
Abstract
We investigate different types of potential parameters for the graphene-nitrogen interaction. Interaction energies calculated at DFT level are fitted with the semi-emperical Improved Lennard-Jones potential. Both a pseudo-atom potential and a full atomistic potential are considered. Furthermore, we consider the influence of the electrostatic part on the parameters using different charge schemes found in the literature as well as optimizing the charges ourselves. We have obtained parameters for both the nitrogen dimer and the graphene-nitrogen system. For the former, the four-charges Cracknell scheme reproduces with high precision the CCSD(T) interaction energy as well as the experimental diffusion coefficient in both the pseudo-atom and full atomstic potential. In the second case, the atom-atom model provides an average interaction energy of 2.3 kcal/mol, comparable with the experimental graphene-\(\text {N}_{\text {2}}\) interaction of 2.4 kcal/mol.
You have full access to this open access chapter, Download conference paper PDF
Similar content being viewed by others


1 Introduction
\(\text {N}_{\text {2}}\) gas is considered a contaminating part of natural gas [1], an energy source believed to be a more environmental friendly alternative for fossil fuels [2]. The efficient use of natural gas requires the separation of, among other components, \(\text {N}_{\text {2}}\) from the combustible parts [3]. Furthermore, also in other pre- and post-combustion processes, \(\text {N}_{\text {2}}\) needs to be separated from other components [4,5,6].
Separation of small gases is often achieved via selective adsorption in porous materials exhibiting strong van der Waals interactions [7]. A lot of possible candidates are currently being investigated [8]. At both the theoretical and experimental level, one of the first characterization steps of an adsorbent candidate is the adsorption of \(\text {N}_{\text {2}}\) gas at 77 K [9, 10]. Because of its inert properties it is assumed that the pore volume of the material can thus be measured [11]. Ohba et al. have even suggested that the amount of graphene layers in a sheet could be determined via \(\text {N}_{\text {2}}\) adsorption [12].
Molecular modelling of processes like gas adsorption on different materials has received increasing interest because of the access to as-yet unsynthesized materials and the microscopic view it offers [13]. If modelling wants to be predictive for experimental research, accurate models are needed, giving results that are directly comparable to experiment [14]. It is thus important to obtain accurate models for the \(\text {N}_{\text {2}}\) molecule [15].
Graphene and derived materials have shown great promise in the adsorption of small gases, among which \(\text {N}_{\text {2}}\), through van der Waals interactions [16, 17]. These interactions, because relatively weak, are not easy to determine from an experimental setup and, therefore, the theoretical calculations that can reproduce them play an important role in the determination of the structure and energies of the adsorbed molecules. It is for this reason that we are aiming to provide a set of interaction potentials to model the graphene-\(\text {N}_{\text {2}}\) system in a simple yet accurate fashion, suitable for direct implementation in molecular dynamics. The circumcoronene(ccn)-\(\text {N}_{\text {2}}\) system has been chosen as a model for quantum chemical study. The potentials have been derived from adequate DFT calculations in order to describe the interactions present in this system, mainly of dispersive origin. These contributions are essentially an effect of the electronic correlation and therefore of quantum mechanical origin. Their precise determination for systems of great size with approximate methods, is still today considered one of the most difficult problems for theoretical chemistry.
The potential we have chosen for the evaluation of the dispersive interactions between molecules is the Improved Lennard-Jones (ILJ) potential [18, 19], a modified version of the Lennard-Jones (LJ) potential which has shown to improve the latter without introducing too much complexity [20,21,22,23].
In addition to dispersion interactions there are electrostatic contributions to the force field that often play a significant role in the interactions. In this context, we will investigate the influence of different charge schemes in the \(\text {N}_{\text {2}}\) molecule. The electrostatic interaction is routinely added to potentials representing the dispersion interaction via the Coulomb summation [24, 25]. The charges, which have no true physical meaning, are often chosen more or less arbitrarily as to represent a molecular property of choice, usually the first non-zero multipole present in the molecule [26]. In the case of \(\text {N}_{\text {2}}\) this is a quadrupole.
In Sect. 2 we cover the computational details of this work. Section 3 discusses the potential energy models that we considered. The potentials are then tested in Sect. 4 by calculating the diffusion coefficient. Finally in Sect. 5, we draw some conclusions.
2 Computational Details
The \(\text {N}_{\text {2}}\) and ccn monomers were optimized at the B3LYP/6-31G** [27, 28] level and assumed rigid in further calculations. The bond distance in the \(\text {N}_{\text {2}}\) dimer was 1.106 Å, while the average C-C and C-H distances in ccn resulted to be 1.421 Å and 1.088 Å respectively.
All the DFT interaction energy calculations were performed at the B97-D/TZV2P [29, 30] level using the Gaussian 09 program [52]. The counterpoise correction was used in all cases to diminish the basis set superposition error [31]. The B97-D functional recovers the attractive forces of dispersion through a correction of empirical nature designed to adequately describe the correlation of long-range origin. The representation of non-covalent systems, including many van der Waals pure complexes, is good, reaching on average the CCSD(T) precision [20, 29, 32,33,34], and, unlike the latter, allows the theoretical characterization of large molecular systems. The combination of a small computational cost and reasonable precision makes it an efficient quantum chemical method for the study of systems involving polycyclic aromatic hydrocarbons (PAHs). Similarly, the use of the split-valence triple-zeta basis supplemented with two polarization functions TZV2P has also been found sufficient to represent the polarization effects in similar systems.
For the \(\text {N}_{\text {2}}\) dimer, 99 different relative configurations were generated at random and for each of those a scan over the distance was done between 3.4 Å and 20 Å. Close to the equilibrium distance, more points were sampled, while at longer distances, less points were included. This led to a total of 44 distances that were calculated for all of the 99 relative orientations.
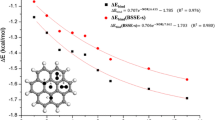
Since graphene is virtually an infinite system, ccn (\(\text {C}_{\text {54}}\text {H}_{\text {18}}\)), was used as a model for DFT calculations. Ten different \(\text {N}_{\text {2}}\) orientations were generated with respect to the ccn molecule at random positions above the ccn plane whereby the \(\text {N}_{\text {2}}\) was not allowed to take positions above the outer benzene rings to avoid edge effects. For these geometries a scan was done over distances between 2.4 Å and 20 Å. Again, care was taken to include more points in the equilibrium region than at large distances. A total of 30 distances were included. Three of the randomly generated \(\text {N}_{\text {2}}\) molecule orientations are shown in Fig. 1 together with a ccn molecule. This approach has proven its worth previously [20, 34]. To optimize the parameters specifying the potentials for the \(\text {N}_{\text {2}}\)-\(\text {N}_{\text {2}}\) and the ccn-\(\text {N}_{\text {2}}\) interactions, we fitted them to the respective interaction energies at DFT level.

Side- and top-view of a ccn molecule with three of the randomly generated positions for the \(\text {N}_{\text {2}}\) molecule.
The molecular dynamics simulations were done using the DL\(\_\)POLY v.2.2 [35] program in the microcanonical (NVE) ensemble using periodic boundary conditions in the x, y and z-direction at standard conditions (273 K and 1 atm). All simulations were run for 5 000 000 time steps with 3 000 000 equilibration steps and a time step of 1 fs. This implies a total run time of 5 ns. Equilibration was verified via monitoring of relevant properties through time, such as energy and temperature. Van der Waals and Coulombic cutoffs were set to 18 Å. As a starting position 100 \(\text {N}_{\text {2}}\), were randomly positioned in a cubic cell with sides of 159.82 Å to ensure a density of 1.139 \(\text {kg/m}^{\text {3}}\), the density of \(\text {N}_{\text {2}}\) at standard conditions.
The diffusion coefficient, D, for a single particle is defined by the Einstein equation
where d is the dimension of the system, \(\overrightarrow{r}(\tau )\) is the position of the particle at time \(\tau \) and the angle brackets denote the average over time origins. The quantity \(\langle [\overrightarrow{r}(\tau ) - \overrightarrow{r}(0)]^2 \rangle \) is denoted as the mean squared displacement (MSD) of the particle and is obtained from the output of DL_POLY v.2.2. Plotting this MSD against time gives a linear relation from which the slope gives the diffusion coefficient after dividing by twice the dimension of the system as shown in (1).
To ensure good statistics, it is important to get a sufficient amount of ensembles to average the diffusion coefficient [36]. Different ensembles can either be taken over time or over space. We have chosen a procedure where the production run is divided in half. A time average is then taken from step 1 to the time step halfway the production run. A second time average is taken from step two to the time step halfway the production run + 1 and so on. This way, we ensure an ensemble of 1 000 000 time-averaged MSD values that we plot as a function of the timestep. We repeated this procedure five times, starting from different initial configurations to ensure averaging over space as well.
3 Interaction Potential Models
To describe the total interaction potential energy, we assume it can be divided into a nonelectrostatic part and an electrostatic contribution. The non electrostatic part is represented by the semi-empirical ILJ potential developed by Pirani et al. [18] while the electrostatic part is calculated as a simple Coulombic sum
It is known that the Lennard-Jones potential performs well in the equilibrium region, but suffers from shortcomings in both the short- and longe range-part of the potential. The Improved Lennard-Jones potential has shown extensively to lower these problems via the introduction of an extra parameter controlling the R-dependence of the repulsive term [19, 37,38,39]. The ILJ potential has the following form
with
Here m takes the value of 6 for neutral systems. \(\epsilon \) represents the well depth, while \(r_0\) represents its location. Both are directly related to the polarizability of the molecules of interest. \(\beta \) is a dimensionless parameter usually restricted between 7 and 9 that allows for fine tuning of the potential, especially in the long range attractive region. It is related to the hardness of the interacting partners and allows overcoming possible deficiencies of the electrostatic part of the potential.
The ILJ potential is a site to site pairwise interaction potential and thus needs the allocation of dispersion centres within the system of interest. In the case of the \(\text {N}_{\text {2}}\) molecule it is possible to place one interaction centre in the centre of mass of the \(\text {N}_{\text {2}}\), thus reducing the molecule to a pseudo-atom based on the total molecular polarizability. We will refer to this type of potential as CM-CM potential from now on. A second possibility is to consider an interaction centre on each N-atom, thus taking into account the effects of atomic polarizabilities by means of separate atom-atom ILJ terms. In that case the dispersion energy of the \(\text {N}_{\text {2}}\) dimer is described as a 4-term potential
where i and j indicate the nitrogen atoms of \(\text {N}_{\text {2}}\) molecules A and B respectively within the dimer. This type of potential will be referred to as atom-atom potential.
For the simulation of small gases, often partial charges are introduced in the system to calculate the electrostatic interaction. Although they do not have an actual physical meaning, they are usually chosen such as to represent a molecular property relevant for the interaction; often the lowest non-zero multipole is chosen. We optimize the parameters for the ILJ potential using different charge schemes found in the literature and try to further optimize the charges ourselves.
For the \(\text {N}_{\text {2}}\) gas, usually a three site or a four site charge system is proposed [15]. The three-charge system has negative charges on the N atoms and a balancing charge on the centre of mass of double the magnitude to get a neutrally charged molecule. Although this contradicts the chemical structure of the \(\text {N}_{\text {2}}\) molecule—where the negative charge is concentrated in the triple bond—this model reproduces the quadrupole of the molecule if the charges are chosen appropriately. The four site charge system, on the other hand, positions a positive and negative charge on either side of the N-atoms. The positive charges are positioned at a distance of 1.694 Å of each other, while the negative charges are separated by a distance of 2.088 Å. Schematic representations are shown in Fig. 2.

Representation of the three and four site charge schemes showing the location of the respective charges in the \(\text {N}_{\text {2}}\) molecule
An electrostatic energy contribution is then calculated as the Coulombic sum
where i and j are the respective point charges present in the \(\text {N}_{\text {2}}\) dimer and follow the order rule stated before (see (5)).
Various charge schemes found in the literature were used. For the three-charge model, we have used values proposed by Stone [40], the charge scheme proposed by Murthy et al. (MOM) [41], and the TraPPE scheme proposed by Potoff and Siepmann [42]. For the four-charge model, we have used the charges proposed by Cracknell et al. [43]. Aside from using previously defined charge schemes, we have also refitted the charges ourselves within the three-charge model. The negative charge was then considered as an extra parameter within the fitting. The value for the positive charge followed naturally from the negative one to assure a neutral \(\text {N}_{\text {2}}\) molecule.
For the ccn molecule, we always consider an ILJ interaction centre on every C atom, to make sure the \(\text {N}_{\text {2}}\) molecule interacts strongest with the closest carbon atoms. Furthermore the electrostatic interactions are considered negligible for the graphene-\(\text {N}_{\text {2}}\) interaction. This means that for the ccn-\(\text {N}_{\text {2}}\) system, after placing an interaction centre on both N atoms, we can write the potential as
where i runs over the 54 carbon atoms within the ccn molecule and j runs over the 2 nitrogen atoms within the \(\text {N}_{\text {2}}\) molecule.
It matters to point out that each of the aforementioned charge schemes belongs to a force field that carries its own parameters for a standard Lennard-Jones potential. Here, however, we use the charges of each force field and then refit all the other parameters of the ILJ potential to obtain new force fields.
We have fitted the models described in this section for both the \(\text {N}_{\text {2}}\) and ccn-\(\text {N}_{\text {2}}\) systems to a set of interaction energies calculated at the DFT level by optimizing the parameters of the ILJ potential and, in specific cases, the partial charges in the Coulombic sum.
3.1 \(\text {N}_{\text {2}}\) Dimer
Firstly we are considering the case of a single interaction centre on the centre of mass of the \(\text {N}_{\text {2}}\) molecule. The interaction parameters can be found in Table 1 with their respective partial charges. It is clear from the table that explicitly including the electrostatic part by means of the Coulombic sum only affects the ILJ parameters when large charges are used. This observation applies for both the three- and four-charge model.
In the three-charge model, increasing the charges, increases the \(\epsilon \) while decreasing \(r_0\) and \(\beta \). Reversed trends are seen for the four-charge model. Increasing the charge, induces a decrease in \(\epsilon \) and an increase in \(r_0\) and \(\beta \). For the three-charge model, the optimized charges are higher than the ones found in the literature.
We can compare the \(\epsilon \) and \(r_0\) values for the Improved Lennard-Jones potential to the respective values of the Lennard-Jones potential since the equilibrium regions of both behave equally. Ravikovitch et al. have reported values of 0.202 kcal/mol for \(\epsilon \) and 4.058 Å for \(r_0\) for a LJ CM-CM model without charges [44]. The \(\epsilon \) is higher than ours, while the \(r_0\) is smaller, but in reasonable agreement taking into account that they were acquired via very different methods.
Atom-atom potential parameters can be found in Table 2. Introduction of the electrostatic part changes the parameters to a smaller extent than in the CM-CM model. For the three-charge model, the \(\epsilon \) and \(r_0\) increase slightly with increasing charges, while \(\beta \) decreases. The optimized charges are again slightly higher than the ones from the literature. For the four-charge model, \(\epsilon \) decreases, while \(r_0\) and \(\beta \) increase.
Again we can compare the \(\epsilon \) and \(r_0\) parameters of the atom-atom model directly to the TraPPE and MOM Lennard-Jones alternatives for the three-charge model [41, 42]. The \(\epsilon \) and \(r_0\) for both the MOM and the TraPPE potential are 0.072 kcal/mol and 3.730 Å respectively which compares reasonably well with our values. Furthermore, we can compare to ILJ parameters using a three-charge model proposed by Bramastya et al. [45]. They propose values of 0.081 kcal/mol, 3.770 Å and 9.000 for \(\epsilon \), \(r_0\) and \(\beta \) respectively while using a charge of −0.515 e on the N atom. The LJ \(\epsilon \) and \(r_0\) proposed by Cracknell using a four-charge model are 0.075 kcal/mol and 3.724 Å respectively, again well comparable with our parameters.
In order to examine the performances of the different approaches, the interaction energies of some highly symmetrical configurations of the \(\text {N}_{\text {2}}\)-dimer have been calculated with the different interaction potentials fitted. The values are presented in Table 3. Three representative configurations, in T-shape, parallel and linear, were taken into account and were compared with the interaction energies computed at the CCSD(T)/CBS [46] level, which is a well-recognized standard for evaluating the accuracy of other computational methods.
From Table 3 it can be seen that the interaction energies of \(\text {N}_{\text {2}}\)-dimers calculated with the different fittings are in general in good agreement with the CCSD(T) results for the considered noncovalent interaction. Indeed, the proposed potentials correctly reproduce the stability sequence for \(\text {N}_{\text {2}}\) dimer, T-shape > parallel > linear. However, they are not able to accurately reproduce the very small interaction energy of the linear configuration. Still, the fittings of B97-D results predict the dimer to be bound in this geometry, unlike M11, \(\omega \) B97X-D or B3LYP-D3 among others [46].
In the CM-CM model the charge schemes giving in average the best results are those using four charges, with absolute errors of 0.004–0.008 kcal/mol. The three-charge system and no-charge model perform worse. For the atom-atom potential, a similar trend is seen so that the four-charge model is to be preferred over the schemes with three or zero charges. The quantitative error in interaction energies relative to CCSD(T) is a little bigger but is in tolerable range. As for the bond distances, the proposed potentials give higher values than those calculated with CCSD(T), with differences around 0.25 Å.
3.2 ccn-\(\text {N}_{\text {2}}\) System
In Table 4 the interaction parameters can be found for the ccn-\(\text {N}_{\text {2}}\) system using the CM and one site per atom model for \(\text {N}_{\text {2}}\). In the literature parameters of this type are usually obtained via the Berthelot mixing rules from the separate parameters for the \(\text {N}_{\text {2}}\) molecule and the C atom within the surface, graphene in this case. This makes a direct comparison with our parameters difficult. Consequently, to validate our fitted potentials for the ccn-\(\text {N}_{\text {2}}\) system, a study similar to that of the \(\text {N}_{\text {2}}\) dimer has been carried out. Furthermore, we also considered the interaction of \(\text {N}_{\text {2}}\) with benzene (Bz), although our parameterization of the ILJ potential is specifically designed for larger molecules. We have, then, evaluated the equilibrium distance and binding energy for both ccn-\(\text {N}_{\text {2}}\) and Bz-\(\text {N}_{\text {2}}\) complexes by means of the ILJ potentials fitted to reproduce B97-D/TZV2P numbers. The obtained results are collected in Table 5.
From the analysis of data in Table 5, it can be seen that the CM-model consistently underestimates the binding energies with respect to experimental values. Just by construction, this model is not able to reproduce the influence of orientation of \(\text {N}_{\text {2}}\) over the aromatic surface on the computed energies, so giving too much weight to less bonded contributions and, consequently, providing too low interactions.
This problem does not appear in the atom-atom model and, in fact, this model can give account of the different stability of different conformations. The obtained interaction energies represent lower and upper limits containing the experimental value and providing an arithmetic mean value of 2.3 kcal/mol. Nevertheless, since the probability of the parallel conformation is larger than that of the perpendicular one, the experimental value of 2.398 kcal/mol is closer to the upper limit. A good agreement is also encountered for the equilibrium distances.
Notably, despite the limitations introduced by its own design, a good agreement between experimental and theoretical results is also found in the Bz-\(\text {N}_{\text {2}}\) interaction. Again, the CM model gives too low binding energies and the experimental value is closer to the upper limit. Finally, there is also a reasonable agreement between our values and those computed by the rigid-body diffusion Monte Carlo (RBDMC) method [49].
4 Diffusion Coefficients
The force fields described above were used to calculate the diffusion coefficients of \(\text {N}_{\text {2}}\) gas at standard conditions (273 K and 1 atm). 100 molecules were simulated in a simulation box with a size coinciding with the gas density of \(\text {N}_{\text {2}}\) at standard conditions.
The calculated diffusion coefficients can be seen in Table 6 along with the experimental value. Neither the CM-CM model nor the atom-atom model perform consistently better than the other over all the different charge schemes.
For the CM-CM model, the three-charge model performs worse upon increasing the charges. The smallest charges of the MOM scheme, give already an absolute error (\(E_{\text {a}}\)) of \(0.341\cdot 10^{-5}\) m\(^2\)/s compared to the experimental value. The four-charge model performs better, with an \(E_{\text {a}}\) of \( -0.081\cdot 10^{-5}\) m\(^2\)/s for the Cracknell charges. Again, the \(E_{\text {a}}\) becomes larger upon increasing the charges. For the CM-CM model, it seems better not to include charges at all which gives rise to an \(E_{\text {a}}\) of only \(0.023\cdot 10^{-5}\) m\(^2\)/s. This result matters because for massive calculations, simplicity in the potential can lead to significant production time savings.
For the atom-atom potential, a similar trend is seen. Increasing the charges worsens the performance. However in this case, the four-charge model of Cracknell (\(\text {E}_{\text {a}}\,= -0.108\cdot 10^{-5}\) m\(^2\)/s) is to be preferred over a model without charges (\(\text {E}_{\text {a}}\,= -0.216\cdot 10^{-5}\) m\(^2\)/s). However, in case of large simulations, leaving out the Coulombic sum altogether seems justified to save computing time. The difference in performance between the no-charge model and the best performing three-charge model (MOM) is a lot smaller than for the CM-CM case, an \(E_{\text {a}}\) of \(-0.216\cdot 10^{-5}\) m\(^2\)/s versus \(0.304\cdot 10^{-5}\) m\(^2\)/s.
As a comparison we calculated the diffusion coefficient using the parameters proposed by Bramastya et al. [45], to obtain a result of \(0.900\cdot 10^{-5}\) m\(^2\)/s, leading to an \(E_{\text {a}}\) of \(0.650\cdot 10^{-5}\) m\(^2\)/s.
5 Conclusions
In this work we have fitted the Improved Lennard-Jones potential to interaction energies at DFT level for the \(\text {N}_{\text {2}}\) dimer and the ccn-\(\text {N}_{\text {2}}\) system. We have considered the \(\text {N}_{\text {2}}\) molecule as a pseudo-atom, but we also obtained parameters for the N atoms within the molecule. No systematic performance difference was found between the centre of mass or the atom representation.
The influence of adding the Coulombic sum to this potential was investigated for the \(\text {N}_{\text {2}}\) dimer by use of different charge schemes found in the literature. Both three- and four-charge models were used. It was shown that raising the charges within the charge schemes, has a progressive influence on the other parameters. The specific trends depend on the model used. The potentials that we have constructed have shown to reproduce well the interaction energies and equilibrium distances found experimentally. It is worth to note that the Cracknell scheme provides a very accurate interaction energy compared to the CCSD(T) reference values [39]. Furthermore, for the graphene-\(\text {N}_{\text {2}}\) system, the atom-atom potential predicts an average interaction energy of ca 2.3 kcal/mol, only 0.1 kcal/mol below the experimental determination [40, 41].
We have tested the developed force field by calculating the diffusion coefficient for the \(\text {N}_{\text {2}}\) gas. Generally speaking, it was observed that increasing the charges within a certain model, worsens the results. Furthermore, we found that the Cracknell four-charge model gives very good results overall, although the CM-CM model without charges performs the best of all the considered models.
References
Cristancho, D., Akkutlu, I.Y., Criscenti, L.J., Wang, Y.: Gas storage in model kerogen pores with surface heterogeneities. In: SPE Europec featured at 78th EAGE Conference and Exhibition (2016)
Chen, J.J., Li, W.W., Li, X.L., Yu, H.Q.: Improving biogas separation and methane storage with multilayer graphene nanostructure via layer spacing optimization and lithium doping: a molecular simulation investigation. Environ. Sci. Technol. 46, 10341 (2012)
Cavenati, S., Grande, C.A., Rodrigues, A.E.: Adsorption equilibrium of methane, carbon dioxide, and nitrogen on zeolite 13X at high pressures. J. Chem. Eng. Data 49(4), 1095–1101 (2004)
Bahamon, D., Vega, L.F.: Systematic evaluation of materials for post-combustion \(\text{ CO }_{\text{2 }}\) capture in a Temperature Swing Adsorption process. Chem. Eng. J. 284, 438–447 (2016)
Lombardi, A., Pirani, F., Laganà, A., Bartolomei, M.: Energy transfer dynamics and kinetics of elementary processes (promoted) by gas-phase \(\text{ CO }_{\text{2 }}\)-\(\text{ N }_{\text{2 }}\) collisions: selectivity control by the anisotropy of the interaction. J. Comput. Chem. 37(16), 1463–1475 (2016)
Lombardi, A., Faginas-Lago, N., Gaia, G., Federico, P., Aquilanti, V.: Collisional energy exchange in CO\(_2\)–N\(_2\) gaseous mixtures. In: Gervasi, O., et al. (eds.) ICCSA 2016. LNCS, vol. 9786, pp. 246–257. Springer, Cham (2016). https://doi.org/10.1007/978-3-319-42085-1_19
Gómez-Gualdrón, D.A., Wilmer, C.E., Farha, O.K., Hupp, J.T., Snurr, R.Q.: Exploring the limits of methane storage and delivery in nanoporous materials. J. Phys. Chem. C 118(13), 6941–6951 (2014)
Morris, R.E., Wheatley, P.S.: Gas storage in nanoporous materials. Angew. Chem. - Int. Ed. 47(27), 4966–4981 (2008)
Do, D.D., Do, H.D., Nicholson, D.: Effects of surface structure and temperature on the surface mediation, layer concentration and molecular projection area: adsorption of argon and nitrogen onto graphitized thermal carbon black. Adsorpt. Sci. Technol. 25(6), 347–364 (2007)
Peng, Y., Krungleviciute, V., Eryazici, I., Hupp, J.T., Farha, O.K., Yildirim, T.: Methane storage in metal-organic frameworks: current records, surprise findings, and challenges. J. Am. Chem. Soc. 135(32), 11887–11894 (2013)
Prosenjak, C., Nabais, J.M.V., Laginhas, C.E., Carrott, P.J.M., Carrott, M.M.L.R.: Simulations of phenol adsorption onto activated carbon and carbon black. Adsorpt. Sci. Technol. 28(8–9), 797–806 (2010)
Ohba, T., Takase, A., Ohyama, Y., Kanoh, H.: Grand canonical Monte Carlo simulations of nitrogen adsorption on graphene materials with varying layer number. Carbon N. Y. 61, 40–46 (2013)
Ohno, H., Mukae, Y.: Machine learning approach for prediction and search: application to methane storage in a metalorganic framework. J. Phys. Chem. C 120(42), 23968 (2016)
Wongkoblap, A., Intomya, W., Somrup, W., Charoensuk, S., Junpirom, S., Tangsathitkulchai, C.: Pore size distribution of carbon with different probe molecules. Eng. J. 14(3), 45–56 (2010)
Makrodimitris, K., Papadopoulos, G.K., Theodorou, D.N.: Prediction of permeation properties of CO\(_{\text{2 }}\) and N\(_{\text{2 }}\) through silicalite via molecular simulations. J. Phys. Chem. B 105, 777–788 (2001)
Gadipelli, S., Guo, Z.X.: Graphene-based materials: synthesis and gas sorption, storage and separation. Prog. Mater Sci. 69, 1–60 (2015)
Szczȩśniak, B., Choma, J., Jaroniec, M.: Gas adsorption properties of graphene-based materials. Adv. Colloid Interface Sci. 243, 46–59 (2017)
Pirani, F., Brizi, S., Roncaratti, L.F., Casavecchia, P., Cappelletti, D., Vecchiocattivi, F.: Beyond the Lennard-Jones model: a simple and accurate potential function probed by high resolution scattering data useful for molecular dynamics simulations. Phys. Chem. Chem. Phys. 10(36), 5489–5503 (2008)
Faginas-Lago, N., Lombardi, A., Albertí, M., Grossi, G.: Accurate analytic intermolecular potential for the simulation of Na\({^{+}}\) and K\({^{+}}\) ion hydration in liquid water. J. Mol. Liq. 204, 192–197 (2015)
Yeamin, M.B., Faginas-Lago, N., Alberti, M., Cuesta, I.G., Sanchez-Marin, J., Sanchez de Meras, A.M.J.: Multi-scale theoretical investigation of molecular hydrogen adsorption over graphene: coronene as a case study. RSC Adv. 4(97), 54447–54453 (2014)
Bartolomei, M., Carmona-Novillo, E., Hernández, M.I., Campos-Martínez, J., Pirani, F.: Global potentials for the interaction between rare gases and graphene-based surfaces: an atom-bond pairwise additive representation. J. Phys. Chem. C 117(20), 10512–10522 (2013)
Faginas-Lago, N., Yeamin, M.B., Sánchez-Marín, J., Cuesta, I.G., Albertí, M., Sánchez de Merás, A.: Modelization of the H\({_{2}}\) adsorption on graphene and molecular dynamics simulation. Theoret. Chem. Acc. 136(8), 91 (2017)
Faginas-Lago, N., Yeni, D., Huarte, F., Wang, Y., Alcamí, M., Martin, F.: Adsorption of hydrogen molecules on carbon nanotubes using quantum chemistry and molecular dynamics. J. Phys. Chem. A 120(32), 6451–6458 (2016)
Getman, R.B., Bae, Y.S., Wilmer, C.E., Snurr, R.Q.: Review and analysis of molecular simulations of methane, hydrogen, and acetylene storage in metal-organic frameworks. Chem. Rev. 112(2), 703–723 (2012)
Eggimann, B.L., Sunnarborg, A.J., Stern, H.D., Bliss, A.P., Siepmann, J.I.: An online parameter and property database for the TraPPE force field. Mol. Simul. 40(1–3), 101–105 (2013)
Albertí, M., Pirani, F., Laganá, A.: Carbon dioxide clathrate hydrates: selective role of intermolecular interactions and action of the SDS catalyst. J. Phys. Chem. A 117(32), 6991–7000 (2013)
Becke, A.D.: Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98(7), 5648 (1993)
Hehre, W.J., Ditchfield, R., Pople, J.A.: Self-consistent molecular orbital methods. XII. Further extensions of Gaussian-type basis sets for use in molecular orbital studies of organic molecules. J. Chem. Phys. 56(5), 2257–2261 (1972)
Grimme, S.: Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 27, 1787 (2006)
Schäfer, A., Huber, C., Ahlrichs, R.: Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 100(8), 5829 (1994)
Boys, S., Bernardi, F.: The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 19(4), 553–566 (1970)
Grimme, S.: Accurate description of van der Waals complexes by density functional theory including empirical corrections. J. Comput. Chem. 25(12), 1463–1473 (2004)
Peverati, R., Baldridge, K.K.: Implementation and performance of DFT-D with respect to basis set and functional for study of dispersion interactions in nanoscale aromatic hydrocarbons implementation and performance of DFT-D with respect to basis set and functional for study of dispersi. J. Chem. Theory Comput. 4, 2030–2048 (2008)
Wilson, J., Faginas-Lago, N., Vekeman, J., Cuesta, I.G., Sánchez-Marín, J., SánchezdeMerás, A.: Modeling the interaction of carbon monoxide with flexible graphene: from coupled cluster calculations to molecular-dynamics simulations. ChemPhysChem 19, 774–783 (2018)
Smith, W., Forester, T.R.: DL\(\_\)POLY\(\_\)2.0: a general-purpose parallel molecular dynamics simulation package. J. Mol. Graph. 14(3), 136–141 (1996)
Pranami, G., Lamm, M.H.: Estimating error in diffusion coefficients derived from molecular dynamics simulations. J. Chem. Theory Comput. 11, 4586–4592 (2015)
Bartolomei, M., Pirani, F., Marques, J.M.: Modeling coronene nanostructures: analytical potential, stable configurations and Ab Initio energies. J. Phys. Chem. C 121(26), 14330–14338 (2017)
Faginas Lago, N., Huarte Larranaga, F., Alberti, M.: On the suitability of the ILJ function to match different formulations of the electrostatic potential for water-water interactions. Eur. Phys. J. D 55(1), 75–85 (2009)
Alberti, M., Faginas Lago, N.: Ion size influence on the Ar solvation shells of M\(^+\)-C\(_6\)F\(_6\) clusters (M = Na, K, Rb, Cs). J. Phys. Chem. A 116(12), 3094–3102 (2012)
Stone, A.: The Theory of Intermolecular Forces, vol. 1. Oxford University Press, Oxford (2013)
Murthy, C.S., Singer, K., Klein, M.L., Mcdonald, I.R.: Pairwise additive effective potentials for nitrogen. Mol. Phys. 41(6), 1387–1399 (1980)
Potoff, J.J., Siepmann, J.I.: Vapor-liquid equilibria of mixtures containing alkanes, carbon dioxide, and nitrogen. AIChE J. 47(7), 1676–1682 (2001)
Cracknell, R.F., Nicholson, D., Tennison, S.R., Bromhead, J.: Adsorption and selectivity of carbon dioxide with methane and nitrogen in slit-shaped carbonaceous micropores: simulation and experiment. Adsorption 2(3), 193–203 (1996)
Ravikovitch, P.I., Vishnyakov, A., Neimark, A.V.: Density functional theories and molecular simulations of adsorption and phase transitions in nanopores. Phys. Rev. E 64(1), 20 (2001)
Bramastya, Y., Faginas-Lago, N., Lombardi, A., Evangelisti, S.: Carbon dioxide and nitrogen separation by multilayer graphtriyne membranes: a molecular dynamics study. Carbon N. Y. (2018, in Press)
Lu, T., Chen, F.: Revealing the nature of intermolecular interaction and configurational preference of the nonpolar molecular dimers (H\({_{2}}\))\({_{2}},\) (N\({_{2}}\))\({_{2}},\) and (H\({_{2}}\))(N\({_{2}}\)). J. Mol. Model. 19(12), 5387–5395 (2013)
Vidali, G., Ihm, G., Kim, H.Y., Cole, M.W.: Potentials of physical adsorption. Surf. Sci. Rep. 12(4), 135–181 (1991)
Zhao, J., Buldum, A., Han, J., Lu, J.P.: Gas molecule adsorption in carbon nanotubes and nanotube bundles. Nanotechnology 13(2), 195–200 (2002)
Buch, V.: Treatment of rigid bodies by diffusion Monte Carlo: application to the para-H\({_{2}}\)-H\({_{2}}\)O and ortho-H\({_{2}}\)-H\({_{2}}\)O clusters. J. Chem. Phys. 97(1), 726–729 (1992)
Ernstberger, B., Krause, H., Neusser, H.J.: Atoms, molecules metastable decay and binding energies of van der Waals cluster ions. Zeitschrift für Phys. D 20, 189–192 (1991)
Mostinsky, I.: https://doi.org/10.1615/AtoZ.d.diffusion_coefficient (2011)
Frisch, M.J., Trucks, G.W., Schlegel, H.B., Scuseria, G.E., Robb, M.A., Cheeseman, J.R., Scalmani, V., Barone, V., Mennucci, B., Petersson, G.A., Nakatsuji, H., Caricato, M., Li, X., Hratchian, H.P., Izmaylov, A.F., Bloino, J., Zheng, G., Sonnenberg, J.L., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Vreven, T., Montgomery, J.A., Peralta, J.J.E., Ogliaro, F., Bearpark, M., Heyd, J.J., Brothers, E., Kudin, K.N., Staroverov, V.N., Kobayashi, R., Normand, J., Raghavachari, K., Rendell, A., Burant, J.C., Iyengar, S.S., Tomasi, J., Cossi, M., Rega, N., Millam, J.M., Klene, M., Knox, J.E., Cross, J.B., Bakken, V., Adamo, C., Jaramillo, J., Gomperts, R., Stratmann, R.E., Yazyev, O., Austin, A.J., Cammi, R., Pomelli, C., Ochterski, J.W., Martin, R.L., Morokuma, K., Zakrzewski, V.G., Voth, G.A., Salvador, P., Dannenberg, J.J., Dapprich, S., Daniels, A.D., Farkas, Ö., Foresman, J.B., Ortiz, J.V., Cioslowski, J., Fox, D.J.: Gaussian-09 Revision D.01, Wallingford CT (2009)
Acknowledgment
The project leading to this application has received funding from the European Unions Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement No. 642294. N. F.-L. acknowledges financial support from the “Fondazione Cassa di Risparmio di Perugia” (Project code: 2015.0331.021 Scientific and Technological research).
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
<SimplePara><Emphasis Type="Bold">Open Access</Emphasis> This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.</SimplePara> <SimplePara>The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.</SimplePara>
Copyright information
© 2018 The Author(s)
About this paper

Cite this paper
Vekeman, J., Faginas-Lago, N., Cuesta, I.G., Sánchez-Marín, J., De Merás, A.S. (2018). Nitrogen Gas on Graphene: Pairwise Interaction Potentials. In: Gervasi, O., et al. Computational Science and Its Applications – ICCSA 2018. ICCSA 2018. Lecture Notes in Computer Science(), vol 10964. Springer, Cham. https://doi.org/10.1007/978-3-319-95174-4_44
Download citation
DOI: https://doi.org/10.1007/978-3-319-95174-4_44
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-95173-7
Online ISBN: 978-3-319-95174-4
eBook Packages: Computer ScienceComputer Science (R0)