
Abstract
The two-way exchange of trace gases between the ocean and the atmosphere is important for both the chemistry and physics of the atmosphere and the biogeochemistry of the oceans, including the global cycling of elements. Here we review these exchanges and their importance for a range of gases whose lifetimes are generally short compared to the main greenhouse gases and which are, in most cases, more reactive than them. Gases considered include sulphur and related compounds, organohalogens, non-methane hydrocarbons, ozone, ammonia and related compounds, hydrogen and carbon monoxide. Finally, we stress the interactivity of the system, the importance of process understanding for modeling, the need for more extensive field measurements and their better seasonal coverage, the importance of inter-calibration exercises and finally the need to show the importance of air-sea exchanges for global cycling and how the field fits into the broader context of Earth System Science.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others

Keywords
- Dissolve Organic Matter
- Coloured Dissolve Organic Matter
- Cloud Condensation Nucleus
- Marine Boundary Layer
- Ozone Flux
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1.1 Introduction
Despite their seemingly low abundances, short-lived trace gases in the atmosphere critically influence global climate change, stratospheric ozone chemistry, and the oxidative capacity of the atmosphere. Because the ocean–atmosphere interface covers a large extent of the Earth’s surface, the ocean is a major control on the atmospheric budget of many trace gases. Further, the chemical, biological, and physical processes that occur around this interface have a large impact on trace gas cycling between the oceanic and atmospheric reservoirs.
In this chapter we present current knowledge on surface ocean cycling processes, atmospheric reactivity and importance, and the influence of air-sea exchange for a suite of trace gases which generally have shorter atmospheric lifetimes than those discussed in Chap. 3 (i.e. CO2, N2O and CH4). We focus not only on research from the past 10 years, but also on topics where much uncertainty remains. Unfortunately, not all of the important issues can be addressed here. Notably missing is detailed information about trace gas cycling in polar regions and particularly over-ice processes, including the role of frost flowers.
The chemical species discussed in the chapter are intimately related to the topics discussed in Chaps. 2, 4, and 5 of this book. More accurate parameterisations of gas exchange will allow for better calculations of oceanic emissions and uptake (Chap. 2). Many of the gases described, such as sulphur gases and non-methane hydrocarbons, are important for marine boundary layer particle formation and cloud coverage (Chap. 4). And, finally, predictive tools for the impacts of future environmental variability on these gases are required (Chap. 5).
In the chapter we have tried to rationalise the use of units wherever possible. However, different units are used by, for example, the atmospheric and ocean communities, the former often using volume units (e.g. ppt by volume), whereas the latter use mass or molar units (both of which are found widely). So, to avoid confusion, we have not tried to standardise on a common unit for all measurements reported here but have allowed the most common usage to remain. In the case of mass units we have tried to use molar units as much as possible but for instance have allowed mass units for global fluxes, since this is what is routinely done in that community.
1.2 Sulphur and Related Gases
Based largely on his own field measurements, Lovelock et al. (1972) suggested that dimethylsulphide (DMS) represents more than 50 % of the natural sulphur emission to the atmosphere, so closing the previous large gap in the global sulphur budget. This discovery stimulated much research as well as the so-called CLAW hypothesis (Charlson et al. 1987, see Sect. 1.2.3.2), which suggested an important role of DMS in climate regulation. Over the last decades, the biological sources of oceanic DMS and its precursor dimethylsulphoniopropionate (DMSP), the ocean–atmosphere flux of DMS, its atmospheric oxidation and climate relevance have been studied in detail. This section reviews the relevant processes of sulphur cycling, via DMS as well as other sulphur compounds, between the ocean and atmosphere and their potential climatic relevance.
1.2.1 DMS(P) in the Surface Ocean
1.2.1.1 Ecosystem Dynamics
DMS originates from dimethylsulphoniopropionate (DMSP), a compound solely produced by phytoplankton. DMSP is thought to regulate osmotic pressure in the cells of some plankton species and may also serve an anti-grazing function (Kirst et al. 1991). The conversion of DMSP to DMS is enzymatically mediated by algal or bacterial DMSP lyases. With respect to regulation of DMSP biosynthesis, currently two hypotheses exist: the anti-oxidant hypothesis (Sunda et al. 2002) and the overflow-metabolism hypothesis (Stefels 2000). Both hypotheses predict increased DMSP production under high-light conditions, but the difference is associated with UV-radiation (UVR), which would result in increased DMSP production in the view of the anti-oxidant hypothesis, but in a reduction according to the overflow hypothesis. The issue is currently unresolved, since the number of studies that differentiate between conditions with and without UVR is very limited.
In marine ecosystems, DMSP, DMS and its oxidation product dimethylsulphoxide (DMSO) are dynamically linked through a myriad of interactions among biological and chemical parameters (Stefels et al. 2007). As a result, algal biomass parameters often show different temporal transitions than the sulphur compounds. In fact, an overarching correlation was found between DMS surface-water concentration and the average radiation in the surface mixed layer, or solar radiation dose (SRD), and not with phytoplankton biomass (Vallina and Simo 2007). This mismatch between source and end product is called the DMS-summer-paradox and has been observed in large areas of the subtropical and temperate oceans (Dacey et al. 1998; Simo and Pedros-Alio 1999).
The simplicity of the correlation between SRD and DMS concentration makes it a useful parameter in large-scale models, but it cannot be used as a mechanistic explanation of underlying processes. Moreover, in other parts of the North Atlantic Ocean, no significant correlation was found between SRD and seasonal variations in DMS (Belviso and Caniaux 2009; Derevianko et al. 2009). On a global scale, the correlation only exists after removing the variance in the global DMS database through averaging into a single value per SRD range (Derevianko et al. 2009). Other factors may therefore play more important roles in determining the distribution of DMS, whereby interactions with light can take place.
Effects of changes in light condition on ecosystem processes are associated with shifts in species composition. The current view is that bacteria are more susceptible to high radiation than phytoplankton species (Herndl et al. 1993; Simo and Pedros-Alio 1999), but shifts also occur within the phytoplankton realm. The ability to produce DMSP may be of competitive advantage for algae, not only because of its potential protective physiological function under high-light conditions, but also because of adverse effects on grazers. Applying different scenarios of a dynamic model to the Sargasso Sea indicated that solar-radiation-induced DMS exudation by phytoplankton is the major contributor to the DMS summer paradox (Vallina et al. 2008). In a review, with the aim to assist sulphur modellers, Stefels et al. (2007) recommend distinguishing six functional phytoplankton groups, based on their size, DMSP production and conversion ability, and interactions with grazers. Shifts in dominance of these groups in association with shifts in bacterial community will have drastic consequences for the turnover of organic carbon and sulphur compounds.
1.2.1.2 DMS Yield
Given the many conversion pathways that originate from DMSP, it is difficult to assess what fraction of DMSP ultimately ends up as atmospheric DMS. As a general rule, the fraction of DMSP produced that is emitted to the atmosphere as DMS is thought to be between 1 % and 10 % (Bates et al. 1994). This far larger DMSP pool therefore leaves room for a several-fold change in the DMS flux, which could arise when shifts in any of the conversion pathways occur. Unfortunately, there is no database of DMSP concentrations comparable to the one for DMS, which makes it difficult to verify underlying ecosystem pathways (see Chap. 5 for discussion of such databases).
The potential for shifts in the DMS yield from DMSP can be illustrated with four scenarios (Fig. 1.1). The terms “Low” and “High DMSP” refer to variation in the species composition of a system, where one system is comprised of species that do not produce large amounts of DMSP – such as diatoms, prochlorophytes and cyanophytes – and the other of species that do produce DMSP – such as haptophytes, chrysophytes and dinoflagellates (Stefels et al. 2007). The term “High stress” refers to conditions that adversely affect algal or bacterial physiology, such as nutrient limitation and light inhibition.


Schematic representation of the major pathways within the marine sulphur cycle and the impact of four different regimes on the relative contribution of each pathway and ultimately on the fraction of DMSP that is emitted to the atmosphere as DMS. “Low DMSP” and “High DMSP” refer to variation in the species composition of a system under blooming or non-blooming conditions. The stress term refers to conditions that adversely affect algal or bacterial physiology, such as nutrient limitation and light inhibition. Thickness of the arrows represents an estimate of the magnitude of the process
Under low-stress conditions (Fig. 1.1a, b), DMS produced directly by algae is usually found to be low (compare exponentially growing algal cultures that have little DMS), and most of the DMSP is released from algal cells through grazing or viral lysis. DMSP has been found to be a major source of reduced sulphur for bacteria, but there is not always a good match between production and consumption. Although bacteria can also cleave DMSP into DMS, the more preferred metabolic pathway is demethylation, which does not yield DMS. About one-third of surface-ocean bacteria are capable of demethylating DMSP (Howard et al. 2006). It provides more energetic benefits than cleavage and is a relatively economic way to assimilate reduced sulphur. Kiene and co-workers proposed a model in which bacterioplankton will prefer the demethylation over the cleavage pathway at low dissolved DMSP concentrations (Kiene et al. 2000) (Fig. 1.1a). It is suggested that the total sulphur demand of bacteria can be derived in this way and, as such, this pathway can be directly linked to bacterial production. At higher dissolved DMSP concentrations (Fig. 1.1b), the DMSP fraction that is not assimilated would then be available to the cleavage pathway. In other words, the fraction that is converted to DMS depends on the biomass and growth of the bacterial community; if the bacterial sulphur demand were relatively small, a larger portion of DMSP would be converted to DMS, either by bacterial or algal enzymes.
Under high-stress conditions (Fig. 1.1c,d), such as nutrient limitation or light inhibition, the relative amount of DMS produced by algae directly is often higher. If DMS is released under conditions of high light, UV radiation will photo oxidise an important part of DMS into DMSO. High-stress conditions are also often detrimental to bacteria (Herndl et al. 1993), which would then result in a reduced S-demand and hence an increase in the relative contribution of the cleavage pathway that produces DMS. The net result of the different scenarios with respect to DMS emission is with our current knowledge difficult to quantify, but may vary by over an order of magnitude.
1.2.1.3 Predicted Impact of Climate Change
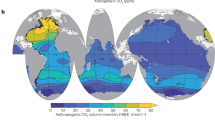
Accurate implementation of DMS cycling in global climate models requires the validation of such models with DMS field measurements. A first global DMS climatology, derived exclusively from field data, was published more than a decade ago (Kettle et al. 1999; Kettle and Andreae 2000). The number of surface DMS data available at that time was approximately 17,000. They are deposited at the Global Surface Seawater (GSS) DMS database maintained by NOAA/PMEL, which is freely accessible at: http://saga.pmel.noaa.gov/dms/. Since the first data collection by Kettle and co-workers, the scientific community was encouraged to upload new data, which increased the number of DMS measurements threefold. Following this increase, a joint initiative of the SOLAS Project Integration, COST Action 735, and EUR-OCEANS was launched to produce an updated DMS climatology (Lana et al. 2011), which is illustrated by the global monthly climatology shown in Fig. 1.2. The DMS climatology is now available and posted for open access at the SOLAS Project Integration website (www.bodc.ac.uk/solas_integration/, see Chap. 5 for further detail).


Monthly climatology of DMS concentrations (nmol L−1). Note that the scale is capped at 15 nmol L−1 to ensure readability of the plots, although only a few specific regions exceed 15 nmol L−1 DMS concentration (Reproduced from Lana et al. (2011) by permission of the American Geophysical Union)
The availability of a DMS climatology allowed verification of model results and hence the inclusion of DMS in global ocean models. A first evaluation of nine models has been published: four global 3D models and five local 1D models (Le Clainche et al. 2010). The (in)ability of the models to predict the summer-DMS maximum that is often observed at low to mid latitudes, appeared to significantly affect estimates of global DMS emissions predicted by the models. A major conclusion of this intercomparison was that prognostic DMS models need to give more weight to the direct impact of environmental forcing (e.g. irradiance) on DMS dynamics to decouple them from ecological processes. Potential controlling effects of light and nutrients on DMSP biosynthesis by algae, resulting in shifts in C:S ratios or species composition, were also found to be key issues.
Global climate change is predicted to result in shallowing of the mixed layer depth at some places with increased storm events at others. This would have drastic impacts on the surface-ocean light environment and nutrient inputs and consequently on plankton development (Schmittner 2005). Predictions of the effects on DMS flux have yielded very different outcomes: Kloster and co-workers predict a 10 % reduction in global DMS flux by the end of the century (Kloster et al. 2007); Vallina et al. (2007) found only a 1 % increase in net global DMS concentrations upon a 50 % increase in atmospheric CO2; Cameron-Smith and co-workers simulated a 50 % increase in DMS flux to the atmosphere over large regions of the Southern Ocean when atmospheric CO2 increases to 970 ppm (Cameron-Smith et al. 2011). The latter result was related to concurrent sea-ice changes and ocean ecosystem composition shifts. In a comparison with previous model outcomes, the authors concluded that increasing model complexity appears to be associated with reduced DMS emission at the equator and increased emissions at high latitudes. The variation in model outcomes illustrates how important it is to improve our understanding of the underlying ecosystem processes.
The different scenarios presented in Fig. 1.1 show that under different environmental conditions the DMS yield from DMSP can shift by possibly an order of magnitude. This can potentially result in changes in DMS emission of several hundreds of percent, an extent that the recent publication from Quinn and Bates (2011), which argues for the retirement of the CLAW hypothesis (see Sect. 1.2.3.2), did not consider possible. Whether such shifts are plausible at the global scale remains to be seen. Since DMS in mechanistic models is dynamically linked to the production of DMSP by phytoplankton, validating this model parameter would help us to understand the underlying controlling factors. Building a DMSP database comparable to the current DMS database would therefore be of great value.
1.2.2 Other Sulphur and Related Gases in the Surface Ocean
1.2.2.1 Carbonyl Sulphide
Carbonyl sulphide (COS) is the most abundant sulphur gas in the atmosphere which has led to the study of air-sea exchange as a source. Measurements in surface seawaters indicate a net flux from the ocean to the atmosphere of approximately 0.1 Tg COS year−1 in the open ocean and 0.2 Tg COS year−1 in the coastal ocean (with considerable uncertainty, Watts 2000; Kettle et al. 2002). COS is produced photochemically in the surface ocean from the interaction of UV light with coloured dissolved organic matter (CDOM) (Uher and Andreae 1997) and its main loss mechanism in the water column is hydrolysis (Andreae and Ferek 1992). Seawater concentrations can vary between 1 and 100 pM (~2 orders of magnitude less than DMS) and typical atmospheric concentrations are about 500 ppt(v) (Kettle et al. 2001). It has been suggested that in situ degradation rates are high in the surface ocean. Diurnal cycling of COS in surface waters has been reported, during which the ocean acts as an atmospheric sink late at night and early in the morning and as a source for the remainder of the day (e.g. Kettle et al. 2001). Approximately 40 % of the total atmospheric source of COS can be accounted for by oceanic emissions.
1.2.2.2 Carbon Disulphide
There are limited published measurements of carbon disulphide (CS2) in the surface ocean. CS2, like COS, is known to have a photochemical source from CDOM (Xie et al. 1998), as well as a biological source (Xie and Moore 1999). There is no significant sink of CS2 in the water column except air-sea exchange, although a small diurnal signal has been observed (Kettle et al. 2001). Surface seawater concentrations between 5 and 150 pM have been reported (highest values in upwelling areas, Kettle et al. 2001). The estimated global oceanic flux to the atmosphere is 0.13–0.24 Tg CS2 year−1 (Xie and Moore 1999). This constitutes between 20 % and 35 % of the total atmospheric source of CS2 (Watts 2000).
1.2.2.3 Hydrogen Sulphide
Hydrogen sulphide (H2S), originally thought to be the dominant volatile sulphur compound in the oceans, makes only a minor contribution to the total marine flux of sulphur to the atmosphere (Watts 2000) and so to the global cycling of sulphur. The ocean source to the atmosphere has been estimated as 1.8 Tg year−1, which is about 25 % of the total atmospheric source of H2S (Watts 2000). In the ocean water column, H2S is produced mainly as part of the hydrolysis of COS (Elliott 1989) and from degradation of particulate organic material. In addition, there is some evidence for a direct algal source (Andreae et al. 1991). H2S is rapidly oxidised in surface waters (2–50 h), with an expected predawn maximum in its diurnal cycle (Andreae 1990). This fast oxidation is made manifest in particular environments, such as upwelled waters off Namibia, where microgranules of elemental sulphur arising from oxidation of sulphide can be detected by remote sensing (Weeks et al. 2002; Brüchert et al. 2009).
1.2.2.4 Methanethiol
Methanethiol (CH3SH), also known as methylmercaptan, is produced from DMSP in a pathway that competes with DMS production and may even be the dominant product in some cases (Kiene et al. 2000). However, CH3SH is thought to be rapidly removed from the water column due to assimilation into proteins by marine bacteria (Kiene 1996) and reaction with dissolved organic matter (Kiene et al. 2000). CH3SH may also have a small photochemical sink in seawater (Flock and Andreae 1996). Seawater concentrations are assumed to be considerably lower than for DMS, although few measurements have been made (Kiene 1996). However, what data there are indicate that levels of CH3SH in the Atlantic Ocean range from 150 to 1,500 pM and are typically about 10 % those of DMS (Kettle et al. 2001). Open ocean values were close to 300 pM, but increased dramatically in upwelling and coastal regions, such that the mean concentration of CH3SH was as high as 20 % that of DMS. In some areas the ratio of CH3SH to DMS was unity. This would make CH3SH the second most dominant volatile sulphur compound in seawater and suggests that CH3SH needs to receive greater consideration in future studies of DMS production or DMSP transformation and in estimates of the flux of biogenic sulphur from the oceans.
1.2.2.5 Dimethyl Selenide
There are considerable similarities between the biogeochemical cycles of the Group VIb elements sulphur and selenium in the marine environment. For example, both can be methylated by micro-organisms to form volatile species which are required to balance their geochemical budgets. However, although DMS as the volatile S form has been extensively studied, there has been much less attention paid to volatile Se. One of the few studies is that of Amouroux et al. (2001), who measured volatile Se in surface waters of the North Atlantic in summer. They found that the dominant forms were DMSe (the direct analogue of DMS) and the mixed S/Se compound DMSeS, and calculate that emissions from this part of the oceans at the period of measurement (6.4 (range 1.4–17.9) nmol m−2 d−1) were of the right order of magnitude to balance the Se budget, if they applied globally. As shown in Fig. 1.3, the positive relationships between seawater concentrations of DMSe and DMS and DMSe and coccolithophore carbon were interpreted to mean that, like DMS, DMSe is an algal product.


Relationship between DMSe and coccolithophorid carbon concentrations (a) and DMSe and DMS (b) in surface waters of the North Atlantic in June 1998 around 60 °N, 20 °W. Error bars represent the relative standard deviation of the reproducibility of the analytical methods on duplicate samples (Reproduced from Amouroux et al. (2001) by permission of Elsevier)
On emission to the atmosphere, the volatile Se compounds will be oxidised and incorporated into particulate phases in pathways which are probably analogous to those discussed in Sect. 1.2.3 for DMS, although the four orders of magnitude smaller flux of volatile Se compared to S means that it’s significance for atmospheric properties will be negligible. However, some of the sea-to-air flux of Se will be dry and wet deposited to the land where it could have implications for human health (Rayman 2000). The pathway of Se from the oceans to land via the atmopshere is supported by measurements of the Se content of mosses in Norway which show decreased concentrations with distance from the sea, indicating a marine source (E. Steinnes, 2003, personal communication).
1.2.3 Atmospheric Sulphur and Related Gases
The recent reviews by Faloona (2009) and Vogt and Liss (2009) provide a detailed picture of the literature regarding sulphur cycling in the marine boundary layer. Here we focus on developments in the last 5–10 years after a few introductory remarks.
Sulphur is present in the atmosphere in both gaseous and particulate forms. It is emitted by natural processes such as from volcanoes, the ocean, vegetation and natural biomass burning as well as anthropogenic processing, mainly combustion of sulphur-containing fuels (oil, coal, anthropogenic biomass burning) and some smeltering processes. Sulphur is emitted in various oxidation states (e.g., -II: COS, H2S, CS2, DMS; +IV: SO2; +VI: H2SO4) and is oxidised in the atmosphere, eventually resulting in sulphate (oxidation number + VI) unless deposited prior to complete oxidation.
The natural marine source is dominated by DMS. In some ocean regions, especially in the northern hemisphere, anthropogenic sulphur emissions from shipping (as SO2 and particulate sulphate) are already of similar magnitude to natural marine sulphur emissions (Corbett et al. 1999) and shipping emissions are expected to increase in the next decades (e.g. Eyring et al. 2010). The estimation of source strengths is not an easy task and comes with large uncertainties. For an oceanic gas, such as DMS, the “bottom-up” flux estimate requires reliable information about the oceanic concentrations (as indicated above) as well as the sea-air flux which depends on its temperature-dependent solubility, wind speed and other poorly quantified factors such as surfactants and sea state. The atmospheric concentration of DMS (typically a few nmol m−3) is often ignored in estimating sea to air fluxes of DMS, since relative to seawater concentrations, it is very small and thereby an error of only a few percent is made. The sea-to-air flux of DMS and similar gases is a non-linear function of wind speed, so it is important to include wind speed variations and possibly even gusts in global estimates of its flux. Kettle and Andreae (2000) showed that depending on the choice of the sea-air-exchange parameterisation, the global flux can vary between 15 and 33 Tg(S) year−1 using the same database for oceanic DMS. Using an approximately threefold larger data set, Lana et al. (2011) estimated the global DMS flux to be 28.1 (17.6–34.4) Tg(S) year−1, which is about 17 % higher than that calculated by Kettle and Andreae (2000).
The “top-down” approach to estimating fluxes involves the balancing of sources and sinks while reproducing atmospheric concentrations. This is usually done with global chemistry transport models. Faloona (2009) summarises the resulting estimates from a series of global models and highlights the uncertainties in the calculation of dry and wet deposition, showing that the largest individual source of variability in the sulphur cycle in global models is the wet deposition of sulphate aerosol, followed by deposition and heterogeneous oxidation of SO2. Nevertheless, the models he cites have a range of DMS flux estimates, from 10.7 to 27.9 Tg(S) year−1, which is roughly within the range of the bottom-up estimates given above. See also Chap. 5 for further information on oceanic DMS climatology and models.
1.2.3.1 Chemistry of Sulphur in the Marine Boundary Layer (MBL)
Once emitted, all reduced sulphur compounds are oxidised in the atmosphere until they are either deposited or reach the highest possible oxidation state for sulphur, +VI (H2SO4, sulphate). In the gas phase, the most important oxidant is the OH radical, which oxidises almost all sulphur compounds, albeit with different rates (see e.g. Atkinson et al. 2007, for a recent compilation of kinetic data). The oxidation of DMS follows either the abstraction pathway (OH at higher temperature, NO3, Cl) or the addition pathway (OH at lower temperature, BrO). Only the abstraction pathway leads to the formation of H2SO4, which is the only sulphur compound with sufficiently low vapour pressure to allow the formation of new aerosol particles (e.g. Kreidenweis and Seinfeld 1988). Many oxidised sulphur compounds (e.g., dimethylsuphoxide (DMSO), methanesulphonic acid (MSA), sulphur dioxide (SO2)) are soluble and can be oxidised further, and often more quickly, in aerosol particles and cloud droplets. The main aqueous oxidants are H2O2, O3, O2 (if metal catalysed) and hypohalous acids (HOCl, HOBr).
The aqueous oxidation of S (IV) by O3 is very pH dependent and only efficient in particles with pH > 6; at high pH, it is orders of magnitude faster than oxidation by H2O2 (see, for example, Fig. 5 in Faloona 2009). This is most relevant for fresh sea-salt particles which initially have the pH of surface ocean water (pH 8.1) or slightly higher due to concentration effects immediately after emission. A particle with a pH 5.6 is in equilibrium with atmospheric CO2 and strong acids, such as H2SO4 and HNO3, often reduce the pH even further. Therefore, aqueous oxidation of S (IV) by O3 is expected to be relevant only during the initial acidification of sea salt particles when the pH is still above the threshold for efficient oxidation by ozone. Depending on the availability of gas phase acidity, sea salt particles are usually acidified within minutes to tens of minutes for sub-micron particles but for super-micron particles this is extended to 1–3 h (von Glasow 2006) allowing for very efficient heterogeneous oxidation of S(IV), as earlier suggested by Sievering et al. (1999) and Chameides and Stelson (1992). Based on laboratory experiments, Laskin et al. (2003) suggested that the surface reaction of OH on sea salt might produce additional alkalinity in sea salt aerosol and hence increase the relevance of the O3 oxidation pathway. However, their extrapolation to ambient conditions was criticised by Sander et al. (2004), Keene and Pszenny (2004), and von Glasow (2006) who all suggested that this pathway might only be minor under atmospheric conditions. Sievering et al. (2004) interpreted their measurements of calcium excesses in coarse aerosol at Baring Head, New Zealand, as evidence for increased buffering of sea salt which would strongly increase the relevance of the aqueous O3 oxidation pathway. A larger scale view of the role of sea salt aerosol alkalinity was presented by Alexander et al. (2005) who measured sulphate aerosol isotopic composition from a cruise in the Indian Ocean that suggest a large role of the ozone pathway for sulphate formation. Using a global 3D model that includes the titration of alkalinity in sea salt aerosol, they managed to reproduce these data. They conclude that the formation of sulphate in sea salt aerosol is limited by the flux of alkalinity and its acidification mainly by HNO3. Alexander et al. (2012) combined oxygen isotope measurements of non-sea salt sulphate from North Atlantic samples with simulations from a global chemical transport model and found that in-cloud oxidation of S (IV) by O3 represents over one-third (36–37 %) of total in-cloud sulphate production on average. They also considered and found the importance of hypohalous acids to be negligible in winter conditions but contributing about 20 % to in-cloud S (VI) formation in summer.
Often it is assumed that OH is the only relevant oxidant for DMS, but this is not necessarily the case. In winter, night-time oxidation by NO3 becomes important (Koga and Tanaka 1993, 1996; von Glasow and Crutzen 2004) and the confirmation of the presence of BrO in the MBL (e.g., Saiz-Lopez et al. 2007b; Read et al. 2008b; Martin et al. 2009) highlights the relevance of this oxidant (see below). The following list is meant to give an idea of DMS lifetimes (at T = 280 K, kinetic data from Atkinson et al. 2007) to reaction with the main gas phase oxidants: τ(OH = 106 molecules cm−3) = 29 h, τ(NO3 = 5 pmol mol−1) = 18 h, τ(BrO = 1 pmol mol−1) = 21 h, τ(Cl = 104 molecules cm−3) = 84 h.
As the formation of SO2 from the intermediates takes additional time, any local increases in DMS concentrations will affect SO2 concentrations only days downwind, which usually equates to hundreds to thousands of kilometres in the MBL (e.g. Woodhouse et al. 2010). It is noteworthy that the oxidation of DMS by BrO is relevant on a global scale even when BrO is present only in mixing ratios of tenths of a pmol mol−1 (e.g. von Glasow et al. 2004; Breider et al. 2010).
The conversion efficiency of DMS to SO2 is important for estimating any climate feedbacks (see Sect. 1.2.3.2). Reaction chamber experiments show the net SO2 yields to be in the range of 0.7–0.8 at room temperature (Barnes et al. 2006 and references therein). However, the measurements were often performed at NOx levels that are much higher than those typically found in the MBL. The laboratory study of NOx oxidation by Patroescu et al. (1999) indicated an increase in the SO2 yield at lower NOx levels. Faloona (2009) compiled the conversion efficiency from many process and global models and found an average conversion efficiency of 71 % and a range of 31–98 %. Estimates of this conversion efficiency in an individual process model cover an even larger range (14–96 %, von Glasow and Crutzen 2004) due to remaining uncertainties in the kinetics, but also due to differences in meteorological, as well as chemical, conditions. The model study by von Glasow and Crutzen (2004) suggests higher conversion efficiencies for tropical regions and lower efficiencies for colder (winter) and cloudy conditions. The conversion efficiency of DMS to SO2 from field studies is also large. Bandy et al. (1992) presented measurements from the northeast Pacific showing a low (but unquantified) DMS to SO2 conversion efficiency. They concluded that SO2 is not an important intermediate in the oxidation of DMS. Measurements on Christmas Island in July and August 1994 indicated a DMS to SO2 conversion efficiency of about 62 % (Bandy et al. 1996), whereas Gray et al. (2010) calculated a DMS to SO2 conversion efficiency of 73 % for more recent measurements at Christmas Island (see below).
The potential of the oxidation of DMS by BrO and Cl has received a lot of attention, initially mainly based on laboratory data and model calculations (e.g., Barnes et al. 1991; Toumi 1994; Ingham et al. 1999; von Glasow et al. 2002) and more recently increasingly based on field measurements. Saiz-Lopez et al. (2004) measured 2–6 pmol mol−1 of BrO at Mace Head, Ireland and stressed its importance for DMS oxidation. Saiz-Lopez et al. (2007b) presented a year-long time series of BrO at Halley, Antarctica showing its presence during most of the year from several pmol mol−1 up to 20 pmol mol−1. Read et al. (2008b) calculated that these BrO levels have a substantial influence on DMS cycling, increasing the production of MSA ninefold. During 8 months of measurements at Cape Verde, Read et al. (2008a) detected on average 2.4 pmol mol−1 of BrO. Mahajan et al. (2010a) showed by comparing model results and observational data that under these conditions BrO would contribute about 50 % to DMS oxidation. Interestingly all published measurements of BrO in the MBL are from the Atlantic, which indicates the need for studies elsewhere. Lawler et al. (2009) presented measurements of Cl2 and HOCl at Cape Verde and their modelling suggested that under these conditions BrO would contribute about 36 % and Cl about 9 % of the DMS oxidation rate, the rest being by OH. For the first time, Lawler et al. (2011) measured HOCl in addition to Cl2 and their surprising results lead to even higher estimates of Cl atom concentrations. The global model study with a 3D aerosol-chemistry model by Breider et al. (2010) suggested a global contribution of BrO to DMS oxidation of about 16 %. Interestingly, they showed that an increase in DMS flux increased the abundance of BrO due to production of additional acidity and resulting enhanced release of bromine from sea salt aerosol. They suggest a possible feedback between DMS, acidity, sea salt and bromine that would act to reduce the sensitivity of the lifetime of DMS to increases in DMS emissions.
Sciare et al. (2000a) concluded, based on observations during a cruise in the Atlantic (October – November 1996), that oxidants other than OH must have been involved in the oxidation of DMS. In contrast, data from Amsterdam Island (Sciare et al. 1998, 2000b, 2001) suggest that additional oxidants for DMS are not needed to close the budget for this location in the southern Indian Ocean. Nowak et al. (2001) discussed observations of DMS and DMSO during the PEM-Tropics B campaign (tropical Pacific, March-April 1999) and show that measured DMSO concentrations are inconsistent with DMS oxidation solely by OH. As the biggest mis-match occurs at night, they discount BrO as a potential reason. Wingenter et al. (2005) concluded, based on hydrocarbon ratios, that an additional oxidant must have been present during their measurements in the equatorial Pacific, equivalent to 6−8 × 104 atoms cm−3 of Cl or 1.3 pmol mol−1 of BrO. However, during the Pacific Atmospheric Sulfur Experiment (PASE, based on Christmas Island, August – September 2007), Conley et al. (2009) concluded that the DMS budget can be closed without invoking oxidants other than OH. Using a 1D, model Gray et al. (2010) also managed to close the DMS budget based on OH oxidation only, but they found that BrO up to levels of about 1 pmol mol−1 would still be consistent with the data. They estimate the conversion efficiency of DMS to SO2 to be 73 %. The presence of 1 pmol mol−1 of BrO would require a 14 % higher ocean flux of DMS, which would also reduce the DMS to SO2 conversion efficiency to 60 %. They found no evidence of relevant non-DMS sources of sulphur to the MBL. Furthermore, they calculated a dry deposition velocity of SO2 that was only 50 % of that used in the global GEOS-Chem model hinting at further unknowns in the global budget of sulphur.
Yang et al. (2011) investigated the sulphur budget during the VOCALS campaign (October–November 2008) in the south-east Pacific (west of Chile). DMS was the predominant source of sulphur to the MBL outside the regions affected by continental outflow. The most important loss for SO2 was in-cloud oxidation. They also found a distinct diurnal cycle in SO42− in the MBL with a rapid rise after sunset and decay for the remainder of the day. The authors suggested that meteorology (night time recoupling of the MBL and precipitation scavenging) was the driving force for this variability
In summary, there appear to be large regional differences in oxidation pathways, which probably is not surprising given the large range of environmental conditions encountered in these different regional studies. In order to reach an improved understanding of the global relevance of the sulphur lifecycle (emission, transformation, deposition), more detailed studies in all seasons are required in the MBL of different oceans.
1.2.3.2 CLAW Hypothesis
Charlson et al. (1987) proposed a potential link between DMS emission from the oceans and production of new aerosol particles (see chapter 4 for a discussion of marine aerosols), resulting in changes in cloud albedo which could influence the temperature in the surface ocean and/or the amount of UV that reaches the ocean. This could, in turn, influence the production of DMS and hence its flux to the atmosphere. In their original paper, they mentioned that the sign of the impact of changes in cloud albedo was uncertain, but a decrease of oceanic DMS production following an increase in cloud albedo would constitute a negative feedback, i.e. a climate stabilising mechanism. This idea is usually referred to as the CLAW hypothesis (using the first letter of the authors’ names, see Fig. 1.4) and has triggered much research since it was first published a quarter of a century ago (Charlson et al. 1987), especially in the field of ocean biogeochemistry and atmospheric particle formation. However, the fact that the hypothesis involves biological, chemical and physical interactions in the ocean and atmosphere over varying time and space scales makes it arguably untestable as a whole. So-called Earth System models that aim to include all components of the earth system (ocean and atmospheric physics and chemistry as well as ocean biogeochemistry) might, in principle, be able to test the CLAW hypothesis. However, many feedbacks are not well understood and are probably non-linear, hence their replication in a global model will be imperfect. Earth System models have shown inconclusive results, some predicting slight increases and others slight decreases in DMS fluxes for future climate simulations (e.g. Bopp et al. 2003; Gabric et al. 2001, 2004; Kloster et al. 2007; Vallina et al. 2007; Cameron-Smith et al. 2011). All studies found large regional differences, so a climate feedback – if present – may not be acting globally. Our limited knowledge of the strong interactions between physics, biogeochemistry and DMS make quantitative modeling of the whole system very challenging.


Schematic of the CLAW hypothesis after Charlson et al. (1987) with additions (in bold) based on von Glasow and Crutzen (2004). Plus (minus) symbols denote positive (negative) feedbacks. Solid arrows indicate ‘leads to’, dotted arrows indicate fluxes (to/from the ocean), whereas dash-dotted arrows indicate uptake to aerosol particles and cloud droplets. Key additions to the CLAW hypothesis are the ‘short-cut’ in the atmospheric sulphur cycle by uptake of DMS and its gas-phase oxidation products on sea salt aerosol with a short lifetime and, therefore, rapid deposition to the ocean; the negative feedback of the growth of existing CCN on cloud albedo; and the importance of sea salt as CCN, thereby possibly reducing the relative role of sulphate aerosol as CCN (Reproduced with permission from von Glasow (2007), copyright CSIRO Publishing)
A crucial link for the CLAW hypothesis to act as a climate stabilising feedback is that an increase of solar radiation at the ocean surface leads to higher DMS concentrations in the ocean. As discussed earlier in this chapter, Vallina and Simó (2007) presented a study where they could in fact show that the “solar radiation dose” (SRD, a measure of the daily integral amount of UV radiation in the surface ocean mixed layer) correlates very well with surface ocean DMS concentrations all over the globe, irrespective of temperature, latitude or plankton biomass. Derevianko et al. (2009) re-examined this relationship and found that only 14 % of the variation in DMS could be explained by SRD, rather than 95 % as found by Vallina and Simó (2007). Derevianko et al. (2009) explain this striking difference by the fact that Vallina and Simó (2007) binned their data in latitude bands, whereas they used the individual data points in their analysis. Additionally, Derevianko et al. (2009) found that most of the correlation between SRD and DMS resulted from locations where the mixed layers deepens, resulting in a reduction of both SRD and DMS, highlighting the importance of ocean physics.
Many studies investigated potential links between DMS, MSA, aerosol and cloud properties especially in the mid and high southern latitudes, as this region is least influenced by anthropogenic activity (see references in Vogt and Liss 2009). These parameters were often found to co-vary but a causal link, as suggested by the CLAW hypothesis, could not be established definitively. Complexities of atmospheric chemistry were pointed out by von Glasow and Crutzen (2004) and von Glasow (2007), stressing that most sulphate derived from atmospheric DMS oxidation will likely be taken up on existing CCN rather than form new ones, which could lead to darker rather than brighter clouds. The role of sea salt as CCN might have also been underestimated in the past (e.g. Lewis and Schwartz 2004; Smith 2007) as well as that of primary organic marine aerosol (e.g. O’Dowd et al. 2004; Leck and Bigg 1999). These effects make detection of any CLAW-related feedbacks even more difficult.
Aerosol indirect effects on climate, by changing cloud reflectivity or lifetime, are potentially very important but are still associated with large uncertainties (e.g. IPCC 2007). Therefore, much research is aimed at elucidating such indirect aerosol effects. These studies have shown an amazing range of complex microphysical and dynamical feedbacks and have also led to the suggestion that on a global scale the aerosol–cloud–precipitation system is buffered (Stevens and Feingold 2009) and might be less sensitive to aerosol perturbations than previously thought. This further complicates the detection of any potential CLAW-like feedback, as the original proposal about a DMS–cloud link was rather simplistic in as much that an increase in DMS would increase cloud albedo. This is not always the case, as cloud dynamics (including heating, mixing with outside air) as well as aerosol size distribution and composition play major roles in the lifecycle of clouds and hence their climatic impacts (e.g. Stevens and Feingold 2009 and reference therein).
A recent study with a global aerosol microphysics model confirmed the importance of DMS in maintaining the aerosol burden (Korhonen et al. 2008). It also highlighted the non-linearity of the processes involved and that the effect of various sulphur sources on aerosol concentrations is not simply additive. They found that DMS emissions increase CCN number between November and April in the latitude band 30–40 °S but at higher latitudes only between December and February, where the effect was also less pronounced.
Wingenter et al. (2007) suggested a CLAW-based climate geo-engineering approach to cool climate by fertilising the ocean and hence increase DMS concentrations. Vogt et al. (2008) pointed out several shortcomings in the argument and calculations and questioned the effectivity of such an approach. Woodhouse et al. (2008) showed, using a global aerosol microphysics model, that an increase in CCN number would be only about 1.4 % for such a fertilised patch. Moreover, they showed how non-regional the impact of this geo-engineering approach would be on CCN number, which is easily explained by the timescales of DMS oxidation and CCN formation and growth, i.e. on the order of days or hundreds to thousands of kilometres. The same group (Woodhouse et al. 2010) used current and future sea surface DMS climatologies to estimate the potential effect of changes in DMS on CCN number. They found a very low sensitivity of CCN number to changes in DMS: a change of only 0.02 % in CCN number per 1 % change in ocean DMS concentrations in the Northern hemisphere, compared to 0.07 % for the Southern hemisphere. Given that expected future changes in ocean DMS concentrations are in the range of 1 %, their study suggests that any CLAW feedback on climate would be very small, similar to interannual variations. The uncertainty of current and future DMS flux estimates has to be noted (see earlier discussion in Sect. 1.2.1.3), but given the very low sensitivity of CCN number to the DMS flux the conclusion is unlikely to be affected.
In a recent review, Quinn and Bates (2011) questioned the impact of DMS on global climate. They discuss all three steps of the feedback loop: 1. DMS as a significant source of CCN to the marine boundary layer; 2. The impact of DMS-derived CCN on cloud albedo; and 3. The response of oceanic DMS production to changes in surface temperature and/or incident solar radiation. Quinn and Bates (2011) conclude that the role of DMS in each step is in fact very limited and that it is time to retire the CLAW hypothesis. They identify significantly more non-DMS sources of marine boundary layer CCN, such as sea salt and organic material. They cite studies indicating that there is little or no theoretical or observational basis for boundary layer nucleation due to DMS derived sulphur species. Finally, they determine that the initial hypothesis was too simple to capture the relationship between aerosols, clouds, and Earth’s albedo. However, Quinn and Bates (2011) do acknowledge that it is possible that a direct link between biological production and climate effects can exist, but that it is more complicated than described in the CLAW hypothesis.
1.3 Halocarbon Gases
Halogenated hydrocarbons (halocarbons) affect the ‘oxidizing capacity’ of the atmosphere, primarily as a result of their influence on ozone concentration in both the troposphere and the stratosphere. Whereas a number of man-made compounds contribute to the chlorine supply, bromine and iodine are supplied to the atmosphere mainly as short-lived natural species. The natural sources exert a stronger influence on ozone now than in the past, since synergetic halogen cycles amplify ozone destruction when coupled with the increased levels of chlorine (Lary 1996; von Glasow 2008).
The temporal and spatial distribution of the highly variable oceanic emissions is uncertain and the oceanic sources of the organic halogen compounds are not well understood. Production and loss processes, both biological and chemical, will influence the oceanic distribution of halogenated compounds and are known to be closely coupled in the marine environment. Other processes affecting halogen distributions include physical transport within the ocean, air–sea exchange, phytoplankton abundance in the open ocean and macroalgae distributions in coastal waters.
Phytoplankton and macroalgae have been identified as sources for halocarbons in seawater. Sea surface temperature, which determines the solubility of the compounds, together with biological production and radiation, is a key factor in controlling seasonal and diurnal variability of concentrations in surface seawater. Anthropogenic sources from the atmosphere and water discharges also contribute to their environmental variability.
The supply of halocarbons to the atmosphere can be altered by changes within the surface ocean associated with climate change, and changes in surface ocean physics, chemistry and biology. Our ability to predict such effects is limited, in part because surface seawater sources and sinks are poorly known and quantified, and the relative importance of production and degradation processes, revealed in laboratory studies, are obscured in the natural environment. It is necessary to investigate the kinetics of photochemical, chemical and biological processes in the water, in organisms, on particles, and in the sea surface microlayer. While in this section we address mainly the oceanic emission of the compounds, it is the case that the background concentrations of many trace gases in ocean surface waters are dictated by the overlying atmospheric mixing ratios. The high atmospheric background concentrations of several gases lead to high surface ocean concentrations, which affects our ability to detect and quantify natural marine sources, particularly for some chlorinated compounds. To improve our understanding of future changes in oceanic emissions and thus their contribution to tropospheric and stratospheric chemistry we need: more measurements in oceanic hot-spot regions; introduction of direct flux measurement techniques; the combining of existing data and intercalibrations within the global database (see Chap. 5 for information on the halocarbon database); and reliable predictive parameterisations. In this section, we outline some of the biogeochemical cycles of halocarbons in surface waters as a prerequisite for understanding future concentration and emission developments.
1.3.1 Chlorinated Compounds
1.3.1.1 Introduction
With the possible exception of methyl chloride (CH3Cl), most of the compounds that contain Cl as the sole halogen do not have a primary ocean source. While CH3Cl is mainly naturally occurring, trichloroethylene (TCE) and perchloroethylene (PCE) are predominantly anthropogenic, and dichloromethane (CH2Cl2) is about 70 % anthropogenic (Cox et al. 2003). The anthropogenic compounds have time varying concentrations in the atmosphere, typically increasing in the mid-twentieth century, and in some cases declining more recently, e.g. CH2Cl2 (at Mace Head, Simmonds et al. 2006), and chloroform CHCl3 (from firn data, Trudinger et al. 2004). The predominance of non-marine sources in the global budget for these gases means that surface water concentrations will be largely controlled by concentrations in the atmosphere. A consequence is that detecting and quantifying natural marine sources of these chlorinated compounds is more challenging than for a gas such as CH3I, which has a very low atmospheric background concentration. Chlorinated gases have widely differing atmospheric lifetimes, ranging from around 7 days for TCE (Simmonds et al. 2006), through 3–4 months for tetrachloroethylene (Olaguer 2002), ca. 5 months for CH2Cl2 (McCulloch and Midgley 1996), ca. 6 months for CHCl3 (O’Doherty et al. 2001), 1–1.5 years for CH3Cl, (Xiao et al. 2010) to ca. 35 years for carbon tetrachloride (CCl4) (Prinn et al. 1999). As the lifetimes are dependent on atmospheric OH, concentrations vary with both season and latitude. Further influences on the latitudinal variation in concentration are the degree to which industrial emissions contribute to the global budget, and the importance of natural terrestrial sources and sinks. Lifetimes of the gases in ocean waters play an additional role in the distributions. Methyl chloride is relatively short-lived on account of microbial consumption, while PCE and TCE are apparently much longer lived in the ocean than in the atmosphere leading to concentrations that increase with depth.
1.3.1.2 Methyl Chloride
Our improving knowledge of sources and sinks of methyl chloride (CH3Cl) has revealed a lesser role for ocean sources than previously thought. Chemical loss in the atmosphere of about 7 × 1010 mol year−1 is matched by a net ocean supply of about 5 % of that amount. It has been found that warm waters tend to be supersaturated with increasing temperature, while cool waters (below about 12 °C) are undersaturated (Moore et al. 1996a). It is clear from decreasing concentrations of CH3Cl with depth in the ocean that CH3Cl sinks exist within the water column. Measurements of isotopically labelled CH3Cl have revealed biological uptake rate constants in the range 0–0.22 d−1 (mean 0.07 d−1) in Southern Ocean surface waters (Tokarczyk et al. 2003a) and up to 0.3 d−1 in coastal waters (annual mean 0.07 d−1, Tokarczyk et al. 2003b). Among the most firmly established marine sources of CH3Cl are those from reactions of CH3I and CH3Br with Cl− in seawater, the rates of which are known to be strongly temperature dependent. There is evidence for more direct biological production of CH3Cl from laboratory studies of algal cultures (e.g. Scarratt and Moore 1996, 1998), though these normally do not uniquely identify the source as the cultured alga rather than associated bacteria. Furthermore, the measured rates are very modest in comparison with estimates of CH3Cl production in ocean waters. For example, taking the most prolific phytoplankton producer of CH3Cl amongst those studied, Phaeocystis, was estimated to be able to account for only 0.5 % of oceanic production (Scarratt and Moore 1998) based on estimates of the abundance of the organism within oceanic blooms and their areal extent. Other limitations of these studies include the major differences that exist between culture conditions and the ocean. Even when production has been demonstrated, it is important to be able to relate it to the growth phase of the organism. Thus, while Ooki et al. (2010) report enhanced concentrations of CH3Cl associated with elevated chlorophyll-a in the NW Pacific (SST 18 °C), the interpretation cannot be more specific than an indication of a biological source, with a wide range of algae, bacteria or other organisms potentially being responsible.
It has more recently been shown that photochemical breakdown of coloured dissolved organic matter in seawater is a source of CH3Cl (Moore 2008). This source is likely to be more significant on a unit area basis in estuarine and coastal waters than in the open ocean, though Ooki et al. (2010) speculate that it could account for highly elevated concentrations in warm, (29 °C) low chlorophyll waters of the subtropical NW Pacific.
1.3.1.3 Dichloromethane
The oceanic distribution of dichloromethane (CH2Cl2) is remarkably different from that of methyl chloride. The short atmospheric lifetime (ca. 5 months) and anthropogenic sources together cause a large asymmetry in atmospheric concentrations between the two hemispheres and, in turn, different atmospherically-supported concentrations in the respective surface waters. Vertical profiles of CH2Cl2 can show increases in concentration with depth, as do the long-lived, anthropogenic Freons (Moore 2004). It appears that CH2Cl2, like Freons, enters the deep ocean at high latitude sites of vertical convection. The lifetime in seawater is apparently long enough to maintain a clear similarity in profile with the very long-lived Freon tracers. Seasonal variations of CH2Cl2 concentration in the atmosphere will tend to supply the compound to the ocean in winter and yield summertime supersaturations in surface waters that do not reflect in situ marine production but rather a recycling of the gas to the atmosphere. While the major atmospheric influences on CH2Cl2 concentration will make smaller natural sources difficult to discern and quantify, it may be predicted that some production occurs in seawater by reactions of chloride with precursors such as chloroiodomethane (CH2ClI), which is both biogenic and formed by light catalysed reaction between di-iodomethane and chloride (Jones and Carpenter 2005). Additionally, the compound may be produced biologically by processes analogous to those involving haloperoxidases that yield dibromomethane. Ooki and Yokouchi (2011) provide evidence for in situ production by quantifying excess saturation anomalies which have had the influence of sea surface temperature rise on CH2Cl2 supersaturation removed, as well as the influence of seasonal change of CH2Cl2 in the atmosphere. Their results provide evidence for efflux to the atmosphere between 10 °S and 40 °S and also for the source being derived from the precursors CH2ClI and CH2Cl2, but their claim for direct biogenic production is less well founded.
1.3.1.4 Tri- and Tetrachloroethylene
There are few published measurements of tri- and tetrachloroethylene (C2HCl3-TCE, C2Cl4-PCE, respectively) in seawater. These two compounds have primarily industrial sources, with production and release to the atmosphere displaying a declining trend in the case of PCE based on measurements at Mace Head (Simmonds et al. 2006). TCE was reported to have relatively constant emissions between 1988 and 1996, as estimated from production data. Surface waters will be influenced by the latitudinal and seasonal variations in atmospheric mixing ratio, tending towards higher and lower concentrations in winter and summer, respectively. Supersaturation has been reported for both compounds in the N. Atlantic during summer (Moore 2001) and it was argued that this is likely to reflect release of gases taken up during winter months. Deep, cooler waters had higher concentrations, increasing with depth, that are likely to result from high latitude ventilation and relative stability of both compounds in seawater. Moore (2001) reported concentration maxima of TCE at a depth of 50 m in the western Atlantic (ca. 21 °N) similar to those frequently seen for biogenic compounds such as isoprene, but physical processes (advection) could provide an alternative explanation. A report of TCE and PCE production by red algae (Abrahamsson et al. 1995) proved controversial with two later studies of the same algae showing no evidence for production (Marshall et al. 2000; Scarratt and Moore 1999). Marshall et al. (2000) suggested that TCE and PCE could potentially be produced in those experiments by dehydrohalogenation of 1,1,2,2-tetrachloroethylene and pentachloroethane, respectively; the significance of any such production is questionable in the absence of data on the precursors in ocean waters. Studies of these compounds in waters of the southern hemisphere, where the atmospherically-supported background concentration is lower, would be especially interesting and useful in identifying and quantifying any marine production.
1.3.1.5 Chloroform
The major sources of chloroform to the atmosphere are soils and anthropogenic emissions, which Trudinger et al. (2004) estimate from firn measurements account for more than half of current emissions. Nightingale et al. (1995) reported that early estimates of the flux of chloroform (CHCl3) from the ocean, based on few measurements and with an assumed high transfer velocity (Khalil et al. 1983), are likely to be too high by about a factor of four. Little has been published on CHCl3 distributions in the ocean, but there is evidence for higher concentrations in both intermediate and deep waters of the Atlantic than near the surface. In the case of TCE, PCE, and CH2Cl2, this has been attributed to influx of the gases in regions of deep convection, coupled with relatively long lifetimes in seawater compared with the atmosphere (Moore 2001, 2004); it is reasonable to interpret the CHCl3 distribution in the same way. Seasonal and spatial fluctuations in both atmospheric concentrations and sea surface temperatures will lead to variations in magnitude, and perhaps direction, of CHCl3 fluxes across the ocean surface.
Nightingale et al. (1995) reported the first direct evidence that some species of macrophytes release CHCl3. It is possible that a chloroperoxidase is responsible, but if the mechanism involves HOCl release, the nature and concentration of halogenated products is evidence that the CHCl3 must be have been produced intracellularly. A macrophyte source would tend to elevate concentrations in coastal waters. Two laboratory studies showed that the benthic microalga, Porphyridium purpureum, produced CHCl3 (Scarratt and Moore 1999; Murphy et al. 2000) by an unidentified mechanism; these findings tell us little directly about producers in ocean waters since the organism studied occurs only in shallow waters.
1.3.2 Brominated Compounds
1.3.2.1 Methyl Bromide
The oceans play an important role in the geochemical cycle of methyl bromide (CH3Br), contributing approximately half of the stratospheric bromine burden. Oceanic emissions contribute roughly 35 % of all known natural and anthropogenic sources to the atmosphere, while industrial production is decreasing due to the Montreal protocol (WMO 2011). The ocean is both a source and a sink of bromine for the atmosphere (0.5 and 0.6 Gmol Br year−1, respectively) in the form of CH3Br (King et al. 2002). Due to declining atmospheric concentrations (Yvon-Lewis et al. 2009), the ocean is now less undersaturated (i.e. closer to equilibrium between the ocean and the atmosphere) with CH3Br than in the period 1996–1998, when atmospheric mixing ratios rose above 10 ppt. Preindustrial mixing ratios of CHBr3 range from 5 to 5.5 ppt, (Saltzman et al. 2008), indicating substantial natural sources. Sea surface temperature (SST) can be used as proxy for the oceanic saturation state of CH3Br (Groszko and Moore 1998; King et al. 2002). While polar and tropical regions are undersaturated the entire year, temperate waters show strong seasonal cycles (Baker et al. 1999), with supersaturations in summer months. Oceanic surface waters with temperatures between 12 °C and 20 °C are, in general, supersaturated. However, Lobert et al. (1995) concluded that coastal and upwelling regions are sources of methyl bromide (CH3Br) to the atmosphere independent of SST, which can possibly be resolved if marine primary productivity is included in the modeling (Anbar et al. 1996). Hydrolysis and reaction with chloride, as well as biological degradation, are the major loss processes from the ocean, and the degradation rates (chemical: 0.04 d−1, biological: 0–0.26 d−1) can match the sea to air exchange rates (Zafiriou 1975; Elliott and Rowland 1995; Yvon-Lewis et al. 2002). Since the consumption reactions are extremely temperature-sensitive, all oceanic temperature variations have large effects on the concentration of CH3Br in seawater and therefore the exchange between the atmosphere and the ocean.
The net flux of CH3Br is also sensitive to variations in the rate of CH3Br production. The natural oceanic production mechanism is unknown but likely phytoplankton in the surface layer are involved. Supersaturations of CH3Br have been observed in coastal waters off Tasmania, especially in the presence of Phaeocystis (Sturrock et al. 2003), possibly due to nutrient limiting conditions. Laboratory studies have revealed CH3Br production from a variety of phytoplankton species (Saemundsdottir and Matrai 1998; Scarratt and Moore 1998) and macroalgae (Gschwend et al. 1985; Manley and Dastoor 1987). However, all observed rates and inferred global estimates are insufficient to support the observed seawater concentrations and global fluxes. The coastal ocean can be a highly productive region for CH3Br, due to enhanced biological processes, and including its emissions may increase the estimates of global oceanic emissions by 1–9 % (Hu et al. 2010). While abiotic production of CH3Br can occur from the degradation of organic matter (Keppler et al. 2000), the relative importance of this process compared to biological generation, likely involving methyl transferases (Wuosma and Hager 1990), is not known.
Future work should include increasing the amount of data representing global coastal regimes and information on seasonal variations. Since the direction and magnitude of CH3Br exchange between the atmosphere and ocean is very sensitive to temperature and marine productivity, future measurements of marine CH3Br, temperature and primary production should be combined with models to determine the relationship between marine biological activity and CH3Br production.
1.3.2.2 CHBr3, CH2Br2 and Other Polybrominated Methanes
The oceans are also a source of reactive bromine to the atmosphere in the form of short-lived brominated methanes, including bromoform (CHBr3) and dibromomethane (CH2Br2). These compounds represent the largest known natural contribution to atmospheric organic bromine and are recognised as an important source of reactive bromine to the troposphere and lower stratosphere, where they may contribute up to 40 % of O3 depletion in mid latitudes (Salawitch et al. 2005; Salawitch 2006; Yang et al. 2005; WMO 2011).
The magnitude of the oceanic emissions is uncertain and ranges from 3 to 22 Gmol Br year−1, with a recurring mean of around 10 Gmol Br year−1 for CHBr3, and from 0.5 to 3.5 Gmol Br year−1 for CH2Br2 (Quack and Wallace 2003; WMO 2011). Elevated atmospheric concentrations have been observed over the tropical oceans (Class and Ballschmiter 1988a; Atlas et al. 1993; Schauffler et al. 1998) and were linked to oceanic supersaturations of the compounds and especially to productive upwelling areas. Equatorial surface waters of the tropical Atlantic were indeed identified as a significant CHBr3 source to the atmosphere, with CHBr3 production occurring in the deep chlorophyll maximum (Quack et al. 2004). Generally the oceanic distributions of the brominated compounds, bromoform, dibromomethane (CH2Br2), dibromochloromethane (CHBr2Cl) and bromodichloromethane (CHBrCl2), are associated with the abundance of phytoplankton in the open ocean and macroalgae in the coastal waters (Baker et al. 1999; Carpenter and Liss 2000; Nightingale et al. 1995; Arnold et al. 2010), with CHBr3 showing an order of magnitude higher concentration compared with the other compounds in source regions. Pronounced seasonality of the brominated compounds has been observed in coastal regions, with elevated concentrations during summer months (Klick 1992; Hughes et al. 2009; Orlikowska and Schulz-Bull 2009). Diurnal variations are also observed with elevated concentrations around midday (Ekdahl et al. 1998; Karlsson et al. 2008).
Laboratory studies have shown CHBr3 production from arctic diatom cultures (Moore et al. 1996b). Since then, high field concentrations of brominated trace gases have generally been found to be associated with diatom-rich open waters, especially upwelling systems (e.g. Baker et al. 2000). Indeed, significant correlations with CHBr3 were found for low concentrations of diatom marker pigments in the Mauritanian upwelling. However, CHBr3 concentrations do not continue to increase with high diatom abundances and chlorophyll a values (Quack et al. 2007). Biological sources of CHBr3 also correlated with degradation pigments and the decay of organic matter. Cyanobacteria have been shown to be a bromocarbon source in the Baltic Sea (Karlsson et al. 2008). The parameterisation of CHBr3 concentration with chlorophyll a and mixed layer depth has been tested (Palmer and Reason 2009) and found in some tropical areas to satisfactorily reproduce observed concentrations. In coastal regions macroalgae are thought to be the major marine sources (Manley et al. 1992; Nightingale et al. 1995; Carpenter and Liss 2000) of bromocarbons, while anthropogenic contamination by industrial or municipal effluents may overwhelm the natural sources in some areas (Quack and Wallace 2003). While advection of different water masses generally influences the oceanic distribution, oceanic supersaturations of the compounds can also be caused by the advection of coastal waters enriched in macroalgal releases (Carpenter and Liss 2000; Raimund et al. 2011) and emissions from floating macroalgae (Moore and Tokarczyk 1993).
The biological production of polybrominated compounds occurs during the enzymatic oxidation of bromine by bromoperoxidases and chloroperoxidases in the presence of hydrogen peroxide, resulting in the halogenation of organic compounds with activated terminal methyl groups (Theiler et al. 1978; Neidleman and Geigert 1986). The formation of CHBr3 during the oxidation of organic matter is chemically favoured over CH2Br2, since intermediate products are stabilised by additional halogen atoms, and thus CHBr3 is the major product in oceanic environments (Wade 1999). The other bromocarbons have been identified as minor products from phytoplankton and macro algae cultures (Theiler et al. 1978; Gschwend et al. 1985; Manley et al. 1992; Tokarczyk and Moore 1994; Nightingale et al. 1995). CHBr3 has generally longer lifetimes compared with CH2Br2 in the oceanic environment under aerobic and anaerobic conditions, though in the case of the reductive hydrogenolysis, CHBr3 reacts faster than CH2Br2 (Bartnicki and Castro 1994; Goodwin et al. 1997). Thus reductive hydrogenolysis of CHBr3 could be a process that generates CH2Br2 in anoxic environments (Vogel et al. 1987; Tanhua et al. 1996). CH2Br2 concentrations have indeed been found to anticorrelate with CHBr3 and increase in deeper waters, suggesting that this process may be possible (Quack et al. 2007). Alternatively, advection of subducted open ocean waters could possibly explain this pattern. Chemical conversion losses from hydrolysis, with half-lives of 183 years for CH2Br2 and 686 years for CHBr3 (Mabey and Mill 1978), and halogen exchange, with a half-live of 5–74 years for CHBr3 (Geen 1992), also serve as sources for other brominated compounds, especially for the mixed bromochloromethanes. The progressive abiotic substitution of bromine in CHBr3 with chloride is suggested as the main oceanic source for dibromochloromethane (CHBr2Cl) and bromodichloromethane (CHBrCl2) in the ocean, and an in situ contributor of chloroform (CHCl3) (Class and Ballschmiter 1988b).
The background concentration ratio of trace gases in the surface ocean is strongly influenced by air-sea exchange with the overlying atmosphere. Since CH2Br2 has a longer lifetime in the atmosphere (WMO 2011) than CHBr3, the concentration ratio between CH2Br2 and CHBr3 in both atmosphere and ocean increases away from coastal source regions towards the open ocean. Thus, remote open ocean surface waters contain relatively more CH2Br2 than CHBr3, giving ratios up to >1, while ratios in source regions are around 0.1. Transport of halocarbons may occur through the atmosphere from coastal source regions to ocean surface waters (Carpenter and Liss 2000).
Future work in this area should include halocarbon measurements in water and air in concert with phytoplankton biomass and productivity, chemical and physical parameters. In this way their distributions and correlations can be evaluated and coupled with numerical modeling of source and sink processes (Hense and Quack 2009).
1.3.3 Iodinated Compounds
Transfer from the ocean reservoir to the atmosphere and then to land is an important pathway in the biogeochemical cycle of iodine, with important implications for human health. The iodocarbons, including volatile mono-, di-, and tri-halogenated organic compounds, are produced in seawater and are believed to play an important role in mediating this transfer.
1.3.3.1 Iodomethane
One volatile organic iodine compound that has generated much interest is iodomethane (CH3I) with estimates of the sea-to-air transfer rate ranging from 0.9 to 9.2 Gmol year−1 (reviewed by Bell et al. 2002). These fluxes are far in excess of those reported for other sources of CH3I including biomass burning (<0.1 Gmol year−1, Andreae et al. 1996), rice paddies (0.1–0.5 Gmol year−1, Muramatsu and Yoshida 1995), peatlands (< 0.1 Gmol year−1, Dimmer et al. 2001) and wetlands (< 0.1 Gmol year−1, Dimmer et al. 2001), making the oceans the dominant source of CH3I to the atmosphere.
CH3I production has been observed in experimental incubations of marine seaweeds (Nightingale et al. 1995; Carpenter et al. 2000), phytoplankton (Moore et al. 1996b; Manley and de la Cuesta 1997), cyanobacteria (Smythe-Wright et al. 2006; Brownell et al. 2010; Hughes et al. 2011), bacteria (Amachi et al. 2001; Fuse et al. 2003), detrital aggregates (Hughes et al. 2008b) and in irradiated sterile seawater, suggesting photochemical formation (Moore and Zafiriou 1994; Richter and Wallace 2004). The relative importance of each of these sources to the oceanic inventory of CH3I is currently unknown. Estimates of global CH3I production by seaweeds (0.4 × 10−2 Gmol year−1, Manley and Dastoor 1988) and phytoplankton (0.8 × 10−2 Gmol year−1, Manley and de la Cuesta 1997) suggest that these organisms do not contribute significantly. However, the calculations on which these estimates are based rely on the extrapolation of results obtained from surveys of a limited number of organisms under laboratory conditions which may not be a true reflection of the natural situation. This is an important consideration as algal CH3I production has been found to vary depending on the species and environmental conditions, such as nutrient-limitation (Smythe-Wright et al. 2010), grazing (Smythe-Wright et al. 2010), light-levels (Moore et al. 1996b; Laturnus et al. 1998), mechanical damage (Nightingale et al. 1995) and desiccation (Nightingale et al. 1995). The only sources which some studies have suggested could contribute significantly to global or regional CH3I production in the marine environment are photochemistry and cyanobacteria. Richter and Wallace (2004) suggest that 50 % of the average daily flux of CH3I from the tropical Atlantic surface layer could be due to photochemistry.
The extrapolation of laboratory production rates suggests that the marine cyanobacterium Prochlorococcus could contribute significantly to CH3I production in the eastern tropical Atlantic (Hughes et al. 2011) and calculations presented in Smythe-Wright et al. (2006) suggest that this organism is also an important global source producing 4.3 Gmol year1. However, orders of magnitude variability in the CH3I production rate have been observed in laboratory cultures of Prochlorococcus between the three studies on this topic published to date (Smythe-Wright et al. 2006; Brownell et al. 2010; Hughes et al. 2011). Hughes et al. (2011) suggest that CH3I production by Prochlorococcus is strongly dependent on cell physiological state. Therefore, the different incubation conditions used in these three studies could explain this variability and should be considered when extrapolating laboratory production rates to the natural environment. Other sources such as bacteria (Amachi et al. 2001; Fuse et al. 2003) and detrital aggregates (Hughes et al. 2008b) are not understood sufficiently to allow accurate global flux estimates to be made.
Several mechanisms for CH3I production in seawater have been proposed:
-
1.
A photochemical CH3I source has been suggested to involve a radical recombination pathway (Eq. 1.1, Moore and Zafiriou 1994) but more information on the mechanism is needed.
-
2.
Proposed pathways by which biological activity produces CH3I include methyl group transfer to iodide via S-adenosyl-L-methionine (SAM)-dependent methyltransferases (Eq. 1.2, Amachi et al. 2001; Ohsawa et al. 2001), the production of methylating agents such as methylcobalamin or methyl vitamin B12 (Eq. 1.3, Manley 1994) and the breakdown of higher molecular weight organic compounds (Fenical 1982).
Loss processes for CH3I in seawater include gas exchange in surface waters (Liss and Slater 1974), nucleophilic (SN2) substitution with Cl− (Elliott and Rowland 1993), and possibly bacterial breakdown (Bell et al. 2002). Hydrolysis is generally an order of magnitude lower than nucleophilic substitution (Moelwyn-Hughes 1938) and the nature of the absorbance cross-section of CH3I means that photolysis is not significant (Zika et al. 1984). The relative importance of gas exchange and the reaction with Cl− varies depending on windspeed and seawater temperature, but under certain conditions loss rates due to these processes can be comparable (Bell et al. 2002). A wide range of methanotrophic and nitrifying bacteria have been found to be capable of breaking down CH3I (McDonald et al. 2002). Methyl halides are molecular analogues of methane (CH4) and ammonium (NH4+) and therefore act as competitive inhibitors of methane monooxygenase (MMO) and ammonium monooxygenase (AMO). To account for discrepancies between modelled and measured CH3I concentrations in mid to high latitude ocean waters, Bell et al. (2002) propose that the bacterial breakdown rate is 0.24 d−1. However, the mean non-chemical loss rate observed in incubations of North Atlantic seawater by Moore (2006) using 13CH3I as a tracer was 7 % d−1, suggesting that this process is less significant.
1.3.3.2 Other Mono-Iodinated Iodocarbons
Other volatile mono-halogenated iodocarbons including iodoethane (C2H5I), 1- and 2-iodopropane (C3H7I) and the iodobutanes (C4H9I) have been detected in seawater (Klick and Abrahamsson 1992) but their generally low concentrations and their relatively higher solubility means that they make a much smaller contribution to sea-to-air iodine flux than CH3I. Their production has been observed alongside CH3I in incubations of marine seaweeds (Carpenter et al. 2000), phytoplankton (Moore et al. 1996b) and detrital aggregates (Hughes et al. 2008b).
1.3.3.3 Di- and Tri-Halogenated Compounds
The marine environment is also known to be a source of reactive di- and tri-halogenated iodocarbons including chloroiodomethane (CH2ClI), di-iodomethane (CH2I2), and bromoiodomethane (CH2BrI), which, due to their relatively short atmospheric lifetimes, play a more important role in the chemistry of the marine boundary layer (MBL) than the mono-iodinated organics. Martino et al. (2009) have measured iodoform in a laboratory study but, as far as we are aware, there are no published measurements in marine waters. Recent work has also examined the relative importance of organic versus inorganic iodine (i.e. I2) flux to the marine boundary layer with varying results (e.g. Jones et al. 2010; Mahajan et al. 2010b). The production of the di- and tri-halogenated iodocarbons has been seen in laboratory experiments involving marine seaweeds (Carpenter et al. 2000), phytoplankton (Moore et al. 1996b), bacteria (Fuse et al. 2003) and ozone (O3) deposition (Martino et al. 2009). Although they share common sources, the mechanisms of formation for the mono-, di- and tri-halogenated iodocarbons are different. The biological formation of reactive iodocarbons is known to be catalysed by haloperoxidase enzymes which have been detected in a wide range of marine organisms and catalyse the oxidation of halide ions by hydrogen peroxide (H2O2) and formation of HOI/I2 (Butler and Walker 1993). The O3 deposition pathway identified by Martino et al. (2009) involves the reaction between O3 and I− at the sea surface to form I2/HOI (Eq. 1.4a, b). Another mechanism by which the reactive iodine precursor of di- and tri-halogenated iodocarbons could be formed in the marine environment is the reaction between reactive oxygen species (e.g. H2O2) and I− released during oxidative stress, as was observed for the kelp Laminaria by Kupper et al. (2008). In all cases the I2/HOI formed reacts with dissolved organic substrates (Truesdale and Luther 1995) to yield the di-/tri-halogenated organics.
Losses of the more reactive iodocarbons in seawater are dominated by gas exchange and photolysis. Photolysis is an important breakdown pathway in surface waters for compounds such as CH2I2 (Jones and Carpenter 2005; Martino et al. 2005). For example, Martino et al. (2005) report photolytic lifetimes of CH2I2 and CH2ClI in surface waters (at 15 °C and a solar zenith angle of 0 °) are on the orders of minutes and hours, respectively. The photolytic breakdown of CH2I2 is also an iodocarbon source as this generates CH2ClI with a yield of up to 30 % (Martino et al. 2005). As with all trace gases, the rate of exchange with the atmosphere can be quantified using known formulations (Liss and Slater 1974) and parameterisations (e.g. Nightingale et al. 2000). In general, published rates of sea-air transfer consider only the water-phase resistance to transfer (kw). However, recent work (Archer et al. 2007; Johnson 2010) suggests that the air-phase resistance to transfer (ka) is also an important control on the sea-to-air flux of the more soluble reactive iodocarbons, and that the total resistance to transfer (Kw) should be used in the calculation of emission rates. For example, the flux rate of CH2I2 is reduced by 40 % at 5 °C, and 20 % at 30 °C when Kw is used to calculate the flux instead of kw (Johnson 2010). Further information on air-sea gas transfer rates can be found in Chap. 2.
1.3.4 Halogens in the Marine Atmospheric Boundary Layer
This section describes recent advances in understanding the impacts of halogen chemistry in the marine boundary layer. During the past decade there has been an explosion of interest in iodine chemistry, which is described here in some detail. For the chemistry of bromine and chlorine, on which there has been rather less work in the past 5 years and which is discussed in less detail here, the interested reader is referred to the review by von Glasow and Crutzen (2007). One point of note in chlorine chemistry is that the discovery during the last 3 years of the widespread abundance of nitryl chloride (ClNO2) is rekindling interest in this halogen (Thornton et al. 2010).
The potential importance of iodine to the gas-phase chemistry of the troposphere was noted over 30 years ago (Chameides and Davis 1980), starting with its potential for changing the atmospheric oxidising capacity, both via ozone (see Sect. 1.5) destruction and changes to the chemistry of hydrogen oxide (OH and HO2 = HOx) and nitrogen oxide (NO and NO2 = NOx) radicals. However, one common conclusion of the early modeling studies on iodine chemistry in the atmosphere was an absence of kinetic data and sparseness of information concerning oceanic iodine sources, which made reliable assessments of the importance of iodine chemistry difficult. Furthermore, apart from measurements of iodine enrichment in sea-salt aerosol (Moyers and Duce 1972), no iodine species had actually been observed in the atmosphere.
During the past decade, the gas-phase iodine species atomic iodine (I), molecular iodine (I2), iodine monoxide (IO) and iodine dioxide (OIO) have all been detected in the atmosphere for the first time, in locations ranging from Antarctica to the equatorial marine boundary layer. These observations have been made using the techniques of differential optical absorption spectroscopy (DOAS) (Alicke et al. 1999; Allan et al. 2000; Saiz-Lopez and Plane 2004), cavity ring-down spectroscopy (Wada et al. 2007), laser induced fluorescence (Whalley et al. 2007), resonance fluorescence (Bale et al. 2008; Gómez Martín et al. 2011), inductively coupled plasma mass spectrometry (Saiz-Lopez et al. 2006a), and atmospheric pressure chemical ionisation with tandem mass spectrometry (Finley and Saltzman 2008). Measurements of IO have very recently been extended to satellite-based DOAS (Saiz-Lopez et al. 2007a; Schönhardt et al. 2008). The resulting confirmation of active and widespread iodine chemistry has produced a huge increase of interest in this halogen.
These discoveries have been accompanied by improved knowledge regarding the fluxes and atmospheric concentrations of short-lived organic and inorganic iodine source compounds, as well as significant developments in laboratory and quantum theory studies of the reaction kinetics, photochemistry and heterogeneous chemistry of iodine species (Saiz-Lopez et al. 2012). In the past 5 years there has been active laboratory research on IO, OIO, IONO2 and the higher iodine oxides (I2Ox, x = 2–5) (Dillon et al. 2008; Gómez Martín et al. 2009; Gravestock et al. 2005; Joseph et al. 2007; Kaltsoyannis and Plane 2008; Plane et al. 2006).
Figure 1.5 is a schematic diagram of our current understanding of tropospheric iodine chemistry. The elucidation of important reaction pathways has emerged from atmospheric models (containing the new laboratory reaction rate coefficients) that have been used to reproduce measured concentrations of I, IO, OIO, I2 and observed new particle formation, as well as shifts in the observed ratios of OH/HO2 and NO/NO2. These modelling studies have been reviewed in detail recently (Saiz-Lopez et al. 2012). The major source of atmospheric iodine is through the evasion from the ocean of a range of compounds (see Sect. 1.3.3): monohalogenated organic compounds such as methyl iodide (CH3I), ethyl iodide (C2H5I) and propyl iodide (1- and 2-C3H7I); reactive polyhalogenated compounds such as chloroiodomethane (CH2ICl), bromoiodomethane (CH2IBr) and diiodomethane (CH2I2); and I2 (Saiz-Lopez et al. 2012). These compounds photodissociate in the atmosphere to generate iodine atoms. In the case of I2, which photolyses most rapidly (< 10 s), I atoms are produced just above the ocean surface. The longer-lived compounds, such as CH3I which has a lifetime of about 2 days, provide a source of iodine throughout the troposphere. It should be noted that, in contrast to iodine, the major source of bromine and chlorine in the marine boundary layer is sea-salt aerosol, from which the Br− and Cl− ions are removed to the gas phase by the uptake and subsequent reaction of species such as N2O5, HOI, HOBr and IONO2 (von Glasow and Crutzen 2007).


Schematic diagram of iodine chemistry in the marine boundary layer. For the gas phase reactions shown rate coefficients and photolysis cross-sections have been measured in the laboratory. Reactions that are thermodynamically possible but for which rates and cross-sections have not been measured are indicated with a question mark. The night-time reaction between I2 and NO3 has been omitted for clarity (Figure adapted from Saiz-Lopez et al. (2012))
As shown in Figure 1.5, I atoms produced by photolysis of these precursors mostly react with ozone to form the IO radical. A rich inorganic gas-phase chemistry then ensues, driven by rapid photochemistry and reactions with HOx and NOx species. The iodine-catalysed depletion of O3 occurs via three catalytic cycles (identified so far). First, IO can deplete O3 through its self reaction to form the OIO radical:
Cycle 1
OIO has strong absorption bands in the visible from about 580 to 630 nm. Although the photodissociation of OIO has been a matter of recent controversy, it is now clear that absorption in these bands leads to photolysis to I + O2 with a quantum yield close to 1 (Gómez Martín et al. 2009). It should be noted that OIO also forms with a branching ratio of about 80 % from the reaction of IO and BrO. This cross-reaction therefore links bromine and iodine chemistry, and can significantly increase Br-catalysed ozone loss (Mahajan et al. 2010b; Read et al. 2008a). Cycle 1 is potentially very O3-depleting, but its efficiency is second-order in IO (Eq. 1.5), so that it becomes most important when the IO (or BrO) mixing ratio is more than several parts per trillion (ppt). At lower halogen oxide concentrations, reaction with HO2 radicals becomes important:
Cycle 2
In semi-polluted environments, IONO2 formation can also lead to O3 depletion:
Cycle 3
However, although the quantum yield for photolysis of IONO2 in the near-UV (Eq. 1.11) is close to 1 (Joseph et al. 2007), the major photolysis pathway (~80 %) of NO3 produces NO2 + O (rather than the minor pathway, Eq. 1.12), leading to no overall O3 depletion. Thus, this cycle can only operate at ~20 % efficiency.
The results in Figure 1.5 show that the HOI and IONO2 formed in Eqs. 1.8 and 1.10, respectively, can also be removed from the gas phase by recycling through sea-salt aerosol. In the case of IONO2 this provides an efficient route for converting NOx to NO3− ions in the aerosol (Mahajan et al. 2009a; Stutz et al. 2007). Moreover, the uptake of HOI and IONO2 enhances the release of chlorine and bromine from sea-salt particles into the gas phase, which can then cause further O3 depletion (McFiggans et al. 2002). Two recent studies have revealed high levels of IO and OIO in polluted environments (Mahajan et al. 2009a; Stutz et al. 2007). Subsequent work using quantum chemistry calculations has shown that IONO2 should rapidly recycle back to I2 by reaction with I (Kaltsoyannis and Plane 2008):
The resulting I atoms, from I2 photolysis, will react with IONO2, rather than O3, if the ratio [IONO2]/[O3] is greater than about 0.01 (Kaltsoyannis and Plane 2008). This sequence represents an autocatalytic cycle that will limit the buildup of IONO2, and explains why iodine chemistry is active even in a relatively high NOx environment (Mahajan et al. 2009b).
The ratio of NO2 to NO is controlled principally by the reactions:
In the presence of significant IO (or BrO) concentrations, the balance will be shifted towards NO2 (McFiggans et al. 2000, 2010; Saiz-Lopez et al. 2008):
where the halogen oxide plays a similar role to peroxy radicals such as HO2 and CH3O2. The HO2/OH ratio in the presence of IO will be reduced by the combination of Eqs.1.8 and 1.9 (Bloss et al. 2005; McFiggans et al. 2000; Saiz-Lopez et al. 2008). Compared to the analogous bromine chemistry, this cycle is particularly important because the IO + HO2 reaction is fast and HOI photolyses more readily than BrO (Atkinson et al. 2007).
The source of I atoms shown in Fig. 1.5 is the photolysis of oceanic precursors (see Sect. 1.3.3), which is therefore restricted to daytime. However, nocturnal IO has also been observed (Mahajan et al. 2009b; Saiz-Lopez and Plane 2004). It has been proposed that the source of IO is the reaction between the NO3 radical and I2, to give I atoms:
followed by reaction with O3 (Eq. 1.7). The reaction between IO and NO3 then produces OIO, which has also been observed at night (Mahajan et al. 2009b; Saiz-Lopez and Plane 2004; Saiz-Lopez et al. 2006b):
Equation 1.21 has recently been studied in the laboratory (Dillon et al. 2008); the measured rate constant is close to the estimated value that is required to model the nocturnal concentration of OIO (Saiz-Lopez et al. 2006b).
The presence of fine particles (diameter < 100 nm) in the coastal atmosphere has been long established (Aitken 1895). Initially, it was postulated that the secondary production of these particles (i.e., growth through condensation of vapour, rather than primary emission into the atmosphere) involved sulphuric acid, likely derived from the oxidation of DMS (see Sect. 1.2.3.1). However, strong evidence for the important role of iodine compounds was provided by the correlation of daytime tidal cycles in iodocarbons (Carpenter et al. 1999), IO (Alicke et al. 1999; Allan et al. 2000), and ultrafine particle bursts (O’Dowd et al. 1999; O’Dowd et al. 1998). Probably the most comprehensive characterisation of coastal ultrafine particle properties was carried out during the Particle Formation in the Coastal Environment (PARFORCE) project at Mace Head, Ireland (O’Dowd et al. 2002a). The explosive appearance of particles between 3 and 10 nm diameter (referred to as particle bursts) was commonly observed during daytime low tide (Flanagan et al. 2005). The simultaneous reduction in measured gaseous H2SO4 concentration, and increase in particle surface area and hygroscopicity, indicated that the condensation of H2SO4 plays a role in the subsequent growth of the particles. This has since been confirmed in laboratory experiments (Saunders et al. 2010). Furthermore, particle analysis identified the presence of both iodine and sulphur in the majority of particles (Mäkelä et al. 2002). In the more recent North Atlantic Marine Boundary Layer Experiment (NAMBLEX), observations show that the dominant iodine precursor at Mace Head is actually I2 (Saiz-Lopez and Plane 2004), released from exposed macroalgae (especially Laminaria) at low tide (McFiggans et al. 2004).
Since many coastal regions are semi-polluted, it has been important to establish whether iodine-mediated particles are formed at sites where NOx is above 1 ppb. The Reactive Halogens in the Marine Boundary Layer (RHaMBLe) coastal experiment at Roscoff (France) showed that this was indeed the case: substantial particle bursts with concentrations up to 3 × 105 cm−3 were observed (McFiggans et al. 2010, Fig. 1.6). Importantly, the particles were then seen to grow to sizes at which they were active as cloud condensation nuclei (CCN). This experiment therefore demonstrated that the iodine chemistry involved in particle formation was not shut down in semi-polluted air by the NOx reactions shown in Fig. 1.5 (Mahajan et al. 2009b).


Particle number concentrations and size distribution evolution during the RHaMBLe Roscoff field project (Reprinted with permission from Saiz-Lopez A, Plane JMC, Baker AR, Carpenter LJ, Glasow Rv, Martín JCG, McFiggans G, Saunders RW (2012) Atmospheric chemistry of iodine. Chem Rev 112:1773–1804. Copyright 2012 American Chemical Society)
The observation that iodine-mediated particles can grow to CCN size lends support to an earlier postulate that the iodine-enhanced marine particle number might be sufficient to affect global radiative forcing (O’Dowd et al. 2002b). To test this requires an understanding of the precise role that iodine plays in ultrafine particle formation. Almost uniquely in the atmosphere, iodine oxide vapours are able to form iodine oxide particles (IOPs) spontaneously (i.e. there is no free energy barrier). As shown in Fig. 1.5, laboratory studies have established that the higher iodine oxides I2O3 and I2O4 form from IO and OIO recombination (Saiz-Lopez et al. 2012). Although one laboratory study of IOP formation from the photolysis of CH2I2 in O3 has indirectly inferred the tetra-oxide form (Jimenez et al. 2003), transmission electron microscope analysis of particles generated photochemically from molecular I2 and O3 in dry conditions showed that the IOPs were essentially I2O5 (Saunders and Plane 2005). Saunders and Plane speculated that I2O5 might form in the gas phase through the oxidation of I2O4 by O3, and that this very stable iodine oxide then polymerised to produce IOPs. However, IOPs can also form without O3 being present, almost certainly initiated by the spontaneous polymerisation of I2O3 and I2O4 (Saunders et al. 2010). These particles must then restructure in the solid phase to I2O5 and I2. Under dry conditions, a kinetic study of IOP formation showed that the particles form fractal-like, aggregate structures (Saunders and Plane 2006), which collapse when humidified (Saunders et al. 2010).
One important question concerns the growth of IOPs in the presence of condensable vapours (water, ammonia and both mineral and organic acids) in the marine atmosphere. This is because the supply of iodine oxides is limited, so that once IOPs form they will only grow by condensation of these other vapour species to sizes (> 50 nm diameter) where the particles can have a significant impact on climate either directly (scattering and absorption of solar radiation) or indirectly (enhancement of CCN) (McFiggans et al. 2006). A recent laboratory study has shown that the accommodation of H2SO4 vapour on IOPs is very efficient, particularly at high relative humidities (Saunders et al. 2010). The growth of IOPs in the remote MBL to sizes at which their role as CCN may become important is therefore likely to be governed by the uptake of H2SO4, accompanied by H2O and NH3 to maintain the pH close to neutral (Kulmala and Kerminen 2008). A recent modelling study (Mahajan et al. 2010a) examined the rate of production of iodine-mediated CCN in the MBL, as a function of the mean daytime IO concentration. There is a highly non-linear relationship, as expected from the chemistry of IOP formation (Fig. 1.5). Another critical factor is the background aerosol surface area, because the loss to background aerosols through uptake is usually faster than growth by coagulation and condensation (except at high IO concentrations). The result is that there is an extremely small probability of forming new potential CCN particles when [IO] < 5 ppt, which seems to be the case in the remote open-ocean MBL (Allan et al. 2000; Mahajan et al. 2010a; Read et al. 2008a). However, IOPs may well produce CCN in environments with higher IO concentrations, such as mid-latitude coastal areas (Mahajan et al. 2009b; Saiz-Lopez and Plane 2004; Whalley et al. 2007) and Antarctica (Saiz-Lopez et al. 2007b).
Iodine occurs at the low ng m−3 level in atmospheric aerosols from a wide variety of environments; these concentrations are considerably enriched over the seawater composition, as indicated by I/Cl and I/Na ratios (Baker et al. 2000; Duce et al. 1967). This enrichment most likely arises from the uptake of gas-phase inorganic iodine species and small IOPs (Fig. 1.5). A range of inorganic iodine species are potentially present in the aerosol. These include I−, HOI, I2, ICl, IBr and IO3−. However, because HOI reacts rapidly with halide ions and the resulting IX (X = Cl, Br, I) species are insoluble (and photochemically active), only the ionic species I− and IO3− are expected to accumulate appreciably. Of these, I− participates in halogen activation reactions to yield I2, and so model studies concluded that IO3− should be the only stable iodine species in aerosols (McFiggans et al. 2000; Vogt et al. 1999). However, measurements of I−/IO3− speciation in marine aerosol have shown that the ratio between these two species is highly variable, for reasons that are poorly understood (Saiz-Lopez et al. 2012).
Soluble organic iodine (SOI) in marine aerosol is ubiquitous (Baker 2005). The proportion of SOI relative to IO3− is quite variable, although SOI is frequently found to constitute the major fraction (Gilfedder et al. 2008). There is also an appreciable insoluble fraction of aerosol iodine, which is likely to be either organic or iodine adsorbed to mineral or black carbon surfaces (Baker et al. 2000; Gilfedder et al. 2010; Tsukada et al. 1987; Xu et al. 2010). Hydration of the higher iodine oxides (I2O4, I2O5) associated with the nucleation of IOPs can account for the presence of aerosol iodate. Aerosol SOI may be introduced by primary emissions of iodinated organic matter from the sea surface during bubble bursting (Seto and Duce 1972). The sea surface microlayer is enriched in organic matter and reactions of O3 and I− at the sea surface are known to produce iodinated organic matter (Martino et al. 2009). SOI could also form via the reaction of aerosol organic matter with HOI (Baker 2005). The interactions of iodine and organic matter, and the degree to which aerosol iodine can be recycled to the gas phase, are the most significant unknowns in aerosol iodine chemistry at present.
1.4 Non-Methane Hydrocarbons (NMHCs)
1.4.1 Oxygenated Volatile Organic Compounds (OVOCs)
Oxygenated volatile organic compounds (OVOCs) are a subgroup of non-methane hydrocarbons consisting of alcohols, aldehydes, ketones, and carboxylic acids (e.g. methanol, acetaldehyde, acetone, and formic acid). In this section only short chain OVOCs (<5 C atoms) will be considered. Many of these gases are ubiquitous in the atmosphere (Singh et al. 1995) and can also play a significant role in atmospheric trace gas chemistry. For example, Lewis et al. (2005) found that acetone, acetaldehyde and methanol constituted up to 85 % of total NMHCs and 80 % of the OH radical sink at Mace Head Ireland under maritime conditions. In the following section, we discuss the atmospheric importance and budgets of OVOCs, as well as the role of the ocean as a control on their atmospheric concentrations.
1.4.1.1 Atmospheric Importance of OVOCs
The build-up of biogenic and anthropogenic chemical compounds in the atmosphere is in many cases largely controlled by their reaction with the hydroxyl radical, OH. The abundance of OH depends on a variety of factors, such as light levels, temperature, and the presence of chemical precursors (Spivakovsky et al. 1990), most importantly water vapour (H2O) and ozone (O3). In certain regions of the atmosphere, such as the upper troposphere, water vapour is not present at high enough concentrations to account for the amount of hydroxyl radical observed there. In these regions it has been suggested that OVOCs are important chemical precursors of OH (Singh et al. 1995; Wennberg et al. 1998; Lary and Shallcross 2000, Fig. 1.7). In the case of acetone, Elias et al. (2011) show that there is enhancement of this compound, although variable, in the upper troposphere in convective regions during summer, indicating transport of acetone from surface sources. However, the acetone source of upper tropospheric OH is still poorly quantified (Sprung and Zahn 2010).


(a) Comparison of HOx (OH + HO2) production rates (dashed lines) with model outputs (solid lines). (b) Measured OH concentrations (open symbols) in a vertical atmospheric profile over the Pacific Ocean. Solid lines are model output. In both panels the red line includes acetone photolysis and the blue does not (From Wennberg PO, et al. (1998) Hydrogen radicals, nitrogen radicals, and the production of O3 in the upper troposphere. Science 279:49–53. Reprinted with permission from AAAS)
OVOCs can also be significant sources of RO2 (Eq. 1.23, Monks 2005; Mueller and Brasseur 1999; Singh et al. 1994) and are therefore an important control on tropospheric ozone (See Sect. 1.5), a harmful pollutant when present in large quantities (Prather et al. 2003 and references therein, see Sect. 1.5). The principal reactions involved in stratospheric ozone formation are the Chapman reactions (Lelieveld and Dentener 2000). However, these reactions cannot account for the high concentrations of ozone in some regions of the troposphere, such as urban areas. Initially, the source of tropospheric ozone was believed to be stratospheric-tropospheric exchange. Later it was found that in highly polluted regions ozone is principally formed by chemical reactions involving hydrocarbons and nitrogen oxide chemical species (\( \mathrm{ N}{{\mathrm{ O}}_{\mathrm{ x}}} = \mathrm{ N}\mathrm{ O} + \mathrm{ N}{{\mathrm{ O}}_2} \)),
Additionally, reactions of OVOCs have been shown to form peroxyaceylnitrate (PAN), a reservoir for reactive nitrogen compounds, and to form secondary organic aerosols and cloud condensation nuclei (see Chap. 4), which influence the radiative budget of the atmosphere (Singh et al. 1995; Blando and Turpin 2000).
1.4.1.2 Atmospheric Budget
The atmospheric sources and sinks of OVOCs have only recently begun to be investigated. There are more detailed budgets for certain compounds, such as acetone and methanol (Heikes et al. 2002; Jacob et al. 2002, 2005; Millet et al. 2008, Schade and Goldstein 2006), but not for others, such as C2–C5 aldehydes and carboxylic acids, although a few exist (Razavi et al. 2011-formic acid, Millet et al. 2010 – acetaldehyde, and Naik et al. 2010 – ethanol). Several studies use models and atmospheric measurements from aircraft to determine budgets (e.g. Jacob et al. 2002), while more recently measurements from satellites have become available (e.g. Elias et al. 2011 – methanol, Dufour et al. 2007 – methanol, Razavi et al. 2011 – methanol and formic acid, Rinsland et al. 2006, 2007 – methanol and formic acid; Harrison et al. 2011a, b – acetone). Tables 1.1 and 1.2 present current estimates of acetone and methanol atmospheric budgets, respectively, and it is evident that there is considerable discrepancy in the literature regarding these compounds. In general, the main sources of OVOCs are primary anthropogenic and biogenic emissions, biomass burning, atmospheric oxidation of precursors (such as methane, isoalkanes, monoterpenes and methylbutanol), and plant decay. The main sinks are wet and dry deposition, reaction with OH, and photochemical oxidation. It is possible to produce OVOCs from atmospheric transformations of other OVOCS. For example, acetaldehyde can be produced from methyl ethyl ketone (Nadasdi et al. 2010; Lewis et al. 2005) and ethanol (Millet et al. 2010; Lewis et al. 2005), as well as formaldehyde from methanol (Heikes et al. 2002), and ethanol from propanal (Naik et al. 2010).
The large extent and productivity of the global oceans dictate their potential to be a significant source or sink for these compounds. Remote marine areas are also of interest since they are the main convective regions that cycle chemical species between the upper and lower troposphere. In these regions oceanic sources of OVOCs can be especially important for upper tropospheric O3, OH, and NOx chemistry. Nonetheless, the role of the oceans in the atmospheric budgets of OVOCs is largely unknown and intensely debated.
In the case of acetone, Singh et al. (2001, 2003, 2004) infer that the ocean is both a source and a sink from tropospheric gradient measurements. Sinha et al. (2007) show that the ocean can be both a source and a sink depending on regional biological productivity and light levels. Marandino et al. (2005) directly measured the flux of acetone using the eddy correlation technique (see Chap. 2) over the north and equatorial Pacific Ocean. They found that the flux (F, Fig. 1.8) was always into the ocean (negative) and, when normalised to wind speed (U), is directly proportional to the atmospheric concentration of acetone (Ca). The results of Mao et al. (2006) are in agreement with the findings of Marandino et al. (2005).


Empirical relationship between the acetone air/sea flux (F), horizontal wind speed (U), and the atmospheric concentration of acetone (Ca). Measurements were made over the northern and equatorial Pacific Ocean during the summer season (Reproduced from Marandino et al. (2005) by permission of the American Geophysical Union)
For methanol, the ocean appears to be both a large source and a large sink. Carpenter et al. (2004) use DMS (solely oceanic in origin because it is biogenically produced in the surface ocean, see Sect. 1.2) and wind speed to calculate a net ocean sink. Williams et al. (2004) also calculate an ocean sink for methanol of 66 ± 266 μmol m−2 d−1 in the N. Atlantic, while Millet et al. (2008) agree that the ocean is a net sink for methanol but can be a weak source when background concentrations are low and the sea surface temperature is warm.
For acetaldehyde, Lewis et al. (2005) state that there is no statistically significant relationship between DMS and acetaldehyde, indicating the ocean is probably not a source for acetaldehyde. Millet et al. (2010) find large regional ocean sources for acetaldehyde. More studies, including direct flux measurements, are needed to constrain the role of the ocean in the atmospheric budgets of a suite of OVOCs. Because flux is directly proportional to the concentration gradient, more information is needed regarding surface ocean processes and OVOC cycling.
1.4.1.3 Surface Ocean Processes
There have been limited investigations of the surface ocean concentrations, sources, and sinks of OVOCs. This is mainly due to the fact that it is difficult to reliably measure these compounds in seawater. Measurement problems include loss from seawater sample to the headspace during extraction, contamination of samples from surrounding air (especially in laboratories that extensively use these compounds as solvents), large Milli-Q blanks from UV photolysis, and high solubility of these compounds in humid sample lines (Hudson et al. 2011).
Low molecular weight carbonyl compounds have been detected in seawater as early as 1955 (Mopper and Stahovec 1986 and references therein). Since then the concentration of a variety of <5 C OVOCs have been measured in the Delaware and Biscayne Bays, the Caribbean, Sargasso, and Aegean Seas, and in the Atlantic and Pacific Oceans. These compounds include methanol, ethanol, 1- and 2-propanol, tert-butyl alcohol, formaldehyde, acetaldehyde, propional, butyraldehyde, acetone, and pyruvate (Beale et al. 2010; de Bruyn et al. 2011; Dixon et al. 2011a, b; Kieber and Mopper 1990; Marandino et al. 2005; Mezcua et al. 2003; Mopper and Stahovec 1986; Obernosterer et al. 1999; Williams et al. 2004; Zhou and Mopper 1997). An example of measured acetone atmospheric mixing ratios and surface ocean concentrations from the Pacific Ocean (Marandino et al. 2005) is shown in Fig. 1.9. These findings illustrate that the atmospheric acetone levels increase with latitude (going north) but that the seawater levels remain nearly constant. The atmospheric sources and sinks of acetone are relatively well understood and the measured change in atmospheric levels is probably due to anthropogenic emissions. However, the explanation for the lack of trend in ocean concentrations is unknown and points to the lack of understanding of surface ocean processes that influence acetone concentrations. In the case of methanol, Carpenter et al. (2004) reports that the biggest uncertainty in the methanol atmospheric budget is related to its concentration in seawater, which cannot be accounted for by their calculated air-to-sea flux.


Shipboard measurements of acetone over the equatorial and northern Pacific Ocean during summer, 2004, top) Atmospheric mixing ratios (Ca) and surface ocean (Cw) acetone concentrations, bottom) Computed saturation state from measured acetone values (Figure adapted from Marandino et al. (2005))
Most oceanic source process studies have focused on the production of OVOCs from the photochemical/photosensitized oxidation of dissolved organic matter (DOM) (de Bruyn et al. 2011; Ehrhardt and Weber 1991; Kieber et al. 1990; Mopper and Stahovec 1986; Mopper et al. 1991; Moran and Zepp 1997; Obernosterer et al. 1999; Sinha et al. 2007; Zhou and Mopper 1997). This process has been highlighted as the most likely source of OVOCs in the surface ocean (see also Sect. 1.5). Other measured sources of OVOCs include biological production, air/sea exchange, and oxidation of DOM via radical chemistry (Dixon et al. 2011a, b; Singh et al. 2003b, 2004; Nemecek-Marshall et al. 1995; Mopper and Stahovec 1986). It is most likely that the production of OVOCs in the surface ocean is from a combination of the above processes. de Bruyn et al. (2011) found a combined effect between the photoproduction of acetaldehyde and acetone and the addition of NO3 radicals to seawater and hypothesised that radical production enhances OVOCs production. When they increased oxygen levels in the seawater samples they measured increased apparent quantum yields (AQY) for formaldehyde and acetone. In addition, while de Bruyn et al. (2011) saw an increase in photoproduction of formaldehyde, acetone, and acetaldehyde with increasing coloured DOM (CDOM), they found a non-linear relationship at low CDOM levels (i.e. those more representative of the open ocean). The AQY increased with decreasing CDOM levels indicating that there is enhanced production efficiency with CDOM photobleaching/humic substance breakdown. This is in contrast to Kieber et al. (1990) who report a linear relationship. The Kieber et al. (1990) study included UV radiation while de Bruyn et al. (2011) did not. Beale et al. (2010), although they did not perform direct process studies, found that the ocean may be an important source of ethanol (concentration 2–33 nmol L−1), and 1- and 2-propanol (concentrations 2–22 and 1–19 nmol L−1, respectively) to the atmosphere. They found enhanced concentrations of the propanols in more biologically productive regions (e.g. upwelling areas). Ethanol concentrations followed different trends in different regions. In one area they saw pronounced diurnal cycles with peaks for ethanol at predawn, possibly indicating bacterial production. They propose that when biological activity is low there is a productivity switch to more photochemical control on ethanol production. In the case of methanol (discussed in more detail below), Dixon et al. (2011b) measured concentrations of 151–296 nmol L−1. This is the highest ever reported value for methanol in oligotrophic waters, with the peak in concentration at predawn. They calculated the production rate of methanol needed to sustain these levels and the uptake rates, as discussed below, and concluded that there must be a large in situ source, unrelated to air-sea exchange.
Surface ocean OVOCs sink processes include biological uptake, air/sea exchange, and oxidation (both radical and photochemical) (Obernosterer et al. 1999; Singh et al. 2003b, 2004; Sinha et al. 2007; Sluis and Ensign 1997). Methanol uptake by bacteria has recently been reported by Dixon et al. (2011a, b). They have found that methanol can be used as both an energy and carbon source. Reported uptake rates are 2–146 nmol L−1 d−1 in surface waters, with enhanced uptake in more biologically productive waters. They hypothesise that methanol is an important carbon source for bacteria in oligotrophic waters (up to 54 % and concentrations are a power function of chlorophyll a concentrations when they are lower than 0.2 μg L−1), where there is not enough DOM produced by phytoplanktion to sustain bacterial populations. Beale et al. (2010) also found evidence of removal of alcohol by marine organisms, this time ethanol. They found an anti-correlation between biologically productive regions and ethanol concentrations in the Atlantic Ocean.
Further process studies looking into the role of biology, photochemistry, and air-sea exchange simultaneously are needed for a range of OVOCs to better constrain how the ocean directly impacts the atmospheric concentrations of these trace gases.
1.4.2 Alkanes and Alkenes
Of relevance to the air-sea interface are the small, more volatile, gas phase alkanes (e.g. methane (discussed in Chap. 3), ethane, propane, butane) and alkenes (e.g. ethene, propene, isoprene), (Donahue and Prinn 1993). Global oceanic emissions of light alkanes and alkenes have been estimated to be between 2.5 and 6 TgC year−1 (Plass-Dülmer et al. 1995; Warneck and Williams 2011). This is small in comparison to terrestrial emissions (ca. 1,141 TgC year−1, Guenther et al. 1995), which are dominated by isoprene from tropical plants (ca. 500 TgC year−1). As a comparison, marine isoprene emissions, which were first measured by Bonsang et al. (1992), have been recently estimated to be between 0.3 and 1.9 Tg isoprene year−1 (Arnold et al. 2009). Nonetheless, oceanic inputs of alkanes and alkenes can have a potentially important role in the remote marine boundary layer. In the atmosphere, the alkenes, by virtue of their double bond, generally react faster than alkanes with ambient oxidants (OH, O3, NO3). When marine alkenes such as isoprene are emitted in the vicinity of ship traffic emitting NOx, rapid ozone production can result (Williams et al. 2010, see Sect. 1.5). Surface water concentrations of alkanes and alkenes result from direct biological production (from seaweed or phytoplankton) and from photochemical degradation of dissolved organic matter (DOM) by UV light (Bonsang et al. 1988; Ratte et al. 1993, 1995, 1998; Riemer 2000). Seawater concentrations of isoprene have been shown to vary from 0 to 55 pmol L−1 in the North Sea, peaking in the summer months when light and phytoplankton populations are highest (Broadgate et al. 1997). Diurnal variations in emissions related to irradiance have been observed under clean Southern Ocean conditions (Bonsang et al. 2008). Laboratory based studies of the isoprene emission rates from various phytoplankton monocultures have shown strong emissions from cyanobacteria and weak emissions from diatoms (Shaw et al. 2003; Bonsang et al. 2010). In the Mediterranean, ethene concentrations in excess of 200 pmol L−1 have been reported (Ratte et al. 1995), while in estuarine waters in Florida ethene reached circa 500 pmol L−1 with carbon equivalent alkanes at several times lower concentrations. Alkenes, and to a lesser extent alkanes, in seawater are generally found to be supersaturated with respect to the atmosphere. Production rates of ethene have been shown to vary by more than an order of magnitude, between 0.22 pmol L−1 min−1 in transparent oligotrophic waters and 6.11 pmol L−1 min−1 in estuarine waters (Riemer et al. 2000). Concentrations of both alkanes and alkenes have been observed to decrease with increasing carbon number in surface waters (Plass-Dülmer et al. 1995). Water column measurements by Ratte et al. (1995) in the Mediterranean, North Atlantic and North Sea showed alkanes have pronounced maxima at the bottom of the euphotic zone, whereas alkene concentrations were maximum near the surface
Marine emissions of isoprene have been a focus of renewed interest in recent years following the suggestion that it was the link between observed correlations between phytoplankton and cloudiness in the Southern Ocean (Meskhidze and Nenes 2006). Modelling studies based on in situ measurements from the same region concluded that isoprene plays an insignificant role in modulating marine aerosol (Arnold et al. 2009, see Sect. 1.5). This was based on its modest emission source (0.31–1.9 Tg year−1) and low aerosol production efficiency (ca. 2 %). In contrast, Gantt et al. (2009) found that isoprene significantly impacts sub-micron marine organic carbon and concluded that it therefore could affect shallow marine clouds and thereby influence climate. In 2008, the first marine emissions of monoterpenes were reported (Yassaa et al. 2008). Exploratory global model studies based on this single dataset suggest that monoterpenes may also impact organic aerosol formation (Luo and Yu 2010). A review of marine isoprene and monoterpenes has been provided by Shaw et al. (2010). From this it has been suggested that marine organic emissions should be introduced into regional air quality models for the western United States (Gantt et al. 2010). All of the recent modelling assessments suffer from the current paucity of open ocean isoprene and monoterpene measurements.
1.4.3 Alkyl Nitrates
The oceans are a source of low-molecular weight alkyl nitrates to the troposphere, particularly methyl (MeONO2) and ethyl nitrates (EtONO2) (Atlas et al. 1992, 1993; Blake et al. 1999, 2003; Fischer et al. 2002; Jones et al. 1999; Swanson et al. 2004). Tropospheric measurements indicate that, of the alkyl nitrates in marine air masses, methyl nitrate is dominant and that they exhibit positive correlations between methyl nitrate and ethyl, isopropyl and n-propyl nitrate (Beyersdorf et al. 2010). Both atmospheric and oceanic measurements indicate that the source of tropospheric alkyl nitrates varies with location. The equatorial oceans appear to be a net source region with saturation anomalies as high as 1,500 % (Blake et al. 2003; Chuck et al. 2002; Dahl et al. 2005). Higher latitude areas of the oceans can be a source or a sink; however, saturation anomaly data is sporadic and does not account for seasonality (Chuck et al. 2002; Hughes et al. 2008a). Few shipboard studies of alkyl nitrate concentrations have been carried out. Surface ocean measurements have been made off the eastern U.S. coast (Russo et al. 2010), in the Atlantic Ocean (Chuck et al. 2002; Dahl 2005), in the Pacific Ocean (Dahl et al. 2005) and in the Southern Ocean (Hughes et al. 2008a). Shipboard measurements of alkyl nitrates have indicated that ocean mixed layer alkyl nitrate concentrations can be up to 100 pmol L−1 in equatorial regions (Chuck et al. 2002; Dahl et al. 2007). This represents considerable water supersaturation relative to atmospheric concentrations that were up to 40 ppt during the PEM-Tropics A experiment (Blake et al. 2003).
Using observations from surface ocean measurements in the equatorial Pacific, Neu et al. (2008) estimated an average flux of 26.7 mg MeONO2 m−2 year−1 and 7.18 mg EtONO2 m−2 year−1 for the region. Their model calculations indicated that the oceanic source of alkyl nitrates is important to the global ozone cycle (see Sect. 1.5) and leads to the formation of between 15 % and 20 % of tropospheric ozone in the western Pacific (Neu et al. 2008). This is the first time that oceanic measurements of alkyl nitrates were linked to an atmospheric chemistry model. Given the findings from the model and the lack of oceanic measurements, it is clear that more work is needed to quantify the levels of alkyl nitrate in the oceans as well as how they vary seasonally.
Alkyl nitrates are also present at depth. Depth profiles in the equatorial Pacific show high concentrations of alkyl nitrates in the mixed layer compared to areas outside the equatorial region (Dahl et al. 2007). Depth profiles of alkyl nitrates in an upwelling region of the North Atlantic correlated with chlorophyll, suggesting a biological source (Chuck et al. 2002). Chuck et al. (2002) conducted depth profile measurements of methyl and ethyl nitrate in the Atlantic Ocean and suggested that alkyl nitrate production: (1) may be associated with algal production in some areas; (2) may be linked to nitrate ions; and (3) may be photochemically produced in areas affected by continental run-off (i.e. high nutrient, high DOM). The loss rates of alkyl nitrates in seawater have not been measured, but lifetimes have been estimated to be over a year at 25 °C and to increase with decreasing temperature (Dahl et al. 2007).
Several possible mechanisms for oceanic alkyl nitrate production have been hypothesised (Ballschmiter 2002; Chuck et al. 2002; Dahl 2005; Dahl et al. 2003; Moore and Blough 2002). To date only two mechanisms have been studied: (1) the photochemically initiated reaction of alkyl peroxy radicals with nitric oxide (Dahl 2005; Dahl et al. 2003; Moore and Blough 2002) and (2) biological production by phytoplankton (Dahl et al. 2012b). The reaction of alkyl peroxy radicals with nitric oxide in the aqueous phase is the same mechanism of alkyl nitrate formation in the gas phase.
The yield of this reaction is 25–75 % in the aqueous phase for C1–C3 alkyl nitrates (Dahl et al. 2003) which is much higher than the gas phase yields (< 5 %) (Atkinson et al. 1982). The rate of reaction of organic peroxy radicals with nitric oxide is of the order of 109 L mol−1 s−1 in the aqueous phase (Padmaja and Huie 1993). Studies of this reaction pathway in seawater indicate that it can be initiated by photochemistry (Dahl et al. 2003; Moore and Blough 2002). Alkyl peroxy radicals can be formed in seawater from photolysis of dissolved organic matter (DOM) and NO can be generated in seawater by the photolysis of nitrite (Blough 1997; Zafiriou and MacFarland 1981). The photochemical production of alkyl nitrates in seawater appears to be highly dependent upon the availability of NO (Dahl and Saltzman 2008; Dahl et al. 2003; Moore and Blough 2002). Recent studies have also indicated that alkyl nitrate speciation depends on location and, potentially, sources of DOM (Dahl et al. 2012a).
Biological production of alkyl nitrates is hypothesised to occur as a result of the formation of nitric oxide during normal cell function. It has been suggested that phytoplankton produce nitric oxide under normal growth conditions and as a response to infochemicals from wounded or dying cells (Kim et al. 2006; Vardi et al. 2006; Zhang et al. 2006). Previous studies of the aqueous phase reaction of alkyl peroxy radicals with NO to form alkyl nitrates indicate that they will form readily whenever the precursors occur in solution. Current work indicates that alkyl nitrate concentrations increase when cells are exposed to chemicals that stimulate production of nitric oxide and that cultures of marine phytoplankton are significantly higher (nmol L−1) in alkyl nitrate concentration than in control solutions (pmol L−1 or lower) (Dahl et al. 2012b). In addition, there is likely to be a bacterial source of alkyl nitrates from nitrification/denitrification processes (Hughes et al. 2010).
1.4.4 Hydrogen Cyanide (HCN) and Methyl Cyanide (CH3CN)
HCN and CH3CN (also called acetonitrile) are abundant components of the earth’s atmosphere and have been measured with a variety of in situ methods and observed remotely from satellites (de Gouw et al. 2003; Singh et al. 2003a; Pumphrey et al. 2011). They have been used extensively as potentially specific tracers of terrestrial biomass combustion (Singh et al. 2010). The current consensus is that biomass combustion is the main source of both HCN and CH3CN and the oceans are the primary sink. There are strong seasonal cycles in the abundances of atmospheric CH3CN and HCN that are linked to seasonal cycles of forest fires and other biomass combustion sources (Li et al. 2000). The industrial emissions of both CH3CN and HCN are negligible even though substantial quantities of HCN (≈ 0.40 Tg(N) year−1) are manufactured as a fumigant. There are indications that under certain conditions automobile exhaust is a small source of HCN.
From measured enhancement ratios with respect to CO in biomass burning plumes, global sources of 1 Tg (N) year−1 for HCN and 0.5 Tg (N) year−1 for CH3CN are estimated. Chemical removal processes (such as hydrolysis) have been determined to be quite slow both in the atmosphere and in the oceans (Bange and Williams 2000). Over the marine boundary layer concentrations decline significantly as expected from an oceanic sink (de Gouw et al. 2003; Singh et al. 2003a). With the exception of Williams et al. (2004) for acetonitrile, there are few direct measurements in ocean waters, although calculations suggest that the surface oceans are likely to be substantially under-saturated (Table 1.3). The exact mechanisms by which HCN and CH3CN are broken down in the oceans are not known but a variety of bacteria can degrade these species and biological processes are expected to be important. Both HCN and CH3CN are a small source of nitrogen to the oceans. As shown in Table 1.3, the global budget of HCN and CH3CN can be roughly balanced but many of the key parameters remain poorly quantified. Seawater measurements are critically needed in order to quantify the ocean sink of these compounds.
1.5 Ozone
Ozone in the stratosphere, at parts per million levels, filters out harmful UV radiation, protecting life on earth. In the troposphere, albeit at three orders of magnitude lower concentration, ozone exhibits mostly harmful properties. Here, ozone acts as a potent greenhouse gas. The warming effect of ‘anthropogenic’, i.e. man-caused, ozone is estimated at 0.35 W m−2 (IPCC 2007), which ranks ozone third in the list of gases contributing to anthropogenic greenhouse warming. Ozone is also well known for its oxidising effects, damaging both plant and animal life in terrestrial environments. These detrimental effects, in contrast to most other atmospheric species, are already noticeable at levels 2–3 times the ozone tropospheric background concentration.
Ozone in the lower atmosphere arises from two sources. Transport from the stratosphere provides a 10–15 ppb background mole fraction. Ozone is also formed and destroyed by photochemical reactions in the troposphere. Anthropogenic emissions of nitrogen oxides and volatile organic compounds have altered the ozone natural cycle, and resulted in a 2–3 times increase in lower tropospheric ozone background concentrations (Lamarque et al.2005). Close to precursor emission sources, ozone can be further enhanced. Tropospheric ozone levels are typically higher over land than over the ocean. However, elevated ozone can be observed for days within the marine boundary layer (MBL) in continental outflow (Li et al.2002). In the remote MBL ozone falls to between 15 and 35 ppb, significantly lower than in the marine lower free troposphere (Allen et al. 2011), indicating that the MBL is a net ozone sink. Long-term monitoring and comparison of historical data sets point towards an increase of MBL ozone, albeit at a slower rate than observed over land (Lelieveld et al.2004; Oltmans et al.2006).
The second most important sink of ozone, after photochemical destruction, is surface uptake. The uptake rate, commonly expressed as a deposition velocity, varies greatly over the Earth’s surfaces. Ozone deposition rates are highest over vegetated land, reaching maxima of 2 m s−1 (Wesely and Hicks 2000). Although ozone is soluble in water, it is unreactive with it alone but can react with certain solutes (see below). Overall, oceanic ozone uptake is significantly slower than over land. Nonetheless, due to the large ocean surface area, the world’s oceans are estimated to account for ~ 1/3 of the global ozone deposition sink (Ganzeveld et al.2009).
Ozone deposition to the ocean surface and ozone-seawater chemical interactions have recently received renewed attention. Earlier research was hampered by limitations on approaches for directly measuring the ozone flux to the ocean surface. Because of this, controlling parameters were primarily studied in laboratory experiments. Ambient scale flux measurements have relied on flux gradient measurements from lighthouses (Tiefenau 1973), turbulent flux measurements by eddy covariance (EC) using airplanes (Lenschow et al.1982), and most recently, EC ozone flux measurements from ocean research vessels (Bariteau et al.2010). Ozone concentrations and fluxes over Arctic sea ice have recently been measured by means of automated, self-powered instrument packages (Knepp et al. 2010; Bottenheim et al. 2009).
Ozone-water deposition rates reported in the literature range from 0.01 to 0.15 m s−1, albeit most results fall between 0.01 and 0.05 cm s−1. This uptake is ~ 40 times faster than expected from ozone dissolution in seawater alone. A chemical enhancement mechanism (explained in Chap. 2) has been postulated (Chang et al.2004). Prime candidates for the oceanic reactivity are the iodide ion (I−) as well as organic constituents (Oh et al.2008). The results of a recent laboratory study indicate that these two reactants are about equally important in determining the deposition velocity of ozone (Martino et al. 2012). Most likely, these reactions occur in the ocean surface microlayer or the immediately underlying seawater. Ozone flux data obtained at sea point to greater ozone deposition in coastal waters, likely due to the enhanced ocean surfactants in those regions (Helmig et al.2012). This gradient in marine ozone uptake was also noted in the review of the oceanic ozone flux literature and modeling work (Ganzeveld et al.2009). Ozone uptake appears to increase with wind speed and sea surface temperature (Chang et al.2004; Helmig et al.2012). However, these do not seem to be simple dependencies, but are probably also determined by concentrations and reactivity of the waterside reactants.
The parameterisation developed by Fairall et al. (2007) considers the reaction of ozone with water-soluble reactants, turbulent-molecular transport, a thin penetration depth of ozone and with reactivity being primarily in the surface microlayer. Calculations from this model indicate an enhancement of up to a factor of three in the oceanic ozone uptake rate during high winds. Integration of this parameterisation into a global atmospheric chemistry model showed a surprisingly low spatial and seasonal variability of ozone fluxes to the oceans, due to compensating effects in the main drivers, i.e. atmospheric turbulence, and oceanic chemical and physical properties (Ganzeveld et al.2009). The most fundamental finding of this research is that chemical reactants in the surface ocean are the primary drivers of ozone uptake. This is a well-known mechanism, as ozone is often used as an oxidising agent in domestic, municipal and industrial water disinfection. Those applications have shown that the ozone reaction with organic constituents results in production of soluble oxygenated organics, such as aldehydes, ketones, and acids (von Gunten 2003). Similar processes are expected to arise from ozone deposition to the ocean, suggesting that ozone reactions at or near the ocean surface oxidise organic material and constitute a source of oxygenated organic compounds (see Sect. 1.4.1). At present it is uncertain if and how reactions with ozone affect the oceanic microlayer, dissolved organic material, and ocean biogeochemistry. The increase in the ozone burden seen in the MBL implies that there has been an increase in the ozone ocean uptake rate since pre-industrial times. Following the arguments presented above nurtures speculation that this may have also driven changes in ocean surface chemical processing and possibly aquatic life, though experimental evidence for this effect is lacking thus far.
1.6 Nitric Oxide
Nitric oxide (NO) is important in the atmosphere due to its role in the cycling of ozone (see Sect. 1.5). However, little is known about its transfer across the air-sea interface. There is evidence from measurements in the equatorial Pacific that NO can be formed photochemically in the surface oceans (Zafiriou et al. 1980). The formation of NO is by photolysis of nitrite ions, with the major destruction pathway being reaction with O2−, which can also mop up any NO from the atmosphere entering the surface microlayer (Blough and Zafiriou 1985). Extrapolation of the limited field data to wider areas is problematic since both light of the correct wavelength (295–410 nm) and the presence of nitrite, whose concentration is very variable, are required. The tentative conclusion is that the oceans are likely to be a source of NO to the atmosphere, although as far as we are aware the size of the flux has not been estimated.
1.7 Ammonia and Amines
Ammonia (NH3) is a polar, strongly soluble compound which is ubiquitous in the environment and is important in the atmosphere as the dominant gas phase base (e.g. Quinn et al. 1992). It has been shown to neutralise between 50 % and 100 % of acidity in aerosol in marine air (e.g. Savoie et al. 1993, Johnson and Bell 2008). The amines, derivatives of ammonia (mostly organic, general formula RxNHy) are also basic, soluble and contribute to neutralisation of atmospheric acidity. In the aqueous phase, ammonia and the amines (NHx) are partitioned between their (basic) molecular forms and their protonated (weakly acidic) ionic forms, e.g. ammonium, NH4+ and dimethylammonium, (CH2)2NH2+. The base dissociation constants (pKb) for ammonia and the aliphatic amines tends to be similar and of the order of 5 (with the amines generally somewhat more basic than ammonia); pKb values for aromatic amines can vary substantially, but they tend to be weaker bases than ammonia.
1.7.1 Ammonia
Ammonia is produced as a byproduct of biological activity, e.g. from the breakdown of nitrogen-containing organic compounds. There are natural terrestrial and marine sources of ammonia to the atmosphere (Bouwman et al. 1997; Dentener and Crutzen 1994) and strong anthropogenic sources, including agricultural, domestic and industrial wastes (e.g. Sutton et al. 2008; Dentener et al. 2006). In the atmosphere it readily reacts with acidic gases and particles to enter the particulate phase as NH4+ (either in aqueous or dry particles, see chap. 4 for a discussion of particles), from which it is deposited to land or water surfaces. This is the major sink for NH3, while oxidation by OH radical is relatively slow (Dentener and Crutzen 1994). The half-life of NH3 in the troposphere is dependent on the net acidity of the atmosphere but is thought to be of the order of 1–2 days, whereas the lifetime of NH4+ in aerosol particles and cloud water is of the order of 5 days or more (Dentener and Crutzen 1994).
It is thought that in the past the ocean would have been a net source of NHx (NH3 + NH4+) to the terrestrial environment, but since the industrial revolution this net flux has reversed (Holland et al. 1999; Galloway 2004). Thus, NHx is transported from the terrestrial to the marine atmosphere and deposited on the ocean surface. However, given the relatively short lifetimes, concentrations in the atmosphere decrease rapidly away from terrestrial source regions, so the large anthropogenic flux of NHx probably affects only the coastal and shelf seas significantly. Typically, NH3 concentrations in the marine atmosphere are of the order of 0.1–10 nmol m−3 (mid-to-high ppt concentrations), but they can be orders of magnitude higher in polluted terrestrial environments (Johnson et al. 2008; Dentener et al. 2006).
In open ocean seawater, NHx exists predominantly as ammonium with concentrations in the nmol L−1 range; concentrations of 1 μmol L−1 or higher may occur frequently in some coastal and shelf seas but are uncommon in the open ocean (Johnson et al. 2008). Ammonium is both a nutrient and a waste product. As a nutrient its uptake by both phytoplankton and bacteria is energetically favourable relative to nitrate and it can be released or produced at various stages in the marine microbial nitrogen cycle, e.g. from ‘leaky’ algal cells to heterotrophic breakdown of nitrogen-containing dissolved organic matter (e.g. Capone 2000). As such, during productive periods, it tends to be cycled extremely rapidly (turnover of the entire standing stock on the order of hours to days) and temporary decoupling of uptake from remineralisation can lead to substantial transient increases in NH4+ concentration over wide areas (Johnson et al. 2007). There is some evidence of photochemical production of ammonium from dissolved organics, which could also contribute to the turnover rate (Kitidis et al. 2006, but see also Grzybowski 2003). The amount of NH3 available to transfer across the air-sea interface depends strongly on pH and temperature (Bell et al. 2007), as does the Henry’s law solubility of ammonia in water (Johnson et al. 2008). The partial pressure of NH3 over the open ocean calculated from typical concentrations, pH and temperatures is in the range 0.1–5 nmol m−3, which is of the same order as atmospheric concentrations, demonstrating that ammonia tends to be close to equilibrium and that the magnitude and direction of the flux can be very sensitive to factors controlling the acid dissociation and Henry’s law equilibrium positions. Johnson et al. (2008) find that the major factor controlling the flux of NH3 at a global scale is temperature, with higher seawater concentrations but downward fluxes at high latitudes, and lower seawater concentrations but ocean emissions at low latitudes. However, transient biologically-driven peaks in NH3 concentration in the surface ocean can drive emission events in regions where the flux might typically be into the ocean (Johnson et al. 2007). The finding that the high latitudes appear to be predominantly a sink for ammonia is somewhat at odds with strong circumstantial evidence for a North Atlantic marine ammonia source presented by Jickells et al. (2003). This may be due to advection from lower latitudes or sampling during transient emission periods. However, further simultaneous measurements of concentrations in the ocean and atmosphere are required to resolve this apparent discrepancy, and to improve estimates of the net global emission of ammonia from the ocean, previous estimates of which lie between 5 (Galloway 2004) and 13 (Schlesinger and Hartley 1992) Tg NH3-N year−1. In the light of evidence of strong downward fluxes at high latitudes (Johnson et al. 2008), future estimates are likely to be lower as low latitude emissions will to some extent be balanced by high latitude uptake.
An alternative mechanism controlling NH3 flux has been proposed by Johnson and Bell (2008) to explain the near-constant ratio of ammonium to non-sea-salt sulphate in aerosol particles in the remote marine environment (see Sect. 1.2.1). They invoke titration of acidic DMS oxidation products with gas phase ammonia, the rate of which in inversely proportional to the degree of neutralisation in the particle phase (i.e. to the NH4+: nssSO42− ratio). Thus, the DMS emission (and subsequent oxidation) can control the gas phase ammonia concentration and thus the oceanic emission, given that the atmosphere and surface ocean are generally close to equilibrium with respect to ammonia.
It is important to note that, as a soluble gas, the air-sea exchange of ammonia is controlled by transfer on the air-side of the interface, particularly as its water phase transport is likely to be significantly chemically enhanced by protonation (Johnson et al. 2011) (as further discussed in Chap. 2). The same applies for the majority of the amines, discussed below. This means that calculating the rate of transfer requires application of a ka (air-side) transfer velocity term rather than the typically applied kw (water side) term used for less soluble gases such as CO2, N2O, DMS etc. (e.g. Liss and Slater 1974; Johnson 2010).
1.7.2 Amines
A recent review has identified >150 amine species which have been measured in the atmosphere or are known to be produced (Ge et al. 2011). Many of these have only industrial sources, but a large number are also known to be biogenically produced and are therefore likely to be formed by biological activity in the surface ocean in a similar manner to ammonium. Others have specific natural production pathways, such as hydrazine (N2H4) which is an intermediary species in the anaerobic bacterial oxidation of ammonia with nitrite (Anammox, e.g. Ward 2003).
In both ocean and atmosphere the most abundant volatile amines are the low molecular weight aliphatic amines of carbon number <6 (Ge et al. 2011), typically occurring at between 1 and 3 orders of magnitude lower concentration than ammonia (Gibb et al. 1999) in the ocean and between 2 and 5 orders of magnitude lower in the atmosphere. There is an increasing body of atmospheric concentration data for the low molecular weight alkyl amines (e.g. Müller et al. 2009).
The significance of both ammonia and the amines in particle formation is currently poorly understood (see Chap. 4). For ammonia the literature is contradictory, with some experimental and modelling studies suggesting that ammonia significantly enhances ternary new particle formation from water and sulphuric acid (Coffman and Hegg 1995; Korhonen et al. 1999); and other evidence suggesting little effect (e.g. Yu 2006). However, recent works suggest that ammonia can play an important role in particle formation and growth (Kirkby et al. 2011; Benson et al. 2011). The amines have been postulated as even stronger agents than ammonia in ternary nucleation (Kurten et al. 2008), due to their higher basicity and higher molecular weight; as well as being significant components of the secondary organic aerosol fraction (e.g. Facchini 2008; Wang et al. 2010). In order to better understand the role of the oceans as sources/sinks of ammonia and the amines and their importance in atmospheric chemistry, substantial further observational data is required.
1.8 Hydrogen
There is growing interest in hydrogen (H2) in view of its role in affecting the oxidising capacity of the atmosphere and also because of the potential for increased emissions in a future ‘hydrogen economy’. The production and flux to the atmosphere of H2 formed in the oceans constitutes about 5 % of the total current global production of the gas (IPCC 2007). The majority of the production is from the photochemical oxidation of methane in the atmosphere (see Chap. 3: Sect. 3.4), with deposition to soils being by far the major sink (Rhee et al. 2005).
The near surface waters of the ocean are frequently at, or well above, saturation with the atmosphere, indicating net production. Below the upper tens of meters of the water column, concentrations decline and, in the case of 8 profiles to 1,000 m in the central equatorial Pacific, become strongly undersaturated, indicating net consumption, probably bacterial (Moore et al. 2009). Processes having the potential to account for supersaturations have long been recognised as anaerobic bacterial activity in detrital particles, nitrogen fixation by cyanobacteria, and photo-oxidation of dissolved organic material (Conrad and Seiler 1986). However, there is no definitive answer as to the relative importance of these three processes (or others) for the formation of H2 in surface seawater. In the study by Moore et al. (2009) it was apparent that the highest concentrations observed in the water column coincided with high levels of nitrogen fixation. A more recent study (Moore, 2012, personal communication) including about 3,000 surface measurements along a meridional transect in the Atlantic (cruise AMT 20) showed marked elevations in hydrogen concentrations in surface waters between about 30 °N and 30 °S. A laboratory study by Wilson et al. (2010) showed that diazotrophic bacteria varied greatly in their net release of H2 during nitrogen fixation, with Trichodesmium erythraeum yielding an order of magnitude more H2 than the unicellular cyanobacterium Cyanothece, and about two orders more than Crocosphaera watsonii. With regard to photoproduction in ocean surface waters, Punshon and Moore (2008) showed from a study of lake and coastal waters that hydrogen is produced during irradiation with sunlight, with the rate being dependent on the concentration of coloured dissolved organic matter (CDOM). They concluded that while it is unlikely that photochemistry can account fully for the widespread supersaturation of H2 in low latitude oligotrophic ocean waters, it may be a contributor.
Whatever the operative production processes, the measured supersaturations lead to a net flux of H2 from the oceans to the atmosphere, estimates of which range from 3 to 6 Tg year−1, with an uncertainty on each estimate of 50 % or more (Schmidt 1974; Conrad and Seiler 1986; Novelli et al. 1999; Rhee et al. 2005).
1.9 Carbon Monoxide
Since the pioneering work of Swinnerton et al. (1970), the ocean is known to be a source of carbon monoxide (CO) to the atmosphere. This compound is important for atmospheric chemistry as its oxidation by the hydroxyl radical, OH (its main sink) leads, in presence of nitrogen oxide, to the formation of ozone (see Sect. 1.5), a pollutant and greenhouse gas. Although the evaluation of its global oceanic source led to estimates ranging over two orders of magnitude (from 4 to 600 Tg CO year−1), the more recent estimates lie in the lower part of this range (Bates et al.1995; Stubbins et al.2006b). Therefore, the oceanic source of CO is likely to play a minor role in its global budget (surface emissions > 1,000 Tg CO year−1, Duncan et al. (2007)), which is dominated by fossil fuel, biomass burning and secondary emissions. Nevertheless, in remote regions, oceanic emissions from CO may affect the regional oxidising capacity of the troposphere and therefore deserve to be better characterised and quantified.
More than three decades ago, the main source and sink of oceanic CO were established as photo-oxidation of dissolved organic matter under UV light and microbial consumption, respectively (Conrad and Seiler 1980; Wilson et al.1970). Further work on the main CO source and sink in the ocean has since been performed, allowing a better understanding and quantification of these processes, which are dependent on several biogeochemical or physical parameters, such as UV irradiance at the seawater surface, quantum efficiency of the conversion of DOC to CO, the attenuation coefficient of UV radiation in the water layers in relation to their DOC content, and microbial consumption (Day and Faloona 2009; Johnson and Bates 1996; Stubbins et al.2006a, 2008; Valentine and Zepp 1993; Xie et al.2005; Zafiriou et al.2003; Zhang et al.2006; Ziolkowski and Miller 2007; Zuo and Jones 1995). A biotic source of CO originating from macro-algae was also investigated by different groups (Loewus and Delwiche 1963; Troxler and Dokos 1973), but King (2001) concluded that this source is not a significant fraction of the global oceanic source. Recently, laboratory studies of trace gas emissions by various phytoplankton species have shown that CO is also emitted directly by phytoplankton under photosynthetically available radiation (Gros et al.2009). Cyanobacteria and diatoms were the largest CO emitters and this source could account for up to 20 % of the CO production in seawater. Finally, another source of oceanic CO is the so-called “dark production” which has been reported by several groups (Day and Faloona 2009 and references therein) and which is needed to explain non-zero CO values below the euphotic zone (Kettle 2005).
The first detailed measurements of CO in the mixed layer of the oceanic water column in the Pacific Ocean showed that it varies seasonally and regionally with daily means between 0.1 and 4.7 nmol L−1 (Bates et al.1995). CO showed pronounced diurnal cycles with maxima in the afternoon, in agreement with the photochemical nature of the main source in water. Since then, measurements of CO have been performed only over a few additional areas in the open ocean. Stubbins et al. (2006b) determined values between 0.2 and 2.6 nmol L−1 during an Atlantic Meridional cruise (from 35 °S to 54 °N). Gros et al. (unpublished data) measured values around 1 nmol L−1 in the Southern Atlantic Ocean, with levels rising to 20 nmol L−1 in a phytoplankton bloom near the Argentinian coast. Xie et al. (2009) conducted measurements in the open waters of the South Eastern Beaufort Sea at two different seasons with mean concentrations of 0.45 and 4.7 nmol L−1 in spring and autumn, respectively.
In shelf waters, CO concentrations were also found to be variable but with values in the upper range of those measured in the open ocean. Day and Faloona (2009) found values between 2.7 and 17 nmol L−1 for a Californian upwelling system; Xie et al. (2005) measured values around 1 nmol L−1 in the Beaufort Sea and Gulf Stream and 2–18 nmol L−1 in the coastal NW Atlantic. Yang et al. (2010, 2011) have recently investigated the East China Sea and the Yellow Sea and observed values between 0.13 and 2.3 nmol L−1 (mean of 0.68 nmol L−1) in November and values between 0.12 and 6.99 nmol L−1 (mean of 2.24 nmol L−1) in April.
Sea-to-air CO fluxes have been shown to vary over two orders of magnitude, depending on the source (which is variable geographically and seasonally) and sink strengths, but also strongly dependent on wind speed. Based on concomitant measurements of CO in seawater and the atmosphere over the Pacific Ocean, Bates et al. (1995) calculated zonal average fluxes ranging from 0.25 to 13 μmol CO m−2 day−1and Stubbins et al. (2006b) have estimated fluxes from 0.4 to 12.1 μmol CO m−2 day−1.
New approaches are becoming available to estimate photoproduction of CO in water on a global scale, as shown recently by the study of Fichot and Miller (2011) who derived CO production fluxes from remotely sensed ocean colour and modelled solar irradiance.
Despite the large temporal and spatial variability of the oceanic source of CO, it is usually considered a constant yearly mean in global model studies. Further work combining field measurements (in less documented areas) and new approaches to derive global fluxes will help to refine estimates of the marine CO source.
1.10 Concluding Remarks
In this chapter we have attempted to summarise in some detail present knowledge of the main trace gases whose transfer across the air-sea interface is environmentally important from local to global scales. This has been done with sections for each gas or group of gases. But we are aware that the real system does not follow such arbitrary chemical sectioning. For example, it is clear that in the atmosphere there is much interaction between various gases and also between the gas and particulate phases (both secondary particle formation and reactions on existing particles). We have tried to deal with this by cross-referencing to relevant sections both within the chapter and across to other chapters, but a more integrated approach would have been desirable.
An example of how focusing on just one gas can lead to misunderstanding is DMS and the CLAW hypothesis. Although the idea that evasion of a biologically-derived gas, DMS, might affect the formation of CCN was on its promulgation a quarter of a century ago a hugely powerful one, at that time the complexity of particle formation in the atmosphere was not well appreciated. Now we are aware that several gases and possibly pre-existing particles are likely to be involved in CCN production and so the original concept needs reframing in the light of the complexity of our present understanding (see, for example, Sect. 1.3.4). This is still a very active research area, as is clear from several earlier sections, and we have consciously not tried to come to an overall conclusion to the ongoing discussion.
We have tried to show how air-sea gas fluxes are driven by processes in both the atmosphere and oceans and thereby affect and are affected by the properties of both. In the future, research effort will need to be put into understanding processes at work in both the surface oceans and marine troposphere because the sophisticated modelling discussed in almost every part of the chapter is highly dependent on such understanding.
Another thread that comes out very strongly from our synthesis is the importance of field measurements. The chemical nature and low concentrations of many of the trace gases makes them difficult to measure accurately. Further, in terms of remoteness and difficulty of sampling, the sea surface and overlying atmosphere are some of the most difficult places from which to obtain reliable measurements. This is particularly acute in terms of remote locations, such as the Arctic and Southern Oceans, and the lack of data covering all seasons is often due to the difficulty of fieldwork in winter and under other adverse conditions. Maybe in the future, satellite and other autonomous measurement platforms will lessen the dependence on ships and aircraft and so, increase both the temporal and spatial coverage of our measurements. This requires investment in miniaturising, automating, and development of new sensitive and stable sensors that can be deployed unattended. If for no other reason, environmental measurements are vital for calibrating and validating modelling studies. Other aspects that need attention are the need for international inter-calibration of measurement techniques and the importance of developing the reliability of techniques for direct flux measurements and their application to a greater range of gases than is possible at present.
In the chapter authors have, where possible, indicated how air-sea trace gas fluxes may change under expected future global change, although this is necessarily quite speculative. One area that has come into prominence recently is ocean acidification and there has been some study of how lowered future ocean pH may alter trace gas concentrations in seawater. For example, mesocosm experiments have shown significant downward changes in DMS and DMSP concentrations under doubled CO2 (Avgoustidi et al. 2012; Hopkins et al. 2010), and for DMS only (Archer et al. 2012). However, this is not always the case and increases in concentrations with lowered pH have been reported by Kim et al. (2010) for both DMS and DMSP and by Archer et al. (2012) for DMSP only. Hopkins et al. 2010 found substantial downward changes in concentrations of organo-halogen gases in a mesocosm experiment with seawater under elevated CO2.
Another aspect of global change that may affect air-sea trace gas fluxes is the role of iron and other nutrients depositing from the atmosphere into the ocean (discussed in detail in Chap. 4) or coming into surface waters from below. The 13 in situ iron fertilisation experiments conducted over the last 20 years give some indications of how changing iron amounts from dust or other sources might affect ocean biogeochemistry and so lead to changed air-sea fluxes of trace gases (see Williamson et al. 2012 for a general overview of these experiments and Law 2008 for a review of the effect on trace gases). The best studied gas in this context is DMS where in many of the experiments concentrations of the gas (and its precursor DMSP) have been found to increase substantially (Turner et al. 1996, 2004). But this is not always the case since in the sub-Arctic Pacific fertilisation with iron was found to decrease or show no change in DMS (Levasseur et al. 2006; Nagao et al. 2009). For a variety of other trace gases including organo-halogens, isoprene and NMHCs the situation is similarly mixed (Wingenter et al. 2004; Liss et al. 2005; Hashimoto et al. 2009; Kato et al. 2009; Nagao et al. 2009). This means that predicting the impact of changes in dust inputs on production of these other gases and hence on, for example, atmospheric oxidising capacity and climate is extremely difficult to predict given present understanding of the complex interactions involved. As noted in Liss et al. 2005, this complexity would need to be taken into account in any proposal to ameliorate global climate by fertilising the oceans with iron.
Throughout the chapter we have endeavoured to show how gas transfer between ocean and atmosphere fits both conceptually and quantitatively into the global cycling of elements. In the future, further attention will need to be paid to how air-sea exchange contributes to the concept of Earth System Science, particularly for forecasting how environmental variables (e.g. wind, pH, dust deposition) are likely to alter in the future under the substantial global changes predicted for this century.
References
Abrahamsson KA, Ekdahl J, Collen M, Pedersen M (1995) Marine algae: a source of trichloroethylene and perchloroethylene. Limnol Oceanogr 40:1321–1326
Aitken J (1895) On the number of dust particles in the atmosphere of certain places in Great Britain and on the continent, with remarks on the relation between the amount of dust and meteorological phenomena. Trans Roy Soc Edinb 37:17–49
Alexander B, Park RJ, Jacob DJ, Li QB, Yantosca RM, Savarino J, Lee CCW, Thiemens MH (2005) Sulfate formation in sea-salt aerosols: constraints from oxygen isotopes. J Geophys Res 110, D10307. doi:10.1029/2004JD005659
Alexander B, Allman DJ, Amos HM, Fairlie TD, Dachs J, Hegg DA, Sletten RS (2012) Isotopic constraints on the formation pathways of sulfate aerosol in the marine boundary layer of the subtropical northeast Atlantic Ocean. J Geophys Res 117. doi:10,1029/2011JD016773
Alicke B, Hebestreit K, Stutz J, Platt U (1999) Iodine oxide in the marine boundary layer. Nature 397:572–573
Allan BJ, McFiggans G, Plane JMC, Coe H (2000) Observations of iodine monoxide in the remote marine boundary layer. J Geophys Res 105:14363–14369
Allen G et al (2011) South East Pacific atmospheric composition and variability sampled along 20 degrees S during VOCALS-Rex. Atmos Chem Phys 11:5237–5262
Amachi S, Kamagata Y, Kanagawa T, Muramatsu Y (2001) Bacteria mediate methylation of iodine in marine and terrestrial environments. Appl Environ Microbiol 67:2718–2722
Amouroux D, Liss PS, Tessier E, Hamren-Larsson M, Donard OFX (2001) Role of oceans as biogenic sources of selenium. Earth Planet Sci Lett 5878:1–7
Anbar AD, Yung YL, Chavez FP (1996) Methyl bromide: ocean sources, ocean sinks, and climate sensitivity. Glob Biogeochem Cycle 10:175–190
Andreae MO (1990) Ocean–atmosphere interaction in the global biogeochemical sulfur cycle. Mar Chem 30:1–29
Andreae MO, Ferek RJ (1992) Photochemical production of carbonyl sulfide in seawater and its emission to the atmosphere. Global Biogeochem Cycle 6:173–175
Andreae TW, Cutter GA, Husain RN, Radford-Knoery J, Andreae MO (1991) Hydrogen-sulfide and radon in an over the western North-Atlantic Ocean. J Geophys Res 96:18753–18760
Andreae MO, Atlas E, Harris GW, Helas G, deKock A, Koppmann R, Maenhaut W, Mano S, Pollock WH, Rudolph J, Scharle D, Schebeske G, Welling M (1996) Methyl halide emissions from savanna fires in Southern Africa. J Geophys Res 101:23603–23613
Archer SD, Goldson LE, Liddicoat MI, Cummings DG, Nightingale PF (2007) Marked seasonality in the concentrations and sea-to-air flux of volatile iodocarbon compounds in the western English Channel. J Geophys Res 112, C08009. doi:10.1029/2006JC003963
Archer SD, Kimmance SA, Stephens JA, Hopkins FE, Bellerby RGJ, Schulz KG, Piontek J, Engel A (2012) Contrasting responses of DMS and DMSP to ocean acidification in Arctic waters. Biogeosciences 9:12803–12843
Arnold SR, Spracklen DV, Williams J, Yassaa N, Sciare J, Bonsang B, Gros V, Peeken I, Lewis AC, Alvain S, Moulin C (2009) Evaluation of the global oceanic isoprene source and its impacts on marine carbon aerosol. Atmos Chem Phys 9:1253–1262
Arnold SR, Spracklen DV, Gebhardt S, Custer T, Williams J, Peeken I, Alvain S (2010) Relationships between atmospheric organic compounds and air-mass exposure to marine biology. Environ Chem 7:232–241
Atkinson R, Carter WPL, Winer AM (1982) Kinetics of the gas-phase reactions of OH radicals with alkyl nitrates at 299 ± 2K. Int J Chem Kinet 14:919–926
Atkinson R, Baulch DL, Cox RA, Crowley JN, Hampson RF, Hynes RG, Jenkin ME, Rossi MJ, Troe J (2007) Evaluated kinetic and photochemical data for atmospheric chemistry: vol. III gas phase reactions of inorganic halogens. Atmos Chem Phys 7:981–1191
Atlas EL, Ridley BA, Hubler G, Walega JG, Carroll MA, Montzka DD, Huebert BJ, Norton RB, Grahek FE, Schauffler S (1992) Partitioning and budget of NOy species during the Mauna Loa observatory photochemistry experiment. J Geophys Res 97:10449–10462
Atlas E, Pollock W, Greenberg J, Heidt L, Thompson AM (1993) Alkyl nitrates, nonmethane hydrocarbons and halocarbon gases over the equatorial Pacific-Ocean during Saga-3. J Geophys Res Atmos 98:16933–16947
Avgoustidi V, Nightingale PD, Joint I, Steinke M, Turner SM, Hopkins FE, Liss PS (2012) Decreased marine dimethyl sulfide production under elevated CO2 levels in mesocosm and in vitro studies. Environ Chem 9:399–404
Baker AR (2005) Marine aerosol iodine chemistry: the importance of soluble organic iodine. Environ Chem 2:295–298
Baker JM, Reeves CE, Nightingale PD, Penkett SA, Gibb SW, Hatton AD (1999) Biological production of methyl bromide in the coastal waters of the North Sea and open ocean of the northeast Atlantic. Mar Chem 64:267–285
Baker AR, Thompson D, Campos MLAM, Perry SJ, Jickells TD (2000) Iodine concentration and availability in atmospheric aerosol. Atmos Environ 34:4331–4336
Bale C, Ingham T, Commane R, Heard D, Bloss W (2008) Novel measurements of atmospheric iodine species by resonance fluorescence. J Atmos Chem 60:51–70
Ballschmiter K (2002) A marine source for alkyl nitrates. Science 197:1127–1128
Bandy AR, Scott DL, Blomquist BW, Chen SM, Thornton DC (1992) Low yields of SO2 from dimethyl sulfide oxidation in the marine boundary layer. Geophys Res Lett 19:1125–1127
Bandy AR, Thornton DC, Blomquist BW, Chen S, Wade TP, Ianni JC, Mitchell GM, Nadler W (1996) Chemistry of dimethyl sulfide in the equatorial Pacific atmosphere. Geophys Res Lett 23:741–744
Bange HW, Williams J (2000) New directions: acetonitrile in atmospheric and biogeochemical cycles. Atmos Environ 34:4959–4960
Bariteau L, Helmig D, Fairall CW, Hare JE, Hueber J, Lang EK (2010) Determination of oceanic ozone deposition by ship-borne eddy covariance flux measurements. Atmos Meas Tech 3:441–455
Barnes I, Bastian V, Becker KH, Overath RD (1991) Kinetic studies of the reactions of IO, BrO and ClO with DMS. Int J Chem Kinet 23:579–591
Barnes I, Hjorth J, Mihalopoulos N (2006) Dimethyl sulfide and dimethyl sulfoxide and their oxidation in the atmosphere. Chem Rev 106:940
Bartnicki EW, Castro CE (1994) Biodehalogenation – rapid xxidative-metabolism of monohalomethanes and polyhalomethanes by methylosinus-trichosporium Ob-3b. Environ Toxicol Chem 13:241–245
Bates TS, Kiene RP, Wolfe GV, Matrai PA, Chavez FP, Buck KR, Blomquist BW, Cuhel RL (1994) The cycling of sulfur in surface seawater of the Northeast Pacific. J Geophys Res Ocean 99:7835–7843
Bates TS, Kelly KC, Johnson JE, Gammon RH (1995) Regional and seasonal variations in the flux of oceanic carbon monoxide to the atmosphere. J Geophys Res 100:23093–23101
Beale R, Liss PS, Nightingale PD (2010) First oceanic measurements of ethanol and propanol. Geophys Res Lett 37, L24607. doi:10.1029/2010GL045534
Bell N, Hsu L, Jacob DJ, Schultz MG, Blake DR, Butler JH, King DB, Lobert JM, Maier-Reimer E (2002) Methyl iodide: atmospheric budget and use as a tracer of marine convection in global models. J Geophys Res 107(D17):4340. doi:10.1029/2001JD001151
Bell TG, Johnson MT, Jickells TD, Liss PS (2007) Ammonia/ammonium dissociation coefficient in seawater: a significant numerical correction. Environ Chem 4:183–186. doi:10.1071/EN07032
Belviso S, Caniaux G (2009) A new assessment in North Atlantic waters of the relationship between DMS concentration and the upper mixed layer solar radiation dose. Glob Biogeochem Cycle 23. doi:Gb1014 10.1029/2008gb003382
Benson DR, Yu JH, Markovich A, Lee SH (2011) Ternary homogeneous nucleation of H2SO4, NH3, and H2O under conditions relevant to the lower troposphere. Atmos Chem Phys 11:4755–4766
Beyersdorf AJ, Blake DR, Swanson A, Meinardi S, Rowland FS, Davis S (2010) Abundances and variability of tropsopheric volatile organic compounds at the South Pole and other Antarctic locations. Atmos Environ 44:4565–4574
Blake NJ et al (1999) Aircraft measurements of the latitudinal, vertical, and seasonal variations of NMHCs, methyl nitrate, methyl halides, and DMS during the First Aerosol Characterization Experiment (ACE 1). J Geophys Res 104:21803–21817
Blake NJ, Blake DR, Swanson AL, Atlas E, Flocke F, Rowland FS (2003) Latitudinal, vertical, and seasonal variations of C1–C4 alkyl nitrates in the troposphere over the Pacific Ocean during PEM-Tropics A and B: Oceanic and continental sources. J Geophys Res 108:8242. doi:10.1029/2001JD001444
Blando JD, Turpin BJ (2000) Secondary organic aerosol formation in cloud and fog droplets: a literature evaluation of plausibility. Atmos Environ 34:1623–1632
Bloss WJ, Lee JD, Johnson GP, Sommariva R, Heard DE, Saiz-Lopez A, Plane JMC, McFiggans G, Coe H, Flynn M, Williams P, Rickard AR, Fleming ZL (2005) Impact of halogen monoxide chemistry upon boundary layer OH and HO2 concentrations at a coastal site. Geophys Res Lett 32, L06814
Blough NV (1997) Photochemistry in the sea-surface microlayer. In: Duce R, Liss PS (eds) The sea-surface and global change. Cambridge University Press, Cambridge, UK
Blough NV, Zafiriou OC (1985) Reactions of superoxide with nitric oxide to form peroxonitrate in alkaline aqueous solution. Inorg Chem 24:3502–3504
Bonsang B, Kanakidou M, Lambert G, Monfray P (1988) The marine source of C2–C6 aliphatic hydrocarbons. J Atmos Chem 6:3–20
Bonsang B, Polle C, Lambert G (1992) Evidence for marine production of isoprene. Geophys Res Lett 19:1129–1132
Bonsang B, Al Aarbaour A, Sciare J (2008) Diurnal variation of nonmethane hydrocarbons in the subantarctic atmosphere. Environ Chem 5:16–23. doi:10.1071/EN07018
Bonsang B, Gros V, Peeken I, Yassaa N, Bluhm K, Zoellner E, Sarda-Esteve R, Williams J (2010) Isoprene from phytoplankton monocultures: the relationship with chlorophyll-a, cell volume and carbon content. Environ Chem 7:554–563. doi:10.1071/EN09156
Bopp L, Aumont O, Belviso S, Monfray P (2003) Potential impact of climate change on marine dimethyl sulfide emissions. Tellus 55B:11–22
Bottenheim JW, Netcheva S, Morin S, Nghiem SV (2009) Ozone in the boundary layer air over the Arctic Ocean: measurements during the TARA transpolar drift 2006–2008. Atmos Chem Phys 9:4545–4557
Bouwman A, Lee D, Asman W, Dentener F, Van Der Hoek K, Olivier J (1997) A global high-resolution emission inventory for ammonia. Glob Biogeochem Cycle 11:561–587
Breider TJ, Chipperfield MP, Richards NAD, Carslaw KS, Mann GW, Spracklen DV (2010) Impact of BrO on dimethylsulfide in the remote marine boundary layer. Geophys Res Lett 37, L02807. doi:10.1029/2009GL040868
Broadgate W, Liss PS, Penkett SA (1997) Seasonal emissions of isoprene and other reactive hydrocarbon gases from the oceans. Geophys Res Lett 24:2675–2878
Brownell DK, Moore RW, Cullen JJ (2010) Production of methyl halides by Prochlorococcus and Synechococcus. Glob Biogeochem Cycle 24, GB2002. doi:10.1029/2009GB003671
Brüchert V, Currie B, Peard K (2009) Hydrogen sulphide and methane emissions on the central Namibian shelf. Prog Oceanogr 83:169–179
Butler A, Walker JV (1993) Marine haloperoxidases. Chem Rev 93:1937–1944
Cameron-Smith P, Elliott S, Maltrud M, Erickson D, Wingenter O (2011) Changes in dimethyl sulfide oceanic distribution due to climate change. Geophys Res Lett 38. L07704. doi:10.1029/2011gl047069
Capone DG (2000) The marine microbial nitrogen cycle. In: Kirchman DL (ed) Microbial ecology of the oceans. Wiley, New York
Carpenter LJ, Liss PS (2000) On temperate sources of bromoform and other reactive organic bromine gases. J Geophys Res Atmos 105:20539–20547
Carpenter LJ, Sturges WT, Penkett SA, Liss PS, Alicke B, Hebestreit K, Platt U (1999) Short-lived alkyl iodides and bromides at Mace Head, Ireland: links to biogenic sources and halogen oxide production. J Geophys Res Atmos 104:1679–1689
Carpenter LJ, Malin G, Liss PS, Kupper FC (2000) Novel biogenic iodine-containing trihalomethanes and other short-lived halocarbons in the coastal East Atlantic. Glob Biogeochem Cycle 14:1191–1204
Carpenter LJ, Lewis AC, Hopkins JR, Read KA, Longley ID, Gallagher MW (2004) Uptake of methanol to the North Atlantic Ocean surface. Global Biogeochem Cycle 18, GB4027. doi:10.1029/2004GB002294
Chameides WL, Davis D (1980) Iodine: its possible role in tropospheric photochemistry. J Geophys Res 85:7383–7398
Chameides WL, Stelson AW (1992) Aqueous-phase chemical processes in deliquescent sea-salt aerosols: a mechanism that couples the atmospheric cycles of S and sea salt. J Geophys Res 97:20565–20580
Chang W, Heikes BG, Lee M (2004) Ozone deposition to the sea surface: chemical enhancement and wind speed dependence. Atmos Environ 38:1053–1059
Charlson RJ, Lovelock JE, Andreae MO, Warren SG (1987) Oceanic phytoplankton, atmospheric sulphur, cloud albedo and climate. Nature 326:655–661
Chuck AL, Turner SM, Liss PS (2002) Direct evidence for a marine source of alkyl nitrates. Science 297:1151–1154
Class TH, Ballschmiter K (1988) Chemistry of organic traces in air: sources and distribution of bromo- and bromochloromethanes in marine air and surface water of the Atlantic Ocean. J Atmos Chem 6:35–46
Coffman DJ, Hegg DA (1995) A preliminary study of the effect of ammonia on particle nucleation in the marine boundary layer. J Geophys Res Atmos 100:7147. doi:10.1029/94JD03253
Conley SA, Faloona I, Miller GH, Lenschow D, Blomquist B, Bandy A (2009) Closing the dimethyl sulfide budget in the tropical marine boundary layer during the Pacific atmospheric sulfur experiment. Atmos Chem Phys 9:8745–8756
Conrad R, Seiler W (1980) Photo-oxidative production and microbial consumption of carbon monoxide in seawater. FEMS Microbiol Lett 9:61–64
Conrad R, Seiler W (1986) Exchange of CO and H2 between ocean and atmosphere. In: Buat-Menard P (ed) The role of air-sea exchange in geochemical cycling. Reidel, Dordrecht
Corbett JJ, Fischbeck PS, Pandis SN (1999) Global nitrogen and sulfur inventories for oceangoing ships. J Geophys Res 104:3457–3470
Cox ML, Sturrock GA, Fraser PJ, Siems S, Krummel PB, O’Doherty S (2003) Regional sources of methyl chloride, chloroform and dichloromethane identified from AGAGE observations at Cape Grim, Tasmania, 1998–2000. J Atmos Chem 45:79–99
Dacey JWH, Howse FA, Michaels AF, Wakeham SG (1998) Temporal variability of dimethylsulfide and dimethylsulfoniopropionate in the Sargasso Sea. Deep Sea Res Part I 45:2085–2104
Dahl EE (2005) Photochemical production of oceanic alkyl nitrates. Dissertation, University of California, Irvine, 178pp
Dahl EE, Saltzman ES (2008) Alkyl nitrate photochemical production rates in North Pacific seawater. Mar Chem 112:137–141
Dahl EE, Saltzman ES, de Bruyn WJ (2003) The aqueous phase yield of alkyl nitrates from ROO + NO: implications for photochemical production in seawater. Geophys Res Lett 30:1271
Dahl EE, Yvon-Lewis SA, Saltzman ES (2005) Saturation anomalies of alkyl nitrates in the tropical Pacific Ocean. Geophys Res Lett 32, L20817
Dahl EE, Yvon-Lewis SA, Saltzman ES (2007) Alkyl nitrate (C1–C3) depth profiles in the tropical Pacific Ocean. J Geophys Res 112, C01012
Dahl EE, Heiss EM, Murawski K (2012a) The effects of dissolved organic matter on alkyl nitrate production during GOMECC and laboratory studies. Mar Chem 142–144:11–17. doi:10.1016/j.marchem.2012.08.001
Dahl EE, Kellogg D, Escobar C (2012b) Are diatoms a source of oceanic alkyl nitrates?. SOLAS Open Science Conference, Cle Elum Washington, May 7–10
Day DA, Faloona I (2009) Carbon monoxide and chromophoric dissolved organic matter cycles in the shelf waters of the Northern California upwelling system. J Geophys Res Ocean 114, CO1006. doi:10.1029/2007JC004590
de Bruyn WJ, Clark PL, Takehara C (2011) Photochemical production of formaldehyde, acetaldehyde, and acetone from chromophoric dissolved organic matter in coastal waters. J Photoch Photobio A 226:16–22
de Gouw JA, Warneke C, Parrish DD, Holloway JS, Trainer M, Fehsenfeld FC (2003) Emission sources and ocean uptake of acetonitrile (CH3CN) in the atmosphere. J Geophys Res 108(D11):4329. doi:10.1029/2002JD002897
Dentener FJ, Crutzen PJ (1994) A three-dimensional model of the global ammonia cycle. J Atmos Chem 19:331–369. doi:10.1007/BF00694492
Dentener F et al (2006) Nitrogen and sulfur deposition on regional and global scales: a multimodel evaluation. Global Biogeochem Cycle 20, GB4003. doi:10.1029/2005GB002672
Derevianko GJ, Deutsch C, Hall A (2009) On the relationship between ocean DMS and solar radiation. Geophys Res Lett 36, L17606. doi:10.1029/2009GL039412
Dillon TJ, Tucceri ME, Sander R, Crowley JN (2008) LIF studies of iodine oxide chemistry Part 3. Reactions IO + NO3 -> OIO + NO2, I + NO3 -> IO + NO2, and CH2I + O2 -> (products): implications for the chemistry of the marine atmosphere at night. Phys Chem Chem Phys 10:1540–1554
Dimmer CH, Simmonds PG, Nickless G, Bassford MR (2001) Biogenic halomethanes from Irish peatland ecosystems. Atmos Environ 35:321–330
Dixon JL, Beale R, Nightingale PD (2011a) Microbial methanol uptake in the northeast Atlantic waters. ISME J 5:704–716
Dixon JL, Beale R, Nightingale PD (2011b) Rapid biological oxidation of methanol in the tropical Atlantic: significance as a microbial carbon source. Biogeosciences 8:2707–2716
Donahue NM, Prinn RG (1993) Non-methane hydrocarbon chemistry in the remote marine boundary layer. J Geophys Res 95:18387–18411
Duce RA, Woodhouse AH, Moyers JL (1967) Variation of ion ratios with size among particles in tropical oceanic air. Tellus 19:367–379
Dufour G, Szopa S, Hauglustaine DA, Boone CD, Rinsland CP, Bernath PF (2007) The influence of biogenic emissions on uppert-tropospheric methanol as revealed from space. Atmos Chem Phys 7:6119–6129
Duncan BN, Logan JA, Bey I, Megretskaia IA, Yantosca RM, Novelli PC, Jones NB, Rinsland CP (2007) Global budget of CO, 1988–1997: source estimates and validation with a global model. J Geophys Res Atmos 112, D22301. doi:10.1029/2007JD008459
Ehrhardt M, Weber RR (1991) Formation of low molecular weight carbonyl compounds by sensitized photochemical decomposition of aliphatic hydrocarbons in seawater. Fresenius J Anal Chem 339:772–776
Ekdahl A, Pedersen M, Abrahamsson K (1998) A study of the diurnal variation of biogenic volatile halocarbons. Mar Chem 63:1–8
Elias T, Szopa S, Zahn A, Schuck T, Brenninkmeijer C, Sprung D, Slemr F (2011) Acetone variability in the upper troposphere: analysis of CARIBIC observations and LMDz-INCA chemistry-climate model simulations. Atmos Chem Phys 11:8053–8074
Elliott S (1989) The effect of hydrogen peroxide on the alkaline hydrolysis of carbon disulfide. Environ Sci Technol 24:264–267
Elliott S, Rowland FS (1993) Nucleophilic substitution rates and solubilities for methyl halides in seawater. Geophys Res Lett 20:1043–1046
Elliott S, Rowland FS (1995) Methyl halide hydrolysis rates in natural-waters. J Atmos Chem 20:229–236
Eyring V, Isaksen ISA, Berntsen T, Collins WJ, Corbett JJ, Endresen O, Grainger RG, Moldanova J, Schlager H, Stevenson DS (2010) Transport impacts on atmosphere and climate: shipping. Atmos Environ 44:4735–4771
Facchini MC (2008) Important source of marine secondary organic aerosol from biogenic amines. Environ Sci Tech 42:9116–9121. doi:10.1021/es8018385
Fairall CW, Helmig D, Ganzefeld L, Hare J (2007) Water-side turbulence enhancement of ozone deposition to the ocean. Atmos Chem Phys 7:443–451
Faloona I (2009) Sulfur processing in the marine atmospheric boundary layer: a review and critical assessment of modeling uncertainties. Atmos Environ 43:2841–2854
Fenical W (1982) Natural products chemistry in the marine environment. Science 215:923–928
Fichot CG, Miller WL (2011) An approach to quantify depth-resolved marine photochemical fluxes using remote sensing: application to carbon monoxide (CO) photoproduction. Rem Sens Environ 114:1363–1377
Finley BD, Saltzman ES (2008) Observations of Cl2, Br2, and I2 in coastal marine air. J Geophys Res 113, D21301
Fischer R, Weller R, Jacobi HW, Ballschmiter K (2002) Levels and pattern of volatile organic nitrates and halocarbons in the air at Neumayer Station (70°S) Antarctica. Chemosphere 48:981–992
Flanagan RJ, Geever M, O’Dowd CD (2005) Direct measurements of new-particle fluxes in the coastal environment. Environ Chem 2:256–259
Flock OR, Andreae MO (1996) Photochemical and non-photochemical formation and destruction of carbonyl sulfide and methyl mercaptan in ocean waters. Mar Chem 54:11–26
Fuse H, Inoue H, Murakami K, Takimura O, Yamaoko Y (2003) Production of free and organic iodine by Roseovarius spp. FEMS Microbiol Lett 229:189–194
Gabric A, Gregg W, Najjar R, Erickson D, Matrai P (2001) Modeling the biogeochemical cycle of dimethylsulfide in the upper ocean: a review. Chemosphere 3:377–392
Gabric AJ, Simo R, Cropp RA, Hirst AC, Dachs J (2004) Modeling estimates of the global emission of dimethylsulfide under enhanced greenhouse conditions. Global Biogeochem Cycle 18, GB2014. doi:10.1029/2003GB002183
Galloway JN (2004) Nitrogen cycles: past, present and future. Biogeochemistry 70:153–226. doi:10.1007/s10533-004-0370-0
Gantt B, Meskhidze N, Kamykowski D (2009) A physically based quantification of marine isoprene and primary organic aerosol emissions. Atmos Chem Phys 9:4915–4927
Gantt B, Meskhidze N, Carlton AG (2010) The contribution of marine organics to the air quality of the western United States. Atmos Chem Phys 10:7415–7423
Ganzeveld L, Helmig D, Fairall CW, Hare J, Pozzer A (2009) Atmosphere–ocean ozone exchange: a global modeling study of biogeochemical, atmospheric, and waterside turbulence dependencies. Glob Biogeochem Cycle 23, GB4021. doi:10.1029/2008GB003301
Ge X, Wexler AS, Clegg SL (2011) Atmospheric amines – part I. A review. Atmos Environ 45:524–546. doi:16/j.atmosenv.2010.10.012
Geen CE (1992) Selected marine sources and sinks of bromoform and other low molecular weight organobromines. Dalhousie University, Halifax, 109pp
Gibb SW, Mantoura RFC, Liss PS (1999) Ocean–atmosphere exchange and atmospheric speciation of ammonia and methylamines in the region of the NW Arabian Sea. Glob Biogeochem Cycle 13:161–178
Gilfedder BS, Lai S, Petri M, Biester H, Hoffmann T (2008) Iodine speciation in rain, snow and aerosols. Atmos Chem Phys 8:6069–6084
Gilfedder BS, Chance R, Dettmann U, Lai SC, Baker AR (2010) Determination of total and non-water soluble iodine in atmospheric aerosols by thermal extraction and spectrometric detection (TESI). Anal Bioanal Chem 398:519–526
Gómez Martín JC, Ashworth SH, Mahajan AS, Plane JMC (2009) Photochemistry of OIO: laboratory study and atmospheric implications. Geophys Res Lett 36, L09802. doi:09810.01029/02009GL037642
Gómez Martín JC, Blahins J, Gross U, Ingham T, Goddard A, Mahajan AS, Ubelis A, Saiz-López A (2011) In situ detection of atomic and molecular iodine using Resonance and Off-Resonance Fluorescence by Lamp Excitation: ROFLEX. Atmos Meas Tech 4:29–45
Goodwin KD, Lidstrom ME, Oremland RS (1997) Marine bacterial degradation of brominated methanes. Environ Sci Tech 31:3188–3192
Gravestock T, Blitz MA, Heard DE (2005) Kinetics study of the reaction of iodine monoxide radicals with dimethyl sulfide. Phys Chem Chem Phys 7:2173–2181
Gray BA, Wang Y, Gu D, Bandy A, Mauldin L, Clarke A, Alexander B, Davis DD (2010) Sources, transport, and sinks of SO2 over the equatorial Pacific during the Pacific atmospheric sulfur experiment. J Atmos Chem. doi:10.1007/s10874-010-9177-7
Gros V, Peeken I, Bluhm K, Zollner E, Sarda-Esteve R, Bonsang B (2009) Carbon monoxide emissions by phytoplankton: evidence from laboratory experiments. Environ Chem 6:369–379
Groszko W, Moore RM (1998) Ocean–atmosphere exchange of methyl bromide: NW Atlantic and Pacific Ocean studies. J Geophys Res Atmos 103:16737–16741
Grzybowski W (2003) Are data on light-induced ammonium release from dissolved organic matter consistent? Chemosphere 52:933–936
Gschwend PM, Macfarlane JK, Newman KA (1985) Volatile halogenated organic-compounds released to seawater from temperate marine macroalgae. Science 227:1033–1035
Guenther A, Hewitt N, Erickson D, Fall R, Geron C, Graedel T, Harley P, Klinger L, Lerdau M, Mckay WA, Pierce T, Scholes B, Steinbrecher R, Tallamraju R, Taylor J, Zimmerman P (1995) A global model of natural volatile organic compound emissions. J Geophys Res 100:8873–8892
Harrison JJ, Allen NDC, Bernath PF (2011a) Infrared absorption cross sections for acetone (propanone) in the 3 μm region. J Quant Spectrosc Rad 112:53–58
Harrison JJ, Humpage N, Allen NDC, Waterfall AM, Bernath PF, Remedios JJ (2011b) Mid-Infrared absorption cross sections for acetone (propanone). J Quant Spectrosc Rad 112:457–464
Hashimoto S et al (2009) Production and air-sea flux of halomethanes in the western subarctic Pacific in relation to phytoplankton pigment concentrations during the iron fertilization experiment (SEEDS II). Deep Sea Res II 56:2928–2935
Heikes BG et al (2002) Atmospheric methanol budget and ocean implication. Glob Biogeochem Cycle 16:1133. doi:10.1029/2002GB001895
Helmig D, Lang EK, Bariteau L, Boylan P, Fairall CW, Ganzeveld L, Hare JE, Hueber J, Pallandt M (2012) Atmosphere–ocean ozone fluxes during the TexAQS 2006, STRATUS 2006, GOMECC 2007, GasEX 2008, and AMMA 2008 cruises. J Geophys Res 117, D04305. doi:10.1029/2011JD015955
Hense I, Quack B (2009) Modelling the vertical distribution of bromoform in the upper water column of the tropical Atlantic Ocean. Biogeoscience 6:535–544
Herndl GJ, Mullerniklas G, Frick J (1993) Major role of ultraviolet-B in controlling bacterioplankton growth in the surface-layer of the ocean. Nature 361:717–719
Holland EA, Dentener FJ, Braswell BH, Sulzman JM (1999) Contemporary and pre-industrial global reactive nitrogen budgets. Biogeochemistry 46:7–43. doi:10.1007/BF01007572
Hopkins FE, Turner SM, Nightingale PD, Steinke M, Bakker D, Liss PS (2010) Ocean acidification and marine trace gas emissions. Proc Natl Acad Sci USA 107:760–765
Howard EC, Henriksen JR, Buchan A, Reisch CR, Buergmann H, Welsh R, Ye WY, Gonzalez JM, Mace K, Joye SB, Kiene RP, Whitman WB, Moran MA (2006) Bacterial taxa that limit sulfur flux from the ocean. Science 314:649–652
Hu L, Yvon-Lewis SA, Liu Y, Salisbury JE, O’Hern JE (2010) Coastal emissions of methyl bromide and methyl chloride along the eastern Gulf of Mexico and the east coast of the United States. Glob Biogeochem Cycle 24, GB1007. doi:10.1029/2009GB003514
Hudson ED, Ariya PA, Gelinas Y (2011) A method for the simultaneous quantification of 23 C1–C9 trace aldehydes and ketones in seawater. Environ Chem 8:441–449
Hughes C, Chuck AL, Turner SM, Liss PS (2008a) Methyl and ethyl nitrate saturation anomalies in the Southern Ocean (36–65°S, 30–70°W). Environ Chem 5:11–15
Hughes C, Chuck AL, Rossetti H, Mann PJ, Turner SM, Clarke A, Chance R, Liss PS (2009) Seasonal cycle of seawater bromoform and dibromomethane concentrations in a coastal bay on the western Antarctic Peninsula. Glob Biogeochem Cycle 23:2024. doi:10.1029/2008GB003268
Hughes C, Malin G, Turley CM, Keely BJ, Nightingale PD, Liss PS (2008b) The production of volatile iodocarbons by biogenic marine aggregates. Limnol Oceanogr 53:867–872
Hughes C, Kettle AJ, Unazi GA, Weston K, Jones MR, Johnson MT (2010) Seasonal variations in the concentrations of methyl and ethyl nitrate in a shallow freshwater lake. Limnol Oceanogr 55:305–314
Hughes C, Franklin D, Malin G (2011) Iodomethane production by two important marine cyanobacteria; Prochlorococcus marinus (CCMP 2389) and Synechococcus sp. (2370). Mar Chem 125:19–25
Ingham T, Bauer D, Sander R, Crutzen PJ, Crowley JN (1999) Kinetics and products of the reactions BrO + DMS and Br + DMS at 298 k. J Phys Chem A 103:7199–7209
IPCC, Climate Change (2007) The physical science basis. Contribution of working group I to the fourth assessment, report of the intergovernmental panel on climate change, Cambridge University Press, Cambridge,UK/New York
Jacob DJ, Field BD, Jin EM, Bey I, Li QB, Logan JA, Yantosca RM, Singh HB (2002) Atmospheric budget of acetone. J Geophys Res 107(D10). doi:10.1029/2001JD000694
Jacob DJ, Field BD, Li QB, Blake DR, de Gouw J, Warneke C, Hansel A, Wisthaler A, Singh HB, Genther A (2005) Global budget of methanol: constraints from atmospheric observations. J Geophys Res 110, D08303. doi:10.1029/2004JD005172
Jickells TD, Kelly SD, Baker AR, Biswas K, Dennis PF, Spokes LJ, Witt M, Yeatman SG (2003) Isotopic evidence for a marine ammonia source. Geophys Res Lett 30. doi: 10.1029/2002GL016728
Jimenez JL, Bahreini R, Cocker DR III, Zhuang H, Varutbangkul V, Flagan RC, Seinfeld JH, O’Dowd CD, Hoffmann T (2003) New particle formation from photooxidation of diiodomethane (CH2I2). J Geophys Res 108:4733. doi:10.1029/2003JD004249
Johnson MT (2010) A numerical scheme to calculate temperature and salinity dependent air-water transfer velocities for any gas. Ocean Sci 6:913–932. doi:10.5194/os-6-913-2010
Johnson JE, Bates TS (1996) Sources and sinks of carbon monoxide in the mixed layer of the tropical South Pacific Ocean. Glob Biochem Cycle 10:347–359
Johnson MT, Bell TG (2008) Coupling between dimethylsupfide emissions and the ocean–atmosphere exchange of ammonia. Environ Chem 5:259–267. doi:10.1071/EN08030
Johnson MT, Sanders R, Avgoustidi V, Lucas MI, Brown L, Hansell DA, Moore CM, Gibb SW, Liss PS, Jickells TD (2007) Ammonium accumulation during a silicate-limited diatom bloom indicates the potential for ammonia emission events. Mar Chem 106:63–75. doi:10.1016/j.marchem.2006.09.006
Johnson MT, Liss PS, Bell TG, Lesworth TJ, Baker AR, Hind AJ, Jickells TD, Biswas KF, Woodward EMS, Gibb SW (2008) Field observations of the ocean–atmosphere exchange of ammonia: fundamental importance of temperature as revealed by a comparison of high and low latitudes. Glob Biogeochem 22, GB1019. doi:10.1029/2007GB003039
Johnson MT, Hughes C, Bell TG, Liss PS (2011) A Rumsfeldian analysis of uncertainty in air-sea gas exchange. In: Komori S, McGillis W, Kurose R (eds) Gas transfer at water surfaces 2010. Kyoto University Press, Kyoto
Jones CE, Carpenter LJ (2005) Solar photolysis of CH2I2, CH2ClI and CH2BrI in water, saltwater and seawater. Environ Sci Technol 39:6130–6138
Jones AE, Weller R, Minikin A, Wolff EW, Sturges WT, Mcintyre HP, Leonard SR, Schrems O, Bauguitte S (1999) Oxidized nitrogen chemistry and speciation in the Antarctic troposphere. J Geophys Res 104:21355–21366
Jones CE, Hornsby KE, Sommariva R, Dunk RM, von Glasow R, McFiggans G, Carpenter LJ (2010) Quantifying the contribution of marine organic gases to atmospheric iodine. Geophys Res Lett 37, L18804. doi:10.1029/2010GL043990
Joseph DM, Ashworth SH, Plane JMC (2007) On the photochemistry of IONO2: absorption cross section (240–370 nm) and photolysis product yields at 248 nm. Phys Chem Chem Phys 9:5599–5607
Kaltsoyannis N, Plane JMC (2008) Quantum chemical calculations on a selection of iodine-containing species (IO, OIO, INO3, (IO)2, I2O3, I2O4 and I2O5) of importance in the atmosphere. Phys Chem Chem Phys 10:1723–1733
Karlsson A, Auer N, Schulz-Bull D, Abrahamsson K (2008) Cyanobacterial blooms in the Baltic – a source of halocarbons. Mar Chem 110:129–139
Kato S, Watiri M, Nagao I, Uematsu M, Kajii Y (2009) Atmospheric trace gas measurements during SEEDS-II over the northwesternPacific. Deep Sea Res Part II: Topical Stud Oceanography 56:2918–2927. doi:http://dx.doi.org/10.1016/j.dsr.2009.07.002
Keene WC, Pszenny AAP (2004) Comment on: Laskin, et al., reactions at interfaces as a source of sulfate formation in sea-salt particles. Science 303:628a–628b
Keppler F, Eiden R, Niedan V, Pracht J, Scholer HF (2000) Halocarbons produced by natural oxidation processes during degradation of organic matter. Nature 403:298–301
Kettle AJ (2005) Diurnal cycling of carbon monoxide (CO) in the upper ocean near Bermuda. Ocean Model 8:337–367
Kettle AJ, Andreae MO (2000) Flux of dimethylsulfide from the oceans: a comparison of updated data sets and flux models. J Geophys Res 105:26793–26808
Kettle AJ, Andreae MO, Amouroux D, Andreae TW, Bates TS, Berresheim H, Bingemer H, Boniforti R, Curran MAJ, DiTullio GR, Helas G, Jones GB, Keller MD, Kiene RP, Leck C, Levasseur M, Malin G, Maspero M, Matrai P, McTaggart AR, Mihalopoulos N, Nguyen BC, Novo A, Putaud JP, Rapsomanikis S, Roberts G, Schebeske G, Sharma S, Simo R, Staubes R, Turner S, Uher G (1999) A global database of sea surface dimethylsulfide (DMS) measurements and a procedure to predict sea surface DMS as a function of latitude, longitude, and month. Glob Biogeochem Cycle 13:399–444
Kettle AJ, Rhee TS, von Hobe M, Poulton A, Aiken J, Andreae MO (2001) Assessing the flux of different volatile sulfur gases from the ocean to the atmosphere. J Geophys Res 106:12193–12209
Kettle AJ, Kuhn U, von Hobe M, Kesselmeier J, Liss PS, Andreae MO (2002) Comparing forward and inverse models to estimate the seasonal variation of hemisphere-integrated fluxes of carbonyl sulfide. Atmos Chem Phys 2:343–361
Khalil A, Rasmussen RA, Hoyt SD (1983) Atmospheric chloroform (CHCl3): ocean-air exchange and global mass balance. Tellus 35B:226–274
Kieber RJ, Mopper K (1990) Determination of picomolar concentrations of carbonyl compounds in natural waters, including seawater, by liquid chromatography. Environ Sci Technol 24:1477–1481
Kieber RJ, Zhou X, Mopper K (1990) Formation of carbonyl compounds from UV induced photodegradation of humic substances in nature waters: fate of riverine carbon in the sea. Limnol Oceanogr 35:1503–1515
Kiene RP (1996) Production of methanethiol from dimethylsulfoniopropionate in marine surface waters. Mar Chem 54:69–83
Kiene RP, Linn LJ, Bruton JA (2000) New and important roles for DMSP in marine microbial communities. J Sea Res 43:209–224
Kim D, Yamaguchi K, Oda T (2006) Nitric oxide synthase-like enzyme mediated nitric oxide generation in harmful red tide phytoplankton Chattonella marina. J Plankton Res 28:613–620
Kim J-M, Lee K, Yang EJ, Shin K, Noh JH, Park K-T, Hyun B, Jeong H-J, Kim J-K, Kim KY, Kim M, Kim H-C, Jang P-G, Jang M-C (2010) Enhanced production of oceanic dimethylsulfide resulting from CO2-induced grazing activity in a high CO2 world. Environ Sci Technol 44:8140. doi:10.1021/ES102028K
King GM (2001) Aspects of carbon monoxide production and oxidation by marine macroalgae. Mar Ecol Prog Ser 224:69–75
King DB, Butler JH, Yvon-Lewis SA, Cotton SA (2002) Predicting oceanic methyl bromide saturation from SST. Geophys Res Lett 29:2199. doi:10.1029/2002GLO16091
Kirkby J et al (2011) Role of sulphuric acid, ammonia and galactic cosmic rays in atmospheric aerosol nucleation. Nature 476:429–433. doi:10.1038/nature10343
Kirst GO, Thiel C, Wolff H, Nothnagel J, Wanzek M, Ulmke R (1991) Dimethylsulfoniopropionate (DMSP) in ice-algae and its possible biological role. Mar Chem 35:381–388
Kitidis V, Uher G, Upstill-Goddard RC, Mantoura RFC, Spyres G, Woodward EMS (2006) Photochemical production of ammonium in the oligotrophic Cyprus Gyre (Eastern Mediterranean). Biogeosciences 3:439–449. doi:10.5194/bg-3-439-2006
Klick S (1992) Seasonal-variations of biogenic and anthropogenic halocarbons in seawater from a coastal site. Limnol Oceanogr 37:1579–1585
Klick S, Abrahamsson K (1992) Biogenic volatile iodated hydrocarbons in the ocean. J Geophys Res 97(C8):12683–12687. doi:10.1029/92JC00948
Kloster S, Six KD, Feichter J, Maier-Reimer E, Roeckner E, Wetzel P, Stier P, Esch M (2007) Response of dimethylsulfide (DMS) in the ocean and atmosphere to global warming. J Geophys Res Biogeosci 112 (G3), G03005. doi:10.1029/2006jg000224
Knepp TN, Bottenheim J, Carlsen M, Donohoue D, Friederich G, Matrai PA, Netcheva S, Perovich DK, Santini R, Shepson PB, Simpson W, Stehle T, Valentic T, Williams C, Wyss PJ (2010) Development on an autonomous sea ice tethered buoy for the study of ocean–atmosphere-sea ice-snow pack interaction: the O-buoy. Atmos Meas Tech 3:249–261
Koga S, Tanaka H (1993) Numerical study of the oxidation process of dimethylsulfide in the marine atmosphere. J Atmos Chem 17:201–228
Koga S, Tanaka H (1996) Simulation of seasonal variations of sulphur compounds in the remote marine atmosphere. J Atmos Chem 23:163–192
Korhonen P, Kulmala M, Laaksonen A, Viisanen Y, McGraw R, Seinfeld JH (1999) Ternary nucleation of H2SO4, NH3, and H2O in the atmosphere. J Geophys Res 104(D21):26349. doi:10.1029/1999JD900784
Korhonen H, Carslaw KS, Spracklen DV, Mann GW, Woodhouse MT (2008) Influence of oceanic dimethyl sulfide emissions on cloud condensation nuclei concentrations and seasonality over the remote Southern Hemisphere oceans: a global model study. J Geophys Res 113, D15204. doi:10.1029/2007JD009718
Kreidenweis SM, Seinfeld JH (1988) Nucleation of sulfuric acid-water solution particles: implications for the atmospheric chemistry of organosulphur species. Atmos Environ 22:283–296
Kulmala M, Kerminen V-M (2008) On the formation and growth of atmospheric nanoparticles. Atmos Res 90:132–150
Kurten T, Loukonen V, Vehkkamaki H, Kulmala M (2008) Amines are likely to enhance neutral and ion-induced sulfuric acid-water nucleation in the atmosphere more effectively than ammonia. Atmos Chem Phys 8:4095–4103
Küpper FC, Carpenter LJ, McFiggans GB, Palmer CJ, Waite TJ, Boneberg EM, Woitsch S, Weiller M, Abela R, Grolimund D, Potin P, Butler A, Luther GW III, Kurtén T, Loukonen V, Vehkamäki H, Kulmala M (2008) Amines are likely to enhance neutral and ion-induced sulfuric acid-water nucleation in the atmosphere more effectively than ammonia. Atmos Chem Phys 8:4095–4103. doi:10.5194/acp-8-4095-2008
Lamarque JF, Hess P, Emmons L, Buja L, Washington W, Granier C (2005) Tropospheric ozone evolution between 1890 and 1990. J Geophys Res Atmos 110, D08304. doi:10.1029/2004JD005537
Lana A, Bell TG, Simo R, Vallina SM, Ballabrera-Poy J, Kettle AJ, Dachs J, Bopp L, Saltzman ES, Stefels J, Johnson JE, Liss PS (2011) An updated climatology of surface dimethlysulfide concentrations and emission fluxes in the global ocean. Glob Biogeochem Cycle 25, GB1004. doi:10.1029/2010GB003850
Lary DJ (1996) Gas phase atmospheric bromine photochemistry. J Geophys Res 101:1505–1516
Lary DJ, Shallcross DE (2000) Centrol role of carbonyl compounds in atmospheric chemistry. J Geophys Res 105:19771–19778
Laskin A, Gaspar DJ, Wang W, Hunt SW, Cowin JP, Colson SD, Finlayson-Pitts BJ (2003) Reactions at interfaces as a source of sulfate formation in sea-salt particles. Science 301:340–344
Laturnus F, Wiencke C, Adams FC (1998) Influence of light conditions on the release of volatile halocarbons by Antarctic macroalgae. Mar Environ Res 45:285–294
Law CS (2008) Predicting and monitoring the effects of large-scale ocean iron fertilization on marine trace gas emissions. Mar Ecol Progr Ser 364:283–288
Lawler MJ, Finley BD, Keene WC, Pszenny AAP, Read KA, von Glasow R, Saltzman ES (2009) Pollution-enhanced reactive chlorine chemistry in the eastern tropical Atlantic boundary layer. Geophys Res Lett 36, L08810. doi:10.1029/2008GL036666
Lawler MJ, Sander R, Carpenter LJ, Lee JD, von Glasow R, Sommariva R, Saltzman ES (2011) HOCl and Cl2 observations in marine air. Atmos Chem Phys 11:7617–7628
Le Clainche Y, Vezina A, Levasseur M, Cropp RA, Gunson JR, Vallina SM, Vogt M, Lancelot C, Allen JI, Archer SD, Bopp L, Deal C, Elliott S, Jin M, Malin G, Schoemann V, Simo R, Six KD, Stefels J (2010) A first appraisal of prognostic ocean DMS models and prospects for their use in climate models. Glob Biogeo Cycles 24, Gb3021. doi:10.1029/2009gb003721
Leck C, Bigg EK (1999) Aerosol production over remote marine areas – a new route. Geophys Res Lett 26:3577–3581
Lelieveld J, Dentener FJ (2000) What controls tropospheric ozone? J Geophys Res 105:3531–3551
Lelieveld J, Van Aardenne J, Fisher H, De Reus M, Williams J, Winkler P (2004) Increasing ozone over the Atlantic Ocean. Science 304:1483–1487
Lenschow DH, Pearson R, Stankov BB (1982) Measurements of ozone vertical flux to ocean and forest. J Geophys Res Ocean Atmos 87(NC11):8833–8837
Levasseur M, Scarratt MG, Michaud S, Merzouk A, Wong CS, Arychuk M, Richardson W, Rivkin RB, Hale M, Wong E, Marchetti A, Kiyosawa H (2006) DMSP and DMS dynamics during a mesoscale iron fertilization experiment in the Northeast Pacific – Part 1: temporal and vertical distributions. Deep Sea Res Part II 53:2353–2369
Lewis ER, Schwartz SE (2004) Sea salt aerosol production, vol 152, Geophysical monograph. American Geophysical Union, Washington, DC
Lewis AC, Hopkins JR, Carpenter LJ, Stanton J, Read KA, Pilling MJ (2005) Sourcs and sinks of acetone methanol, and acetaldehyde in North Atlantic marine air. Atmos Chem Phys 5:1963–1974
Li Q, Jacob DJ, Bey I, Yantosca RM, Zhao Y, Kondo Y, Notholt J (2000) Atmospheric hydrogen cyanide (HCN): biomass burning source, oceanic sink? Geophys Res Lett 27:357–360
Li QB et al (2002) Transatlantic transport of pollution and its effects on surface ozone in Europe and North America. J Geophys Res Atmos 107:4166. doi:10.1029/2001JD001422
Liss PS, Slater PG (1974) Flux of gases across the air-sea interface. Nature 247:181–184. doi:10.1038/247181a0
Liss PS, Chuck A, Bakker D, Turner S (2005) Ocean fertilization with iron: effects on climate and air quality. Tellus 57B:269–271
Lobert JM, Butler JH, Montzka SA, Geller LS, Myers RC, Elkins JW (1995) A net sink for atmospheric CH3Br in the East Pacific-Ocean. Science 267(5200):1002–1005
Loewus MW, Delwiche CC (1963) Carbon monoxide production by algae. Plant Physiol 38:371–374
Lovelock JE, Maggs RJ, Rasmussen RA (1972) Atmospheric dimethyl sulphide and the natural sulphur cycle. Nature 237:452–453
Luo G, Yu F (2010) A numerical evaluation of global oceanic emissions of pinene and isoprene. Atmos Chem Phys 10:2007–2015
Lupu A, Kaminski JW, Neary L, McConnell JC, Toyota K, Rinsland CP, Bernath PF, Walker KA, Boone CD, Nagaham Y, Suzuki K (2009) Hydrogen cyanide in the upper troposphere: GEM-AQ simulation and comparison with ACE-FTS observations. Atmos Chem Phys 9:4301–4313
Mabey W, Mill T (1978) Critical-review of hydrolysis of organic-compounds in water under environmental-conditions. J Phys Chem Ref Data 7:383–415
Mahajan AS, Oetjen H, Saiz-Lopez A, Lee JD, McFiggans GB, Plane JMC (2009) Reactive iodine species in a semi-polluted environment. Geophys Res Lett 36, L16803. doi:16810.11029/12009GL038018
Mahajan AS, Plane JMC, Oetjen H, Mendes L, Saunders RW, Saiz-Lopez A, Jones CE, Carpenter LJ, McFiggans GB (2010a) Measurement and modelling of tropospheric reactive halogen species over the tropical Atlantic Ocean. Atmos Chem Phys 10:4611–4624
Mahajan AS, Shaw M, Oetjen H, Hornsby KE, Carpenter LJ, Kalescheke L, Tian-Kunze X, Lee JD, Moller SJ, Edwards P, Commane R, Ingham T, Heard DE, Plane JMC (2010b) Evidence of reactive iodine chemistry in the Arctic boundary layer. J Geophys Res 115, D20303. doi:10.1029/2009JD013665
Mäkelä JM, Hoffmann T, Holzke C, Väkevä M, Suni T, Mattila T, Aalto PP, Tapper U, Kauppinen EI, O’Dowd CD (2002) Biogenic iodine emissions and identification of end-products in coastal ultrafine particles during nucleation bursts. J Geophys Res 107:8110
Manley SL (1994) The possible involvement of methylcobalamin in the production of methyl iodide. Mar Chem 46:361–369
Manley SL, Dastoor MN (1987) Methyl halide (CH3X) production from the giant-kelp, Macrocystis, and estimates of global CH3X production by kelp. Limnol Oceanogr 32:709–715
Manley SL, Dastoor MN (1988) Methyl iodide production by kelp and associated microbes. Mar Biol 88:447–482
Manley SL, de la Cuesta J (1997) Methyl iodide production from marine phytoplankton cultures. Limnol Oceanogr 42:142–147
Manley SL, Goodwin K, North WJ (1992) Laboratory production of bromoform, methylene bromide, and methyl-iodide by macroalgae and distribution in nearshore Southern California waters. Limnol Oceanogr 37:1652–1659
Mao H, Talbot R, Nielsen C, Sive B (2006) Controls on methanol and acetone in the marine and contintental atmospheres. Geophys Res Lett 33, L02803. doi:10.1029/2005GL024810
Marandino CA, de Bruyn WJ, Miller SD, Prather MJ, Saltzman ES (2005) Oceanic uptake and the global atmospheric acetone budget. Geophys Res Lett 32, L15806. doi:10.1029/2005GL023285
Marshall RA, Hamilton RTJ, Dring MJ, Harper DB (2000) The red alga Asparagopsis taxiformis/Falkenbergia hillebradiii – a possible source of trichloroethylene and perchloroethylene? Limnol Oceanogr 45:516–519
Martin M, Pohler D, Seitz K, Sinreich R, Platt U (2009) BrO measurements over the eastern North-Atlantic. Atmos Chem Phys 9:9545–9554
Martino M, Liss PS, Plane JMC (2005) The photolysis of dihalomethanes in surface seawater. Env Sci Technol 39:7097–7101. doi:10.1021/es048718s
Martino M, Mills GP, Woeltjen J, Liss PS (2009) A new source of volatile organoiodine compounds in surface seawater. Geophys Res Lett 36, L01609. doi:10.1029/2008GL036334
Martino M, Lézé B, Baker AR, Liss PS (2012) Chemical controls on ozone deposition to water. Geophys Res Lett 39, L05809. doi:10.1029/2011GL050282
McCulloch A, Midgley PM (1996) The production and global distribution of emissions of trichloroethene, tetrachloroethene and dichloromethane over the period 1988–1992. Atmos Environ 30:601–608
McDonald IR, Warner KL, Mcanulla C, Woodall CA, Oremland RS, Murrell JC (2002) A review of bacterial methyl halide degradation: biochemistry, genetics and molecular ecology. Environ Microbiol 4:193–203
McFiggans G, Plane JMC, Allan BJ, Carpenter LJ, Coe H, O’Dowd C (2000) A modeling study of iodine chemistry in the marine boundary layer. J Geophys Res Atmos 105:14371–14385
McFiggans G, Cox RA, Mössinger JC, Allan BJ, Plane JMC (2002) Active chlorine release from marine aerosols: roles for reactive iodine and nitrogen species. J Geophys Res Atmos 107:4271. doi:10.1029/2001JD000383
McFiggans G, Coe H, Burgess R, Allan J, Cubison M, Alfarra MR, Saunders R, Saiz-Lopez A, Plane JMC, Wevill DJ, Carpenter LJ, Rickard AR, Monks PS (2004) Direct evidence for coastal iodine particles from Laminaria macroalgae – linkage to emissions of molecular iodine. Atmos Chem Phys 4:701–713
McFiggans G, Artaxo P, Baltensperger U, Coe H, Facchini MC, Feingold G, Fuzzi S, Gysel M, Laaksonen A, Lohmann U, Mentel TF, Murphy DM, O’Dowd CD, Snider JR, Weingartner E (2006) The effect of physical and chemical aerosol properties on warm cloud droplet activation. Atmos Chem Phys 6:2593–2649
McFiggans G, Bale CSE, Ball SM, Beames JM, Bloss WJ, Carpenter LJ, Dorsey J, Dunk R, Flynn MJ, Furneaux KL, Gallagher MW, Heard DE, Hollingsworth AM, Hornsby K, Ingham T, Jones CE, Jones RL, Kramer LJ, Langridge JM, Leblanc C, LeCrane JP, Lee JD, Leigh RJ, Longley I, Mahajan AS, Monks PS, Oetjen H, Orr-Ewing AJ, Plane JMC, Potin P, Shillings AJL, Thomas F, von Glasow R, Wada R, Whalley LK, Whitehead JD (2010) Iodine-mediated coastal particle formation: an overview of the Reactive Halogens in the Marine Boundary Layer (RHaMBLe) Roscoff coastal study. Atmos Chem Phys 10:2975–2999
Meskhidze N, Nenes A (2006) Phytoplankton and cloudiness in the Southern Ocean. Science 314:1419–1423
Mezcua M, Aguera A, Hernando MD, Piedra L, Fernandez-Alba AR (2003) Determination of methyl tert.-butyl ether and ter.-butyl alcohol in seawater samples using purge-and-trap enrichment coupled to gas chromatography with atomic emission and mass spectrometric detection. J Chromatogr A 999:81–90
Millet DB, Jacob DJ, Custer TG, de Gouw JA, Goldstein AH, Karl T, Singh HB, Sive BC, Talbot RW, Warneke C, Williams J (2008) New constraints on terrestrial and oceanic sources of atmospheric methanol. Atmos Chem Phys 8:6887–6905
Millet DB, Guenther A, Siegel DA, Nelson NB, Singh HB, de Gouw JA, Warneke C, Williams J, Eerdekens G, Sinha V, Karl T, Flocke F, Apel E, Riemer DD, Palmer PI, Barkley M (2010) Global atmospheric budget of acetaldehyde: 3-D model analysis and constraints from in-situ and satellite observations. Atmos Chem Phys 10:3405–3425
Moelwyn-Hughes EA (1938) The hydrolysis of the methyl halides. Proc R Soc A164:295–306
Monks PS (2005) Gas-phase radical chemistry in the troposphere. Chem Soc Rev 34:376–395
Moore RM (2001) Trichloroethylene and tetrachloroethylene in Atlantic waters. J Geophys Res 106:135–227
Moore RM (2004) Dichloromethane in North Atlantic waters. J Geophys Res 109, C09004. doi:10.1029/2004JC002397
Moore RM (2006) Methyl halide production and loss rates in seawater from field incubation experiments. Mar Chem 101:213–219
Moore RM (2008) A photochemical source of methyl chloride in saline waters. Environ Sci Technol 42(6):1933–1937. doi:10.1021/es071920l
Moore RM, Blough NV (2002) A marine source of methyl nitrate. Geophys Res Lett 29:1–4
Moore RM, Tokarczyk R (1993) Volatile biogenic halocarbons in the Northwest Atlantic. Glob Biogeochem Cycle 7:195–210
Moore RM, Zafiriou O (1994) Photochemical production of methyl iodide in seawater. J Geophys Res 99:16415–16420
Moore RM, Groszko W, Niven S (1996a) Ocean–atmosphere exchange ofmethyl chloride: results from N.W. Atlantic and Pacific Ocean studies. J Geophys Res 101:28529–28538
Moore RM, Webb M, Tokarczyk R, Wever R (1996b) Bromoperoxidase and iodoperoxidase enzymes and production of halogenated methanes in marine diatom cultures. J Geophys Res 101:20899–20908
Moore RM, Punshon S, Mahaffey C, Karl D (2009) The relationship between dissolved hydrogen and nitrogen fixation in ocean waters. Deep Sea Res I 56:1449–1458
Mopper K, Stahovec WL (1986) Sources and sinks of low molecular weight organic carbonyl compounds in seawater. Mar Chem 19:305–321
Mopper K, Zhou X, Kieber RJ, Kieber DJ, Sikorski RJ, Jones RD (1991) Photochemical degradation of dissolved organic carbon and its impact on the oceanic carbon cycle. Nature 353:60–62
Moran MA, Zepp RG (1997) Role of photoreactions in the formation of biologically labile compounds from dissolved organic matter. Limnol Oceanogr 42:1307–1316
Moyers JL, Duce RA (1972) Gaseous and particulate iodine in the marine atmosphere. J Geophys Res 77:5229–5238
Mueller J-F, Brasseur G (1999) Sources of upper tropospheric HOx: a three-dimensional study. J Geophys Res 104(D1):1705–1715
Müller C, Iinuma Y, Karstensen J, Van Pinxteren D, Lehmann S, Gnauk T, Herrmann H (2009) Seasonal variation of aliphatic amines in marine sub-micrometer particles at the Cape Verde islands. Atmos Chem Phys 9:9587–9597
Muramatsu Y, Yoshida S (1995) Volatilization of methyl iodide from the soil plant system. Atmos Environ 29:21–25
Murphy CD, Moore RM, White RL (2000) An isotopic labeling method for determining production of volatile organohalogens by marine microalgae. Limnol Oceanogr 45:1868–1871
Nadasdi R, Zuegner GL, Farkas M, Dobe S, Maeda S, Morokuma K (2010) Photochemistry of methyld ethyl ketone: quantum yields and S1/S0-diradical mechanism of photodissociation. Chem Phys Chem 11:3883–3895
Nagao I, Hashimoto S, Suzuki K, Toda S, Narita Y, Tsuda A, Saito H, Kudo I (2009) Responses of DMS in the seawater and atmosphere to iron enrichment in the subarctic western North Pacific (SEEDS-II). Deep Sea Res Part II 56:2899–2917
Naik V, Fiore AM, Horowitz LW, Singh HB, Wiedmeyer C, Guenther A, de Gouw JA, Millet DB, Goldan PD, Kuster WC, Goldstein A (2010) Obsevational constraints on the global atmospheric budget of ethanol. Atmos Chem Phys 10:925–945
Neidleman SL, Geigert J (1986) Biohalogenation: principles basic roles and applications. Ellis Horwood, Chichester
Nemecek-Marshall M, Wojciechowski C, Kuzma J, Silver GM, Fall R (1995) Marine Vibrio species produce the volatile organic compound acetone. Appl Environ Microbiol 61:44–47
Neu JL, Lawler MJ, Prather MJ, Saltzman ES (2008) Oceanic alkyl nitrates as a natural source of tropospheric ozone. Geophys Res Lett 35(L13814)
Nightingale PD, Malin G, Liss PS (1995) Production of chloroform and other low-molecular weight halocarbons by some species of marine algae. Limnol Oceanogr 40:680–689
Nightingale PD, Malin G, Law CS, Watson AJ, Liss PS, Liddicoat MI, Boutin J, Upstill-Goddard RC (2000) In situ evaluation of air-sea gas exchange parameterizations using novel conservative and volatile tracers. Glob Biogeochem Cycle 14:373–387
Novelli PC, Lang PM, Masarie KA, Hurst DM, Myers R, Elkins JW (1999) Molecular hydrogen in the troposphere: global distribution and budget. J Geophys Res 104:30427–30444
Nowak JB, Davis DD, Chen G, Eisele FL, Mauldin RL, Tanner DJ, Cantrell C, Kosciuch E, Bandy A, Thornton D, Clarke A (2001) Airborne observations of DMSO, DMS and OH at marine tropical latitudes. Geophys Res Lett 28:2201–2204
O’Doherty S et al (2001) In situ chloroform measurements at AGAGE atmospheric research stations from 1994–1998. J Geophys Res 106:20429–20444
O’Dowd CD, Geever M, Hill MK, Smith MH, Jennings SG (1998) New particle formation: nucleation rates and spatial scales in the clean marine coastal environment. Geophys Res Lett 25:1661–1664
O’Dowd C, McFiggans G, Creasey DJ, Pirjola L, Hoell C, Smith MH, Allan BJ, Plane JMC, Heard DE, Lee JD, Pilling MJ, Kulmala M (1999) On the photochemical production of new particles in the coastal boundary layer. Geophys Res Lett 26:1707–1710
O’Dowd CD, Hämeri K, Mäkelä JM, Pirjola L, Kulmala M, Jennings SG, Berresheim H, Hansson H-C, de Leeuw G, Kunz GJ, Allen AG, Hewitt CN, Jackson A, Viisanen Y, Hoffmann T (2002a) A dedicated study of New Particle Formation and Fate in the Coastal Environment (PARFORCE): overview of objectives and achievements. J Geophys Res 107:8108. doi:10.1029/JD000555
O’Dowd CD, Jimenez JL, Bahreini R, Flagan RC, Seinfeld JH, Hameri K, Pirjola L, Kulmala M, Jennings SG, Hoffmann T (2002b) Marine aerosol formation from biogenic iodine emissions. Nature 417:632–636
O’Dowd CD, Facchini MC, Cavalli F, Ceburnis D, Mircea M, Decesari S, Fuzzi S, Yoon YJ, Putaud J-P (2004) Biogenically driven organic contribution to marine aerosol. Nature 431:676–680
Obernosterer I, Kraay G, de Ranitz E, Herndl GJ (1999) Concentrations of low molecular weight carboxylic acids and carbonyl compounds in the Aegean Sea (Eastern Mediterranean) and the turnover of pyruvate. Aquat Microb Ecol 20:147–156
Oh IB, Byun DW, Kim HC, Kim S, Cameron B (2008) Modeling the effect of iodide distribution on ozone deposition to seawater surface. Atmos Environ 42:4453–4466
Ohsawa N, Tsujita M, Morikawa S, Itoh N (2001) Purification and characterisation of a monohalomethane-producing enzyme S.adenosyl-L-methionine: halide ion methyltransferase from a marine microalga, Pavlova pinguis. Biosci Biotechnol Biochem 65:2397–2404
Olaguer EP (2002) The distribution of the chlorinated solvents dichloromethane, perchloroethylene, and trichloroethylene in the global atmosphere. Environ Sci Pollut Res 9:175–182
Oltmans SJ et al (2006) Long-term changes in tropospheric ozone. Atmos Environ 40:3156–3173
Ooki A, Yokouchi Y (2011) Dichloromethane in the Indian Ocean: evidence for in-situ production in seawater. Mar Chem 124:119–124
Ooki A et al (2010) Methyl halides in surface seawater and marine boundary layer of the northwest Pacific Source. J Geophys Res Ocean 115, C10013. doi:10.1029/2009JC005703
Orlikowska A, Schulz-Bull DE (2009) Seasonal variations of volatile organic compounds in the coastal Baltic Sea. Environ Chem 6:495–507
Padmaja S, Huie RE (1993) The reaction of nitric oxide with organic peroxyl radicals. Biochem Bioph Res Co 195:539–544
Palmer CJ, Reason CJ (2009) Relationship of surface bromoform concentrations with mixed layer depth and salinity in the tropical oceans. Glob Biogeochem Cycle 23, doi:10.1029/2008GB003338
Patroescu IV, Barnes I, Becker KH (1999) FT-IR product study of the OH-initiated oxidation of DMS in the presence of NOx. Atmos Environ 33:25–35
Plane JMC, Vondrak T, Broadley S, Cosic B, Ermoline A, Fontijn A (2006) Kinetic study of the reaction Ca+ + N2O from 188 to 1207 K. J Phys Chem A 110:7874–7881
Plass-Dülmer C, Koppmann R, Ratte M, Rudolph J (1995) Light nonmethane hydrocarbons in seawater. Global Biogeochem Cycle 9:79–100
Prather M et al (2003) Fresh air in the 21st century? Geophys Res Lett 30:1100. doi:10.1029/2002GL016285
Prinn RG et al (1999) Long-lived ozone-related compounds in scientific assessment of ozone depletion: 1998. World Meteorological Organization, Geneva
Pumphrey HC, Santee MJ, Livesey NJ, Schwartz MJ, Read WG (2011) Microwave Limb Sounder observations of biomass-burning products from the Australian bush fires of February 2009. Atmos Chem Phys 11:6285–6296
Punshon S, Moore RM (2008) Photochemical production of molecular hydrogen in lake water and coastal seawater. Mar Chem 108:215–220
Quack B, Wallace DWR (2003) Air-sea flux of bromoform: controls, rates, and implications. Global Biogeochem Cycle 17:1023. doi:10.1029/2002GB001890
Quack B, Atlas E, Petrick G, Stroud V, Schauffler S, Wallace DWR (2004) Oceanic bromoform sources for the tropical atmosphere. Geophys Res Lett 31, L23S05. doi:10.1029/2004GL020597
Quack B, Peeken I, Petrick G, Nachtigall K (2007) Oceanic distribution and sources of bromoform and dibromomethane in the Mauritanian upwelling. J Geophys Res Ocean 112, C10006. doi:10.1029/2006JC003803
Quinn PK, Bates TS (2011) The case against climate regulation via oceanic phytoplankton sulphur emissions. Nature 480:51–56
Quinn PK, Asher WE, Charlson RJ (1992) Equilibria of the marine multiphase ammonia system. J Atmos Chem 14:11–30. doi:10.1007/BF00115219
Raimund S, Quack B, Bozec Y, Vernet M, Rossi V, Garcon V, Morel Y, Morin P (2011) Sources of short-lived bromocarbons in the Iberian upwelling system. Biogeosciences 8:1551–1564
Ratte M, Plass-Dülmer C, Koppmann R, Rudolph J, Denga J (1993) Production mechanism of C2–C4 hydrocarbons in seawater: field measurements and experiments. J Glob Biogeochem Cycle 7:369–378
Ratte M, Plass-Dülmer C, Koppmann R, Rudolph J (1995) Horizontal and vertical profiles of light hydrocarbons in sea water related to biological, chemical and physical profiles. Tellus B 47:607–623
Ratte M, Bujok O, Spitzy A, Rudolph J (1998) Photochemical alkene formation in seawater from dissolved organic carbon: results from laboratory experiments. J Geophys Res 103:5707–5717
Rayman MP (2000) The importance of selenium to human health. Lancet 356:233–241
Razavi A, Karagulian F, Clarisse L, Hurtmans D, Coheur PF, Clerbaux C, Mueller JF, Stavrakou T (2011) Global distributions of methanol and formic acid retrieved for the first time from the IASI/MetOp thermal infrared sounder. Atmos Chem Phys 11:857–872
Read KA, Mahajan AS, Carpenter LJ, Evans MJ, Faria BVE, Heard DE, Hopkins JR, Lee JD, Moller SJ, Lewis AC, Mendes L, McQuaid JB, Oetjen H, Saiz-Lopez A, Pilling MJ, Plane JMC (2008a) Extensive halogen-mediated ozone destruction over the tropical Atlantic Ocean. Nature 453:1232–1235
Read KA, Lewis AC, Bauguitte S, Rankin AM, Salmon RA, Wolff EW, Saiz-Lopez A, Bloss WJ, Heard DE, Lee JD, Plane JMC (2008b) DMS and MSA measurements in the Antarctic boundary layer: impact of BrO on MSA production. Atmos Chem Phys 8:2985–2997
Rhee TS, Brenninkmeijer CAM, Rockmann T (2005) The overwhelming role of soils in the global atmospheric hydrogen cycle. Atmos Chem Phys Discuss 5:11215–11248
Richter U, Wallace DWR (2004) Production of methyl iodide in the tropical Atlantic Ocean. Geophys Res Lett 31, L23S03. doi:10.1029/2004GL020779
Riemer DD, Milne P, Zika RG, Pos WH (2000) Photoproduction of nonmethane hydrocarbons (NMHC) in seawater. Mar Chem 71:177–198
Rinsland C, Boone C, Bernath P, Mahieu E, Zander R, Dufour G, Clerbaux C, Turquety S, Chiou L, Mc-Connel J, Neary L, Kaminski JW (2006) First space-based observations of formic acid (HCOOH): atmospheric chemistry experiment austral spring 2004 and 2005 Southern Hemisphere tropical-midlatitude upper tropospheric measurements. Geophys Res Lett 33, L23804. doi:10.1029/2006GL027128
Rinsland C, Dufour G, Boone C, Bernath P, Chiou L, Coheur P, Turquety S, Clerbaux C (2007) Satellite boreal measurements over Alaska and Canada during June-July 2004: simulataneous measurements of upper tropospheric CO, C2H6, HCN, CH3Cl, CH4, C2H2, CH3OH, HCOOH, OCS, and SF6 mixing ratios. Glob Biogeochem Cycle 21, GB3008. doi:10.1029/2006GB002795
Russo RS, Zhou Y, Haase KB, Wingenter OW, Frinak EK, Mao H, Talbot RW, Sive BC (2010) Temporal variability, sources, and sinks of C1–C5 alkyl nitrates in coastal New England. Atmos Chem Phys 10:1865–1883
Saemundsdottir S, Matrai PA (1998) Biological production of methyl bromide by cultures of marine phytoplankton. Limnol Oceanogr 43:81–87
Saiz-Lopez A, Plane JMC (2004) Novel iodine chemistry in the marine boundary layer. Geophys Res Lett 31, L04112
Saiz-Lopez A, Plane JMC, Shillito JA (2004) Bromine oxide in the mid-latitude marine boundary layer. Geophys Res Lett 31. doi: 10.1029/2003GL018956
Saiz-Lopez A, Plane JMC, McFiggans G, Williams PI, Ball SM, Bitter M, Jones RL, Hongwei C, Hoffmann T (2006a) Modelling molecular iodine emissions in a coastal marine environment: the link to new particle formation. Atmos Chem Phys 6:883–895
Saiz-Lopez A, Shillito JA, Coe H, Plane JMC (2006b) Measurements and modelling of I2, IO, OIO, BrO and NO3 in the mid-latitude marine boundary layer. Atmos Chem Phys 6:1513–1528
Saiz-Lopez A, Chance K, Liu X, Kurosu TP, Sander SP (2007a) First observations of iodine oxide from space. Geophys Res Lett 34, L12812
Saiz-Lopez A, Mahajan AS, Salmon RA, Bauguitte SJ-B, Jones AE, Roscoe HK, Plane JMC (2007b) Boundary layer halogens in coastal Antarctica. Science 317:348–351
Saiz-Lopez A, Plane JMC, Mahajan AS, Anderson PS, Bauguitte SJ-B, Jones AE, Roscoe HK, Salmon RA, Bloss WJ, Lee JD, Heard DE (2008) On the vertical distribution of boundary layer halogens over coastal Antarctica: implications for O3, HOx, NOx and the Hg lifetime. Atmos Chem Phys 8:887–900
Saiz-Lopez A, Plane JMC, Baker AR, Carpenter LJ, Glasow Rv, Martín JCG, McFiggans G, Saunders RW (2012) Atmospheric chemistry of iodine. Chem Rev 112:1773–1804. doi: http://dx.doi.org/10.1021/cr200029u
Salawitch RJ (2006) Atmospheric chemistry – biogenic bromine. Nature 439:275–277
Salawitch RJ, Weisenstein DK, Kovalenko LJ, Sioris CE, Wennberg PO, Chance K, Ko MKW, McLinden CA (2005) Sensitivity of ozone to bromine in the lower stratosphere. Geophys Res Lett 32, L05811. doi:10.1029/2004GL021504
Saltzman ES, Aydin M, Tatum C, Williams MB (2008) 2,000-year record of atmospheric methyl bromide from a South Pole ice core. J Geophys Res Atmos 113, D05304. doi:10.1029/2007JD008919
Sander R, von Glasow R, Crutzen PJ (2004) Comment on: Laskin, et al. Reactions at interfaces as a source of sulfate formation in sea-salt particles. Science 303:628c
Saunders RW, Plane JMC (2005) Formation pathways and composition of iodine oxide ultra-fine particles. Environ Chem 2:299–303
Saunders RW, Plane JMC (2006) Fractal growth modelling of I2O5 nanoparticles. J Aerosol Sci 37:1737
Saunders RW, Mahajan AS, Gómez Martín JC, Kumar R, Plane JMC (2010) Studies of the formation and growth of aerosol from molecular iodine precursor. Z Phys Chem (Munich) 224:1095–1117
Savoie DL, Prospero JM, Larsen RJ, Huang F, Izaguirre MA, Huang T, Snowdon TH, Custals L, Sanderson CG (1993) Nitrogen and sulfur species in Antarctic aerosols at Mawson, Palmer Station, and Marsh (King George Island). J Atmos Chem 17:95. doi:10.1007/BF00702821
Scarratt MG, Moore RM (1996) Production of methyl bromide and chloride in laboratory cultures of marine phytoplankton. Mar Chem 54:263–272
Scarratt MG, Moore RM (1998) Production of methyl bromide and chloride in laboratory cultures of marine phytoplankton II. Mar Chem 59:311–320
Scarratt MG, Moore RM (1999) Production of chlorinated hydrocarbons by the red microalga, Porphyridium purpureum. Limnol Oceanogr 44:703–707
Schade GW, Goldstein AH (2006) Seasonal measurements of acetone and methanol: abundances and implications for atmospheric budgets. Global Biogeochem Cycle 20, GB1011. doi:10.1029/2005GB002566
Schauffler SM, Atlas EL, Flocke F, Lueb RA, Stroud V, Travnicek W (1998) Measurements of bromine containing organic compounds at the tropical tropopause. Geophys Res Lett 25:317–320
Schlesinger WH, Hartley AE (1992) A global budget for atmospheric NH3. Biogeochemistry 15:191–211. doi:10.1007/BF00002936
Schmidt U (1974) Molecular hydrogen in the atmosphere. Tellus 26:78–90
Schmittner A (2005) Decline of the marine ecosystem caused by a reduction in the Atlantic overturning circulation. Nature 434:628–633
Schönhardt A, Richter A, Wittrock F, Kirk H, Oetjen H, Roscoe HK, Burrows JP (2008) Observations of iodine monoxide columns from satellite. Atmos Chem Phys 8:637–653
Sciare J, Baboukas E, Hancy R, Mihalopoulos N, Nguyen BC (1998) Seasonal variation of dimethylsulfoxide in rainwater at Amsterdam Island in the Southern Indian Ocean; Implications on the biogenic sulfur cycle. J Atmos Chem 30:229–240
Sciare J, Baboukas E, Kanakidou M, Krischke U, Belviso S, Bardouki H, Mihalopoulos N (2000a) Spatial and temporal variability of atmospheric sulfur-containing gases and particles during the Albatross campaign. J Geophys Res 105:14433–14448
Sciare J, Kanakidou M, Mihalopoulos N (2000b) Diurnal and seasonal variation of atmospheric dimethylsulfoxide at Amsterdam Island in the southern Indian Ocean. J Geophys Res 105:17257–17265
Sciare J, Baboukas E, Mihalopoulos N (2001) Short-term variability of atmospheric DMS and its oxidation products at Amsterdam Island during summer time. J Atmos Chem 39:281–302
Seto FYB, Duce RA (1972) A laboratory study of iodine enrichment on atmospheric sea-salt particles produced by bubbles. J Geophys Res 77:5339–5349
Shaw SL, Chisholm SW, Prinn RG (2003) Isoprene production by Prochlorococcus, a marine cyanobacterium, and other phytoplankton. Mar Chem 80:227–245
Shaw SL, Gantt B, Meskhidze N (2010) Production and emissions of marine isoprene and monoterpenes: a review. Adv Meteor 2010:4048696. doi:10.1155/2010/408696
Shim C, Wang Y, Singh HB, Blake DR, Guenther AB (2007) Source characteristics of oxygenated volatile organic compounds and hydrogen cyanide. J Geophys Res 112, D10305. doi:10.1029/2006JD007543
Sievering H, Lerner B, Slavich J, Anderson J, Posfai M, Cainey J (1999) O3 oxidation of SO2 in sea-salt aerosol water: size distribution of non-sea-salt sulfate during the first aerosol characterization experiment (ACE 1). J Geophys Res 104:21707–21717
Sievering H, Cainey J, Harvey M, McGregor J, Nichol S, Quinn P (2004) Aerosol non-sea-salt sulfate in the remote marine boundary layer under clear-sky and normal cloudiness conditions: ocean-derived biogenic alkalinity enhances sea-salt sulfate production by ozone oxidation. J Geophys Res 109, D19317. doi:10.1029/2003JD004315
Simmonds PG et al (2006) Global trends, seasonal cycles, and European emissions of dichloromethane, trichloroethene, and tetrachloroethene from the AGAGE observations at Mace Head, Ireland, and Cape Grim, Tasmania. J Geophys Res 111, D18304. doi:10.1029/2006JD007082
Simo R, Pedros-Alio C (1999) Role of vertical mixing in controlling the oceanic production of dimethyl sulphide. Nature 402:396–399
Singh HB, Ohara D, Herlth D, Sachse W, Blake DR, Bradshaw JD, Kanakidou M, Critzen PJ (1994) Acetone in the atmosphere: distribution, sources, and sinks. J Geophys Res 99:1805–1819
Singh HB, Kanakidou M, Crutzen PJ, Jacob DJ (1995) High concentrations and photochemical fate of oxygenated hydrocarbons in the global troposphere. Nature 378:50–54
Singh H, Chen Y, Staudt A, Jacob D, Blake D, Heikes B, Snow J (2001) Evidence from the Pacific troposphere for large global sources of oxygenated organic compounds. Nature 410:1078–1081
Singh HB et al (2003a) In situ measurements of HCN and CH3CN over the Pacific Ocean: sources, sinks and budgets. J Geophys Res 108:8795. doi:10.1029/2002JD003006
Singh HB, Tabazadeh A, Evans MJ, Field BD, Jacob DJ, Sachse G, Crawford JH, Sette R, Brune WH (2003b) Oxygenated volatile organic chemicals in the oceans: inferences and implications based on atmospheric observations and air-sea exchange models. Geophys Res Lett 30:1862–1866
Singh HB et al (2004) Analysis of the atmospheric distribution, sources, and sinks of oxygenated volatile organic chemicals based on measurements over the Pacific during TRACE-P. J Geophys Res 109, D15D07. doi:10.1029/2003JD003883
Singh HB et al (2010) Pollution influences on atmospheric composition and chemistry at high northern latitudes: Boreal and California forest fire emissions. Atmos Environ 44:4553–4564
Sinha V, Williams J, Meyerhoefer M, Riebesell U, Paulino AI, Larsen A (2007) Air-sea fluxes of methanol, acetone, acetaldehyde, isoprene, and DMS from a Norwegian fjord following a phytoplankton bloom in a mesocosm experiment. Atmos Chem Phys 7:739–755
Sluis MK, Ensign SA (1997) Purification and characterization of acetone carboxylase from Xanthobacter strain Py2. Proc Natl Acad Sci USA 94:8456–8461
Smith MH (2007) Sea-salt particles and the CLAW hypothesis. Environ Chem 4:391–395
Smythe-Wright D, Boswell M, Breithaupt P, Davidson RD, Dimmer CH, Eiras-Diaz LB (2006) Methyl iodide production in the ocean: implications for climate change. Glob Biogeochem Cycle 20, GB3003. doi:10.1029/2005GB002642
Smythe-Wright D, Peckett C, Boswell S, Harrison R (2010) Controls on the production of organohalogens by phytoplankton: effect of nitrate concentration and grazing. J Geophys Res 115, G03020. doi:10.1029/2009JG001036
Spivakovsky CM, Yevich R, Logan JA, Wofsy SC, McElroy MB (1990) Tropospheric OH in a three-dimensional chemical tracer model: an assessment based on observations of CH3CCl3. J Geophys Res 95:18441–18471
Sprung D, Zahn A (2010) Acetone in the upper troposphere/lowermost stratosphere measured by the CARIBIC passenger aircraft: distribution, seasonal cycle, and variability. J Geophys Res 115, D16301. doi:10.1029/2009JD012099
Stefels J (2000) Physiological aspects of the production and conversion of DMSP in marine algae and higher plants. J Sea Res 43:183–197
Stefels J, Steinke M, Turner S, Malin G, Belviso S (2007) Environmental constraints on the production and removal of the climatically active gas dimethylsulphide (DMS) and implications for ecosystem modeling. Biogeochemistry 83:245–275
Stevens B, Feingold G (2009) Untangling aerosol effects on clouds and precipitation in a buffered system. Nature 461:607–613
Stubbins A, Uher G, Law CS, Mopper K, Robinson C, Upstill-Goddard RC (2006a) Open-ocean carbon monoxide photoproduction. Deep Sea Res II 53:1695–1705
Stubbins A, Uhera G, Kitidis V, Law CS, Upstill-Goddard RC, Woodward EMS (2006b) The open-ocean source of atmospheric carbon monoxide. Deep Sea Res II 53:1685–1694
Stubbins A, Hubbard V, Uher G, Law CS, Upstill-Goddard RC, Aiken GR, Mopper K (2008) Relating carbon monoxide photoproduction to dissolved organic matter functionality. Environ Sci Technol 42:3271–3276
Sturrock GA, Reeves CE, Mills GP, Penkett SA, Parr CR, McMinn A, Corno G, Tindale NW, Fraser PJ (2003) Saturation levels of methyl bromide in the coastal waters off Tasmania. Global Biogeochem Cycle 17:1107. doi:10.1029/2002GB002024
Stutz J, Pikelnaya O, Hurlock SC, Trick S, Pechtl S, von Glasow R (2007) Daytime OIO in the Gulf of Maine. Geophys Res Lett 34, L22816
Sunda W, Kieber DJ, Kiene RP, Huntsman S (2002) An antioxidant function for DMSP and DMS in marine algae. Nature 418:317–320
Sutton MA, Erisman JW, Dentener F, Möller D (2008) Ammonia in the environment: from ancient times to the present. Environ Poll 156:583–604. doi:10.1016/j.envpol.2008.03.013
Swanson AL, Davis DD, Arimooto R, Robert P, Atlas EL, Flocke F, Meinardi S, Rowland FS, Blake DR (2004) Organic trace gases of oceanic origin observed at South Pole during ISCAT 2000. Atmos Environ 38:5462–5472
Swinnerton J, Linnenbom V, Lamontagne R (1970) Ocean: a natural source of carbon monoxide. Science 167:984–986
Tanhua T, Fogelqvist E, Basturk O (1996) Reduction of volatile halocarbons in anoxic seawater, results from a study in the Black Sea. Mar Chem 54:159–170
Theiler R, Cook JC, Hager LP (1978) Halohydrocarbon synthesis by bromoperoxidase. Science 202:1094–1096
Thornton JA, Kercher JP, Riedel TP, Wagner NL, Cozic J, Holloway JS, Dube WP, Wolfe GM, Quinn PK, Middlebrook AM, Alexander B, Brown SS (2010) A large atomic chlorine source inferred from mid-continental reactive nitrogen chemistry. Nature 464:271–274
Tiefenau HK (1973) The specific ozone destruction rate of the ocean surface and its dependence on horizontal wind velocity. Pure Appl Geophys 106–108:1116–1123
Tokarczyk R, Moore RM (1994) Production of volatile organohalogens by phytoplankton cultures. Geophys Res Lett 21:285–288
Tokarczyk R, Goodwin KD, Saltzman ES (2003a) Methyl chloride and methyl bromide degradation in the Southern Ocean. Geophys Res Lett 30:1808. doi:10.1029/2003GL017459
Tokarczyk R, Saltzman ES, Moore RM, Yvon-Lewis SA (2003b) Biological degradation of methyl chloride in coastal seawater. Global Biogeochem Cycle 17:1057. doi:10.1029/2002GB001949
Toumi R (1994) BrO as a sink for dimethylsulphide in the marine atmosphere. Geophys Res Lett 21:117–120
Troxler RF, Dokos JM (1973) Formation of carbon-monoxide and bile pigment in red and blue-green-algae. Plant Phys 51:72–75
Trudinger CM, Etheridge DM, Sturrock GA, Fraser PJ, Krummel PB, McCulloch A (2004) Atmospheric histories of halocarbons from analysis of Antarctic firn air: methyl bromide, methyl chloride, chloroform, and dichloromethane. J Geophys Res 109, D22310. doi:10.1029/2004JD004932
Truesdale VW, Luther GW III (1995) Molecular iodine reduction by natural and model organic substances in seawater. Aquat Geochem 1:89–104
Tsukada H, Hara H, Iwashima K, Yamagata N (1987) The iodine content of atmospheric aerosols as determined by the use of a Fluoropore filter for collection. Bull Chem Soc Japan 60:3195–3198
Turner SM, Nightingale PD, Spokes LJ, Liddicoat MI, Liss PS (1996) Increased dimethyl sulphide concentrations in sea water from in situ iron enrichment. Nature 383:513–517
Turner SM, Harvey MJ, Law CS, Nightingale PD, Liss PS (2004) Iron-induced changes in oceanic sulfur biogeochemistry. Geophys Res Lett 31:doi:10.1029/2004GL020296
Uher G, Andreae MO (1997) Photochemical production of carbonyl sulfide in North Sea water: a process study. Limnol Oceanogr 42:432–442
Valentine RL, Zepp RG (1993) Formation of carbon-monoxide from the photodegradation of terrestrial dissolved organic-carbon in natural-waters. Environ Sci Technol 27:409–412
Vallina SM, Simo R (2007) Strong relationship between DMS and the solar radiation dose over the global surface ocean. Science 315:506–508
Vallina SM, Simo R, Manizza M (2007) Weak response of oceanic dimethylsulfide to upper mixing shoaling induced by global warming. Proc Natl Acad Sci USA 104:16004–16009
Vallina SM, Simo R, Anderson TR, Gabric A, Cropp R, Pacheco JM (2008) A dynamic model of oceanic sulfur (DMOS) applied to the Sargasso Sea: Simulating the dimethylsulfide (DMS) summer paradox. J Geophys Res-Biogeosciences 113 (G1). doi:10.1029/2007JG000415
Vardi A, Formiggini F, Casotti R, De Martino A, Ribalet F, Miralto A, Bowler C (2006) A stress surveillance system based on calcium and nitric oxide in marine diatoms. PLoS Biol 4:411–419
Vogel TM, Criddle CS, McCarty PL (1987) Transformations of halogenated aliphatic-compounds. Environ Sci Tech 21:722–736
Vogt M, Liss PS (2009) Dimethylsulfide and climate. In: Le Quéré C, Saltzman ES (eds) Surface ocean-lower atmosphere processes. American Geophysical Union, Washington, DC, pp 197–232
Vogt R, Sander R, Glasow RV, Crutzen PJ (1999) Iodine chemistry and its role in halogen activation and ozone loss in the marine boundary layer: a model study. J Atmos Chem 32:375–395
Vogt M, Vallina S, von Glasow S (2008) New directions: Correspondence on “Enhancing the natural cycle to slow global warming”. Atmos Environ 42:4803–4805
von Glasow R (2006) Importance of the surface reaction OH + Cl- on sea salt aerosol for the chemistry of the marine boundary layer – a model study. Atmos Chem Phys 6:3571–3581
von Glasow R (2007) A look at the CLAW hypothesis from an atmospheric chemistry point of view. Environ Chem 4:379–381. http://www.publish.csiro.au/nid/188/paper/EN07064.htm, http://www.publish.csiro.au/nid/188/paper/EN07064.htm
Von Glasow R (2008) Sun, sea and ozone destruction. Nature 453:1195–1196
von Glasow R, Crutzen PJ (2004) Model study of multiphase DMS oxidation with a focus on halogens. Atmos Chem Phys 4:589–608
von Glasow R, Crutzen PJ (2007) Tropospheric halogen chemistry. In: Heinrich DH, Karl KT (eds) Treatise on geochemistry. Pergamon, Oxford
von Glasow R, Sander R, Bott A, Crutzen PJ (2002) Modeling halogen chemistry in the marine boundary layer. 1. Cloud-free MBL. J Geophys Res 107:4341
von Glasow R, von Kuhlmann R, Lawrence MG, Platt U, Crutzen PJ (2004) Impact of reactive bromine chemistry in the troposphere. Atmos Chem Phys 4:2481–2497
von Gunten U (2003) Ozonation of drinking water: Part I. Oxidation kinetics and product formation. Water Res 37:1443–1467
Wada R, Beames J, Orr-Ewing A (2007) Measurement of IO radical concentrations in the marine boundary layer using a cavity ring-down spectrometer. J Atmos Chem 58:69–87
Wade LG (1999) Organic chemistry, 4th edn. Prentice-Hall, Upper Saddle River
Wang L, Lal V, Khalizov AF, Zhang R (2010) Heterogeneous chemistry of alkylamines with sulfuric acid: implications for atmospheric formation of alkylaminium sulfates. Environ Sci Tech 44:2461–2465. doi:10.1021/es9036868
Ward BB (2003) Significance of anaerobic ammonium oxidation in the ocean. Trends Microbiol 11:408–410. doi:10.1016/S0966-842X(03)00181-1
Warneck P, Williams J (2011) The atmospheric chemist’s companion, 1st edn. Springer, Dordrecht. ISBN 10: 9400722745
Watts SF (2000) The mass budgets of carbonyl sulfide, dimethyl sulfide, carbon disulfide and hydrogen sulfide. Atmos Environ 34:761–779
Weeks SJ, Currie B, Bakun A (2002) Massive emissions of toxic gas in the Atlantic. Nature 415:493–494
Wennberg PO et al (1998) Hydrogen radicals, nitrogen radicals, and the production of O3 in the upper troposphere. Science 279:49–53
Wesely ML, Hicks BB (2000) A review of the current status of knowledge on dry deposition. Atmos Environ 34:2261–2282
Whalley L, Furneaux K, Gravestock T, Atkinson H, Bale C, Ingham T, Bloss W, Heard D (2007) Detection of iodine monoxide radicals in the marine boundary layer using laser induced fluorescence spectroscopy. J Atmos Chem 58:19–39
Williams J, Holzinger R, Gros V, Xu X, Atlas E, Wallace DWR (2004) Measurements of organic species in air and seawater from the tropical Atlantic. Geophys Res Lett 31, L23S06. doi:10.1029/2004GL020012
Williams J, Custer T, Riede H, Sander R, Jöckel P, Hoor P, Pozzer A, Wong-Zehnpfennig S, Hosaynali-Beygi Z, Fischer H, Gros V, Colomb A, Bonsang B, Yassaa N, Peeken I, Atlöas EL, Waluda CM, van Aardenne JA, Lelieveld J (2010) Assessing the effect of marine isoprene and ship emissions on ozone, using modeling and measurements from the South Atlantic Ocean. Environ Chem 7:171–182. doi:10.1071/EN09154
Williamson P, Wallace DWR, Law CS, Boyd BW, Collos Y, Croot P, Denman K, Riebesell U, Takeda S, Vivian C (2012) Ocean fertilization for geoengineering: a review of effectiveness, environmental impacts and emerging governance. Process Saf Environ Prot. doi:10.1016/j.psep.2012.10.007
Wilson DF, Swinnerton J, Lamontagne R (1970) Production of carbon monoxide and gasesous hydrocarbons in seawater – relation to dissolved organic carbon. Science 168:1576–1577
Wilson ST, Foster RA, Zehr JP, Karl DM (2010) Hydrogen productioin b Trichodesmium erhthraeum Cyanothece sp. and Crocosphaera watsonii. Aquat Microb Ecol 59:197–206
Wingenter OW, Haase KB, Strutton P, Friederich G, Meinardi S, Blake DR, Rowland FS (2004) Changing concentrations of CO, CH4, C5H8, CH3Br, CH3I and dimethyl sulfide during the southern ocean iron enrichment experiments. Proc Natl Acad Sci USA 101:8537–8541
Wingenter OW, Sive BC, Blake NJ, Blake DR, Rowland FS (2005) Atomic chlorine concentrations derived from ethane and hydroxyl measurements over the Equatorial Pacific Ocean: implication for dimethyl sulfide and bromine monoxide. J Geophys Res 110, D20308. doi:10.1029/2005JD005875
Wingenter OW, Elliot SM, Blake DR (2007) New directions: enhancing the natural sulfur cycle to slow global warming. Atmos Environ 41:7373–7375
WMO (2011) Scientific assessment of ozone depletion: 2010, Global ozone research and monitoring project-report no. 52 Rep. World Meteorological Organization, Geneva
Woodhouse MT, Mann GW, Carslaw KS, Boucher O (2008) New directions: the impact of oceanic iron fertilization on cloud condensation nuclei. Atmos Environ 42:5728–5730
Woodhouse MT, Carslaw KS, Mann GW, Vallina SM, Vogt M, Halloran PR, Boucher O (2010) Low sensitivity of cloud condensation nuclei to changes in the sea-air flux of dimethyl-sulphide. Atmos Chem Phys 10:7545–7559
Wuosma AM, Hager PL (1990) Methylchloride transerfrase. A carbocation route for biosynthesis of halometabolites. Science 249:160–162
Xiao X et al (2010) Optimal estimation of the surface fluxes of methyl chloride using a 3-D global chemical transport model. Atmos Chem Phys 10:5515–5533
Xie HX, Moore RM, Miller WL (1998) Photochemical production of carbon disulphide in seawater. J Geophys Res 103:5635–5644
Xie HX, Moore RM (1999) Carbon disulfide in the North Atlantic and Pacific Ocean. J Geophys Res 104:5393–5402
Xie HX, Zafiriou OC, Umile TP, Kieber DJ (2005) Biological consumption of carbon monoxide in Delaware Bay, NW Atlantic and Beaufort Sea. Mar Ecol Prog Ser 290:1–14
Xie HX, Belanger S, Demers S, Vincent WF, Papakyriakou TN (2009) Photobiogeochemical cycling of carbon monoxide in the southeastern Beaufort Sea in spring and autumn. Limnol Oceanogr 54:234–249
Xu S, Xie Z-Q, Li B, Sun L, Kang H, Yang H, Zhang P (2010) Iodine speciation in marine aerosols along a 15000-km round-trip cruise path from Shanghai, China, to the Arctic Ocean. Environ Chem 7:406–412
Yang M, Huebert BJ, Blomquist BW, Howell SG, Shank LM, McNaughton CS, Clarke AD, Hawkins LN, Russell LM, Covert DS, Coffman DJ, Bates TS, Quinn PK, Zagorac N, Bandy AR, de Szoeke SP, Zuidema PD, Tucker SC, Brewer WA, Yang X, Cox RA, Warwick NJ, Pyle JA, Carver GD, O’Connor FM, Savage NH (2005) Tropospheric bromine chemistry and its impacts on ozone: a model study. J Geophys Res Atmos 110, D23311. doi:10.1029/2005JD006244
Yang GP, Wang WL, Lu XL, Ren CY (2010) Distribution, flux and biological consumption of carbon monoxide in the Southern Yellow Sea and the East China Sea. Mar Chem 122:74–82
Yang GP, Ren CY, Lu XL, Liu CY, Ding HB (2011) Distribution, flux, and photoproduction of carbon monoxide in the East China Sea and Yellow Sea in spring. J Geophys Res Ocean 116, CO2001. doi:10.1029/2010JC006300
Yassaa N, Peeken I, Zöllner E, Bluhm K, Arnold S, Spracklen D, Williams J (2008) Evidence for marine production of monoterpenes. Environ Chem 5:391–401. doi:10.1071/EN08047
Yu F (2006) Effect of ammonia on new particle formation: a kinetic H2SO4-H2O-NH3 nucleation model constrained by laboratory measurements. J Geophys Res D111, D01204. doi:10.1029/2005JD005968
Yvon-Lewis SA, Butler JH, Saltzman EH, Matrai PA, King DB, Tokarczyk R, Moore RM, Zhang JZ (2002) Methyl bromide cycling in a warm-core eddy of the North Atlantic Ocean. Glob Biogeochem Cycle 16:1141. doi:10.1029/2002GB001898
Yvon-Lewis SA, Saltzman ES, Montzka SA (2009) Recent trends in atmospheric methyl bromide: analysis of post-montreal protocol variability. Atmos Chem Phys 9:5963–5974
Zafiriou OC (1975) Reaction of methyl halides with seawater and marine aerosols. J Mar Res 33:75–81
Zafiriou OC, MacFarland M (1981) Nitric oxide formation from nitrite photolysis in the central equatorial Pacific. J Geophys Res 86:3173–3182
Zafiriou OC, McFarland M, Bromund RH (1980) Nitric oxide in seawater. Science 207:637–639
Zafiriou OC, Andrews SS, Wang W (2003) Concordant estimates of oceanic carbon monoxide source and sink processes in the Pacific yield a balanced global “blue-water” CO budget. Glob Biogeochem Cycle 17. doi:10.1029/2001GB001638
Zhang Z, Liu C, Wu Z, Xing L, Li P (2006) Detection of nitric oxide in culture media and studies on nitric oxide formation by marine microalgae. Med Sci Monit 12:BR75–BR85
Zhou X, Mopper K (1997) Photochemical production of low-molecular-weight carbonyl compounds in seawater and surface microlayer and their air-sea exchange. Mar Chem 56:201–213
Zika RG, Gidel LT, Davis DD (1984) A comparison of photolysis and substitution decomposition rates of methyl iodide in the ocean. Geophys Res Lett 11:353–356
Ziolkowski L, Miller W (2007) Variability of the apparent quantum efficiency of CO photoproduction in the Gulf of Maine and Northwest Atlantic. Mar Chem 105:258–270
Zuo Y, Jones RD (1995) Formation of carbon monoxide by photolysis of dissolved marine organic material and its significance in the carbon cycling of the oceans. Naturwissenschaften 82:472–474
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is distributed under the terms of the Creative Commons Attribution Noncommercial License, which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Copyright information
© 2014 The Author(s)
About this chapter
Cite this chapter
Liss, P.S. et al. (2014). Short-Lived Trace Gases in the Surface Ocean and the Atmosphere. In: Liss, P.S., Johnson, M.T. (eds) Ocean-Atmosphere Interactions of Gases and Particles. Springer Earth System Sciences. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-25643-1_1
Download citation
DOI: https://doi.org/10.1007/978-3-642-25643-1_1
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-25642-4
Online ISBN: 978-3-642-25643-1
eBook Packages: Earth and Environmental ScienceEarth and Environmental Science (R0)