
Zusammenfassung
Nach der Marseille-Rom-Klassifikation ist die chronische Pankreatitis gegenwärtig definiert als kontinuierliche entzündliche Erkrankung, die zu einer fortschreitenden oder permanenten exokrinen und/oder endokrinen Funktionseinschränkung führt. Morphologisch findet sich eine unregelmäßige Sklerosierung des Organs mit fokaler, segmentaler oder diffuser Zerstörung des exokrinen Gewebes. Ätiologische Faktoren wurden bei dieser Klassifikation nicht berücksichtigt. In der Marseille-Rom-Klassifikation wurde die akute Pankreatitis „nicht als Krankheit, sondern als Spektrum entzündlicher Läsionen“ definiert, wobei der Schweregrad von einer leichten interstitiellödematösen bis zu einer schweren hämorrhagisch-nekrotisierenden Entzündung variieren kann. Auch stimmten die meisten Teilnehmer der Konferenz in Rom darin überein, dass die chronische Pankreatitis eine Ursache der akuten Pankreatitis, aber nicht deren Folge ist und dass die akute Pankreatitis nur selten in eine chronische Entzündung mündet. Nach diesem Konzept stellen akute und chronische Pankreatitis zwei separate Krankheitsentitäten dar, die nur selten zusammenfließen.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


1 Ätiologie, klinisches Bild und Pathogenese
Die in den letzten Jahren erhobenen genetischen Befunde untermauern das Konzept, dass ein Ungleichgewicht von Proteasen und ihren Inhibitoren wesentlich an der Pathogenese der Pankreatitis beteiligt ist. Die Identifizierung von Mutationen im kationischen Trypsinogen (PRSS1) bei Patienten mit hereditärer Pankreatitis hat das pathophysiologische Verständnis der Erkrankung entscheidend beeinflusst. Der Nachweis von SPINK1-, CTRC- ,CFTR- und PRSS1-Mutationen bei Patienten ohne Familienanamnese für eine Pankreatitis deutet darauf hin, dass auch die idiopathische Pankreatitis genetisch determiniert ist. Neben genetischen Faktoren sind systemische und metabolische Erkrankungen, die zystische Fibrose, anatomische Anomalien, Gallensteine, Traumata und Infektionen pathogenetisch relevant.
Definition
Nach der Marseille-Rom-Klassifikation ist die chronische Pankreatitis gegenwärtig definiert als kontinuierliche entzündliche Erkrankung, die zu einer fortschreitenden oder permanenten exokrinen und/oder endokrinen Funktionseinschränkung führt. Morphologisch findet sich eine unregelmäßige Sklerosierung des Organs mit fokaler, segmentaler oder diffuser Zerstörung des exokrinen Gewebes. Ätiologische Faktoren wurden bei dieser Klassifikation nicht berücksichtigt. In der Marseille-Rom-Klassifikation wurde die akute Pankreatitis „nicht als Krankheit, sondern als Spektrum entzündlicher Läsionen“ definiert, wobei der Schweregrad von einer leichten interstitiell-ödematösen bis zu einer schweren hämorrhagisch-nekrotisierenden Entzündung variieren kann. Auch stimmten die meisten Teilnehmer der Konferenz in Rom darin überein, dass die chronische Pankreatitis eine Ursache der akuten Pankreatitis, aber nicht deren Folge ist und dass die akute Pankreatitis nur selten in eine chronische Entzündung mündet. Nach diesem Konzept stellen akute und chronische Pankreatitis zwei separate Krankheitsentitäten dar, die nur selten zusammenfließen.
Die derzeitige Definition ist jedoch mit der klinischen Präsentation der erblichen Pankreatitis unvereinbar, die durch in der Kindheit beginnende, wiederkehrende Pankreatitisschübe gekennzeichnet ist. Zeichen einer funktionellen oder morphologischen Pankreasschädigung sind initial nicht vorhanden. Mit den Jahren entwickeln jedoch viele Patienten mit akuter rekurrierender Pankreatitis eine exokrine oder endokrine Funktionseinschränkung sowie Gangveränderungen und Kalzifizierungen. Demzufolge sind akute und chronische Pankreatitis als unterschiedliche Stadien eines dynamischen Krankheitsprozesses zu begreifen. Die verschiedenen Konzepte der chronischen Pankreatitis in der pädiatrischen und internistischen Literatur spiegeln die Tatsache wider, dass der Pädiater mit dem frühen Erkrankungsstadium und der internistische Gastroenterologe häufig mit dem Endstadium konfrontiert wird. Die Existenz gleicher genetischer Defekte bei verschiedenen Formen der akuten und chronischen Pankreatitis unterstützt zudem das alte Konzept, dass alle Zwischenstadien zwischen akuter und chronisch-kalzifizierender Pankreatitis bestehen, wie es ursprünglich von Comfort und Mitarbeitern postuliert wurde.
Epidemiologie
Zur Inzidenz und Prävalenz der akuten und chronischen Pankreatitis im Kindesalter existieren keine epidemiologischen Daten. Die Inzidenz der akuten Pankreatitis im Erwachsenenalter wird für die USA und Europa mit 5–50 und die Inzidenz der chronischen Pankreatitis mit 3,5–10 Neuerkrankungen auf 100.000 Einwohner und Jahr angegeben. Allerdings sind die akute wie auch die chronische Pankreatitis im Erwachsenenalter zu 70–80 % biliär (Gallensteine) oder ethyltoxisch bedingt. Obwohl einige weitere gut charakterisierte Faktoren, wie metabolische Störungen, anatomische Anomalien oder Traumata, bekannt sind, findet sich bei etwa 10–30 % der Patienten keine auslösende Ursache; diese Form der Pankreatitis wird als idiopathisch bezeichnet. Ungefähr 5–10 % der Patienten werden aufgrund einer positiven Familienanamnese als hereditär klassifiziert.
Da Alkoholabusus im Kindesalter pathogenetisch nicht bedeutsam ist, sind die zur Verfügung stehenden epidemiologischen Daten nicht verwertbar. Nach eigenen Schätzungen dürfte in Deutschland der Anteil der sog. idiopathischen Form bei 20–40 % und der Anteil der hereditären Form bei 10–20 % liegen. Weitere häufige Ursachen im Kindesalter stellen anatomische Anomalien, Traumata, Sepsis oder Schock, die zystische Fibrose und chronisch-entzündliche Darmerkrankungen dar.
Pathogenese
Vor über einem Jahrhundert entwickelte Chiari die Hypothese, dass die Pankreatitis Folge einer Selbstverdauung des Organs ist. Die zellulären Mechanismen blieben aber für lange Zeit ungeklärt. Es wurde postuliert, dass eine übermäßige Trypsinaktivität im Pankreasparenchym mit konsekutiver Aktivierung anderer Enzyme für den Entzündungsprozess verantwortlich ist. Genetische Studien unterstützen die Theorie eines intrapankreatischen Ungleichgewichts zwischen Verdauungsenzymen und ihren Inhibitoren. Das Verdauungsenzym Trypsin nimmt im pankreatischen Proteasensystem eine Schlüsselrolle ein. Die Serinprotease Trypsin vermag sowohl sich selbst als auch alle anderen proteolytischen Proenzyme des Pankreas zu aktivieren. Das Pankreas synthetisiert und sezerniert Trypsin als inaktives Trypsinogen (Zymogen). Erst im Darm erfolgt durch Abspaltung des Aktivierungspeptids mit Hilfe des Enzyms Enteropeptidase (Enterokinase) die Umwandlung des Trypsinogens zu Trypsin. Geringe Mengen an Trypsinogen werden auch im normalen Pankreasgewebe durch Autolyse zu aktivem Trypsin umgewandelt. Zwei Mechanismen schützen das Pankreas vor einer überschießenden Trypsinaktivität und Selbstverdauung:
-
Zum einem wird Trypsin durch den Serinproteaseinhibitor, Kazal-Typ 1 (SPINK1) komplexiert. SPINK1, auch als pankreatischer sekretorischer Trypsin-Inhibitor (PSTI) bezeichnet, ist ein wichtiger intrapankreatischer Trypsininhibitor, der Trypsin durch Bildung einer kovalenten Bindung zwischen dem katalytischen Serin der Protease und einem Lysin im reaktiven Zentrum von SPINK1 inhibiert.
-
Zum anderen werden Trypsin und weitere Pankreasproteasen durch Trypsin und trypsinähnliche Enzyme wie Chymotrypsin C (CTRC) degradiert (◘ Abb. 22.1).
Abb. 22.1 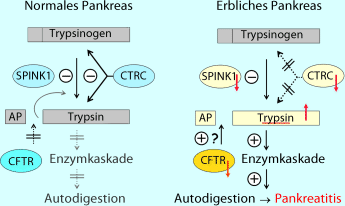
Modell der erblichen Pankreatitis. AP Aktivierungspeptid; CFTR „cystic fibrosis transmembrane conductance regulator“; CTRC Chymotrypsinogen C; SPINK1 Serinproteaseinhibitor Kazal-Typ 1


Ätiologie
Nach neueren Erkenntnissen ist ein erheblicher Prozentsatz der idiopathischen Pankreatitis im Kindesalter genetisch bedingt. Daher werden im Folgenden die idiopathische und hereditäre Form als primäre Pankreatitis zusammengefasst in Abgrenzung zu den durch Stoffwechseldefekte, Noxen, anatomische Anomalien oder systemische Grunderkrankungen bedingten sekundären Formen.
Primäre Pankreatitis
Die klassische Form der hereditären Pankreatitis folgt einem autosomal-dominanten Erbgang und wurde zum ersten Mal 1952 von Comfort und Steinberg beschrieben. Vor Kurzem gelang es, einen Genort für die hereditäre Pankreatitis auf dem langen Arm des Chromosoms 7 (7q35) zu lokalisieren. Wenig später wurde eine Mutation im kationischen Trypsinogen (PRSS1) als Erkrankungsursache identifiziert. Bei fünf untersuchten Familien fand sich ein Arginin-Histidin-Austausch an Position 122 des Proteins (R122H). Inzwischen wurden etliche weitere Mutationen im PRSS1-Gen beschrieben. Studien bei pädiatrischen Patienten zeigten, dass Trypsinogenmutationen auch bei Patienten ohne Familienanamnese für eine Pankreatitis nachweisbar sind. Es wird angenommen, dass die Trypsinogenmutationen zu einer vermehrten Selbstaktivierung und teilweise zu einem geringeren Abbau der aktiven Enzyme im Pankreasgewebe führen.
Auch Mutationen im Serinproteaseinhibitor, Kazal-Typ 1 (SPINK1) sind mit Pankreatitis assoziiert. Eine Punktmutation im Exon 3, die zu einem Asparagin-Serin-Austausch an Position 34 des Proteins führt (N34S), ist mit 80–90 % aller SPINK1-Mutationen die wichtigste Veränderung. Die N34S-Mutation findet sich vornehmlich bei Patienten ohne Familienanamnese: 20–40 % der Patienten mit sog. idiopathischer chronischer Pankreatitis tragen auf einem oder beiden Allelen diese Mutation. Eine Heterozygotie für N34S dürfte allerdings pathogenetisch nicht ausreichend sein, da etwa 1–2 % der Bevölkerung N34S-Träger sind. Wahrscheinlich führt erst die Kombination mit anderen Gendefekten oder Umweltfaktoren zum Ausbruch der Erkrankung. Neben N34S wurden mehrere weitere SPINK1-Mutationen identifiziert, deren Bedeutung in den meisten Fällen zurzeit noch unklar ist. Im Weiteren finden sich Mutationen im Chymotrypsinogen C (CTRC) ca. 4-mal häufiger bei Patienten mit chronischer Pankreatitis. CTRC ist eine pankreatische Protease, die aktives Trypsin spalten und damit inaktivieren kann. Die CTRC-Mutationen führen zu einer verminderten Enzymsekretion und/oder -aktivität und konsekutiv zu einer vermehrten Trypsinaktivität im Pankreas. SPINK1- und CTRC-Mutationen finden sich außer bei der sog. idiopathischen Pankreatitis auch vermehrt bei alkoholinduzierter chronischer Pankreatitis und bei tropischer Pankreatitis.
Nach neuesten Daten finden sich insbesondere bei sehr jungen Patienten vermehrt Mutationen in der Carboxypeptidase A1 (CPA1 ): Über 10 % der Patienten, die vor dem 10. Lebensjahr eine Pankreatitis entwickeln, weisen CPA1-Mutationen auf, die zu einem Funktionsverlust des Enzyms führen („loss of function“). Warum CPA1-Mutationen zu einer Pankreatitis disponieren, ist zurzeit noch unklar.
Auch eine Assoziation zwischen genetischen Veränderungen im Zystische-Fibrose-Gen („cystic fibrosis transmembrane conductance regulator“; CFTR ) und idiopathischer chronischer Pankreatitis ist beschrieben worden. Mehrere Arbeiten zeigten, dass heterozygote Träger einer CFTR-Mutation ein erhöhtes Erkrankungsrisiko besitzen. Das CFTR-Gen kodiert für einen Chloridkanal. Möglicherweise begünstigen eine veränderte Viskosität des Pankreassaftes und/oder eine pH-Wert-Änderung infolge eines gestörten Ionentransportes bei heterozygoten CFTR-Trägern die Autoaktivierung von Trypsinogen und damit die Krankheitsentstehung. Warum allerdings die überwiegende Mehrheit der heterozygoten Merkmalsträger keine Pankreatitis entwickelt, ist zum gegenwärtigen Zeitpunkt unbekannt.
Kleinere Studien beschrieben eine Assoziation zwischen einem α1-Antitrypsin-Mangel sowie Zytokeratin-8-(KRT8-)Mutationen und chronischer Pankreatitis, die in nachfolgenden Studien allerdings nicht bestätigt wurden.
Die genetischen Studien der letzten Jahre haben das Verständnis der erblichen Pankreatitis entscheidend verändert. Lange Zeit galt die erbliche Pankreatitis als seltene Erkrankung. Der Nachweis von PRRS1-, SPINK1-, CTRC- und CFTR-Mutationen bei Patienten mit sog. idiopathischer Pankreatitis zeigt jedoch, dass erbliche Fälle der chronischen Pankreatitis weitaus häufiger sind als bislang vermutet. Diese Befunde stellen zugleich die Unterscheidung zwischen „erblicher“ und „idiopathischer“ Pankreatitis in Frage. Verschiedene Mutationen in unterschiedlichen Genen können zu einem unterschiedlichen Phänotyp und Vererbungsmuster führen und selbst dieselbe Mutation in einem Gen kann in Abhängigkeit des individuellen genetischen Hintergrundes und Umweltfaktoren verschiedene Konsequenzen haben.
So ist die primäre chronische Pankreatitis eine genetisch heterogene Erkrankung, die in Abhängigkeit von den defekten Genen bzw. den zugrunde liegenden Mutationen einem autosomal-dominanten, einem autosomal-rezessiven oder einem komplexen Erbgang folgt.
In Zukunft werden voraussichtlich Defekte in weiteren Genen identifiziert werden, da Veränderungen im PRSS1-, SPINK1-, CTRC-, CPA1- und CFTR-Gen nur bei etwa 60 % der Patienten mit primärer chronischer Pankreatitis zu finden sind. Die Assoziation von SPINK1- und CTRC-Mutationen mit tropischer kalzifizierender Pankreatitis und alkoholischer Pankreatitis verwischt weiter die Grenzen zwischen den einzelnen Pankreatitisunterformen. In den nächsten Jahren wird sich wahrscheinlich zeigen, dass sehr komplexe Interaktionen zwischen Umwelteinflüssen und zahlreichen genetischen Faktoren bestehen mit fließenden Übergängen zwischen den einzelnen Subtypen. Diese Hypothese wird durch die Beobachtung von SPINK1-Mutationen bei Patienten mit metabolischen Erkrankungen oder anatomischen Anomalien untermauert.
Ursachen der chronischen Pankreatitis
-
Hereditär/„idiopathisch“:
-
Kationisches Trypsinogen (PRSS1)
-
Carboxypeptidase A1 (CPA1)
-
SerinproteaseiInhibitor, Kazal-Typ 1 (SPINK1)
-
Chymotrypsinogen C (CTRC)
-
„Cystic fibrosis transmembrane conductance regulator“ (CFTR)
-
Weitere Gendefekte?
-
-
Systemische Erkrankungen:
-
Schock
-
Chronisch-entzündliche Darmerkrankungen
-
Primär sklerosierende Cholangitis (PSC)
-
Systemischer Lupus erythematodes
-
Rheumatoide Arthritis
-
Panarteriitis nodosa
-
M. Behçet
-
Hämolytisch-urämisches Syndrom
-
Sichelzellanämie
-
Autoimmune Pankreatitis
-
-
Metabolisch:
-
Hypertriglyceridämie
-
Hyperkalzämie
-
Zystische Fibrose
-
Dystrophie
-
Malnutrition
-
Niereninsuffizienz
-
Diabetische Ketoacidose
-
-
Medikamentös/toxisch:
-
Medikamente
-
Toxine
-
-
Mechanisch/strukturell:
-
Anatomische Anomalien
-
Obstruktion (Gallensteine, Tumoren, Parasiten)
-
Trauma
-
-
Infektiös:
-
Viral
-
Bakteriell
-
Parasitär
-
Sekundäre Pankreatitis
Jede schwere Einschränkung der Herz-Kreislauf-Situation, die zu einer verminderten Oxygenierung oder zu einer reduzierten Blutzufuhr des Pankreas führt, wie z. B. ein Schockzustand, kann eine Pankreatitis provozieren.
Eine Verbindung zwischen Pankreatitis und Kollagenosen wie systemischer Lupus erythematodes, rheumatoider Arthritis, Polyarteriitis nodosa und M. Behçet ist berichtet worden. Vermutlich ist die Pankreatitis Folge einer Vaskulitis. Inwieweit immunologische Mechanismen im Sinne einer Autoimmunpankreatitis an der Pathogenese beteiligt sind, ist zurzeit ungeklärt. Die Verbindung zwischen chronisch-entzündlichen Darmerkrankungen und einer Pankreatitis war lange Zeit Gegenstand von Kontroversen. Ein lokales Entzündungsgeschehen im Duodenum, eine begleitende primär sklerosierende Cholangitis oder die medikamentöse Therapie wurden als Ursache diskutiert. Neuere Studien legen jedoch nahe, dass die Pankreatitis unabhängig von diesen genannten Faktoren entsteht und den klinischen Zeichen einer chronisch-entzündlichen Darmerkrankung um Jahre vorausgehen kann. Eine Assoziation mit der primär sklerosierenden Cholangitis (mit oder ohne chronisch-entzündliche Darmerkrankung) besteht ebenfalls. Inwieweit die Pankreatitis durch Obstruktion des gemeinsamen pankreatikobiliären Gangs oder durch andere Mechanismen bedingt ist, ist zurzeit ungeklärt. Die Mehrheit der Patienten mit einem hämolytisch-urämischen Syndrom zeigen transiente Pankreasenzymerhöhungen im Rahmen einer akuten Begleitpankreatitis. In seltenen Fällen kann es zur Ausbildung einer chronischen Pankreatitis kommen. Auch bei Patienten mit Sichelzellanämie wird vermehrt eine Pankreatitis beobachtet. Es ist unklar, ob die Pankreatitis biliärer Genese, d. h. durch Gallensteine, oder durch vasookklusive Krisen bedingt ist.
In den letzten Jahren wurden zunehmend Fälle mit sog. autoimmuner Pankreatitis beschrieben. Diese Entität wurde zuerst in Japan bei erwachsenen Patienten beobachtet und geht mit einer mit IgG4-Erhöhung einher. Viele aus Europa berichtete Fälle sind allerdings eher als Pankreasbeteiligung, d. h. als extraintestinale Manifestation einer chronisch-entzündlichen Darmerkrankung zu werten.
Primäre und sekundäre Hyperlipoproteinämien, die mit hohen Plasmatriglyceridwerten einhergehen (Lipoproteinlipasemangel , Apolipoprotein-CII-Mangel ) können zu rezidivierenden Pankreatitiden führen. Der Mechanismus ist bislang ungeklärt. Eine Hyperkalzämie (z. B. bei primärem Hyperparathyreoidismus, Vitamin-D-Intoxikation oder exzessiver iatrogener Kalziumzufuhr) vermag – wahrscheinlich aufgrund eines kalziumvermittelten Sekretionsstimulus – eine akute Pankreatitis auszulösen. Obwohl die Pankreasinsuffizienz bei zystischer Fibrose Folge einer entzündlichen Zerstörung der Drüse ist, wurde die zystische Fibrose nicht in die Marseille-Rom-Klassifikation aufgenommen. Etwa 1–2 % der Patienten mit zystischer Fibrose zeigen klinisch das Bild einer rezidivierenden Pankreatitis. Diese Verlaufsform wird meistens bei pankreassuffizienten Patienten gefunden.
In nichtindustrialisierten Ländern wird die Malnutrition als wichtiger Auslöser einer Pankreatitis angesehen. Die Ursache ist weitgehend ungeklärt. Eine Proteinmangelernährung, ein Mangel an Spurenelementen wie Zink oder Selen oder ein Vitaminmangel werden als ursächlich diskutiert. Neuere Daten legen jedoch zumindest für die chronisch-kalzifizierende Pankreatitis eine genetische Grundlage (SPINK1- und CTRC-Mutationen) nahe. Einzelne Fallberichte beschrieben eine Pankreatitis bei chronischer Niereninsuffizienz bzw. nach Nierentransplantation. Es ist unklar, ob die Pankreatitis bei Nierentransplantierten Folge der durch die Niereninsuffizienz bedingten metabolischen Veränderungen oder Folge immunsuppressiver Medikamente (Tacrolimus etc.) ist. Bei bis zu 2 % der Patienten mit einer diabetischen Ketoacidose findet sich als Komplikation eine akute Pankreatitis.
Obwohl unzählige Fallberichte über medikamentös induzierte Pankreatitiden existieren, ist der kausale Zusammenhang nur für wenige Medikamente gesichert, da in vielen Fällen nach Absetzen des verdächtigten Agens keine Reexposition erfolgte (◘ Tab. 22.1). Häufig lässt sich nicht unterscheiden, ob die Pankreatitis im Rahmen einer – medikamentös behandelten – Grunderkrankung oder durch das Medikament selbst hervorgerufen wurde. Beispielhaft seien hier die Steroide erwähnt, die lange Zeit als pathogenetisch relevant angesehen wurden. Die Latenzzeit zwischen erstmaliger Einnahme und Auftreten einer Pankreatitis kann in Abhängigkeit vom Medikament beträchtlich differieren: Während unter Azathioprintherapie eine Pankreatitis meist schon im ersten Behandlungsmonat auftritt, sind für Valproat Latenzzeiten von über 10 Jahren beschrieben worden.
Anatomische Anomalien wie Choledochuszysten, ein Pancreas anulare oder Pankreasgangduplikationen sind etablierte Risikofaktoren für eine Pankreatitis, während die Assoziation zwischen Pancreas divisum und Pankreatitis bis heute kontrovers diskutiert wird. Eine Obstruktion des Pankreasgangs durch Gallensteine, Tumoren oder Parasiten (insbesondere Ascaris lumbricoides) stellt eine weitere, häufigere Ursache für eine akute Pankreatitis dar. Abdominale Traumata verursachen eine selbstlimitierende akute Pankreatitis, die in seltenen Fällen, insbesondere bei Pseudozystenbildung oder Ruptur des Pankreasgangs, rezidivieren kann.
Unter den vielen Erregern, die eine akute Pankreatitis auslösen, sind insbesondere die Enteroviren (Coxsackie-B- und Echoviren) sowie Mumpsviren, Yersinien und Ascariden hervorzuheben.
Infektionserkrankungen, die mit einer Pankreatitis assoziiert sind
-
Virusinfektionen:
-
Coxsackie-B-Virus-Infektion
-
Infektion durch Echoviren
-
Hepatitis A, B und E
-
Infektion mit Herpesviren (Zytomegalie-, Epstein-Barr-, Herpes-simplex-, Varizella-Zoster-Virus)
-
HIV-Infektion
-
Masern
-
Mumps
-
Röteln
-
-
Bakterielle Infektionen durch folgende Erreger:
-
Campylobacter
-
Escherichia coli (assoziiert mit hämolytisch-urämischem Syndrom, HUS)
-
Legionellen
-
Leptospiren
-
Mykoplasmen
-
Salmonellen
-
Yersinien
-
-
Infektionen mit folgenden Parasiten:
-
Ascaris lumbricoides
-
Clonorchis sinensis
-
Cryptosporidium parvum – vorwiegend bei Immunkompromittierten (AIDS)
-
Echinococcus granulosus
-
Fasciola hepatica
-
Toxoplasma gondii – vorwiegend bei Immunkompromittierten (AIDS)
-
Klinik
Leitsymptome
Leitsymptom im Kindesalter sind plötzlich auftretende abdominale Schmerzen. Die Schmerzen sind meistens im Oberbauch lokalisiert, können aber auch in andere Körperregionen wie Unterbauch oder Rücken projiziert werden. Weitere Befunde sind Übelkeit, Erbrechen, abdominaler Druckschmerz und verminderte Darmgeräusche. Zusätzlich können leichtes Fieber, Tachykardie und Hypotension auftreten. Bei der chronischen Pankreatitis entwickelt ein Teil der Patienten im Verlauf der Erkrankung eine exokrine und/oder endokrine Pankreasinsuffizienz mit Maldigestion und einem Insulinmangeldiabetes. Die Maldigestion äußert sich klinisch mit massigen, stinkenden Stuhlentleerungen, Steatorrhö und Gewichtsabnahme.
Komplikationen
Im Rahmen der entzündlichen Reaktion können pankreatische und extrapankreatische Komplikationen auftreten, die in der folgenden ▶ Übersicht zusammengefasst sind. Nicht selten bilden sich Pankreaspseudozysten aus. Die Pseudozysten können zu einer Kompression des Ductus choledochus, des Duodenums oder der Milzvene führen, sich infizieren oder in die Bauchhöhle bzw. ins Retroperitoneum rupturieren. Weitere Komplikationen sind Verkalkungen und Nekrosen mit oder ohne Ausbildung eines Pankreasabszesses. Bei schweren Verläufen können systemische Komplikationen wie eine metabolische Acidose, Stoffwechselentgleisungen (Hyperglykämie, Hyperkaliämie, Hypokalzämie), Schock, Organversagen (Herz-Kreislauf-System, Lunge, Niere) und eine disseminierte intravasale Gerinnung (DIC) auftreten. In einigen Fällen finden sich extrapankreatische Komplikationen wie Pleuraerguss, Aszites, portale Hypertension, Ulkus mit gastrointestinaler Blutung oder eine Gallengangobstruktion.
Nach neueren Studien besteht bei der erblichen Pankreatitis ein erhöhtes Risiko für die Entwicklung eines Pankreaskarzinoms.
Komplikationen der Pankreatitis
-
Pankreatische Komplikationen:
-
Pseudozysten
-
Nekrosenbildung
-
Pankreasabszess
-
Verkalkungen
-
Exokrine Insuffizienz
-
Endokrine Insuffizienz
-
Karzinom
-
-
Extrapankreatische Komplikationen:
-
Pleuraerguss
-
Aszites
-
Portale Hypertension
-
Ulkus/gastrointestinale Blutung
-
Gallengangobstruktion
-
Systemisch (Acidose, DIC, Elektrolytentgleisungen, Schock)
-
Diagnostik
Der Nachweis einer akuten Pankreatitis erfolgt anhand der klinischen Symptomatik sowie anhand der Bestimmung der Lipase im Serum und der sonographischen Untersuchung des Abdomens. In der ätiologischen Abklärung ist ein Schweißtest zum Ausschluss einer zystischen Fibrose unerlässlich. Insbesondere bei rezidivierenden Pankreatitisschüben oder bei einer chronischen Pankreatitis sollte eine gezielte klinisch-chemische, serologische, genetische und bildgebende Abklärung erfolgen.
Neben der klinischen Symptomatik nehmen laborchemische und bildgebende Verfahren einen wichtigen Stellenplatz in der Diagnostik der Pankreatitis ein.
Laborparameter
Richtungsweisend bei entsprechender klinischer Symptomatik ist die Bestimmung der Lipase im Serum. Bei Erhöhungen über das 3-Fache des oberen Referenzbereiches ist eine Pankreatitis sehr wahrscheinlich. Die Lipasekonzentration im Blut steigt innerhalb weniger Stunden nach Erkrankungsbeginn an.
Die Lipase ist der Amylase an Sensitivität und Spezifität überlegen; insbesondere einige Tage nach Beginn der Erkrankung ist die Sensitivität und Spezifität der Amylase gering. Eine zusätzliche Bestimmung der Amylase bringt keine weitere diagnostische Information. Die Urinamylase ist von sehr geringer Aussagekraft und sollte nicht bestimmt werden.
Die Bestimmung der Isoenzyme (Pankreasamalyse) gibt nur selten zusätzliche Information und hat sich in der klinischen Routinediagnostik nicht durchsetzen können.
Die Enzymwerte korrelieren nicht mit dem klinischen Schweregrad.
Blande Verläufe oder diagnostische Prozeduren wie eine endoskopische retrograde Cholangiopankreatikographie (ERCP) können mit Enzymerhöhungen um mehr als das 20-Fache einhergehen, während bei Patienten mit einer nur leichten Enzymerhöhung von weniger als dem 3-Fachen des oberen Referenzbereichs durchaus eine schwere nekrotisierende Pankreatitis vorliegen kann. Die Plasmakonzentration der Pankreasenzyme im Serum wird durch Mahlzeiten nicht beeinflusst.
Bei akuter Pankreatitis oder bei Verschlüssen im Bereich des Ductus pancreaticus werden die normalerweise in den Dünndarm sezernierten Enzyme vermehrt in die Blutbahn abgegeben. Da die Amylase aufgrund des niedrigen Molekulargewichts die Glomerulusmembran passieren kann und nur partiell tubulär resorbiert wird, ist sie auch im Urin bestimmbar. Die Lipase wird in der Niere vollständig tubulär resorbiert und kann daher im Gegensatz zur Amylase nicht im Urin nachgewiesen werden.
Erhöhte Lipase- und Amylasewerte finden sich bei akuter Pankreatitis bzw. dem akuten Schub einer chronischen Pankreatitis sowie nach einer ERCP (▶ folgende Übersicht). Abdominalprozesse mit Beteiligung der Bauchspeicheldrüse wie z. B. Ileus, Cholezystitis und Cholezystolithiasis, Lebererkrankungen, eine Niereninsuffizienz, virale und bakterielle Infektionen können ebenfalls zu erhöhten Werten führen.
Sehr niedrige Werte können bei zystischer Fibrose und bei einigen Patienten mit chronischer Pankreatitis gefunden werden. Dieser Befund ist jedoch diagnostisch irrelevant.
Die Enzymbestimmung erfolgt photometrisch. Die Referenzbereiche für beide Enzyme sind abhängig von der eingesetzten Bestimmungsmethode und unterliegen somit breiten Schwankungen. In den ersten Lebensjahren sind die Werte niedriger als bei Erwachsenen.
Eine Makroamylasämie (Immunkomplexe mit IgA oder IgG) führt zu Erhöhungen bis zum 3- bis 4-Fachen der Norm. Infusionen von Hydroxyethylstärke (HES) führen zu einer Hyperamylasämie (3–5 Tage dauernd) durch Komplexbildung und dadurch bedingter Ausscheidungsverzögerung. Im Gegensatz zur Makroamylasämie ist eine Makrolipasämie sehr selten.
Neben der Lipase und Amylase sind weitere Serummarker der Pankreatitis beschrieben worden wie z. B. die pankreatische Elastase, das Trypsinogen, Aktvierungspeptide des Trypsinogens (TAP) oder das pankreatitisassoziierte Protein (PAP). Alle diese Parameter konnten sich allerdings in der Routinediagnostik nicht durchsetzen.
Erkrankungen, die eine Erhöhung der Amylase oder Lipase im Serum verursachen
-
Erhöhung der Amylase und Lipase:
-
Akute Pankreatitis
-
Akuter Schub einer chronischen Pankreatitis
-
Zystische Fibrose
-
Nach ERCP
-
Niereninsuffizienz – verminderte renale Ausscheidung der Enzyme
-
Enzymwerterhöhungen durch die Mitbeteiligung der Bauchspeicheldrüse:
-
Akutes Abdomen (Ileus, perforiertes Ulkus, Cholezystitis)
-
Diabetische Ketoacidose
-
Virushepatitis
-
Mumps (Parotitis epidemica)
-
Sarkoidose
-
-
-
Erhöhung der Amylase:
-
Parotitis – bei Mumps mit Pankreasbeteiligung auch Lipaseerhöhung
-
Akute Alkoholintoxikation
-
Tumoren (paraneoplastisch)
-
Leberzirrhose
-
Chronisch-entzündliche Darmerkrankungen
-
Anorexia nervosa
-
Zerebrales Trauma
-
Makroamylasämie – im Gegensatz zur Amylase sind Makrolipasen extrem selten
-
Bildgebende Verfahren
Da die Lipase nicht nur bei Erkrankungen des Pankreas erhöht sein kann, sollte die Pankreatitis durch ein bildgebendes Verfahren bestätigt werden. In erster Linie sollte hierfür die Sonographie eingesetzt werden. Sonographisch zeigt sich ein vergrößertes und echoarmes Organ. Gleichzeitig können am Organrand reflexarme Formationen zu sehen sein, welche auf Fettgewebsnekrosen hinweisen. Zusätzlich können Veränderungen des Pankreas- oder Gallengangsystems, Pankreassteine, Kalzifikationen oder Pseudozysten nachgewiesen werden.
Wenn das Pankreas sonographisch nicht ausreichend darstellbar ist (z. B. aufgrund von Luftüberlagerung) oder der Verdacht auf eine nekrotisierende Pankreatitis besteht, sollte eine Magnetresonanztomographie (MRT) bzw. eine kontrastmittelverstärkte Computertomographie (CT) durchgeführt werden (◘ Abb. 22.2). Wegen der Strahlenbelastung sollte der MRT der Vorzug gegeben werden. Abszesse lassen sich allerdings nur mit einer CT sicher nachweisen.


Computertomographie. a Massiv erweiterter Pankreasgang mit Verkalkungen; b Pankreaspseudozyste und intrapankreatische Verkalkungen. Zur Verfügung gestellt von Herrn Dr. Christian Bassir, Charité, Berlin. (Aus Witt 2002)
Es ist zu bedenken, dass sich aus einer initial milden Form eine schwere, tödlich verlaufende Erkrankung entwickeln kann und somit das tatsächliche Ausmaß der Nekrosen in den ersten 48 h nach Krankheitsbeginn mittels kontrastmittelverstärkter CT falsch eingeschätzt werden kann.
Kontrolluntersuchungen
Während des akuten Schubes einer Pankreatitis sollten initial täglich folgende Blutwerte kontrolliert werden:
-
Lipase,
-
Blutzellenstatus (Hämatokrit),
-
C-reaktives Protein (CRP),
-
Blutgase,
-
Kalzium, Phosphat, Glukose, Harnstoff, Kreatinin,
-
Laktatdehydrogenase (LDH) und Alaninaminotransferase (ALAT).
Die Lipasewerte korrelieren nicht mit dem Schweregrad der Entzündung. Bei einem CRP-Wert >12 mg/dl ist von einer nekrotisierenden Pankreatitis auszugehen. Das CRP ist aber erst nach 48 h ein prognostischer Indikator. Ein hoher initialer Hämatokrit deutet als erste klinisch-chemische Kenngröße auf eine nekrotisierende Pankreatitis hin. Bisher existieren für das Kindesalter keine Studien über die prognostische Wertigkeit der einzelnen Laborparameter.
Bei nekrotisierender Pankreatitis sollten venöse Blutkulturen angelegt werden. Bei Verdacht auf eine disseminierte intravasale Gerinnung (DIC) erfolgt eine Kontrolle des Gerinnungsstatus. Erhöhte Cholestaseparameter (alkalische Phosphatase [AP] und γ-Glutamyltranspeptidase [GT]) finden sich bei Kompression des Ductus choledochus.
Im Weiteren sollten sonographische Verlaufskontrollen bzw. bei entsprechender Klinik MRT- oder notfalls auch CT-Kontrollen erfolgen.
Diagnostik der exokrinen und endokrinen Insuffizienz
Zur Beurteilung der exokrinen Pankreasfunktion stehen direkte und indirekte Verfahren zur Verfügung. Alle indirekten Testverfahren können bei nur geringer Pankreasfunktionseinschränkung normale Resultate liefern. Die Pankreasfunktionstests sind ausführlich ▶ Abschn. 3.9 beschrieben.
Die Kontrolle der endokrinen Funktion erfolgt durch ein Glukosetagesprofil oder durch die Messung der Uringlukose mittels Streifentest. Als Langzeitparameter für die vorangegangenen 2–3 Monate steht das glykierte Hämoglobin (HbA1c) zur Verfügung. In Zweifelsfällen ist ein oraler Glukosetoleranztest (oGTT) indiziert.
Neben einer gründlichen Anamnese nehmen klinisch-chemische, molekulargenetische und bildgebende Untersuchungen eine bedeutende Rolle bei der ätiologischen Abklärung ein.
Anamnese
Die gewissenhafte Anamnese ist richtungsweisend für die zu veranlassende weiterführende Diagnostik. Eine positive Familienanamnese für eine Pankreatitis ist durch Befragung beider Elternteile auszuschließen. Dabei sollte nach rezidivierenden Oberbauchschmerzen wie auch nach einem Pankreaskarzinom oder einem Diabetes mellitus bei Angehörigen gefragt werden. Das alleinige Vorhandensein eines Diabetes mellitus, insbesondere eines Diabetes vom Typ 2, ist allerdings bei der hohen Diabetesprävalenz kein hinreichender Anhalt für eine erbliche Pankreatitis.
Eine Anamnese auf Medikamenteneinnahme, vorangegangene Bauchtraumata und auf Symptome anderer Grunderkrankungen (Kollagenosen, chronisch-entzündliche Darmerkrankungen etc.) sollte ebenfalls erfolgen (▶ Übersicht „Ursachen der Pankreatitis“).
Klinisch-chemische Diagnostik
Die Triglycerid- und Kalziumbestimmung zum Ausschluss metabolischer Ursachen gehört zur Basisdiagnostik. Bei Hinweisen auf eine Kollagenose oder Vaskulitis erfolgt die Bestimmung der entsprechenden Autoantikörper (antinukleäre Antikörper, extrahierbare nukleäre Antikörper, Rheumafaktor und Antikörper gegen neurophile Granulozyten). Im Weiteren sind virale, bakterielle und parasitäre Infektionen wie Mumps oder eine Ascariasis auszuschließen (▶ Abschn. Ätiologie).
Da sich die zystische Fibrose klinisch als rezidivierende Pankreatitis manifestieren kann, ist die Durchführung eines Schweißtests unerlässlich.
Genetische Diagnostik
Nach derzeitigen Erkenntnissen findet sind nicht nur bei Patienten mit Familienanamnese (sog. hereditäre Form) sondern auch bei Patienten mit sog. idiopathischer Pankreatitis eine genetische Disposition (▶ Abschn. Ätiologie). Eine Genanalyse auf Mutationen im kationischen Trypsinogen (PRSS1), im Serinprotease-Inhibitor, Kazal-Typ 1 (SPINK1), im Chymotrypsinogen C (CTRC) und in der Carboxypeptidase A1 (CPA1) sollte bei Patienten mit einer akuten oder chronischen Pankreatitis und einer positiven Familienanamnese sowie bei Patienten mit chronischer Pankreatitis ohne positive Familienanamnese nach Ausschluss anderer Ursachen (chronisch-entzündliche Darmerkrankungen, Hyperlipidämie etc.) veranlasst werden. Bei 50 % der Patienten lässt sich jedoch in keinem der oben genannten Gene eine Mutation nachweisen. Die genetische Untersuchung nichtsymptomatischer Familienangehöriger (sog. prädiktive Testung) sollte aufgrund der fehlenden therapeutischen Konsequenzen nur nach ausführlicher Aufklärung erfolgen. Die Untersuchung nicht betroffener Kinder ist nach dem neuen Gendiagnostikgesetz (GenDG) nicht mehr zulässig.
Da auch eine zystische Fibrose klinisch als rezidivierende Pankreatitis imponieren kann, ohne dass das Vollbild einer Mukoviszidose vorliegt, ist bei Patienten mit sog. idiopathischer chronischer Pankreatitis eine Analyse auf CFTR-Mutationen zu erwägen. Es ist allerdings zu bedenken, dass bei diesen Patienten häufig „atypische“ CFTR-Varianten vorliegen, die mit herkömmlichen genetischen „CF-Kits“ nur mangelhaft erfasst werden. Von einigen Zentren wird auch eine Pränataldiagnostik auf PRSS1-Mutationen angeboten. Nach persönlicher Auffassung des Verfassers ist die erbliche Pankreatitis keine Indikation für eine pränatale Diagnostik. Da genetische Untersuchungen kostenintensiv und zeitaufwendig sind, sollte bei Patienten mit einer akuten Pankreatitis und negativer Familienanamnese keine genetische Analytik erfolgen.
Endoskopische und radiologische Diagnostik
Die endoskopische retrograde Cholangiopankreatikographie (ERCP) erlaubt Aussagen über zugrunde liegende anatomische Anomalien und Veränderungen des Pankreasgangsystems wie Stenosierungen oder Erweiterungen (◘ Abb. 22.3). Somit dient die ERCP nicht nur der Diagnosebestätigung bei einer chronischen Pankreatitis, sondern auch der Identifizierung angeborener oder erworbener Anomalien des Pankreas- oder Gallengangsystems und der präoperativen Diagnostik bei chirurgisch korrigierbaren Veränderungen wie Strikturen oder Zysten. Zudem kann während der Untersuchung auch eine therapeutische Intervention wie eine Papillotomie, eine Stenteinlage oder eine Steinextraktion erfolgen.


Endoskopische retrograde Cholangiopankreatikographie (ERCP): massiv erweiterter Pankreasgang und Darstellung einer Pankreaspseudozyste. Zur Verfügung gestellt von Herrn Dr. Christian Bassir, Charité, Berlin. (Aus Witt 2002)
Die Magnetresonanz-Cholangiopankreatikographie (MRCP) hat sich zunehmend als nichtinvasives diagnostisches Alternativverfahren zur ERCP etabliert. Aufgrund der fehlenden Strahlenbelastung sollte im Kindesalter die MRCP der ERCP vorgezogen werden. Eine MRCP ist insbesondere indiziert, wenn die Patienten ein hohes Risiko für die Entwicklung einer Post-ERCP-Pankreatitis besitzen oder wenn aufgrund vorangegangener Operationen der Pankreasgang unzugänglich ist. Eine gleichzeitige therapeutische Intervention ist bei dieser Untersuchung im Gegensatz zur ERCP nicht möglich.
2 Therapie der akuten und chronischen Pankreatitis
2.1 Akute Pankreatitis
Kinder mit einer akuten Pankreatitis sollten immer stationär behandelt werden, in schweren Fällen auf einer Intensivtherapiestation, da der Verlauf und mögliche Komplikationen nicht vorhersehbar sind. Die Behandlung ist symptomatisch, eine spezifische medikamentöse Therapie existiert bislang nicht. Eine auslösende Ursache sollte nach Möglichkeit ausgeschlossen werden. Die in Studien getesteten Präparate (Gabexat-Mesilat, Aprotinin, Glukagon, Somatostatin, Octreotid, Immunmodulatoren, gereinigte Plasmaderivate) sowie Plasmapherese und Peritoneallavage sind nicht oder wenig wirksam. Platelet-activating-factor-(PAF)-Antagonisten haben in Studien positive Ergebnisse gezeigt, für eine generelle Anwendungsempfehlung ist es noch zu früh.
Die basistherapeutischen Maßnahmen umfassen:
-
intensivmedizinische Überwachung (klinische Untersuchung; Kontrolle von Blutdruck, Puls, zentralem Venendruck, Serumelektrolyten, Blutgasen, Blutglukose und Nierenfunktion),
-
Kreislaufstabilisierung,
-
Schmerzbehandlung,
-
enterale und parenterale Ernährung,
-
prophylaktische Antibiotikatherapie bei Hinweisen auf eine nekrotisierende Pankreatitis.
Wichtigste Sofortmaßnahme bei einer akuten Pankreatitis ist ein adäquater Flüssigkeitsersatz, d. h. eine suffiziente intravenöse Infusion zur Verhinderung eines drohenden Kreislaufschocks, um insbesondere ein akutes Nierenversagen und eine Ischämie des Pankreas zu vermeiden.
Gegebenenfalls ist die Gabe von Plasmavolumenexpandern notwendig. Großzügig sollten Infusionen mit Albumin verabreicht werden. Albumin ist nicht nur kreislaufstabilisierend, sondern wirkt möglicherweise auch durch Bindung von freien Fettsäuren, Toxinen, Entzündungs- und Schockmediatoren. Bei Lage eines zentralvenösen Katheters sollte der zentrale Venendruck auf 6–10 cm Wassersäule eingestellt werden.
Schmerztherapie
In leichten Fällen ist ein Versuch mit Paracetamol oder Metamizol gerechtfertigt. Die Schmerztherapie mit einem Pethidin-Perfusor über 24 h bei starker Schmerzsymptomatik hat sich bewährt, bei Bedarf in Kombination mit Metamizol.
Cave! Opiate verstärken meist den paralytischen Ileus und können zum Spasmus des Sphincter Oddi führen.
Enterale und parenterale Ernährung
Eine adäquate Energiezufuhr ist bei dem hyperkatabolen Zustand durch die akute Pankreatitis essenzieller Bestandteil der Therapie. Es muss eine mukosale Ischämie und Atrophie der Dünndarmmukosa vermieden werden. Dadurch wird die Mukosabarriere stabilisiert, die intestinale Integrität erhalten und damit das Risikos von infizierten Pankreasnekrosen, die den wichtigsten Letalitätsfaktor darstellen, vermindert.
Um dies zu erreichen, wird neuerdings bei Erwachsenen, selbst bei einer schweren, nekrotisierenden Pankreatitis, eine frühzeitige enterale Ernährung als praktikabel und sicher eingeschätzt. Bei einem solchen Vorgehen kommt es offenbar zu keiner wesentlichen Stimulation der Pankreassekretion, und die Trophik des Darmes bleibt erhalten. Die meisten Studien bestätigen, dass es bei einer frühzeitigen enteralen Ernährung seltener zu Infektionen und chirurgischen Interventionen kommt, die Dauer des Krankenhausaufenthaltes verkürzt ist und die Behandlungskosten geringer sind.
Für das Kindesalter existieren diesbezüglich keine Studien, so dass man sich an den Ergebnissen bei Erwachsenen orientieren muss.
Die orale Ernährung sollte so früh als möglich, nach einer etwa 24-stündigen Nahrungskarenz begonnen werden, jedoch nicht bei Schmerzzuständen, Ileussymptomatik und Brechneigung. Nach Gabe von Tee mit Glukose und Zwieback zur Überprüfung der Verträglichkeit, kann ab dem 2.–3.Tag Wunschkost verabreicht werden.
Bei einer schweren akuten Pankreatitis mit Inappetenz und Brechneigung ist eine enterale Ernährung über eine nasogastrale oder transpylorische Sonde indiziert. Es sollte mit einer Elementarkost begonnen werden, wobei eine nährstoffdefinierte Sondenkost ebenfalls möglich ist. Die Osmolalität der Sondenkost sollte unter 400 mosm/l liegen. Immunstimulierende Sondennahrungen sind noch in der Erprobung.
Eine nasogastrale Sonde zur Magenentleerung ist routinemäßig nicht notwendig, sondern nur bei einer Ileussymptomatik und anhaltendem Erbrechen, sowie bei beeinträchtigter Bewusstseinslage.
Bei einem paralytischen Ileus ist eine totale parenterale Ernährung indiziert. Zur Deckung des erhöhten Kalorienbedarfs können auch Lipide verabreicht werden; Ausnahmen: Hypertriglyceridämie, manifester Kreislaufschock und respiratorische Insuffizienz. Die Substitution von Vitaminen, Mineralien und Spurenelementen ergänzt die Kalorienzufuhr.
Eine Glutaminsupplementation scheint einen günstigen Effekt hinsichtlich der Stabilität der Dünndarmmukosa und damit der Verhinderung von Komplikationen zu haben.
Antibiotikatherapie
Gefürchtet ist die Infektion von Parenchymnekrosen mit nachfolgender Sepsis. Infektionserreger sind meist aszendierende aerobe (E. coli, Pseudomonaden, Bakteroides, Klebsiellen, Proteus) und anaerobe Darmkeime, aber auch Staphylococcus aureus und Candidapilze. Infektions- bzw. Keimnachweise sollten durch eine CT-gestützte transkutane Feinnadelpunktion versucht werden. Infizierte Nekrosen sind operativ anzugehen. Ansonsten ist eher Zurückhaltung bei operativen Maßnahmen im Rahmen einer akuten Pankreatitis geboten. Auch wenn der Vorteil einer prophylaktischen antibiotischen Therapie bei (noch) sterilen Nekrosen umstritten ist, sollte diese im Kindesalter bei Verdacht auf eine nekrotisierende Pankreatitis, d. h. wenn das C-reaktive Protein (CRP) über 100 mg/l angestiegen ist und entsprechende Hinweise in der Bildgebung bestehen, durchgeführt werden. Bei dem zu erwartenden Keimspektrum bietet sich eine Behandlung mit Imipenem oder Meropenem oder die Kombination eines Gyrasehemmers (Ciprofloxazin, Ofloxazin) mit Metronidazol an. Trotz des gelegentlichen Candidapilznachweises wird die zusätzliche Verabreichung eines Antimykotikums bisher nicht empfohlen.
Behandlung von Komplikationen
Komplikationen treten nur bei der hämorrhagisch-nekrotisierenden akuten Pankreatitis auf und sind in der Frühphase vasoaktiv-toxischer und später septischer Natur.
Lokale Komplikationen
Außer der sterilen oder infizierten Nekrose ist eine Hämorrhagie, Abszedierung oder Pseudozystenbildung zu fürchten. Die infizierte Nekrose erfordert ein chirurgisches Debridement; Hämorrhagie und Abszedierung sind ebenfalls operativ anzugehen.
Im Gegensatz zu den meist angeborenen, mit Epithel ausgekleideten echten Zysten sind Pseudozysten flüssigkeitsgefüllte und enzymhaltige Hohlräume ohne Epithelauskleidung (◘ Abb. 22.4). Sie stellen eine Komplikation einer akuten (akute Pseudozyste; meist parapankreatisch gelegen) oder chronischen Pankreatitis (chronische Pseudozyste; meist intrapankreatisch gelegen) dar. Man unterscheidet zwischen akuter Pseudozyste und akuter Flüssigkeitsansammlung als Vorstufe der Pseudozyste. Einziges Unterscheidungsmerkmal ist der Wall aus Granulations- oder Bindegewebe um die Pseudozyste, der bei der akuten Flüssigkeitsansammlung fehlt. Von einer akuten Pseudozyste kann deshalb in der Regel erst 4 Wochen nach Beginn der Erkrankung gesprochen werden.


Magnetresonanz-Cholangio-Pankreatikographie (MRCP): Gangunregelmäßigkeiten und Darstellung einer Pankreaspseudozyste. Zur Verfügung gestellt von Herrn Dr. Christian Bassir, Charité, Berlin. (Aus Witt 2002)
Pseudozysten können rupturieren, einbluten, sich infizieren oder auch Nachbarorgane komprimieren. Meist bilden sich die Pseudozysten spontan zurück oder verkleinern sich. Bestehen sie länger als 6 Wochen und sind sie größer als 5 cm, ist kaum mit einer Spontanheilung zu rechnen. Dann sollte aktiv in Form von Punktionen oder Drainagen nach innen oder außen vorgegangen werden.
Systemische Komplikationen
In der folgenden ▶ Übersicht sind die möglichen extrapankreatischen Komplikationen aufgeführt.
Neben dem hypovolämischen Schock und der Infizierung von Pankreasparenchymnekrosen sind systemische Komplikationen bis hin zum Multiorganversagen gefürchtet. Die systemischen Komplikationen sind sehr wahrscheinlich auf aktivierte Pankreasenzyme, freigesetzte vasoaktive und toxische Substanzen und eine systemische Entzündungsreaktion zurückzuführen. Auch oxidativer Stress aufgrund eines Mangels an Radikalfängern und damit der Präsenz gewebstoxischer freier Sauerstoffradikale wird für die systemischen Komplikationen verantwortlich gemacht. Bei der Organmitbeteiligung sind das akute Nierenversagen, die respiratorische Insuffizienz, eine dissiminierte intravasale Gerinnung sowie die pankreatische Enzephalopathie besonders gefürchtet. Für Letztere ist fast immer ein Alkoholabusus Voraussetzung, so dass diese Komplikation im Kindesalter kaum zu erwarten ist. Eine passagere, milde Hyperglykämie (bis etwa 15 mmol/l) ist nicht behandlungspflichtig, höhere Werte werden vorsichtig mit Insulin behandelt, da es rasch zur Hypoglykämie kommen kann. Eine Hypokalzämie wird gezielt durch intravenöse Gaben von Kalziumglukonat ausgeglichen.
Folgende Parameter weisen im Kindesalter auf systemische Komplikationen und damit auf eine ernste Prognose hin:
– Systolischer Blutdruck | <90 mmHg |
– Tachykardie | >130/min |
– Arterieller Partialdruck (paO2) | <8 kpa |
– Urinproduktion | <1 ml/kg KG/h |
– Serumkalzium | <2 mmol/l |
– Serumalbumin | <32 g/l |
– CRP | >100 mg/l |
– α2-Makroglobulin | <1,5 g/l |
– Blutzucker | >12 mmol/l |
– Antithrombin III | erniedrigt |
Extrapankreatische Komplikationen bei akuter Pankreatitis
-
Sepsis
-
Lunge:
-
Hypoxämie
-
Ödem, Atelektase, Infiltration
-
„Adult respiratory distress syndrome“ (ARDS)
-
Pleuraerguss (meist linksseitig)
-
-
Niere: akutes Nierenversagen
-
Abdomen:
-
Gastrointestinale Blutungen (Entzündung, Erosion, Ulzeration)
-
Paralytischer Ileus
-
Aszites
-
-
Herz: Veränderungen der Funktion (Tachykardie, Arrhythmie, EKG-Veränderungen)
-
Kreislauf: Hypovolämie, Hypotonie, Schock
-
ZNS: Enzephalopathie
-
Gerinnung: Gefäßthrombosen, disseminierte intravasale Gerinnung
-
Stoffwechsel:
-
Hypokalzämie
-
Hyperglykämie, Hypoglykämie
-
Acidose
-
Hyperlipidämie
-
Akutes Nierenversagen
Um dieser Komplikation vorzubeugen, ist ein rascher Flüssigkeitsersatz und die Behandlung einer hypotonen Kreislaufsituation mit Adrenergika und dopaminergen Substanzen besonders wichtig. Zur Überprüfung der Nierenfunktion sind die Kontrolle der Urinausscheidung und tägliche Serumkreatininbestimmungen erforderlich. Nur wenn mit Kreislaufstabilisierung und ggf. Diuretikagabe die Nierenfunktion gestört bleibt, sind Hämodialyse oder Hämofiltration zu erwägen.
Respiratorische Insuffizienz
Bei Hinweisen auf eine Lungenbeteiligung (Tachy-/Dyspnoe, respiratorische Alkalose, Erniedrigung des Sauerstoffpartialdrucks [pO2]) ist ein (meist linksseitiger) Pleuraerguss auszuschließen oder zu behandeln. Bei zunehmender respiratorischer Insuffizienz bis hin zum ARDS sind Sauerstoffinsufflation über eine Sauerstoffsonde oder -brille oder eine maschinelle Beatmung notwendig.
Prophylaxe
Zur Vermeidung einer akuten bzw. rezidivierenden Pankreatitis sind prädisponierende Faktoren wie Gallensteine, Malformationen (Choledochuszysten, Pankreas divisum), Stoffwechselstörungen (Hypertriglyceridämie, Hyperkalzämie), Mukoviszidose, bestimmte Medikamente u. a. nach Möglichkeit auszuschalten.
2.2 Chronische Pankreatitis
Die Therapie bei Kindern und Jugendlichen mit einer chronischen Pankreatitis ist praktisch immer konservativ. Chirurgisch- oder endoskopisch-interventionelle Maßnahmen (Stenteinlage, Drainage von Pseudozysten, Operation von Ductus choledochus- oder Duodenumstenosen, Resektion eines entzündlichen Pankreaskopftumors) sind in dieser Altersgruppe kaum indiziert.
Die konservative Therapie umfasst die Schmerzbekämpfung, diätetische Maßnahmen, die Pankreasenzymsubstitution und ggf. eine Diabetesbehandlung.
Schmerztherapie
Da die schmerzauslösenden Mechanismen komplex sind (diskutiert werden ein erhöhter Parenchymdruck durch Fibrosierung, eine Organischämie, ein erhöhter intraduktaler Druck, Nervenalterationen, Stenose des Gallengangs und Duodenums) und alle Ursachen noch gar nicht bekannt sind, ist eine gezielte Schmerztherapie kaum möglich.
Die Schmerzen können selten, häufig, intermittierend oder persistierend auftreten, mild bis stark sein, an- oder abschwellenden Charakter haben. Häufig sind sie krampfartig und strahlen gürtelförmig vom Oberbauch in den Rücken aus.
Die medikamentöse Therapie beginnt man in der Regel mit Spasmolytika, ehe bei ungenügendem Erfolg nichtsteroidale Antirheumatika unter gleichzeitiger Gabe von Protonenpumpenhemmern zum Einsatz kommen. Erst dann besteht eine Indikation für Opioide (z. B. Tramadol).
Pankreasenzyme haben sehr wahrscheinlich keinen Einfluss auf den Schmerz, wenn man die Bauchschmerzen durch Meteorismus bei exokriner Pankreasinsuffizienz und ungenügender Enzymsubstitution ausklammert.
Diätetische Maßnahmen
Sie unterstützen einerseits die Schmerzbehandlung, andererseits sollen sie die Verdauung erleichtern und damit die gerade im Kindesalter ganz besonders wichtige normale körperliche und Pubertätsentwicklung garantieren.
Zu empfehlen sind gehäufte kleine Mahlzeiten mit anfangs normaler Zusammensetzung der Kalorienträger und Vermeidung blähender Speisen. Im Verlauf wird meist eine fettreduzierte, kohlenhydratreiche Kost notwendig. Eine Vitaminsubstitution sollte gezielt erfolgen. Pankreasabhängige Defizienzen sind bei den fettlöslichen Vitaminen A, D, E und K sowie beim Vitamin B12 zu erwarten. Vitamin B12 ist erst nach Abspaltung und Inaktivierung des R-Proteinkomplexes durch pankreatische Proteasen resorbierbar.
Beizeiten sollte auf das strikte Alkohol- und Rauchverbot hingewiesen werden.
Therapie der exokrinen Pankreasinsuffizienz
Mit zunehmender Krankheitsdauer entwickelt sich eine behandlungsbedürftige exokrine Pankreasinsuffizienz, die eine Indikation für die Substitution mit Pankreasenzymen ist. Kriterium für die Behandlungsbedürftigkeit ist eine Steatorrhö von mehr als 7 g Stuhlfettausscheidung in 24 h bzw. eine verminderte Fettresorption von weniger als 93 %. Der Fettresorptionskoeffizient lässt sich aus den Parametern Fettaufnahme mit der Nahrung und Stuhlfettausscheidung nach folgender Formel berechnen:

Die höchste Effektivität besitzen säuregeschützte, mikrosphärische darmlösliche Präparate (Mikrotabletten oder -pellets). Die Mikrotabletten bzw. -pellets sollten einen Durchmesser von höchstens 1,8 mm haben, damit sie zeitgleich mit dem Speisebrei den Magen verlassen. Der Säureschutz löst sich oberhalb eines pH-Wertes von 5,5 – also erst im Duodenum. Bei eingeschränkter pankreatogener Bicarbonatsekretion ist diese Voraussetzung nicht immer gegeben. Dann sollten zeitlich begrenzt Magensäureblocker versucht werden, zumal Patienten mit einer exokrinen Pankreasinsuffizienz zur Hyperacidität im Magen neigen.
Die Dosierung von Pankreasenzympräparaten sollte sich am besten nach dem Fettverzehr richten, was aber für den Patienten weniger praktikabel ist.
Als Richtwerte für die Dosierung von Pankreasenzympräparaten gelten 1000–2000 (bis höchstens 4000) Lipaseeinheiten für 1 g Nahrungsfett bzw. 8.000 bis 10.000 Lipaseeinheiten/kg KG und Tag. Für das Kindesalter kann eine tägliche Aufnahme von 4–5 g Fett/kg KG kalkuliert werden.
Literatur zu Abschn. 22.1
Bhatia E, Choudhuri G, Sikora SS et al. (2002) Tropical calcific pancreatitis: strong association with SPINK1 trypsin inhibitor mutations. Gastroenterology 123: 1020–1025
Eigler A, Eigenbrod S, Endres S (2003) Medikamentös induzierte Pankreatitis. Dtsch Med Wochenschr 128: 366–369
Enriquez G, Vazquez E, Aso C et al. (1998) Pediatric pancreas: an overview. Eur Radiol 8: 1236–1244
Keim V, Witt H, Bauer N et al. (2003) The course of genetically determined chronic pancreatitis. JOP 4: 146–154
Merkle EM, Nüssle K, Glasbrenner B et al. (1998) MRCP – eine aktuelle Bestandsaufnahme. Z Gastroenterol 36: 215–224
Rinderknecht H (1986) Activation of pancreatic zymogens. Normal activation, premature intrapancreatic activation, protective mechanisms against inappropriate activation. Dig Dis Sci 31: 314–321
Robertson MA (1996) Acute and chronic pancreatitis. In: Walker WA, Durie PR, Hamilton JR, Walker-Smith JA, Watkins JB (eds) Pediatric gastrointestinal disease: pathophysiology, diagnosis, management, 3rd edn. Mosby, St. Louis, pp 1321–1344
Rosendahl J, Witt H, Szmola R et al. (2008) Chymotrypsin C (CTRC) variants that diminish activity or secretion are associated with chronic pancreatitis. Nat Genet 40: 78–82
Sarles H, Adler G, Dani R et al. (1989) The pancreatitis classification of Marseilles-Rome 1988. Scand J Gastroenterol 24: 641–442
Sharer N, Schwarz M, Malone G et al. (1998) Mutations of the cystic fibrosis gene in patients with chronic pancreatitis. New Engl J Med 339: 645–652
Whitcomb DC, Gorry MC, Preston RA et al. (1996) Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nat Genet 14:141–145
Witt H (2002) Chronische Pankreatitis. Monatsschr Kinderheilkd 150: 87–99
Witt H (2003) Chronic pancreatitis and cystic fibrosis. Gut 52 (Suppl 2): ii31–41
Witt H, Becker M (2002) Genetics of chronic pancreatitis. J Pediatr Gastroenterol Nutr 34: 125–136
Witt H, Luck W, Hennies H C et al. (2000) Mutations in the gene encoding the serine protease inhibitor, Kazal type 1 are associated with chronic pancreatitis. Nat Genet 25: 213–216
Yadav D, Agarwal N, Pitchumoni CS (2002) A critical evaluation of laboratory tests in acute pancreatitis. Am J Gastroenterol 97: 1309–1318
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Witt, H., Henker, J. (2013). Pankreatitis. In: Rodeck, B., Zimmer, KP. (eds) Pädiatrische Gastroenterologie, Hepatologie und Ernährung. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-24710-1_22
Download citation
DOI: https://doi.org/10.1007/978-3-642-24710-1_22
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-24709-5
Online ISBN: 978-3-642-24710-1
eBook Packages: Medicine (German Language)