
Abstract
Current research in Life Sciences without doubt has been established as a Big Data discipline. Beyond the expected domain-specific requirements, this perspective has put scalability as one of the most crucial aspects of any state-of-the-art bioinformatics framework. Sequence alignment and construction of phylogenetic profiles are common tasks evident in a wide range of life science analyses as, given an arbitrary big volume of genomes, they can provide useful insights on the functionality and relationships of the involved entities. This process is often a computational bottleneck in existing solutions, due to its inherent complexity. Our proposed distributed framework manages to perform both tasks with significant speed-up by employing Grid Computing resources provided by EGI in an efficient and optimal manner. The overall workflow is both fully automated, thus making it user friendly, and fully detached from the end-users terminal, since all computations take place on Grid worker nodes.
You have full access to this open access chapter, Download conference paper PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Over the last decade, the amount of available data in the life sciences domain has increased exponentially and is expected to keep growing at an ever accelerating pace. This significant increase in data acquisition leads to a pressing need for scalable methods that can be employed to interpret them; a scaling that cannot be met by traditional systems as they cannot provide the necessary computational power and network throughput required. Several efforts are evident in recent literature towards developing new, distributed methods for a number of bioinformatics workflows through the use of HPC systems and paradigms, such as MapReduce [6]. However most, if not all, of these efforts necessitate the setup of a rather complex computing system, as well as the expertise to manage and update an independent software project, since most implementations radically differ from their vanilla counterparts. This is in stark contrast with the situation for most life science researchers who lack the expertise needed to use and manage those systems. As a result, and despite the overall advantages of these frameworks, their ultimate use is fairly limited.
In order to overcome these issues, while at the same time providing the much needed computing power for complex analyses, we developed a bioinformatics framework on top of a Grid architecture that is able to perform common comparative genomics workflows at a massive scale using EGI resources. Special care has been taken to make the framework as automated as possible, increasing the user friendly factor in order to further facilitate wider use by the scientific community. Moreover, every major submodule in the framework utilizes the latest vanilla version available to the community, in order to ensure that the framework can always stay up-to-date through automatically consuming updates of its individual vanilla parts.
The rest of this paper is structured as follows; Sect. 2 provides an overview of the concepts and technologies used throughout this work. Section 3 outlines the proposed framework, with particular focus on the requirements driving the current implementation. Section 4 establishes the technical aspects of the implementation, including the different modes of operations and the expected input and output. In Sect. 5, the efficiency of the framework is validated for all supported execution modes and finally, Sect. 6 provides some insights towards future steps in this direction.
2 Background
2.1 Comparative Genomics
BLAST Algorithm. BLAST has become the industry and research standard algorithm for sequence alignment. An open source implementation is provided by the NCBI organization which produces all alignments, as well as a number of parameters defining the significance of the alignment. A comprehensive list of the available parameters, as well as an in-depth description of the algorithm can be found in the online manual of the application. Our framework relies in particular on one of these parameters, namely the e-value e for each match. The e-value refers to the number of alignments expected to happen by chance. Therefore, a low e-value indicates a high statistical significance of an alignment.
Phylogenetic Profiles. Although the BLAST algorithm has been designed to readily identify and quantify sequence similarity, it will inherently miss any information that is not directly tied to this aspect. In particular, given a pair of protein sequences that may exhibit a high functional correlation (e.g. similar domains, active sites etc.) while at the same time do not expose any significant similarity in their composition, BLAST will fail to detect their interesting relationship. An alternative way to detect and quantify such relations, is to observe the joint presence or absence of sequences across a common set of genomes, for example protein sequences which only appear in the same family of genomes are highly likely to be related to that familys distinct functionality.
Phylogenetic profiles are vectors that characterize each sequence. Specifically, they indicate the sequence’s homologs with every genome found in a given dataset. Each element in the vector corresponds to presence (denoted as the number 1) or absence (denoted as the number 0) of any homologue of the protein sequence under study in the respective genome. Beyond the standard (binary) phylogenetic profiles, an extended version may be constructed by replacing the presence/absence attribute by the actual number of homologs between that sequence and a target genome.
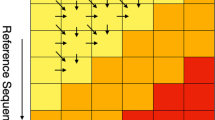
An even stronger evolutionary link between two sequences can be inferred through the use of best bidirectional hits (BBH). By definition, the best hit of a given sequence \(seq_A\) derived from genome A to a target genome B, is the sequence \(seq_B\) in genome B that represents the best match, i.e. a homologue scoring lower e-value e than all other hits produced. This particular match is also bidirectional if the \(seq_A\) is also the best hit for \(seq_B\). A bidirectional best hit represents a very strong similarity between two sequences, and is considered evidence that the genes may be orthologs arising from a common ancestor [6]. A graphical representation of the two cases of phlyogenetic profiles is show in Fig. 1.

Graphical overview of the general hit and bidirectional hit profiles, given a set of example sequences.
Grid Computing. A computer Grid is a system that coordinates resources that are not subject to centralized control using standard, open, general-purpose protocols and interfaces to deliver non-trivial quality of service [3]. A grid computing architecture can bring massive processing power to bear on a problem, as SETI (Search for ExtraTerrestrial Intelligence) and other similar projects have shown [1, 2]. An additional abstraction layer, called middleware, makes creating and controlling grids easier.
The European Grid Infrastructure (EGI) is the result of pioneering work that has, over the last decade, built a collaborative production infrastructure of uniform services through the federation of national resource providers that supports multidisciplinary science across Europe and around the world. Our framework is deployed on the HellasGrid infrastructure, part of EGI, which offers high performance computing services to Greek universities and research institutes. HellasGrid is also the biggest infrastructure for Grid computing in the area of South-East Europe.
3 Framework Description
We designed and implemented a framework capable of performing sequence alignment and phylogenetic profiling in a time-efficient and user-friendly manner, by utilizing EGI resources. These processes were both scaled to resources and automated due to the fact that they comprise an essential part in a plethora of biological analysis pipelines including, but not limited to: species identification, DNA mapping and domain localization [5, 7]. The overall design and ultimate implementation of our framework has been dictated by the following requirements:
-
Optimal efficiency
-
Fully automated and robust against system failures
-
Reusable output for building on top of previous results
-
Reuse of existing, optimized and tested submodules
Workflow efficiency was maximized through an optimal use of EGI resources, minimizing queue delays as well as potential load failures that lead to resubmission of computational jobs [8]. The workflow does not require any interaction from the user after the initial submission phase; instead everything runs automatically in the background thus releasing the end-users’ machine and allowing for better utilization of the local resources until the analysis is finished. Possible system errors are also handled automatically, with the possible exception of fatal errors. The final output is presented in a simple, widely accepted format that can be either consumed as a final output or used as an intermediate step to the next steps of any analysis pipeline. Finally, instead of reinventing the wheel, our framework utilizes well documented and maintained vanilla implementations of its submodules when available in order to accommodate both updates as well as support any further development of the framework through targeted extensions.
3.1 Program Flow
As outlined earlier in the requirements of the framework, the workflow is fully automated. A Grid job is submitted for each file in the query directory corresponding to a genome to be examined, meaning that the number of submitted jobs is equal to the number of genomes we are interested in. Job submission is in reverse order to the size of the query file, thus ensuring that the most computationally expensive jobs are submitted first. Given that the total time needed for our analysis is equal to the worst time of its parallel counterparts, minimizing the delay of the lengthier jobs results in a notable improvement in efficiency, namely 20–30 h for large queries as shown in the experiments performed (Sect. 5). In order to minimize the uploading time from the user’s UI, as well as be able to handle an arbitrarily large database file, we exploit the Grid’s storage services. Specifically, we upload the database only once to a Storage Element (SE), and provide each worker node with its qualified name. The worker node is thus able to retrieve the database directly downloading from the SE. Finally, each job runs BLAST to align its specific genome file with the database (which is common for all jobs).
The alignments produced by BLAST are then used to construct the phylogenetic profile of any type requested and for each sequence in the query genome. Since the computation at each node is completely independent of other nodes, no blocking occurs which qualifies the procedure as “embarrassingly parallel”. Finally, the output of every job is returned to the users terminal and combined in a way that facilitates visualization as well as post processing. An instance of a job handler script is launched in the background for each of those jobs in order to monitor their state and resubmit them in case of failure caused by stability or load issues that the Grid might face at a given time (Fig. 2).

(A) Diagram of the workflow from the users point of view (B) Diagram of the workflow taking place at each WN.
4 Technical Overview
4.1 Modes of Operation
The driving use case for the proposed framework is to provide users with the means to run an independent analysis. Specifically, the user specifies both the sequences under study as well as the set of genomes against which the sequences are going to be aligned and consequently profiled. Beyond this information and during the initialization phase, additional parameters can be configured such as the type of phylogenetic profiles to be constructed (BBH or plain, extended or binary), the type of the input sequence data type (nucleotides or aminoacid) etc.
Moreover, our framework provides support for building upon previous results; due to the frequent appearance of such analyses in typical biological workflows, the framework provides the ability to combine new results (i.e. new genomes) with any previously computed results. Therefore, and instead of running the whole process from scratch, the framework can expand the previous query without recalculating all involved alignments and phylogenetic profiles’ elements.
4.2 Expected Input
The input for every analysis comprises of the database file, including the sequences of every genome in our dataset, and a set of files corresponding to the genomes under study. The former files will be referred to as query files and should reside in a common directory, namely the query directory. It is important to note that both the query files and the database file must comply to the FASTA specification. The user must also supply a text file mapping each genome found in his database with a unique identifier. Finally, the user fills in a properties file in which the exact mode of operation and its parameters are configured.
4.3 Framework Output
The output is comprised of the BLAST alignment results and the phylogenetic profile for each sequence. The framework also includes a library of usual post processing utilities that the user might use to visualize the output. These include (a) filters that are able to isolate the most significant lines of the output, such as alignments with a top e-value score or profiles with specific matches, as well as (b) reducers to collapse profiles at the genome level, i.e. present the phylogenetic profile of a whole genome instead of a profile at the sequence level.
5 Experiments and Results
The framework has been implemented within a standard UNIX environment in mind, i.e. any Debian based version of Linux. This restriction has been placed as a requirement of the underlying Grid infrastructure. The basic workflow is comprised of scripts to exploit the machines native calls, whereas complementary functionalities are designed in an object oriented manner using the Java programming language. In order to validate our framework for scalability we employed a test case, performing an all-vs-all analysis, since this is the most computationally demanding mode of operation. The total of 30 genomes used comprised a BLAST database including 544,538 sequences.
Our experiment involves the creation of BBH profiles for a total of 30 Genomes (all-vs-all operation). It can be seen in Fig. 3 that the total time for each job is comprised of 3 parts. First, there is the submission delay. As explained before, we attempt to minimize the number potential job failures, by inserting a 1-hour delay between consecutive submissions. We take special care to ensure that bigger jobs are scheduled first, thus minimizing the total time for the lengthier jobs. Since the total query time equals that of its most demanding job, this method results in a huge efficiency boost as shown above. The green part corresponds to the scheduling time, which is usually minimal. However, there exist situations where a job fails and has to be resubmitted multiple times, resulting in extended waiting time such as the case of job number 17. Lastly, we can see the running time on each WN. This demonstrates significant variance, caused not only by input anomalies, but also by the heterogeneity of hardware resources employed by the Grid infrastructure.

Execution times in a 30 genome all-vs-all BBH profile construction query.
Finally, the produced phylogenetic profiles can be readily used for any further analysis, including functional correlations, pangenome approaches and evolutionary studies. Figure 4 provides some preliminary results through the use of BBH phylogenetic profiles.

Visual representation of the BBH profiles produced by the all-vs-all comparison of 30 genomes. Each cell corresponds to the number of BBH that have been identified in the respective pair of genomes. Moreover, the matrix has been hierarchically clustered in order to produce evolutionary meaningful clusters of genomes, as evident by the highly intra-correlated groups (bottom left, middle and top right).
6 Discussion and Conclusions
The ever increasing biological big data that need to be processed and interpreted in almost every modern bioinformatics workflow, require extreme computational power, storage capability and network throughput that only large-scale distributed systems can offer. The proposed framework exploits the native nature of the Grid infrastructure to achieve parallelization at the data layer instead of the software layer, allowing us to utilize well known vanilla implementations of comparative genomics tools. This allowed the framework to achieve significant speed-up without requiring any user effort, or introducing additional maintenance and setup concerns for the end user. This aspect, along with the fact that the main functionality offered poses a significant role in a wide range of diverse biological workflows, can potentially provide an easy solution to researcher in the wider scientific community.
Finally, the modular nature of the frameworks inner workings not only allows for the automatic updating of its submodules, but can also facilitate the addition of new features, such as a user friendly GUI, through integration with existing visualization platforms like Galaxy [4].
References
Anderson, D.P.: Boinc: A system for public-resource computing and storage. In: Proceedings of the Fifth IEEE/ACM International Workshop on Grid Computing, pp. 4–10. IEEE (2004)
Anderson, D.P., Cobb, J., Korpela, E., Lebofsky, M., Werthimer, D.: Seti@ home: an experiment in public-resource computing. Commun. ACM 45(11), 56–61 (2002)
Foster, I., Kesselman, C., Tuecke, S.: What is the grid?-A three point checklist. Gridtoday (6), July 2002
Giardine, B., Riemer, C., Hardison, R.C., Burhans, R., Elnitski, L., Shah, P., Zhang, Y., Blankenberg, D., Albert, I., Taylor, J., et al.: Galaxy: a platform for interactive large-scale genome analysis. Genome Res. 15(10), 1451–1455 (2005)
Marcotte, E.M., Xenarios, I., Van der Bliek, A.M., Eisenberg, D.: Localizing proteins in the cell from their phylogenetic profiles. Proc. Nat. Acad. Sci. 97(22), 12115–12120 (2000)
Overbeek, R., Fonstein, M., Dsouza, M., Pusch, G.D., Maltsev, N.: The use of gene clusters to infer functional coupling. Proc. National Acad. Sci. 96(6), 2896–2901 (1999)
Pellegrini, M.: Using phylogenetic profiles to predict functional relationships. Bacterial Molecular Networks: Methods and Protocols, pp. 167–177 (2012)
Vrousgou, O.T., Psomopoulos, F.E., Mitkas, P.A.: A grid-enabled modular framework for efficient sequence analysis workflows. In: Iliadis, L., et al. (eds.) EANN 2015. CCIS, vol. 517, pp. 47–56. Springer, Heidelberg (2015). doi:10.1007/978-3-319-23983-5_5
Acknowledgements
This work used the European Grid Infrastructure (EGI) through the National Grid Infrastructure - HellasGrid.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 IFIP International Federation for Information Processing
About this paper
Cite this paper
Stergiadis, E., Kintsakis, A.M., Psomopoulos, F.E., Mitkas, P.A. (2016). A Scalable Grid Computing Framework for Extensible Phylogenetic Profile Construction. In: Iliadis, L., Maglogiannis, I. (eds) Artificial Intelligence Applications and Innovations. AIAI 2016. IFIP Advances in Information and Communication Technology, vol 475. Springer, Cham. https://doi.org/10.1007/978-3-319-44944-9_39
Download citation
DOI: https://doi.org/10.1007/978-3-319-44944-9_39
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-44943-2
Online ISBN: 978-3-319-44944-9
eBook Packages: Computer ScienceComputer Science (R0)