
Abstract
We performed the genome-wide association study of estimated breeding values for milk production traits in Russian Holstein and black-and-white cattle population. The join dairy cows’ population of Moscow and Leningrad regions was used to create a common reference group of animals to obtain the genomic breeding values. We identified breeding and genetic parameters for milk yield for 305 days of lactation, milk fat and protein content, milk fat and protein yield. We found several high-significant conservative mutations associated with milk fat content (e.g., DGAT1, P = 6.8 × 10−22), as well as a 1.5 Mb locus on BTA14. Our results will be used to develop a genomic evaluation programs, aimed to improve economically important traits in dairy cattle in Russia.
You have full access to this open access chapter, Download conference paper PDF
Similar content being viewed by others


Keywords
- Estimated breeding value
- Genome-wide association studies (GWAS)
- Milk production
- Single nucleotide polymorphism (SNP)
- Quantitative trait loci
- Reference population
Introduction
Improving the productive qualities of farm animals is the main goal of the most of breeding programs. The methods under development are targeted to collect and to accumulate not only phenotypic information from direct records of animal’s productive qualities, but also the data of whole genome scanning. Presently, genomic evaluation assay was developed to increase the accuracy of estimated breeding values (EBVs) for productive and other economically important traits [5, p. 321]. In Russia, as well as in the world dairy farming, genomic prediction methods have been applied in cattle breeding, specifically, in the most numerous populations of Holstein and holsteinized black-and-white breed. The genome-wide associations studies (GWAS) performed on cows’ population of Moscow region revealed several high-significant associations between 14 SNP localized on chromosomes 1, 5, 9, 13, 14, 17, 20, and 27 and an additive effect of more than 9% was observed [11, p. 188]. One of the validation tests for genes, responsible for a quantitative traits of dairy cattle, is the availability of accurate phenotypic data records, i.e., there are mutations that have been fixed under selection pressure and may explain about 40% of genetic variation [4, p. 387]. Respectively, sufficient number of animals, reliable information, considering the level of trait heritability should improve accuracy of the mapping of quantitative trait loci and thus the estimated genomic breeding values. Ongoing research on the analysis of genome-wide associations among different dairy cattle populations of the USA, Germany, the Netherlands, Australia and China identified a pool of common reference genes responsible for milk production traits and for milk fat content. Single nucleotide polymorphisms in DGAT1, SCD1, GHR, EPS8, GPAT4 genes, and casein cluster genes (Hapmap24184-BTC-070077) were characterized by the highest significant values of the associations and an additive effects [10, p. 6; 16, p. 2; 6, p. 6519; 1, p. 869]. It is worth noting that pleiotropic effect of DGAT1 gene on milk protein percentage and content, milk yield was observed in Holstein cattle population of Chinese origin [3, p. 6].
The GWAS for traits with low heritability (fertility, health) and strongly influenced by the genotype environment factors (exterior) are of special interest. Thus, the large number of SNP-effects for type traits was detected on chromosomes BTA11, BTAX, BTA10, BTA5, and BTA26 [2, p. 7]. Regarding fertility, the search is focused on the single semi-lethal mutations, which might be observed or be fixed, because only metabolic analysis can characterize the complex interaction of genotype and phenotype under the hormonal regulation [8, p. 6431]. The main restricting factor of wide implementation of genomic methods in animal breeding is often an insufficient population size or various objectives in breeding programs in different countries. For example, the join of Jersey cattle reference populations of Denmark and the USA are considered [12, p. 2] in one case, and a merger of the herds of different sizes, being bred under various environmental conditions, at the level of a country’s regions is examined in the other case. In addition, issues of influence of single nucleotide mutations on the level of retirement and milk cow productivity are debated [14, p. 5804]. As part of a national program for dairy cattle improvement in New Zealand, it was indicated that the use of SNP data, which were more closely associated with milk production and reproduction traits, improved an accuracy of predictions for proven bulls by 1–2% [17, p. 663].
In this regard, studies, performed in Russian Holstein cattle population, are of some interest for an understanding the selection process commonality on a par with global peers, as well as to create own dairy cattle reference population.
The purpose of the study was to evaluate the genome-wide associations with estimated breeding values for milk yield and milk components in Holstein and black-and-white bulls from the different regions of Russia
Materials and Methods
Medium Density Bovine SNP50K v2 BeadChip (Illumina Inc., USA) was used for genotyping 477 individuals of Holstein and holsteinized Black-and-White breeds, which daughters had been lactating in 138 herds. The sample included 256 sires from the population of Moscow region, and 221 sires from the population of Leningrad region. The total number of daughters (primiparous) was 119,106 individuals. The following phenotyping traits were used for GWAS analysis: 305-day milk yield (MY), milk fat content (FC), and milk protein content (PC). Quality control for genotyping was carried out using Plink 1.9 software [9, p. 562]. After quality check, 466 bulls and 40279 polymorphic SNPs were selected for the analysis.
BLUP Sire Model was used for EBV calculation. The following equation was used:
where Y ijk is the k-th heifer trait index; μ is population constant; HYS i is fixed effect of the i-th «herd-year-season» calving; (i = 1, …, 3917 factors); b 1 and b 2 are linear regression coefficients; A k is first calving age of the k-th heifer; DO k is days open of the k-th heifer; sire j is randomized effect of the j-th bull with normal distribution with a mean of 0, and a variance of Aσa 2, where A is additive relationship matrix (j = 1, …, 466 individuals); e ijk is unaccounted factor effect (0, σe 2).
EBV calculations were conducted using BLUPF90 software. Estimation of variance components was performed by the method of restricted maximum likelihood (REML), with the inclusion of additional features to the model: fat yield (FY), protein yield (PY), breedings per conception (BC), and days open (DO) [7, p. 21]. Estimation of genomic relationship matrix (G) was performed according to the algorithm developed by P.M. VanRaden [15, p. 4416] in the R programming language environment. The matrix consisted of elements presented by homozygous and heterozygous loci estimations: AA = 1, AB = 0, BB = −1. GEBVs were calculated as combination of SNP direct genomic value (DGV) and EBV (Parent Average) according to the GBLUP approach.
To identify associations of SNP-markers with milk production, traits regression analysis with pseudo-phenotypes or GEBV assessments implemented in Plink 1.90 were used (flags: --assoc --qt-means --adjust). To confirm the significant impact of SNPs and identify significant regions in the genome of cattle, several tests were used to check for null hypotheses by Bonferroni (threshold P < 1.24 × 10−6, \({\raise0.7ex\hbox{${0.05}$} \!\mathord{\left/ {\vphantom {{0.05} {40279}}}\right.\kern-0pt} \!\lower0.7ex\hbox{${40279}$}}\)).
To search for the genes closely associated with economic traits, the National Center for Biotechnology Information (NCBI) database was used. Functional gene identification was performed using the Discover EggNOG 4.1 database (http://eggnogdb.embl.de/#/app/home). Data visualization was conducted using the qqman package and R programming language [13].
Results
The analysis of genetic differences between populations of bulls from Moscow and Leningrad regions shows their close relationship according to the fixation index (Fst = 0.00356). It was found that the heritability (h2) of milk yield was = 0.180, indicating a relatively low proportion of additive genetic variation in the population of holsteinized Black-and-White breed (CVa = 3.4%). The highest value of heritability was observed for milk fat yield—0.221, while for milk protein yield it was comparable to the total production of milk components (0.173). Heritability of the reproductive traits ranged from 0.015 to 0.039 and was largely due to paratypic (environmental or technological) factors (Table 1).
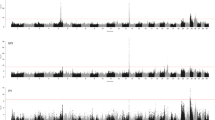
GWAS analysis for milk productive traits showed the following results of SNP associations that have significant impact on the additive value of a sire in terms of the daughter’s milk yield (Fig. 1).

Distributions of significance regression coefficients for sires’ EBV of daughter’s milk traits
The highest number of highly significant SNPs for MY was observed on chromosomes 1, 2, 3, 11, 17, and 23, for FP—on chromosomes 9 and 14, and for PP—on chromosomes 9, 17, 20, and 23. Regression analysis revealed that 425 mononucleotide substitutions had significant effect on the assessment of the sires’ EBV for MY, that actually corresponds to the lower threshold of reliability for genomic research (p ≤ 1.2 × 10−6). In total, 77 significant mutations were detected for FP, 34 of which were found on BTA14 that indicates a high probability of QTL detection in this region of genome with size of 1.42 Mb. The number of SNPs, which were significantly (P = 8.8 × 10−6) associated with PP was lower (14 SNPs). The impact of 17 highly significant mutations is shown in Table 2.
The GWAS results for milk production traits showed the presence of 13 significantly associated polymorphisms, which is localized in functional genes. It was found that along with North American and European Holstein populations the SNPs, associated with quantitative traits were identified on chromosomes 2, 3, 6, 9, 14, 17, and 23. The inheritance complexity of such a comprehensive trait as the milk yield did not allow to identify unequivocal polymorphisms by its effect, but several of them were found to have molecular influence in the following substitutions in ECI2, SPOPL, HNMT, MACF1,and DTX1 genes. It is known that percentage and content of milk fat are mostly influenced by DGAT1 gene expression that was confirmed in our studies. In addition, the region, responsible for lipid synthesis and metabolic exchange, was detected in a quantitative trait locus on chromosome 14, at 253 kb between polymorphisms of DGAT1 and PLEC (LOC786966) genes. Association analysis of milk protein content showed that this trait was determined by a small number of genes because of low variability as well as of complex nature of inheritance and synthesis of milk proteins. The surrounding polymorphisms in UBE3D and BOD1L1 genes, responsible for protein metabolism and posttranslational modification of amino acid compounds on a par with cell control of development, are worth to be highlighted. Besides the mutation in IL15 gene was associated with proliferation of T-lymphocytes and transduction mechanisms.
Conclusions
In general, we can state that significant influence of the reference mutations, responsible for metabolic processes of synthesis of lipids and proteins of milk, was confirmed on the basis of Holstein and holsteinized black-and-white cattle populations in the join reference groups of two regions of Russia. Regarding milk yield, we identified polymorphisms, influencing complex nature of metabolic processes: from histidine metabolism to posttranslational modification of cell structures. Our results show that further association studies of milk yield and milk protein content are required. The data will be used to improve the genetic evaluation of cattle in Russia.
References
Chamberlain, A.J., Hayes, B.J., Savin, K., Bolormaa, S., McPartlan, H.C., Bowman, P.J., Van Der Jagt, C., MacEachern, S., Goddard, M.E.: Validation of single nucleotide polymorphisms associated with milk production traits in dairy cattle. J. Dairy Sci. 95. 864–875
Cole, J.B., Wiggans, G.R., Ma, L., Sonstegard, T.S., Lawlor, T.J.Jr., Crooker, B.A., Van Tassell, C.P., Yang, J., Wang, S., Matukumalli, L.K., Da, Y.: Genome-wide association analysis of thirty one production, health, reproduction and body conformation traits in contemporary U.S. Holstein cow. BMC Genomics 12(408) (2011)
Fang, M., Fu, W., Jiang, D., Zhang, Q., Sun, D., Ding, X., Liu, J.: A multiple-SNP approach for genome-wide association study of milk production traits in Chinese Holstein cattle. PLoS ONE 9(8) (2014) doi:10.1371/journal.pone.0099544
Goddard, M.E., Hayes, B.J.: Mapping genes for complex traits in domestic animals and their use in breeding programmes. Nat. Rev. Genet. 10, 381–391 (2009). doi:10.1038/nrg2575
Gondro, C., Van der Werf, J., Hayes, B.: Genome-Wide Association Studies and Genomic Prediction, p. 566. Humana Press, Australia (2013)
Maurice-Van Eijndhoven, M.H.T., Bovenhuis, H., Veerkamp, R.F., Calus, M.P.L.: Overlap in genomic variation associated with milk fat composition in Holstein Friesian and Dutch native dual-purpose breeds. J. Dairy Sci. 98, 6510–6521. http://dx.doi.org/10.3168/jds.2014-9196
Misztal, I., Tsuruta, S., Strabel, T., Auvray, B., Druet, T., Lee, D.H.: BLUPF90 and related programs (BGF90). In Proceedings of the 7th World Congress on Genetics Applied to Livestock Production, 28, 21–22 (2002)
Parker Gaddis, K.L., Null, D.J., Cole, J.B.: Explorations in genome-wide association studies and network analyses with dairy cattle fertility traits. J. Dairy Sci. 99. 6420–6435. http://dx.doi.org/10.3168/jds.2015-10444
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M., Bender, D., Maller, J., Sklar, P., de Bakker, P., Daly, M.J., Sham, P.C.: PLINK: A toolset for whole-genome association and population-based linkage analysis. Am. J. Hum. Genet. 81(3), 559–575 (2007) (doi:10.1086/519795)
Qanbari, S., Pimentel, E.C.G., Tetens, J., Thaller, G., Lichtner, P., Sharifi, A.R., Simianer, H.: A genome-wide scan for signatures of recent selection in Holstein cattle. Anim. Genet. (2010). doi:10.1111/j.1365-2052.2009.02016.x
Sermyagin, A.A., Gladyr’, E.A., Kharitonov, S.N., Ermilov, A.N., Strekozov, N.I., Brem, G., Zinovieva, N.A.: Genome-wide association study for milk production and reproduction traits in Russian Holstein cattle population. Agric. Biol. 51(2), 182–193 (2016). (Sel’skokhozyaistvennaya Biologiya). doi:10.15389/agrobiology.2016.2.182eng
Su, G., Nielsen, U.S., Wiggans, G., Aamand, G.P., Guldbrandtsen, B., Lund, M.S.: Improving genomic prediction for Danish Jersey using a joint Danish-US reference population. In Proceedings, 10th World Congress of Genetics Applied to Livestock Production (2014)
The R Project for Statistical Computing: Free software environment for statistical computing and graphics. http://www.r-project.org
Tsuruta, S., Lourenco, D.A.L., Misztal, I., Lawlor, T.J.: Genotype by environment interactions on culling rates and 305-day milk yield of Holstein cows in 3 US regions. J. Dairy Sci. 98, 5796–5805. http://dx.doi.org/10.3168/jds.2014-9242
Van Raden, P.M.: Efficient methods to compute genomic predictions. J. Dairy Sci. 91, 4414–4423 (2008) (doi:10.3168/jds.2007-0980)
Wang, X., Wurmser, C., Pausch, H., Jung, S., Reinhardt, F., Tetens, J., Thaller, G., Fries, R.: Identification and dissection of four major QTL affecting milk fat content in the German Holstein-Friesian population. PLoS ONE 7(7) (2012). doi:10.1371/journal.pone.0040711
Winkelman, A.M., Johnson, D.L., Harris, B.L.: Application of genomic evaluation to dairy cattle in New Zealand. J. Dairy Sci. 98, 659–675. http://dx.doi.org/ 10.3168/jds.2014-8560
Acknowledgements
Studies were performed under the financial support of the Ministry of Education and Science of Russia, agreement No 14.604.21.0062. Unique identifier of the project: RFMEFI60414X0062.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
This chapter is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
Copyright information
© 2018 The Author(s)
About this paper

Cite this paper
Sermyagin, А.А. et al. (2018). Genome-Wide Association Studies for Milk Production Traits in Russian Population of Holstein and Black-and-White Cattle. In: Anisimov, K., et al. Proceedings of the Scientific-Practical Conference "Research and Development - 2016". Springer, Cham. https://doi.org/10.1007/978-3-319-62870-7_62
Download citation
DOI: https://doi.org/10.1007/978-3-319-62870-7_62
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-62869-1
Online ISBN: 978-3-319-62870-7
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)