
Abstract
Blood Disorders covers disorders of red blood cells, white blood cells, platelets, and coagulation including congenital and acquired disorders. Neoplastic Disorders covers both Hematopoietic and Solid Tumors with information on epidemiology, clinical presentation, diagnosis, and treatment of these disorders.
You have full access to this open access chapter, Download chapter PDF
Abbreviations
- DIC:
-
disseminated intravascular coagulation
- LCH:
-
Langerhans cell histiocytosis
- MCV:
-
mean cell volume
- TIBC:
-
total iron binding capacity
- RBCs:
-
red blood cells
- WBCS:
-
white blood cells
- SLE:
-
systemic lupus erythematosus
- RA:
-
rheumatoid arthritis
- IL:
-
interleukin
- TNF:
-
tumor necrosis factor
- PCR:
-
polymerase chain reaction
- PNH:
-
paroxysmal nocturnal hemoglobinuria
- SS:
-
homozygous sickle cell genes
- SC:
-
heterozygous sickle cell and C genes
- CXR:
-
chest x-ray
- ACS:
-
acute chest syndrome
- PK:
-
pyruvate kinase
- EBV:
-
Epstein-Barr virus
- SDS:
-
-
- GCSF:
-
granulocyte colony stimulating factor
- HSM:
-
hepatosplenomegaly
- MDS:
-
myelodysplastic syndrome
- AML:
-
acute myelogenous leukemia
- ASD:
-
atrial septal defect
- VSD:
-
ventricular septal defect
- PDA:
-
patent ductus arteriosus
- TOF:
-
tetralogy of Fallot
- CoA:
-
coarctation of the aorta
- ITP:
-
idiopathic thrombocytopenic purpura
- HUS:
-
hemolytic uremic syndrome
- MPV:
-
mean platelet volume
- IVIg:
-
intravenous immunoglobulin
- DDAVP:
-
desmopressin
- VWD:
-
von Willebrand disease
- PTT:
-
partial thromboplastin time
- GI:
-
gastrointestinal
- CMP:
-
complete metabolic panel
- ESR:
-
erythrocyte sedimentation rate
- CBC:
-
complete blood count
- JPA:
-
juvenile pilocytic astrocytoma
- CNS:
-
central nervous system
- US:
-
ultrasound
- KUB:
-
kidney, ureter, and bladder x-ray
- U/A:
-
urinalysis
Blood Disorders
Blood disorders generally fall into four categories:
-
Red cell disorders
-
Anemia
-
Erythrocytosis
-
-
White cell disorders
-
Neutropenia
-
Abnormal white cells
-
-
Platelet disorders
-
Thrombocytopenia
-
Abnormal platelets
-
-
Coagulation disorders
Red Cell Disorders
Anemia
-
Incidence of anemia in childhood (Fig. 1)
-
Iron deficiency anemia (IDA), 60–70 %
-
Hemolytic anemia, 15–20 %
-
Hypoproliferative anemia, 10 %
-
Maturation abnormalities, 7–8 %
Fig. 1 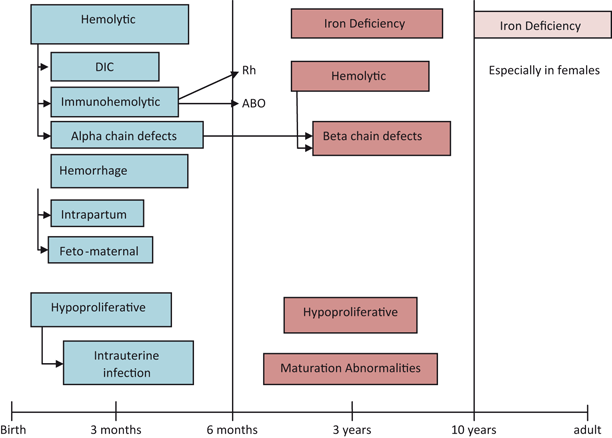
Prevalence of anemia in different age groups
-

Anemia in the Newborn (Fig. 2)
Hemolysis
Congenital
-
Hemoglobinopathies
-
Chain defects more common
-
-
Red cell membrane defects
-
Hereditary spherocytosis
-
Hereditary elliptocytosis
-
Hereditary stomatocytosis
-
-
Red cell enzyme defects
-
G6PD
-
PK
-
Acquired
-
Nonimmune
-
Vitamin E deficiency
-
◦ Hemolytic anemia
-
◦ Edema
-
◦ Thrombocytosis
-
-
Infantile pyknocytosis
-
-
Immune
-
ABO
-
RH
-
-
Infections
-
DIC
-
Bacterial
-
Viral
-
Blood loss
-
Prenatal
-
Fetomaternal
-
Twin–twin transfusion
-
-
Placental
-
Umbilical
-
Postnatal
-
Plasma factor deficiencies
-
Platelets––deficiency or dysfunction
-
Abnormal platelet function
-
Decreased red cell production
-
Pure red cell aplasia
-
Anemia of prematurity
-
Early
-
Late
-
Iatrogenic
-
-
Infection
-
Infiltration
-
Congenital leukemia
-
Neuroblastoma
-
LCH
-
Osteopetrosis
Fig. 2 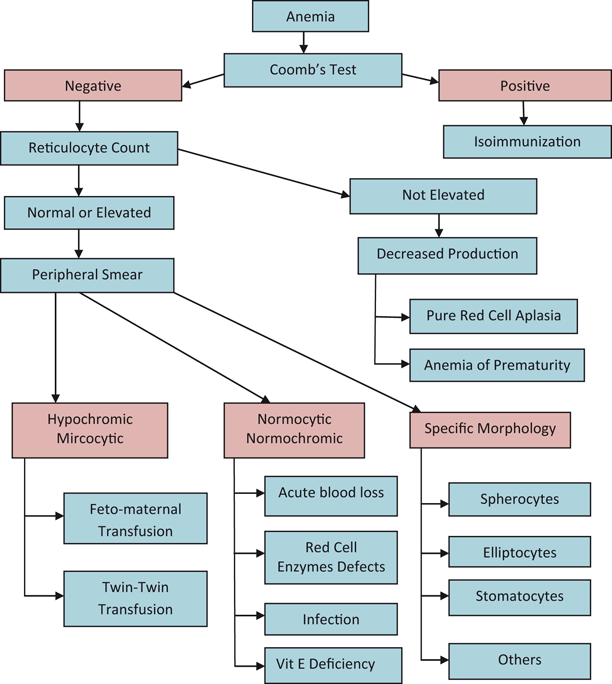
An approach to the diagnosis of anemia in the newborn infant
-

Approach to Diagnosis of Anemia in Older Child


Microcytic Anemia
Iron Deficiency Anemia (IDA)
-
Most common hematologic disease in infancy and childhood
Etiology
-
Nutritional
-
Low birth weight
-
Rapid growth
-
Consumption of large amount of cow’s milk (> 32 oz whole cow’s milk/day)
-
-
Impaired absorption
-
Primary iron deficiency
-
Malabsorption syndrome
-
-
Blood loss
-
Gastrointestinal
-
Primary iron deficiency
-
Cow’s milk allergy or exudative enteropathy
-
Lesions: Meckel’s, vascular malformations
-
Parasites: hookworms
-
Genitourinary
-
Menstrual
-
Hemoglobinuria
-
Hemosiderinuria
-
Pulmonary
-
Goodpasture’s syndrome
-
Pulmonary hemosiderosis
-
Clinical Presentation
-
Pallor
-
Pagophagia: desire to eat unusual substance as ice, dirt, etc.
-
If Hgb level falls < 5 g/dL
-
Irritability
-
Anorexia
-
Tachycardia
-
Systolic murmur
-
Laboratory
-
Low serum ferritin (depleted iron stores)
-
Low serum iron—may fluctuate
-
Increased TIBC (serum transferrin)
-
RBCs become more microcytic, hypochromic, and increased poikilocytosis as disease progresses (Fig. 5)
Fig. 5 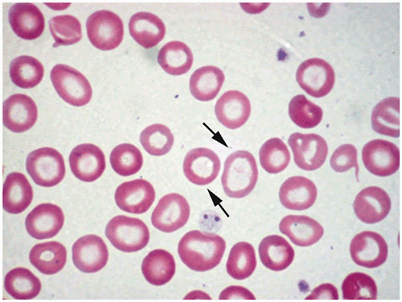
Peripheral blood smear example of hypochromic/microcytic anemia. Notice the variability in the sizes of red blood cells. The arrows point to hypochromic erythrocytes with large central hollow. (Courtesy of Dr. Nawar Hakim)
-
Increased RBC distribution width (RDW)
-
Normal WBCs
-
Thrombocytosis; occasionally marked (600,000–1 million/mm3)
-
Low reticulocyte count
-
Mentzer index > 13 (MCV/RBCs)

Treatment
-
Response to iron therapy is diagnostic and therapeutic.
-
Oral administration of ferrous salts at dose of 4–6 mg/kg of elemental iron in three divided doses .
-
Very inexpensive.
-
Downsides; taste, Gl irritability, and constipation (more water and fiber can solve this problem).
-
-
Rapid correction of anemia with transfusion may precipitate heart failure.
-
In severely anemic children (< 4 gm/dl) transfusions can be administered at a very slow rate (2–3 ml/kg).
-
If there is evidence of heart failure present, a modified exchange transfusion using fresh PRBCs can be considered.
-
Changes after treatment with iron.
-
Within 12–24 h: irritability decreases, increased appetite.
-
36–48 h: initial bone marrow response with erythroid hyperplasia.
-
48–72 h: reticulocytosis, peaking at 5–7 days.
-
1–3 months: repletion of stores.
-
-
Hgb may increase by 0.5 g/dl/day.
-
Iron therapy should be continued for at least 2 months after the Hgb normalizes to replenish iron stores.
-
Limit cow’s milk to less than 500 cc/day .
Anemia of Chronic Disease
Associations
-
Chronic systemic diseases
-
Chronic inflammatory process, e.g., SLE, RA
-
Chronic pyogenic infection
Etiology
-
Release of inflammatory cytokines: IL-6,IL-1,TNF
-
Hepcidin released from the liver decreases intestinal iron absorption, also block release of iron from the macrophages
Laboratory
-
Hgb concentration usually 6–9 g/dL
-
Normal-to-low MCV
-
Often normochromic anemia with progression to hypochromia
-
Low serum iron
-
Normal-to-low TIBC
-
Elevated serum ferritin
Treatment
-
Treatment of the cause
-
Recombinant EPO may increase the Hgb level and improve well-being in patients with cancer
Lead Poisoning
-
High serum lead level
-
Markedly elevated free erythrocyte protoporphyrin
-
Basophilic stippling of RBCs
-
Ringed sideroblasts in bone marrow
Thalassemias
Alpha Thalassemia
-
Healthy individuals have 4 alpha globin genes, 2 on each chromosome 16
-
Alpha globin production is reduced to absent
-
Seen more frequently in those of southeast Asian and African ancestry
-
Diagnosis: clinically or with alpha globin chain analysis
-
Excess beta chains lead to beta 4 chains (Hemoglobin H, HbH)
-
Excess gamma chains lead to gamma 4 chains (Hemoglobin Barts, Hb Barts)
Alpha Thalassemia Syndromes
Silent trait
-
Deletion or dysfunction of one gene
-
Asymptomatic
-
1–2 % Hb Barts on neonatal electrophoresis
-
Normal Hgb electrophoresis
Alpha thalassemia trait
-
Deletion or dysfunction of two genes
-
Mild hypochromic microcytic anemia
-
3–10 % Hb Barts on neonatal electrophoresis
-
Laboratory
-
Mentzer Index < 13
-
Hgb > 9 g/dl
-
Normal Hgb electrophoresis
-
Often misdiagnosed as IDA
-
Hemoglobin H disease
-
Deletion of three genes
-
Mild-to-moderate hypochromic microcytic anemia
-
Splenomegaly
-
Jaundice
-
Cholelithiasis (pigment stones)
-
Anemia exaggerated by infection, pregnancy, exposure to oxidizing drugs
-
> 25 % Hgb Barts on neonatal electrophoresis
Alpha thalassemia major
-
Deletion of four genes
-
Fetal hydrops-fatal disease
-
Predominant Hb Barts
Beta Thalassemia
-
Healthy individuals have 2 beta globin genes, 1 on each chromosome 11
-
Beta globin production is reduced to absent
-
Multiple possible genetic mutations or deletions
-
More clinical overlap
-
Seen more frequently in those of Mediterranean, southeast Asian ancestry
-
Also seen in African Americans but generally have a milder course
-
Relative alpha chain excess leads to shortened red cell survival and variable splenic sequestration
-
Diagnosed by hemoglobin electrophoresis or beta globin chain analysis
-
Cannot be diagnosed by electrophoresis in the neonate
-
Iron, folate, and B12 must be repleted to have an accurate hemoglobin electrophoresis
Beta Thalassemia Syndromes
-
Beta thalassemia minor—silent or near silent trait (heterozygous β0 or β +)
-
Asymptomatic
-
Smear can be normal
-
Occasional microcytosis, hypochromia, target cells, basophilic stippling
-
Often normal indices or decreased MCV
-
Normal to slightly elevated HgbA2 on electrophoresis
-
Thalassemia intermedia
-
More symptomatic than thalassemia trait
-
Refers to a clinical phenotype with diverse genetic explanations
-
Laboratory
-
Microcytosis, hypochromia, target cells, and basophilic stippling on smear
-
Mentzer Index < 13
-
Hgb usually between 7 and 10 g/dl
-
Elevated HgbA2 and HgbF on electrophoresis
-
Thalassemia major (Cooley’s anemia)
-
Variable reduction of beta globin gene production
-
Homozygous or double heterozygous forms (β0, β + variants)
-
Excess alpha globin chains result in increased destruction of RBCs and ineffective erythropoiesis
-
Shortened red cell life span and splenic trapping
-
Clinical presentation
-
General
-
◦ Dependent on amount of HgbF
-
◦ Severe anemia with increased iron absorption and subsequent toxicity
-
◦ Pallor, jaundice, fatigue
-
◦ Hepatosplenomegaly
-
-
Skeletal
-
◦ Typical facial features with maxillary hyperplasia, flat nasal bridge, frontal bossing
-
◦ Pathological bone fractures
-
-
Endocrine dysfunction
-
◦ Hypothyroidism
-
◦ Hypoparathyroidism
-
◦ Diabetes mellitus
-
-
Cardiovascular
-
◦ Congestive heart failure
-
◦ Cardiac arrhythmias
-
-
-
Laboratory
-
Severe anemia
-
Few reticulocytes < 8 % compared to degree of anemia
-
Microcytosis with no normal appearing RBCs on the smear
-
Numerous nucleated RBCs
-
Target cells
-
Mentzer index(MCV/RBCs) is < 9
-
Indirect (unconjugated) bilirubin is elevated
-
-
Treatment
-
Chronic transfusion therapy
-
Before chronic transfusion is initiated diagnosis of beta thalassemia must be confirmed first
-
Deferoxamine for iron chelation
-
Newer chelating agent, deferasirox (Exjade, Novartis), is oral and more tolerable but long term data still being accumulated
-
Other Hemoglobinopathies
-
Hemoglobin E
-
Hemoglobin Lepore
-
Hemoglobin Koln
Rare Disorders
-
Sideroblastic anemia
-
May be microcytic
-
Ineffective erythropoiesis caused by iron deposition in erythroblasts
-
Mild-to-moderate hemolysis
-
Ringed sideroblasts in bone marrow
-
-
Protein calorie malnutrition-microcytosis without IDA
-
Metabolic abnormalities of iron absorption and metabolism
Macrocytic Anemia (MCV > 100 in Child Older than 2)
Folic Acid Deficiency
Etiology
-
Nutritional
-
Sources—leaves; vegetable; fruits; animal organs, for example, liver and kidneys
-
Body stores for folic acid is limited 2–3 months on folate-free diet
-
-
Inadequate intake—during pregnancy, growth in children, and hemolytic anemia
-
Goat milk consumption
-
Decreased folic acid absorption—removal of ileum or IBD
-
Anticonvulsant medication, for example, phenytoin, primidone
-
Congenital dihydrofolate reductase deficiency
-
Drug-induced abnormal metabolism—Methotrexate
Clinical presentation
-
Megaloblastic anemia
-
Irritability
-
Inadequate weight gain
-
Chronic diarrhea
-
Hemorrhage from thrombocytopenia in severe cases
Laboratory
-
Macrocytic anemia (MCV> 100; Fig. 6)
Fig. 6 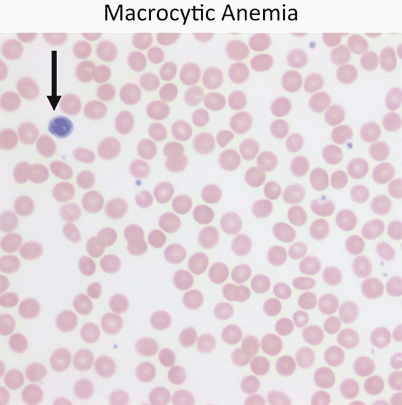
Red cells are usually approximately the size of a small lymphocyte nucleus (arrow). In this case the red cells are slightly larger than the lymphocyte nucleus on average. Macrocytic anemia is most often a result of folate or vitamin B12 deficiency
-
Megaloblastic changes including hypersegmented neutrophils (> 5 lobes)
-
Elevated LDH
-
Hypercellular bone marrow

Treatment
-
Rule out B12 deficiency before starting folic acid therapy
-
Folic acid 0.5–1 mg/day IV or oral
-
Hematologic response can occur within 72 h (diagnostic test as well)
-
Treatment continued for only 3–4 weeks
-
Maintenance dose is 0.2 mg daily
Vitamin B12 Deficiency
-
Vitamin B12 stores last for 3–5 years
-
Sources—animal products
Etiology
-
Inadequate B12 intake (strict vegan)
-
Exclusively breast fed and maternal vegan diet
-
Removal of terminal ileum
-
Inflammatory bowel disease
-
Fish tapeworm (Diphylobothrium latum)
-
Absence of Vitamin B12 transport protein and stomach intrinsic factor (IF)
Clinical presentation
-
Weakness
-
Fatigue
-
Failure to thrive
-
Irritability
-
Pallor
-
Glossitis
-
Vomiting
-
Diarrhea
-
Icterus
-
Neurologic symptoms
-
Paresthesias
-
Developmental regression
-
Neuropsychiatric changes
-
Laboratory
-
Macrocytic anemia (MCV > 100; see Fig. 6)
-
Megaloblastic changes including hypersegmented neutrophils (> 5 lobes)
-
Elevated LDH
-
Normal iron and folic acid levels
-
Increased methylmalonic acid in urine
-
Increased homocysteine
-
Low reticulocyte count for degree of anemia
-
Antiparietal cell antibody positive in pernicious anemia
-
Less than 10 % of cases present under age 40
-
-
Classic Schilling test is no longer regarded as the diagnostic test
Treatment
-
Parenteral administration of Vitamin B12 1 mg daily
-
With neurologic involvement continue for minimum of 2 weeks
-
-
Reticulocytosis in 2–4 days unless concurrent inflammatory disease
-
Maintenance of monthly IM Vitamin B12
Pearson Marrow–Pancreas Syndrome
-
Variant of sideroblastic anemia
Clinical presentation
-
Macrocytic anemia in neonatal period
-
Elevated level of alpha fetoprotein
-
Neutropenia
-
Thrombocytopenia
-
Failure to thrive
-
Pancreatic fibrosis
-
Insulin dependent diabetes mellitus
-
Exocrine pancreatic deficiency
-
Muscle and neurologic impairment
Laboratory
-
Bone marrow
-
Ringed sideroblast
-
Vacuolated erythroblast and myeloblast
-
-
Often confused with Diamond–Blackfan anemia and transient erythroblastopenia of childhood
Diamond–Blackfan Anemia (Congenital Hypoplastic Anemia)
-
Primary defect in the erythroid progenitors
Clinical presentation
-
Profound anemia manifested by 2–6 months of age
-
More than 50 % have congenital anomalies
-
Short stature
-
Craniofacial dysmorphism (snub nose, wide-set eyes, thick upper lip)
-
Triphalangeal thumbs
-
Bifid, subluxed, absent, or supernumerary thumbs
-
Laboratory
-
Macrocytic RBCs with no hypersegmentation of neutrophils
-
Normal B12 and folate
-
Increased adenosine deaminase activity in most patients
-
Decreased RBCs precursor in bone marrow
-
Elevated serum iron
-
Normal bone marrow chromosomal studies
-
Normal to low reticulocyte count
-
Negative PCR for Parvovirus B19
Treatment
-
Steroids
-
Iron chelating agents (if transfusion dependent)
-
Stem cell transplantation for non respondents to corticosteroids, and after several years of RBC transfusions
Prognosis
-
Median survival > 40 years
Normocytic Anemia (MCV > 70 + Age and < 100 in Child Older Than 2; Fig. 7)

Approach to normocytic anemia
Transient Erythroblastopenia Childhood
Background
-
Most common acquired red cell aplasia in childhood
-
More common than Diamond–Blackfan anemia (congenital hypoplastic anemia)
Etiology
-
Transient suppression of RBC production
-
Often noted after a viral infection
-
No evidence of Parvovirus B19
Clinical presentation
-
Age—3 months to 3 years of age, most > 12 months
-
More common in males
Laboratory
-
MCV normal for age
-
Hemoglobin can be as low as 2.2 g/dl
-
Reticulocytes decreased
-
Bone marrow biopsy rarely needed but erythroid suppression seen
-
Normal adenosine deaminase (ADA)
Treatment
-
Reassurance
-
Recover within 2–3 months
-
Occasionally transfusion is necessary
Hereditary Spherocytosis
Background
-
Autosomal-dominant inheritance
-
Less frequently can be autosomal recessive
-
-
25 % of patients have no family history
-
Most common molecular defects are in spectrin or ankyrin, major components of the RBC cytoskeleton
Clinical presentation
-
May be asymptomatic into adulthood
-
Anemia
-
Hyperbilirubinemia sufficient to require exchange transfusion in newborn period
-
Pallor
-
Jaundice
-
Fatigue
-
Exercise intolerance
-
Splenomegaly
-
Pigment gallstones may form as early as 4–5 years of age
-
Susceptible to aplastic crisis as a result of parvovirus B19 infections
-
Erythroid marrow failure may result rapidly in profound anemia HCT < 10 %, high cardiac output failure, hypoxia, cardiovascular collapse, and death; platelet may also fall
-
Laboratory
-
Reticulocytosis
-
Indirect hyperbilirubinemia
-
High LDH
-
Low haptoglobin
-
Normal MCV
-
Elevated MCHC
-
High percentage of spherocytes on smear (Fig. 8)
Fig. 8 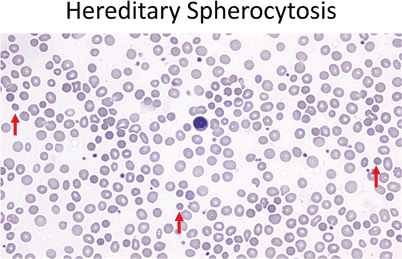
Red cells should be similar in size to the small lymphocyte nucleus (center). In hereditary spherocytosis the red cells are small and hyperchromatic, lacking central pallor (40 ×). Red arrows point out a few of the examples in this field
-
Can be confirmed with osmotic fragility test or flow cytometry
-

Treatment
-
Folic acid 1 mg po daily to prevent deficiency and the resultant decrease in erythropoiesis
-
Splenectomy indications:
-
Hgb < 10 g/dl
-
Reticulocytosis
-
Aplastic crisis
-
Poor growth
-
Cardiomegaly
-
-
Some do not recommend splenectomy in patients with hemoglobin > 10 g/dl and reticulocytes < 10 %
-
Vaccination for encapsulated organism Haemophilus influenza, meningococcus, pneumococcus should be given before splenectomy, then prophylactic penicillin V 125 mg BID < 5 years and 250 BID for > 5 years
-
Partial splenectomy is useful in children < 5 years
Hereditary Elliptocytosis
-
Less common than hereditary spherocytosis (HS)
Clinical presentation
-
Presentation same as in HS
Laboratory
-
Red blood cells shows various degree of elongation, may be rod shaped
-
Other abnormal shapes may be present microcytosis, spherocytes, poikilocytosis
-
Treatment
-
No treatment necessary unless hemolysis present
-
Otherwise same as in HS
Paroxysmal Nocturnal Hemoglobinuria
Background
-
Most often caused by an acquired (rather than inherited) intrinsic defect in the cell membrane
-
Deficient membrane associated protein include decay-accelerating factor, C8-binding protein
Clinical presentation
-
Nocturnal and morning hemoglobinuria
-
Thrombosis and thromboembolic phenomena is a very serious complication
-
Aplastic anemia may precede the episodes of PNH
Laboratory
-
Red blood cells shows various degree of elongation; may be rod shaped
-
Evidence of hemolysis—elevated LDH, elevated bilirubin, low haptoglobin
-
Negative direct antiglobulin test
-
Flow cytometry for CD55 and CD59
-
Positive results on acidified serum hemolysis Ham test, or sucrose lysis test (historical)
-
Markedly decreased acetylcholinesterase activity and decay accelerating factor is found
Treatment
-
Acute
-
Transfusion to suppress production of PNH cells
-
Glucocorticoids 2 mg/kg/24 h (controversial)
-
-
Chronic
-
Eculizumab prevents complement binding and decreases hemolysis
-
Warfarin to prevent thrombotic complications
-
May need supplemental iron to offset losses from hemoglobinuria
-
Sickle Cell Disease (SCD)
Background
-
Hemoglobin S is the result of a mutation resulting in a substitution of valine for glutamic acid at sixth position in beta globin chain
-
Autosomal recessive inheritance
Clinical presentation
-
Usually diagnosed on neonatal screen
-
Manifestations of clinical symptoms can be as early as 6 months of age
-
Crises
-
Splenic sequestration
-
Pain crisis
-
Aplastic crisis
-
Parvovirus B19 frequent cause of aplastic crises
-
-
Infections
-
Bacterial sepsis is the greatest cause of morbidity and mortality
-
Bacterial infection by encapsulated organisms is the most common at all ages
-
-
Functional asplenia as early as 6 months, by age 5 in most children
Laboratory
-
Anemia
-
Sickle cells on smear (Fig. 9)
Fig. 9 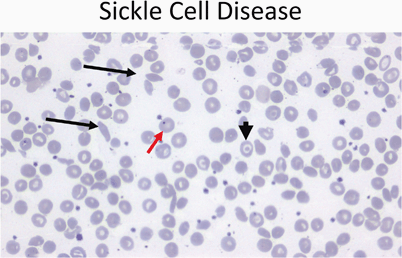
Peripheral smear (40 ×) from a patient with sickle cell disease showing sickle cells (black arrows), target cells (arrowhead), and a Howell–Joley body (red arrow)
-
Positive sickle prep
-
Hemoglobin electrophoresis—SS, SC, SD

Management
-
Fever
-
Medical emergency due to high risk of severe bacterial infection and high fatality
-
Parenteral IV third generation cephalosporin (Cefotaxime)
-
Penicillin VK oral prophylaxis until 5 years of age
-
125 mg PO BID until 3 years
-
250 mg PO BID until 5 years
-
Continue past 2 years of age if history of infection with encapsulated organism
-
Osteomyelitis—frequently staph or salmonella
-
Pain crisis
-
Hydroxyurea
-
Increases level of hemoglobin F and total hemoglobin
-
Decreases the pain crises by 50 %
-
Side effect is myelosuppression, but reversible
-
If begun in infancy may preserve the splenic function, improve the growth, and decrease ACS
-
Initial dose is 15–20 mg/kg increase gradually by 2.5–5 mg/kg up to max of 35 mg/kg/day
-
Monitor for toxicity
-
Aplastic crisis
-
RBC lifespan is between 10 and 20 days in patient with SCD
-
Cessation of RBC production for 10–14 days can lead to profound anemia
-
Clinical presentation
-
Pallor
-
Fatigue
-
Decreased activity
-
Poor feeding
-
Altered mentation
-
-
Laboratory
-
Severe anemia
-
Reticulocytopenia
-
Occasional thrombocytopenia
-
-
Management
-
Transfusion support as needed until reticulocyte recovery has occurred.
-
-
Dactylitis (hand–foot disease)
-
Often the first manifestation of pain in children
-
Occurs in 50 % of children by 2 years of age
-
Unilateral can be confused with osteomyelitis
-
Treatment
-
◦ Pain medications (e.g.,acetaminophen with co-deine)
-
-
Splenic sequestration
-
Etiology unknown
-
30 % of children with sickle cell anemia have episodes of significant sequestration
-
Clinical presentation
-
Increase in size of spleen
-
Evidence of hypovolemia
-
Decline in hemoglobin of at least 2 g/dl from the base line
-
-
Treatment
-
Maintenance of hemodynamic stability
-
Isotonic fluid
-
Blood transfusion
-
-
Prognosis
-
Repeat sequestration is very common
-
Parents should be taught how to palpate the spleen
-
Vaso-occlusive crisis
-
Disruption of blood flow in microvasculature by sickle cells
-
Risk factors exposure to cold, hypoxia, and acidosis
-
Clinical presentation
-
Pain which can affect any part of the body
-
Pain most often in chest, back, abdomen, and extremities
-
-
Management
-
Pain medications
-
Acetaminophen up to IV morphine depending on the severity
-
IV hydration does not relieve the pain
-
Blood transfusion does not prevent or relieve the pain
-
Concern about opioids dependency must not be a reason not treat a child with pain
-
Priapism
-
Penile erection lasts > 30 min
-
Pain medication
-
Sitz bath
-
-
Penile erection lasts > 4 h
-
May result in sexual dysfunction
-
Aspiration of blood from corpora cavernosa
-
Followed by irrigation with diluted epinephrine will cause immediate relief
-
Neurological complications
-
11–20 % will have either overt or silent stroke
-
Overt stroke means presence of focal neurological deficit > 24 h and or cerebral infarct by T2-weighted MRI
-
Silent stroke means absence of focal neurological lesions > 24 h with cerebral infarct on T2-weighted MRI
-
-
Clinical presentation
-
Headache
-
Seizures
-
Cerebral venous thrombosis
-
Reversible posterior leukoencephalopathy syndrome (RPLS)
-
-
Treatment
-
Oxygen to maintain saturation > 96 %
-
Transfusion within 1 h to increase Hgb level to max of 11 g/dL
-
CT to exclude cerebral hemorrhage
-
-
Primary prevention of stroke
-
TCD (transcranial Doppler) to measure blood velocity
-
If blood velocity is > 200 cm/s prophylactic transfusion is indicated to decrease the Hgb S to < 30 %
-
Can start as early as 2–3 years of age
-
-
Secondary prevention
-
Transfusion therapy after initial stroke
-
Maintain the Hgb S < 30 %
-
Complications:
-
-
20 % have second stroke in the first year after first stroke
-
Iron overload (200 mg of iron/unit RBCs):
-
Iron-chelating agents
-
Phlebotomy
-
Erythrocytapheresis (expensive and complicated)
-
Acute chest syndrome
-
Clinical presentation
-
Fever
-
Respiratory distress
-
Chest pain
-
New radiodensity on CXR
-
All patients with fever should have CXR even in absence of respiratory symptoms
-
-
Treatment
-
Oxygen
-
Simple exchange transfusion indications:
-
◦ Decreasing oxygen saturation
-
◦ Increasing work of breathing
-
◦ Rapid change in respiratory effort
-
-
Most common episode preceding ACS is pain crisis treated with opioids, especially morphine
-
Overlap between pneumonia and ACS requires use of macrolide and third-generation cephalosporin
-
Most common organism in ACS: S. pneumoniae,Mycopl asma, Chlamydia pneumoniae
-
-
Pulmonary hypertension
-
PH is a major risk of death in adult with sickle cell anemia
-
Renal disease
-
Gross hematuria
-
Papillary necrosis
-
Nephritic syndrome
-
Renal infarcts
-
Pyelonephritis
-
Renal medullary necrosis
-
Treatment
-
ACE inhibitors beneficial for patients with proteinuria
-
General considerations
-
High risk of academic failure, poor high school graduation rate
-
1/3 of children have cerebral infarcts
-
Other complication of sickle cell anemia
-
Delayed puberty
-
Vascular necrosis of femoral head
-
Retinopathy
-
Surgical procedures—complications include pain and ACS post operatively
-
Blood transfusion before surgery to keep the hemoglobin approximately 10 g/dl
-
Methemoglobinemia (congenital or acquired)
-
Decrease ability to release o2 to tissues
-
Methemoglobin of 15 % associated with visible cyanosis
-
Methemoglobin of 70 % is lethal
-
Methemoglobin colors the blood brown
-
Exposure to 100 % oxygen will change the color
-
Triggers
-
Rotavirus infection
-
Gastroenteritis
-
Water high nitrites
-
Aniline teeth gel
-
-
Treatment: methylene blue
Pyruvate Kinase Deficiency
Background
-
Active enzyme in Embden–Meyerhof pathway
-
Deficiency leads to defective red cell glycolysis and decrease ATP production
-
Red cells are rigid and deformed, metabolically and physically vulnerable with decreased red cell survival
Clinical presentation
-
Varies from severe neonatal hemolytic anemia to mild well compensated hemolysis
-
Severe jaundice and anemia and can occur during neonatal period
-
Splenomegaly
-
Aplastic crisis with parvovirus B19 infection
Laboratory
-
Reduced RBC PK enzyme level
-
Elevated reticulocyte count
-
Smear with polychromatophilia, macrocytosis, ovalocytes, acanthocytes, or pyknocytes
Treatment
-
Exchange transfusion may be indicated for hyperbilirubinemia in newborn
-
Blood transfusion as necessary
-
Folic acid supplementation
-
Splenectomy should be performed if frequent transfusion after age 5–6 years
Glucose-6-Phosphate Dehydrogenase
Pathophysiology
-
First enzyme in the pentose phosphate pathway of glucose metabolism
-
Activity falls rapidly as red cell ages
-
Decreased glucose metabolism with impaired elimination of oxidants and subsequent loss of red cell membrane integrity
-
-
Severity of hemolysis depends on the quantity and type of G6PD deficiency and nature of hemolytic agent (usually an oxidation mediator) (Table 1)
Table 1 WHO classification of G6PD deficiency
Genetics
-
X-linked recessive
-
Variable intermediate expression shown by heterozygous females
-
More common in African American and Mediterranean ancestry
Clinical presentation: episodes of hemolysis produced by:
-
Drugs
-
Antioxidant drugs include:
-
◦ Aspirin
-
◦ Sulfonamides
-
◦ Antimalarials
-
-
Usually 24–48 h after exposure
-
Hemoglobin usually normal between episodes
-
Occasionally need additional stress of infection or neonatal state
-
-
Fava beans
-
Acute life-threatening, often leading to acute renal failure
-
Associated with Mediterranean and Canton varieties
-
Blood transfusions usually required
-
-
Infection
-
Neonatal jaundice
-
Associated with Mediterranean and Canton varieties
-
Occasional exposure to naphthalene, aniline dyes, marking ink, or other drug
-
Infants may present with pallor, jaundice, dark urine
-
Jaundice may be hepatic in origin
-
Often no known exposure to drugs
-
-
Chronic nonspherocytic hemolytic anemia
-
Mainly in northern Europeans
-
Reticulocytosis
-
Increased autohemolysis with only partial correction by glucose
-
Slight jaundice
-
Mild splenomegaly
-
Laboratory
-
Anemia
-
Heinz bodies seen in unstained red blood cells due to hemoglobin precipitation
-
Diagnosis demonstrated by reduced G6PD activity in RBCs should be few weeks after the hemolytic episode
Treatment
-
Avoidance of agents
-
Transfusion as needed
-
Folic acid supplementation
-
Chronic nonspherocytic hemolytic anemia
-
Consider chronic transfusion to keep Hgb at approximately 8 g/dl
-
Iron chelation as needed
-
-
Splenectomy
-
Severe chronic anemia
-
Hypersplenism
-
Splenomegaly with physical impediment
-
Other Enzyme Deficiencies
-
Hexokinase deficiency
-
Glucose phosphate isomerase deficiency
-
Aldolase deficiency
-
Diphosphoglycerate deficiency
-
Adenosine triphosphate deficiency
-
Enloase deficiency
-
Phosphofructokinase deficiency
-
Myopathy
-
Associated with type VII glycogen storage disease
-
Common in Ashkenazi Jews
-
-
Triosephosphate isomerase deficiency
-
Cardiac anomalies
-
Recurrent infections
-
Progressive neuromuscular disease with generalized spasticity
-
-
Phosphoglyercate kinase deficiency
-
First ATP generating enzyme
-
Sex-linked recessive
-
Intellectual disability (ID)
-
Seizures
-
Behavioral disorders
-
Autoimmune Hemolytic Anemia
Etiology
-
Antibodies against antigens on RBCs surface
-
IgG against Rh complex is the most common in children
-
IgM cold antibodies usually associated with infections, for example, Mycoplasma and EBV
Clinical presentation
-
Pallor
-
Jaundice
-
Pyrexia
-
Hemoglobinuria
-
Splenomegaly
Laboratory
-
Profound anemia
-
Reticulocytosis
-
Positive direct antiglobulin (Coombs) test
-
Polychromasia
-
Spherocytosis
-
High cold agglutinin titre
Treatment
-
Supportive treatment for mild cases
-
Corticosteroids for IgG mediated disease
-
Blood transfusion (blood unit with the least reaction by Coomb’s technique)
-
IVIg
-
Splenectomy in persistent cases
Prognosis of acute form
-
Response to glucocorticoids
-
Low mortality rate
-
Full recovery
Hemolytic Anemia Secondary to Extracellular Factors
-
Mechanical injury
-
HUS
-
Kasabach-Merrit syndrome: hemangioma and thrombocytopenia
-
-
Thermal injury
-
Renal disease
-
Liver disease
-
Change in cholesterol to phosphlipid level which affects the membrane of RBC
-
-
Toxins and venom
-
Streptococcus, haemophilus influenzae, staphylococcus and clostridium infection
-
Cobras, rattlesnakes, have phospholipids in their venom—cause spherocytic hemolysis
-
Erythrocytosis
Definition
-
RBCs 25 % > upper normal value
Clinical presentation
-
Hypertension, headache , shortness breath, neurologic symptoms, thrombocytosis may cause hemorrhage and thrombosis
Primary (polycythemia vera)
-
Major criteria
-
Increased red cell mass
-
Arterial oxygen saturation > 92 %
-
Palpable spleen
-
-
Minor criteria
-
Platelet count > 400,000
-
Leukocytosis > 12,000
-
Increased leukocyte alkaline phosphatase
-
Increased vitamin B12 > 900 pg/ml, binding capacity > 2200 pg/ml
-
Secondary
-
Increase HCT > 65 %
-
Clinical presentation
-
Hyperviscosity, headache, hypertension
-
-
Etiology
-
Familial
-
◦ Hemoglobinopathy
-
-
Hypoxia
-
◦ Altitude
-
◦ Cardiac disease
-
◦ Lung disease
-
◦ Central hypoventilation
-
-
Hormonal
-
◦ Adrenal
-
◦ Anabolic
-
-
Renal
-
◦ Tumor/cysts
-
◦ Renal artery stenosis
-
◦ Hydronephrosis
-
-
Liver
-
◦ Dysfunction
-
◦ Hepatoma
-
-
Metabolic
-
◦ 2,3 diphosphoglycerate deficiency
-
-
Neonatal
-
◦ Normal intrauterine environment
-
◦ Twin-Twin transfusion
-
◦ Diabetic mother
-
◦ IUGR
-
◦ Trisomies
-
◦ Congenital adrenal hyperplasia
-
◦ Thyrotoxicosis
-
-
Treatment
-
Periodic phlebotomy for hematocrit > 65–70 % or hemoglobin > 23 g/dl
Fanconi Anemia
Genetics
-
Autosomal recessive
Clinical presentation
-
Skin abnormalities in 65 % of cases
-
Hyperpigmentation of the trunk and intertriginous areas, café-au-lait spots, vitiligo
-
-
Short stature—60 %
-
Upper limb anomalies—50 %
-
Absent thumbs
-
Triphalangeal thumbs
-
Congenital hip dysplasia
-
-
Male genitalia—40 %
-
Underdeveloped penis
-
Undescended testes
-
Hypogonadism
-
-
Female genitalia
-
Malformation of vagina, uterus
-
-
Facial anomalies
-
Microcephaly , small eyes, epicanthal folds, abnormal shape ears, or absent ears
-
-
Intellectual disability (ID)—10 %
-
Kidney abnormalities
-
Horseshoe kidney, absent, or duplicate kidney
-
Laboratory
-
Macrocytic anemia
-
Variable progression to full-blown pancytopenia due to aplasia
Complications
-
Acute leukemia
-
Carcinoma of head and neck, and upper esophagus
Shwachman–Diamond Syndrome
-
Rarest form of pancytopenia
Genetics
-
Autosomal recessive
Clinical presentation
-
Failure to thrive
-
Exocrine pancreatic insufficiency—50 %
-
Fat malabsorption—absence of steatorrhea does not exclude SDS
-
-
Skeletal abnormalities
-
Short stature—Metaphyseal chondrodysplasia
-
Abnormal digits—syndactyly, clinodactyly, or supernumerary metatarsals
-
-
Abnormal facies
-
Bifid uvula, short, or cleft palate
-
Dental dysplasia
-
Hypertelorism
-
Microcephaly
-
-
Retinitis pigmentosa
-
Recurrent bacterial infections
Laboratory
-
Abnormal pancreatic enzymes and steatorrhea
-
Neutropenia
-
Bone marrow showing myeloid hypoplasia
-
Pancytopenia—60 %
Diagnosis
-
Mutation analysis for SBDS is definitive in 90 %
Complications —increase with age, usually after 10 years of age
-
Aplastic anemia
-
Myelodysplastic syndrome
-
Acute myelogenous leukemia
Treatment
-
Androgen with low-dose prednisone
White Cell Disorders
Neutropenia
Acute
-
Viral infection
-
Epstein–Barr virus
-
Respiratory syncytial virus
-
Influenza A and B
-
Hepatitis
-
Human herpesvirus 6 (HHV 6) infections
-
-
Bacterial infection
-
Hypersplenism
-
Drug-induced—recovery after medication cessation
-
Antimicrobials—sulfonamides, penicillin
-
Antirheumatics—gold, phenylbutazone, penicillamine
-
Anticonvulsants—phenothiazine
-
Analgesic and anti-inflammatory—ibuprofen
-
Chronic
-
Cyclic neutropenia
-
Clinical presentation
-
◦ Approximately 21-day cycles with changing neutrophil counts with neutropenia spanning 3–6 days
-
◦ Nadir may be in severe range
-
◦ Fever and oral ulceration often during nadir
-
◦ Gingivitis, pharyngitis, skin infections during nadir
-
◦ Occasionally more serious infections—pneumonia, necrotizing enterocolitis with peritonitis, and Escherichia coli or Clostridium sepsis.
-
◦ Count may be recovering when brought to medical attention
-
-
Laboratory
-
◦ Counts 2–3/week for 6 weeks
Treatment
-
◦ Prophylactic GCSF during nadir in some cases
-
◦ Immediate attention with fevers
-
-
-
Chronic benign neutropenia
-
No specific abnormality found
-
No serious infections
-
No treatment necessary except attention for fevers
-
Congenital
-
Kostmann syndrome (severe congenital neutropenia)
-
Autosomal recessive
-
Clinical presentation
-
◦ Mouth ulcers
-
◦ Gingivitis
-
◦ Otitis media
-
◦ Cellulitis
-
◦ Respiratory infections
-
◦ Skin infections and abscesses—most common
-
◦ Pneumonia and deep tissue abscesses—often life threatening
-
◦ Mild HSM
-
◦ Progress to MDS/AML
-
-
Treatment
-
◦ GCSF
-
◦ Stem cell transplant for MDS/AML
-
-
-
Cartilage hair hypoplasia
-
Chédiak–Higashi syndrome
-
Fanconi anemia
Immune
-
Autoimmune neutropenia
-
Neonatal alloimmune neutropenia
-
Dysgammaglobulinemia
-
Hyper IgM syndrome
-
HIV
-
PNH
Nutritional
-
B12 and folic acid deficiency—ineffective erythropoiesis with neutropenia
Bone marrow infiltration
-
Malignancy
-
MDS
-
Lymphoproliferative disorders
Platelet Disorders (Fig. 10)

Approach to platelet disorders
Thrombocytopenia
Decreased Production
Amegakaryocytic Thrombocytopenia
Genetics
-
Autosomal recessive
Clinical presentation
-
Rash, bruising, or bleeding at birth
-
Most common anomalies:
-
Neurologic—cerebellar and cerebral atrophy are frequent
-
Cardiac findings—ASD, VSD, PDA, TOF, CoA
-
-
Other anomalies
-
Abnormal hips, feet, kidney, eye, and palate malformation
-
Diagnosis
-
Initially absent megakaryocytes then pancytopenia
-
If beyond neonatal periods, bone marrow aspirate, and biopsy will confirm the diagnosis
Thrombocytopenia Absent Radius Syndrome (TARS)
Clinical presentation
-
Thrombocytopenia
-
Absent radius
-
Congenital heart disease—TOF, ASD, VSD
-
Others
-
Eosinophilia
-
Leukemoid reaction
-
Intellectual disability (ID)
-
Increased Destruction
-
Normal to increased megakaryocytes in bone marrow
-
Platelet destruction
-
Immune
-
◦ ITP
-
◦ Drugs
-
-
Non-Immune
-
◦ TTP
-
◦ HUS
-
◦ DIC
-
◦ Infection
-
◦ Cardiac
-
-
Idiopathic Thrombocytopenic Purpura (ITP)
Etiology
-
Antiplatelet antibody
-
Often a few weeks after infection
Clinical presentation
-
Petechiae, ecchymoses, epistaxis
-
Variable symptoms, but usually healthy appearing child
Laboratory
-
Thrombocytopenia
-
Normal to increased size of platelets (MPV)
-
Normal RBCs and WBCs
Treatment
-
Observation
-
IVIg
-
Steroids
-
WinRho
-
Platelet transfusion is contraindicated unless life threatening bleeding is present
-
Splenectomy if > 4 years of age with severe ITP longer than 1 year
-
Neonatal immune thrombocytopenias
-
Autoimmune
-
Alloimmune
-
Erythroblastosis fetalis
-
-
Secondary
-
Viral
-
Bacterial
-
Drug induced
-
Posttransfusion purpura
-
SLE
-
Hyperthyroidism
-
Lymphoproliferative disorders
-
Hemolytic uremic syndrome
Background
-
Non-immune
-
Microangiopathic hemolytic anemia
-
E. coli O157:H7 is a very common cause
-
Shigella dysenteriae type I is a another cause
Clinical presentation
-
Usually children between 4 months and 2 years
-
Infection with gastrointestinal symptoms—vomiting and often bloody diarrhea
-
Development of oliguria, hypertension, renal failure
Laboratory
-
Thrombocytopenia
-
Microangiopathic hemolytic anemia
-
Helmet cells, schistocytes, burr cells, spherocytes
-
Elevated BUN and creatinine
-
Reduced large multimers of von Willebrand factor (VWF)
-
Decreased immunoglobulins in some patients
-
Decreased prostaglandin 12 (PG12) in some patients
Treatment
-
Aggressive management of renal failure
-
Correction of anemia with transfusion
-
Avoid platelet transfusion if possible
Thrombotic Thrombocytopenic Purpura (TTP)
Background
-
Nonimmune
-
Microangiopathic hemolytic anemia
Etiology
-
Idiopathic
-
Acute
-
◦ Autoantibody, ADAMTS13 IgG inhibitor
-
-
Chronic
-
◦ ADAMTS13 mutation
-
◦ Mutation of HF gene
-
-
Sporadic
-
◦ Gene mutations may be less severe
-
-
Secondary
-
Autoimmune disease
-
Malignancy
-
Infection
-
Drugs
-
Stem cell transplantation
-
Bacterial endocarditis
-
-
Clinical presentation
-
Fever
-
Headache
-
Malaise
-
Abdominal/chest pain
-
Arthralgia/myalgia
-
Nausea/vomiting
-
Pallor
-
Purpura
-
Jaundice
-
Fluctuating neurologic signs and symptoms
-
Progressive renal failure
Laboratory
-
Thrombocytopenia
-
DIC
-
Blood smear with polychromasia, basophilic stippling, schistocytes, microspherocytes, and nucleated RBCs (Fig. 11)
Fig. 11 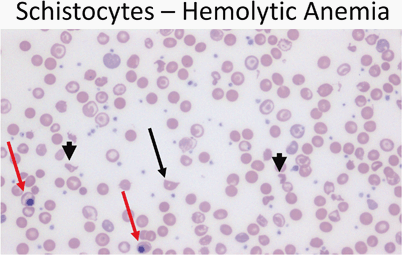
Peripheral smear (40 ×) from a patient with hemolytic anemia showing a schistocyte (arrow) as well as fragmented cells (arrowheads). Note the presence of nucleated red cells (red arrows)
-
Elevated VWF antigen
-
Reduced haptoglobin
-
Hemoglobinuria and hemosiderinuria
-
Increased unconjugated bilirubin
-
Increased LDH
-
Widespread hyaline microthrombi in the microvasculature in biopsy specimens
-
Other disorders with consumption thrombocytopenia
-
DIC
-
Virus associated hemophagocytic syndrome
-
Hemangioma (Kasabach–Merritt syndrome)
-
Cyanotic heart disease
-

Abnormal Platelets
Wiskott–Aldrich Syndrome
-
Thrombocytopenia
-
Tiny platelet
-
Eczema
-
Recurrent infection
Bernard–Soulier Syndrome
-
Absence or deficiency of VWF receptors on the platelet membrane
-
Markedly prolonged bleeding time
Glanzmann’s Thrombasthenia
-
Severe platelet dysfunction that yield prolonged bleeding time
-
Normal platelet count
-
Aggregation studies show abnormal or absent aggregation
-
Prolonged bleeding time
Coagulation Disorders
Hemophilia
-
X-linked recessive
-
Factor VIII (hemophilia A)—85 %
-
Factor IX (hemophilia B)—10–15 %
-
-
Bleeding may start from birth or even fetus
Clinical presentation
-
Easy bruising
-
Intramuscular (deep) hematomas—localized pain and swelling
-
Hemarthroses
-
Hallmark of hemophilia
-
Ankle most common
-
Knee and elbow increasing frequency with age
-
Laboratory
-
PTT is usually 2–3 times upper limit of normal
-
PT, bleeding time, platelet count normal
-
Specific assay for factor VIII or IX will confirm the diagnosis
Classifications
-
Severe hemophilia < 1 %
-
Moderate hemophilia 1–5 %
-
Mild hemophilia > 5 %
Treatment
-
Factor replacement
-
Mild to moderate bleeding—raise factor to 35–50 %
-
Severe or life threatening hemorrhage—raise level to 100 %
-
-
Lifelong prophylaxis usually started with first joint hemorrhage
-
DDAVP may be sufficient in mild forms of hemophilia
-
Avoidance of high risk behavior
Complications
-
Severe hemorrhage
-
Arthropathy
Von Willebrand Disease
Etiology
-
VWF is a carrier protein for factor VIII
-
VWF stored in platelets and endothelial cell
-
VWF adheres to exposed the subendothelial matrix after vascular damage causing platelets to adhere via glycoprotein IB receptors on the VWF
Clinical presentation
-
VWD usually have symptoms of mucocutaneous hemorrhage
-
Excessive bruising, epistaxis, menorrhagia, post-operative bleeding (e.g., tonsillectomy, wisdom teeth extraction)
-
Females more commonly diagnosed than males secondary to menorrhagia
-
Any menstruating female with iron deficiency, should have a detailed history of bruising and other bleeding symptoms
-
Stress doubles or triples level of VWF
-
Laboratory
-
No single assay to rule out or diagnose VWF
-
Bleeding time or PFA
-
PTT—often prolonged but frequently normal in type 1 VWD
-
VWF antigen
-
VWF Ristocetin cofactor activity
-
Plasma factor VIII activity
-
VWF multimers
-
Platelet count
-
Treatment
-
Based on subtype and trial of DDAVP
-
Type 1 usually treated with DDAVP
-
DDAVP 0.3 microgram/kg increases the level of VWF and factor VIII 3–5 fold
-
Type 2B and 3 primarily treated with FVIII:VWF concentrates
-
Platelet type treated with platelet transfusions
-
Disseminated Intravenous Coagulopathy
Etiology
-
Widespread intravascular consumption of platelets and plasma clotting factors and deposition of fibrin
Clinical presentation
-
Bleeding (e.g., from venipuncture sites)
-
Petechiae, ecchymoses
-
Clot formation
-
Associated conditions
-
Tissue injury
-
◦ Trauma, especially cranial
-
◦ Burns
-
◦ Venom
-
◦ Malignancy
-
◦ Obstetric emergencies
-
-
Endothelial cell injury or abnormal vascular surfaces
-
◦ Infection/sepsis
-
◦ Immune complexes
-
◦ Eclampsia
-
◦ Oral contraceptives
-
◦ Giant hemangioma
-
◦ Respiratory distress syndrome (ARDS)
-
◦ Malignancy
-
-
Platelet, leukocyte, or red cell injury
-
◦ Incompatible blood transfusion
-
◦ Infection
-
◦ Allograft rejection
-
◦ Hemolytic syndromes
-
◦ Drug hypersensitivity
-
◦ Malignancy
-
-
Laboratory
-
Prolonged PT and PTT
-
Decreased fibrinogen
-
Decreased platelets
-
Increased fibrin degradation products and D-dimers
-
Presence of helmet cells, schistocytes
-
Increased PF4 (platelet factor 4)
-
Increased FPA (fibrinopeptide A)
-
Decreased factor V, VIII, XIII
Treatment
-
Treatment of underlying disorder
-
Replacement therapy of components as indicated
Neoplastic Disorders
Acute Leukemia
Epidemiology (Table 2 )
-
25–30 % of all childhood cancer
-
Peak age 2–5 years
Clinical presentation
-
Anorexia
-
Fatigue
-
Fever
-
Bone and joint pain (especially lower extremities)
-
Pallor
-
Petechiae, ecchymoses, epistaxis
-
Extramedullary spread
-
Lymphadenopathy
-
Hepatosplenomegaly
-
Cough, orthopnea
-
CNS disease—5 %—cranial nerve palsies
-
Testicular involvement—20 %—testicular enlargement
-
Ovarian involvement—30 %
-
Skin lesions
-
Gingival hypertrophy
-
Laboratory
-
Cytopenias
-
Thrombocytopenia—90 %
-
Anemia—80 %
-
Neutropenia
-
95 % have two cytopenias
-
4 % have only one cytopenia
-
1 % have a normal CBC
-
-
50 % with elevated WBC
-
Usually see blasts if WBC > 5000
-
-
Flow cytometry diagnosis
Peripheral blood
-
ALL: Peripheral blood usually shows leukocytosis with a population of large mononuclear cells (Fig. 12)
Fig. 12 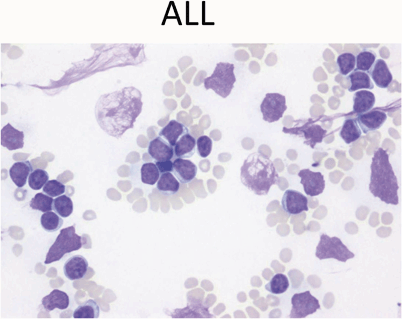
Peripheral blood showing leukocytosis with a population of large mononuclear cells with high nuclear-cytoplasmic ratio, scant blue cytoplasm, and fine chromatin with occasional nucleoli. These are features of lymphoblasts. Note scattered smudge cells, another feature often seen in peripheral smears with leukemia
-
AML: Peripheral blood usually shows myeloblasts with a high ratio of nucleus to cytoplasm (Fig. 13)
Fig. 13 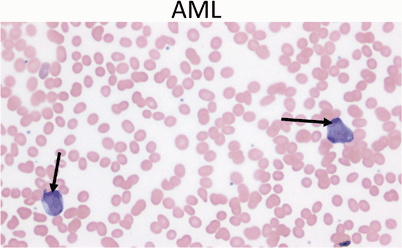
Peripheral blood showing two myeloblasts with a high ratio of nucleus to cytoplasm, finely dispersed chromatin and one or more large nucleoli (arrows). Acute myeloid leukemia represents only 20 % of childhood leukemia


Treatment
-
Per local or national protocols
Associated syndromes/risk factors
-
ALL
-
Down’s syndrome.
-
◦ Acute leukemia is 34 times more common in children with Down’s syndrome.
-
◦ 20–30 % will develop leukemia by age 3 years.
-
◦ Ratio of ALL and AML is the same as the general population.
-
◦ AML has a better outcomes in children with Down’s syndrome.
-
◦ 10 % of neonates with Down’s syndrome may develop a transient leukemia or myelodysplastic syndrome.
-
▪ Characterized by a high leukocyte count, blast cells, anemia , thrombocytopenia and hepatosplenomegaly.
-
▪ Resolve within days to weeks from initial presentation.
-
-
-
Ataxia-telangiectasia.
-
Bloom’s syndrome.
-
◦ Immunodeficiency, progeria, growth retardation.
-
◦ Chromosome fragility/breakage.
-
◦ Predisposition to cancer.
-
-
Fanconi anemia.
-
◦ Pancytopenia, radial bone abnormalities, kidney, skin, or GI abnormalities .
-
◦ Chromosome fragility/breakage.
-
-
-
AML
-
Ionizing radiation
-
Organic solvents
-
Paroxysmal nocturnal hemoglobinuria
-
Down’s syndrome
-
Fanconi anemia
-
Bloom’s syndrome
-
Kostmann syndrome
-
◦ Severe congenital neutropenia
-
◦ High mortality rate—70 %
-
-
Shwachman–Diamond syndrome
-
◦ Congenital neutropenia
-
◦ Metaphyseal chondrodysplasia
-
◦ Exocrine pancreatic deficiency
-
-
Diamond–Blackfan syndrome
-
◦ Congenital pure red cell aplasia
-
◦ Increased erythrocyte adenosine deaminase
-
◦ Short stature
-
◦ Developmental delay
-
◦ Thumb malformations
-
◦ Craniofacial anomalies
-
◦ Urogenital anomalies
-
◦ Increased MCV on CBC
-
-
Neurofibromatosis
-
◦ Bone marrow failure
-
◦ Predisposition to cancers, especially AML and neuroblastoma
-
-
-
Chronic myelogenous leukemia (CML)
-
99 % characterized by specific translocation known as the Philadelphia chromosome t(9;22)
-
Lymphadenopathy
Causes of lymphadenopathy according to location
-
Cervical
-
Oropharyngeal infections, for example, EBV
-
Mycobacterial lymphadenitis
-
Cat scratch disease
-
Kawasaki disease
-
-
Supraclavicular
-
Right side—Malignancy or infection in the mediastinum
-
Left side—Malignancy or infection from the abdomen
-
Lymphoma
-
Tuberculosis
-
-
Hilar
-
Tuberculosis
-
Histoplasmosis
-
Leukemia
-
Lymphoma
-
Sarcoidosis
-
-
Axillary
-
Cat scratch disease
-
Arm or chest infection
-
Leukemia
-
Lymphoma
-
-
Abdominal
-
Malignancy
-
Mesenteric adenitis
-
Clinical approach to lymphadenopathy
-
History
-
Associated other systemic symptoms
-
-
Age
-
Lymph node enlargement in children less than 5 years most likely infectious
-
Histiocytosis can cause lymphadenopathy in children < 3 years
-
Large lymph node in neonate most likely related to congenital infection
-
Likelihood of malignant lymphoma increases in adolescents
-
-
Location
-
Supraclavicular lymphadenopathy is always abnormal and the chances of malignancy are high
-
-
Size
-
Size of the enlarged lymph node aids in determining the need for further evaluation
-
Axillary and cervical > 1 cm
-
Inguinal > 1.5 cm
-
Epitrochlear > 0.5 cm
-
Anywhere > 2 cm
-
-
Characteristics
-
Usually develops over weeks or months.
-
Nontender, discrete, firm, rubbery, often immobile
-
Biopsy criteria
-
Size
-
> 2 cm
-
Increasing over 2 weeks
-
No decrease in size after 4 weeks
-
-
Location
-
Supraclavicular
-
-
Consistency
-
Hard
-
Matted
-
Rubbery
-
-
Associated features
-
Abnormal CXR
-
Fever
-
Weight loss
-
Hepatosplenomegaly
-
Hodgkin Lymphoma
Hodgkin disease (HD)
-
Rare in children < 10 years
-
15 % of cancers in persons between 15 and 19 years
-
Bimodal peaks of incidence from 15–35 years of age and at 55 years of age
-
Infectious agents may be involved
-
EBV
-
HHV6
-
CMV
-
-
Reed–Sternberg cell is the hallmark of HD (Fig. 14)
Fig. 14 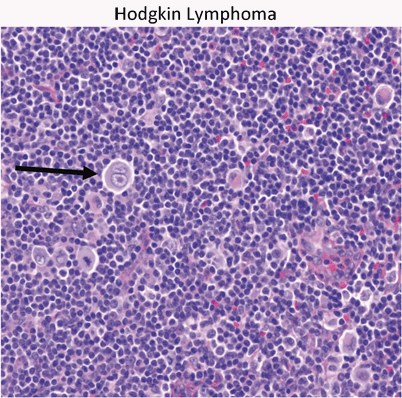
Hodgkin’s lymphoma presents as a localized or regional lymphadenopathy. The characteristic cell in Hodgkin’s is the Reed–Sternberg cell (arrow)

Clinical presentation
-
Painless lymphadenopathy
-
Airway obstruction
-
Pleural dysfunction
-
Pericardial dysfunction
-
Hepatocellular dysfunction
-
Bone marrow infiltration
-
Systemic symptoms (B symptoms)
-
Fever > 39 C
-
Weight loss > 10 % of body weight
-
Night sweats
-
Diagnosis
-
CXR
-
CT abdomen and pelvis
-
PET scan
-
CBC, CMP, ESR, ferritin
Treatment
-
Chemotherapy and radiotherapy are very effective
-
Chemotherapy regimens
-
COPP (cyclophosphamide, vincristine, procarbazine, and prednisone)
-
ABVD (Doxorubicin (adriamycin) bleomycin, vinblastine, and dacarbazine)
-
Prognosis
-
Early stage disease have event free survival 85–90 %, overall survival at 5 years of 95 %
-
Poor prognostic features
-
Bulky tumor
-
Advanced stage at diagnosis
-
B symptoms
-
-
Patient who relapse > 12 months after chemotherapy alone or combined modality have good retrieval response
Non-Hodgkin Lymphoma
-
60 % of all lymphomas in children
-
Burkitt lymphoma is the most common
-
Most children have de novo disease (no underlying condition)
-
Related diseases
-
Severe combined immunodeficiency (SCID)
-
Wiskott–Aldrich syndrome
-
Ataxia telangiectasia
-
Bloom’s syndrome
-
HIV
-
EBV
-
Clinical presentation
-
Rapidly growing tumors with symptoms based on size and location
-
Burkitt lymphoma of abdomen (sporadic type) more common in the USA
-
Burkitt lymphoma of head and neck (endemic type) more common in Africa
-
Superior vena cava (SVC) syndrome—chest involvement
-
Intestinal obstruction—abdominal mass
-
Paraplegia with spinal cord involvement
-
Tumor lysis syndrome
-
Hyperkalemia, hyperuricemia, hyperphosphatemia, hypocalcemia
-
Diagnosis
-
CXR
-
CT abdomen and pelvis
-
CBC, CMP, Mg, Phos, Uric Acid, LDH
-
EBV
Biopsy
-
Classic “starry sky” appearance of Burkitt lymphoma (Fig. 15)
Fig. 15 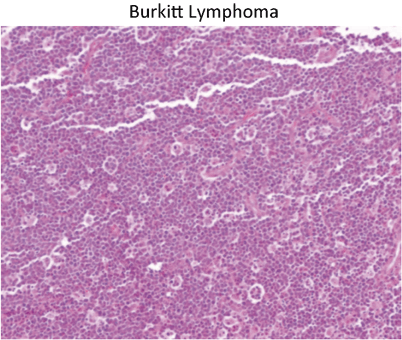
Classic “starry sky” appearance of Burkitt lymphoma. The stars are actually macrophages that are phagocytosing apoptotic Burkitt cells. This example presented as a colonic mass with intussusception


Medulloblastoma (40x) is a so-called “small round blue” cell tumor of childhood. Medulloblastoma is a posterior fossa tumor and the second most common brain tumor of childhood

Pilocytic astrocytoma is composed of bipolar cells with frequent microcystic spaces. Juvenile pilocytic astrocytoma is the most common childhood primary brain tumor

Neuroblastoma is one of the small round blue cell tumors of childhood. A majority are at least poorly differentiated with the presence of some neuropil (black arrow) often in association with Homer- Wright rosettes (10x)

Wilms’ Tumor is a triphasic tumor composed of blastemal (black arrow), epithelial (arrowhead) and mesenchymal components (red arrow). Most are diagnosed before 6 years of age

At low power (10x) alveolar rhabdomyosarcoma has a vaguely alveolar growth pattern with neoplastic cells lining thin fibrous septae. At higher power pink cytoplasmic material is evident (arrows) showing early myogenic differentiation

Osteosarcoma is composed of a pleomorphic cell population of ovoid and frankly bizarre cells with focal osteoid formation (black arrow)

Left – Gross photo showing the white tumor mass filling twothirds of the posterior chamber of the eye. Right – Retinoblastoma is another “small round blue” cell tumor of childhood

Hepatoblastoma is composed of epithelial components – fetal, embryonal, or a mixture of the two – and occasionally mesenchymal components. The image here is of fetal epithelial type hepatoblastoma with a classic “light and dark” appearance
Treatment
-
Chemotherapy
Prognosis
-
Excellent in most of children
-
90–100 % survival rate with localized disease
Brain Tumors
Epidemiology
-
Almost 20 % of all pediatric cancers
-
Peak age 0–4 years
-
Most common cancer mortality in children
-
25 % of all deaths from cancer
-
Clinical presentation
-
Based on location, size, growth rate and age
-
Increased intracranial pressure
-
Headache
-
Vomiting (often mornings)
-
Mental changes, irritability
-
Visual disturbances
-
◦ Diplopia
-
◦ Papilledema
-
◦ Parinaud’s
-
-
Gait disturbances
-
-
Failure to thrive
-
Cranial nerve abnormalities
-
Focal neurologic deficits
-
Seizures
Pathologic diagnosis
-
Based on cell of origin
-
Can occur at multiple locations in the CNS
-
Infratentorial—60 %
-
Supratentorial—40 %
-
-
Common in children
-
Astrocytoma (Fig. 17)
-
◦ 40 % of all CNS tumors
-
◦ Juvenile pilocytic astrocytoma—most common subtype in children
-
◦ Classic site for JPA is cerebellum, but can occur anywhere in CNS
-
-
Treatment
-
◦ Surgery—primary treatment
-
◦ Chemotherapy
-
◦ Radiation therapy
-
-
Medulloblastoma (Fig. 16)
-
◦ 20 % of all brain tumors (second most common)
-
◦ 90 % of embryonal tumors
-
◦ Arises in cerebellum and fourth ventricle
-
◦ May metastasize down spinal cord and rarely outside CNS
-
◦ Similar cell type to primitive neuroectodermal tumor (PNET)
Treatment
-
▪ Surgery—prognosis based on extent of resection
-
▪ Chemotherapy
-
▪ Radiation therapy
-
-
-
Ependymoma
-
◦ Derived from the ependymal lining of the ventricles
-
◦ 70 % occur in the posterior fossa
-
-
Pineal tumors
-
◦ Germ cell tumors
-
▪ Germinoma
-
▪ Yolk sac tumor
-
▪ Mixed germ cell tumor
-
-
◦ Pineoblastoma
-
◦ PNET
-
-
Craniopharyngioma
-
◦ 7–10 % of childhood brain tumors
-
▪ Suprasellar location
-
◦ Solid and cystic components
-
◦ Associated with panhypopitutarism and visual loss
-
▪ Tumor related
-
▪ Treatment related
-
-
-
-
Syndromes associated with brain tumors
-
Neurofibromatosis type 1: optic glioma, astrocytoma, neurofibroma, malignant nerve sheath tumor
-
NF type 2: vestibular schwannomas, meningiomas, spinal cord ependymoma, spinal cord astrocytoma
-
Von Hippel–Lindau: Hemangioblastoma, angiomatosis, pheochromocytoma, renal cell carcinoma, pancreatic cyst
-
Li–Fraumeni: astrocytoma
-
Cowden syndrome: multiple hamartomas including the brain; dysplastic gangliocytoma of the cerebellum
-
Turcot syndrome: medulloblastoma and colon polyps
-
Neuroblastoma (Fig. 18)
Epidemiology
-
Third most common pediatric cancer
-
8 % of childhood malignancy
-
Most commonly diagnosed neoplasm in infants (28–39 % of neonatal malignancies)
-
Mean age is 2 years
Clinical presentation
-
Fever, failure to thrive
-
Paraneoplastic symptoms
-
Secretory diarrhea
-
Increased sweating
-
Hypertension
-
Opsoclonus, myoclonus (dancing eyes and dancing feet)
-
-
Most cases arise in abdomen
-
Abdominal pain
-
Distended abdomen, mass
-
-
Thoracic tumors
-
Occasional Horner’s syndrome
-
-
Spinal tumors
-
Paraplegias
-
-
Metastatic disease
-
Bone pain (bone mets)
-
Cytopenias (bone marrow infiltrate)
-
Orbital proptosis and ecchymosis-“raccoon eyes” (retro-orbital soft tissue infiltrate)
-
Bluish subcutaneous nodules (skin infiltrate)
-
Diagnosis
-
CT/MRI scans often show calcifications
-
Tumor markers
-
Urine homovanillic acid (HVA), vanillylmandelic acid (VMA)
-
-
Poor prognostic factors on pathology
-
N-myc proto-oncogene (MYCN) amplification
-
DNA hyperdiploidy (if less than 1 year of age)
-
Treatment
-
Chemotherapy
-
Radiation therapy
-
Stem cell transplant
-
New vaccines/antibodies
-
Retinoic acid
Associated syndromes/risk factors
-
Hirschsprung’s disease
-
Pheochromocytoma in family
-
Fetal hydantoin syndrome
-
Fetal alcohol syndrome
-
Nesidioblastosis
Wilms Tumor (Fig. 19)
-
WT-1 gene located on 11p13
Epidemiology
-
Peak incidence 2–5 years of age
-
8 cases/million children < 15 years
Clinical presentation
-
Abdominal mass often noted first by parents
-
Abdominal pain, vomiting, hematuria in 12–25 %
-
Hypertension
-
Anomalies and syndromes associated with Wilms tumor
-
Beckwith-Wiedemann (organomegaly, macroglossia, omphalocele, hemihypertrophy)
-
WAGR (aniridia, genitourinary abnormalities, intellectual disability (ID), del 11p13)
-
Denys-Drash (early onset renal failure with renal mesangial sclerosis, male pseudohermaphroditism)
-
Diagnosis
-
US, KUB, CT, and/or MRI
-
U/A
Treatment
-
Surgery, chemotherapy, and radiotherapy
-
Poor prognostic factor
-
Large tumor > 500 g
-
Advanced stage (III or IV)
-
Unfavorable histologic type
-
Rhabdomyosarcoma
Epidemiology
-
Most common soft tissue sarcoma
-
3.5 % of childhood tumors
-
Increased frequency with neurofibromatosis
-
Peak incidence 1–5 years
-
10 % occur in the first year of life
-
70 % appear within first decade
Clinical presentation
-
Anatomic distribution
-
Head and neck—40 %
-
GU—20 %
-
Trunk—10 %
-
Retroperitoneal and others
-
Specific histologic types
-
Embryonal: 60 %, intermediate prognosis
-
Alveolar type: 15 %, most in trunk and extremeties, poor prognosis (Fig. 20)
-
Botryoid type: 6 %, “bunch of grapes”, most in vagina, uterus, bladder, nasopharynx, and middle ear, good prognosis
-
Pleomorphic form: 1 %, adult type
Osteosarcoma
Epidemiology
-
Most common primary malignant bone tumor in children
-
Most present in second decade
-
More common in males
Clinical presentation
-
Local pain, swelling, often history of injury
-
Associated syndromes/risk factors
-
Retinoblastoma , Li–Fraumeni syndrome, Paget disease, radiotherapy
-
Diagnosis
-
Pathologic findings (Fig. 21)
-
Spindle to epithelioid cells producing osteoid (bone forming)
-
-
Radiologic findings
-
Scelerotic destruction (sunburst)
-
Lytic lesion less common
-
Differential diagnosis
-
Ewing sarcoma
-
Osteomyelitis
Metastasis
-
Lung and bone
Treatment
-
Chemotherapy
-
Surgical resection
-
Amputation
-
Prosthesis
-
Ewing Sarcoma
Epidemiology
-
Second decade
-
More common in males
Clinical presentation
-
Local pain, swelling, fever
-
Location
-
Diaphysis of long bone, flat bones
-
Diagnosis
-
Pathology
-
Undifferentiated small round cell tumor
-
-
Radiologic findings
-
Primarily lytic lesions (onion ring appearance)
-
Treatment
-
Chemotherapy
-
Radiation therapy
-
+/ − surgery
Prognosis
-
Localized—60 % survival
-
Metastatic—20–30 % survival
Osteoid Osteoma
-
Small benign bone tumor
Epidemiology
-
Occurs in patients from 2–50 years of age
-
Male are more common than females
Clinical presentation
-
Gradually increasing pain
-
Often worse at night and relieved by aspirin
-
-
Lower extremity lesion may develop limp, atrophy, or weakness
-
Palpation and range of motion may not alter the discomfort
-
Vertebral lesions may cause scoliosis
-
Most common in proximal tibia and femur, can involve any bone
Diagnosis
-
Radiologic findings
-
Round, oval metaphyseal or diaphyseal lucency surrounded by sclerotic bone
-
Central lucency or nidus shows intense uptake of bone scan
-
25 % only visualized by CT
-
Not seen on MRI
-
Treatment
-
Removal of the lesion and ablation of nidus
-
Treat pain with aspirin
Retinoblastoma
Epidemiology
-
Arises following mutation of both Rb genes at 13q14
-
Hereditary form associated with germline inactivating mutation of one copy of RB1 gene
-
Need “second hit”, somatic mutation to second RB1 gene to develop tumor
-
80–90 % with germline mutation get a second hit and develop retinoblastoma
-
-
Sporadic cases involve 2 somatic mutations to RB1 gene
-
60 % sporadic, 40 % familial
-
30 % bilateral
-
90 % of familial tumors are bilateral
-
-
May be present congenitally
-
Most present between 6 months and 2 years of age
Clinical presentation
-
Leukocoria—white pupillary reflex
-
Strabismus —usually the initial presenting complaint
-
Orbital inflammation, proptosis, hyphema , irregular pupils with advanced disease
-
Pain if secondary glaucoma develops
Diagnosis
-
Exam by ophthalmologist under anesthesia
-
CT or MRI
-
Metastatic workup for larger lesions
Treatment
-
Chemotherapy
-
Focal laser photocoagulation
-
Radiation therapy in severe cases
-
Enucleation of unresponsive cases, especially if loss of vision (Fig. 22)
Prognosis
-
95 % cure rate in US
Hepatoblastoma (Fig. 23)
Epidemiology
-
Children < 3 years
-
Can be congenital
-
90 % occur by age of 5 years, 70 % by age of 2 years
-
Male predominance
-
Prevalence of 1 per 120,000 (1 per 1 million children under age 15 years)
-
Associated syndromes/risk factors
-
Familial adenomatous polyposis (APC gene mutation)
-
Glycogen storage disease
-
Beckwith–Wiedemann syndrome
-
Li–Fraumeni syndrome
-
Low birth weight infants
-
Wilms tumor
-
Clinical presentation
-
Large asymptomatic mass
-
Right lobe more common
-
Weight loss, anorexia, vomiting, or abdominal pain
Diagnosis
-
US, KUB, CT, and/or MRI
-
Bilirubin and liver enzymes are usually normal
-
Alpha-fetoprotein is elevated in all hepatoblastomas
-
Anemia and thrombocytosis are common
-
Hepatitis B and C serologies {usually negative}
Treatment
-
Chemotherapy
-
Tumor resection
-
As much as 85 % of liver can be resected
-
Hepatic regeneration noted within 3–4 months of surgery
Suggested Readings
Boxer LA. Neutrophil abnormalities. Pediatr Rev. 2003;24:52–62.
Donadieu J, Fenneteau O, Beaupain B, Mahlaoui N, Chantelot CB. Congenital neutropenia: diagnosis, molecular bases and patient management. Orphanet J Rare Dis. 2011;6:26.
Gaston MH, Verter JI, Woods G, et al. Prophylaxis with oral penicillin in children with sickle cell anemia. A randomized trial. N Engl J Med. 1986;314:1593–9.
Knight PJ, Mulne AF, Vassy LE. When is lymph node biopsy indicated in children with enlarged peripheral nodes? Pediatrics. 1982;69:391–6.
Rogers ZR. Priapism in sickle cell disease. Hematol Oncol Clin N Am. 2005;19:917–28.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Bryson, S., Mulne, A. (2015). Blood and Neoplastic Disorders. In: Naga, O. (eds) Pediatric Board Study Guide. Springer, Cham. https://doi.org/10.1007/978-3-319-10115-6_16
Download citation
DOI: https://doi.org/10.1007/978-3-319-10115-6_16
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-10114-9
Online ISBN: 978-3-319-10115-6
eBook Packages: MedicineMedicine (R0)