
Abstract
Structural variations (SVs) are one of the genetic markers in the human genome and detecting them by using ultra high-throughput genome sequencing techniques has vital significance for genetic and evolutionary studies. In recent decades, bioinformatics techniques based on next-generation sequencing (NGS) have become a research focus owing to its high resolution and accuracy. Moreover, NGS devices are becoming cheaper. In this survey, we will summarize current methods based on next-generation sequencing algorithms for SVs detection and discuss the impacts of them. We also analyze the problems and give an outlook for the future research directions.
You have full access to this open access chapter, Download conference paper PDF
Similar content being viewed by others


Keywords
1 Introduction
In recent years, studies have shown that a diverse array of genetic variation occur in the human genome. Studies on these variations will not only help to reveal a large number of complex diseases and to find the genetic mechanism associated with individuals but also speed up the pace of personalized medicine. The genetic variation is simply classified into two kinds: single nucleotide polymorphism (SNP) and structural variation (SV). Since the last quarter of the twentieth century, SNP has long been regarded as the most common genetic variation in human genomic and widely studied [1], and identified by traditional PCR-based methods. On the other hand, several types of genetic variations, including insertions or deletions and copy number variations (CNVs) are also widespread in human genomes [2], which have more significance in several areas of biology such as the studies of obesity diseases [3], cancer genome [4–6], and molecular evolution [7, 8]. Especially, the discovery of CNVs, present in the human genome, has changed dramatically our focus on structural variations and phenotype association with diseases. Recent review has revealed that SVs, especially CNVs have extended along over 1000 genes; in addition to, CNVs often encompass a large proportion of the genome, to a great extent than SNPs, ranging from ~ 12 % of the human reference genome [9, 10]. This proves that CNVs is more responsible for genetic diversity and evolution between human populations. CNVs are just one class of SVs, which are defined in terms of the size of insertions and deletions (> 1 kb). The recognized types of SVs (are shown in Fig. 1) contain indels, inversions, copy-number variations (CNVs) and translocations. Term “indels” means that the amount of inserted or deleted genes is smaller than 1 kb. If the amount is larger than 1 kb, these types of structural variations are referred to as copy number variations, that is, the large genomic segment of duplications or deletions.


Several types of structural variations [17].
Until recently, the methods used to detect SVs mainly have microarray-based technology [11], the fluorescent hybrid technology [12], Multiple PCR technology [13] and Sequencing-based technology [14]. The earliest methods are based on micro-array platforms such as the oligonucleotide-based microarray comparative genomic hybridization (array-CGH) [15] and bacterial artificial chromosome (BAC) [16]. Although these computational approaches based on array data, were successfully used to identify CNVs and other types of SVs like translocations. They also have some limitations. For example, Array-CGH method cannot detect chromosomal translocations or inversions owing to a limited dynamic range; furthermore, the prediction of breakpoint resolution is controlled by the density of the array. Sequencing-based method is emerging recently like Sanger sequencing, which was in place to identify genomic variants in the human genome, while the objectionable feature of Sanger sequencing is too expensive and time-consuming. Compared with the conventional microarray-based method and Sanger sequencing, NGS has an overriding strength on cost-effective and high-throughput. It has driven the development of NGS-based technologies for detecting genetic variations in the human genome.
There are many NGS-based detection algorithms for SVs springing up, have enabled extensive SVs detection. It basically contains the following mainstream algorithms: PEM-based method, read-depth method, split-read method and sequence assembly method. The split-read method generally was combined with PEM-based method and used in various detecting SVs tools. In general, the identification and detecting for SVs involve these steps: Firstly, aligning the short sequencing reads to a given reference genome; then finding the interest regions that different from the reference which is likely being a potential SV; finally, verifying these variations by some strategies.
In this survey, we will describe currently algorithms for detecting SVs by using next-generation sequencing, which have roughly classified into three types. And then, we will discuss the strength and the weakness of these methods, lastly we will provide an outlook for the future research development and make a summary of this article.
2 Algorithms for Structural Variations Detection
Paired-end reads, and mate-pair reads are two distinct reads generated by sequencing-based technologies and two disparate strategies at a known distance. The difference between them is that the length of fragments of the paired-end reads is shorter than those of mate pairs. The length of these read is restricted by the space of the slide, so we can distinguish according to the length of them. The first strategy is the circularization of the DNA segments within the sequencing process; the generated reads with a long insert size are better for detecting a large SV. Another way is obtained from both ends of a segment of DNA, whose sizes are approximately known; that is, the insert size. This method can obtain a high resolution during detecting a small SV. In this survey, these two reads are called “pairs-read” unified.
PEM-based methods identify SV breakpoints by aligning the short paired-end reads to the reference genome to find the ‘discordant’ with the reference genome, which is probably being one class of SVs. The paired-end reads comes from a sequencing library which contains plenty of fragments with a known length. The ‘discordant’ paired-end reads are either the expected distance or orientation divergence. In the aligning or the examining strategy, a mapping signature can be produced, which indicate the presence of SVs. So we should discuss the mapping signature first.
2.1 Signatures Based on PEM
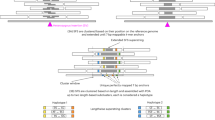
The earliest two signatures were insertions and deletions (Fig. 2a and b). Figure 2 shows two types of read signatures; one is the paired-end reads, and the other is split-reads. Since most of the current methods use the fusion of these two reads to detect SVs, we simplify to categorize it into the PEM-based methods.


Interpretation of PEM signatures [18].
Term “ref” means that an original genome. A ref case often is used as a control genome; similarly, term “donor” represents a group genome, which comes from the sequencing process. An inversion pairs-read, spanning either of breakpoint of an inversion, will map with an orientation opposite to the reference (Fig. 2c). Figure 2d is the most obvious discordant paired-end read, which occurs within a chromosome. In a case that two mapped reads beyond its expected distance, and the other read of both appear on another chromosome (Fig. 2e). Note that cases from Fig. 2e to Fig. 2i are more complicate than the simple deletions and insertions.
Tandem duplication is the ordinary case of SVs; these pairs-read were linked from the end of the duplication part to its beginning (Fig. 2f). A linking case is that the two distant reads of the ref genome are very close lying on the donor, in other words, comparing with the ref the orientation and the order of these pairs-read remain un-changed, while the distance between them after mapping is fewer than the distance on the ref genome (Fig. 2g). A somewhat akin to case Fig. 2g while more complex situation is that a distant mobile element or segment was inserted into a donor genome, resulting in a linked insertion and the distance between pairs-read closer than before (Fig. 2h). Sometimes a long segment was embedded in a donor genome, longer than the insert size; a hanging insertion signature is formed with one read unmapped (Fig. 2i). A lengthy piece was inserted into the ref, resulting in a hanging deletion signature akin to Fig. 2i with another read of both pairs read unmapped on the donor genome (Fig. 2j).
Split-read mapping also has multiple types of SVs. For an insertion, the prefix and suffix of split-read are mapped to a neighbor location, whereas the intermediate region is an inserted segment (Fig. 2l). In a case of deletion, the prefix and suffix of split-read are mapping around the breakpoint neighbor with each other (Fig. 2k). An interspersed duplication is the case that a segment from another location of the donor genome shifts to one end of split-read and link (Fig. 2o). A similar case is the mobile-element insertion (Fig. 2m). And contrary to Fig. 2m, the orientation of mobile-element is opposite to its original orientation in a reference genome (Fig. 2n). A tandem duplication case is similar to the case described in Fig. 2f (Fig. 2p).
2.2 Methods Using PEM
In the process of technology development, many algorithms and tools based-on PEM have been proposed and designed for SVs (Table 1). There are two classes of strategies were utilized to detect SVs, the distribution-based method and the clustering-based method. The main step of clustering-based method is to label the concordant and discordant pairs, and next to call the underlying SVs by using current clustering approaches. Only if the orientation of pairs-read is same as the reference genome and the distance of pairs-read match the expected distance, it can be defined as the concordant, otherwise is discordant. For example, Mateo et al. [19] used a SVM model to cluster the local pattern of mapping read and after that predicted the position of SVs. Korbel et al. [20] and Tuzun et al. [21] first labeled the signature of PEM and then clustered the discordant together, only if the number of clusters is higher than a specified value, it can be identified as potential SVs.
These methods are related to two parameters: the number of standard deviations which can determine whether a pairs-read is discordant and the minimum number of pairs-read to define a cluster. These factors are interconnected and associated to the coverage, in other words, the coverage and the number of pairs-read or the number of standard deviations is an inverse relationship.
One of the weaknesses of the clustering approach is ignoring the case that many multiple mapping sites can match the pairs-read, so detecting the signatures within the repeat regions in the genome is a tough work. However, the region of repeat is strongly associated with the duplication read, so various methods were designed to address this issue. The adopted optimization processes are to select a ‘good’ cluster with the max support for each pairs-read.
Another deficiency is the clustering method used an unchanged critical value for the number of standard deviations after a signature of PEM is considered as a discordant. When the threshold of discordance changes mapped distance of PEM signatures from 2 s.d to 1 s.d, spanning the same breakpoint, there are no clusters be set up. While Lee et al. [22] successfully solves this problem by proposing a distribution-based method, which allows the distribution of all the map-ping around a known breakpoint to be visualizing. If the mapping distribution corresponds with the distribution of expected insert size, while the orientation is opposite, then an indel cluster was set up. Despite this method is good at detecting much smaller indels than the clustering-based method, it also leads to other problems such as the rare variants appeared between homozygous and heterozygous, the power of detecting is not always reliable.
2.3 Signatures Based on Depth of Coverage
Unlike the signature of PEM, there are only two cases happened on the based-depth of coverage. One case is the copy-number duplication; that is, the frequency of read fragment in the donor in some region is higher than in the ref genome; another situation is the copy-number deletion. The density in this region is lower than in the ref. These two cases are shown in the Fig. 3.


The illustrations of DOC signatures [31]
2.4 Methods Using Read-Depth
Most of read-depth methods usually partition a genome into the tag count windows and non-overlapping windows. The general procedure of these methods is to determine the region which tags counts are notably different from the normal counts in the genome. These strategies have got an admirable accuracy in detecting large CNVs. Despite the strength of sensitivity and specificity of these methods arising with the size of CNVs, they only customized for the dosage changing SVs; in other words, the range of these methods can be detected not include translocations and inversions, just CNVs and indels.
There are numerous investigations on the based-depth of coverage, and a number of tools have been developed (Table 2). For instance, Xie et al. [32] proposed one method to segregate the genome of a small fixed size window and then find out that the window of the case genome which notably distinct from the reference genome. The resolution of these methods is related to the size of the window. That is to say, too small will weaken the detecting power; too large may lose the resolution.
The deficiency of these methods is the factor resulting in abnormal tag count is uncertainty. For instance, the sequencing error rate of NGS such as the poor or rich region of GC is lower than the average GC; it may cause a potential loss or gain of read; moreover, a read mapped by mistake will make the discovery of signature of DOC more complicate.
2.5 Methods Using Sequence Assembly
As the signatures are more and more complicate, people are attaching much importance to the local de novo. Recently, local de novo has been rapid development, just as its name implies, it just finds out the local region which differs from the reference genome, and then will be cut out to reassemble from the set of reads. Comparing the de novo assembly by using all the reads, local de novo methods have enabled the computational time reduced greatly. There also have been several types of SVs, and we will give an illustration in the following (Fig. 4).


The illustrations of assembly signature [31]
Given a case that a larger insertion reads lies on the donor genome, the number of matching bases is fewer, or a larger deletion occurs on the donor genome, the mapping signature will more complicate as we described on the signature of PEM. As we point out, PEM-based method is difficult in detecting these cases, since the matched bases are fewer and no enough evidence to use for detecting. Although in later some “soft clip” of PEM-based methods can tackle with this problem, but result is inefficient. So we should consider another alternative way to recover them for detecting. With the development of next-generation sequencing, some reassembly methods for variation detection are emerging to become a popular alternative method such as the micro-assembly methods. Its main idea is that to perform localized de novo, to detect the region encompasses potential SVs and perform assembly, finally to remap a contig, created by assembly from the reads set, to the reference genome. Table 3 shows the recent developed tools that based on local de novo. In this table, signal ‘*’ means that there are no published literature about this tool, but we can learn more detail on its website on guide section.
These methods are roughly similar to each other except the way to handle the cycle in the graph. For example, Scalpel has a high accuracy in detecting repeat region via utilizing a self-tuning k-mer size approach, and a deeper analysis of the rich repeat region used by Scalpel can avoid the cycle path generated. GATK Haplotype-Caller is akin to Scalpel to improve the accuracy of indels detection through larger the k-mer size gradually, while the detected accuracy is weaker than Scalpel on account of ignoring the approximately matching repeat sequences. SOAPindel uses another strategy to form a non-cycle path from unused reads by reducing k-mer sizes. The k-mer size of TIGRA can be specified by the user, and this tool is just designed for breakpoint detection other than finding out repeat regions. ABRA has the same technique processing with Scalpel, which utilizing an increasing k-mer size to generate a non-repeat path except the k-mer reached the upper bound, while the scope of the assembly is no more than 2 kb. The scope of Platypus can assemble is 1.5 kb, smaller than ABRA. Different from the above-mentioned methods, Bubbleparse adopts a Cortex framework to implement indels detection, but the result of a high false-positive rate is not satisfactory.
3 Discussion
Recent studies have shown that SVs are prevalent as SNPs in the genome. SV has become a hot area of biomedical researchers, and the precise identification of SV will accelerate the research of mechanisms related to human genetics or complex diseases. It is virtually certain that some new algorithms and experimental schemes for detecting SV will continuously arise in the future. The NGS-based method has provided numerous opportunities to mutation detection. Although these methods described above have some strength, they also have their scope of application. For instance, read-depth methods can achieve good accuracy in detecting CNVs and indels, but the power of detecting dosage-unchanged mutation is poor. PEM-based methods may be difficult in looking for the precise position of breakpoint; furthermore, its performance is dependent on the completeness of the pairs-read. For example, if a larger insertion or deletion emerges around the breakpoint and the matched bases are fewer, the accuracy of SVs detection will decrease. The PEM-based method and read-depth method have their respective strengths, so the combination of them may have a more satisfactory result. So we can consider the fusion of multiple methods or strategies, because a single method or strategy is too plain for detecting composite variation, and the fusion can utilize more information. Now some tools also comprehensively use two or more strategies such as algorithm in BreakDancer which has combined clustering-based strategy and distribution-based method, and SVseq has been fused with the PEM-based method and split-read method. Integrating various detection algorithms is becoming a popular strand for SVs.
Another difficulty of SV detection is that the optimal value of the parameter is hard to ascertain. For instance, the PEM-based method has two parameters, the standard deviation to determine whether the pairs-read is discordant and the minimum number of the pairs-read. The first parameter is associated with the distance mean, which is a fixed value and too reliant on the experience. It is vital that to make the parameter self-tuning based on some adaptive technology.
With the development of sequencing techniques, the structural variation algorithm is suffering from the problem that how to adapt to the new characteristics of the sequenced data. Regardless of the fact that short data generated by NGS-based technology can well be utilized for detecting SVs, some large insertions or deletions cases also happen in the genome. Moreover, other technologies such as Sanger and Roche have generated a long read data. To use a long read is becoming a feasible strategy on many platforms in the future. So the related research is also required.
References
Altshuler, D.: A haplotype map of the human genome. J. Nat. 437, 1299–1320 (2005)
Check, E.: Human genome: patchwork people. J. Nat. 437, 1084–1086 (2005)
McCarroll, S.A., Altshuler, D.M.: Copy-number variation and association studies of human disease. J. Nat. Genet. 39, S37 (2007)
Parkin, D.M., Bray, F., Ferlay, J., Pisani, P.: Global Cancer Statistics, 2002. J CA: A Cancer J. Clin. 55, 74–108 (2005)
Parkin, D.M., Pisani, P., Ferlay, J.: Global cancer statistics. J CA: A Cancer J. Clin. 49, 33–64 (1999)
Jemal, A., Siegel, R., Ward, E., Murray, T., Xu, J., Smigal, C., Thun, M.J.: Cancer statistics 2006. J CA: A Cancer J. Clin. 56, 106–130 (2006)
Dover, G.A., Linares, A.R., Bowen, T., Hancock, J.M.: Detection and quantification of concerted evolution and molecular drive. J. Methods Enzymol. 224, 525–541 (1993)
Nei, M.: Human evolution at the molecular level. J. Popul. Genet. Mol. Evol. (Mishima, 1984), pp. 41–64 (1985)
Stankiewicz, P., Lupski, J.R.: Structural variation in the human genome and its role in disease. J. Annu. Rev. Med. 61, 437–455 (2010)
Bickhart, D.M., Liu, G.E.: The challenges and importance of structural variation detection in livestock. J. Front. Genet. 5, 37 (2014)
Durbin, R.M., Abecasis, G.R., Altshuler, D.L., Auton, A., Brooks, L.D., Gibbs, R.A., Hurles, M.E., McVean, G.A.: A map of human genome variation from population-scale sequencing. J. Nat. 467, 1061–1073 (2010)
Bauman, J.G.J., Wiegant, J., Borst, P., van Duijn, P.: A new method for fluorescence microscopical localization of specific DNA sequences by in situ hybridization of fluorochromelabelled RNA. J. Exp Cell Res. 128, 485–490 (1980)
Cheng, Z., Sharp, A.J., Eichler, E.E.: Structural variation of the human genome. J Annu. Rev. Genomics Hum. Genet. 7, 477 (2006)
Redon, R., Ishikawa, S., Fitch, K.R., Feuk, L., Perry, G.H., Andrews, T.D., Fiegler, H., Shapero, M.H., Carson, A.R., Chen, W.: Global variation in copy number in the human genome. J. Nat. 444, 444–454 (2006)
Carter, N.P.: Methods and strategies for analyzing copy number variation using DNA microarrays. J. Nat. Genet. 39, S16–S21 (2007)
Ylstra, B., van den, IJssel, P., Carvalho, B., Brakenhoff, R.H., Meijer, G.A.: BAC to the future! or oligonucleotides: a perspective for micro array comparative genomic hybridization (array CGH). J Nucleic Acids Res. 34, 445–450 (2006)
Yong, L: Survey on structural variants detection algorithms for next generation sequencing technology. J. Appl. Res. Comput. 31(2), 328–332 (2014)
Medvedev, P., Stanciu, M., Brudno, M.: Computational methods for discovering structural variation with next-generation sequencing. J. Nat Meth. 6, S13–S20 (2009)
Chiara, M., Horner, D.S., Pesole, G., Chiara, M., Horner, D.S.: SVM2: an improved paired-end-based tool for the detection of small genomic structural variations using high-throughput single-genome resequencing data. J. Nucleic Acids Res. 40, 727–739 (2012)
Korbel, J.O., Abyzov, A., Mu, X.J., Carriero, N., Cayting, P., Zhang, Z., Snyder, M., Gerstein, M.B.: PEMer: a computational framework with simulation-based error models for inferring genomic structural variants from massive paired-end sequencing data. J. Genome Biol. 10, R23–R23 (2009)
Tuzun, E., Sharp, A.J., Bailey, J.A., Kaul, R., Morrison, V.A., Pertz, L.M., Haugen, E., Hayden, H., Albertson, D., Pinkel, D.: Fine-scale structural variation of the human genome. J. Nat. Genet. 37, 727–732 (2005)
Lee, S., Hormozdiari, F., Alkan, C., Brudno, M.: MoDIL: detecting small indels from clone-end sequencing with mixtures of distributions. J. Nat. Methods. 6, 473–474 (2009)
Hormozdiari, F., Hajirasouliha, I., Dao, P., Hach, F., Yorukoglu, D., Alkan, C., Eichler, E.E., Sahinalp, S.C.: Next-generation variationHunter: combinatorial algorithms for transposon insertion discovery. J. Bioinform. 26, i350–i357 (2010)
Chen, K., Wallis, J.W., McLellan, M.D., Larson, D.E., Kalicki, J.M., Pohl, C.S., McGrath, S.D., Wendl, M.C., Zhang, Q., Locke, D.P.: BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. J. Nat Methods. 6, 677–681 (2009)
Zeitouni, B., Boeva, V., Janoueix-Lerosey, I., Loeillet, S., Legoix-né, P., Nicolas, A., Delattre, O., Barillot, E.: SVDetect: a tool to identify genomic structural variations from paired-end and mate-pair sequencing data. J. Bioinform. 26, 1895–1896 (2010)
Hormozdiari, F., Hajirasouliha, I., McPherson, A., Eichler, E.E., Sahinalp, S.C.: Simultaneous structural variation discovery among multiple paired-end sequenced genomes. J. Genome Res. 21, 2203–2212 (2011)
Handsaker, R.E., Korn, J.M., Nemesh, J., McCarroll, S.A.: Discovery and genotyping of genome structural polymorphism by sequencing on a population scale. J. Nat. Genet. 43, 269–276 (2011)
Qi, J., Zhao, F.: inGAP-sv: a novel scheme to identify and visualize structural variation from paired end mapping data. J. Nucleic Acids Res. 39, W567–W575 (2011)
Zhang, J., Wu, Y.: SVseq: an approach for detecting exact breakpoints of deletions with low-coverage sequence data. J. Bioinform. 27, 3228–3234 (2011)
Ye, K., Schulz, M.H., Long, Q., Apweiler, R., Ning, Z.: Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. J. Bioinform. 25, 2865–2871 (2009)
Public Library of Bioinformatics. http://www.plob.org/2014/03/08/6794.html
Xie, C.: Martti T Tammi: CNV-seq, a new method to detect copy number variation using high-throughput sequencing. J. BMC Bioinform. 10, 883–890 (2009)
Chiang, D.Y., Getz, G., Jaffe, D.B., O’Kelly, M.J., Zhao, X., Carter, S.L., Russ, C., Nusbaum, C., Meyerson, M., Lander, E.S.: High-resolution mapping of copy-number alterations with massively parallel sequencing. J. Nat Methods. 6, 99–103 (2009)
Miller, C.A., Hampton, O., Coarfa, C., Milosavljevic, A.: readdepth: a parallel r package for detecting copy number alterations from short sequencing reads. J. PLOS ONE. 6, e16327 (2011)
Yoon, S., Xuan, Z., Makarov, V., Ye, K., Sebat, J.: Sensitive and accurate detection of copy number variants using read depth of coverage. J. Genome Res. 19, 1586–1592 (2009)
Li, J., Lupat, R., Amarasinghe, K.C., Thompson, E.R., Doyle, M.A., Ryland, G.L., Tothill, R.W., Halgamuge, S.K., Campbell, I.G., Gorringe, K.L.: CONTRA: copy number analysis for targeted resequencing. J. Bioinform. 28(7), 1307–1313 (2012)
McKernan, K.J., Peckham, H.E., Costa, G.L., McLaughlin, S.F., Fu, Y., Tsung, E.F., Clouser, C.R., Duncan, C., Ichikawa, J.K., Lee, C.C.: Sequence and structural variation in a human genome uncovered by short-read, massively parallel ligation sequencing using two-base encoding. J. Genome Res. 19, 1527–1541 (2009)
Abyzov, A., Urban, A.E., Snyder, M., Gerstein, M.: CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. J. Genome Res. 21, 974–984 (2011)
Narzisi, G., Schatz, M.C.: The challenge of small-scale repeats for indel discovery. J. Front Bioeng Biotechnol. 3, 8 (2015)
Narzisi, G., O’Rawe, J.A., Iossifov, I., Fang, H., Lee, Y.H., Wang, Z., Wu, Y., Lyon, G.J., Wigler, M., Schatz, M.C.: Accurate de novo and transmitted indel detection in exome-capture data using microassembly. J. Nat Methods 11, 1033–1036 (2014)
Li, S., Li, R., Li, H., Lu, J., Li, Y., Bolund, L., Schierup, M.H., Wang, J.: SOAPindel: efficient identification of indels from short paired reads. J. Genome Res. 23, 195–200 (2013)
Rimmer, A., Phan, H., Mathieson, I., Iqbal, Z., Twigg, S.R., Consortium, W.G.S., Wilkie, A.O., McVean, G.: Integrating mapping-, assembly- and haplotype-based approaches for calling variants in clinical sequencing applications. J. Nat Genet. 46, 912–918 (2014)
Mose, L.E., Wilkerson, M.D., Hayes, D.N., Perou, C.M., Parker, J.S.: ABRA: improved coding indel detection via assembly based re-alignment. J. Bioinform. 30, 2813–2815 (2014)
Chen, K., Chen, L., Fan, X., Wallis, J., Ding, L., Weinstock, G.: TIGRA: a targeted iterative graph routing assembler for breakpoint assembly. J. Genome Res. 24, 310–317 (2014)
Leggett, R.M., MacLean, D.: Reference-free SNP detection: dealing with the data deluge. J. BMC Genomics. 15, 246–253 (2014)
Acknowledgements
This work was supported by Shenzhen Municipal Science and Technology Innovation Council (Grant No. CXZZ20140904154910774, Grant No.JCYJ20140417172417174, Grant No.JCYJ20140904154645958, Grant No.JCYJ20130329151843309) and China Postdoctoral Science Foundation funded project (Grant No.2014M560264).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this paper
Cite this paper
Wei, ZL. (2015). Recent Progress of Structural Variations Detection Algorithms Based on Next-Generation Sequencing: A Survey. In: Zhang, YJ. (eds) Image and Graphics. Lecture Notes in Computer Science(), vol 9219. Springer, Cham. https://doi.org/10.1007/978-3-319-21969-1_13
Download citation
DOI: https://doi.org/10.1007/978-3-319-21969-1_13
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-21968-4
Online ISBN: 978-3-319-21969-1
eBook Packages: Computer ScienceComputer Science (R0)
