
Abstract
Dysregulation of the stress response is implicated in many psychopathologies. Data gathered over the past two decades have proposed a rather dualistic view of the central corticotropin-releasing factor (CRF)-urocortin system. Originally, it was thought that CRF/CRF receptor type 1 (CRFR1) signaling mediated stress-initiated effects and increased anxiety-like behavior, whereas activation of urocortins/CRFR2 ensured adequate recovery from stress and restoration of homeostasis. However, this view was based on data gained from genetically modified mouse models and pharmacological approaches; now, with the emergence of new and more specific biological tools, it has become clear that this is an over-simplistic proposal. It is becoming apparent that the function of the CRF-urocortin system’s components relies profoundly on the spatial and temporal patterns of activity of the CRF family members. Here, we provide an overview of recent work that proposes a more dynamic, modulatory role for the CRF system’s central pathways in the modulation of stress-linked behaviors. Recent findings suggest that the CRF system’s actions are brain-region specific and dependent on the type of neuronal cell involved.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
In 1955, Hans Seyle recognized that the hypothalamic-pituitary-adrenal (HPA) axis orchestrates the stress response (Selye 1955). However, it took until 1981 for the structure of its principal regulatory peptide, corticotropin-releasing factor (CRF), to be characterized (Vale et al. 1981). Neurons in the paraventricular nucleus (PVN) of the hypothalamus secrete CRF, which binds to receptors in the pituitary, which, in turn, secretes adrenocorticotropic hormone (ACTH) into the circulation. The circulating ACTH binds at the adrenal cortex stimulating the synthesis and secretion of glucocorticoids: cortisol in humans and corticosterone in rodents. Glucocorticoids are the downstream biological effector of the neuroendocrine stress response and also provide negative feedback in the HPA axis.
The CRF family of peptides includes the 41 amino acid peptide CRF and the more recently discovered urocortin (UCN) 1, 2 and 3. As shown in Fig. 1, this family of peptides binds to two distinct receptors: the CRF receptor type 1 (CRFR1) and type 2 (CRFR2). CRF is a high-affinity ligand for the CRFR1, whereas UCN1 binds with equal affinity to both receptors. UCN2 and UCN3 preferentially bind the CRFR2. However, specificity seems to be lost at higher concentrations of the ligand, with CRF activating CRFR2. CRFR1 and CRFR2 are produced from distinct genes and have numerous splice variants (CRFR1α,β, and CRFR2α,β,γ), some of which are nonfunctional (Perrin and Vale 1999; Grammatopoulos and Chrousos 2002).

The CRF family of neuropeptides and their action on the CRFR1 and CRFR2 (Kuperman and Chen 2008, with permission). Colored arrows indicate the receptors with which each ligand preferably interacts; dashed lines indicate a lower affinity binding. CRFR2 has two apparent membrane-bound splice variants in rodents, resulting in two receptor proteins, CRFR2α and CRFR2β
CRF receptors are Gs protein-coupled receptors. They stimulate a cascade of events, firstly adenylate cyclase, which subsequently activates cyclic AMP (cAMP). Cyclic AMP stimulates protein kinase A (PKA) to phosphorylate substrates such as the cAMP responsive-element–binding (CREB) protein, thereby inducing transcription of downstream target genes. In addition, cAMP binds to exchange proteins, which then activate the extracellular signal-regulated kinase-mitogen-activated protein kinase (ERK-MAPK) cascade (Gutknecht et al. 2009). The ERK/MAPK pathway regulates synaptic plasticity, including dendrite stabilization, ion channel transmission, transcription of CREB and other genes, and receptor scaffolding, trafficking, and crosstalk. Coupling to Gs is the dominant mechanism for stimulating intracellular calcium mobilization by CRFR1 and CRFR2. However, CRF receptors also interact with other G protein systems, including Gq, Gi, Go, Gil/2, and Gz (Grammatopoulos et al. 2001). Coupling to Gq activates PLCβ, which cleaves the phospholipid phosphatidylinositol 4,5-bisphosphate into diacyl glycerol (DAG) and inositol 1,4,5-trisphosphate (IP3). IP3 then diffuses through the cytosol to bind to IP3 receptors to increase intracellular calcium levels, whereas DAG activates protein kinase C (PKC), which activates ERK1/2 via Ras, Raf and MEKs. Gi/0 mediated activation of Gβγ subunits activates the ERK1/2 by PI-3 kinase and induces PLCγ activation, which increases intracellular calcium release. Thus, depending on their localization and cellular context, CRF receptors are able to modulate a wide variety of signaling pathways (Hauger et al. 2006, 2009) and kinases [including PKA, protein kinase B (PKB), PKC (Gutknecht et al. 2009), mitogen activated protein (MAP) kinases (e.g., ERK1/2), and intracellular Ca2+ concentrations]. CRF receptors activate these various G-protein systems in a concentration-dependent manner (Grammatopoulos and Chrousos 2002).
Besides the activated pathway, the desensitization and internalization following receptor activation seems to depend on the specific ligand bound. Desensitization of CRFR2 cAMP signaling was shown to occur more rapidly and to a greater extent in response to UCN2 binding compared to UCN3, whereas CRF is a relatively weak desensitizing agonist (Gutknecht et al. 2008). Additionally, the internalization of CRFR2 was shown to be greater upon exposure to UCN2 (Markovic et al. 2008).
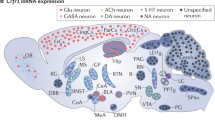
In situ hybridization histochemical studies (and recently generated reporter mice; Justice et al. 2008; Kuhne et al. 2012) have shown that CRFR1 is widely distributed throughout the brain. The highest expression levels are seen in neocortical, limbic, brain stem regions and the cerebellum (Fig. 2). Moderate levels are found in the dorsal and median raphe nuclei, and only low levels are found in the PVN of the hypothalamus (Chalmers et al. 1995; Van Pett et al. 2000). CRFR1 is expressed in diverse neuronal subpopulations. They have been found on cell bodies, dendritic shafts and dendritic spines of hippocampal neurons (Chen et al. 2004a,b, 2010). Compared to CRFR1, CRFR2 mRNA has a much more localized distribution pattern throughout the brain, which is virtually confined to subcortical structures. The highest levels of CRFR2 expression are found in the lateral septum, the ventromedial hypothalamic nucleus, and the choroid plexus (Fig. 2). Moderate levels are seen in the olfactory bulb, nuclei of the extended amygdala, hippocampus, the PVN and supraoptic nuclei of the hypothalamus, the inferior colliculus and the raphe nucleus (Chalmers et al. 1995; Van Pett et al. 2000). Only a limited amount of data is available on brain region-specific modulation of CRFR2 signaling. Among the most investigated regions are the extended amygdala, hippocampus, lateral septum, medial prefrontal cortex, and brain stem nuclei. Additionally, all four CRF family neuropeptides have been detected in the periphery, in particular UCN2 and UCN3, which have been recognized as novel modulators of centrally and peripherally controlled metabolic function (Chen et al. 2004a; Kuperman and Chen 2008).

The distribution of CRFR1 (yellow) and CRFR2 (pink) mRNA expression in the rodent brain. AMYG, amygdala; BST, bed nucleus of the stria terminalis; RAPHE, raphe nucleus; SEPT, lateral septum; VMH, ventromedial hypothalamic nucleus
Alterations in HPA axis function, attributed to centrally elevated CRF levels, were observed in depressed patients (Nemeroff et al. 1984; Holsboer 1999; Reul and Holsboer 2002) and in patients with post-traumatic stress disorder (PTSD; Bremner et al. 1997; McEwen 2002). These findings stimulated huge interest into the functioning of the CRF family. CRFR1 and CRFR2 seem to modulate anxiety-like behavior in a brain region-dependent manner. One obvious explanation for the differential behavioral effects of CRF receptor activation is that distinct brain regions simply serve distinct behavioral functions, and their activation would thus induce a distinct behavioral phenotype. Activation of brain regions implicated in emotional arousal, such as the extended amygdala, would be expected to increase anxiety, whereas activation of cognitive control regions, such as the medial prefrontal cortex, would contribute to stress coping and reduce anxiety. However, both the behavioral and electrophysiological effects of CRF receptor activation implicate a more complex underlying mechanism of region dependency and suggest specificity due to the cell type and ligand concentration.
Since specific CRF receptor antibodies are lacking, much about the action of the CRF family has been learned from transgenic mice, overexpressing or knocked out for the various members. Increased central levels of CRF [induced by either CRF administration (Britton et al. 1986; Dunn and File 1987; Dunn and Berridge 1990) or its overexpression in transgenic mice (Stenzel-Poore et al. 1994; Heinrichs et al. 1997; van Gaalen et al. 2002; Binneman et al. 2008)] produces anxiogenic behavior, whereas suppression induces anxiolytic effects (Skutella et al. 1994) and reduced stress-induced anxiety (Skutella et al. 1994). This action was attributed to CRFR1 activation specifically, since CRFR1 blockage by antisense treatment (Skutella et al. 1998; Liebsch et al. 1999) or selective antagonists (i.e., antalarmin; Habib et al. 2000; Valdez et al., 2002) prevented CRF/stress-induced anxiety. Furthermore, CRFR1 knock-out mice displayed reduced anxiety-like behavior (Smith et al. 1998; Timpl et al. 1998; Contarino et al. 1999; Muller et al. 2003). Taken together, these data suggested a critical role for CRFR1 activation in eliciting stress-induced anxiety. However, although these findings suggest a causative role for CRFR1 over-activation in stress-related psychopathologies, clinical trials of CRFR1 antagonists as potential next-generation anxiolytics/antidepressants have unfortunately met with little success.
In contrast to CRFR1, the role of CRFR2 in the mediation of psychopathologies is less clear, and two prominent theories are currently circulating that explain its role. The most popular view is that CRFR2 activation is responsible for ensuring homeostasis and counteracts the stress response-provoking effects and anxiety-like behavior induced by CRFR1 activation (Coste et al. 2001; Bale and Vale 2004; Heinrichs and Koob 2004; Muller and Holsboer 2006). This theory is primarily based on the increased corticosterone stress response observed in CRFR2 KO mice (Bale et al. 2000; Coste et al. 2000), and the anxiogenic phenotype observed in some (but not all; Coste et al., 2000) of the CRFR2 KO mouse lines (Bale et al. 2000; Kishimoto et al. 2000). However, the increased levels of anxiety and stress-related behaviors observed in a mouse line displaying (chronic) overexpression of UCN3 (Neufeld-Cohen et al. 2012), as well as the observed reduction in shock-induced freezing in response to reduced CRFR2 expression (by administration of CRFR2 mRNA antisense oligonucleotides; Ho et al. 2001) contradict this theory.
UCN1 neurons are mainly localized in the Edinger Westphal nucleus where they constitute the centrally projecting part of the nucleus to the lateral septum. UCN1 mRNA is also found in the lateral olivary and supraoptic nucleus (Bittencourt et al. 1999). UCN2 expression has been shown in several regions that are involved in the physiological and behavioral responses to stress, such as the PVN, locus coeruleus, and it partly overlaps with CRF and UCN1 expression in the hypothalamus and brainstem. UCN2 is thought to be the primary ligand for CRFR2s in the bed nucleus of the stria terminalis (BNST), PVN, central amygdala, parabrachial nucleus and nucleus tractus solitarii (Reyes et al. 2001). UCN3 also shows a distinct expression pattern and is mainly found in the medial preoptic area, rostral perifornical area, the posterior part of the BNST and the medial amygdala, with projections observed in the intermediate lateral septum (Lewis et al. 2001) and in the BNST (Deussing et al. 2010). However, although the sites of mRNA expression of these ligands are pretty well established, it is still largely unknown at which exact sites (i.e., axonal, dendritic, synaptic) the ligands are in fact released.
UCN1 neurons are recruited following chronic stress exposure and stay active for a prolonged period of time, suggesting that this peptide plays a prominent role in the later, adaptive phase of the stress response (Korosi et al. 2005; Xu et al. 2010; Ryabinin et al. 2012). The UCN family members’ involvement in stress-related behavior was assessed in UCN1/UCN2 (dKO) double and UCN1/UCN2/UCN3 (tKO) triple knockout mice. Although the dKO knockout mice displayed no changes in basal HPA axis activity, they had elevated corticosterone levels following acute stress exposure (Neufeld-Cohen et al. 2010b). HPA axis function was unchanged in tKO mice compared to controls (Neufeld-Cohen et al. 2010a). The dKO knockout mice displayed decreased anxiety-like behavior under basal and acute stress conditions, which was accompanied by elevated serotonin concentrations in a number of brain regions, including the dorsal raphe nucleus, hippocampus, basolateral amygdala and subiculum (Neufeld-Cohen et al. 2010b). In contrast, tKO mice exhibited increased anxiety-like behavior, but only 24 h after restraint stress. Moreover, tKO mice displayed an increased stress-induced startle response (Neufeld-Cohen et al. 2010b). As opposed to dKO mice, the behavioral phenotype in tKO mice was associated with decreased serotonergic metabolism in regions such as the septum, central and basolateral amygdala (Neufeld-Cohen et al. 2010a). Again, the effect of compensatory changes in CRF expression on emotional behavior cannot be excluded in many of the UCN mouse models, as shown in dKO mice (Neufeld-Cohen et al. 2010b). Overall, the data suggest that the UCN’s binding to CRFR2 is able to regulate specific aspects of stress-related emotional behavior, complementing the effects of CRF to CRFR1.
Another great advance in the understanding of the CRF system’s actions can be accredited to the specificity of genetic or viral-mediated loss-of-function approaches. For example, deletion of the CRFR1 in glutamatergic neurons reduces anxiety-related behavior, whereas deletion in dopaminergic neurons increases anxiety-related behavior (Refojo et al. 2011). Lentiviral-mediated knockdown of CRF in the central amygdala attenuated stress-induced anxiety-like behavior and altered HPA axis activity, reinforcing a role for amygdalar CRF in the modulation of fear and anxiety (Regev et al. 2011). Lentiviral knockdown of CRFR1 in the basolateral amygdala was also shown to decrease anxiety-like behavior and mimicked the anxiolytic effect of environmental enrichment (Sztainberg et al. 2010). Using stereotactic delivery of lentiviruses into the paraventricular nucleus, Elliott et al. (2010) targeted CRF by using lentiviral constructs that carried shRNA against CRF. This approach enabled the site-specific manipulation of gene expression. The knockdown attenuated chronic stress-induced social avoidance, which was shown to result from demethylation of the CRF promoter (Elliott et al. 2010).
The BNST is thought to be involved in the regulation of anxiety. It is positioned as a relay center between the limbic structures and mediates anticipatory stress. Lentiviral-mediated knockdown of CRFR2 in the BNST increased anxiety-related behavior both immediately and 24 h after restraint stress. Studying the knockdown and overexpression of CRFR2 specifically in the BNST revealed an important role of this receptor in PTSD-like behavior (Lebow et al. 2012).
In some of our most recent work, we have used optogenic regulation of posterior BNST CRFR2 neurons using bilateral fiber optic implantation. CRFR2 Cre mice were crossed with conditional channelrhodopsin (ChR2) mice, in which blue light activates CRFR2-expressing neurons specifically, or with conditional halorhodopsin (NpHR2) mice, in which yellow light inhibits CRFR2-expressing neurons specifically. Using this tool, we found that specific activation of CRFR2 neurons in the posterior BNST reduced anxiety-related behavior and, conversely, their suppression increased anxiety-related behavior (Henckens et al. 2016). Similar viral-mediated loss-of-function tools were used to knockdown CRFR1 in the globus pallidus, revealing a previously unknown anxiolytic effect of the receptor in this brain region, which was further confirmed with site-specific CRFR1 antagonist administration (Sztainberg et al. 2011). The relevance of CRFR1 receptors in addiction and reward processes was investigated in the ventral tegmental area using lentiviral-mediated knockdown. Knockdown of CRFR1 reduced cue-induced and acute food deprivation, stress-induced cocaine seeking, but had no effect on self-administering behavior. CRFR1 signalling in the ventral tegmental area presents a target for convergent effects of both cue- and stress-induced cocaine-seeking pathways (Chen et al. 2014).
In summary, recent advances in methodological approaches have begun elucidating the site-specific actions of the CRF system. Most notably, they suggest that, via the CRFR2, UCNs might be the central system involved in coping with stress. Improved understanding of the CRF system holds the promise of better treatment of stress-related psychopathologies.
References
Bale TL, Vale WW (2004) CRF and CRF receptors: role in stress responsivity and other behaviors. Annu Rev Pharmacol Toxicol 44:525–557
Bale TL, Contarino A, Smith GW, Chan R, Gold LH, Sawchenko PE, Koob GF, Vale WW, Lee KF (2000) Mice deficient for corticotropin-releasing hormone receptor-2 display anxiety-like behaviour and are hypersensitive to stress. Nat Genet 24:410–414
Binneman B, Feltner D, Kolluri S, Shi Y, Qiu R, Stiger T (2008) A 6-week randomized, placebo-controlled trial of CP-316,311 (a selective CRH1 antagonist) in the treatment of major depression. Am J Psychiatry 165:617–620
Bittencourt JC, Vaughan J, Arias C, Rissman RA, Vale WW, Sawchenko PE (1999) Urocortin expression in rat brain: evidence against a pervasive relationship of urocortin-containing projections with targets bearing type 2 CRF receptors. J Comp Neurol 415:285–312
Bremner JD, Licinio J, Darnell A, Krystal JH, Owens MJ, Southwick SM, Nemeroff CB, Charney DS (1997) Elevated CSF corticotropin-releasing factor concentrations in posttraumatic stress disorder. Am J Psychiatry 154:624–629
Britton KT, Lee G, Vale W, Rivier J, Koob GF (1986) Corticotropin releasing factor (CRF) receptor antagonist blocks activating and 'anxiogenic' actions of CRF in the rat. Brain Res 369:303–306
Chalmers DT, Lovenberg TW, De Souza EB (1995) Localization of novel corticotropin-releasing factor receptor (CRF2) mRNA expression to specific subcortical nuclei in rat brain: comparison with CRF1 receptor mRNA expression. J Neurosci 15:6340–6350
Chen Y, Brunson KL, Adelmann G, Bender RA, Frotscher M, Baram TZ (2004a) Hippocampal corticotropin releasing hormone: pre- and postsynaptic location and release by stress. Neuroscience 126:533–540
Chen Y, Bender RA, Brunson KL, Pomper JK, Grigoriadis DE, Wurst W, Baram TZ (2004b) Modulation of dendritic differentiation by corticotropin-releasing factor in the developing hippocampus. Proc Natl Acad Sci U S A 101:15782–15787
Chen Y, Rex CS, Rice CJ, Dube CM, Gall CM, Lynch G, Baram TZ (2010) Correlated memory defects and hippocampal dendritic spine loss after acute stress involve corticotropin-releasing hormone signaling. Proc Natl Acad Sci U S A 107:13123–13128
Chen NA, Jupp B, Sztainberg Y, Lebow M, Brown RM, Kim JH, Chen A, Lawrence AJ (2014) Knockdown of CRF1 receptors in the ventral tegmental area attenuates cue- and acute food deprivation stress-induced cocaine seeking in mice. J Neurosci 34:11560–11570
Contarino A, Dellu F, Koob GF, Smith GW, Lee KF, Vale W, Gold LH (1999) Reduced anxiety-like and cognitive performance in mice lacking the corticotropin-releasing factor receptor 1. Brain Res 835:1–9
Coste SC, Kesterson RA, Heldwein KA, Stevens SL, Heard AD, Hollis JH, Murray SE, Hill JK, Pantely GA, Hohimer AR, Hatton DC, Phillips TJ, Finn DA, Low MJ, Rittenberg MB, Stenzel P, Stenzel-Poore MP (2000) Abnormal adaptations to stress and impaired cardiovascular function in mice lacking corticotropin-releasing hormone receptor-2. Nat Genet 24:403–409
Coste SC, Murray SE, Stenzel-Poore MP (2001) Animal models of CRH excess and CRH receptor deficiency display altered adaptations to stress. Peptides 22:733–741
Deussing JM, Breu J, Kuhne C, Kallnik M, Bunck M, Glasl L, Yen YC, Schmidt MV, Zurmuhlen R, Vogl AM, Gailus-Durner V, Fuchs H, Holter SM, Wotjak CT, Landgraf R, de Angelis MH, Holsboer F, Wurst W (2010) Urocortin 3 modulates social discrimination abilities via corticotropin-releasing hormone receptor type 2. J Neurosci 30:9103–9116
Dunn AJ, Berridge CW (1990) Is corticotropin-releasing factor a mediator of stress responses? Annu NY Acad Sci 579:183–191
Dunn AJ, File SE (1987) Corticotropin-releasing factor has an anxiogenic action in the social interaction test. Horm Behav 21:193–202
Elliott E, Ezra-Nevo G, Regev L, Neufeld-Cohen A, Chen A (2010) Resilience to social stress coincides with functional DNA methylation of the Crf gene in adult mice. Nat Neuroci 13:1351–1353
Grammatopoulos DK, Chrousos GP (2002) Functional characteristics of CRH receptors and potential clinical applications of CRH-receptor antagonists. Trends Endocrinol Metab 13:436–444
Grammatopoulos DK, Randeva HS, Levine MA, Kanellopoulou KA, Hillhouse EW (2001) Rat cerebral cortex corticotropin-releasing hormone receptors: evidence for receptor coupling to multiple G-proteins. J Neurochem 76:509–519
Gutknecht E, Hauger RL, Van der Linden I, Vauquelin G, Dautzenberg FM (2008) Expression, binding, and signaling properties of CRF2(a) receptors endogenously expressed in human retinoblastoma Y79 cells: passage-dependent regulation of functional receptors. J Neurochem 104:926–936
Gutknecht E, Van der Linden I, Van Kolen K, Verhoeven KF, Vauquelin G, Dautzenberg FM (2009) Molecular mechanisms of corticotropin-releasing factor receptor-induced calcium signaling. Mol Pharmacol 75:648–657
Habib KE, Weld KP, Rice KC, Pushkas J, Champoux M, Listwak S, Webster EL, Atkinson AJ, Schulkin J, Contoreggi C, Chrousos GP, McCann SM, Suomi SJ, Higley JD, Gold PW (2000) Oral administration of a corticotropin-releasing hormone receptor antagonist significantly attenuates behavioral, neuroendocrine, and autonomic responses to stress in primates. Proc Natl Acad Sci U S A 97:6079–6084
Hauger RL, Risbrough V, Brauns O, Dautzenberg FM (2006) Corticotropin releasing factor (CRF) receptor signaling in the central nervous system: new molecular targets. CNS Neurol Disord Drug Targets 5:453–479
Hauger RL, Risbrough V, Oakley RH, Olivares-Reyes JA, Dautzenberg FM (2009) Role of CRF receptor signaling in stress vulnerability, anxiety, and depression. Annu NY Acad Sci 1179:120–143
Heinrichs SC, Koob GF (2004) Corticotropin-releasing factor in brain: a role in activation, arousal, and affect regulation. J Pharmacol Exp Ther 311:427–440
Heinrichs SC, Min H, Tamraz S, Carmouche M, Boehme SA, Vale WW (1997) Anti-sexual and anxiogenic behavioral consequences of corticotropin-releasing factor overexpression are centrally mediated. Psychoneuroendocrinology 22:215–224
Henckens MJAG, Printz Y, Shamgar U, Lebow M, Drori Y, Kuehne C, Kolarz A, Deussing JM, Justice NJ, Yizhar O, Chen A (2016) The posterior bed nucleus of the stria terminalis critically contributes to stress recovery. Mol Psychiatry (in press)
Ho SP, Takahashi LK, Livanov V, Spencer K, Lesher T, Maciag C, Smith MA, Rohrbach KW, Hartig PR, Arneric SP (2001) Attenuation of fear conditioning by antisense inhibition of brain corticotropin releasing factor-2 receptor. Brain Res Mol Brain Res 89:29–40
Holsboer F (1999) The rationale for corticotropin-releasing hormone receptor (CRH-R) antagonists to treat depression and anxiety. J Psychiatr Res 33:181–214
Justice NJ, Yuan ZF, Sawchenko PE, Vale W (2008) Type 1 corticotropin-releasing factor receptor expression reported in BAC transgenic mice: implications for reconciling ligand-receptor mismatch in the central corticotropin-releasing factor system. J Comp Neurol 511:479–496
Kishimoto T, Radulovic J, Radulovic M, Lin CR, Schrick C, Hooshmand F, Hermanson O, Rosenfeld MG, Spiess J (2000) Deletion of crhr2 reveals an anxiolytic role for corticotropin-releasing hormone receptor-2. Nat Genet 24:415–419
Korosi A, Schotanus S, Olivier B, Roubos EW, Kozicz T (2005) Chronic ether stress-induced response of urocortin 1 neurons in the Edinger-Westphal nucleus in the mouse. Brain Res 1046:172–179
Kuhne C, Puk O, Graw J, Hrabe de Angelis M, Schutz G, Wurst W, Deussing JM (2012) Visualizing corticotropin-releasing hormone receptor type 1 expression and neuronal connectivities in the mouse using a novel multifunctional allele. J Comp Neurol 520:3150–3180
Kuperman Y, Chen A (2008) Urocortins: emerging metabolic and energy homeostasis perspectives. Trends Endocrinol Metab 19:122–129
Lebow M, Neufeld-Cohen A, Kuperman Y, Tsoory M, Gil S, Chen A (2012) Susceptibility to PTSD-like behavior is mediated by corticotropin-releasing factor receptor type 2 levels in the bed nucleus of the stria terminalis. J Neurosci 32:6906–6916
Lewis K, Li C, Perrin MH, Blount A, Kunitake K, Donaldson C, Vaughan J, Reyes TM, Gulyas J, Fischer W, Bilezikjian L, Rivier J, Sawchenko PE, Vale WW (2001) Identification of urocortin III, an additional member of the corticotropin-releasing factor (CRF) family with high affinity for the CRF2 receptor. Proc Natl Acad Sci U S A 98:7570–7575
Liebsch G, Landgraf R, Engelmann M, Lorscher P, Holsboer F (1999) Differential behavioural effects of chronic infusion of CRH 1 and CRH 2 receptor antisense oligonucleotides into the rat brain. J Psychiatr Res 33:153–163
Markovic D, Punn A, Lehnert H, Grammatopoulos DK (2008) Intracellular mechanisms regulating corticotropin-releasing hormone receptor-2beta endocytosis and interaction with extracellularly regulated kinase 1/2 and p38 mitogen-activated protein kinase signaling cascades. Mol Endocrinol 22:689–706
McEwen BS (2002) The neurobiology and neuroendocrinology of stress. Implications for post-traumatic stress disorder from a basic science perspective. Psychiatr Clin North Am 25(469-494):ix
Muller MB, Holsboer F (2006) Mice with mutations in the HPA-system as models for symptoms of depression. Biol Psychiatry 59:1104–1115
Muller MB, Zimmermann S, Sillaber I, Hagemeyer TP, Deussing JM, Timpl P, Kormann MS, Droste SK, Kuhn R, Reul JM, Holsboer F, Wurst W (2003) Limbic corticotropin-releasing hormone receptor 1 mediates anxiety-related behavior and hormonal adaptation to stress. Nat Neurosci 6:1100–1107
Nemeroff CB, Widerlov E, Bissette G, Walleus H, Karlsson I, Eklund K, Kilts CD, Loosen PT, Vale W (1984) Elevated concentrations of CSF corticotropin-releasing factor-like immunoreactivity in depressed patients. Science 226:1342–1344
Neufeld-Cohen A, Tsoory MM, Evans AK, Getselter D, Gil S, Lowry CA, Vale WW, Chen A (2010a) A triple urocortin knockout mouse model reveals an essential role for urocortins in stress recovery. Proc Natl Acad Sci U S A 107:19020–19025
Neufeld-Cohen A, Evans AK, Getselter D, Spyroglou A, Hill A, Gil S, Tsoory M, Beuschlein F, Lowry CA, Vale W, Chen A (2010b) Urocortin-1 and -2 double-deficient mice show robust anxiolytic phenotype and modified serotonergic activity in anxiety circuits. Mol Psychiatry 15(426-441):339
Neufeld-Cohen A, Kelly PA, Paul ED, Carter RN, Skinner E, Olverman HJ, Vaughan JM, Issler O, Kuperman Y, Lowry CA, Vale WW, Seckl JR, Chen A, Jamieson PM (2012) Chronic activation of corticotropin-releasing factor type 2 receptors reveals a key role for 5-HT1A receptor responsiveness in mediating behavioral and serotonergic responses to stressful challenge. Biol Psychiatry 72:437–447
Perrin MH, Vale WW (1999) Corticotropin releasing factor receptors and their ligand family. Annu NY Acad Sci 885:312–328
Refojo D, Schweizer M, Kuehne C, Ehrenberg S, Thoeringer C, Vogl AM, Dedic N, Schumacher M, von Wolff G, Avrabos C, Touma C, Engblom D, Schutz G, Nave KA, Eder M, Wotjak CT, Sillaber I, Holsboer F, Wurst W, Deussing JM (2011) Glutamatergic and dopaminergic neurons mediate anxiogenic and anxiolytic effects of CRHR1. Science 333:1903–1907
Regev L, Neufeld-Cohen A, Tsoory M, Kuperman Y, Getselter D, Gil S, Chen A (2011) Prolonged and site-specific over-expression of corticotropin-releasing factor reveals differential roles for extended amygdala nuclei in emotional regulation. Mol Psychiatry 16:714–728
Reul JM, Holsboer F (2002) Corticotropin-releasing factor receptors 1 and 2 in anxiety and depression. Curr Opin Pharmacol 2:23–33
Reyes TM, Lewis K, Perrin MH, Kunitake KS, Vaughan J, Arias CA, Hogenesch JB, Gulyas J, Rivier J, Vale WW, Sawchenko PE (2001) Urocortin II: a member of the corticotropin-releasing factor (CRF) neuropeptide family that is selectively bound by type 2 CRF receptors. Proc Natl Acad Sci U S A 98:2843–2848
Ryabinin AE, Tsoory MM, Kozicz T, Thiele TE, Neufeld-Cohen A, Chen A, Lowery-Gionta EG, Giardino WJ, Kaur S (2012) Urocortins: CRF's siblings and their potential role in anxiety, depression and alcohol drinking behavior. Alcohol 46:349–357
Selye H (1955) Stress and disease. Science 122:625–631
Skutella T, Criswell H, Moy S, Probst JC, Breese GR, Jirikowski GF, Holsboer F (1994) Corticotropin-releasing hormone (CRH) antisense oligodeoxynucleotide induces anxiolytic effects in rat. Neuroreport 5:2181–2185
Skutella T, Probst JC, Renner U, Holsboer F, Behl C (1998) Corticotropin-releasing hormone receptor (type I) antisense targeting reduces anxiety. Neuroscience 85:795–805
Smith GW, Aubry JM, Dellu F, Contarino A, Bilezikjian LM, Gold LH, Chen R, Marchuk Y, Hauser C, Bentley CA, Sawchenko PE, Koob GF, Vale W, Lee KF (1998) Corticotropin releasing factor receptor 1-deficient mice display decreased anxiety, impaired stress response, and aberrant neuroendocrine development. Neuron 20:1093–1102
Stenzel-Poore MP, Heinrichs SC, Rivest S, Koob GF, Vale WW (1994) Overproduction of corticotropin-releasing factor in transgenic mice: a genetic model of anxiogenic behavior. J Neurosci 14:2579–2584
Sztainberg Y, Kuperman Y, Tsoory M, Lebow M, Chen A (2010) The anxiolytic effect of environmental enrichment is mediated via amygdalar CRF receptor type 1. Mol Psychiatry 15:905–917
Sztainberg Y, Kuperman Y, Justice N, Chen A (2011) An anxiolytic role for CRF receptor type 1 in the globus pallidus. J Neurosci 31:17416–17424
Timpl P, Spanagel R, Sillaber I, Kresse A, Reul JM, Stalla GK, Blanquet V, Steckler T, Holsboer F, Wurst W (1998) Impaired stress response and reduced anxiety in mice lacking a functional corticotropin-releasing hormone receptor 1. Nat Genet 19:162–166
Valdez GR, Inoue K, Koob GF, Rivier J, Vale W, Zorrilla EP (2002) Human urocortin II: mild locomotor suppressive and delayed anxiolytic-like effects of a novel corticotropin-releasing factor related peptide. Brain Res 943:142–150
Vale W, Spiess J, Rivier C, Rivier J (1981) Characterization of a 41-residue ovine hypothalamic peptide that stimulates secretion of corticotropin and beta-endorphin. Science 213:1394–1397
van Gaalen MM, Stenzel-Poore MP, Holsboer F, Steckler T (2002) Effects of transgenic overproduction of CRH on anxiety-like behaviour. Eur J Neurosci 15:2007–2015
Van Pett K, Viau V, Bittencourt JC, Chan RK, Li HY, Arias C, Prins GS, Perrin M, Vale W, Sawchenko PE (2000) Distribution of mRNAs encoding CRF receptors in brain and pituitary of rat and mouse. J Comp Neurol 428:191–212
Xu L, Bloem B, Gaszner B, Roubos EW, Kozicz T (2010) Stress-related changes in the activity of cocaineand amphetamine-regulated transcript and nesfatin neurons in the midbrain non-preganglionic Edinger-Westphal nucleus in the rat. Neuroscience 170:478–488
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, duplication, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, a link is provided to the Creative Commons license and any changes made are indicated.
The images or other third party material in this chapter are included in the work's Creative Commons license, unless indicated otherwise in the credit line; if such material is not included in the work's Creative Commons license and the respective action is not permitted by statutory regulation, users will need to obtain permission from the license holder to duplicate, adapt or reproduce the material.
Copyright information
© 2016 The Author(s)
About this chapter
Cite this chapter
Chen, A. (2016). Genetic Dissection of the Neuroendocrine and Behavioral Responses to Stressful Challenges. In: Pfaff, D., Christen, Y. (eds) Stem Cells in Neuroendocrinology. Research and Perspectives in Endocrine Interactions. Springer, Cham. https://doi.org/10.1007/978-3-319-41603-8_6
Download citation
DOI: https://doi.org/10.1007/978-3-319-41603-8_6
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-41602-1
Online ISBN: 978-3-319-41603-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)