr oder weniger unvermittelte Entstehung grundsätzlich neuartiger Organismen postuliert. Solche dramatischen Veränderungen waren zwar sicher viel seltener als die kleinen Mutations- und Rekombinationsereignisse, aber folgenreicher. Sie entsprechen den zukunftsträchtigen Bifurkationen im deterministischen Chaos der Evolution und markieren die großen Verzweigungen der Stammbäume. Allerdings bleiben solche Vorgänge wegen ihrer Seltenheit der direkten Beobachtung entzogen, die zugrunde liegenden Mechanismen sind einer experimentellen Erforschung nur schwer zugänglich. Eine mögliche Erklärung liefert das Konzept der phylogenetischen Großübergänge ( major evolutionary transitions ). Grundaussage ist, dass sich in der biologischen Evolution immer wieder Fortpflanzungseinheiten, die sich zunächst selbstständig entwickelt hatten, zu komplexeren Einheiten zusammengeschlossen haben. Die so entstandenen Systeme konnten dann zu Ausgangspunkten für völlig neue Entwicklungslinien werden.
Ein Beispiel für einen solchen Großübergang ist die evolutive Entstehung von Vielzellern aus Einzellern. Bei Einzellern repräsentiert die einzelne Zelle einen ganzen Organismus, im Vielzeller ist sie nur mehr eines von vielen Elementen eines einzigen Lebewesens. Die einzelnen Zellen büßen mit dem Einbau in das größere System, das ihnen stabile Lebensbedingungen gewährt, viel von ihrer Selbstständigkeit ein. Steuernde Einflüsse des Gesamtsystems diktieren z. B. die Teilungstätigkeit und entscheiden über Leistungen und Lebensdauer der einzelnen Zellen im übergeordneten Funktionsgefüge. Damit ist ein wichtiges Charakteristikum solcher Systeme angesprochen: sie sind arbeitsteilig. Im vielzelligen Organismus können sich die einzelnen Zellen auf bestimmte Teilaufgaben spezialisieren, andere sind ihnen dafür abgenommen. Dadurch können nicht nur Teilprozesse wichtiger Stoffwechselvorgänge bzw. bestimmte Funktionen mit höherer Effizienz ausgeführt werden, sondern auch Synergiepotenziale voll ausgenützt werden. Zusätzlich können die so differenzierten Zellen bzw. Gewebe während der weiteren Phylogenese ohne große genetische Veränderungen vermehrt oder vermindert, verschoben und wie Module im Gesamtsystem neu kombiniert werden. Dank dieser Kombinatorik können mit relativ wenigen unterschiedlichen Elementen fast beliebig viele verschiedene Systeme aufgebaut werden. Dem ist die enorme Arten- und Formenfülle an Makroorganismen in der Biosphäre mit zu verdanken.
Komplexe Vielzeller haben sich nur bei den Eukaryoten entwickelt. Mit steigender Komplexität der Organismen nimmt die für die systemgerechte Steuerung der einzelnen Zellen, Gewebe und Organe und ihrer Entwicklung in der Ontogenese erforderliche Informationsmenge zu. Die Information kann jedoch in verschiedenen Ebenen gespeichert und abgerufen werden. In der Tat findet sich eine gewisse Korrelation zwischen Komplexität eines Organismus und der Anzahl der in seinem Genom codierten Gene, die bei Eukaryoten im Gegensatz zu den meisten Bakterien in mehreren Chromosomen lokalisiert sind. Jedoch können homologe Gene in unterschiedlichen Organismen verschieden exprimiert werden, was zur Ausbildung neuer Einheiten führen kann. Neben dem Übergang vom Einzeller- zum Vielzellerstatus gibt es weitere Möglichkeiten für evolutive Großübergänge. Eine besonders bedeutsame ergibt sich aus Symbiosen, dem intimen Zusammenleben artverschiedener Organismen (◘ Abb. 3.13).
3.2.7.2.1.1.3 Endocytobiose
Unter Endocytobiose versteht man den Einbau artfremder Zellen in größeren Wirtszellen. Solche intrazellulären Symbiosen stellen den engsten Symbiosebezug dar, der überhaupt denkbar ist. In der rezenten Organismenwelt finden sich zahlreiche Beispiele für mutuelle Endocytobiosen. Als Endocytobionten treten dabei vielfach Bakterien auf, so die N2-fixierenden Knöllchenbakterien (Rhizobium, Bradyrhizobium) der Leguminosen oder die Photosynthese betreibenden Cyanobakterien im Erdpilz Geosiphon (s. unten). In vielen Fällen finden sich eukaryotische Einzeller in Wirtszellen eingebaut, z. B. einzellige Grünalgen („Zoochlorellen“) in bestimmte Amöben, Paramecien (◘ Abb. 3.14) und Hydren, oder Dinophyceen („Zooxanthellen“) in Foraminiferen und in die Polypenzellen von Riffkorallen. Dabei gibt es neben vorübergehenden, fakultativen Endocytobiosen stabilere Verbindungen, deren Partner in der Natur zwar nur gemeinsam auftreten, nach künstlicher Trennung aber auch einzeln zu überleben vermögen. Dagegen können in obligaten Endocytobiosen die Partner ohne einander nicht mehr überleben.
3.2.7.2.1.1.4 Symbiogenese
Schon 1905 hat der russische Biologe Constantin Mereschkowsky postuliert, dass die Etablierung stabiler, mutualistischer Endocytobiosesysteme neue phyletische Entwicklungen einleiten kann. Sie entspricht dann einem evolutiven Großübergang. Er hat dergleichen als Symbiogenesis bezeichnet und damit nach seiner Überzeugung „eine neue Lehre von der Entstehung der Organismen“ entwickelt. Tatsächlich erzwingt ja der dauerhafte Einbau artfremder Zellen in eine Wirtszelle eine besonders enge Koevolution der Partner. Vor allem müssen das Teilungsverhalten beider Teilzellen aufeinander abgestimmt und der gegenseitige Stoffaustausch optimiert werden. Den Startpunkt einer symbiogenetischen Entwicklung markiert der ursprüngliche Vereinigungsprozess ungleichartiger Zellen, die intertaxonische Kombination (ITC). Dabei ist wesentlich, dass es zwar zu einer stabilen Vereinigung, aber nicht zu einer Fusion der Partner kommt. Echte Zellfusionen, d. h. die Vermischung bisher getrennter Zellplasmen, sind nur zwischen artgleichen Zellen möglich. Beispiele dafür sind die Gametenverschmelzung bei Syngamie oder die Bildung vielkerniger quergestreifter Skelettmuskelfasern aus einkernigen Myoblasten. Von artfremden Zellen können die natürlichen Fusionsbarrieren offenbar nicht durchbrochen werden. Solche Zellen können zwar durch Phagocytose- bzw. Endocytose-ähnliche Mechanismen in Wirtszellen aufgenommen werden, sie bleiben aber in Membran-umschlossenen Strukturen und damit abgegrenzt gegen das Cytoplasma der Wirtszelle. Das entspricht in vielen Fällen der Situation bei rezenten Endocytobiosen, die auch bei fortdauernder Symbiogenese beibehalten werden kann (◘ Abb. 3.13).
Die Kombination vorher selbstständiger Partner bei ITC und nachfolgender Symbiogenese hat nur dann auf dem Prüfstand der Selektion Bestand, wenn sie sich in mindestens einer Hinsicht entscheidend ergänzen können. Denn nur dann weist das neue Übersystem emergente Eigenschaften auf, die den einzelnen Partnern nicht zukamen. Bei erfolgreichen Endocytobiosen ist Arbeitsteiligkeit, wie sie für Großübergänge typisch ist, bereits vorgegeben. Und da beide Partner ihre eigene genetische Information mit einbringen, ist auch der Gehalt an genetischem Material im Gesamtsystem von vornherein entsprechend höher. Dazu zwei konkrete Beispiele:
-
Geosiphon: Dieser unscheinbare Pilz ohne Chloroplasten, der zu den Glomeromycota gezählt wird, bildet unter geeigneten Bedingungen etwa 1 mm große, zartwandige Blasenzellen und nimmt in diese durch Phagocytose Cyanobakterien (Nostoc punctiforme) auf. Die endocytobiotischen Nostoc-Zellen betreiben Photosynthese, sie spielen im Pilz die Rolle von Chloroplasten. Das Endocytobiosesystem ist dadurch phototroph und vermag auch Luftstickstoff zu assimilieren.
-
Paramecium bursaria: Das „Grüne Pantoffeltierchen“ (◘ Abb. 3.14a) enthält mehrere hundert Chlorella-Zellen, die ihren heterotrophen Wirt mit Produkten der Photosynthese – Maltose, Glucose und Sauerstoff – versorgen und von ihm mit anorganischen Ionen und CO2 beliefert werden. Die Verdauung der eukaryotischen Symbionten durch den Wirt wird durch das Lektin Concanavalin A verhindert, ein Mannose und Glucose bindendes Oberflächenprotein der Chlorellen. Chlorellen, die dieses Lektin nicht bilden können, werden im Paramecium wie andere Nahrungspartikel abgebaut. Das Grüne Pantoffeltierchen ist im Gegensatz zu anderen Paramecien phototaktisch, es bringt also seine „Gäste“ in günstige Lichtbedingungen für optimale Photosynthese. Das gesamte Endocytobiosesystem kann bei Licht in rein anorganischen Medien kultiviert werden. Die Paramecium-Chlorella-Symbiose ist nicht obligatorisch, beide Partner sind unter geeigneten Umständen auch zu selbstständigem Leben befähigt. Doch wird dank seinem Selektionsbonus in freier Natur nur das komplette Symbiosesystem gefunden.
3.2.7.2.1.1.5 Die Endosymbiontentheorie
Schon vor über hundert Jahren hatte Andreas Schimper nachgewiesen, dass Plastiden in den Eizellen der Pflanzen nicht neu gebildet, sondern in ununterbrochener Folge über die Eizellen von den Mutterpflanzen auf die Nachkommen übertragen werden. Zeitgleich hatte Friedrich Schmitz für die Plastiden von Algen gezeigt, dass sie nicht aus dem Zellplasma neu gebildet werden können, sondern immer nur durch Teilung aus ihresgleichen hervorgehen. Entsprechendes wurde bald auch für Mitochondrien vermutet.
C. Mereschkowsky hat dann eine bereits von Schimper (und nach ihm noch mehrfach) geäußerte Vermutung zu einer konsequenten Hypothese großer Tragweise ausgebaut. Danach geben Plastiden stammesgeschichtlich auf endosymbiotische Cyanobakterien zurück; die verschiedenen Algen und grünen Pflanzen verdanken ihre Befähigung zur Photosynthese einem Symbiogeneseprozess. Tatsächlich weisen Plastiden viele Typenmerkmale von Protocyten auf (◘ Tab. 3.7) und verfügen über ein eigenes genetisches System. Nach RNA- und DNA-Sequenzvergleichen stammen sie aus dem Bereich der Cyanobakterien, wie schon Mereschkowsky postuliert hatte.
Für die Mitochondrien, die Atmungsorganellen der Eucyten, haben Sequenzvergleiche gezeigt, dass sie dem Bereich der α-Proteobakterien entstammen. Somit gehen auch alle atmenden Eukaryoten (das sind die allermeisten) auf einen phylogenetischen Großübergang zurück, der auf ITC und Symbiogenese beruht. So haben sich also jene beiden Organellen, die bei den meisten Eukaryoten den zellulären bzw. organismischen Energiebedarf befriedigen, aus endocytierten Prokaryoten evoluiert. Es ist ein Verdienst von Lynn Margulis, die Endosymbiontenhypothese in den 70er und 80er Jahren des letzten Jahrhunderts so popularisiert zu haben, dass sie heute zum Lehrbuchwissen zählt.
Bei den heute lebenden Organismen verfügen nun allerdings weder Plastiden noch Mitochondrien über genügend eigene genetische Information, um alle ihre Proteine selbst synthetisieren zu können. Während der Koevolution (Symbiogenese) von Symbiont und Wirt ist ein Großteil des Symbiontengenoms durch intrazellulären Gentransfer in den Kern der eukaryoten Wirtszelle verlagert worden. Dadurch wurde die Steuerung wesentlicher Leistungen der Endocytobionten zentralisiert. Die Symbionten wurden zu Xenosomen reduziert, die außerhalb der Wirtszelle nicht mehr auf Dauer überleben können. Die Vereinigung der taxonomisch so unterschiedlichen Zellen bzw. Genome ist damit unauflöslich. Eucyten sind genetische Chimären, nicht eigentlich Einzelzellen, sondern Mosaikzellen. Dennoch verhalten sie sich wie einheitliche Zellen.
Unter den Algen gibt es die kleine, heterogene Gruppe der Glaucocystophyten, die alle wesentlichen Aussagen der Endosymbiontentheorie gut illustrieren kann. Die Vertreter dieser Gruppe waren durch ihre blaugrün gefärbten Plastiden aufgefallen, die schon im Lichtmikroskop an Cyanobakterien erinnern (◘ Abb. 3.14). Sie werden daher als Cyanellen bezeichnet, die stabilen Endocytobiosesysteme als Endocyanome. Inzwischen haben elektronenmikroskopische, biochemische und molekularbiologische Untersuchungen gezeigt, dass es sich bei den Cyanellen tatsächlich um Abkömmlinge aufgenommener Cyanobakterien handelt, die sich zu Plastiden evolviert haben. So ließen sich zwar an den Cyanellen noch Reste prokaryotischer Zellwände nachweisen, aber ihr Genom ist bereits großenteils in den Kern der Wirtszelle verlagert wie bei Chloroplasten. Dementsprechend ist bei den Glaucocystophyten – im Gegensatz zu Geosiphon – die Endocytobiose obligatorisch, die Partner können nicht getrennt voneinander kultiviert werden.
3.2.7.2.1.1.6 Sekundäre Endocytobiose: Eucyten in Eucyten
Die Plastiden der höheren Pflanzen sowie der Glaucocystophyten, Rot- und Grünalgen sind von zwei Membranen umgeben (◘ Abb. 3.13). Die Plastiden aller übrigen Algen haben allerdings mehr als zwei Hüllmembranen. Solche „komplexe“ Plastiden mit drei Hüllmembranen finden sich bei den Euglenen und den meisten Dinoflagellaten, solche mit vier Membranen sind charakteristisch für alle Heterokonten von den Xantho- und Chrysophyceen bis zu den Kiesel- und Braunalgen, ferner für die Haptophyten, Cryptomonaden und Chlorarachniophyten sowie für einige Alveolaten (s. unten). Die evolutive Entstehung komplexer Plastiden kann durch sekundäre Endocytobiose erklärt werden (◘ Abb. 3.13). In diesem Fall waren nicht nur die Wirtszellen, sondern auch die Endosymbionten eukaryote Zellen (und nicht Prokaryoten, wie bei der primären Endocytobiose). Bei den Algen, die Plastiden mit vier Hüllmembranen haben, war der sekundäre Endosymbiont ursprünglich offenbar ein phototropher Protist, dessen Plastoplasma vom Cytoplasma der Wirtszelle nun durch vier Membranen getrennt ist. In einem solchen System aus zwei artverschiedenen Mosaikzellen finden sich viele einander entsprechende Zellstrukturen und Gene sowohl im Wirt wie auch im Symbionten. Man kann nun annehmen, dass der Symbiont während der Symbiogenese durch Eliminierung überflüssiger Zellorganellen immer weiter reduziert wurde, bis im Extremfall schließlich nur seine Plastiden übrig blieben als die einzigen Organellen, die der Wirtszelle gefehlt hatten. Dies scheint allerdings nur die Spitze des Eisbergs zu sein, die ansatzweise auch morphologisch nachweisbar ist. Denn neue genomische und bioinformatische Daten lassen den Schluss zu, dass die Wirtszelle einiger sekundär evolvierter Organismen bereits phototroph war, d. h. bereits eine Plastide besaß. Somit wäre eine bereits vorhandene Plastide mittels Endosymbiose durch eine komplexe ausgetauscht, ein Szenario, das den Mosaikcharakter dieser Zellen entscheidend vergrößert.
Komplexe Plastiden geben ihre Abstammung aus einem phototrophen, endosymbiontischen Eucyten in den meisten Fällen nur noch durch die vier oder drei umhüllenden Membranen morphologisch zu erkennen. Nun konnte allerdings bei zwei Algengruppen überzeugend gezeigt werden, dass sie ihre Plastiden tatsächlich durch sekundäre Endocytobiose erworben haben. Sowohl bei den einzelligen, biflagellaten Cryptomonaden als auch bei den Chlorarachniophyten liegen die beiden Membranpaare ihrer komplexen Plastiden nicht wie sonst unmittelbar aneinander, sondern sind durch schmale Cytoplasmasäume voneinander getrennt. Diese sind sicher eukaryotischen Ursprungs, weil sie 80S Ribosomen enthalten und einen kleinen Zellkern, das Nucleomorph (◘ Abb. 3.13, 3.14). Das in den Nucleomorphen enthaltene Genom besteht in beiden Fällen aus drei sehr kleinen, linearen Chromosomen, deren rRNA-Gensequenzen sich eindeutig von denen der Wirtszellkerne unterscheiden. Die Pigmente der Plastiden und Sequenzvergleiche haben gezeigt, dass die sekundären Endosymbionten der Cryptomonaden von Rotalgen abstammen, jene der Chlorarachniophyten von Grünalgen.
Die Erforschung phyletisch sekundärer Endocytobiosen kann übrigens auch für die Medizin wichtig sein. Sowohl Toxoplasma gondii, der Erreger der Toxoplasmose, wie auch der Malaria-Parasit Plasmodium falciparum (beide zum Stamm der Apicomplexa gehörend) enthalten komplexe Plastiden („Apicoplasten“), die von vier Membranen umgeben sind. Die Apicoplasten sind photosynthetisch inaktiv, dennoch zeigen Genvergleiche, dass sie auf internalisierte Rotalgen zurückgehen. Somit zeigt dies den Übergang von einer photosynthetisch aktiven Zelle hin zu einem intrazellulären Parasiten. Neben diesen wichtigen evolutionären Erkenntnissen eröffnet dieses Wissen die Möglichkeit, diese gefährlichen Parasiten durch Pharmaka auszuschalten, die spezifisch auf den Stoffwechsel von Plastiden einwirken, was die Gefahr ungünstiger Nebenwirkungen mindert.
Für sekundäre Endocytobiosen mit zwei eukaryoten Partnern gibt es rezente Beispiele, bei denen die Mosaik-Natur der resultierenden Zellen deutlich hervortritt, weil sie noch nicht durch sekundäre Veränderungen während einer langen Symbiogenese verschleiert ist. So lässt sich etwa beim Grünen Paramecium (◘ Abb. 3.14) klar erkennen, dass fünf verschiedenartige Zellen ineinander geschachtelt sind: die Wirtszelle (1) mit ihren Mitochondrien (2), sowie die endocytischen Chlorellen (3) mit ihren Plastiden (4) und Mitochondrien (5).
3.2.7.2.1.1.7 Die Herkunft der Eukaryoten
Nach der Endosymbiontentheorie sind also die Mitochondrien und Plastiden, die hauptsächlichen Energielieferanten der Eucyten, als Prokaryoten in die Zellen urtümlicher Eukaryoten endocytiert worden. Dabei besteht über die phyletische Herkunft der Endocytobionten heute Klarheit. Offen ist aber die Frage, woher ihre Wirte kamen, jene urtümlichen Einzeller („Ur-Karyoten“ oder „Protoeukaryoten“), die weder Mitochondrien noch Plastiden besaßen und aus denen sich die modernen Eucyten entwickeln konnten.
Wenn alles Leben dieser Erde auf einen gemeinsamen Ahnen zurückgeht (Cenancestor bzw. „LUCA“, Last Universal Cellular Ancestor), dann legen eklatante Unterschiede in den rRNA-Sequenzen das Drei-Domänen-Konzept nahe (◘ Abb. 3.16). Die Stämme der Archaeen, Bakterien und Eukaryoten sollten sich danach sehr frühzeitig voneinander getrennt haben. Nun weisen die Archaeen in den Genen für Replikation, Transkription und Translation auffällige Ähnlichkeiten zu den Eukaryern auf. Daher wird angenommen, dass sich die Bakterien noch vor der Trennung der Stammlinien von Archaeen und Eukarya vom zunächst gemeinsamen Stammbaum abgetrennt und ihre eigene Entwicklung eingeschlagen haben. Die Hoffnung, durch weitere Sequenzvergleiche diese Vorstellung konkretisieren zu können, hat sich allerdings nicht erfüllt – im Gegenteil. Je mehr DNA-, RNA- und Proteinsequenzen bekannt geworden sind, desto verwirrender wurde das Bild. Der intertaxonische, horizontale Gentransfer (HGT) hat sich durch den Vergleich ganzer Genomsequenzen als sehr weit verbreitet erwiesen. Der Stammbaum des Lebens nimmt immer mehr den Charakter eines Netzwerkes an. Die in ◘ Abb. 3.16a eingezeichnete gemeinsame Wurzel aller Stammbäume ist jedenfalls hinsichtlich ihrer Positionierung ganz unsicher. Es ist sogar fraglich, ob das gesamte irdische Leben nur eine einzige Wurzel hat. Die Bildung erster Zellen (Progenoten) aus den damals verfügbaren Bausteinen hätte theoretisch mehrfach unabhängig voneinander erfolgt sein können. Gegen jene Vorstellung und für den gemeinsamen Ursprung allen Lebens spricht jedoch die Universalität des genetischen Codes sowie die universelle Verwendung von L-Aminosäuren beim Aufbau der Proteine am Ribosom.
Gibt es heute noch – als lebende Fossilien – urtümliche eukaryotische Einzeller, an denen klärende Untersuchungen vorgenommen werden könnten? Eine Zeit lang war angenommen worden, dass es tatsächlich rezente Protoeukaryoten in Gestalt der mitochondrienlosen und plastidenfreien Archaezoen gäbe. Zu diesen wurden einst die protozoischen Diplomonaden, Trichomonaden, Hypermastiginen und Microsporidien gezählt. Diese Gruppen weisen nämlich von den übrigen Eukaryern relativ stark abweichenden DNA-, RNA- und Proteinsequenzen auf, die eine frühzeitige Abspaltung der genannten Gruppen im Stammbaum der Eukaryoten nahelegen. Aber abweichende Sequenzen können sowohl auf eine basale phylogenetische Stellung als auch auf stark abgeleitete Merkmale spät abzweigender Gruppen hindeuten. Bei diesen Organismen handelt es sich nämlich um obligate Parasiten, die auf funktionelle Mitochondrien nicht angewiesen sind. Auch hat sich gezeigt, dass ihre Zellen DNA-freie, von Doppelmembranen umhüllte Organellen enthalten (Mitosomen bzw. Hydrogenosomen, s. unten). Schließlich fanden sich in ihrer Kern-DNA Sequenzen, die nur von Mitochondrien stammen können. Auch andere Molekulardaten sprechen dafür, dass die ehemals als Archaezoen bezeichneten Organismen extrem abgeleitete Einzeller sind, die ursprünglich Mitochondrien besaßen.
Durch diese Befunde ist das Stammbaumschema von ◘ Abb. 3.16 in Frage gestellt, alternative Vorstellungen wurden entwickelt. Eine Hypothese, die sich unter anderem auf die besonderen Stoffwechselverhältnisse bei den Trichomonaden und Diplomonaden stützt, ist die Wasserstoffhypothese. Ihr Basispostulat ist, dass bereits die ersten Eucyten das Produkt einer zellulären Symbiose waren. Die Partner jener Symbiose waren demnach H2-abhängige, methanogene Archaeen als Wirtszelle und zur H2-Produktion befähigte α-Proteobakterien als Mitochondrien. Die Eukaryer wären durch Symbiogenese entstanden.
α-Proteobakterien sind fakultative Anaerobier, sie können auch ohne O2 leben und gewinnen das erforderliche ATP dann nicht durch Atmung, sondern – wenn auch weniger effizient – durch Gärung. Dabei wird in vielen Vertretern Pyruvat in H2, CO2 und Essigsäure gespalten. Dieselbe Reaktion führen auch Hydrogenosomen aus, die nach Größe und Form Mitochondrien entsprechen, wie diese eine doppelte Membranhülle besitzen und sich durch Querteilung vermehren, aber in der Regel keine DNA enthalten. Das Kerngenom von Zellen mit Hydrogenosomen enthält Gene für mitochondriale Hitzeschockproteine. Hydrogenosomen sind also offenbar anaerobe Formen der Mitochondrien (und damit letztlich Abkömmlinge vom gleichen α-proteobakteriellen Endosymbionten, der die aeroben Mitochondrien hervorbrachte). Die allermeisten Hydrogenosomen haben nicht nur einen Teil, sondern ihre gesamte DNA an den Kern der Wirtszelle abgegeben. Eine wichtige Ausnahme ist jedoch beim Ciliaten Nyctotherus ovalis anzutreffen, dessen Hydrogenosomen noch ein kleines Genom beibehalten haben, das Genom eines Ciliaten-Mitochondriums, was die phylogenetische Identität der Hydrogenosomen und der Mitochondrien belegt.
Viele rezente anaerobe Protozoen, die Hydrogenosomen enthalten (z. B. der Ciliat Plagiopyla frontata, aber eben auch Trichomonaden und Diplomonaden), bergen zusätzlich methanogene Archaeen als Endocytobionten. Diese erzeugen aus H2 und CO2 Methan (CH4) und H2O und bilden dabei ATP. Hydrogenosomen und Methanogene sind in den sie enthaltenden Zellen verständlicherweise eng assoziiert. Im Sinne der Wasserstoffhypothese kann man in solchen Assoziationen Abbilder der Entstehung der Eucyten sehen: Methanogene Archaeen lagerten sich unter anaeroben Bedingungen an gärende α-Proteobakterien an, deren H2-Ausscheidung sie unabhängig machte von abiotischen Wasserstoffquellen. Im Zuge der weiteren Symbiogenese konnten diese Assoziationen dadurch gefestigt werden, dass die Methanogenen ihre H2-Lieferanten schließlich ganz umwuchsen und sie sich damit total einverleibten – die primäre Endocytobiose. Die stabile intrazelluläre Lebensweise der α-proteobakteriellen Endosymbionten erforderte jedoch Gentransfer vom Symbionten zum Wirt. Durch den Transfer von Genen für Transportproteine der Zellmembran vom Bakteriengenom in das Archaeengenom konnte ja auch die zur Wirtszelle gewordene Archaeenzelle die organischen Substrate für die Gärung ihrer Symbionten aus der Umwelt aufnehmen und sie an diese weitergeben. Zugleich wurde durch diesen Gentransfer die zur Endocytobiose avancierte Assoziation unauflöslich. Nebenbei: Gerade die methanogenen Archaeen verfügen – wie sonst nur Eukaryoten – über Histone und vermögen dementsprechend ihre DNA in Nucleosomen zu kompaktieren.
Sobald Sauerstoff zur Verfügung stand, konnten die bakteriellen Endocytobionten ihre Atmungskette als Mitochondrien verwenden, unter anaeroben Bedingungen blieb die Fermentationen der Hydrogenosomen. Der Wasserstoffhypothese nach stammen alle Eukaryoten aus mitochondrienhaltigen Vorfahren ab, Eukaryoten ohne Mitochondrien wären somit allesamt als abgeleitete Formen einzuordnen, was sich soweit mit den Befunden bis dato deckt. Rezente Übergangsformen zwischen Mitochondrien und Hydrogenosomen, die sogenannten anaeroben Mitochondrien sind bekannt. Der geforderte Transfer von Genen aus dem Genom der Mitochondrien und deren Integration im Genom der Wirtszelle ist in der Natur ein ganz gewöhnlicher Vorgang, wie die Analyse eukaryotischer Kerngenome belegt. Einige Eukaryoten besitzen weder Mitochondrien noch Hydrogenosomen. Zumindest bei den gut untersuchten Vertretern konnte allerdings ein weiteres Kompartiment nachgewiesen werden, dass wie die Mitochondrien und Hydrogenosomen von zwei Membranen umgeben und α-proteobakteriellen Ursprungs ist, die Mitosomen. Diese liefern der Wirtszelle allerdings kein ATP. Ihre essenzielle Funktion ist in der Synthese von Eisen-Schwefel-Clustern zu suchen, wichtigen Komponenten einiger Proteine, die in Mitochondrien, Hydrogenosomen oder Mitosomen synthetisiert und dem Cytoplasma der Wirtszelle zur Verfügung gestellt werden.
Der allgemeine Stammbaum muss nach dieser Hypothese umgezeichnet werden (◘ Abb. 3.16). Nach ihr gab es zunächst nur zwei Domänen, die Eukarya wären erst später aus einer intertaxonischen Vereinigung bestimmter Vertreter dieser Domänen hervorgegangen. Schon die urtümlichsten Eukarya hätten außerdem bereits Proteobakterien enthalten und brauchten sie sich (entgegen der entsprechenden Aussage der Endosymbiontentheorie) nicht erst nachträglich einzuverleiben. Doch wären auch nach dieser Vorstellung zwei unterschiedliche, zunächst selbstständige Organismen mit unterschiedlichen, sich aber ergänzenden metabolischen Fähigkeiten zu einem komplexeren System zusammengetreten und hätten damit einen fundamental neuen, überaus erfolgreichen Evolutionsprozess gestartet.
3.2.7.2.1.1.8 Offene Probleme
Von den geschilderten, noch weiter zu prüfenden Hypothesen werden andere wesentliche Aspekte der Eucytenevolution nicht berührt. Wie bildeten sich die zahlreichen Endomembranen (endoplasmatisches Reticulum und Kernhülle, Golgi-Dictyosomen, Vakuolen usw.)? Sind sie, wie zu vermuten ist, letztlich Abkömmlinge der Plasmamembran? Wie entstanden aus einem einzigen DNA-Ring mehrere lineare Chromosomen mit besonderen Telomeren und zahlreichen Startpunkten der Replikation?
Trotz dieser und anderer noch offener Fragen steht heute fest, dass es in der Evolution nicht nur zu ständigen Verzweigungen von Stammbäumen (Kladogenese) gekommen ist, sondern gelegentlich auch zu folgenschweren Vernetzungen. Durch ITC und Symbiogenese können sich weit getrennte Zweigenden evolutiver Stammbäume zu Startpunkten für neue Entwicklungslinien verbinden. Die Formierung stabiler intertaxonischer Kombinationen ist damit neben Mutation, genetischer Rekombination und horizontalem Gentransfer ein wichtiger Motor der Evolution, zumal er auch evolutive Großübergänge zu provozieren vermag.
3.3 Veränderlichkeit und Vererbung der genetischen Information
Darwin schrieb 1872: „variations, … if they be in any degree profitable to the individuals of a species, … will tend to the preservation of such individuals, and will generally be inherited by the offspring … I have called this principle, by which each slight variation, if useful, is preserved, by the term Natural Selection“. („Variationen, die auf irgendeine Weise einem Individuum einer Art nützen, werden zum Überleben dieser Individuen beitragen und an die nachkommenden Generationen weitergegeben …. Ich habe dieses Prinzip, durch das jede kleine Variation, falls nützlich, beibehalten wird, Natürliche Selektion genannt.“). Damit kommt der Frage, auf welche Weise Variabilität entstehen kann, eine entscheidende Bedeutung zu.
In diesem Kapitel werden wir erörtern, wie die Variabilität der genetischen Information zustande kommen kann.
3.3.1 Mutationen
Die Struktur der DNA muss relativ stabil sein und DNA muss nahezu fehlerfrei repliziert und transkribiert werden, um als verlässlicher Informations- und Erbträger dienen zu können. Andererseits müssen Erbänderungen (Mutationen) zugelassen werden, um die notwendige Variabilität für die natürliche Selektion zu schaffen. Offensichtlich haben sich die Mutationsraten bei allen Organismen im Laufe der Evolution auf ein Niveau eingependelt, das eine Weiterentwicklung der Organismen ermöglicht und positive und negative Mutationseffekte in ein Gleichgewicht bringt.
Unter einer Mutation versteht man Veränderungen der DNA, die einzelne Basen oder auch längere Sequenzbereiche auf den Chromosomen betreffen können. Mutationen können spontan oder nach Induktion (z. B. durch mutagene Substanzen oder ionisierende Strahlung) auftreten. Punktmutationen liegen vor, wenn nur einzelne oder wenige Nucleotide ausgetauscht wurden (◘ Abb. 3.19). Man spricht von single nucleotide polymorphisms (SNPs), wenn Punktmutationen in einer Population an identischen Stellen in einem DNA-Abschnitt auftreten. Man schätzt, dass sich jeder Mensch von einem anderen Menschen durch 1–10 Mio. Punktmutationen unterscheidet. Erfolgen Mutationen innerhalb von Transkriptionseinheiten, sprechen wir von Genmutationen; sind mehrere Gene, Chromosomenabschnitte oder mehrere Chromosomen betroffen, handelt es sich um Chromosomenmutationen. Zahlenmäßige Abweichungen im Gesamtbestand der Chromosomen (z. B. Polyploidie; s. Abschn. 3.4.2) werden als Genommutationen bezeichnet. Nucleotide oder DNA-Abschnitte können herausgeschnitten (Deletion), eingefügt (Insertion oder Translokation), verdoppelt (Duplikation) oder in ihrer Orientierung umgedreht wurden (Inversion). Die Auswirkungen von Punktmutationen oder dem Herausschneiden von Sequenzelementen sind in ◘ Abb. 3.19 und ◘ Abb. 3.20 erläutert. Wird der Leserahmen des genetischen Codes in einem Protein-codierenden Gen durch Deletionen und Insertionen einzelner Basenpaare verändert, entstehen häufig Frame-shift-Mutationen; d. h. die nachfolgende Basenreihenfolge ist zwar identisch, jedoch wurde das Leseraster um ein oder zwei Basen verschoben, so dass jetzt andere Codons entstehen. Dadurch geht die Funktionalität des betreffenden Proteins verloren.

Schematische Darstellung der Auswirkung von Punkt- und Genmutationen
Schematische Darstellung der Auswirkung von Punkt- und Genmutationen

a, b. Schematische Übersicht über die Mechanismen von Chromosomen-Rearrangements durch homologe Rekombination. Die Zahlen in den farblich markierten Kästchen deuten definierte DNA-Abschnitte in DNA-Doppelsträngen. a Es können direkte Chromosomenbrüche auftreten, die anschließend wieder verbunden werden. b Zwischen repetitiven DNA-Elementen (z. B. Mikrosatelliten; s. Abschn. 4.1.2) kann es zur homologen Rekombination und Crossing over kommen; dies ermöglicht den Austausch von DNA-Abschnitten. 1. Deletion von DNA-Abschnitten innerhalb eines Chromosoms. 2. Inversion innerhalb eines Chromosoms. 3. Austausch von Chromosomenstücken zwischen homologen Chromosomen durch reziproke Translokation, die jeweils mit einer Sequenzdeletion und Sequenzduplikation einhergeht. 4. Austausch von Chromosomenstücken zwischen verschiedenen Chromosomen durch reziproke Translokation, die jeweils mit einer Sequenzdeletion und Sequenzinsertion einhergeht. 5. Inversion von DNA-Abschnitten innerhalb eines Chromosoms infolge von Crossing over zwischen benachbarten repetitiven Elementen. 6. Rekombination zwischen unterschiedlichen Chromosomen an repetitiven Elementen führt zur reziproken Translokationen von Chromosomenabschnitten
a, b. Schematische Übersicht über die Mechanismen von Chromosomen-Rearrangements durch homologe Rekombination. Die Zahlen in den farblich markierten Kästchen deuten definierte DNA-Abschnitte in DNA-Doppelsträngen. a Es können direkte Chromosomenbrüche auftreten, die anschließend wieder verbunden werden. b Zwischen repetitiven DNA-Elementen (z. B. Mikrosatelliten; s. Abschn. 4.1.2) kann es zur homologen Rekombination und Crossing over kommen; dies ermöglicht den Austausch von DNA-Abschnitten. 1. Deletion von DNA-Abschnitten innerhalb eines Chromosoms. 2. Inversion innerhalb eines Chromosoms. 3. Austausch von Chromosomenstücken zwischen homologen Chromosomen durch reziproke Translokation, die jeweils mit einer Sequenzdeletion und Sequenzduplikation einhergeht. 4. Austausch von Chromosomenstücken zwischen verschiedenen Chromosomen durch reziproke Translokation, die jeweils mit einer Sequenzdeletion und Sequenzinsertion einhergeht. 5. Inversion von DNA-Abschnitten innerhalb eines Chromosoms infolge von Crossing over zwischen benachbarten repetitiven Elementen. 6. Rekombination zwischen unterschiedlichen Chromosomen an repetitiven Elementen führt zur reziproken Translokationen von Chromosomenabschnitten
Punktmutationen, bei denen es sich um den Austausch (Substitution) einzelner Basen handelt, werden in zwei große Klassen unterteilt (◘ Abb. 3.21):

Schematische Darstellung der Transition und Transversion von Nucleotiden. A: Adenin; G: Guanin; C: Cytosin, T: Thymin
Schematische Darstellung der Transition und Transversion von Nucleotiden. A: Adenin; G: Guanin; C: Cytosin, T: Thymin
-
Transitionen: Darunter versteht man die Substitution einer Pyrimidinbase durch eine andere, d. h. T → C oder umgekehrt, oder einer Purinbase durch eine andere, d. h. A → G oder umgekehrt (◘ Abb. 3.21). Transitionen stellen die häufigste Klasse der Punktmutationen dar (u. a. durch Fehlpaarung tautomerer Basen hervorgerufen; ◘ Abb. 3.22). Betrachtet man homologe Gene nah verwandter Arten, so machen Transitionen ca. 90 % und mehr der Substitutionen aus. Zwischen entfernt verwandten Arten geht der Anteil der Transitionen durch multiple Substitutionen, d. h. mehrfacher Austausch einer Base an derselben Position, auf unter 35 % zurück.
Abb. 3.22 Abb. 3.22 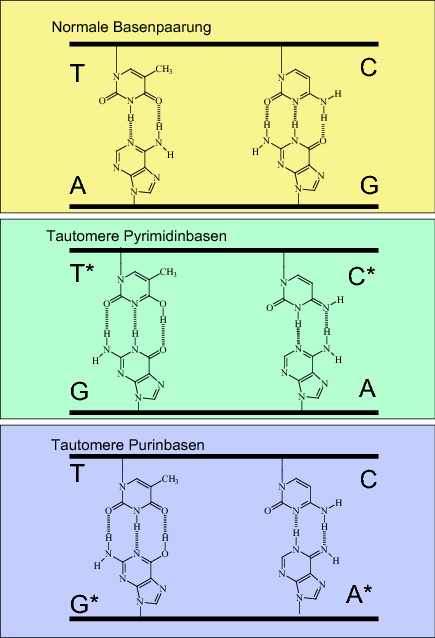
Auslösung von Punktmutationen (Transitionen): Prinzip der Basenfehlpaarung tautomerer Nucleotide, die durch intramolekulare Umlagerung der „normalen“ Basen entstehen. A: Adenin; G: Guanin; C: Cytosin, T: Thymin; A*: seltene Iminoform des Adenins; G*: seltene Enolform des Guanins; C*: seltene Iminoform des Cytosins; T*: seltene Enolform des Thymins
Auslösung von Punktmutationen (Transitionen): Prinzip der Basenfehlpaarung tautomerer Nucleotide, die durch intramolekulare Umlagerung der „normalen“ Basen entstehen. A: Adenin; G: Guanin; C: Cytosin, T: Thymin; A*: seltene Iminoform des Adenins; G*: seltene Enolform des Guanins; C*: seltene Iminoform des Cytosins; T*: seltene Enolform des Thymins
-
Transversionen: Darunter versteht man den Austausch einer Purinbase durch eine Pyrimidinbase (◘ Abb. 3.2), d. h. A → C oder T und G → C oder T oder umgekehrt (◘ Abb. 3.21). Transversionen sind deutlich seltener als Transitionen.

3.3.1 Wie entstehen Mutationen?
DNA-Basen können spontan und zufällig desaminiert, depuriniert und oxydiert werden. In eukaryotischen Zellen treten Basen-Desaminierungen mit einer Rate von 100 Desaminierungen pro Tag und Zelle auf (◘ Abb. 3.23). Durch Desaminierung von Cytidin entsteht Uridin. Werden solche Mutationen nicht repariert (s. unten), paart sich bei der nachfolgenden Replikation U mit A statt mit G, wie es das ursprüngliche C getan hätte. Dadurch ist das CG-Paar letztlich durch ein TA-Paar ersetzt worden (also eine Transition) (◘ Abb. 3.24). Da viele Gene durch Methylierung in ihrer Expression reguliert werden (abgeschaltete Gene sind häufig stark methyliert), führt die spontane oder induzierte Desaminierung von 5-Methylcytosin zu Thymin. Bei der nachfolgenden Replikation würde jetzt T mit A anstelle von C mit G paaren (Transition). Als Beispiel für eine chemisch induzierte Mutation sei auf die Wirkung von salpetriger Säure oder Disulfit hingewiesen, die zu einer oxidativen Desaminierung von Cytosin und 5-Methylcytosin führt.

Ursachen für Punktmutationen: Prinzip und Auswirkung der Desaminierung, Depurinierung, Oxidation und Dimerisierung von DNA-Basen. Durch Desaminierung kann aus Cytosin Uracil, aus 5-Methylcytosin Thymin und aus Adenin Hypoxanthin entstehen. Durch Depurinierung können sowohl Guanosin als auch Adenosin verändert werden. Guanosin wird zu 8-Oxoguanosin oxidiert. Dimerisierung aufgrund von UV-Strahlung tritt besonders bei Thymidin auf, ist aber auch bei Cytosin festgestellt worden
Ursachen für Punktmutationen: Prinzip und Auswirkung der Desaminierung, Depurinierung, Oxidation und Dimerisierung von DNA-Basen. Durch Desaminierung kann aus Cytosin Uracil, aus 5-Methylcytosin Thymin und aus Adenin Hypoxanthin entstehen. Durch Depurinierung können sowohl Guanosin als auch Adenosin verändert werden. Guanosin wird zu 8-Oxoguanosin oxidiert. Dimerisierung aufgrund von UV-Strahlung tritt besonders bei Thymidin auf, ist aber auch bei Cytosin festgestellt worden

Auswirkungen von Desaminierung, Depurinierung und Oxidation. Wurde eine durch Desaminierung, Depurinierung oder Oxidation hervorgerufene Punktmutation nicht durch Reparaturenzyme rückgängig gemacht, so wirken sie sich in den nachfolgenden Replikationen aus: Während der nicht-mutierte Strang (blau) identisch repliziert wird, führt der mutierte Strang (grün) dazu, dass auf dem komplementären neuen Strang nach Desaminierung oder Oxidation ein verändertes Nucleotid eingebaut wird oder aber nach Depurinierung eine Deletion erfolgt oder eine neue Base zufällig ersetzt wird. A: Adenin; G: Guanin; C: Cytosin, T: Thymin, U: Uracil
Auswirkungen von Desaminierung, Depurinierung und Oxidation. Wurde eine durch Desaminierung, Depurinierung oder Oxidation hervorgerufene Punktmutation nicht durch Reparaturenzyme rückgängig gemacht, so wirken sie sich in den nachfolgenden Replikationen aus: Während der nicht-mutierte Strang (blau) identisch repliziert wird, führt der mutierte Strang (grün) dazu, dass auf dem komplementären neuen Strang nach Desaminierung oder Oxidation ein verändertes Nucleotid eingebaut wird oder aber nach Depurinierung eine Deletion erfolgt oder eine neue Base zufällig ersetzt wird. A: Adenin; G: Guanin; C: Cytosin, T: Thymin, U: Uracil
Das Genom ist permanent oxidativem Stress, z. B. durch Sauerstoffradikale (reactive oxygen species; ROS) ausgesetzt. Die Base Guanosin ist besonders anfällig für oxidativen Stress, da sie das geringste Oxidationspotenzial besitzt. Durch Oxidation kann aus Guanin 8-Oxoguanin entstehen (8-OxoG). 8-OxoG paart nicht länger mit Cytosin sondern mit Adenosin. Wird eine solche Mutation nicht durch Reparaturenzyme entfernt, kommt es nach einer Replikation zu einer Transversion, d. h. das GC-Paar wird durch ein TA-Paar ausgetauscht (◘ Abb. 3.24). Man nimmt an, dass Alterungsprozesse unter anderem durch Mutationen entstehen, die durch Oxidation von Guanosin durch ROS hervorgerufen werden. Daher wird den Antioxidanzien, die in vielen Nahrungsmitteln vorkommen, eine besondere Bedeutung für eine gesunde Ernährung zugesprochen.
Die Purinreste Guanin und Adenin können spontan durch Hydrolyse aus der DNA entfernt werden (◘ Abb. 3.23). Solche Depurinierungen zählen zu den häufigsten spontanen Veränderungen; über 5000–10.000 Purinbasen werden täglich in jeder menschlichen Zelle depuriniert. Werden depurinierte Basen nicht repariert (s. unten), so kommt es bei der nachfolgenden Replikation entweder zur Deletion einer einzelnen Base (◘ Abb. 3.24) oder es wird anstelle des depurinierten Guanins jede beliebige Base nach dem Zufallsprinzip eingebaut. Solche Substitutionen können entweder zu einer Transition aber auch Transversion führen.
Durch UV-Bestrahlung können benachbarte Thymin- oder Cytosinreste aktiviert werden, die dann jeweils Dimere ausbilden (◘ Abb. 3.23). Solche Dimere führen unrepariert zu Deletionen oder wenn sie häufig vorhanden sind, zum programmierten Zelltod (Apoptose). Solche Dimerisierungen treten verstärkt auf, wenn natürlich vorkommende Mutagene, wie z. B. die in Apiaceen und Rutaceen vorkommenden Furanocumarine (s. Abschn. 4.3.3), mit der Nahrung oder als Arzneimittel aufgenommen wurden. Denn diese Sekundärstoffe werden in der Haut abgelagert. Setzt man sich intensiver Sonnen- und damit UV-Strahlung aus, so werden die DNA-interkalierenden Furanocumarine photochemisch angeregt und können mit der DNA von Hautzellen in der Epidermis reagieren und diese quervernetzen. Interkalierende Substanzen sind planar und lipophil. Sie lagern sich zwischen die Basenstapel der DNA-Doppelhelix ein und führen bei der Replikation zur Deletion oder zur Insertion eines Basenpaars. Dadurch wird der Leserahmen um eine Base verschoben, es entstehen Frame-shift-Mutationen, die sich meist negativ auf das zu codierende Protein auswirken (◘ Abb. 3.19). Interkalierende Substanzen kommen auch in der Natur vor; z. B. haben diverse Sekundärstoffe, die Pflanzen zur Abwehr von Fraßfeinden und Mikroorganismen einsetzen, solche Eigenschaften (s. Abschn. 4.3.3).
Depurinierung, Interkalation und Alkylierung können Einzel- und Doppelstrangbrüche in der DNA auflösen, die zum Zelltod oder Chromosomenmutationen führen können.
In seltenen Fällen können die DNA-Basen durch intramolekulare Umlagerungen tautomere Formen einnehmen (◘ Abb. 3.22), die zu Fehlpaarungen bei der Replikation führen. Normalerweise liegen die Basen in der Keto-Form vor und nehmen selten die Enol-Form ein. Tautomeres Adenin paart mit Cytosin statt mit Thymin, und tautomeres Thymin mit Guanin statt mit Adenin und umgekehrt. Die durch Einbau tautomerer Basen bewirkten Nucleotidsubstitutionen fallen alle in die Klasse der Transitionen (◘ Abb. 3.21).
Die meisten primären Veränderungen (Desaminierung, Depurinierung, Oxidation, Dimerisierung) werden von Reparaturenzymen (u. a. AP-Endonuclease; DNA-Glycosylasen) erkannt und herausgeschnitten (solange nicht auch der zweite DNA-Strang beschädigt wurde) und durch DNA-Polymerase und DNA-Ligase repariert. Aber auch Alkyltransferasen, Photolyasen und Fehlpaarungsreparatur- und Rekombinationsreparatursysteme, die vor der Replikation aktiv werden, sind vorhanden. Die Anzahl der an Reparaturvorgängen beteiligten Enzyme liegt bei weit über 50. Eine Reparatur ist aber nur möglich, wenn die Zelle eine korrekte Kopie besitzt. Darin liegt der große Vorteil der Doppelhelix, in der genetische Information komplementär als Kopie gespeichert ist. Selbst wenn die Information auf einem Strang verloren geht, ist sie auf dem komplementären Strang noch vorhanden und kann genutzt werden, um eine entsprechende Korrektur durchzuführen. In diploiden Organismen kommt zudem eine zweite Genkopie vor, die mittels Rekombination und Genkonversion (s. Abschn. 3.4.2) nutzbar wird, falls das Original geschädigt wurde.
In einer Keimbahnzelle, z. B. beim Menschen, kommt es dank der Effektivität der Reparatursysteme nur zu 10–20 Basensubstitutionen pro Jahr bezogen auf die vorhandenen 3 × 109 Basenpaare. Die Bedeutung der Reparatursysteme lässt sich gut bei Menschen erkennen, die an Xeroderma pigmentosum, einer seltenen autosomal-rezessiven neurocutanen Krankheit, erkrankt sind. Bei ihnen sind einzelne Elemente des Reparatursystems ausgefallen, die bei durch UV-Strahlung verursachter DNA-Schädigung benötigt werden. Als Folge der UV-Strahlung des Sonnenlichts treten neben zahlreichen neurologischen und psychischen Symptomen eine starke Hautfleckenbildung und Hautkrebs auf, die sich nur durch vollständige Vermeidung von Sonnenlichtexposition verhindern lassen. Inzwischen sind weitere vererbbare Störungen von Reparaturenzymen und die dadurch hervorgerufenen Krankheitsbilder bekannt geworden, die belegen, wie wichtig Reparaturenzyme sind (◘ Tab. 3.8).
Mutationen treten in allen Organismen spontan und zufällig auf; die Häufigkeit dieser Spontanmutationen (auch „Hintergrundmutationen“ genannt) ist organismenspezifisch. Die meisten Mutationen, soweit sie nicht durch Reparaturenzyme beseitigt werden, beobachtet man in somatischen Zellen, die mit dem Tod des Individuums untergehen (somatische Mutationen). Somatische Mutationen sind für das Auftreten vieler Krankheiten, u. a. Tumorerkrankungen, verantwortlich. Nur Mutationen in Keimbahnzellen und Gameten (Gametenmutationen oder generative Mutationen) werden an Nachkommen weitergegeben. Mutationen, die in einer Population fixiert sind (d. h. mehrere Individuen tragen dieses Merkmal), werden zu einem evolutionären Ereignis und unterliegen der Selektion sowie Zufallsprozessen (genetische Drift).
Natürliche Mutationshäufigkeiten, die zum Ausfall eines Gens führen, schätzt man bei Bakterien auf 10–5 bis 10–6 Mutationen pro Genlocus (darunter versteht man den Ort eines Gens auf dem Chromosom) und Generation. Bei Eukaryoten sind diese Häufigkeiten nur schwer zu bestimmen, dürften aber in derselben Größenordnung liegen. Auf die einzelne Nucleotidposition bezogen, beträgt die Mutationshäufigkeit bei Bakterien 10–9 bis 10–10 Substitutionen pro Generation. Beim Menschen kommt auf 100.000–200.000 Genreplikationen eine Mutation. Bei etwa 20.000 Genloci pro haploidem Genom finden wir entsprechend weniger als 1 mutiertes Allel pro Genom, das bei den jeweiligen Eltern nicht vorhanden war. Bei Bakterien und Viren mit hoher Teilungsrate und Individuenzahl kann man die Auswirkung von Mutationen direkt beobachten. Vermehrt man Bakterien in Anwesenheit eines starken Selektionsfaktors, z. B. Antibiotika, so werden die meisten von ihnen abgetötet oder im Wachstum gehemmt. Gelingt es aber einer einzigen Bakterienzelle, durch Mutation eine Resistenz gegen das Antibiotikum auszubilden, so wird sie sich vermehren und – zumal es alle 20 Minuten zu einer Zellteilung kommt – bald hohe Dichten erreichen. Interessanterweise sind die Gene für Antibiotikaresistenz offenbar bereits sehr alt (> 30.000 Jahre) und wurden von Bakterien evolviert bevor Antibiotika medizinisch eingesetzt wurden (Wright u. Poinar 2012). Auch die schnelle Veränderlichkeit der Viren ist medizinisch bekannt und gut belegt. Viren, deren Erbinformation auf einzelsträngiger RNA gespeichert ist (z.B Retroviren) zeichnen sich durch besonders hohe Veränderlichkeit aus. Man denke an die Variabilität von HIV, dem Erreger von AIDS, der sich so schnell ändert, dass es bislang nicht möglich war, einen wirksamen Impfstoff zu entwickeln. An der schnellen Entwicklung von Resistenzen bei Bakterien und Viren kann man erkennen, dass auch heute noch evolutive Prozesse ablaufen, die wir im Gegensatz zu den Prozessen in der fernen Vergangenheit unmittelbar untersuchen können. Bei den Tieren und Pflanzen mit längerer Generationszeit und meist geringen Nachkommenzahlen dauert es entsprechend länger, bis man den Effekt einer Mutation feststellen kann.
Mutationen können durch Behandlung mit energiereichen Strahlen (UV-Strahlung, radioaktive Strahlung) oder mutagenen Agentien vermehrt ausgelöst werden (induzierte Mutationen). Die meisten Mutagene bewirken eine Modifikation einzelner Basen (Alkylierung, Desaminierung, Depurinierung), werden als Basenanaloge eingebaut, hemmen Reparaturenzyme oder DNA-Topoisomerasen oder führen zu Strangbrüchen und Um- sowie Neuordnungen (rearrangements). Chemische Alkylantien (z. B. Dimethylsulfat) methylieren u. a. Guanin zu O6-Methylguanin, das nicht länger mit Cytosin ein Basenpaar bilden kann. Ähnliche Reaktionen können auch durch natürlich vorkommende Substanzen, wie z. B. S-Adenosylmethionin, hervorgerufen, werden. In der Natur sind etliche Sekundärstoffe bekannt, die nach metabolischer Aktivierung in der tierischen Leber zu starken Alkylantien werden, Beispiele sind Pyrrolizidinalkaloide, Aristolochiasäure oder Aflatoxine. Weitere mutagene Effekte werden in unseren Zellen durch ROS (Wasserstoffperoxid, Sauerstoff- und Superoxidradikale) (s. oben) hervorgerufen.
Bedingt durch den redundanten genetischen Code (s. Abschn. 3.2.5) führt bei weitem nicht jede Punktmutation in einem Gen zu einer Veränderung der Aminosäuresequenz. 134 (24,4 %) der 549 theoretisch möglichen Substitutionen sind synonym; 23 (4,2 %) führen zu Stoppcodons und 392 (71,4 %) zu Aminosäureaustauschen (◘ Tab. 3.9). Wie aus ◘ Abb. 3.7 und ◘ Tab. 3.9 ersichtlich, hat aber die Nucleotidsubstitution in der dritten Codonposition in ca. 69 % der Fälle (126 von 183) keine Veränderung zur Folge (man spricht von stiller, neutraler Mutation oder silent mutation). Eine solche Mutation kann demnach beibehalten werden, ohne dass der Träger des Merkmals negative Auswirkungen erfährt. Wie ◘ Tab. 3.9 andeutet, sind unter den 549 Mutationsformen theoretisch 392 Aminosäure-ändernde (nicht-synonyme) Mutationen möglich; davon ändern 184 (47 %) die Polaritätsklasse der Aminosäuren und 128 (33 %) die Ladung, während 273 (70 %) zu einer Aminosäure einer anderen Klasse führen. Doch selbst konservierte Aminosäuresubstitutionen führen nur selten zum Totalausfall eines Proteins oder Enzyms (man spricht von neutraler Substitution), es sei denn, das aktive Zentrum wäre von einer Mutation betroffen oder ein Stoppcodon wäre eingefügt. Bei den Mutationen, die sich etablieren konnten, überwiegen Aminosäureaustausche, die keinen oder nur geringen negativen Selektionswert aufweisen. Wenn auch positive Veränderungen selten sind, so führt die natürliche Auslese im Verlauf der Jahrmillionen doch zu einer Optimierung der Genfunktionen. Träger von Austauschen mit negativen Eigenschaften sind geschwächt (man denke an Patienten mit neuromuskulärer Dysfunktion, Thalassämie und anderen Erbkrankheiten, bei denen einzelne kritische Aminosäuren mutiert sind) oder sterben.
Insertionen/Deletionen oder Translokation längerer Sequenzelemente (Chromosomenmutationen) treten mit einer gewissen Häufigkeit auf und werden u. a. durch Transposons (s. Abschn. 3.4.3) oder Viren hervorgerufen. Strangbrüche und Fehlpaarungen der DNA während der Meiose, so z. B. im Bereich repetitiver Sequenzelemente (◘ Abb. 3.20), können ungleiches Crossing over, fehlerhafte Replikation und Reparatur zur Folge haben und führen meist zu einem größeren Nucleotidaustausch als einfache Punktmutationen (◘ Abb. 3.19).
Außerhalb von codierenden oder regulatorischen DNA-Abschnitten wirken sich Mutationen nicht oder nur geringfügig aus. Wenn ein oder zwei Nucleotide oder größere Einheiten innerhalb eines Gens entfernt oder hinzugefügt werden, kommt es zu einer Verschiebung des Leserasters (Frame-shift-Mutation) und damit zu gänzlich anderen Proteinen (◘ Abb. 3.19), die fast immer funktionslos sind (loss-of-function), in seltenen Fällen aber zu Proteinen mit neuen Funktionen (gain-of-function) führen. Die meisten Mutationen sind neutral oder negativ, und nur in seltenen Fällen vermag ein mutiertes Gen oder Allel seinen Träger besser an seine Umwelt anzupassen und dadurch den Fortpflanzungserfolg der Nachkommen zu steigern. Wenn wir demnach DNA-Sequenzen oder Genomstrukturen der heute lebenden Organismen analysieren, so sehen wir im Wesentlichen nur Mutationen, die entweder neutral waren oder einen positiven Selektionswert hatten, da die Träger von schädlichen Mutationen keine nachhaltige Überlebenschance hatten, sondern im Verlauf der Evolution herausgefiltert wurden.
3.3.2 Mitose und Meiose
In Bakterien liegt die DNA als ringförmiges, in Eukaryoten jedoch als lineares Molekül vor. Eukaryotische Chromosomen bestehen aus einem Centromer, an dem die Mikrotubuli während der Zellteilung angreifen, diversen Replikationsstarts (origin of replication) und Telomersequenzen an den Enden. Die DNA liegt in den Chromosomen nicht als freier Faden, sondern mit basischen Histonproteinen verbunden vor. Vier Histonproteine (H2 A, H2B, H3, H4), die viele positiv geladen Lysinreste aufweisen, bilden octamere Zylinder aus, um die sich die DNA (Nukleosomen mit ca. 145 Basenpaare DNA) windet. Hierbei spielen ionische Wechselwirkungen zwischen den positiven geladenen Lysinresten und den negativ geladenen Phosphatgruppen der DNA eine wichtige Rolle. Zwischen zwei benachbarten Nucleosomen befindet sich meist ein linearer DNA-Abschnitt von ca. 80 Basenpaaren, an dem sequenzspezifische Proteine binden.
Im Gegensatz zu den haploiden Bakterien und primitiven Eukaryoten weisen die meisten höheren Eukaryoten einen diploiden Chromosomensatz auf. Im diploiden Genom sind von jedem Chromosom zwei Exemplare vorhanden, deren Länge und Struktur (z. B. Sitz des Centromers, lineare Anordnung der Genloci) identisch sind; man spricht deshalb von homologen Chromosomen. Jeweils eins der homologen Chromosomen lässt sich auf das haploide väterliche, das andere auf das haploide mütterliche Genom zurückführen, denn bei der Befruchtung werden die jeweils haploiden Chromosomensätze der männlichen und weiblichen Gameten in der Zygote vereinigt und bleiben in den somatischen Zellen als diploider Chromosomensatz erhalten. In ◘ Tab. 3.10 ist eine Übersicht von haploiden Chromosomenzahlen einiger Organismen zusammengestellt. Wie man leicht erkennt, gibt es keinen einheitlichen Trend hinsichtlich der evolutionären Entwicklungshöhe und der Chromosomenzahl. Die Grünalge Chlamydomonas hat bereits 18 Chromosomen, der Seidenspinner 28, während der Mensch (Homo sapiens) 23 Chromosomen im haploiden Chromosomensatz aufweist.
Man unterscheidet paarweise auftretende Autosomen und Geschlechtschromosomen (Gonosomen); letztere sind beim heterogametischen Geschlecht nicht als identisches Paar (◘ Abb. 3.25) vorhanden. Beim Menschen finden wir im diploiden Chromosomensatz (auch als Karyotyp bezeichnet) bekanntlich 46 Chromosomen, darunter 2 Geschlechtschromosomen, die bei Frauen, dem homogametischen Geschlecht mit XX, bei Männern, dem heterogametischen Geschlecht mit XY bezeichnet werden (◘ Abb. 3.25). Nicht bei allen Organismen ist das männliche Geschlecht heterogametisch; bei Vögeln und Schmetterlingen, z. B., sind die Weibchen heterogametisch und besitzen WZ-Geschlechtschromosomen, die homogametischen Männchen dagegen ZZ-Chromosomen (s. Abschn. 4.3).

a–c. Schematische Illustration der menschlichen Chromosomen während der Metaphase. a Darstellung der Metaphase-Präparation einer Frau mit einer Trisomie 21 (Chromatinregionen farblich markiert) und b eines Mannes (schwarze Umrissdarstellung). Von jedem Chromosom sind zwei homologe Partner vorhanden; nur die X- und Y-Chromosomen treten beim Manne einzeln auf. Jedes einzelne Chromosom liegt verdoppelt vor, wobei die Schwesterchromatiden durch ein gemeinsames Centromer zusammengehalten werden. c Lokalisation wichtiger Gene, die bei genetisch bedingten Krankheiten eine Rolle spielen, auf den jeweiligen Chromosomen
a–c. Schematische Illustration der menschlichen Chromosomen während der Metaphase. a Darstellung der Metaphase-Präparation einer Frau mit einer Trisomie 21 (Chromatinregionen farblich markiert) und b eines Mannes (schwarze Umrissdarstellung). Von jedem Chromosom sind zwei homologe Partner vorhanden; nur die X- und Y-Chromosomen treten beim Manne einzeln auf. Jedes einzelne Chromosom liegt verdoppelt vor, wobei die Schwesterchromatiden durch ein gemeinsames Centromer zusammengehalten werden. c Lokalisation wichtiger Gene, die bei genetisch bedingten Krankheiten eine Rolle spielen, auf den jeweiligen Chromosomen
Die Anzahl und Form der in einer Zelle vorhandenen Chromosomen ist in der Regel artkonstant. Bei einigen Organismen gibt es Ausnahmen, indem die Chromosomenzahl innerhalb einer Art schwanken kann (z. B. Silbergrüner Bläuling, Polyommatus coridon). Bei einer Zellteilung muss auch die Tochterzelle das identische Genom der Mutterzelle erhalten; diesen Prozess nennt man Mitose. Bei der mitotischen Zellteilung wird die gesamte DNA zunächst verdoppelt (Replikation); es entstehen dabei aus einem Chromosom jeweils zwei parallel liegende identische Schwesterchromatiden, die über ein gemeinsames Centromer eng zusammenhängen (◘ Abb. 3.25, 3.26). Die kondensierten Chromatiden werden über den Spindelapparat so auseinander gezogen, dass jede neue Zelle einen kompletten diploiden Chromosomensatz erhält (s. EXKURS 3.2 Abschn. 3.3.3).

Schematische Darstellung des Verlaufs von Meiose, Mitose und Cytokinese. Bei der Mitose entstehen stets identische Tochterzellen. Bei der Meiose kommt es durch Crossing over zu einem Austausch von DNA-Abschnitten zwischen väterlichen und mütterlichen Chromosomen. Die haploiden Gameten erhalten eine Zufallsmischung von jeweils väterlichen und mütterlichen Chromatiden
Schematische Darstellung des Verlaufs von Meiose, Mitose und Cytokinese. Bei der Mitose entstehen stets identische Tochterzellen. Bei der Meiose kommt es durch Crossing over zu einem Austausch von DNA-Abschnitten zwischen väterlichen und mütterlichen Chromosomen. Die haploiden Gameten erhalten eine Zufallsmischung von jeweils väterlichen und mütterlichen Chromatiden
Die Gameten sind haploid. Der Prozess, der vom diploiden zum haploiden Chromosomensatz führt, wird als Meiose bezeichnet. Bei der Meiose wird der verdoppelte Chromosomensatz halbiert (Reduktionsteilung) und somit wieder in haploide Genome zurückgeführt (s. EXKURS 3.2). Die Meiose ist bei diploiden Organismen die Voraussetzung für die geschlechtliche Fortpflanzung. Wären die Gameten diploid, so würde jede neue Zygotenbildung eine Verdopplung der Chromosomensätze mit sich bringen. Nur durch haploide Gameten lässt sich dieses Dilemma lösen.
Über die Evolution und Bedeutung der Geschlechter gibt es unterschiedliche Ansichten. Fest steht, dass die geschlechtliche Fortpflanzung die genetische Variabilität der Individuen erhöht. Jeder Gamet erhält in der Meiose eine unterschiedliche Mischung der ursprünglich mütterlichen und väterlichen Chromosomen (s. EXKURS 3.2); beim Menschen mit 23 Chromosomen sind 223 Kombinationen möglich; d. h. theoretisch kann jeder Mensch über 8 Mio. genetisch unterschiedliche Gameten bilden. Da bei der Zygote zwei Gameten verschmelzen, liegt die Zahl der Chromosomenkombinationen bei (8 × 106)2 = 64 × 1012. Wie in Abschn. 3.3.3 gezeigt, kommt es bei der Meiose außerdem zu einem Crossing over der homologen Chromosomen und damit zu einem zusätzlichen Genaustausch zwischen den jeweils homologen Chromosomen. Dadurch stellt bereits jedes einzelne Chromosom in einem Gameten ein Mosaik aus jeweils homologen mütterlichen und väterlichen Genen dar. Deshalb ist die Wahrscheinlichkeit, dass zwei Kinder eines Elternpaares identisch sind, extrem klein. Nur eineiige Zwillinge sind weitgehend gleich.
Bei der sexuellen Fortpflanzung finden also ein starker Austausch und eine Rekombination der elterlichen Gene statt; damit wird die notwendige genetische Variabilität erzeugt, an der die natürliche Selektion ansetzen kann. Die Entwicklung der sexuellen Fortpflanzung war demnach ein extrem wichtiger Evolutionsschritt, ohne den die Weiter- und Höherentwicklung der Organismen nicht hätte stattfinden können.
3.3.3 Rekombination
Unter Rekombination verstehen wir den Austausch von homologen DNA-Sequenzen. Mehrere Formen der Rekombination werden unterschieden:
-
Homologe Rekombination tritt zwischen homologen DNA-Sequenzen, z. B. während der Meiose in der Prophase I (s. EXKURS 3.2) auf, die meist auf den homologen männlichen und weiblichen Chromosomen lokalisiert sind. In geringem Umfang findet Rekombination auch während der Mitose statt.
-
Ortsspezifische Rekombination durch spezifische Basenpaarung wird bei Viren beobachtet, deren DNA in die Wirtszell-DNA eingefügt wird.
-
Transpositionale Rekombination ist durch Einfügen von Transposons an Stellen ohne Sequenzhomologie gekennzeichnet.
Für die Evolution sind die Rekombinationsereignisse während der meiotischen Reduktionsteilung am wichtigsten. Während der Meiose (s. EXKURS 3.2) kommt es durch Crossing over regelmäßig zu einer gemischten Basenpaarung der jeweils homologen Gensequenzen homologer Chromosomen (◘ Abb. 3.27). Werden die Stränge dabei durchschnitten, kann ein DNA-Strang aus dem ursprünglich mütterlichen Erbgut nun mit dem homologen väterlichen Strang hybridisieren (Breakage-and-reunion-Hypothese). Dabei können große, aber auch kleine Chromosomenabschnitte (z. B. Allele) ausgetauscht werden (◘ Abb. 3.20; ◘ Abb. 3.27). Bei geschlechtlicher Vermehrung werden also Allele neu kombiniert und nach dem Zufallsprinzip auf die Chromosomen verteilt. Auf diese Weise entstehen neue Genotypen. Aus evolutionärer Sicht ist ein einzelnes Chromosom demnach nur ein Übergangszustand mit einer zeitweiligen Assoziation bestimmter Allele.

a, b. Schematische Darstellung der homologen Rekombination. a Austausch von Chromosomenabschnitten durch Crossing over und Rekombination; Beispiel Chromosomentetrade in der meiotischen Prophase I. b Crossing over und Rekombination auf der DNA-Ebene
a, b. Schematische Darstellung der homologen Rekombination. a Austausch von Chromosomenabschnitten durch Crossing over und Rekombination; Beispiel Chromosomentetrade in der meiotischen Prophase I. b Crossing over und Rekombination auf der DNA-Ebene
Fehlerhafte Hybridisierung während der Rekombination, insbesondere im Bereich repetitiver DNA (◘ Abb. 3.20), erhöht deren Variabilität. Die daraus bedingte individuelle Architektur der Genome nutzt man beim DNA-Fingerprinting oder bei der Mikrosatelliten-Analyse (s. Abschn. 4.1.2) zur Untersuchung von Paternität oder von Genfluss in Populationen.
3.4 Veränderung des Genoms während der Evolution
Aus relativ einfach gebauten Genomen, wie man sie heute noch bei einigen Bakterien sieht und in denen nahezu alle DNA-Abschnitte funktionell von Bedeutung sind, entwickelten sich im Verlauf der Evolution bei den Eukaryoten durch genetische Rekombination (s. Abschn. 3.3.3) und Mutationen (s. Abschn. 3.3.1) sehr große und komplex aufgebaute Genome (◘ Abb. 3.28), die umfangreiche nicht-codierende Bereiche aufweisen (z. B. Introns, Promotor- und Enhancerbereiche, Pseudogene und repetitive DNA). Die Erforschung der Genomstruktur ist die Domäne eines eigenen Forschungszweiges, der strukturellen oder vergleichenden Genomforschung.

Übersicht über die wichtigsten Elemente des Genoms einer tierischen Zelle
Übersicht über die wichtigsten Elemente des Genoms einer tierischen Zelle
3.4.1 Eukaryotengene mit regulatorischen Sequenzabschnitten und Intron-Exon-Struktur
3.4.1.1 Bedeutung der Exon-Intron-Struktur der Eukaryotengene
Evolutionär von Bedeutung ist die Entwicklung der Mosaikstruktur der Eukaryotengene mit Introns und Exons (◘ Abb. 3.29). Prokaryoten besitzen in der Regel keine Introns. Eine Ausnahme stellen die Archaea dar, bei denen nur wenige Gene, wie z. B. die 23S rDNA, Introns aufweisen. Wie man ◘ Abb. 3.30 entnehmen kann, steigt die Zahl der Introns, die in einem Gen vorkommen, in etwa mit der Entwicklungshöhe der Organismen.

Struktur und Transkription eukaryotischer Mosaikgene, Intron-Exon-Struktur eukaryotischer Gene, Transkription und alternatives Spleißen der mRNA
Struktur und Transkription eukaryotischer Mosaikgene, Intron-Exon-Struktur eukaryotischer Gene, Transkription und alternatives Spleißen der mRNA

Prozentualer Anteil der Zahl der Exons pro Gen bei Hefen, Insekten und Säugetieren
Prozentualer Anteil der Zahl der Exons pro Gen bei Hefen, Insekten und Säugetieren
Introns weisen eine Länge von über 1000 Nucleotiden auf, während Exons mit nur 100–300 Nucleotiden (entsprechend 30–100 Aminosäuren) deutlich kürzer sind. Durch die Intron-Exon-Struktur erhalten Eukaryotengene häufig eine Gesamtlänge von 5–40 kB. Einzelne Gene haben sogar eine Länge von mehr als 2 Mio. Basenpaaren (2000 kB): das Dystrophie-Gen (eine mutierte Form ist für die Duchenne-Muskeldystrophie verantwortlich) weist 50 Introns auf, während die eigentliche Transkriptlänge 17.000 Nucleotide beträgt. Die Intronbereiche werden nach der Transkription herausgeschnitten (RNA-splicing) (◘ Abb. 3.29). Der Spleißprozess verläuft nicht in allen Zellen nach demselben Muster: Durch alternatives Spleißen werden nicht nur die Introns, sondern auch einige der Exons entfernt. Auf diese Weise können aus einem Gen nicht nur ein einziges Protein, sondern mehrere Proteine erzeugt werden, die sich in der Zusammensetzung der Exons unterscheiden. Die Zahl der Protein-codierenden Gene liegt beim Menschen bei ca. 20.000; man schätzt jedoch, dass die Zahl der Proteine, bedingt durch den alternativen Spleißprozess zwischen 50.000 und 200.000 liegt. Durch diesen Prozess wird die phänotypische Variabilität und Plastizität der Eukaryoten zusätzlich erhöht.
Die funktionelle Analyse der Proteine hat gezeigt, dass man in einem Protein häufig mehrere funktionelle Bereiche, die Domänen, unterscheiden kann, z. B. Domänen für aktive katalytische Zentren, für Transmembranbereiche oder Bindungsstellen. Ein Protein wird in der Regel von mehreren Exons codiert, wobei die Exons häufig speziellen funktionellen Domänen entsprechen. Die nicht-codierenden Introns fungieren demnach als Spacer. Bei einigen Genen (unterschiedlicher Organismen) findet man Introns an identischen Genpositionen, so z. B. im Triosephosphat-Isomerase-Gen von Mensch und Mais. Dieses Phänomen deutet daraufhin, dass Introns recht alte genetische Erfindungen sein müssen, denn bei späterer Insertion hätte man Introns an unterschiedlichen Stellen erwarten müssen.
Man nimmt heute an, dass im Verlauf der frühen Evolution zunächst einmal die Struktur einzelner Domänen optimiert wurde, die nur jeweils eine oder wenige Funktionen ausübten. Diese Grundbausteine (Module) wurden in der späteren Evolution bei den Eukaryoten offensichtlich so miteinander kombiniert, dass zusammengesetzte Proteine, Enzyme oder Rezeptoren mit neuen und mehreren Funktionen entstanden (◘ Abb. 3.31). Dieser Prozess wurde von W. Gilbert 1979 als Exon-Shuffling bezeichnet. Die mannigfaltigen neuen Möglichkeiten, die sich aus einer solchen Kombinatorik ergeben, lassen sich gut am Beispiel der Antikörpergene der Säugetiere (über 1 Mio. Antikörper können in einem einzigen Individuum erzeugt werden) oder der variablen Oberflächenantigene der Trypanosomen und anderer Parasiten erkennen, mit denen das Immunsystem der Wirte überlistet wird.

Beispiele für Domänenkombinatorik. In bakteriellen lac- und cro-Repressorproteinen findet man typische DNA-Bindungsdomänen, die ebenfalls als Modul im bakteriellen Katabolit-Aktivator-Protein (CAP-Protein) vorkommen. Zusätzlich enthält das CAP-Protein auch eine Bindungsdomäne für cyclisches AMP (cAMP), die bei Proteinkinasen mehrfach vorkommen kann. Chymotrypsin ist eine einfache Protease mit einer Serinprotease-Domäne; diese Domäne kommt als Modul in spezialisierten eukaryotischen Proteasen, wie der Urokinase, dem Faktor IX und im Plasminogen vor. Diese Proteasen enthalten zusätzliche Module, z. B. des epidermalen Wachstumsfaktor (EGF), von Calcium-Bindungsstellen und des kringle-Proteins, das drei interne Disulfidbrücken trägt
Beispiele für Domänenkombinatorik. In bakteriellen lac- und cro-Repressorproteinen findet man typische DNA-Bindungsdomänen, die ebenfalls als Modul im bakteriellen Katabolit-Aktivator-Protein (CAP-Protein) vorkommen. Zusätzlich enthält das CAP-Protein auch eine Bindungsdomäne für cyclisches AMP (cAMP), die bei Proteinkinasen mehrfach vorkommen kann. Chymotrypsin ist eine einfache Protease mit einer Serinprotease-Domäne; diese Domäne kommt als Modul in spezialisierten eukaryotischen Proteasen, wie der Urokinase, dem Faktor IX und im Plasminogen vor. Diese Proteasen enthalten zusätzliche Module, z. B. des epidermalen Wachstumsfaktor (EGF), von Calcium-Bindungsstellen und des kringle-Proteins, das drei interne Disulfidbrücken trägt
Man kann das Exon-Shuffling mit dem Fertighausprinzip vergleichen. Wenn man alle Elemente eines Hauses vorgefertigt hat, ist es relativ einfach und wenig zeitaufwendig, eine Vielzahl von unterschiedlichen Häusern zu bauen. Fängt man dagegen mit einzelnen Ziegeln an, so ist der Prozess wesentlich aufwendiger und dauert länger. Ähnlich war es wohl in der Evolution: Nur durch Abänderung eines ursprünglichen Gens durch Punktmutationen hätte es sehr lange gedauert, bis ein neues Gen entstanden wäre, das für ein Protein mit mehreren funktionellen Domänen codiert. Durch Verwendung und Kombination der „vorgefertigten“ Exonmodule konnten sprunghaft Gene mit neuen Eigenschaften geschaffen werden.
3.4.1.2 Bedeutung regulierbarer Genaktivitäten
Bereits auf der Stufe der Prokaryoten sind die meisten Strukturgene mit regulierbaren Promotoren ausgestattet, die es einem Organismus erlauben, Gene nur dann anzuschalten, wenn die Genprodukte benötigt werden. Bei den Eukaryoten ist die Genregulation noch wesentlich stärker weiterentwickelt. Von den vielen vorhandenen Genen des Genoms ist nur ein kleiner Teil in allen Zellen aktiv (house-keeping genes). Viele Gene werden nicht nur zell- und gewebespezifisch exprimiert, sondern auch noch entwicklungsspezifisch. Nur das korrekte An- und Abschalten der Gene während der Embryogenese (Epigenetik, s. EXKURS 5.9 Abschn. 5.7), der Gewebe- und Zelldifferenzierung, der Jugendzeit und im Erwachsenstadium führt zu Individuen mit angepasstem und artspezifischem Phänotyp. Die Steuerung erfolgt über spezifische Transkriptionsfaktoren und enhancer- bzw. silencer-Proteine (◘ Abb. 3.6). Nur wenn diese Proteine an die regulatorischen Genabschnitte binden, kann die RNA-Polymerase die zugehörigen Gene transkribieren (◘ Abb. 3.4).
Viele Gene der heutigen Organismen entstanden bereits in der frühen Evolution der Pro- und Eukaryoten. Sie umfassen sowohl Gene, die Enzyme, Rezeptoren und andere Elemente des Zellstoffwechsels und der Zellstruktur codieren, als auch Bauplan- und Entwicklungsgene, die eine entwicklungsspezifische Morphogenese steuern (s. EXKURS 3.3).
Einmal entstandene Gene oder Genkomplexe wurden im Verlauf der Evolution meist beibehalten (wenn auch nicht notwendigerweise exprimiert) und standen als Bausteine in späteren Evolutionsphasen zur Verfügung, da die Genome ja von Generation zu Generation weitervererbt werden. Der vorhandene Genpool lässt sich durch Kombinatorik insbesondere auf der Ebene der Genregulation vielfältig variieren. Die DNA-Sequenz im Promotorbereich eines Gens kann durch Punkt- oder andere Mutationen so geändert werden, dass andere oder neue Transkriptionsfaktoren binden können. Auf diese Weise würde dieses Gen zu einem Zeitpunkt oder in einem Organ aktiviert, wo es früher abgeschaltet gewesen wäre. Ebenso können Sequenzabschnitte, an die spezifische Transkriptionsfaktoren binden, als DNA-Elemente vor vorhandene Gene inseriert werden (z. B. durch Transposons) und so neue Expressionsmuster hervorrufen. Auch auf diese Weise wird die phänotypische Variabilität und Plastizität gefördert.
Das Arbeiten mit Modulen oder vorgefertigten Bausätzen findet man in der Evolution an vielen Stellen; selbst die Ausbildung komplexer morphologischer Strukturen ist offenbar modular geordnet und durch wenige Schritte, z. B. durch An- oder Abschalten von Kontroll- oder Mastergenen ursprünglich angelegter Gencluster regulierbar. Beispielsweise führt bei der Ackerschmalwand (Arabidopsis thaliana) oder beim Löwenmäulchen der Austausch eines einzigen Mastergens dazu, dass die Blüte von einer radiären zu einer zygomorphen Form oder umgekehrt verändert wird. Bei A. thaliana zeigen die homöotischen apetala-Mutanten unterschiedliche Maße der Ausbildung von Staub-, Kron- und Fruchtblättern. Diese Kombinationsebene, d. h. das Umschalten von einem komplexen Phänotyp zu einem anderen durch An- und Abschalten weniger Kontroll- und Bauplangene (Morphogene), wird experimentell bald in einem größeren Maße zugänglich sein als heute. Vermutlich werden wir dann besser verstehen können, wie durch „Makromutationen“ komplexe Strukturen relativ schnell entstehen konnten.
Auch bei künstlicher Selektion, wie sie bei Haustieren und Kulturpflanzen stattfindet, lässt sich diese Evolutionsebene erkennen. Man denke an die vielen, oft ungewöhnlichen Formen von Hunden, Katzen, Tauben oder Hühnern, die in vergleichsweise kurzer Zeit (meist innerhalb der letzten 5000–10.000 Jahre) selektiert wurden (s. Abschn. 5.8.3), oder an die verschiedenen Kohlsorten, die aus der Wildform Brassica oleracea entstanden (◘ Abb. 3.32).

a–i. Sprunghafte Veränderung einer Wildform durch Züchtung, am Beispiel von Kohl Brassica oleracea, den man auch heute als Wildform noch an der deutschen Nordseeküste finden kann, z. B. auf Helgoland. Schematische Illustration der Merkmalsvariation in den kultivierten Kohlsorten: a Wildform; b Weißkohl, c Rotkohl, d Grünkohl, e Blumenkohl, f Brokkoli, g Markstammkohl, h Kohlrabi, i Rosenkohl
a–i. Sprunghafte Veränderung einer Wildform durch Züchtung, am Beispiel von Kohl Brassica oleracea, den man auch heute als Wildform noch an der deutschen Nordseeküste finden kann, z. B. auf Helgoland. Schematische Illustration der Merkmalsvariation in den kultivierten Kohlsorten: a Wildform; b Weißkohl, c Rotkohl, d Grünkohl, e Blumenkohl, f Brokkoli, g Markstammkohl, h Kohlrabi, i Rosenkohl
Fände man die verschiedenen Haustier- und Kulturpflanzenvarietäten in einer Museumssammlung ohne eine Kenntnis ihrer Vorgeschichte, so würde man vermutlich eine größere Anzahl von distinkten Arten beschreiben und nicht auf die Idee kommen, dass es sich nur um junge Varietäten weniger Arten handelt. Charles Darwin hatte eine große Sammlung von verschiedenen Taubenrassen angelegt. Als Charles Lyell ihn im April 1856 besuchte, legte ihm Darwin Bälge von 15 Taubenrassen vor, die so unterschiedlich aussahen, dass „three good genera, and about 15 species according to the received mode of species and genera making of the best ornithologists“ (drei gute Gattungen und mindestens 15 Arten entsprechend dem Art- und Gattungskonzept der besten Ornithologen) plausibel gewesen wären.
Ein solches Variieren von Bauplan- und Entwicklungsgenen (insbesondere von Hox-Genen und Genen des Wnt-Signalweges) hat sicherlich zu einer schnellen Evolution auf morphologischer Ebene beigetragen (man könnte sie auch „Makromutationen“ nennen) (s. EXKURS 3.3). Wenn ein schneller Wechsel der Morphologie in erdgeschichtlich neuerer Zeit erfolgte, ist er auf der Ebene der Markergen-Sequenzen (s. Abschn. 4.1.2) meist nicht zu sehen. Mit anderen Worten, bei jungen Ereignissen kann sich das sichtbare Tempo zwischen morphologischer und molekularer Evolution deutlich unterscheiden. In anderen Fällen beobachtet man eine Konstanz der Baupläne über viele Jahrmillionen hinweg, obwohl diese Arten weiterhin der molekularen Evolution unterlagen. Diese werden als „lebende Fossilien“ bezeichnet (s. Abschn. 2.4.2).
Umformung und Neukombination von Vorhandenem ist offenbar ein wichtiges Evolutionsprinzip, das von dem französischen Molekularbiologen François Jacob als tinkering (spielen, basteln) bezeichnet wurde. Durch diese Prozesse verlief die Evolution natürlich schneller als durch Veränderung der Proteine über einfache Punktmutationen. Man kann sich so eher jene sprunghaften morphologischen Veränderungen vorstellen, die bei Fossilien oft beobachtet wurden.
3.4.1.2.1 EXKURS 3.3
3.4.1.2.1.1 Evo-Devo-Forschung
Detlev Arendt und Thomas Holstein (Heidelberg)
Ontogenie und Phylogenie
Die Evolution vielzelliger Organismen erschließt sich durch die vergleichende Analyse ihrer Entwicklung. Dieser fundamentale Zusammenhang zwischen Evolution und Entwicklung spiegelt sich in der Bezeichnung einer sehr lebhaften neuen Forschungsrichtung – „Evo-devo“ – wieder (Gould 1977; Riedl 1977; Raff u. Kaufman 1983; Schlosser u. Wagner 2004; Kirschner u. Gerhart 2007). In seiner Bedeutung und Tragweite zuerst erfasst wurde dieser Zusammenhang in der „biogenetischen Grundregel“ von Ernst Haeckel.
„Die Ontogenie rekapituliert die Phylogenie“ erläuterte Haeckel 1874 am Beispiel der Gastrula, die er als Rekapitulation einer urtümlichen Metazoen-Stammart, der Gastraea, verstand (◘ Abb. 3.33). Wie erklärt sich dieser Zusammenhang? In Anlehnung an Haeckel verändern sich Tierarten in der Evolution durch eine Abwandlung ihrer Entwicklung, entweder durch Hinzufügen neuer Entwicklungsschritte an das bestehende Programm oder durch Veränderung desselben. Ersteres erklärt das Phänomen der „Rekapitulation“, letzteres macht begreiflich, warum nicht jeder Weg (und Umweg) der Evolution in der heutigen Entwicklung Niederschlag findet: „In der Tat existiert immer ein gewisser Parallelismus (zwischen Entwicklung und Evolution). Aber dieser wird dadurch verwischt, dass meistens in der ontogenetischen Entwicklungsfolge vieles fehlt und verloren gegangen ist, was in der phylogenetischen Entwicklungskette früher existiert und wirklich gelebt hat“ (Haeckel 1874).

Ontogenie und Phylogenie. Die obere Reihe zeigt die hypothetische Entstehung eines Gastrea-ähnlichen Organismus in der frühen Evolution der Metazoen. Hier kam es zu einer Trennung von somatischen und reproduktiven Zellen und zur Entstehung eines eingesenkten Verdauungsraums (Urdarm). Die untere Reihe zeigt die Rekapitulation dieses frühen Stadiums in der Evolution, die von allen Tiere in mehr oder weniger stark abgewandelter Form in der Gastrulation durchlaufen wird (Stern 2004, Bildrechte liegen bei CSHL Press)
Ontogenie und Phylogenie. Die obere Reihe zeigt die hypothetische Entstehung eines Gastrea-ähnlichen Organismus in der frühen Evolution der Metazoen. Hier kam es zu einer Trennung von somatischen und reproduktiven Zellen und zur Entstehung eines eingesenkten Verdauungsraums (Urdarm). Die untere Reihe zeigt die Rekapitulation dieses frühen Stadiums in der Evolution, die von allen Tiere in mehr oder weniger stark abgewandelter Form in der Gastrulation durchlaufen wird (Stern 2004, Bildrechte liegen bei CSHL Press)
3.4.1.2.1.1.1 Homologie
Wie lässt sich unterscheiden, welche Entwicklungsschritte die Evolution rekapitulieren und welche im Verlauf derselben abgewandelt worden sind? Diese Unterscheidung ist Ziel der vergleichenden Evolutionsforschung, welche die Entwicklungsgänge möglichst vieler Arten vergleicht und nach Gemeinsamkeiten sucht. Wenn sich zwei Arten in bestimmten Entwicklungsschritten im Detail ähneln, besteht der Verdacht, dass diese Ähnlichkeit auf Vererbung beruht und als Homologie gedeutet werden kann. Homologe Merkmale zweier Arten lassen sich auf dasselbe Vorläufermerkmal in der letzten gemeinsamen Stammart zurückführen und sind am besten an morphologischen und/oder molekularen Gemeinsamkeiten zu erkennen, die sich plausibel nur durch ein gemeinsames Erbe erklären lassen. Die Evo-devo-Forschung ist also zu einem großen Teil Homologieforschung, die Entwicklungsgänge verschiedener Organismen vergleicht und nach Gemeinsamkeiten und Unterschieden abklopft. Darüber hinaus stellt sie einen Ansatz dar, um die Mechanismen der Makro-Evolution (Evolution von Bauplänen) aufzuklären (Schlosser u. Wagner 2004). Ihre momentane Blüte erklärt sich dadurch, dass den vergleichenden Entwicklungsbiologen ein ganzer neuer Werkzeugkasten in die Hand gedrückt wurde, der ihnen völlig neue Ebenen des Vergleichs erschlossen hat: Evo-devo ist heute im Wesentlichen eine molekulare Disziplin, welche die Methoden der Gentechnik für sich nützt. Haeckels Ziele werden heute mit molekularen Werkzeugen verfolgt.
3.4.1.2.1.1.2 Orthologe und paraloge Gene
Grundlage eines jeden Vergleichs auf molekularer Ebene ist die Analyse der Verwandtschaft der beteiligten Gene.
Vergleicht man die Entwicklung von Organismen auf molekularer Ebene, muss zunächst die Homologie der beteiligten Gene untersucht werden. Somit ist der bioinformatische Vergleich von Molekülen ein Grundpfeiler moderner Evo-devo-Forschung. Durch das Alinieren der DNA- und Proteinsequenzen wird zunächst ermittelt, ob eine Verwandtschaft zwischen Molekülen besteht. Als nächstes wird geprüft, welcher Art diese Verwandtschaft ist. Mit Hilfe der Sequenzalinierungen und durch das Errechnen von Genstammbäumen werden Gene und Proteine in Familien und „Überfamilien“ eingeteilt. Genfamilien sind in der Evolution durch Genduplikation, also durch die Verdoppelung einzelner Gene, Chromosomenabschnitte oder ganzer Genome, entstanden. Oftmals findet man viele Duplikate eines ursprünglichen Gens innerhalb eines Genoms. Beim Vergleich von Genen ist es daher besonders wichtig, zunächst die „serielle Homologie“ duplizierter Gene innerhalb eines Genoms zu erkennen und zu berücksichtigen. Paraloge Gene sind durch Genduplikation und anschließende Diversifizierung entstanden. Man findet sie innerhalb einer Evolutionslinie; wenn das Duplikationsereignis jedoch weit genug zurückreicht, existieren dieselben verdoppelten paralogen Gene auch in den Genomen weiterer Organismen, welche sie ebenfalls geerbt haben. Orthologe Gene hingegen werden stets zwischen zwei Spezies identifiziert. Es handelt sich um Gene, die auf dasselbe Vorläufergen im letzten gemeinsamen Vorfahren zurückzuführen sind, deren Sequenzdivergenz also im Wesentlichen auf phylogenetischer Distanz beruht. Die Unterscheidung von paralogen und orthologen Gene kann man am besten mit Hilfe von Genstammbäumen verstehen, wie in ◘ Abb. 3.34 illustriert. Eine Komplikation ergibt sich, wenn zwei zwischen zwei Spezies nahe verwandte Gene auf zwei unterschiedliche Paraloge (Schwestergene) des letzten gemeinsamen Vorfahren zurückgehen. Diese Gene sind nicht ortholog!

Paraloge und orthologe Gene. Am Beispiel der Evolution der Opsin-Genfamilie wird der Unterschied von paralogen Genen (z. B. GPR op11 und 12) und von orthologen Genen (z. B. r-Opsin und c-Opsine) deutlich (für weitere Details siehe Text)
Paraloge und orthologe Gene. Am Beispiel der Evolution der Opsin-Genfamilie wird der Unterschied von paralogen Genen (z. B. GPR op11 und 12) und von orthologen Genen (z. B. r-Opsin und c-Opsine) deutlich (für weitere Details siehe Text)
3.4.1.2.1.1.3 Die Bedeutung konservierter Genfunktionen
Einen wichtigen Einblick in die Entwicklung und die organismische Komplexität unserer Vorfahren erhält man bereits durch die Rekonstruktion ihres Genoms und ihres Proteoms (der Gesamtheit der exprimierten Proteine) sowie der damit verbundenen Molekülfunktionen.
Ein zunehmend wichtiger Aspekt der vergleichenden Genomforschung ist die Rekonstruktion von Genom (und Proteom) bestimmter Schlüsselarten, wie z. B. der letzten gemeinsamen Stammart der Bilaterier und der Cnidarier (Eumetazoa), durch die vergleichende Analyse sequenzierter Genome. Beispielweise wissen wir heute durch den Vergleich des Genoms des Anthozoenpolyps Nematostella mit Wirbeltier- und Polychaetengenomen, dass die Stammart der Eumetazoa bereits 12 paraloge Liganden der Wnt-Familie besaß (Kusserow et al. 2005; Holstein 2012) (◘ Abb. 3.35a). Diese Tatsache allein ist von großer Wichtigkeit für unser Verständnis dieser Stammart, denn diese unterschiedlichen Liganden stehen für verschiedene Induktions- und Signaltransduktionsereignisse während der Entwicklung, was auf eine unerwartet hohe Komplexität des Polypen und damit der Stammart der Eumetazoa hinweist. Einen Einblick in die möglicherweise ursprünglichen Funktionen dieser Wnt-Gene liefert die Analyse ihrer gestaffelten Expression entlang der Körperlängsachse des Cnidarierpolypen (◘ Abb. 3.35b).

Evolution und basale Funktion der Wnt-Genfamilie. Vergleiche zwischen den an der Basis der Metazoenevolution stehenden Cnidaria und den restlichen Metazoen zeigen, dass es in der Evolution zu einem Genverlust in mehreren Großgruppen kam. Die Vertebraten zeichnen sich dagegen durch einen kompletten Satz von Wnt-Genen aus, die sie mit den Cnidariern und den Ur-Eumatazoen, den gemeinsamen Vorfahren aller Tiere, teilen. Die Wnt-Gene werden während der Gastrulation am Blastoporus in der Planulalarve in einer gestaffelten Abfolge vom oralen zum aboralen Pol exprimiert („Wnt-Code“) (aus: Kusserow et al. 2005, Bildrechte liegen bei Nature)
Evolution und basale Funktion der Wnt-Genfamilie. Vergleiche zwischen den an der Basis der Metazoenevolution stehenden Cnidaria und den restlichen Metazoen zeigen, dass es in der Evolution zu einem Genverlust in mehreren Großgruppen kam. Die Vertebraten zeichnen sich dagegen durch einen kompletten Satz von Wnt-Genen aus, die sie mit den Cnidariern und den Ur-Eumatazoen, den gemeinsamen Vorfahren aller Tiere, teilen. Die Wnt-Gene werden während der Gastrulation am Blastoporus in der Planulalarve in einer gestaffelten Abfolge vom oralen zum aboralen Pol exprimiert („Wnt-Code“) (aus: Kusserow et al. 2005, Bildrechte liegen bei Nature)
Die Entdeckung eines Östrogenrezeptormoleküls beim Seehasen Aplysia, einer Gattung der Weichtiere, ist ein weiteres Beispiel für die Aussagekraft von Proteomvergleichen (Thornton et al. 2003). Aus der Existenz dieses Moleküls im Seehasen und seiner (noch unvollständigen) funktionellen Analyse können wir schließen, dass die Stammart der Bilaterier, der Urbilater, bereits einen solchen Steroidhormonrezeptor besaß. Das bedeutet wiederum, dass ein östrogenähnliches Hormon bereits vorhanden und wirksam war, was man bislang für eine Spezialität der Wirbeltiere gehalten hatte. Das Östrogensystem kann in der Entwicklung oder in der adulten Physiologie dieser Stammart eine Rolle gespielt haben.
Der Transkriptionsfaktor Pax6 ist das wohl berühmteste Beispiel für ein innerhalb der Bilaterier konserviertes Protein und für aus der vergleichenden funktionellen Analyse gewonnene spektakuläre Schlussfolgerungen. Das Pax6-Gen ist durch die Augenmutante Aniridia in der Maus zuerst als Schlüsselgen der Augenentwicklung bei Wirbeltieren identifiziert worden, bis man herausfand, dass der Verlust der Genfunktion des orthologen Gens bei der Taufliege Drosophila zum vollständigen Verlust der Augen führt (Quiring et al. 1994). Bei Drosophila lässt sich durch die ortsfremde (ektopische) Expression von Pax6 auch an anderen Körperstellen ein Auge erzeugen, was die zentrale Rolle dieses Gens bei der Augenentwicklung noch unterstreicht. Diese Ergebnisse erlauben den Schluss, dass der Urbilater bereits ein Pax6-Gen besaß, und sie machen es sehr wahrscheinlich, dass die Funktion des urtümlichen Pax6-Transkriptionsfaktors auch dort bereits mit der Augenentwicklung zu tun hatte. Letzteres wiederum setzte voraus, dass der Urbilater bereits Augen besaß – die eigentliche Überraschung dieser Studie.
3.4.1.2.1.1.4 Konservierte Signalkaskaden bei der Bildung der Körperachsen im Tierreich
Die wichtigste Entdeckung der vergangenen Jahrzehnte ist die gemeinsame Verwendung homologer Signalkaskaden bei der embryonalen Achsenbildung völlig unterschiedlicher Tiergruppen, von Wirbeltieren bis zu Insekten oder Polypen, gewesen. Diese überraschende Gemeinsamkeit ermöglicht den Vergleich von Körperachsen durch das gesamte Tierreich und erlaubt Rückschlüsse auf die Evolution dieser Achsen vor Hunderten von Jahrmillionen.
Nachdem Lewis in einer bahnbrechenden Arbeit die Musterbildung entlang der Körperlängsachse der Taufliege durch die Aktivität der Hox-Gene erklärt hatte, kam es einer Revolution gleich, dass dieselbe homologe Genkassette auch bei Wirbeltieren entlang der Körperlängsachse aktiv sein sollte. Zum ersten Mal ergab sich die Möglichkeit, die Bildung von Körperachsen und damit auch die Körperachsen selbst zu homologisieren. Die konservierte Rolle der Hox-Gene bei Fliege und Maus machte eine Homologie der Körperlängsachse von Fliege und Maus unausweichlich (◘ Abb. 3.36), was im Zootyp-Konzept Niederschlag fand (Slack et al. 1993). Dieses Konzept deutet die Hox-Genkassette als eine Synapomorphie bilateralsymmetrischer Tiere. Auch die Wnt-Gene sind primär in der Entwicklung und Evolution entlang dieser Achse exprimiert (Holstein 2012).

Dorsoventrale Achsenumkehr zwischen Vertebraten und Arthropoden. Der inversen Anlage des Nervensystems (hell-grünlich) und Blutgefäßsystems (schwarz) bei Arthropoden und Vertebraten entlang der Dorsoventral-Achse entspricht die antagonistische Expression und Funktion von dpp (BMP-4) und sog (Chordin) bei Arthropoden und Vertebraten in der Blastula (Details siehe Text) (aus: Wolpert 2002, Bildrechte liegen bei Oxford University Press)
Dorsoventrale Achsenumkehr zwischen Vertebraten und Arthropoden. Der inversen Anlage des Nervensystems (hell-grünlich) und Blutgefäßsystems (schwarz) bei Arthropoden und Vertebraten entlang der Dorsoventral-Achse entspricht die antagonistische Expression und Funktion von dpp (BMP-4) und sog (Chordin) bei Arthropoden und Vertebraten in der Blastula (Details siehe Text) (aus: Wolpert 2002, Bildrechte liegen bei Oxford University Press)
Die Überraschung steigerte sich noch, als auch für die Bildung der Dorsoventralachse (Rücken-Bauch-Achse) der Insekten und Wirbeltiere eine konservierte Genkassette ins Spiel gebracht wurde, die allerdings in beiden Tiergruppen in umgekehrter Orientierung aktiv ist (◘ Abb. 3.36). Unverhofft verhalf dieser Befund einem jahrhundertealten Konzept zum Durchbruch, das die Rückenseite samt Neuralrohr der Wirbeltiere mit der Bauchseite samt Bauchmark der Insekten homologisierte und eine dorsoventrale Achsenumkehr in der frühen Evolution der Wirbeltiervorfahren postulierte (Arendt u. Nübler-Jung 1994). Molekular handelt es sich bei dieser Genkassette um ein System zweier gegenläufig wirkender Signalmoleküle namens Decapentaplegic (Dpp) und Chordin, das inzwischen auch in Polypen identifiziert werden konnte (Holstein et al. 2011) und damit erstmalig erlaubt, die Entstehung der Dorsoventralachse durch das gesamte Tierreich zu verfolgen.
3.4.1.2.1.1.5 Abgleich der molekularen Fingerabdrücke von Zelltypen
Diese jüngste Variante der Evo-devo-Forschung identifiziert homologe Zelltypen zwischen weit entfernten Tiergruppen aufgrund ihres molekularen Fingerabdrucks und zeichnet die Diversifizierung von Zelltypen in der Evolution der Tiere nach.
Ein Zelltyp repräsentiert eine molekular und morphologisch homogene Population von Zellen, in denen eine einzigartige Kombination von Transkriptionsfaktoren und nachgeschalteten Effektorgenen eine zelltypspezifische Differenzierung bewirkt. Diese Kombination zelltypspezifischer Gene wird als molekularer Fingerabdruck bezeichnet. Der Abgleich molekularer Fingerabdrucke zwischen weit entfernten Tierstämmen hat ergeben, dass Zelltypen ein hoch konserviertes Merkmal darstellen, deren Evolution über weite evolutive Distanzen nachgezeichnet werden kann. So konnten konservierte Motoneuronentypen zwischen Insekten, Nematoden und Wirbeltieren identifiziert werden (Thor u. Thomas 2002), was eine entscheidende Voraussetzung für ein tieferes Verständnis funktioneller Gemeinsamkeiten der zentralen Nervensysteme ist (wie z. B. der nervösen Steuerung des Bewegungsapparats). Ein weiteres Beispiel ist die Entdeckung einer besonderen Form von cilientragenden Lichtsinneszellen im Gehirn der Polychaeten, die durch den Abgleich der molekularen Fingerabdrücke als homolog mit den ebenfalls ciliären Stäbchen und Zäpfchen der Wirbeltierretina identifiziert werden konnten (Arendt et al. 2004) (◘ Abb. 3.37). Dieses sind erste Belege für die Tragfähigkeit eines Ansatzes, der die Evolution komplexer Organsysteme der Metazoen wie z. B. des Nervensystems als eine Abfolge von Differenzierungsereignissen von Zelltypen nachzuzeichnen versucht.

Molekularer Fingerabdruck. Am Beispiel ciliärer Lichtsinnesorgane bei Polychaeten kann gezeigt werden, dass Polychaeten (oben) und Vertebraten (unten) homologe cilliäre Zelltypen besitzen (Details siehe Text). Purpur: ciliäre Photorezeptorzellen; gelb: rhabdomere Photorezeptorzellen und retinale Ganglionzellen; grün: photoperiodisch aktive Neuronen; grau: Pigmentzellen. Abkürzungen: ae: adultes Auge; cPRC: ciliärer Photorezeptor; le: larvales Auge; nsc: Nucleus suprachiasmaticus; pin: Pinealorgan; ret: Retina; RGC: retinale Ganglionzelle; rPRC: rhabdomerer Photorezeptor. Sterne zeigen die Lage des Apical organs (aus: Arendt et al. 2004, Bildrechte liegen bei AAAS)
Molekularer Fingerabdruck. Am Beispiel ciliärer Lichtsinnesorgane bei Polychaeten kann gezeigt werden, dass Polychaeten (oben) und Vertebraten (unten) homologe cilliäre Zelltypen besitzen (Details siehe Text). Purpur: ciliäre Photorezeptorzellen; gelb: rhabdomere Photorezeptorzellen und retinale Ganglionzellen; grün: photoperiodisch aktive Neuronen; grau: Pigmentzellen. Abkürzungen: ae: adultes Auge; cPRC: ciliärer Photorezeptor; le: larvales Auge; nsc: Nucleus suprachiasmaticus; pin: Pinealorgan; ret: Retina; RGC: retinale Ganglionzelle; rPRC: rhabdomerer Photorezeptor. Sterne zeigen die Lage des Apical organs (aus: Arendt et al. 2004, Bildrechte liegen bei AAAS)
3.4.1.2.1.1.6 Synthese der Evolutionsforschung: Evo-devo und Populationsbiologie
Neben der Evo-devo-Forschung ist die Populationsgenetik ein weiterer klassischer Ansatz der Evolutionsbiologie. Die Populationsbiologie betont vor allem die Variationen innerhalb einer Art. Sie untersucht, unter welchen Bedingungen sich manche dieser Variationen unter natürlicher Selektion effektiv vermehren (◘ Abb. 3.38). Sie erklärt nicht, wie es zur Entstehung neuer Strukturen kommt.

Evo-devo. Aus Populationsgenetik und Entwicklungsgenetik ist es in den letzten Jahren zu einer neuen evolutionsbiologischen Synthese gekommen (mod. nach Gilbert 2003)
Evo-devo. Aus Populationsgenetik und Entwicklungsgenetik ist es in den letzten Jahren zu einer neuen evolutionsbiologischen Synthese gekommen (mod. nach Gilbert 2003)
Die Entwicklungsgenetik untersucht hingegen, wie es durch Expression von regulatorischen Genen zu Morphogenese, Organbildung und Zelldifferenzierung kommt. Dieser Ansatz erklärt die Entstehung neuer Strukturen im Kontext von Zwängen, welche die Variation einschränken (evolutionary novelties und constraints). Gemeinsam eröffnen beide Ansätze einen umfassenden genetischen Ansatz, um die Mechanismen der Evolution zu verstehen.
3.4.2 Genomduplikationen und Evolution von Multigenfamilien
Die Genomgröße der Tiere hat sich im Verlauf der Evolution vermutlich durch wiederholte Duplikationen des Genoms stark erhöht (◘ Abb. 3.12). Protostomia und die Deuterostomia-Vorfahren (s. Abschn. 4.2) enthielten in der Regel nur eine Ausführung eines Gens, während man in den Genomen der Chordaten meist mehrere Kopien eines Gens vorfindet. Es wird deshalb angenommen, dass die Chordatengenome mindestens zweimal verdoppelt wurden (1-2-4-Regel). Die erste Genomduplikation während der Evolution der Chordaten erfolgte bereits vor der kambrischen Explosion (s. Abschn. 2.2.1), während die zweite und nächste Verdopplung (also auf vier Kopien) im frühen Devon stattfand. In der Evolution der Fische erfolgte im späten Devon, nachdem sich bereits die Sarcopterygii abgetrennt hatten, eine weitere Verdopplung des Genoms auf acht Kopien (1-2-4-8-Hypothese) (Wittbrodt et al. 1998, Robinson-Rechavi et al. 2004). Zu den Sarcopterygii gehören die Quastenflosser und Lungenfische. Aus ihnen gingen alle Landwirbeltiere (Amphibien, Reptilien, Vögel und Säuger) hervor ( Kap. 2 und Abschn. 4.2).
Auch bei der Evolution der Angiospermen spielen Genomduplikationen (Polyploidisierung) eine sehr große Rolle (Soltis u. Soltis 2009). Man unterscheidet zwischen Allopolyploidisierung (nach Hybridisierung verschiedener Arten) und Autopolyploidisierung (Verdopplung ohne vorherige Artkreuzung). Polyploidisierung trat zu Beginn der Angiospermenentwicklung auf und wiederholte sich in vielen Pflanzenfamilien. Eine Polyploidisierung kann durch Wegfall der meiotischen Reduktionsteilung auftreten, indem diploide Gameten entstehen, die zu tetraploiden Zygoten verschmelzen (Autopolyploidie). Eine andere Entstehungsweise der Polyploidie beruht auf der Hybridisierung zweier Arten (Allopolyploidie). Hybridisierung war offenbar eine treibende Kraft in der Pflanzenevolution, die zur großen Biodiversität der Landpflanzen geführt hat.
Polyploidisierung findet man häufiger bei Pflanzen als bei Tieren. Bei Pflanzen sieht man in den meisten Familien polyploide Chromosomensätze, die einem Mehrfachen des diploiden Chromosomensatzes entsprechen. Viele Tiere sind diploid: Ausnahmen im Tierreich sind zwittrige Oligochaeten, Turbellarien und Gastropoden sowie parthenogenetische Arten (einige Coleopteren, Lepidopteren und Garnelen). Aber auch innerhalb eines ansonsten diploiden Organismus können einzelne Zelltypen polyploid sein: Beispiel für polyploide Zellen beim Menschen sind die Megakaryocyten und Urothelzellen (nur obere Zellschichten). Da in den polyploiden Zellen alle Chromosomen funktionell sind, erhöht sich in diesen Zellen die Transkriptionsrate, da mehr Genkopien vorhanden sind als im diploiden Genom.
Polyploidie stört besonders, wenn ein XY-XX-Mechanismus der genotypischen Geschlechtsbestimmung vorliegt, da die XY-Gleichgewichte in der Zygote gestört werden. Bei Fischen und Amphibien, deren Mechanismus der Geschlechtsbestimmung weniger streng ist, tritt Polyploidisierung regelmäßig auf. Bei einigen Amphibien existieren heute noch diploide und tetraploide Arten in derselben Gattung nebeneinander. Selbst unter Säugetieren existieren polyploide Arten, z. B. bei der argentinischen Kammratte (Familie Octodontidae). Auch die stark schwankenden Chromosomenzahlen der Vertebraten und Invertebraten (z. B. bei Lycaenidae; Lepidoptera) (◘ Tab. 3.10) deuten darauf hin, dass Chromosomenduplikation und Polyploidisierung in der Evolution auftraten.
Verdoppelte Chromosomensätze (z. B. von Hybriden) tragen aber eine verdoppelte genetische Information, die evolutionär genutzt werden kann. Einzelne Allele werden durch Mutation abgeschaltet (sogenannte Null-Allele), oder aber die duplizierten Gene durchlaufen eine divergente Evolution und erwerben neue Eigenschaften. Meist werden sie dann entwicklungs- und gewebsspezifisch unterschiedlich exprimiert. Letztlich wird die Funktionalität des diploiden Chromosomensatzes durch diese Mechanismen wiederhergestellt (Diploidisierung).
Die wiederholten Gen- und Genomverdopplungen eröffneten die Möglichkeit, mit den zusätzlich geschaffenen Genkopien neue Wege zu gehen, da diese nicht unmittelbar zum Überleben gebraucht wurden. Aus den Genkopien entwickelten sich durch Punktmutationen und Genmutationen Gene mit neuen Eigenschaften, die für Proteine mit meist ähnlichen Eigenschaften codierten. Auf diese Weise entstanden in der Evolution Genfamilien. In ◘ Abb. 3.39 ist ein molekularer Stammbaum der Proteinkinasefamilie dargestellt, der zeigt, wie aus einem ursprünglichen Protein eine Vielzahl von funktionellen Proteinkinasen entstehen konnte, die sich heute in diverse Unterfamilien aufteilen.

a–f. Nicht gewurzelter molekularer Stammbaum der Proteinkinasefamilie rekonstruiert über Aminosäuresequenzen der zugehörigen Proteine. Mehrere Unterfamilien können definiert werden: a durch Mitogene aktivierte Proteinkinasen (MAP-Kinase); b Proteinkinasen, die Tyrosinreste phosphorylieren; c Proteinkinasen, die Serinreste phosphorylieren; d Calcium/Calmodulin-abhängige Proteinkinasen; e die im Stoffwechsel verbreiteten Proteinkinasen, die cAMP oder cGMP als allosterischen Aktivator benötigen; f Cyclin-abhängige Proteinkinasen
a–f. Nicht gewurzelter molekularer Stammbaum der Proteinkinasefamilie rekonstruiert über Aminosäuresequenzen der zugehörigen Proteine. Mehrere Unterfamilien können definiert werden: a durch Mitogene aktivierte Proteinkinasen (MAP-Kinase); b Proteinkinasen, die Tyrosinreste phosphorylieren; c Proteinkinasen, die Serinreste phosphorylieren; d Calcium/Calmodulin-abhängige Proteinkinasen; e die im Stoffwechsel verbreiteten Proteinkinasen, die cAMP oder cGMP als allosterischen Aktivator benötigen; f Cyclin-abhängige Proteinkinasen
In Multigenfamilien lassen sich besondere Phänomene beobachten. In vielen Fällen sind die Mitglieder von Genfamilien in Genclustern auf den Chromosomen angeordnet und nicht zufällig irgendwo im Genom verteilt, sondern benachbart. Man könnte diese Cluster auch als Supergen bezeichnen. Markant und nicht ohne weiteres erklärbar ist die häufig beobachtete Sequenzähnlichkeit zwischen den Genen einer Familie, da sich die Sequenzen innerhalb von Multigenfamilien häufig nicht unabhängig entwickeln, wie man es eigentlich erwarten würde.
Der zugrunde liegende Evolutionsprozess wird als konzertierte oder horizontale Evolution bezeichnet. Diese Prozesse (◘ Abb. 3.40) führen nicht nur dazu, dass die DNA-Sequenzen aller Mitglieder einer Genfamilie angeglichen werden, sondern auch dazu, dass sich Varianten in der Genfamilie ausbreiten können und letztlich in der Population fixiert werden. Dieses Phänomen ist für rRNA-Gene, Globin-, Immunoglobin-, HLA-, Histon- und Hitzeschockgene sowie für repetitive DNA genauer untersucht worden. Eine wichtige Multigenfamilie stellen die rRNA-Gene dar, die in der Grundeinheit als Genkassette vorliegen (◘ Abb. 3.9). Zunächst wird die ganze Kassette transkribiert, später erfolgt das Herausschneiden der 5S, 18S und 28S rRNAs. Beim Krallenfrosch (Xenopus) findet man 400–600 tandemartige Wiederholungen der RNA-Kassette auf einem einzigen Chromosom. Beim Menschen sind ca. 300 Kopien auf fünf Chromosomen verteilt. Trotz hoher Kopienzahl sind die Nucleotidsequenzen in allen repeats fast immer identisch; dies ist eine wichtige Voraussetzung für die Verwendung der rRNA-Gene in der molekularen Phylogenieforschung (s. Abschn. 4.2); die Sequenz eines einzelnen rDNA-Gens ist meist repräsentativ für die vielen Kopien, die im Genom vorhanden sind.

a–c. Sequenzangleichung (Homogenisation) von tandemartig angeordneten DNA-Sequenzen durch ungleiches Crossing over und Genkonversion. a In einer Familie von tandemartig angeordneten DNA-Elementen kommt es zu einem regelmäßigen Gewinn oder Verlust von Einzelelementen durch ungleiches Crossing over, denn homologe Genelemente sind Orte erhöhter genetischer Rekombination. b Mutationen können sich durch ungleiches Crossing over in einer Genfamilie durchsetzen, indem das mutierte Element bevorzugt dupliziert wird, während die Wildtypallele verloren gehen. c Ausbreitung von Mutationen in einer Genfamilie durch Genkonversion. Dabei dient eine Genkopie als Matrize und überführt ihre Information (und Mutation) auf andere Genkopien. Dieser Prozess verläuft bei höheren Eukaryoten nur bei Genkopien, die nebeneinander auf einem Chromosom lokalisiert sind
a–c. Sequenzangleichung (Homogenisation) von tandemartig angeordneten DNA-Sequenzen durch ungleiches Crossing over und Genkonversion. a In einer Familie von tandemartig angeordneten DNA-Elementen kommt es zu einem regelmäßigen Gewinn oder Verlust von Einzelelementen durch ungleiches Crossing over, denn homologe Genelemente sind Orte erhöhter genetischer Rekombination. b Mutationen können sich durch ungleiches Crossing over in einer Genfamilie durchsetzen, indem das mutierte Element bevorzugt dupliziert wird, während die Wildtypallele verloren gehen. c Ausbreitung von Mutationen in einer Genfamilie durch Genkonversion. Dabei dient eine Genkopie als Matrize und überführt ihre Information (und Mutation) auf andere Genkopien. Dieser Prozess verläuft bei höheren Eukaryoten nur bei Genkopien, die nebeneinander auf einem Chromosom lokalisiert sind
Für Um- und Neuordnungen (rearrangements) der Kern-DNA werden zwei Prozesse verantwortlich gemacht, das ungleiche Crossing over (unequal crossing-over) und die Genkonversion (◘ Abb. 3.40). Beide Mechanismen beruhen auf Fehlpaarungen homologer DNA. Crossing over ist ein wesentlicher Prozess in der Meiose (s. EXKURS 3.2 Abschn. 3.3.3), bei dem homologe Abschnitte der Chromatiden ausgetauscht werden. Beim ungleichen Crossing over, das insbesondere im Bereich repetitiver Sequenzen auftritt (◘ Abb. 3.20), werden Sequenzbereiche oder vollständige Gene verdoppelt, d. h. Genzahl und Genlänge verändern sich. Bei repetitiver DNA (z. B. Minisatelliten-DNA; s. Abschn. 3.4.3) ist dieses Phänomen am auffälligsten.
Genkonversion tritt dagegen sowohl in der Mitose als auch in der Meiose auf, wenn bereits durch Genduplikation mehrere Kopien eines Gens vorliegen. Bei der Genkonversion bleibt die Zahl der Gene und ihre Länge unverändert (◘ Abb. 3.40c ). Genkonversion beginnt mit einer Heteroduplexbildung von DNA-Einzelsträngen verwandter, aber nicht gleicher Gene, die dann durch Mismatch-Repair in vollständig komplementäre Stränge umgewandelt werden. Ausgangspunkt der Heteroduplexbildung sind identische (häufig repetitive) Sequenzbereiche, z. B. in der Intronregion oder in anderen Sequenzbereichen. Durch Genkonversion werden Sequenzen einander angeglichen oder neue Sequenzkombinationen erzeugt. Bei Säugetieren liegen rDNA-Repeats auf fünf Chromosomen und weisen alle die gleiche Sequenz auf. Demnach erfolgt Genkonversion nicht nur innerhalb eines Chromosomenpaares, sondern auch zwischen nicht-homologen Chromosomen. Nicht immer werden komplette Gene umgewandelt, manchmal sind es auch nur begrenzte Genbereiche, die dieses Merkmal zeigen, z. B. findet man bei zwei humanen Cytochrom p450-Genen eine Sequenzidentität im 5´-Ende, aber eine Variabilität an 36 Positionen im 3´-Ende. Ein anderes Beispiel betrifft die beiden Alpha-Globin-Gene (α1 und α2) des Hämoglobins, die an beiden Loci bei verschiedenen Organismen identische Mutationen aufweisen, obwohl eine Trennung beider Gene vermutlich bereits vor 300 Mio. Jahren erfolgte.
Genkonversion ist also nur bei DNA-Abschnitten möglich, die eine Sequenzhomologie aufweisen; ihre Häufigkeit ist proportional zur Sequenzhomologie. Sind die Mitglieder von Genfamilien durch Alu-Sequenzen (s. Abschn. 3.4.4) getrennt, so tritt eine Genkonversion nicht oder seltener ein; wir beobachten in solchen Fällen, dass die Einzelgene getrennt evolvierten, d. h. unterschiedliche Sequenzen aufweisen.
Unklar ist der Mechanismus, über den eigentlich eine Sequenzkonstanz in den vielen Kopien der Organell-DNA (Mitochondrien und Chloroplasten) erreicht wird, da hier Rekombinationsprozesse (angeblich) fehlen. Ob bei ihnen ebenfalls extra-chromosomale Elemente, wie z. B. Plasmide, eine Rekombination ermöglichen, wie bei haploiden Prokaryoten angenommen wird, bleibt zu prüfen.
3.4.3 Nicht-codierende repetitive DNA
Der vermutlich größte Teil des Genoms (über 50 % bei vielen höheren Eukaryoten) wird nicht transkribiert und ist teilweise vermutlich funktionslos. Wichtige Elemente stellen Pseudogene und repetitive DNA-Sequenzen dar (◘ Abb. 3.28).
3.4.3.1 Pseudogene
Durch Verdopplung oder Vervielfachung von Genen und Genomen entwickelten sich meist, aber nicht immer, neue funktionelle Gene (s. Abschn. 3.4.2). Im Gegensatz dazu stehen Pseudogene, bei denen es sich um nicht translatierbare Kopien von ehemals aktiven Genen handelt, die Frame-shift-, Nonsense-Mutationen, Deletionen und Insertionen aufweisen. Pseudogene haben nicht mehr die ursprüngliche Genfunktion. Auch sie unterliegen der horizontalen Evolution, d. h. sie können durch ungleiches Crossing over und durch Genkonversion (s. Abschn. 3.4.2) verändert werden.
Bei Pseudogenen kann man zwei Gruppen unterscheiden: Die erste entsteht durch Genduplikation und anschließender Geninaktivierung. Die zweite Gruppe mit sogenannten Retropseudogenen (oder prozessierten Pseudogenen) durch Retrotransposons. Im diesem Falle werden Gene transkribiert, die mRNAs prozessiert und nach Rückübersetzung mittels reverser Transcriptase an einer neuen Stelle im Genom als cDNA-Kopie inseriert. Diese Retropseudogene haben keine Introns, aber häufig Poly-A-Schwänze und liegen nicht in Nachbarschaft zum Ursprungsgen, wie dies bei Pseudogenen, die durch Duplikation entstanden sind, der Fall ist.
Erstaunlicherweise kann die Natur es sich leisten, diese Sequenzen, die früher aufgrund mangelnden Wissens als „Müll“ (junk-DNA) bezeichneten Sequenzen, mit jeder Generation weiter zu vermehren, obwohl die Replikation ein energieaufwendiger Prozess ist. Aber vielleicht sind ja diese DNA-Abschnitte, die heute funktionslos erscheinen, in einer späteren Evolutionsphase (als molekulares „Ersatzteillager“) wieder von Nutzen. Unter anderen befinden sich in diesen DNA-Abschnitten Sequenzen für RNAi, die bei der Genregulation eine wichtige Rolle spielen (s. Abschn. 3.2.4).
3.4.3.2 Repetitive DNA
Wird ein DNA-Bereich verdoppelt und neben dem ursprünglichen Gen positioniert, so sprechen wir von Tandem-Repeat. Diese Tandem-Repeats sind der Ausgangspunkt für weitere DNA-Amplifikationen, hervorgerufen durch ungleiches Crossing over (◘ Abb. 3.20 ; ◘ Abb. 3.40). Mengenmäßig bedeutsam ist die repetitive DNA, die man in mittelrepetitive DNA (umfasst Transposons und Retroelemente) und hochrepetitive DNA unterteilen kann. Die letzte Klasse umfasst kurze Nucleotidsequenzen, die tandemartig in großer Anzahl in den Chromosomen vorkommen (◘ Abb. 3.28; ◘ Abb. 3.41). Man unterscheidet Telomer-, Satelliten-, Minisatelliten- und Mikrosatelliten-DNA.

Schematische Darstellung der Anordnung von Protein-codierenden Genen, VNTR, LINE und SINE auf einem Chromosom: VNTR (variable number tandem repeats), SINE (short interspersed elements), LINE (long interspersed elements)
Schematische Darstellung der Anordnung von Protein-codierenden Genen, VNTR, LINE und SINE auf einem Chromosom: VNTR (variable number tandem repeats), SINE (short interspersed elements), LINE (long interspersed elements)
An den Enden der Chromosomen findet man mehrere Tausend Kopien der tandemartig wiederholten Telomersequenzen, die über weite Organismengruppen konserviert wurden (◘ Tab. 3.11). Telomere verhindern, dass Chromosomenenden vorzeitig durch Nucleasen abgebaut werden. Telomere werden durch spezielle Telomerasen an die Chromosomenenden angeheftet. Die Telomerase, die mit den reversen Transcriptasen von LINEs (s. unten) verwandt ist, ist in Embryonal- und Tumorzellen besonders stark, in somatischen Zellen erwachsener Organismen ist sie dagegen weitgehend inaktiv. Ausgehend von diesem Phänomen wurde die Hypothese aufgestellt, dass Altern und Tod eine Konsequenz des irreversiblen Abbaus der Telomersequenzen und funktionaler Chromosomenabschnitte ist.
Trennt man die Gesamt-DNA von Eukaryoten durch Cäsiumchlorid-Gradientenzentrifugation auf, so findet man häufig zwei Banden, von denen eine kleinere die sogenannte Satelliten-DNA enthält. Diese Satelliten-DNA ist besonders reich an repetitiven Sequenzen und kann auf den Chromosomen bevorzugt im Bereich der Centromeren lokalisiert sein. Bei Insekten und anderen Arthropoden ist die Satelliten-DNA sehr homogen aufgebaut, d. h. ihre Sequenzelemente sind hochkonserviert (◘ Tab. 3.12). Bei Vertebraten sind die bis 1000-fach wiederholten Sequenzeinheiten der Satelliten-DNA deutlich länger und variabler (Länge über 200 Basenpaare); in diesen Elementen findet man variierte Unterelemente, wie z. B. GA5TGA. Durch ungleiches Crossing over (s. Abschn. 3.4.3) ist die Variabilität in der Satelliten-DNA etwa zehnmal höher als bei Genen, die nur in wenigen Kopien vorkommen. Verteilung und Organisation der repetitiven DNA-Elemente in den Centromerenbereichen sind chromosomen- und artspezifisch; vermutlich dient die repetitive Centromeren-DNA dazu, dass sich die homologen Chromosomen während der Meiose erkennen und zusammenlagern können. Sie ist auch der Ansatzpunkt für die Kinetochorenproteine, an denen sich die Mikrotubuli des Spindelapparats anheften können.
Neben der eigentlichen Satelliten-DNA findet man bei Tieren und Pflanzen 5- bis 50-fach wiederholte Sequenzelemente, die jeweils 15–100 Basenpaare umfassen. Die Sequenzelemente lassen sich auf ursprüngliche Sequenzen zurückführen, die durch Punktmutationen variiert wurden. Diese repetitive DNA, die jeweils ca. 500–5000 Nucleotide umfasst, ist wesentlich kürzer als die eigentliche Satelliten-DNA (◘ Tab. 3.13) und wird als Minisatelliten-DNA oder VNTR (variable number tandem repeats) bezeichnet. Sie zeigt eine starke Längenvariabilität an jedem Locus und weist durch ungleiches Crossing over (s. Abschn. 3.4.2) eine besonders hohe Mutationsrate auf (indem z. B. die Zahl und Länge der Repeats verändert wird), die bis zu 5 % pro Gamet betragen kann. Man hat die Minisatelliten-DNA deshalb auch als hot spot der meiotischen Rekombination bezeichnet. Minisatelliten-DNA eignet sich besonders zur Identifizierung von Individuen (z. B. in der forensischen Medizin oder Kriminalistik bei Aufklärung von Sexualstraftaten oder Mord) und zur Aufklärung von Paternität und Homozygotie in einer Population. Viele VNTR-Loci haben in einer Population Dutzende von Allelen, die dominant vererbt werden. Diese Eigenschaft wird im Multilocus-DNA-Fingerprinting (s. Abschn. 4.1.2) ausgenutzt, bei dem Dutzende Loci gleichzeitig analysiert werden. Die Wahrscheinlichkeit, dass zwei nicht verwandte Individuen identische Fingerprints aufweisen, ist kleiner als 1 zu 10 Mio. Diese Voraussetzung ist jedoch nicht immer gegeben, denn gerade in natürlichen Populationen oder bei Haustieren finden wir, dass nah verwandte Individuen in einer Population vorhanden sind (bedingt durch Philopatrie, d. h. bevorzugte Ansiedlung am Geburtsort, und Inzucht). Je höher die innere Verwandtschaft in einer Gruppe oder Population, desto schwieriger kann es sein, über DNA-Fingerprinting eindeutige identitäts- und Vaterschaftsnachweise zu erbringen.
Daneben treten in tierischen und pflanzlichen Genomen noch kürzere repetitive Elemente auf, deren Grundeinheit aus zwei (manchmal bis fünf) Nucleotiden (z. B. (GC)n oder (CA)n) bestehen, die bis zu 100-mal wiederholt sind. Von diesen, als Mikrosatelliten oder short tandem repeats (STR) bezeichneten Elementen, finden wir beim Menschen ca. 30.000 Loci, die für die Erkennung von Individuen, für Paternitäts-, Populationsuntersuchungen und Genomkartierungen von großer Wichtigkeit sind (s. Abschn. 4.1.2). Die Allele eines Mikrosatellitenlocus, die dominant vererbt werden, lassen sich mittels PCR (s. Abschn. 4.1.2) amplifizieren. Zur eindeutigen Identifizierung eines Individuums benötigt man die Allelverteilung von 10–20 STR-Loci. Da die Mikrosatelliten-PCR mit geringsten Mengen an DNA auskommt, ist sie heute für viele forensische und biologische Fragen die Methode der Wahl (s. Abschn. 4.1.2) und hat das Multilocus-Fingerprinting inzwischen weitgehend ersetzt. Die Variabilität von Mikrosatelliten-DNA wird während der Meiose durch ungleiches Crossing over und slippage (man kann dies bildhaft als Stottern der DNA-Polymerase auffassen, die durch die kurzen repetitiven DNA-Elemente „irritiert“ wird) stark erhöht, indem die kurzen Sequenzelemente mutiert, oder in ihrer Anzahl verdoppelt oder verringert werden können (◘ Abb. 3.20; ◘ Abb. 3.40).
Im Genom von Pflanzen und Tieren findet man zusätzlich bis 500 Basen lange DNA-Abschnitte, die short interspersed elements (SINE) (mit über 1,8 Mio. Kopien im Humangenom) oder im Falle von Volllängenelementen ca. 6000 Nucleotide lange long interspersed elements (LINEs), die in vielen Kopien (>1 Mio. im Humangenom) auftreten (jedoch nicht in tandemartigen Wiederholungen) (◘ Abb. 3.41). In tierischen und pflanzlichen Genomen gibt es ferner Tausende Kopien von miniature inverted-repeat transposable elements (MITEs), die an ihren Enden ca. 15 Basen lange inverted repeats tragen (s. EXKURS 3.4).
Zu den SINEs zählen die DNA-Elemente Alu (s. EXKURS 3.4). Im menschlichen Genom umfasst der Anteil dieser Elemente ca. 13 % des Gesamtgenoms. SINEs eignen sich auch als phylogenetische Marker, da sie Merkmalspolarität aufweisen. Diese Elemente, die man auch als mobile genetische Elemente („springende Gene“) oder Retroposons bezeichnet, werden durch Reverse-Transcriptase-Aktivität von intakten LINEs gebildet. Aus evolutionärer Sicht könnte man die Transposons (mit long terminal repeats, LTR oder inverted repeats, IR), Retrotransposons und Retroposons (Transposons ohne LTR) als Beispiele für aktive „egoistische“ Gene (selfish genes) ansehen, die nur ihre Vermehrung „im Sinn haben“. Andererseits führen diese mobilen Elemente zur genetischen Variabilität (u. a. vermehrtes Exon-Shuffling oder Enhancer-Shuffling), die sich langfristig auch positiv auswirken kann. Chromosomen zeigen im Bereich von Alu-Sequenzen erhöhte Raten von Neuorientierung. Wenn Alu-Elemente in aktive Gene springen, so werden diese meist inaktiviert; umgekehrt können „schlafende“ Gene aktiviert werden, indem springende Elemente als Enhancer fungieren. Letztlich werden damit der Selektion neue Merkmale zur Verfügung gestellt. Sexuelle Isolation und Artbildung können durch diese Mechanismen gefördert werden.
Retrovirale Sequenzen sind ebenfalls in vielen eukaryoten Genomen identifizierbar. Bei voller Länge codieren retrovirale Sequenzen verschiedene Proteine, u. a. gag (Kapsidprotein), reverse Transcriptase, Envelope (Hüllprotein) und manchmal Oncoproteine. Sie werden von LTR-Sequenzen flankiert. Die retrovirale RNA wird durch die retrovirale reverse Transcriptase in eine doppelsträngige DNA umgeschrieben und an einer neuen Stelle im Genom als sogenanntes Provirus inseriert.
Der relative Anteil der nicht-repetitiven DNA liegt bei Bakterien bei 100 % und nimmt bei höher entwickelten Eukaryoten ab: 70 % bei Drosophila, ca. 55 % bei Säugetieren und 33 % bei Pflanzen. Der Anteil der repetitiven DNA nimmt entsprechend zu. Bedingt durch ungleiches Crossing over wird der Anteil der repetitiven DNA im Genom der Eukaryoten in der zukünftigen Evolution vermutlich weiter wachsen. Wie bereits oben erwähnt, kennen wir die Funktion von deutlich über 50 % des Genoms nicht, die Funktion und Regulation vieler Gene ist größtenteils oder gänzlich unverstanden. Die repetitive mobile DNA hatte zunächst einmal als „egoistische“ DNA aber entscheidenden Einfluss auf das Genom selbst und trug wesentlich zur Evolution des Genoms bei. Die zukünftige Forschung wird solche Einflüsse detaillierter aufklären.
3.4.3.2.1 EXKURS 3.4
3.4.3.2.1.1 Repetitive, mobile und retrovirale Sequenzen im menschlichen Genom
Jens Mayer (Homburg)
Das haploide menschliche Genom besteht aus ca. 3,2 Mrd. Basenpaaren. Jedoch codiert nur ein kleiner Teil unseres Genoms, ca. 1,5 %, für Proteine, also die den menschlichen Organismus maßgeblich aufbauenden und regulierenden Moleküle. Ein deutlich größerer Teil der DNA des menschlichen Genoms, ca. 45 % besteht aus sogenannten repetitiven Elementen, auch mobile DNA genannt. Dies bedeutet, dass bestimmte Nucleotidsequenzen (mit entsprechend ihrem evolutiven Alter angesammelten Sequenzveränderungen) an vielen Stellen im Genom vorkommen. Alle diese Sequenzen waren oder sind mobil; sie haben sich in unserem Genom über lange Zeiträume hinweg bewegt oder können sich sogar noch bewegen. Man unterscheidet verschiedene Klassen solcher repetitiven, mobilen Elemente, und untersucht, wie sich diese Elemente im Genom beweg(t)en bzw. wie sie entstanden.
Ca. 3 % unseres Genoms sind sogenannte DNA-Transposons. Diese codieren ein Protein (Transposase), mit dem sie ihre eigene DNA-Sequenz aus einem DNA-Strang ausschneiden und an einer anderen Stelle im Genom wieder einsetzen können. Das Intermediat während dieses Vorganges ist also eine DNA (und keine RNA wie bei Retrotransposons). Im menschlichen Genom finden sich jedoch keine aktiven DNA-Transposons mehr, sondern nur noch evolutiv sehr alte, teilweise Hunderte von Millionen Jahren alte DNA-Transposonsequenzen.
Ungefähr 13 % unseres Genoms bestehen aus sogenannten Short Interspersed Elements (SINEs), wobei bisher ca. 1,8 Mio. solcher SINEs in unserem Genom identifiziert werden konnten. Diese wenige hundert Basenpaare langen Elemente haben sich über RNA-Intermediate in unserem Genom ausgebreitet. SINEs können durch eigene Promotorelemente eine RNA von sich herstellen, die an einer anderen Stelle im Genom wieder als DNA integriert werden kann. Da SINE-Sequenzen selbst keine Proteine codieren, profitieren sie quasi als Nebenprodukte von den folgenden mobilen DNA-Elementen.
Ungefähr 17 % unseres Genoms bestehen aus sogenannten Long Interspersed Elements (LINEs), wobei sich in unserem Genom ca. 1,4 Mio. solcher Elemente, welche eine Länge von mehreren Kilobasenpaaren haben, finden. Die im Genom vorkommenden LINEs sind in den meisten Fällen nicht komplett; sehr oft fehlen größere 5‘-Bereiche der Elemente. Volllängenelemente sind ca. 6 kb lang, enthalten in ihrem 5‘-Bereich einen Promotor und codieren zwei Proteine, wobei das eine mit RNA interagieren kann und das andere eine reverse Transcriptase (RT) und eine Endonuclease codiert, also Enzyme, welche einen RNA-Strang in einen DNA-Strang umschreiben und zusätzlich DNA-Stränge durchtrennen können. Volllängen-LINEs können, sofern sie noch entsprechend intakt sind, eine RNA von sich transkribieren, welche von dem codierten RT/EN-Protein an einer anderen Stelle im Genom als DNA wieder integriert wird, ein als Retrotransposition bezeichneter Prozess. Dieser Prozess ist nicht sehr effizient, so dass oft nur ein 3‘-Abschnitt einer LINE-RNA in DNA umgeschrieben wird. Von diesem Mechanismus profitieren auch SINEs, deren RNA ebenfalls von der Enzymmaschinerie von LINEs in DNA umgeschrieben werden kann. Ebenso bildet die LINE-Maschinerie sogenannte prozessierte Pseudogene, quasi cDNA-Kopien (copy oder complementary DNA) die mittels reverser Transcriptase von RNA-Transkripten zellulärer Gene hergestellt wurden.
Im menschlichen Genom findet sich noch eine weitere Klasse von mobilen Elementen, die ursprünglich auf Retroviren zurückgeht und ca. 8 % unseres Genoms einnimmt. Wenn Retroviren einen neuen Wirtsorganismus infizieren, bauen sie ihr retrovirales Genom als Provirus in das Genom bestimmter somatischer Zellen des Wirtes ein. Solche Proviren enthalten in ihrem etwa 9kb langen Genom Elemente zur Steuerung der Transkription, wie Promotoren, Polyadenylierungs- und Spleißsignale sowie Proteine, welche retrovirale Partikel bilden, retrovirale Proteine prozessieren oder sich in die Membran der Wirtszelle integrieren und später die Infektiosität retroviraler Partikel vermitteln können, sowie eine reverse Transcriptase, eine Endonuclease und eine RNase H, welche die Umschreibung eines retroviralen RNA-Genoms in provirale DNA bewerkstelligen. Zu retroviralen Sequenzen in unserem Genom kommt es, wenn sich provirale Sequenzen nicht (nur) in somatischen Zellen unseres Organismus, sondern in Genomen von Zellen der Keimbahn bilden. In einzelnen Fällen werden solchermaßen „infizierte“ Genome (Chromosomen) an die folgende Generation vererbt. Da alle Zellen der Nachkommen aus dem Genom von Keimbahnzellen hervorgehen, werden also die Genome aller Zellen des Nachkommens dieses Provirus enthalten. Das Provirus wurde vertikal an nachfolgende Generationen und über evolutive Zeiträume auf neue Spezies vererbt. Für den Menschen bezeichnet man solche Sequenzen als humane endogene Retroviren (HERV). Weitere Infektionen durch ein bestimmtes Retrovirus A konnten zu weiteren HERV-Elementen im Genom führen. Andere Retroviren B, C, D usw. können ebenfalls Kopien in der Keimbahn bilden. Tatsächlich hinterließen während der Evolution eine ganze Reihe verschiedener Retroviren ihre Spuren in den Genomen der evolutiven Vorläufer des Menschen (wie auch in denen vieler anderer Vertebraten). Es finden sich ca. 35 verschiedene Gruppen von HERVs in unserem Genom. Zusätzlich konnten die einzelnen HERV-Gruppen noch innerhalb des Wirtsgenoms weitere Kopien von sich bilden, so dass manche HERV-Gruppen mit vielen tausend Kopien im Genom vertreten sind. Verschiedene HERV Gruppen bildeten bzw. breiteten sich im Genom z. B. vor 35 oder 55 Mio. Jahren jeweils innerhalb relativ kurzer evolutiver Zeiträume aus. Danach verloren diese Sequenzen durch Mutationen ihre Fähigkeit zur Codierung der retroviralen Proteine. Es gibt aber Ausnahmen, manche HERV-Gruppen bildeten noch nach der evolutiven Abspaltung des Menschen vom gemeinsamen Vorfahren mit dem Schimpansen neue Proviren im menschlichen Genom. Diese Proviren sind noch relativ intakt und können noch retrovirale Proteine codieren.
Warum gibt es mobile Elemente in unserem Genom? Welche Auswirkungen hat diese Mobilität auf unser Genom bzw. Genome allgemein? Zunächst ist hier zu betonen, dass keineswegs nur das menschliche Genom solche mobilen Sequenzen enthält. In den Genomen von praktisch allen eukaryoten Spezies finden sich Spuren von mobilen Elementen, die sich in den Genomen von (gemeinsamen) Vorläuferspezies bewegten, oder aktuell noch in den Genomen dieser Spezies bewegen. Innerhalb der SINEs finden sich im Menschen sogenannte Alu-Elemente, die sich im Verlauf der Evolution der Primaten innerhalb von deren Genomen stark ausbreiteten und deshalb in den Genomen von Primaten zu finden sind. In Nagern verbreiteten sich innerhalb der SINEs dagegen sogenannte B1- und B2-Sequenzen. Die Evolution von LINEs überspannt vermutlich mehrere hundert Millionen Jahre, und man kann Sequenzen entsprechender unterschiedlicher Alter in Genomsequenzen vieler Spezies detektieren. Auch finden sich homologe Sequenzen von im menschlichen Genom vorhandenen HERVs je nach evolutivem Alter auch in anderen Primaten oder gar in Nagern.
Generell sind mobile Elemente (genetische) Moleküle, welche sich in bestimmter Weise bewegen oder vervielfältigen können. Sie nutzen dabei ihre Wirtsgenome als „Lebensraum“, in dem sie sich auf molekularer Ebene vermehren bzw. erhalten. Mobile Elemente sind offensichtlich sehr erfolgreiche genetische Elemente, auch in niederen Eukaryoten und Bakterien findet man mobile Elemente. Dabei mussten vor allem die mobilen Elemente sicherstellen, dass sich durch ihre eigene Aktivität die Genome des Wirtes nicht zu sehr schädigten, also nicht zu stark mutierten. Das ist offensichtlich gut gelungen.
Durch ihre eigene Aktivität innerhalb von Genomen verändern mobile Elemente die Struktur und teilweise auch Funktionalität eines Genoms, und die Auswirkungen solcher Aktivitäten sowie die sich daraus ergebenden sekundären Phänomene sind keinesfalls zu unterschätzen. Wenn sich z. B. die Alu-Elemente innerhalb von 60 Mio. Jahren auf heute ca. 1 Mio. Elemente vermehrten, hatte dies sicherlich Auswirkungen auf die Struktur des Genoms und manche Gene. In der Tat finden sich vielfach Beispiele, in denen die Funktion von Genen durch die Bildung von mobilen Elementen verändert wurde. Retrovirale Elemente konnten z. B. mit ihren eigenen Sequenzen alternative Promotoren oder Spleißsignale einbringen. Die Bildung neuer SINEs oder LINEs konnte während der Evolution Gene verändern oder zerstören. Sowohl SINEs als auch LINEs sind noch heute im menschlichen Genom aktiv und bilden neue Kopien von sich. Es ist deshalb eine ganze Reihe von Fällen genetisch bedingter Erkrankungen bekannt, bei denen das jeweils relevante Gen durch die Bildung eines LINE oder SINE innerhalb des Gens zerstört wurde. Für den Menschen wird deshalb auch wissenschaftlich untersucht, ob mobile Elemente oder die von bestimmten HERVs codierten Proteine an der Entstehung von bestimmten Erkrankungen mit genetischen Ursachen, oder z. B. gar Krebs, beteiligt sein könnten. Zum Beispiel wird für eine evolutiv junge und deshalb noch für verschiedene retrovirale Proteine codierende Gruppe von HERVs (als HERV-K[HML-2] bezeichnet) die Involvierung in bestimmte Formen von Krebs, wie z. B. Hodentumoren (der häufigsten Tumorform des jungen Mannes), Brustkrebs und Hautkrebs diskutiert und grundlagenwissenschaftlich untersucht. Für die HERV-W-Gruppe wird deren Involvierung in Multiple Sklerose erwogen und ebenso untersucht.
Jedoch sind mobile Elemente nicht grundsätzlich schädigend für das Wirtsgenom bzw. den menschlichen Organismus. Mobile Elemente hatten auch durchaus positive Auswirkungen für den Wirtsorganismus. Wenn durch mobile Elemente die Funktion und die Produkte eines Gens verändert wurden, könnte dies im jeweiligen evolutiven Kontext einen Vorteil für die betreffenden Individuen gehabt haben, und diese wurde deshalb selektiert. In Mäusen finden sich z. B. von proviralen Sequenzen abstammende Gene, welche teilweise sehr effektiv gegen die Infektion durch bestimmte exogene Retroviren schützen. Im Menschen und einigen anderen Primaten ist ein von einem endogenen Retrovirus abstammendes Gen bzw. Proteinprodukt anscheinend essenziell in die Entwicklung der Plazenta involviert. Hierbei wurde ein einzelner Locus der HERV-W-Gruppe, insbesondere der für ein Hüllprotein (Envelope Protein) codierende Teil des Locus, zu einem echten Gen (Syncytin) hin selektiert. Syncytin wird während der Bildung von Syncytiotrophoblasten aus Trophoblasten exprimiert. Die Membranen von Trophoblasten-Einzelzellen verschmelzen hierbei miteinander. Die Evolution hat offensichtlich eine für Retroviren während der Infektion von Zellen wichtigen Prozess, nämlich die Verschmelzung von Zellmembranen, welche vom retroviralen Hüllprotein vermittelt wird, für einen körpereigenen Prozess nutzbar gemacht. Syncytin ist nun anscheinend ein im Menschen essenzielles Gen; in bisherigen Studien konnten keine Nonsens-Mutationen innerhalb des Syncytin-Locus identifiziert werden.
Aus wissenschaftlicher Sicht liefern die Sequenzen von HERVs, wie auch die von endogenen Retroviren in anderen Spezies, zudem wichtige Erkenntnisse für ein besseres Verständnis der Evolution von Retroviren. HERVs sind im Prinzip die Sequenzüberreste von Retroviren, welche Vorläuferspezies vor vielen Millionen von Jahren infizierten. Die retroviralen Sequenzen im Menschen beinhalten also Hinweise, welche Proteine diese damaligen Retroviren codierten, inwieweit diese Retroviren z. B. schon spezialisierte Proteine codierten, die sich heute bezüglich der Proteinfunktion auch im Humanen Immundefizienz-Virus (HIV) finden. Man kann hier also gewissermaßen Paläo(retro)virologie betreiben.
3.4.4 Horizontaler Gentransfer und Symbiosen
Die Genome wurden nicht nur durch intraspezifische Duplikationen, Deletionen und Insertionen verändert, sondern auch durch fremde DNA-Bereiche angereichert. Der Transfer von genetischem Material über Art- und Familiengrenzen hinweg wird als horizontaler Gentransfer (HGT) bezeichnet, tritt bei Prokaryoten weit verbreitet auf und ist gut belegt (s. EXKURS 3.5). Ein HGT war offenbar in der frühen Evolution von besonderer Bedeutung und hat dazu geführt, dass im Durchschnitt 6 % eines bakteriellen Genoms auf Import von anderen Organismen zurückgeht. Aber auch heute noch tritt HGT auf: So werden z. B. Antibiotikaresistenzen, die durch Gene auf den Plasmiden codiert werden, durch Plasmidaustausch auch über Artgrenzen hinweg weitergegeben.
Ebenso bekannt ist der Gentransfer von Viren (z. B. Retroviren), die als mobile genetische Elemente angesehen werden können, in tierische Genome. Dieser Prozess hat in der Evolution offenbar vielfach stattgefunden, so dass die Vertebratengenome viele, meist repetitive DNA-Abschnitte enthalten, die auf Retroviren zurückgehen (s. Abschn. 3.4.3). Bei Pflanzen kommt es bei Infektion durch Agrobacterium tumefaciens zu Kronengalltumoren oder durch A. rhizogenes zu hairy roots. Auch hier findet ein Transfer der t-DNA (Transfer-DNA) in die Pflanzenzelle statt, bei dem Gene für die Biosynthese von Wuchsstoffen und für die Opine (nicht-proteinogene Aminosäuren, die von den Agrobakterien als Stickstoffquelle genutzt werden können) übertragen werden. Das gemeinsame Vorkommen von Hämoglobingenen bei Tieren und Leguminosen könnte ebenfalls durch horizontalen Gentransfer verursacht worden sein, nur kennen wir die Übertragungswege nicht.
Ein neu entdeckter Fall von HGT wurde für Wolbachia beschrieben, eine Gattung von parasitisch lebenden gram-negativen Bakterien, die offenbar sehr viele Insekten, einige Spinnen- und Nematodenarten befallen. Wolbachia lebt intrazellulär und manipuliert offenbar die Fortpflanzung ihrer Wirte. Man kennt inzwischen mehrere Fälle, dass Teile des Wolbachia-Genoms auf die Chromosomen der Wirtsarten (Insekten, Nematoden) übertragen und dort zu einem geringen Teil sogar exprimiert wurden.
HGT wurde inzwischen bei einigen Pilzen, insbesondere bei Hefen nachgewiesen und scheint für die Evolution der Pilzgenome wichtig zu sein. Pflanzen leben mit pilzlichen Endophyten zusammen. In den letzten Jahren wurden Endophyten entdeckt, welche dieselben Sekundärstoffe produzieren, wie ihre Wirtspflanze (◘ Abb. 4.46). Es wird spekuliert, dass die Endophyten durch HGT Sekundärstoffgene in Pflanzen übertragen haben. Die laufenden und zukünftigen Genomprojekte werden sicher noch viel mehr Evidenzen für horizontalen Gentransfer aufdecken, als wir heute vermuten.
Ein klassisches Beispiel für einen besonders wichtigen horizontalen Gentransfer kann man in Eukaryotenzellen sehen: Wie im EXKURS 3.1 in Abschn. 3.2.7 dargestellt, entstanden durch die Aufnahme von α-Proteobakterien in frühe Eucyten Mitochondrien und durch Aufnahme von Cyanobakterien Chloroplasten. Zunächst wurden natürlich die bakteriellen Genome in die Eucyte übertragen und waren vermutlich funktionsfähig. In der späteren Evolution wurden die bakteriellen Gene aus dem mitochondrialen bzw. Chloroplastengenom entfernt und teilweise in den Zellkern der Wirtszelle ausgelagert. Mitochondrien und Chloroplasten können im Verlauf der Evolution auch wieder verloren gehen; dies sieht man bei einigen parasitischen Protozoen (s. Abschn. 4.2.2). Ebenso wurden sekundäre Symbiosen beschrieben, indem Zellen mit ihren Organellen von Wirtszellen aufgenommen wurden (s. Abschn. 4.2.2). W. Ford Doolittle hat den HGT als wichtigsten Mechanismus der Phylogenie angesehen; diese Aussage wird inzwischen auf die frühe Evolution vor 1 Mrd. Jahren eingeschränkt; die meisten Phylogenien, die wir heute sehen, gehen vermutlich mehr auf vertikale Abstammungsprozesse zurück (Kurland et al. 2003).
Symbiosen sind aber nicht auf Mitochondrien oder Chloroplasten beschränkt. Symbiosen, d. h. ein enges Miteinander von Partnern mit gänzlich unterschiedlichen Genomen, treten in vielen Organismengruppen auf. Sie sind aber noch nicht soweit fortgeschritten, wie die Mitochondrien oder Chloroplasten. Beispiele sind (s. EXKURS 1.2 Abschn. 1.1.8):
-
Flechten bestehen aus einer Symbiose von Algen- und Pilzzellen
-
Viele Pflanzen leben mit Endophyten und Mykorrhizen zusammen
-
Riffbildende Korallen enthalten häufig photosynthetisch aktive einzellige Algen als Symbiosepartner
-
Viele Insekten, deren Nahrung nicht alle essenziellen Nahrungsbestandteile enthält, kultivieren in speziellen Mycetomen intrazellulär oder extrazellulär Bakterien, Protozoen und Pilze, die in der Lage sind, die fehlenden Nahrungsbestandteile zu synthetisieren. Bei den intrazellulär lebenden Bakterien liegt eine Endosymbiose vor.
-
In den Därmen kultivieren Tiere eine große Diversität an meist symbiontischen Bakterien, welche zur Verdauung der Nahrung beitragen
-
Leguminosen enthalten in Wurzelknöllchen symbiotische Bakterien, die es vermögen, den Luftstickstoff in organische Verbindungen zu fixieren
-
In Erlenwurzeln sorgen andere Bakterien (Actinomyceten) in analoger Weise für die Verwertung des Luftstickstoffs
Die symbiontischen Beziehungen belegen, dass für die Evolution nicht nur Wettbewerb (survival of the fittest), sondern auch Kooperationen zwischen unterschiedlichen Partnern eine sehr große Rolle gespielt hat (s. EXKURS 1.2 Abschn. 1.1.8).
3.4.4.1 EXKURS 3.5
3.4.4.1.1 Die Bedeutung des horizontalen Gentransfers für die Evolution der Bakterien
Gottfried Wilharm (Wernigerode)
1995 begann mit der Sequenzierung des ersten kompletten Genoms eines Organismus’ – des Bakteriums Haemophilus influenzae – ein neues Zeitalter in den Biowissenschaften: das der Genomforschung. Inzwischen hat uns die Geräteentwicklung dahin gebracht, dass bakterielle Genome in einem einzigen experimentellen Ansatz in wenigen Stunden vollständig sequenziert werden können. So werden inzwischen die Metagenome ganzer Lebensräume studiert und wir lernen, dass der kollektive Genpool der etwa 1013 bis 1014 Darmbakterien eines Menschen etwa hundertfach mehr Gene umfasst als unser eigenes Genom. Gerade für die Evolutionsforschung stellen diese Datensammlungen eine besonders wertvolle Ressource dar. Die Analyse dieser Daten lässt uns vor allem die fundamentale Bedeutung des horizontalen Gentransfers (HGT) für die Phylogenie der Bakterien und Archaebakterien erkennen. HGT bezeichnet den Erwerb von Erbsubstanz außerhalb der normalen Erbfolge. Die am besten verstandenen Mechanismen zum Erwerb neuer Erbsubstanz bei Bakterien sind die Aufnahme nackter DNA aus der Umgebung (Transformation), die Übertragung von Erbsubstanz zwischen Bakterien über sogenannte Sex-Pili (Konjugation) sowie die Übertragung durch Infektion mit Bakteriophagen (Transduktion). Man weiß inzwischen, dass alle funktionellen Kategorien von Genen dem HGT unterliegen können, wenn auch mit unterschiedlicher Frequenz. Selbst die taxonomisch so wichtigen rRNA-Operons bilden hier keine grundsätzliche Ausnahme. Aus diesen Erkenntnissen folgt nicht nur die Erosion der gebräuchlichen Taxonomie sondern vor allem der Abschied von einer rein Stammbaum-artigen Betrachtung der Phylogenie. Stattdessen werden Netzwerk-artige Betrachtungen der Entwicklungsgeschichte der Bakterien und auch der anderen Lebensformen erforderlich. Die entwicklungsgeschichtliche Bedeutung des horizontalen Gentransfers soll hier anhand einiger Beispiele illustriert werden.
Während sich die Akquisition neuer Gene in Eukaryoten vor allem durch Genduplikation innerhalb einer Entwicklungslinie abzuspielen scheint, ist für die Entwicklung prokaryotischer Genome vor allem der horizontale Gentransfer (HGT) zwischen verschiedenen Entwicklungslinien entscheidend. Immerhin weist aber auch das menschliche Genom mehr als 100 Gene auf, die wahrscheinlich über HGT Eingang in unsere Erbmasse gefunden haben und von Bakterien stammen. Umgekehrt hat auch menschliches Erbgut vereinzelt Einzug in bakterielle Genome gehalten. So findet man etwa in einigen Isolaten des Gonorrhoe-Erregers Neisseria gonorrhoeae einen Genabschnitt menschlichen Ursprungs. Deutlich stärker sind jedoch die meisten bakteriellen Genome durch HGT untereinander bzw. durch Austausch mit Archaebakterien geprägt. Dies führt dazu, dass auch nah verwandte Taxa sich vor allem auf Grund von HGT dramatisch im Gengehalt unterscheiden können. Die Art Escherichia coli als Beispiel umfasst neben kommensalisch lebenden Darmbewohnern auch verschiedene Pathovare wie enterohämorrhagische E. coli (EHEC), enteroaggregative E. coli (EAEC) oder extraintestinal pathogene E. coli (ExPEC). Ein Genomvergleich zwischen dem gebräuchlichen Laborstamm E. coli K12 und einem EHEC zeigt, dass diese nur 70 % Gemeinsamkeit in der Genausstattung aufweisen. Vergleicht man nun alle bekannten E.-coli-Genome, so kommt man bei einer Genomgröße der verschiedenen Vertreter von etwa 4200–5500 Genen nur auf etwa 2000 Gene, die bei allen Vertretern anzutreffen sind (das sogenannte Kerngenom). Demgegenüber ist das Pangenom von E. coli, also die Gesamtheit aller nicht-orthologen Gene, die man in dieser Spezies finden kann, mit mehr als 18.000 Genen fast zehnmal so groß wie der Kernbestand der Art. Diese enorme Plastizität bakterieller Genome spielt unter anderem für die Entwicklung vieler bakterieller Krankheitserreger eine entscheidende Rolle.
Ein aktuelles Lehrstück, das diese Plastizität illustriert, stellt ein Lebensmittel-assoziierter Infektionsausbruch im Jahr 2011 in Deutschland dar, bei dem ein E. coli vom Serotyp O104:H4 fast 4000 Durchfallerkrankungen verursachte. Dabei kam es über 900-mal zu der schweren Komplikation eines hämolytisch-urämischen Syndroms (HUS) sowie zu 54 Todesfällen. Die blutigen Durchfallerkrankungen mit der Komplikation HUS deuteten auf enterohämorrhagische E. coli (EHEC) hin, allerdings war der Serotyp O104:H4 für einen EHEC-Erreger ebenso ungewöhnlich wie die hohe Komplikationsrate bei diesem Ausbruchsgeschehen. Die Genomsequenzierung mehrerer Ausbruchsisolate zeigte dann, dass der Erreger die höchste Verwandtschaft mit enteroaggregativen E. coli (EAEC) aufwies, aber zusätzlich ein für EHEC-Erreger typisches Shiga-Toxin erworben hatte, ebenso einige typische Eigenschaften extraintestinal pathogener E. coli (ExPEC) sowie mehrere Antibiotika-Resistenzgene. Zusammengefasst zeigt dies, wie durch HGT aus verschiedenen Erregerpools neue aggressive Erregervarianten entstehen können. Das gleichzeitige Akkumulieren von Antibiotika-Resistenzgenen deutet an, dass die Entstehung dieser Erregervariante zumindest teilweise in einem Umfeld stattgefunden hat, in dem ein Selektionsdruck durch Antibiotika bestand, also z. B. durch human- oder tiermedizinische Intervention.
Die Entwicklung multiresistenter bakterieller Krankheitserreger innerhalb weniger Jahrzehnte seit der therapeutischen Nutzung von Antibiotika, die vor allem in Krankenhäusern zu einem immer größeren Problem wird, fußt vor allen Dingen auf HGT. Antibiotika sind keine Erfindung des Menschen sondern eine alte Erfindung der Natur. Deshalb waren Resistenzgene auch schon lange vor der Entdeckung und therapeutischen Nutzung von Antibiotika in der Natur vorhanden und konnten so unter Selektionsdruck schnell aus verschiedensten Genpools mobilisiert werden. Zu den bekanntesten Krankenhauserregern gehören Methicillin-resistente Staphylococcus aureus (MRSA). Für eine bestimmte Gruppe von MRSA, den klonalen Komplex 398 (CC398), der zunehmend häufig von Nutztieren auf den Menschen übertragen wird, konnte man durch phylogenetische Untersuchungen an Hand einer Vielzahl von ermittelten Genomen die jüngere Entwicklungslinie nachzeichnen. Ursprünglich handelte es sich um Methicillin-sensitive Kolonisierer des Menschen, die dann auf Nutztiere wie Schweine übertragen wurden. Dort erwarben sie Resistenzgene gegen die Antibiotika Methicillin und Tetracyclin und von dort werden sie nun in immer schwerer zu behandelnder Form wieder zurück auf den Menschen übertragen. Es wird hier offensichtlich, dass durch den Einsatz von Antibiotika in der Tiermast die Evolution von Krankheitserregern befeuert wird.
Erscheinen diese Entwicklungen schon rasant gemessen an den in der Evolutionsbiologie sonst üblicherweise betrachteten Zeiträumen, so kann man in Krankenhäusern Evolution wie im Zeitraffertempo erleben. So konnte man nacheinander in einem einzelnen Patienten die zur Familie der Enterobacteriaceae gehörigen Erreger Klebsiella pneumoniae, E. coli und Serratia marcescens isolieren, die alle das gleiche Resistenzplasmid trugen. Auch bei Klinikausbrüchen mit einer Vielzahl von Patienten konnte man schon die Weitergabe von Resistenzplasmiden über Art-, Gattungs-, Familien- und sogar Ordnungsgrenzen hinweg nachweisen. Zwischen Krankenhausproblemkeimen aus der Familie der Enterobacteriaceae und entfernter verwandten Gram-negativen Erregern wie Pseudomonas aeruginosa und Acinetobacter baumannii bilden sich regelrechte genetische „Austauschgemeinschaften“.
Dass durch den Gebrauch von Antibiotika in Human- und Tiermedizin, aber auch in der Pflanzenproduktion, vielfältige Selektionsdrücke auf bakterielle Populationen aufgebaut werden und so deren Evolution beeinflusst wird, ist unmittelbar einsichtig. Das wahre Ausmaß dieser anthropogenen Einflüsse ist jedoch bislang kaum abzuschätzen. Bakterien haben Mechanismen entwickelt, die eine genetische „Auffrischung“ unter Stressbedingungen fördern. Dazu gehört nicht nur die Erhöhung von Mutations- und Rekombinationsraten sondern auch die Stimulation von HGT-Ereignissen. Besonders bedrohlich ist nun die Beobachtung, dass bereits unter dem Einfluss von subinhibitorischen Antibiotikakonzentrationen die Frequenz von HGT-Ereignissen erhöht werden kann und somit der Resistenzentwicklung weiter Vorschub geleistet wird. So reicht die Bedeutung des HGT weit über die Evolution der Bakterien hinaus und betrifft den Menschen ganz unmittelbar.
3.5 Vererbung, Populationsgenetik und Artbildung
3.5.1 Allel- und Genotypenfrequenz und Vererbungsregeln
Auf den Chromosomen befinden sich linear angeordnet die Gene. Genorte mit einer spezifischen Erbinformation bezeichnet man als einen Genlocus (Plural: Genloci) mit jeweils einem Allel auf den beiden homologen elterlichen Chromosomen. Ihre Vererbung erfolgt demnach biparental. Ist die Nucleotidsequenz identisch, sprechen wir davon, dass die Individuen homozygot sind; unterscheiden sie sich, z. B. weil in einem Allel eine Punktmutation oder anderweitige Veränderung aufgetreten ist, dann sind sie heterozygot. In einem diploiden Individuum gibt es natürlich für jeden Locus nur zwei Allele (Ausnahme sind multiple Kopien eines Gens). Anders kann es in der Gesamtheit einer Population aussehen. Wenn man für einen Locus mehrere Allele findet spricht man von Genpolymorphismus. In der Praxis sucht man nach Polymorphismen, deren Frequenz im häufigsten Allel kleiner als 0,99 bis 0,95 ist. Den Gesamtbestand an Genen und Allelen in einer Population zu einem bestimmten Zeitpunkt bezeichnet man als Genpool der Population.
Der Genotyp eines Individuums umfasst alle Gene des Genoms, während der Phänotyp nur durch die exprimierten Gene zustande kommt. Auf der Stufe der Allele kann die Expression ausfallen, wenn ein oder beide Allele durch Mutationen verändert und die abgeleiteten Proteine funktionsunfähig geworden sind (◘ Abb. 3.42). Ein Funktionsausfall wird auch als loss of function bezeichnet. Solche Fälle kann man zuweilen schon makroskopisch analysieren. Wir wollen von dem einfachsten Fall ausgehen, dass in einer Population das Allel A und seine mutierte Form a vorkommen. In der Population finden wir demnach Individuen mit den Allelverteilungen AA, Aa und aa. AA und aa bezeichnen wir als homozygot und Aa als heterozygot. A soll ein Gen darstellen, das für ein Enzym codiert, welches einen schwarzen Farbstoff bildet. AA-Tiere wären dann schwarz gefärbt, während aa-Tiere, die dieses Pigment nicht bilden, weiß aussähen (in der Wirklichkeit benötigen wir meist eine Serie von Biosynthesegenen, um einen Farbstoff zu bilden, die von einem oder mehreren Mastergenen reguliert werden; unsere Betrachtung könnte demnach auf ein Kontrollgen zurückgehen, das einen Biosyntheseweg an- oder abschaltet). Treten heterozygote Individuen auf, so sind diese entweder intermediär grau gefärbt oder entsprechen dem homozygoten schwarzen Phänotyp. Im letzteren Fall würden wir A (man beachte die Großschreibung!) als dominant bezeichnen. Das entsprechende rezessive Allel wird mit a gekennzeichnet (man beachte die Kleinschreibung!). In welchem Grade die Dominanz ausgeprägt ist, kann von Genlocus zu Genlocus variieren, und der Phänotyp der Heterozygoten kann irgendwo zwischen dem der beiden Homozygoten liegen, in unserem Falle auch also alle Graustufen umfassen (wenn z. B. ein Enzym der Biosynthesekette von der Mutation betroffen wäre). Die meisten Merkmale sind jedoch nicht disjunkt sondern kontinuierlich, da eine Vielzahl an Genen zur Ausprägung des Merkmals beiträgt. Die formale Analyse solcher Merkmale ist daher wesentlich komplexer als in unserem einfachen Beispiel angenommen wurde.

Auswirkung von Mutationen, die zur Funktionslosigkeit von Proteinen in diploiden Organismen führen. a Wildtyp Genotyp AA; b heterozygoter Genotyp Aa, c homozygoter rezessiver Genotyp aa. A codiert für das funktionsfähige Protein, a für das funktionslos gewordene Protein
Auswirkung von Mutationen, die zur Funktionslosigkeit von Proteinen in diploiden Organismen führen. a Wildtyp Genotyp AA; b heterozygoter Genotyp Aa, c homozygoter rezessiver Genotyp aa. A codiert für das funktionsfähige Protein, a für das funktionslos gewordene Protein
Das in der Population am stärksten vertretene Allel wird häufig als Wildtyp-Allel bezeichnet. Bei diploiden Organismen wird das Wildtyp-Allel meist dominant, das mutierte Allel dagegen rezessiv vererbt. Unvollständige, partielle Dominanz oder Kodominanz liegen vor, wenn die Heterozygoten intermediäre Merkmale ausbilden, z. B. sind die Nachkommen aus Kreuzungen zwischen rotblühenden mit weißblühenden Löwenmäulchen rosafarben; in der F2-Generation erhalten wir rote, rosa und weiße Pflanzen im 1:2:1-Verhältnis. Werden beide Allele gleich stark und unabhängig vererbt, so sprechen wir von Kodominanz.
In vielen Fällen finden wir Nucleotidsubstitutionen in den Allelen eines Genlocus, durch die sich die Aminosäuresequenz des resultierenden Proteins aufgrund des degenerierten Codes nicht ändert oder im Falle einer Veränderung der Aminosäuresequenz dessen Funktion nicht oder nicht wesentlich verändert wird. Dieser Allelpolymorphismus ist makroskopisch nicht zu erkennen und die Grundlage der Allozymanalyse oder DNA-Analytik (s. Abschn. 4.1.2). Häufig unterscheiden sich die Proteine eines Locus durch ein bis zwei Aminosäuren in ihrer Sequenz, was zu einem veränderten Laufverhalten in der Elektrophorese führt. So verändert sich etwa durch den Austausch einer basischen Aminosäure durch eine saure Aminosäure die elektrophoretische Eigenschaft des Proteins (d. h. die Gesamtladung bei definiertem pH-Wert), so dass sich hierdurch auch die Wanderungsgeschwindigkeit im elektrischen Feld ändert. Als 1966 die Enzymelektrophorese in die Populationsgenetik eingeführt wurde, hatte dies eine „elektrophoretische Revolution“ zur Folge, denn nun konnten äußerlich nicht sichtbare Allele eines Individuums bestimmt werden. Heute werden in diesem Zusammenhang zunehmend DNA-Analysen wie beispielsweise DNA-Fingerprinting, AFLPs (amplified fragment length polymorphisms) und Mikrosatelliten-Analysen verwendet (s. Abschn. 4.1.2), da sie den direkten Zugriff auf die variablere DNA-Sequenzebene erlauben.
Über die Allozym- und Mikrosatelliten-Analyse lässt sich auch der Heterozygotiegrad einer Population bestimmen. Die Heterozygotie des einzelnen Locus h ergibt sich aus folgender Berechnung:

Wobei xi die Frequenz des i-ten Allels darstellt.
In einer bisexuellen Population mit Zufallspaarung entspricht h dem Anteil der an einem einzelnen Locus heterozygoten Tiere und H dem durchschnittlichen Anteil heterozygoter Loci. In natürlichen Populationen weicht der beobachtete Anteil der Heterozygoten H obs oft von der erwarteten Größe H exp ab. Inzucht und Selbstbefruchtung verringern H obs , d. h. der Anteil der Homozygoten nimmt zu. H variiert in natürlichen Populationen zwischen 0,1 und 0,35. Die Wahrscheinlichkeit, dass zwei Individuen in solchen Populationen an z Loci die identische Allelverteilung aufweisen, kann über
W = (1–H)Z
ermittelt werden.
Bei H = 0,1 und z = 104 ergibt sich W = 10–458; d. h. die Wahrscheinlichkeit identischer Allelmuster ist extrem gering; nur bei eineiigen Zwillingen sind die Muster nahezu identisch. Diese Erkenntnis ist die Grundlage der Anwendung von DNA-Fingerprinting und Mikrosatelliten-Analysen zum Erkennen von Individuen und für Vaterschaftsanalysen (s. Abschn. 4.1.2).
3.5.2 Mendelsche Vererbungsregeln
Diese Betrachtung führt uns zu den allgemeinen Mendelschen Vererbungsregeln, die für die moderne Evolutionstheorie extrem wichtig sind, denn ohne diese Basis bleibt die Darwinsche Evolutionstheorie unvollständig. Diese Regeln erklären die Häufigkeit der Genotypen bei Nachkommen von Eltern, deren Genotyp genau definiert ist.
Für die formale Erläuterung der Mendelschen Regeln bleiben wir bei den Genotypen AA, Aa und aa, wobei A dominant und a rezessiv ist (◘ Abb. 3.43). In unserem Beispiel sind die Nachkommen von AA × AA und AA × aa (oder vice versa) zu 100 % schwarz gefärbt, da sie alle das dominante A-Allel besitzen. (1. Mendelsche Regel [Uniformitätsregel], nach der die Nachkommen reziproker Kreuzungen reiner Linien einen einheitlichen Phänotyp besitzen). Sind beide Eltern Aa-heterozygot (Ergebnis der F1-Kreuzung AA × aa), so erhalten wir 25 % AA, 50 % Aa und 25 % aa-Genotypen. Wir beobachten ein Phänotypenverhältnis von 3:1 und ein Genotypenverhältnis von 1:2:1 in der F2-Generation (2. Mendelsche Regel [Spaltungsregel]) (◘ Abb. 3.43). Bei der Kombination AA × Aa entstehen zu 50 % AA und 50 % Aa-Genotypen.

a, b. Illustration der Mendelschen Regeln. a In der Elterngeneration (Parentalgeneration, P0) werden die homozygoten Genotypen AA und aa miteinander gekreuzt. In der Tochtergeneration (Filialgeneration, F1) erhalten wir nur Aa-Genotypen, die das dominante Gen A exprimieren. Kreuzt man die Heterozygoten miteinander, so beobachtet man in der F2-Generation eine Aufspaltung in den AA-, Aa- und aa-Genotyp. b Intermediäre Vererbung der Blütenfarbe bei der Wunderblume Mirabilis jalapa (Nyctaginaceae)
a, b. Illustration der Mendelschen Regeln. a In der Elterngeneration (Parentalgeneration, P0) werden die homozygoten Genotypen AA und aa miteinander gekreuzt. In der Tochtergeneration (Filialgeneration, F1) erhalten wir nur Aa-Genotypen, die das dominante Gen A exprimieren. Kreuzt man die Heterozygoten miteinander, so beobachtet man in der F2-Generation eine Aufspaltung in den AA-, Aa- und aa-Genotyp. b Intermediäre Vererbung der Blütenfarbe bei der Wunderblume Mirabilis jalapa (Nyctaginaceae)
Betrachtet man die Kreuzung in einem dihybriden Erbgang mit zwei unabhängigen Merkmalen (z. B. AaBb × AaBb) so erhält man ein 9:3:3:1 Phänotypenverhältnis. Dieses Ergebnis belegt, dass die Allele frei kombiniert werden (3. Mendelsche Regel [independent assortment], Prinzip der unabhängigen Segregation). Kreuzungsschemata lassen sich auch für trihybride Erbgänge mit drei unabhängigen Merkmalen usw. aufstellen. Komplizierter wird der Erbgang, wenn multiple Allele für einen Locus in der Population vorhanden sind; ein bekanntes Beispiel dafür ist die Ausprägung der Blutgruppen AB0. Viele Merkmale variieren jedoch nicht diskontinuierlich, wie in ◘ Abb. 3.46 dargestellt, sondern kontinuierlich. Für ihre Vererbung gelten die Regeln der quantitativen Genetik.
Auf einem typischen Säugerchromosom liegen ca. 1000 Gene. Bei der Vererbung segregieren einige Gene/Allele nicht unabhängig voneinander, sondern zusammen. Wir bezeichnen die Zusammengehörigkeit solcher Gene, die häufig benachbart liegen, als Kopplung. Dieses Phänomen hat man zuerst bei der Vererbung von Puffmustern auf den Drosophila-Riesenchromosomen beobachtet. Als Maß wurden die Morgan-Distanzen ermittelt. Heute kann durch Erstellung von Kopplungskarten (linkage maps) mittels RFLP-, AFLP- oder STR-Markern (s. Abschn. 4.1.2) die Verteilung der verschiedenen Gene auf einzelnen Chromosomen und Chromosomenabschnitten und damit die Architektur des Genoms ermitteln. Auch der Vergleich solcher Kopplungskarten verschiedener Organismen kann uns interessante Information über die Phylogenie dieser Taxa liefern.
3.5.3 Grundlagen der Populationsgenetik
Die Betrachtung der Allelvariabilität führt unmittelbar zur Frage der Populationsgenetik, die herauszufinden versucht, wie und warum gewisse genetische Varianten in der Population erhalten bleiben, während andere abnehmen oder mit der Zeit gänzlich verschwinden. Variation, Vererbung und natürliche Selektion sind die wichtigsten Prozesse.
Dieser Bereich der sogenannten Mikroevolution ist zum Verstehen des Artbildungsprozesses und damit für das Gesamtverständnis der Evolutionsvorgänge wichtig. Die Grundlagen der Populationsgenetik, die bereits in den ersten Jahrzehnten des 20. Jahrhunderts erarbeitet wurden, sind im EXKURS 3.6 erläutert.
Am Beispiel der Gefiedermorphen des Eleonorenfalken (Falco eleonorae) lassen sich die Mendelschen Regeln und das Hardy-Weinberg-Gesetz gut erläutern. Diese Falkenart brütet in großen Kolonien auf Felsinseln des Mittelmeers und überwintert in gemischten Schwärmen zusammen mit dem Schieferfalken (Falco concolor), mit dem der Eleonorenfalke nah verwandt ist (s. Abschn. 4.4), auf Madagaskar. Beim Eleonorenfalken kann man eine helle und eine dunkle Gefiedermorphe eindeutig erkennen, während beim Schieferfalken nur die dunkle Morphe vorkommt (◘ Abb. 3.45). In einer vereinfachten Betrachtung kann man etwa 29 % der Altvögel des Eleonorenfalken in einer ägäischen Kolonie der dunklen und 71 % der hellen Morphe zurechnen (in Wirklichkeit treten aber auch Übergänge auf). ◘ Tabelle 3.14 zeigt die Gefiedermorphen der Jungvögel in Relation zu den Morphen der Eltern.

Häufigkeit der Genotypen AA, Aa und aa in Abhängigkeit zur Frequenz des Allels a
Häufigkeit der Genotypen AA, Aa und aa in Abhängigkeit zur Frequenz des Allels a

a–d. Gefiedermorphen des Eleonorenfalken (Falco eleonorae) (nach Ristow et al. 1998). a Männchen und b Weibchen der hellen Morphe; c heterozygotes Männchen der dunklen Morphe; d homozygoter Jungvogel der dunklen Morphe. Bildrechte liegen bei Ornithological Society of the Middle East
a–d. Gefiedermorphen des Eleonorenfalken (Falco eleonorae) (nach Ristow et al. 1998). a Männchen und b Weibchen der hellen Morphe; c heterozygotes Männchen der dunklen Morphe; d homozygoter Jungvogel der dunklen Morphe. Bildrechte liegen bei Ornithological Society of the Middle East
Demnach treten bei Paaren der dunklen Morphe sowohl helle (in geringer Zahl) als auch dunkle Nachkommen (überwiegend) auf, während bei Paaren der hellen Morphe ausschließlich helle und keine dunklen Jungvögel beobachtet wurden. Diese Verteilung lässt sich erklären, wenn das Merkmal „dunkel“ (D) dominant und „hell“ (d) rezessiv ist. Bei DD × DD-, DD × dd- und DD × Dd-Paaren hätten 100 % der Nachkommen den dunklen Phänotyp, während bei dd × dd-Paaren alle Nachkommen hell sind. Ebenso sind bei der Kombination von zwei mischerbig dunklen Falken sowohl helle als auch dunkle Nachkommen zu erwarten (◘ Tab. 3.14). Wäre das Merkmal d dagegen dominant, so hätte man in heterozygoten d × d-Kombinationen auch dunkle Morphen bei den Nachkommen finden müssen, was aber nicht der Fall ist.
Da wir den Anteil der hellen Falken, d. h. des homozygot rezessiven Genotyps kennen, können wir über das Hardy-Weinberg-Gesetz (s. EXKURS 3.6) die Häufigkeit der DD- und Dd-Genotypen ermitteln. Da q2 = 0,71, ist nach der Formel p + q = 1, p = 0,157. Demnach ergibt sich eine Genotypfrequenz für DD von 2,5 % und für Dd von 26,5 %; dieses Ergebnis hätte man grafisch auch aus ◘ Abb. 3.44 entnehmen können. Im Phänotyp lassen sich homozygote DD-Falken nur bei genauem Hinsehen von heterozygoten Dd-Genotypen unterscheiden (◘ Abb. 3.45); sie sind lediglich stärker schwarz gefärbt und weisen einen dunklen Schnabel auf. Diese Merkmale lassen sich bereits bei Falken im Jugendkleid erkennen. Da Paarungen zwischen hellen und dunklen Falken rein stochastisch erfolgen und da der Anteil der dunklen Morphe von Jahr zu Jahr zwar schwankt, aber im Trend weder zu- noch abnimmt, kann man annehmen, dass sich die Population im Hardy-Weinberg-Gleichgewicht befindet. Unklar ist, warum sich der Anteil der dunklen dominanten Morphe auf 29 % eingependelt hat.
In vielen Fällen liegt jedoch nicht das einfache Vererbungs- und Populationsmodell vor, wie wir es in den vorangehenden Abschnitten besprochen haben. In der Natur finden wir wesentlich häufiger Merkmale, die genetisch von mehreren Loci gesteuert werden. Ein Beispiel dafür ist der Farbpolymorphismus bei den Schmetterlingen Papilio glaucus in Nordamerika (mimetischer Polymorphismus) oder Papilio memnon in Südostasien, bei denen nicht die Männchen, wohl aber die Weibchen extrem variable und polymorph sind. Gut untersucht ist der Polymorphismus in der südamerikanischen Schmetterlingsgattung Heliconius, bei der die Morphen geographisch getrennt vorkommen.
Wenn nur ein Genlocus pro Merkmal vorkommt, ermittelt die Populationsgenetik Allelfrequenzen; liegt ein Multilocussystem vor, dann werden Haplotyphäufigkeiten bestimmt. Unter einem Haplotyp versteht man eine Variante einer Nucleotidsequenz in einem bestimmten Chromosomenabschnitt oder in der mitochondrialen oder plastidären DNA. Wenn in einem Multilocussystem die Loci auf einem Chromosom liegen, kann man Kopplung beobachten; analog zum Hardy-Weinberg-Gleichgewicht kann man bei Multilocussystemen ein Kopplungsgleichgewicht ( linkage equilibrium ) bestimmen. Liegt kein Kopplungsgleichgewicht vor, so ist dies häufig ein Hinweis darauf, dass die Partnerwahl nicht zufällig erfolgt oder dass die Population sehr klein ist.
Neben diskreten Verteilungen von Merkmalen findet man in der Natur häufig eine kontinuierliche Variation eines Merkmals. In diesen Fällen geht man davon aus, dass mehrere Gene für die Expression eines Merkmals zuständig sind. Ein klassisches Beispiel für dieses Phänomen kann man bei den Darwinfinken auf den Galapagos-Inseln beobachten: 14 Darwinfinkenarten wurden als eigenständige Arten beschrieben, die sich in ihrer Körpergröße, insbesondere aber in der Schnabellänge und -größe unterscheiden, je nachdem, welche Nahrungsnische genutzt wird. Große Finken haben „Nussknacker-Schnäbel“ und können entsprechend große Samen öffnen, während sich Arten mit kleinen Schnäbeln auf kleine Samen spezialisiert haben. Die Schnabelgröße ist vererbbar. Das ist daran zu erkennen, dass Eltern mit größeren Schnäbeln im statistischen Mittel auch Nachkommen mit größeren Schnäbeln produzieren (◘ Abb. 3.46). Für die Vererbung kontinuierlicher Merkmale (Gewicht, Größe, Farben), die in allen Organismen häufig vorkommen, gelten die Regeln der quantitativen Genetik. Die zugehörigen Loci werden als QTL ( quantitative trait loci) bezeichnet. Es würde den Rahmen des Buches sprengen, auf die Genetik von QTLs ausführlicher einzugehen (s. hierzu Camp u. Cox, 2002).

Beispiel für die Vererbung eines kontinuierlichen Merkmals am Beispiel der Schnabelgröße des Galapagosfinken (Geospiza fortis). Eltern mit großen Schnäbeln erzeugen meist auch Nachkommen mit großen Schnäbeln. Da die Nahrungsbedingungen von Jahr zu Jahr schwanken, wirkt sich die Schnabellänge unterschiedlich auf die Fitness und letztlich auf das Reproduktionsverhalten aus
Beispiel für die Vererbung eines kontinuierlichen Merkmals am Beispiel der Schnabelgröße des Galapagosfinken (Geospiza fortis). Eltern mit großen Schnäbeln erzeugen meist auch Nachkommen mit großen Schnäbeln. Da die Nahrungsbedingungen von Jahr zu Jahr schwanken, wirkt sich die Schnabellänge unterschiedlich auf die Fitness und letztlich auf das Reproduktionsverhalten aus
Aus Zwillingsforschung und Ethologie kann man heute sicher sagen, dass auch viele Verhaltensmerkmale der Tiere und des Menschen genetische Komponenten aufweisen und vererbbar sind. Nur in wenigen Fällen wird ein Verhaltensmerkmal auf einem Genlocus liegen, eher müssen wir QTLs erwarten. Über die Struktur der Verhaltensgene und ihre Veränderung in der Evolution können wir heute meist noch keine Aussage machen. Nicht vergessen werden darf in diesem Zusammenhang, dass Verhaltensmerkmale zwar genetisch determiniert sein können, aber häufig plastisch sind, d. h. durch Umwelteinflüsse, Erziehung und Lernen (s. EXKURS 5.9 Abschn. 5.7) gestaltet werden.
3.5.4 Selektion und Mikroevolution
Charles Darwin hat als erster klar erkannt, dass man die Entstehung neuer Arten nur unter Annahme der natürlichen Selektion durch Umweltfaktoren plausibel erklären kann. Es geht dabei nicht um das Überleben des Stärksten, sondern um den Fortpflanzungserfolg. Dabei spielen Kooperation und Altruismus ebenso eine Rolle wie eine körperliche Fitness. Im Unterschied dazu steht die künstliche Selektion, bei der ein Züchter den Fortpflanzungserfolg jener Individuen fördert, die Eigenschaften besitzen, welche vom Züchter gewünscht werden. Am Beispiel der Rassen von Haustieren hatten wir bereits gesehen, dass die künstliche Selektion relativ schnell zu morphologisch unterschiedlichen Rassen kommen kann.
Selektion greift nicht an einzelnen Genen (Genselektion) sondern am ganzen Organismus (Individualselektion) an; nicht nur die Fähigkeit zu überleben entscheidet, sondern der reproduktive Erfolg (Fitness), d. h. die Frage, wie viele Nachkommen ein erfolgreiches Individuum produzieren kann. Doch der evolutionäre Einfluss der natürlichen Auslese zeigt sich nur, wenn man die Entwicklung einer Population (Gruppenselektion) über längere Zeiträume hin verfolgt.
Selektionsvorgänge auf der Stufe von Populationen werden diskutiert, um die Evolution von gewissen Verhaltensmerkmalen (z. B. Altruismus, Kooperation) und den Zusammenhalt in sozialen Gruppen zu erklären (Clutton-Brock 2009). Während die Theorie der Gruppenselektion nach Vero Wynne-Edwards (Wynne-Edwards 1962) heute meist abgelehnt wird, wird heute die Theorie der Verwandtenselektion (kin selection), die von John Maynard Smith und William D. Hamilton entwickelt wurde, eher akzeptiert. Richard Dawkins hat dieses Konzept durch seine Theorie vom „egoistischen Gen“ erweitert und popularisiert. Auf Robert Trivers geht das Konzept des reziproken Altruismus zurück (Trivers 1971). Die Theorie der Multilevelselektion erklärt, dass Selektion nicht nur auf der Gen- und Individualebene auftritt, sondern auch in sozialen Gruppen und Populationen wirkt (Wilson 1975, Wilson u. Wilson 2007).
Ein wichtiges Thema in diesem Zusammenhang ist die sexuelle Selektion. Schon Charles Darwin hatte erkannt, dass die Männchen vieler Arten energieaufwendige morphologische Strukturen (Geweihe, Federn beim Pfau) oder Verhaltensmerkmale (Balz, Schaukämpfe) aufweisen, die für die Fitness oft eher nachteilig sein können. Diese Merkmale erleichtern jedoch den Weibchen, ein Männchen zu wählen, das die besten Gene für die Nachkommen liefert und/oder sich um die Ernährung des Weibchens und der Brut besonders gut kümmern wird. Die sexuelle Selektion ist zum Verständnis des Sexualdimorphismus, zur Erklärung des Geschlechterverhältnis in Populationen und für die Interpretation des Verhaltens und von Sozialsystemen wichtig.
3.5.4.1 Formen der Selektion
Innerhalb der Selektion kann man drei Formen unterschieden:
-
stabilisierende Selektion
-
gerichtete Selektion
-
disruptive Selektion
Stabilisierende Selektion: Liegen konstante Umweltbedingungen vor, so haben Individuen, deren Merkmale nahe dem Mittelwert der Population liegen, eine höhere Fitness als Individuen, die extreme Merkmale aufweisen. Durch die stabilisierende Selektion ist die phänotypische Variabilität herabgesetzt.
Gerichtete (oder transformierende, dynamische oder verschiebende) Selektion: Ändern sich die Umweltbedingungen, so haben Individuen, deren Merkmale vom Mittelwert der Population abweichen unter Umständen einen Überlebensvorteil, wenn sie besser angepasst sind (z. B. Birkenspanner, Biston betularia, s. unten).
Disruptive Selektion: Wenn Individuen, deren Merkmale dem Mittelwert einer Population entsprechen, bevorzugt von Parasiten, Pathogenen oder Fressfeinden dezimiert werden, werden unter Umständen Phänotypen bevorzugt, die unterschiedliche Merkmale ausgebildet haben.
Ein anderes Selektionsregime ist die frequenzabhängige Selektion, bei der ein Phänotyp in Abhängigkeit seiner Häufigkeit in der Population ausgelesen wird. Steigt die Fitness mit der Häufigkeit des Phänotyps, liegt eine positiv-frequenzabhängige Selektion vor. Ein bekanntes Beispiel betrifft das Auftreten einer Warnfärbung. Je mehr Tiere in einer Population warnfarben sind, desto höher der Schutz vor Fressfeinden, die schneller das Signal erlernen und beachten. Bei der negativ-frequenzabhängigen Selektion sinkt die Fitness in Abhängigkeit zur Häufigkeit eines Phänotyps. Ein Beispiel wäre das Auftreten von Nachahmern in Populationen von warnfarbenen Arten (Mimikry). Je häufiger der Nachahmer, desto geringer der Abschreckungseffekt, denn Fressfeinde werden den Unterschied schnell lernen. Weitere Beispiele sind Wirt-Parasit-Beziehungen bei Schnecken und bei Daphnien.
Auf der Gen- und Allelebene kann durch Selektion die Allelfrequenz negativ oder positiv beeinflusst werden. Unter negativer Selektion (purifying selection) versteht man die Entfernung nachteiliger Allele in einer Population. Im Gegensatz dazu führt die positive Selektion zur Auswahl bestimmter Allele. Bei der gerichteten positiven Selektion (directional selection) werden die Träger einzelner Allele bevorzugt, so dass dieses Allel in einer Population häufiger vorkommt. Dadurch werden Allele in einer Population fixiert und die Variabilität (Polymorphismus) vermindert. Bei der ausgleichenden Selektion (balancing selection) werden Allele aufgrund ihrer Häufigkeit (Allelfrequenz) unterschiedlich ausgelesen. Wird ein Allel in der Population häufiger, wird es benachteiligt, wird es seltener so wird es bevorteilt. Damit erhält die ausgleichende Selektion den Polymorphismus in einer Population. Wenn für einen bestimmten Genlocus heterozygote Individuen gegenüber homozygoten bevorzugt werden, spricht man von Überdominanz (overdominance). Ein Beispiel wäre der Erhalt des Polymorphismus der MHC-Gene, die für die Vielfältigkeit der Immunantwort wichtig sind. In diese Gruppe fällt auch die Sichelzellanämie, bei der heterozygote Individuen gegenüber Malaria resistent sind (s. unten).
Ursprünglich wurde angenommen, dass Richtung und Geschwindigkeit der Evolution ausschließlich von der Selektion abhängen. Als man in den 60er Jahren des 20. Jahrhunderts den ungeheuren Allozym-Polymorphismus und später die Häufigkeit der synonymen Nucleotidsubstitution (s. Abschn. 4.1.2) in Populationen und die Grundlagen der molekularen Uhr erkannte, suchte man nach neuen Erklärungsmöglichkeiten. Kimura (1983) entwickelte die Neutrale Theorie der molekularen Evolution, die eine Gegenposition zum Denkmodell der Evolution durch natürliche Selektion darstellen sollte. Beide Standpunkte sind zwischenzeitlich konträr diskutiert worden. Heute erkennt man jedoch, dass sich die Argumente beider Seiten ergänzen und keineswegs so gegensätzlich sind, wie zunächst angenommen. Weder ist die neutrale Evolution so wichtig, noch die natürliche Selektion so unwichtig, wie Kimura vermutete. Die molekulare Evolutionsforschung zeigt, dass Kimuras Vorstellung der neutralen Evolution am ehesten für die häufigen synonymen Mutationen gilt (die zu keinem Wechsel des Phänotyps führen), während die nicht-synonymen Substitutionen den Regeln der natürlichen Selektion unterliegen. Der ursprüngliche Titel „Non-Darwinian Evolution“, (King u. Jukes 1969) entfachte eine Diskussion darüber, ob die Neutrale Theorie und die molekulare Genetik den Darwinismus in Frage stellen. Jedoch erweitern gerade die Erkenntnisse der Molekularbiologie die darwinistische Evolutionstheorie. Sie belegen lediglich, dass nicht alle Mutationen (man denke an die synonymen Substitutionen) der Selektion, sondern der freien Drift unterliegen. Um zwischen Selektion und neutraler Evolution zu entscheiden, kann man den McDonald-Kreitman- und den Hudson-Kreitman-Aguade-Test (HKA-Test) einsetzen.
Die natürliche Selektion wird Individuen bevorzugen, die über eine höhere Fitness verfügen. Fitness-steigernde Allele werden daher in einer Population im Vergleich zu neutralen Allelen zunehmen. Neutrale Mutationen, die mit solchen Fitness-steigernden Allelen gekoppelt sind, werden ebenfalls bevorzugt; man nennt dies genetic hitchhiking. Durch einen selective sweep wird die genetische Variabilität in DNA-Abschnitten in Nachbarschaft zu Fitness-steigernden Allelen reduziert oder eliminiert.
3.5.4.2 Modellierung von Selektionsvorgängen
Die Auswirkungen der Selektion zugunsten oder zuungunsten eines Genotyps lassen sich einfach berechnen. Liegt keine Selektion vor, d. h. wenn der Selektionskoeffizient s = 0 ist, dann befindet sich die Population im Hardy-Weinberg-Gleichgewicht. Ein Selektionskoeffizient von 0,1 gegen aa würde z. B. bedeuten, dass bei gleicher Häufigkeit der Allele A und a in der Parentalgeneration in der ersten Filialgeneration auf 100 AA- 90 aa-Genotypen kommen. Unter diesen Bedingungen beobachten wir (wie in ◘ Tab. 3.15 dargestellt) über vergleichsweise wenige Generationen eine Abnahme von aa und eine Zunahme von AA in der Population.
Kennt man die Veränderung der Allelfrequenzen in zwei aufeinanderfolgenden Generationen, dann lässt sich der Selektionskoeffizient s wie folgt berechnen:
s = Δp / p'· q2 ;
dabei ist p die Allelhäufigkeit in Generation 1 und p´ diejenige in Generation 2.
Bei starker Selektion wird einer der Genotypen eliminiert und der andere in der Population fixiert (gerichtete Selektion). Ein aufschlussreiches Beispiel für eine starke Selektion ist die Zunahme der melanistischen Morphe des Birkenspanners (Biston betularia) in England. In der vor- und frühindustriellen Zeit, d. h. vor 1850, lag der Anteil der hellen Morphe (typica) bei annähernd 100 % (◘ Abb. 3.47a ). Durch zunehmende Umweltverschmutzung hatten sehr seltene dunkle Phänotypen (carbonaria) den Selektionsvorteil, nicht so leicht von Prädatoren, z. B. Vögeln, erkannt zu werden (◘ Abb. 3.47b ). Bereits 1878 lag der Anteil dunkler Spanner bei 45 %, 1908 bei 90 % und 1938 bei 96 %. Aus diesen Daten lässt sich ein Selektionsvorteil von s = 0,33 berechnen; d. h. nach weniger als 100 Generationen kann sich eine Morphe in der Population quantitativ durchsetzen. Inzwischen ist die Luftverschmutzung in England aber deutlich zurückgegangen und parallel dazu hat sich die helle Morphe typica wieder stärker ausgebreitet. Die Birkenspanner-Ergebnisse, die Kettlewell in den 1950er Jahren durchführte, wurden von Majerus in den neunziger Jahren überprüft und wegen der Methodik kritisiert. Da einige der experimentellen Befunde nicht reproduzierbar waren, wurde von Seiten der antievolutionären Kritik die gesamte Biston-Forschung als fragwürdig dargestellt, während Majerus die Schlussfolgerungen im Prinzip für richtig einschätzte. Aus dem Hunsrück, wo die Luftverschmutzung als gering angesehen werden kann, ist bekannt, dass die carbonaria-Form eine Häufigkeit von ca. 50 % hat. Hier gibt es aber viele Buchen, die dunkle Stämme aufweisen und damit den dunklen Birkenspannern einen Überlebensvorteil liefern (T. Schmitt, Universität Trier). Andere Beispiele für aktuelle Selektionsprozesse sind das Auftreten von resistenten Insekten nach längerer Insektizidbehandlung, wie man es z. B. bei der Anopheles-Mücke nach DDT-Behandlung feststellte, oder resistenter pathogener Bakterien nach Antibiotika-Applikation.

a, b Farbmorphen von Biston betularia. a Illustration der Tiere; Form typica (links), Form carbonaria (rechts). b Verteilung der Formen in Großbritannien. Die carbonaria-Mutante überwiegt in Industriegebieten mit starken Rußemissionen (aus: Sperlich 1988)
a, b Farbmorphen von Biston betularia. a Illustration der Tiere; Form typica (links), Form carbonaria (rechts). b Verteilung der Formen in Großbritannien. Die carbonaria-Mutante überwiegt in Industriegebieten mit starken Rußemissionen (aus: Sperlich 1988)
Ein weiteres gut untersuchtes Beispiel zur ausgleichenden Selektion ist die Sichelzellanämie, die in Gebieten mit Malaria vermehrt auftritt. Bei der Sichelzellanämie tritt eine Mutation im Hämoglobingen auf, indem in der Position 6 Glutamin durch Valin ausgetauscht wird. Dies bewirkt, dass das Hämoglobin in den Erythrozyten nicht länger globulär, sondern als fibrilläres Netzwerk vorliegt. Entsprechende Erythrozyten sind sichelförmig und nicht länger als Sauerstoffträger funktionsfähig. Plasmodien, die Erreger der Malaria, überleben in Sichelzellen deutlich schlechter als in normalen Erythrozyten. Deshalb haben Menschen, die für die Sichelzellmutation heterozygot sind, in Malariagebieten einen Vorteil, während Homozygote zwar auch keine Malaria bekommen, aber unter starker Anämie leiden und frühzeitig sterben. Aus diesem Grunde ist der heterozygote Genotyp in Malariagebieten Afrikas deutlich erhöht. In Asien ist die Sichelzellmutation vermutlich unabhängig entstanden (◘ Abb. 3.48).

a, b Potenzielles Verbreitungsgebiet der Malaria tropica und b Häufigkeit des Sichelzellallels (HbS). Nach Bodmer u. Cavalli-Sforza (1976; Bildrechte liegen bei Freeman)
a, b Potenzielles Verbreitungsgebiet der Malaria tropica und b Häufigkeit des Sichelzellallels (HbS). Nach Bodmer u. Cavalli-Sforza (1976; Bildrechte liegen bei Freeman)
3.5.5 Genfluss und genetische Drift
Bisher haben wir die Allelverteilung bzw. den Polymorphismus in einer uniformen Population betrachtet. In der Natur finden wir jedoch häufig Populationen, die geographisch isoliert sind. Am auffälligsten ist dieses Phänomen bei Inselpopulationen, die mit Festlandspopulation nicht länger im Austausch stehen. Jedoch bilden sich auch auf dem zusammenhängenden Festland lokale Verbreitungsinseln, die meistens geographisch z. B. durch physische Barrieren (Gebirge, breite Flüsse, große Wälder, Wüsten) oder zeitliche Trennung bedingt sind. Da viele Tierarten dazu neigen, sich an ihrem Geburtsort anzusiedeln (Philopatrie), wird der „Verinselungseffekt“ weiter verstärkt und ist wesentlich häufiger, als meist allgemein angenommen wird.
In Inselpopulationen oder anderen ähnlich isolierten Populationen können sich die Allelfrequenzen durch Zufallseffekte deutlich verändern. Diese Zufallseffekte, die insbesondere bei kleinen Populationen auftreten, werden als genetische Drift bezeichnet. Sie sind besonders häufig bei neutralen Allelen, die dem Träger weder Vor- noch Nachteil bringen. In kleinen und mittelgroßen Populationen bewirkt der Einfluss der Gendrift, dass neutrale Mutationen häufig durch Zufall fixiert werden.
Wird von einer Art ein neues Areal besiedelt, z. B. eine bisher unbewohnte Insel, so hängt es von der Größe der Gründerpopulation ab, ob alle Genotypen vertreten sind und ob sich die gleiche Allelfrequenz wie in der Ursprungspopulation entwickelt. Da Gründerpopulationen meist klein sind (im Minimum ein trächtiges Weibchen bei Tieren oder ein Samen bei Pflanzen), kommt es meist kurzfristig zu einer genetischen Verarmung (Flaschenhalseffekt), und eine Zunahme der Homozygoten wird beobachtet. Dieser Trend kann leicht erkannt werden, da sich diese Populationen nicht länger im Hardy-Weinberg-Gleichgewicht befinden.
Betrachten wir einmal zwei isolierte Inselpopulationen, deren Allelfrequenzen sich stark unterscheiden. Kommt es aber zu einem Genfluss zwischen beiden Inseln (dazu reicht bereits der Austausch von ein bis zwei Individuen pro Generation aus), so verändern sich die Allelfrequenzen bald wieder in Richtung des Hardy-Weinberg-Gleichgewichtes. Man beobachtet dann wieder eine Zunahme der Heterozygoten und eine Abnahme von Erbkrankheiten, die bei homozygot rezessiven Individuen vermehrt auftreten können.
Die Veränderung der Allelfrequenzen durch Genfluss lässt sich nach folgender Formel berechnen:
Δp = m(pm–p)
dabei ist p die Frequenz von A in der Hauptpopulation und pm die Allelfrequenz von A bei den Einwanderern. Δp beschreibt die Veränderung nach einer Generation; m ist der Einwanderungskoeffizient (Verhältnis der Einwanderer zur Populationsgröße pro Generation).
Über die Allelverteilung kann man die Struktur von Populationen und zugrunde liegende Effekte (z. B. Wahlund-Effekt, FST, Coalescence) ermitteln. Die Populationsgenetik ist ein umfangreiches und schwieriges Gebiet, das sich nicht im Rahmen des vorliegenden Buches abhandeln lässt (näheres in Gillespie 2004; Hartl u. Clark 2007; Hamilton 2009; Hedrick 2009).
Populationsgröße und Selektion beeinflussen demnach signifikant die evolutionären Veränderungen und den Polymorphismus. Ist eine Population klein und die Selektion niedrig, so herrscht im Wesentlichen die genetische Drift vor. In großen Populationen mit deutlicher Selektion beobachtet man dagegen besonders die Effekte der natürlichen Auslese.
Die Paarung erfolgt unter natürlichen Bedingungen nicht immer nach dem Zufallsprinzip, sondern häufig gerichtet: Positives assortative mating liegt vor, wenn verwandte oder ähnliche Partner, negatives assortative mating, wenn nicht verwandte, ungleiche Partner gewählt werden. Ein Spezialfall ist die Inzucht (inbreeding), bei der sich nah verwandte Individuen paaren. Als Konsequenz fortgesetzter Inzucht wird man in einer Population eine Zunahme an Homozygoten und eine Abnahme an Heterozygoten feststellen. Insbesondere wird der Anteil der Nachkommen mit nachteiligen homozygot rezessiven Genen zunehmen; man spricht von Inzuchtdepression. Der inbreeding-Koeffizient F nach Sewall Wright beschreibt die Wahrscheinlichkeit, dass zwei Allele eines Gens in einem Individuum von einem gemeinsamen Gen des Vorfahren abstammen.
Auch in großen natürlichen Populationen tritt Inzucht auf. Da die Jugendmortalität in freier Natur meist sehr hoch liegt, kommt bei fast allen Organismen nur ein kleiner Teil der Nachkommen zur Fortpflanzung. Daher werden sich nachteilige rezessive Homozygote kaum auswirken, da sie meist nicht zur Fortpflanzung kommen. Anders sieht die Situation bei Haus- oder Zootieren aus. Hier fällt die natürliche Jugendmortalität weitgehend fort, so dass man die negativen Inzuchteffekte schon nach wenigen Generationen bemerkt. Durch Mikrosatelliten-Analyse lässt sich der Homozygotiegrad einer Zuchtpopulation leicht ermitteln (s. Abschn. 3.5.1 und 4.1.2). Die erhaltenen Daten geben dem Züchter die Möglichkeit, möglichst wenig verwandte Tiere zur Nachzucht einzusetzen und damit Inzuchtdepression zu vermeiden.
Mutation, Selektion, Drift, Migration und Kopplung gehören demnach zu den Mechanismen, welche die Allel- und Genotypfrequenzen in einer Population beeinflussen. Dieser Evolutionsbereich wird auch als Mikroevolution bezeichnet, die den Wandel in der genetischen Ausstattung einer Population widerspiegelt.
3.5.6 Artbildung (Speziation)
Eine neue Art (zum Thema Artkonzepte s. Abschn. 1.2.3) kann entstehen, wenn Angehörige einer Fortpflanzungsgemeinschaft in zwei getrennte Populationen aufgeteilt werden, deren Mitglieder sich nur noch innerhalb, aber nicht mehr zwischen den Populationen fortpflanzen. Man unterscheidet zwischen allopatrischer Artbildung (d. h. Populationen entwickeln sich in geographischer Isolierung), parapatrischer (d. h. Populationen grenzen aneinander) und sympatrischer (d. h. Populationen überlappen sich) Artbildung (◘ Abb. 3.49) (Coyne u. Orr 2004; Butlin u. Ritchie 2009; Schluter 2009; Sobel et al. 2010).

Schematische Darstellung der verschiedenen Speziationsmodelle
Schematische Darstellung der verschiedenen Speziationsmodelle
3.5.6 Allopatrische Artbildung
Bei den meisten Vertebraten geht man von einer allopatrische Artbildung (Artbildung durch räumliche Trennung) aus. Viele Arten, insbesondere wenn sie sessil leben, zeigen erhebliche geographische Variabilität; bei einer räumlichen Trennung einzelner variabler Populationen können diese sich zu selbstständigen Arten entwickeln. Allopatrische Speziation kann durch Teilung der Ausgangspopulation oder durch Abtrennung peripherer Teilpopulationen zustande kommen. Ein gut belegtes Beispiel ist die Besiedlung vulkanischer Inseln, die in den Ozeanen neu entstanden. Wenn Individuen oder Teilpopulationen vom Festland auf diese Inseln verdriftet werden, können dort neue Arten entstehen, die sich von der Ausgangsart oft deutlich unterscheiden. Auf den Makaronesischen Inseln, zu denen die Azoren, Madeira, die Kanaren und die Kapverden zählen, wird ein erheblicher Anteil der Arten als endemisch angesehen, d. h. sie kommen nur hier vor: von 2400 Pflanzenarten gelten 1120 Arten als endemisch, unter den 105 Brutvogelarten, die wesentlich mobiler sind, liegt der Anteil der Endemiten bei 13 %. Ozeanische Inseln sind daher zur Untersuchung von Artbildungsprozessen besonders gut geeignet.
Als Beispiel ist in ◘ Abb. 3.50 und ◘ Abb. 3.51 die molekulare Phylogeographie der Geckos der Gattung Tarentola und von Rotkehlchen, Goldhähnchen und Blaumeisen dargestellt. Bei den Geckos kann man vermuten, dass Tarentola mauritanica vom afrikanischen (Kanarentiere) oder europäischen Festland (Madeiratiere) auf die Inseln verdriftet wurde (z. B. mit Holzstämmen). Es scheinen mehrere unabhängige Kolonisierungen stattgefunden zu haben. So lassen sich alle Gecko-Haplotypen der Ostinseln, die als T. angustimentalis abgetrennt werden, von einer gemeinsamen Stammart ableiten. Selbst die vorgelagerten kleinen Inseln sind durch Geckos besiedelt, die bereits inselspezifische Haplotypen ausgebildet haben. Die Besiedlung der Westinseln erfolgte in einer zweiten Phase. Betrachtet man die Astlängen in den Phylogrammen, die mit den Divergenzzeiten korreliert sind, erhalten wir einen Anhaltspunkt dafür, wann die Besiedlung der einzelnen Inseln erfolgte. Da die Kanaren vulkanischen Ursprungs sind, kann man ihr Alter gut datieren. Alle Inseln entstanden in den letzten 20 Mio. Jahren (◘ Abb. 3.50a ). Die Astlängen in den Phylogrammen der Geckos von den Westinseln sind größer als die der Ostinseln. Da die Westinseln ein Alter von 10–12 Mio. Jahren und weniger aufweisen, kann man folgern, dass die Geckos frühestens vor 5–10 Mio. Jahren dort ankamen, während die Besiedlung der Ostinseln vermutlich erst später erfolgte.

a, b. a Lage und Alter der Kanarischen Inseln und Madeiras. b Phylogeografie der Geckos (Gattung Tarentola) auf den Makaronesischen Inseln; rekonstruiert über Nucleotidsequenzen mitochondrialer DNA (Cytochrom b, 12S rRNA) (nach Nogales et al. 1998). Im Phylogramm entsprechen die Astlängen genetischen Distanzen. Bildrechte liegen bei Wiley
a, b. a Lage und Alter der Kanarischen Inseln und Madeiras. b Phylogeografie der Geckos (Gattung Tarentola) auf den Makaronesischen Inseln; rekonstruiert über Nucleotidsequenzen mitochondrialer DNA (Cytochrom b, 12S rRNA) (nach Nogales et al. 1998). Im Phylogramm entsprechen die Astlängen genetischen Distanzen. Bildrechte liegen bei Wiley

Phylogeographie der Rotkehlchen, Goldhähnchen und Blaumeisen auf den Makaronesischen Inseln; rekonstruiert über Nucleotidsequenzen des mitochondrialen Cytochrom-b-Gens. Im Phylogramm entsprechen die Astlängen genetischen Distanzen (nach Dietzen et al. 2006, Bildrechte liegen beim Autor)
Phylogeographie der Rotkehlchen, Goldhähnchen und Blaumeisen auf den Makaronesischen Inseln; rekonstruiert über Nucleotidsequenzen des mitochondrialen Cytochrom-b-Gens. Im Phylogramm entsprechen die Astlängen genetischen Distanzen (nach Dietzen et al. 2006, Bildrechte liegen beim Autor)
Auf den Kanaren leben weitverbreitet Rotkehlchen (Erithacus rubecula), die teilweise eigenen Unterarten, wenn nicht sogar Arten angehören (◘ Abb. 3.51a ): Auf Teneriffa und Gran Canaria konnten aufgrund von DNA-Sequenzen zwei eigenständige Taxa erkannt werden, E. r. superbus auf Teneriffa und E. r. marionae auf Gran Canaria. Die Rotkehlchen der übrigen kanarischen Inseln haben Haplotypen (also Sequenzvarianten), die denen der Rotkehlchen auf dem europäischen Festland entsprechen (E. r. rubecula). Man kann die Unterarten auf Teneriffa und Gran Canaria auch an der intensiveren Rotfärbung und den abweichenden Gesängen unterscheiden; aufgrund der genetischen Distanzen und der übrigen Unterschiede könnte diese Besiedlung schon bald nach der Entstehung dieser Inseln vor max. 11,6 bzw. 16 Mio. Jahren erfolgt sein, während die übrigen Inseln deutlich später vom europäischen Festland her besiedelt wurden. Von einigen Autoren werden die beiden Kanaren-Rotkehlchen als eigenständige Arten angesehen.
Ein ähnliches Szenario ergibt sich für den Blaumeisenkomplex (Cyanistes caeruleus) der Makaronesischen Inseln (◘ Abb. 3.51c ), die gut definierbare Inselunterarten ausgebildet haben, welche sich durch Gefiederfärbung und Rufe gut differenzieren lassen. Auf der molekularen Ebene unterscheiden sich die Blaumeisen der Westinseln El Hierro und La Palma (C. c. ombriosus und C. c. palmensis) klar von denen der mittleren (Teneriffa und Gran Canaria) und östlichen Inseln (Fuerteventura und Lanzarote). Aus genetischer Sicht lassen sich die Bewohner von Teneriffa und Gran Canaria, die bislang zur derselben Unterart gerechnet werden, ähnlich wie die Rotkehlchen, in zwei eigenständige Arten unterteilen. Ausgehend von den Blaumeisen des europäischen Festlands gab es möglicherweise mehrere Kolonisationsereignisse, wobei die erdzeitlich jungen Westinseln offenbar zuerst besiedelt wurden.
Spannend ist auch die Phylogeographie der Wintergoldhähnchen (Regulus regulus), die nur wenige der Kanareninseln besiedeln. Von Teneriffa wird eine eigene Unterart (R. r. teneriffae) beschrieben, die sich überraschenderweise von den Goldhähnchen der Westinseln El Hierro und La Palma (R. r. ellenthaleri) unterscheidet. Die Bewohner der Westinseln sind mit den Wintergoldhähnchen der Azoren und des Festlandes offenbar näher verwandt als mit den Bewohnern Teneriffas. Das Wintergoldhähnchen kommt jedoch nicht auf Madeira vor, sondern wird dort durch einen Vertreter aus dem Sommergoldhähnchen-Komplex (R. ignicapillus) ersetzt.
Für andere Archipele (z. B. die Galapagosinseln und Hawaii) lassen sich ähnliche Beispiele aufführen. Als Fazit kann man feststellen, dass sich auf den meisten landfernen Inseln inselspezifische (endemische) Haplotypen und Spezies/Subspezies erkennen lassen, die sowohl genetisch als auch morphologisch (bei Vögeln meist auch akustisch) differenziert sind. Da endemische Inselarten meist auf kleinem Raum vorkommen und kleine Bestände aufweisen, sind viele von ihnen durch Habitatverluste oder andere menschliche Eingriffe viel stärker bedroht als weit verbreitete Festlandarten. Es ist daher nicht verwunderlich, dass unter den nachweislich ausgestorbenen Arten und unter den hochgradig gefährdeten Arten besonders viele Inselendemiten zu finden sind.
3.5.6 Parapatrische Artbildung
Wenn sich die Umweltbedingungen im einem geographisch abgrenzbaren Teilbereich einer Population ändern, kommt es durch natürliche Selektion zur Ansiedlung von Individuen, die an die neuen Bedingungen besser angepasst sind, als die Ursprungsart. Bleiben die veränderten Umweltbedingungen konstant, so können sich zunächst zwei separate Entwicklungslinien herausbilden, die zunächst als Unterarten, später als Arten abzutrennen sind. Da deren Areale aneinander grenzen, findet man häufig eine Hybridzone. Isolationsmechanismen und Hybridisierungsschranken folgen später. Isolationsmechanismen können präzygotisch oder postzygotisch erfolgen. Präzygotische Isolation kann durch Unterschiede im Timing des Brutgeschehens, in Bruthabitaten, Balzverhalten, Morphologie oder gametischer Inkompatibilität hervorgerufen werden. Eine postzygotische Isolation tritt erst nach der Befruchtung ein und reduziert die Vitalität oder Fertilität der hybriden Nachkommen.
Diese Art der Artbildung wird auch als ökologische Artbildung bezeichnet. Ein Beispiel aus der heimischen Fauna ist das Auftreten von Raben- und Nebelkrähe (Corvus corone und C. cornix), die früher in zwei Unterarten untergliedert wurden. In Westdeutschland treten Rabenkrähen auf, während östlich der Elbe die Nebelkrähen überwiegen. Wo beide Taxa aneinander stoßen, bilden sich Hybridzonen aus. Auf der Ebene von DNA-Sequenzen des mitochondrialen Cytochrom-b-Gens unterscheiden sich beide Taxa noch nicht, ein Hinweis darauf, dass immer noch ein Genfluss stattfindet und die Trennung erst vor erdgeschichtlich kurzer Zeit erfolgte. Demnach ist die Artbildung noch nicht abgeschlossen. Erst wenn keine Hybridzone mehr vorhanden ist, können wir von „guten“ Arten sprechen. Dies gilt z. B. für Schwesterartenpaar Sprosser (Luscinia luscinia) und Nachtigall (Luscinia megarhynchos), die sich geographisch ersetzen und nur in Ausnahmefällen hybridisieren. Viele nah verwandte und junge Arten unterscheiden sich morphologisch nur wenig; solche „kryptischen“ Arten lassen sich am ehesten durch DNA-Marker auseinanderhalten (s. Abschn. 4.1.2).
Bei parapatrischen Arten kennt man das Phänomen des character displacements. In den Bereichen, wo zwei nah verwandte parapatrische Arten sich nicht begegnen, ähneln sich beide Arten meist sehr. Dagegen findet man in den Überlappungsbereichen eine starke Ausprägung von unterscheidenden Merkmalen im Aussehen, Verhalten oder ökologischen Ansprüchen.
3.5.6 Sympatrische Artbildung
Wenn Arten innerhalb desselben Verbreitungsgebietes entstehen, so liegt Sympatrie vor. Da sympatrische Arten regelmäßig aufeinander treffen können, sind bei jungen Arten Hybridisierungen nicht selten. Sympatrische Artbildung ist schwerer nachzuweisen und bei Arten mit stabilen Polymorphismen denkbar, bei denen sich eine Verpaarung zwischen speziellen Genotypen bevorzugt einstellt. Insbesondere bei Cichliden, Anolis-Leguanen, Insekten und Palmen gibt es klare Belege für diese Form der Artbildung, wie dies in den Labors von A. Meyer, B. Dieckmann, M. Doebeli, D. Tautz und U. Schlieven eindrucksvoll gezeigt werden konnte. Bei Vögel und Säugetieren ist die sympatrische Artbildung bislang nur selten belegt; ein Beispiel dafür ist die Differenzierung der Witwenvögel, bei denen ein Wirtswechsel zu einer anderen Prachtfinkenart zur generativen Isolierung und damit zur Artbildung führen kann (Dieckmann et al. 2004, Pennisi 2006).
Wie schon in Abschn. 3.4.2 diskutiert spielen die Hybridisierung und Polyploidisierung für Pflanzen (sowohl Allopolyploidisierung als auch Autopolyploidisierung) und sicher auch für einige Tiergruppen eine wichtige Rolle bei der sympatrischen Artbildung, da sie zu Genfluss und komplexen Genomreorganisationen führen: einige Wissenschaftler vermuten, dass 50 % der höheren Pflanzen durch Hybridisierung entstanden. Die Genomevolution von Polyploiden und Hybriden wird durch vielfältige genetische Interaktionen gesteuert, z. B. Rekombination, Genkonversion, konzertierte Evolution, innergenomische Chromosomenaustausche, cytonukleäre Stabilisation und Genstilllegungen. Durch diese Reorganisationen können neue Genkombinationen zustande kommen, die zu neuen Funktionen führen und adaptive Radiationsprozesse erleichtern.
Normalerweise sind Hybride steril. Durch Polyploidisierung (s. Abschn. 3.4.2) kann die Fertilität jedoch u. U. wieder restauriert werden, so dass fertile neue Arten entstehen. Ein Beispiel für Artbildung durch natürliche Hybridisierung ist der Hohlzahn Galeopsis tetrahit (Familie Lamiaceae), der aus G. pubescens und G. speciosus entstand (◘ Abb. 3.52 ). Ein anderes Beispiel stellen Minzen da, die nah verwandt sind und in der Natur oder in Kultur leicht hybridisieren. Viele der Hybride sind steril oder haben eine reduzierte Fertilität; sie können sich jedoch vegetativ vermehren und bleiben daher erhalten. Die bekannte Pfefferminze (Mentha × piperita), die zur Gewinnung von ätherischem Öl angebaut wird, stellt einen sterilen Tripel-Bastard dar, mit M. aquatica und M. spicata als Ausgangsarten, wobei M. spicata selbst ein Hybrid zwischen M. suaveolens und M. longifolia darstellt (◘ Abb. 3.53). In der Pflanzenzucht wird diese Strategie häufig genutzt, indem man Arten kreuzt, anschließend die sterile Hybride durch Colchicin polyploidisiert und fertile Mutanten selektiert; z. B. ist so die beliebte Zierpflanze Primula kewensis (2n = 36) aus P. verticillata (2n = 18) und P. floribunda (2n = 18) gezüchtet worden. In der Natur sind Arthybride besonders häufig in einigen Gattungen der Asteraceae, Brassicaceae, Lamiaceae, Asphodelaceae (Gattung Aloe) und Orchidaceae zu finden.

a–c. Sympatrische Artbildung durch Hybridisierung, am Beispiel der Gattung Galeopsis. Aus den diploiden Ausgangsarten Galeopsis speciosa (a) und G. pubescens (c) entstand eine neue tetraploide Art, G. tetrahit (b)
a–c. Sympatrische Artbildung durch Hybridisierung, am Beispiel der Gattung Galeopsis. Aus den diploiden Ausgangsarten Galeopsis speciosa (a) und G. pubescens (c) entstand eine neue tetraploide Art, G. tetrahit (b)

Hybridisierung bei Minzen. Aus den Ausgangsarten Mentha suaveolens (2n = 24 Chromosomen) und M. longifolia (2n = 24 Chromosomen) entstand M. spicata (2n = 36 und 48 Chromosomen), die mit den Ausgangsarten rückkreuzbar ist. Durch Hybridisierung von M. spicata mit M. aquatica (2n = 96) entstand die sterile M. x. piperita (2n = 66 und 72 Chromosomen)
Hybridisierung bei Minzen. Aus den Ausgangsarten Mentha suaveolens (2n = 24 Chromosomen) und M. longifolia (2n = 24 Chromosomen) entstand M. spicata (2n = 36 und 48 Chromosomen), die mit den Ausgangsarten rückkreuzbar ist. Durch Hybridisierung von M. spicata mit M. aquatica (2n = 96) entstand die sterile M. x. piperita (2n = 66 und 72 Chromosomen)
3.5.7 EXKURS 3.2
3.5.7.1 Mitose, Meiose und Cytokinese
Mitose (Kern- und Zellteilung; Karyokinese und Cytokinese)
Grundsätzlich werden bei den meisten Eukaryoten mehrere Phasen der Mitose unterschieden: Prophase, Prometaphase, Metaphase, Anaphase und Telophase (◘ Abb. 3.26).
Nachdem die Chromosomen im Interphasekern (in der S-Phase) verdoppelt (repliziert) wurden, erfolgt in der Prophase eine Verdichtung des Chromatins zu diskreten Chromosomen, die aus zwei identischen Schwesterchromatiden bestehen (s. oben). Am Ende der Prophase und vor Beginn der Prometaphase lösen sich Kernmembran und Nucleoli auf; die Kernspindel (bestehend aus polaren Mikrotubuli und Kinetochorenmikrotubuli), die in den Zentrosomen (Zentriolen) verankert ist, bildet sich aus. In der Prometaphase heften sich die Mikrotubuli über spezielle Proteinkomplexe (Kinetochoren) an die Centromeren der Chromosomen an. Die Chromosomen werden über die Mikrotubuli zum Zelläquator hin bewegt. In der Metaphase liegen die Chromosomen in der Äquatorialebene aufgereiht vor und Mikrotubuli verbinden die Centromere mit den beiden Spindelpolen. In der Anaphase I verkürzen sich die Kinetochorenmikrotubuli; dadurch werden die Schwesterchromatiden zu den jeweiligen Spindelpolen gezogen. In der darauffolgenden Anaphase II verlängern sich die polaren Mikrotubuli, so dass die Zelle sich zu strecken beginnt. In der Telophase befinden sich die Schwesterchromatiden an den jeweiligen Spindelpolen; die Kernmembran bildet sich wieder aus; auch werden die Nucleoli sichtbar. Gleichzeitig schieben die polaren Mikrotubuli die Zellen weiter auseinander. In der anschließenden Cytokinese (Zellteilung) wird die Mutterzelle in der Mitte ein- und durchgeschnürt. Damit ist die Mitose abgeschlossen und zwei Tochterzellen mit jeweils fast identischen Chromosomensätzen (bis auf Änderungen durch Rekombination) sind entstanden.
3.5.7.1.1 Meiose
Der wesentliche Unterschied zur Mitose liegt in der Paarung homologer Chromosomen und der anschließenden Reduktion des Chromosomensatzes. Es werden zwei Teilungen unterschieden: die 1. und 2. Reduktionsteilung (oder Meiose I und Meiose II) (◘ Abb. 3.26).
In der Prophase der 1. Reduktionsteilung unterscheidet man fünf Stadien: Leptotän, Zygotän, Pachytän, Diplotän und Diakinese. Nach der Verdopplung der DNA der Chromosomen in jeweils zwei Schwesterchromatiden werden diese im Leptotän als kondensierte Chromosomen sichtbar. Im Zygotän beginnt die Paarung der homologen mütterlichen und väterlichen Schwesterchromatiden-Paare (Synapsis). Die jeweils gepaarten Bereiche entsprechen sich auch auf der Sequenzebene, wodurch Crossing over und Rekombination möglich werden. Im Pachytän wird die Paarung der homologen Schwesterchromatiden-Paare abgeschlossen. Im darauf folgenden Diplotän trennen sich die Schwesterchromatiden-Paare, haften aber an den Stellen, an denen ein Crossing over stattgefunden hat, zusammen (Chiasmata, Singular: Chiasma). In dieser Phase sind die Chromosomen entspiralisiert und transkriptorisch aktiv. In der Diakinese hört die Transkription auf und die Chromosomen kondensieren erneut.
In der folgenden Metaphase I werden Kernmembran und Nucleoli aufgelöst und der Spindelapparat bildet sich aus. Die Schwesterchromatiden-Paare ordnen sich in der Äquatorialplatte an, wobei die jeweiligen Centromeren zu den Spindelpolen ausgerichtet sind. Die Kinetochorenmikrotubuli setzen jedoch nicht an den Centromeren einzelner Chromatiden (wie in der Mitose) an, sondern an einem gemeinsamen Centromer jedes Chromatidenpaares. In der meiotischen Anaphase I werden die Schwesterchromatiden-Paare über die sich verkürzenden Kinetochorenmikrotubuli zu den Zellpolen auseinander gezogen. An den Chiasmata trennen sich die rekombinierten Chromosomenbereiche. Die Schwesterchromatiden bleiben dabei über ihr Centromer vereint.
Nach einer kurzen Interphase setzt die 2. Reduktionsteilung ein. Die Chromosomen werden wieder in einer Metaphase, der Metaphase II, angeordnet. Die Chromatiden werden über Kinetochorenmikrotubuli zu den Zellpolen auseinander gezogen. Dieser Vorgang wird mit der Anaphase II abgeschlossen. Nach der Telophase II und Cytokinese liegen vier haploide Zellen (Meiosporen oder Meiogameten) vor, die jeweils über einen haploiden Chromosomensatz verfügen. Auf diese Weise erreicht die Meiose zwei Ziele:
-
Zurückführung des diploiden Genoms auf das haploide Genom und
-
eine intensive Mischung der väterlichen und mütterlichen Gene durch Rekombination.
Die Meiose verläuft bei Pflanzen und Tieren, ebenso bei Männchen und Weibchen grundsätzlich nach dem hier schematisch dargestellten Muster ab. Dies deutet schon daraufhin, dass diese Mechanismen früh in der Evolution entstanden sein müssen. Im Speziellen gibt es jedoch etliche Unterschiede, z. B. in der männlichen und weiblichen Gametogenese.
Veränderung | Varietät |
|---|---|
Gehemmte Knospenentfaltung | Kopfkohl, Weißkohl |
Vergrößerung der Blätter; Anthocyanbildung | Rotkohl |
Verdickung des Stiels und Vergrößerung des Blütenstandes | Broccoli |
Vergrößerung des Blütenstandes; Stauchung der Blütenstandachsen | Blumenkohl |
Verlängerung und Verdickung des Stiels | Markstammkohl |
Starke Verdickung des Stiels | Kohlrabi |
Vergrößerung der Blätter | Grünkohl |
Vergrößerung und Vermehrung der Achselknospen mit Kopfbildung | Rosenkohl |
Gehemmte Knospenentfaltung | Kopfkohl, Weißkohl |
Vergrößerung der Blätter; Anthocyanbildung | Rotkohl |
Verdickung des Stiels und Vergrößerung des Blütenstandes | Broccoli |
Vergrößerung des Blütenstandes; Stauchung der Blütenstandachsen | Blumenkohl |
Verlängerung und Verdickung des Stiels | Markstammkohl |
Starke Verdickung des Stiels | Kohlrabi |
Vergrößerung der Blätter | Grünkohl |
Vergrößerung und Vermehrung der Achselknospen mit Kopfbildung | Rosenkohl |
3.5.8 EXKURS 3.6
3.5.8.1 Grundlagen der Populationsgenetik
Aus dem einfachen Genmodell mit einem Locus und zwei Allelen kann man die Grundlagen der Populationsgenetik ableiten. Zwei Kenngrößen spielen hier eine besondere Rolle:
-
die Gen- oder Allel-Häufigkeit (oder -Frequenz),
-
die Genotypen-Häufigkeit (oder -Frequenz).
In einer Modellpopulation mit den Allelen A und a werden z. B. folgende Genotypen Aa, AA, aa, aa, AA, Aa, AA, Aa beobachtet. Daraus leitet sich die Genotypenhäufigkeit P (für AA), Q (für Aa) und R (für aa) ab; wobei P + Q + R = 1,0 ergibt: P: AA(3/8) = 0,375; Q: Aa(3/8) = 0,375 und R: aa(2/8) = 0,25.
Als Allelfrequenz p für A und q für a ergibt sich: p: A(9/16) = 0,56; q: a(7/16) = 0,44; wobei p + q = 1,0 ist. Aus den Genotypenhäufigkeiten kann man leicht die Allelfrequenz ermitteln:
p = P + 1/2Q und q = R + 1/2Q.
Wenn man die Häufigkeit der Genotypen und Allele in einer Population kennt, so möchte man in der Populationsgenetik wissen, wie sich diese Parameter in nachfolgenden Populationen entwickeln, denn durch natürliche Auslese könnten einzelne Genotypen unterschiedlich gut überleben oder sich fortpflanzen. Falls in großen Populationen Paarungen zufällig und nicht selektiv verlaufen und wenn keine Selektion gegen einen Genotyp eintritt, gelten die Regeln des Hardy-Weinberg-Gesetzes (nach den Wissenschaftlern Godfrey Harold Hardy [englischer Mathematiker] und Wilhelm Robert Weinberg [Arzt aus Stuttgart] benannt, die das Gesetz unabhängig voneinander 1908 erkannten).
Es lautet:
(p + q)2 = p2 + 2pq + q2,
wobei p2 für AA, pq für Aa und q2 für aa stehen.
Aus ◘ Abb. 3.44 kann man, wenn diese Voraussetzung erfüllt ist, die Relation zwischen der Genotypenfrequenz und der Allelfrequenz bestimmen, auch wenn man nicht alle Größen kennt. Das Hardy-Weinberg-Gesetz hat drei wichtige Eigenschaften. Es besagt:
-
Über die Allelfrequenz lässt sich die Genotypfrequenz vorhersagen.
-
Allel- und Genotypfrequenzen ändern sich nicht von Generation zu Generation, wenn ein Hardy-Weinberg-Gleichgewicht vorliegt.
-
Das Hardy-Weinberg-Gleichgewicht stellt sich bereits nach einer Generation ein, wenn die Partnerwahl zufällig erfolgt.
Als Voraussetzungen für die Gültigkeit des Hardy-Weinberg-Gesetzes gelten:
-
Die Population ist so groß, dass Probenfehler und Zufallseffekte keine Rolle spielen.
-
Die Partnerwahl in der Population erfolgt zufällig.
-
Alle Genotypen sind gleich lebensfähig und fertil; d. h. sie haben keinen selektiven Vorteil gegenüber anderen Genotypen.
-
Mutation, Migration und zufällige Gendrift spielen keine Rolle.
Über das Hardy-Weinberg-Gesetz lässt sich auch die Frequenz der Heterozygoten berechnen (◘ Abb. 3.44). Beispiel: Das Merkmal Albinismus ist autosomal rezessiv; d. h. Träger des Merkmals (ihren Epidermiszellen fehlt durch einen Gendefekt das Enzym Tyrosinase, so dass kein Melanin gebildet werden kann) müssen homozygot sein. Wenn die Häufigkeit von albinotischen Individuen in einer Population bei 1:10.000 liegt, so ergibt sich die Frequenz des rezessiven Allels als
 ;
;
q = 0,01 oder 1:100. Da p + q = 1,
ergibt sich für p die Häufigkeit 1 – q = 0,99. Die Frequenz der Heterozygoten ist 2 pq; also
2 × (0,99 × 0,01) = 0,02 oder 2 %.
Man kann das Hardy-Weinberg-Gleichgewicht natürlich auch für Populationen anwenden, in denen drei und mehr Allele in einem Genlocus beobachtet werden. Liegt in einer Population kein Hardy-Weinberg-Gleichgewicht vor, so findet entweder natürliche Auslese statt oder es erfolgt eine gerichtete Verpaarung. Wenn die Population sehr klein ist und stochastische Effekte an Bedeutung gewinnen, kann das Hardy-Weinberg-Gesetz verletzt sein. In diesen Fällen lohnt es sich, den zugrunde liegenden Variablen weiter nachzugehen.
Literatur
Arendt D, Nübler-Junk K (1994) Inversion of dorsoventral axis? Nature 371: 26
Arendt D, Tessmar-Raible K, Snyman H, Dorresteijn AW, Wittbrodt J (2004) Ciliary photoreceptors with a vertebrate-type Opsin in an invertebrate brain. Science 306: 869–871
Bodmer WF, Cavalli-Sforza LL (1976) Genetics, evolution and man. Freeman, San Francisco
Brown WM, George M, Wilson AC (1979) Rapid evolution of animal mitochondrial DNA. Proc Natl Acad Sci USA 76: 1967–1971
Butlin RK, Ritchie MG (2009) Genetics of speciation. Heredity 102: 1–3
Camp NJ, Cox A (2002) Quantitative Trait Loci: Methods and Protocols. Reihe: Methods in Molecular Biology. Band 195. Humana Press, Totowa
Clutton-Brock T (2009) Cooperation between non-kin in animal societies. Nature 462: 51–57.
Coyne JA, Orr HA (2004) Speciation. Sinauer Associates, Sunderland
Cracraft J (2005) Phylogeny and evo-devo: Characters, homology, and the historical analysis of the evolution of development. Zoology 108: 345–356
Dawkins D (1999) The Selfish Gene. Oxford University Press, Oxford
Dawkins R (2000) The Blind Watchmaker. Neuaufl. Penguin, London
Dawkins R (2009) The Greatest Show on Earth. Free Press, New York
de Duve C (1994) Ursprung des Lebens. Präbiotische Evolution und die Entstehung der Zelle. Spektrum, Heidelberg
de Duve C (1995) Aus Staub geboren. Spektrum, Heidelberg (Vgl. auch: Die Herkunft der komplexen Zellen (1996) Spektrum Wiss, S 60–68
Dieckmann U, Doebeli M, Metz JAJ, Tautz D (2004) Adaptive speciation. Cambridge Univ Press, Cambridge
Dietzen C, Garcia-del-Rey E, Delgado Castro G, Witt HH, Wink M (2006) Molecular phylogeography of passerine bird species on the Atlantic Islands. Poster IOC Berlin
Doolittle WF (2000) Stammbaum des Lebens. Spektrum Wiss, S 52–57
Doyle JJ, Gaut BS (2000) Evolution of genes and taxa: a primer. Plant Mol Biol 42: 1–23
Eigen M, Winkler R (1975) Das Spiel. Naturgesetze steuern den Zufall. Pieper, München
Finnerty JR, Pang K, Burton P, Paulson D, Martindale MQ (2004) Origins of bilateral symmetry: Hox and dpp expression in a sea anemone. Science 304: 1335–1337
Futuyma DJ (1990) Evolutionsbiologie. Birkhäuser, Basel
Gilbert SF (2003) Developmental Biology, 7. Aufl. Sinauer, Sunderland
Gillespie JH (2004) Population Genetics: A Concise Guide, 2. Aufl. Johns Hopkins Press
Gobert V, Moja S, Colson M, Taberlet P (2002) Hybridization in the section Mentha (Lamiaceae) inferred from AFLP markers. Am J of Botany 89: 2017–2023
Gobert V, Moja S, Taberlet P, Wink M. (2006) Heterogeneity of three molecular data partition phylogenies of mints related to M. × piperita (Mentha, Lamiaceae). Plant Biol 8: 470–485
Gould SJ (1977) Ontogeny and Phylogeny. Harvard Univ Press, Cambridge/MA
Haeckel E (1884) Die Gastraea-Theorie, die phylogenetische Classification des Thierreiches und die Homologie der Keimblätter. Jena Z Naturwiss 8: 1–55
Hamilton M (2009) Population Genetics. Wiley, New York
Harley RM, Brighton CA (1977) Chromosome numbers in the genus Mentha L. Botanical J of the Linnaean So 74: 71–96
Hartl DL, Clark AG (2007) Principles of Population Genetics, 4. Aufl. Palgrave Macmillan, Houndmills
Hedrick PW (2009) Genetics of Populations, 4. Aufl. Jones & Bartlett, Burlington
Hennig W (1998) Genetik, 2. Aufl. Springer, Heidelberg
Holstein TW (2012) The evolution of the wnt pathway. Cold Spring Harb Perspect Biol. 4 pii: a007922. doi: 10.1101/cshperspect.a007922
Holstein TW, Watanabe H, Ozbek S (2011) Signaling pathways and axis formation in the lower metazoa. Curr Top Dev Biol 97: 137–77
Keeling PC (2004) Diversity and evolutionary history of plastids and their hosts. Amer J Bot 91: 1481–1493
Kimura M (1983) The Neutral Theory of Molecular Evolution. Cambridge Univ Press, Cambridge
King JL, Jukes TH (1969) „Non-Darwinian Evolution“. Science 164: 788–798
Kirschner MW, Gerhart JC (2007) Die Lösung von Darwins Dilemma. rororo Reinbek
Kurland CG, Canback B, Berg OG (2003) Horizontal gene transfer: A critical view. PNAS 100: 9658–9662
Kusserow A et al (2005) Unexpected complexity of the Wnt gene family in a sea anemone. Nature 433: 156–160
Kutschera U (2008) Evolutionsbiologie. 3. Aufl. Ulmer, Stuttgart
Kutschera U, Niklas KJ (2005) Endosymbiosis, cell evolution, and speciation. Theory Biosci 124: 1–24
Li WH, Wu CI, Luo CC (1985) A new method for estimating synonymous and non-synonymous rates of nucleotide substitution considering relative likelihood of nucleotide and codon changes. Mol Biol Evol 2: 150–174
Maier U-G, Douglas SE, Cavalier-Smith T (2000) The nucleomorph genomes of Cryptophytes and Chlorarachniophytes. Protist 151: 103–109
Margulis L (1981) Symbiosis in Cell Evolution. Freeman, New York
Martin W, Koonin EV (2006) Introns and the origin of nucleus-cytosol compartmentalization. Nature 440: 41–45
Martin W, Müller M (1998) The hydrogen hypothesis for the first eukaryote. Nature 392: 37–41 (vgl. auch Spektrum Wiss (1998) 18–20)
Mayr E (1967) Artbegriff und Evolution. Parey, Hamburg
Mayr E (2005) Das ist Evolution. Goldmann, München
Meyer A, Schartl M (1999) Gene and genome duplications in vertebrates: The one-to-four (-to-eight in fish) rule and the evolution of novel gene functions. Curr Opin Cell Biol 11: 699–704
Mindell DP (1997) Avian molecular evolution and systematics. Academic Press, San Diego
Monod J (1971) Zufall und Notwendigkeit. Pieper, München
Müller WA, Hassel M (1999) Entwicklungsbiologie, 2. Aufl. Springer, Heidelberg
Nogales LM, Jimenez-Asensio J, Larruga J M, Hernandez M, Gonzalez P (1998) Evolution and biogeography of the genus Tarentola (Sauria: Gekkonidae) in the Canary islands, inferred from mitochondrial DNA sequences. J Evol Biol 11: 481–494
Pennisi E (2006) Speciation standing in place. Science 311: 1372–1374
Plomin R, DeFries JC, McClearn GE, Rutter M (1997) Behavioral Genetics, 3. Aufl. Freeman, New York
Quadbeck-Seeger, HJ (2008) Die Struktur der DNA – ein Modell-Projekt. Chemie i u Zeit 42: 292–294
Quiring R, Walldorf U, Kloter U, Gehring WJ (1994) Homology of the eyeless gene of Drosophila to the small eye gene in mice and aniridia in humans. Science 265: 785–789
Raff RA, Kaufman TC (1983) Embryos, genes, and evolution. Macmillan, New York
Riedl R (1977) A systems-analytical approach to macroevolutionary phenomena. Quart Rev Biol 52: 351–370
Riedl R (1978) Order in living organisms: A systems analysis of evolution. Wiley, New York
Rieseberg LH, Baird, SJE, Gardner KA (2000) Hybridization, introgression, and linkage evolution. Plant Molecular Biology 42: 205–224
Ristow D, Wink C, Wink M, Scharlau W (1998) Colour polymorphism in Eleonora’s Falcon, Falco eleonorae. Sandgrouse 20: 56–64
Rivera MC, Lake JA (2004) The ring of life provides evidence for a genome fusion origin of eukaryotes. Nature 431: 152–155 (Vgl auch: Groß M (2005) Der Ring des Lebens schließt sich. Spektrum Wiss, S 20–22)
Robinson-Rechavi M, Boussau B, Laudet V (2004) Phylogenetic dating and characterisation of gene duplications in vertebrates: The cartilaginous fish reference. Mol Biol Evol 21: 580–586
Sarich VM, Wilson AC (1973) Generation time and genome evolution in primates. Science 179: 1144–1147
Schlosser G, Wagner GP (Hrsg) (2004) Modularity in development and evolution. Chicago Univ Press, Chicago/IL
Schluter D (2009) Evidence for ecological speciation and ist alternatives. Science 323: 737–741
Seehausen O (2004) Hybridization and adaptative radiation. Trends Ecol Evol 19: 198–207
Slack JM, Holland PW, Graham CF (1993) The zootype and the phylotypic stage. Nature 361: 490–492
Sobel JM, Chen GF, Watt LR, Schemske DW (2010) The biology of speciation. Evolution 64: 295–315
Soltis DE, Soltis PS (1995) The dynamic nature of polyploid genomes. Proc Natl Acad Sci USA 92: 8089–8091
Soltis DE, Soltis PS (1999) Polyploidy: recurrent formation and genome evolution. Trends Ecol Evol 14: 348–352
Soltis PS, Soltis DE (2009) The role of hybridisation in plant speciation. Annu Rev Plant Biol 60: 561–588
Soltis PS, Soltis DE, Doyle JJ (1992) Molecular systematics of plants. Chapman & Hall, New York
Sperlich D (1988) Populationsgenetik, 2. Aufl. Fischer, Stuttgart
Szathmary E, Maynard Smith J (1995) The major evolutionary transitions. Nature 374: 227–232
Thoms SP (2005) Ursprung des Lebens. Fischer, Frankfurt am Main
Thor S, Thomas J (2002) Motor neuron specification in worms, flies and mice: conserved and ‚lost‘ mechanisms. Curr Opin Genet Dev 12: 558–564
Trivers RL (1971) The evolution of reciprocal altruism. Quarterly Revue of Biology 46: 35–57
Turmel M, Otis C, Lemieux C (1999) The complete chloroplast DNA sequence of the green alga Nephroselmis olivacea: insights into the architecture of ancestral chloroplast genomes. Proc Natl Acad Sci USA 96: 10248–10253
Wendel JF (2000) Genome evolution in polyploids. Plant Mol Biol 42: 225–249
Wilson DS (1975) A theory of group selection. Proc Natl Acad Sci USA 72: 143–146
Wilson DS, Wilson EO (2007) Rethinking the theoretical foundations of sociobiology. Quart Rev Biol 82: 327–348
Wink M, Wink C, Ristow D (1978) Biologie des Eleonorenfalken (Falco eleonorae). 2. Zur Vererbung der Gefiederphasen (hell-dunkel). J Ornithol 119: 421–428
Wittbrodt J, Meyer A, Schartl M (1998) More genes in fish? Bioessays 20: 511–515
Wolpert L (2002) Principles of Development, 2. Aufl. Oxford University Press, Oxford
Wright GD, Poinar H (2012) Antibiotic resistance is ancient: implications for drug discovery. Trends Microbiol 20: 157–159
Woese CR, Kandler O, Wheelis ML (1990) Towards a natural system of organisms: Proposal for the domains Archaea, Bacteria, and Eukarya. Proc Natl Acad Sci USA 87: 4576–4579
Wynne-Edwards V (1962) Animal Dispersion in Relation to Social Behaviour. Oliver & Boyd, Edinburgh
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Copyright information
© 2013 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Storch, V., Welsch, U., Wink, M. (2013). Evolution – genetische und zellbiologische Grundlagen. In: Evolutionsbiologie. Springer Spektrum, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-32836-7_3
Download citation
DOI: https://doi.org/10.1007/978-3-642-32836-7_3
Published:
Publisher Name: Springer Spektrum, Berlin, Heidelberg
Print ISBN: 978-3-642-32835-0
Online ISBN: 978-3-642-32836-7
eBook Packages: Life Science and Basic Disciplines (German Language)