
Abstract
The need for a framework to justify that a model has sufficient credibility to be used as a basis for internal or external (typically regulatory) decision-making is a primary concern when using modelling and simulation (M&S) in healthcare. This chapter reviews published standards on verification, validation, and uncertainty quantification (VVUQ) as well as regulatory guidance that can be used to establish model credibility in this context, providing a potential starting point for a globally harmonised model credibility framework.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others

4.1 Introduction
The lack of a framework to justify that a model has sufficient credibility to be used as a basis for internal or external (typically regulatory) decision-making is a primary concern when using modelling and simulation (M&S) in healthcare. Verification, validation, uncertainty quantification (VVUQ), and applicability assessment are processes used to establish the credibility of a computational model. Verification establishes that a computational model accurately represents the underlying mathematical model and its solution. In contrast, validation establishes whether the mathematical model accurately represents the reality of interest. Uncertainty quantification helps to identify potential limitations in the modelling, computational, or experimental processes due to inherent variability (aleatoric uncertainty) or lack of knowledge (epistemic uncertainty). Finally, applicability assesses the relevance of the validation evidence to support using the model for a specific Context of Use (CoU) (Pathmanathan et al., 2017).
Various global organisations have formalised some of these concepts in guidance documents or technical standards for specific use cases. Annexe 1 systematically reviews this existing body of knowledge for various computational model types, ranging from QSAR to ABMs to physics-based models. And given the increasing interest in in silico methodologies, various global standards bodies (e.g., ICH, ISO) are currently developing or revising guidelines and standards in this field.
This chapter outlines concepts related to model credibility assessment in a way that is agnostic to the nature of the computational model type or medical product, extracting the relevant concepts common to the aforementioned standards and guidance documents. To be as inclusive as possible, a level of granularity has been chosen that can incorporate most of the existing knowledge, but which may result in the grouping or omission of steps that do not exist in every standard or guidance. One example is the EMA’s distinction between technical and clinical validation. This was outlined in a letter of support to a request for qualification advice on the use of digital mobility outcomes (DMOs) as monitoring biomarkers,Footnote 1 where it was stated that “The technical validation will verify the accuracy of the device and algorithm to measure a range of different DMOs. […] clinical validation will be obtained in an observational multicentre clinical trial” (see also (Viceconti et al., 2020) for more details). In the follow-up qualification advice, the same authors propose that a DMO is considered clinically validated for a well-defined CoU when one can demonstrate its construct validity, predictive capacity, and ability to detect change (Viceconti et al., 2022). Translating to in silico methodologies, both technical and clinical aspects of model predictions must be evaluated during validation. A clinical interpretation of the model validation should also be provided to support the clinical credibility of the predicted quantities.
Recognizing the lack of a globally harmonised framework, this chapter will describe the concepts related to model credibility, supported by illustrative examples and references to standards, guidance, and additional documents that provide further clarification. We also introduce a hierarchical validation approach that distinguishes between a model's physiological, pathological, and treatment layers.
4.2 Model Credibility in Existing Regulatory Guidelines
Regulatory agencies have provided operational guidelines for assessing a predictive model's credibility (see Annex 1 for a complete list of all guidance and standard documents). For example, a 2003 FDA guideline on exposure–response relationshipsFootnote 2 acknowledged that “The issue of model validation is not totally resolved”. It recommended separating the training set from the validation set of experimental data (implicitly assuming the models are all data-driven). Also, the 2018 EMA guideline on reporting PBPK modelsFootnote 3 recommends validating models against experimental clinical studies of more than 100 patients. It also provides instructions on how the comparison between predictions and experiments should be graphed. While this guideline does not explicitly refer to a risk-based credibility assessment, it states: “The acceptance criteria (adequacy of prediction) for the closeness of the comparison of simulated and observed data depends on the regulatory impact”. At around the same time, a 2018 FDA guidance on PBPK modelsFootnote 4 requests VVUQ evidence in a generalised sense: “To allow the FDA to evaluate the robustness of the models, the sponsor should clearly present results from the methods used to verifyFootnote 5 the model, confirm model results, and conduct sensitivity analyses.” However, this guidance also requests that electronic files related to the modelling software and simulations be submitted along with the PBPK study report, to “allow FDA reviewers to duplicate and evaluate the submitted modeling and simulation results and to conduct supplemental analyses when necessary”. This request may overlook the complexities of reproducing studies involving computational models.
The 2016 FDA guidance on Reporting of Computational Modeling Studies in Medical Device SubmissionsFootnote 6 outlined the importance of providing a complete and accurate summary of computational modelling and simulation evidence that is included in a dossier. This guidance referenced the ASME VV-10:2019Footnote 7 and ASME VV-20:2009 (R2021)Footnote 8 standards, but not ASME VV-40:2018Footnote 9 because it was not yet published. However, the 2023 FDA guidanceFootnote 10 outlines a generalised framework for assessing model credibility that relies heavily upon the ASME VV-40:2018 standard. This guidance proposes eight possible categories of credibility evidence (see Table 4.1). It is important to note that categories 1, 3 and 8 are explicitly within the scope of ASME VV-40:2018, while the others may be considered extensions of the ASME VV-40:2018 framework. While this guidance acknowledges that there are different types of credibility evidence, the issue of different levels of credibility (as proposed in Sect. 2.7) is not considered.
4.3 A Standard Framework: ASME VV-40:2018
The American Society of Mechanical Engineers (ASME) Committee on Verification, Validation, and Uncertainty Quantification in Computational Modeling and Simulation has published the ASME VV-10:2019 and VV-20:2009 (R2021) standards, which outline the processes of verification, validation, and uncertainty quantification for finite element analysis and computational fluid dynamics, respectively. These standards outline VVUQ best practices, but do not provide formalised procedures for steering model validation (and thus the associated model development activities) towards being sufficiently credible for a CoU.
This led to the formation of the ASME VVUQ 40 Subcommittee on Verification, Validation, and Uncertainty Quantification in Computational Modeling of Medical Devices. Through close collaboration between medical device manufacturers, regulatory agencies, and other device industry stakeholders, this subcommittee published the standard “Assessing Credibility of Computational Modeling and Simulation Results through Verification and Validation: Application to Medical Devices” in 2018. This standard introduces a risk-informed credibility assessment framework for physics-based models that can be applied to various scientific, technical, and regulatory questions. And while the standard is written with a focus on medical devices, the framework is general enough to be recast to a variety of applications, including model-informed drug development and physiologically based pharmacokinetic modelling (Kuemmel et al., 2020; Musuamba et al., 2021; Viceconti et al., 2021b).
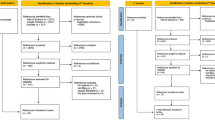
The ASME VV-40:2018 risk-informed credibility assessment framework is shown in Fig. 4.1. Model credibility activities begin by stating the question of interest, which describes the specific question, decision, or concern being addressed (at least in part) by the computational model. The next step is to define the CoU, which aims to describe the role and scope of the model and how it is going to be used in relation to other forms of evidence, e.g., in vitro or in vivo data, to address the question of interest (see Viceconti et al. (2021a) for examples of CoUs). The overall model risk is then assessed for the CoU, where risk is a combination of model influence and decision consequence (see Fig. 4.2). Model influence is defined as the contribution of the computational model relative to other available evidence when answering the question of interest, and decision consequence is the consequence (on the patient, for the clinician, business, and/or regulator) if an incorrect decision is made that is based at least partially on the model. The overall model risk sets the requirements for model credibility, determining the required degrees of model verification, validation, uncertainty quantification, and applicability such that the model has sufficient credibility for the CoU.

Process diagram for the risk-informed credibility assessment framework. Reprinted from ASME VV-40:2018, by permission of The American Society of Mechanical Engineers. All rights reserved

Schematic of how model influence and decision consequence determine overall model risk. Reprinted from ASME VV-40:2018, by permission of The American Society of Mechanical Engineers. All rights reserved
As an example of how the CoU drives risk-informed credibility, a computational model used for a diagnosis that is also supported by medical imaging and clinical assessment would have lower model risk versus a scenario where the diagnosis relies solely on the computational model. As another example, a model used to define the optimal dosing regimen for a phase 3 clinical trial will have a lower risk if it is complemented with exploratory results from an in vivo phase 2 clinical trial than if used alone. Both scenarios illustrate the impact of model influence on risk, where the lack of supporting evidence to answer the question of interest means that the model credibility requirements are greater. As an example of the impact of decision consequence, consider a model used to make decisions about a medical device whose adverse outcome could result in severe patient injury or death. This case will generally be associated with a higher risk than a model used to make decisions regarding a medical device whose adverse outcome would not significantly affect patient safety or health.
Model risk assessment may also be completed with a regulatory impact assessment for certain applications, which describes what evidence would have been provided in the regulatory dossier had it not been for the inclusion of the digital evidence (Musuamba et al., 2020).
4.4 Verification
Verification aims to quantify the part of the predictive error due to the numerical approximations/representations. To effectively separate the three sources of predictive error, the numerical error should be negligible compared to the sum of epistemic and aleatoric errors (see Chap. 2 for details).
There are three possible sources of numerical error in mechanistic models: procedural errors, numerical approximation (round-off) errors, and numerical discretisation errors. The first is explored through code verification, while the second and third are estimated through the calculation verification (Roy & Oberkampf, 2011) (see also ASME VV-10:2019 and ASME VV-20:2009 (R2021)).
4.4.1 Code Verification
Code verification aims to identify and, if possible remove, the source code’s procedural errors and numerical approximation errors in the solution algorithms. Code verification testing should be performed for each computing platform, i.e., hardware configuration and operating system.
Code verification relies on software verification tests such as unit tests, integration tests, and case tests. These tests can be conducted through self-developed or existing automatic software regression suites. In addition, quality control, portability and versioning control should also be considered (see Chap. 2). Code verification also involves developing and implementing a certified software quality assurance (SQA) program to help ensure the integrity of existing code capabilities during development.
In order of increasing rigour, one can use the following code verification methodologies to identify deficiencies: expert judgement, code-to-code comparisons, discretisation error estimation, convergence studies, and calculating the observed order of accuracy, to ensure an appropriate minimisation of numerical approximation errors.Footnote 11 Besides expert judgement and code-to-code comparison, each approach requires comparing code outputs to analytical solutions (or at least mathematical conditions that ensure asymptotic convergence). Traditional engineering problems are one source of analytical solutions, e.g., laminar flow in a pipe or bending of a beam. However, because of their simplicity, these solutions are often limited in their ability to verify the full breadth of the source code. The Method of Manufactured Solutions (MMS) provides a more general source of analytical solutions (Roache, 2019), and the Method of Rotated Solutions (MRS) can be utilised to expand the scope of traditional engineering problems to provide a broader code coverage (Horner, 2021). Documented results from verification studies conducted by the software developer may also serve as a source of data to support code verification; however, since numerical accuracy is also hardware-dependent, it is a good practice to repeat those verification tests on the same hardware that will be used to run the models once in use.
Lastly, it is important to note that the scope of the code verification study must include only those portions of the simulation platform (e.g., model form, element type, solver) that will be accessed as part of validation and model deployment.
4.4.2 Calculation Verification
Calculation verification (also sometimes referred to as solution verification) aims to estimate the upper bound for the numerical approximation error.
A first important step in calculation verification is to estimate the magnitude of numerical errors caused by the discrete formulation of a mathematical model, e.g., due to iterative errors and discretization errors. The purpose of calculation verification is to analyse the numerical solution's spatial and temporal convergence behaviour by refining the discretisation parameters and convergence tolerances and to estimate the numerical errors associated with using a given model.
A sensitivity analysis could also be used to ensure that the calculation does not present a particular combination of input values around which slight variations in the inputs cause significant variations in the outputs (chaoticity). Such occurrence might be due to a software bug or insufficiently robust solver implementations that would have ideally been caught during code verification and to ill-conditioning of the numerical problem due to an unfortunate combination of input values.
Finally, calculation verification should ensure that user errors are not corrupting the simulation outputs. No matter how accurate the calculations, the predictive model would be unreliable if the result is inaccurately transcribed due to a typing error.
4.5 Validation
4.5.1 A General Definition
As outlined in Chap. 2, validation aims to estimate the prediction error and associated uncertainty of a computational model. An essential part of the validation exercise is the evaluation of the model input(s) and output(s) for the various quantities of interest (QoIs) against a comparator, e.g., the experimental data that are used for validation. The comparator should be relevant to the defined CoU and cover a sufficient sample size (“Test samples” in ASME VV-40:2018) as well as the desired range of inputs (“Test conditions” in ASME VV-40:2018). As mentioned in the 2023 FDA guidance (see footnote 10), acceptable forms of comparators include in vitro, ex vivo, or in vivo test data; these tests may be performed ex novo as part of the validation process (e.g., prospective clinical trial) or based upon historical data (e.g., retrospective clinical trial) or real-world evidence. To evaluate the model capacity to predict the QoIs, this comparator cannot be used during model development or the calibration process.
Additionally, uncertainty quantification is critical to validating an in silico study. This includes estimating the uncertainty associated with the comparator inputs and outputs and propagating input uncertainties through the computational model to estimate uncertainty in each QoI.
4.5.2 Definition and Examples
Mechanistic models rely on four distinct elements: governing equations (i.e., the mathematical formulation of the modelled process or phenomenon), system configuration (i.e., the device geometry or in vitro system), system properties (i.e., material properties or physiological parameters) and system conditions (i.e., initial, boundary and loading conditions). Model inputs include initial conditions and parameter values related to model set-up, such as:
-
part geometry specifications,
-
medical imaging settings,
-
material model frameworks and ranges,
-
boundary conditions,
-
specific patient descriptors such as diet, age, weight, or comorbidities for a drug model.
The assessment of the model inputs can be divided into the quantification of sensitivities and uncertainties. Sensitivity analysis is concerned with how variations in input parameters propagate through the simulation and their relative impact on the output(s). The sensitivity study results in a rank-ordered assessment of model input parameters from dominant to negligible impact. On the other hand, quantifying uncertainties enables modelers to propagate known or assumed model input uncertainties to uncertainties in the model predictions. The uncertainty analysis provides an error bar (or confidence interval) associated with each model output. In some scenarios, collecting model inputs is the limiting factor in the credibility assessment. However, the same is often true for all evidence collected by designed experiments or observation.
Typically, validation assessment is framed around comparing the model input(s) and output(s) to experimental data—i.e., the comparator—obtained in a set-up that is well-characterised, well-controlled, and relevant to the CoU. This situation corresponds to category 3 of credibility evidence outlined in the previously discussed FDA guidance (see footnote 10) describing sources of model credibility evidence (Table 4.1). The definition of the comparator should include consideration of both the test samples and the test conditions, where each of these can be defined by their quantity, uncertainty, and other descriptors. An assessment of the validation activities should also be used to establish the similarity of model inputs to those of the comparator and the similarity of the outputs and quality of the output comparison. An example of the various elements of a validation study is provided in Table 4.2.
ASME VV-40:2018 provides a framework to demonstrate that a model captures the physics of a medical device by comparison to a well-controlled benchtop test. However, a model used as part of an in silico clinical trial must be shown to reproduce clinical findings. And while the ASME VV-40:2018 standard refers to clinical trials as possible comparators, detailed considerations are not provided. However, as outlined in the FDA guidance on model credibility evidence, (see footnote 10) a clinical data comparator may be based on in vivo tests performed to support a CoU or population-based data from a clinical study (published, retrospective, or prospective) addressing a similar question of interest. And while the clinical comparator does not have to be targeted to the same device or drug of interest, it should be reasonably similar to ensure appropriate applicability (see Sect. 4.5.2). Examples of validation for a clinical comparator are provided in Tables 4.3 and 4.4.
4.5.3 Validation Layers for in Silico Methodologies
To provide rigorous validation of models used to represent preclinical and clinical studies, the ICH E11(R1) guideline on clinical investigation of medicinal products in the paediatric population suggests starting from “pharmacology, physiology and disease considerations”. As such, the following three layers are suggested:
Physiological layer: This model describes the underlying physiology of a human or animal system, which could model the treatment at the molecular, cellular, organ, to organ systems scales. Associated QoIs are in qualitative or quantitative agreement with an appropriate comparator measured from a healthy system.
Pathological layer: This model describes the disease processes of a human or animal system, which could model the treatment at the molecular, cellular, organ, to organ systems scales. Associated QoIs are in qualitative or quantitative agreement with an appropriate comparator measured from a pathological system.
Treatment layer: This model describes the treatment effect on a physiological or pathological human or animal system (which could model the treatment at the molecular, cellular, organ, to organ systems scales). The model could be used to evaluate whether the produced QoIs are in qualitative or quantitative agreement with an appropriate comparator.
A strict distinction between these layers is not always possible. For example, the layers may be intertwined in the computational model (e.g., physiological and pathological layers) or even non-existent (e.g., a physiological layer doesn’t apply when simulating an in vitro experiment).
Most guidelines recommend describing the assessment of model form in detail, defined in ASME VV-40:2018 as “the conceptual and mathematical formulation of the computational model”. Where present, each of the three layers needs to be described separately and in full detail as appropriate for each modelling activity. But we also recommend providing results of a validation activity following the previously described method for each layer, following the so-called hierarchical validation approach, even if not required by any regulatory guideline or standard. Indeed, there is always the theoretical possibility that the errors of one layer hide those of another layer.
4.5.4 Uncertainty Quantification
The quantitative impact of model uncertainty is estimated by observing how input uncertainties affect the model's outputs. If a particular input is individually measured, the uncertainty is related to the reproducibility of the measurement chain in use; otherwise, it is the distribution of possible values that the input can assume in the reference population. As outlined in Sect. 4.1, performing such analysis is essential within risk-based frameworks.
An elegant theoretical framing of the role of uncertainty quantification in decision-making can be found in (Farmer, 2017) and reviews of numerical methods for sensitivity analysis used in other industrial sectors can be found in (Cartailler et al., 2014; Schaefer et al., 2020). Some early examples are available for pharmacokinetic models (Farrar et al., 1989), cardiac electrophysiology models (Mirams et al., 2016; Pathmanathan & Gray, 2014; Pathmanathan et al., 2015), models for physiological closed-loop controlled devices for critical care medicine (Parvinian et al., 2019), models of intracranial aneurysms (Berg et al., 2019; Sarrami-Foroushani et al., 2016) and in systems biology models (Villaverde et al., 2022).
Uncertainty quantification is a stepwise process (Roy & Oberkampf, 2011) that begins with identifying all sources of uncertainty, followed by quantifying these uncertainties. Uncertainties are then propagated through the model to provide the system response, which can be expressed through probabilities under a given confidence interval. A detailed technical explanation and an illustrative example can be found in (Roy & Oberkampf, 2011). The type of uncertainty quantification method (e.g., intrusive, non-intrusive, via surrogate models, etc.) should be chosen appropriately according to the model under investigation (Nikishova et al., 2019; Smith, 2013).
The discussion so far has implicitly assumed that all model inputs are affected by a quantifiable uncertainty, e.g., due to measurement errors. But in some cases, certain inputs do not refer to an individual but rather to a population, and the uncertainty is dominated by inter-subject variability. In this case, some authors use the term “prediction interval”, which encapsulates quantification uncertainties and inter-subject variabilities (Tsakalozou et al., 2021).
4.5.4.1 Clinical Interpretation of Validation Results
This section is inspired by EMA’s distinction between technical and clinical validation, which is outlined in the guidance for “Qualification of novel methodologies for medicine development”Footnote 12 and suggested in a letter of support to a request for qualification advice on the use of digital mobility outcomes (DMOs) as monitoring biomarkers. This letter stated, “The technical validation will verify the device's accuracy and algorithm to measure a range of different DMOs. […] clinical validation will be obtained in an observational multicentre clinical trial” (see also (Viceconti et al., 2020) for more details). While a letter of support is not the most authoritative source, we are unaware of any official source providing such definitions.
The distinction that some regulators make between technical and clinical validation of new methodologies comes from quantitative biomarkers. Technical validation deals with the accuracy with which the quantification is done (i.e., a metrology problem of accuracy and precision estimation); while clinical validation deals with the validity of using such measurements as evidence in a specific regulatory decision. Traditionally, the accuracy with which quantitative biomarkers are measured is high, so technical validation is considered necessary but not critical. On the other hand, the relationship between a specific biomarker value and the clinical outcome is usually very complex, so clinical validation is considered the challenging part of assessing a new methodology. The complexity of the relationship between biomarker value and the clinical outcome is also an important reason why clinical validation is usually framed in terms of frequentist statistics. The expectation is that prior knowledge about such a relationship is scarcely informative. Thus, the validity of using the biomarker as a predictor of the clinical outcome is qualified only through an extensive induction, where a very large number of clinical experimental validations are required.
A clinical interpretation of the model validation is an assessment of the clinical credibility of the predicted quantities, i.e., in situations where the comparator used in the validation is population-based data collected as part of a clinical trial. This clinical interpretation may be required to satisfy regulatory requirements depending on the CoU. However, there is very little experience and no published guidelines from the regulatory agencies on this topic. For the time being, we propose to extract and interpret key results of the validation process in a manner similar to the one used to demonstrate the regulatory validity of a conventional biomarker used as an outcome measure.
For conventional biomarkers, which are measured experimentally, an outcome measure is considered valid for a well-defined CoU when one can demonstrate its construct validity, its predictive capacity, and its ability to detect change, where:
-
Construct validity is “the extent to which the measure ‘behaves’ in a way consistent with theoretical hypotheses and represents how well scores on the instrument are indicative of the theoretical construct” (Killewo et al., 2010, page 199). Construct validity is typically demonstrated through the evaluation of simulation results (or model behaviour) regarding what is known (either quantitative data or qualitative knowledge).
-
Predictive capacity provides evidence that measures can be used to predict outcomes. It is thus extracted from comparing the model input and output to experimental data obtained in a set-up that is well-characterised, well-controlled, and relevant to the CoU described in the previous section. As written above, experimental data used to demonstrate predictive capability should differ from the data used to develop and calibrate the model.
-
The ability to detect change is the most critical aspect, as it relates directly to the decision-making process at the core of the regulatory process. To demonstrate that a prediction can detect change, it is necessary to demonstrate longitudinal validity, minimal important difference, and responsiveness:
-
Longitudinal validity is the extent to which changes in the prediction will correlate with changes in the outcome over time or with changes in measures that are accepted surrogates for the outcome. Whereas predictive capacity is the correlation of the prediction with the outcome at a given time point, longitudinal validity is the correlation of changes in the predictions with changes in the outcome over time. The relationship between the simulated output and the outcome of interest for the regulatory decision should be evaluated in a manner similar to any other biomarker. This relationship may be obvious when the model output is a clinical endpoint or easily supported if the model predicts a validated biomarker (e.g., a validated surrogate endpoint in the example of tuberculosis vaccine efficacy). However, the model would likely need more supportive evidence if the output does not fall into one of these two categories. This question is mostly treated in the definition of the CoU, where the model’s use to answer the question of interest is described and justified.
-
The Minimal Important Difference (MID) is the smallest change in the outcome identified as important in the patient’s and doctor’s opinion. This requires answering the following question: is the model precise enough to detect the MID for the outcome of interest? The answer to this question is extracted from the uncertainty quantification, representing the prediction confidence interval. The prediction confidence interval cannot include at the same time the MID value and the null value (e.g., absence of difference).
-
Responsiveness to the treatment is the most important attribute for establishing the clinical validity of a predicted biomarker. It can also be described as the model's ability to estimate a clinical benefit. It should be possible to estimate the expected clinical benefit from the estimated predicted change of the simulated result. This last aspect is closely linked to the validation of the treatment layer, where the modelling of the treatment’s impact on the system of interest is evaluated.
-
Reframing the VVUQ results to this entirely different credibility logic poses several challenges. Many in silico methodologies can directly predict the primary clinical outcome or at least a QoI that is already accepted as a valid construct for that specific regulatory decision. In this case, construct validity is already ensured. Otherwise, this evidence needs to be generated using the same approaches used for an experimental QoI; for example, by demonstrating convergent and discriminant validity (see for example the systematic review by Xin and McIntosh (2017)). Predictive capacity and longitudinal validity are two sources of evidence that fit well with the concept of validation according to ASME VV-40:2018. The concept of minimal important difference is also implicit in the VVUQ framework. If the QoI is already accepted in the regulatory practice as a measured value, then there is a good chance that a MID value has already been estimated. Again, an MID study is required if the model predicts a new QoI. The need to demonstrate responsiveness is the one most frequently debated. This typically requires a narrowly defined CoU (specific disease, even a specific range of disease progression, specific class of treatments to be tested) and one or more fully randomised interventional clinical trials, possibly conducted by someone independent from the proponents of the in silico methodology.
But all this makes sense only if we consider an L2 validation (see Chap. 2), where the validation expectation is that the model predicts with sufficient accuracy a central property (e.g., the mean) of the distribution of a certain QoI as observed in a well-defined sub-population. Because the implicit assumption is that the accuracy of the predictor may vary as a function of how we choose the validation sub-population, responsiveness assessment requires validation with a sub-population that is as close as possible to the parameter space used by the in silico methodology (from which the need for a narrow definition of the CoU). But all this would not be valid if the in silico methodology is tested at an L3 level of validity. In this case, each prediction is subject-specific, and the predicted QoI is compared to that measured on the same subject. For such validation, in our opinion, the concept of clinical responsiveness collapses into that Applicability according to the ASME VV-40:2018. Ideally, in an L3 validation study, the predictive accuracy of the in silico methodology is evaluated for the widest possible range of patients, severity of the disease, type of treatments, and even across multiple diseases where this is applicable. This is a critical point that will need to be addressed.
4.6 Applicability of the Validation Activities
Applicability represents the relevance of the validation activities to support using the computational model for the CoU. It includes (i) a systematic review of all validation evidence supporting the use of the model in the CoU, (ii) a precise comparison of the validation context, including both the QoIs and conditions of simulations (e.g., simulated population or experimental conditions and the range of conditions studied) and (iii) a rationale justifying model use despite the potential differences between the validation conditions and the requirements of the CoU. These comparisons are critical since any differences or shortcomings can reduce the overall credibility of the model to answer the question of interest, even in situations where their validation assessment is sufficient. We refer the reader to the framework of Pathmanathan et al. (Pathmanathan et al., 2017), which provides step-by-step instructions for determining validation applicability.
In analogy to what is proposed to evaluate the applicability of the analytical validation activities for biomarkers, we recommend describing and assessing the collection/acquisition, preparation/processing, and storage of the comparator data.Footnote 13
4.7 VVUQ Considerations for Data-Driven Models and Agent-Based Models
The logic behind credibility assessment through VVUQ plus applicability analysis assumes implicitly that the model being assessed is mostly knowledge-driven, and that the causal knowledge used to build the model has resisted extensive falsification attempts; thus, can be considered a scientific truth. Furthermore, the causal knowledge used to build the model is expressed in terms of mathematical equations. In such a case, the credibility assessment aims to quantify the prediction error and decompose it into its components (numerical approximation, aleatoric, and epistemic errors). Then the applicability analysis confirms that the prediction error observed in the validation studies represents an acceptable prediction error across the range of possible input values.
The extension of this reasoning to data-driven models poses some problems. Here we only give a summary; please refer to Chap. 2 for an in-depth discussion. Validation of data-driven models can be performed by calculating the predictive accuracy against one or more annotated datasets (e.g., results of experimental studies where the QoI is measured together with all input values of the model), as long as these datasets were not already used to train the model (test sets). In knowledge-driven models, the epistemic errors are limited to how we implement reliable knowledge in our model; in data-driven models, epistemic errors are not bounded a priori. Numerical approximation errors do not exist when there are no equations to solve; hence some verification aspects may not apply (whereas others, such as software quality assurance, remain). Applicability analysis assumes a certain degree of smoothness in how the prediction error varies over the range of possible input values. While for knowledge-driven models, this assumption descends from the properties of the equations that represent the knowledge, such an assumption is not guaranteed in data-driven models. In principle, an artificial neural network model could have a predictive error of 10% of the measured value for a given set of input values, and an error of 100% for another set of inputs, even if those are quite close to the first set. But the bigger difference is related to the risk of concept drift that all data-driven models face. Data-driven models make predictions by analysing the correlations between inputs and outputs over a set of experimental measurements. Concept drift means the predictive accuracy of a data-driven model decreases over time. This may happen for several reasons, for example, if the data sample used to train the data-driven model is no longer fully representative of the phenomenon being modelled. While there are techniques to reduce this problem, there is never absolute certainty that concept drift will not occur. This is why there is a growing consensus that the credibility of data-driven models must be framed in a Predetermined Change Control Plan, where the predictive accuracy is re-assessed on newly collected data.Footnote 14
Moving to agent-based models (ABM), these are a class of predictive models used in biomedicine. Agent-based models are a generalisation of the concept of cellular automata that was first proposed in the 1940’s. Most ABMs are formulated in terms of rules, through which, at each time step the state transitions of the autonomous agents in the simulation are decided. The key point here is how such rules are defined. If the rules are defined empirically, for example by analysing experimental data, the credibility of that agent-based model should be assessed in a manner similar to data-driven models, with all the implications mentioned above. On the other hand, if the rules descend from quantitative knowledge that has resisted extensive falsification attempts, then the agent-based model should be considered a knowledge-driven model. However, in this second case, some differences apply due to the fact that the knowledge that drives the model is not expressed in terms of mathematical equations, but in terms of rules. This makes the concept of verification more complex to define (see for example (Curreli et al., 2021)).
As this field evolves, more and more sophisticated models will appear. For example, some problems may require a combination of data-driven and knowledge-driven modelling to make accurate predictions. The definition of the correct credibility assessment process for such hybrid models is challenging and cannot be generalised at this time. As a rule of thumb, each model should be classified as predominantly data-driven or as predominantly knowledge-driven, and the credibility assessment process should stem from such classification.
4.8 Final Credibility
Once the credibility assessment is completed, it must be determined if the model is sufficiently credible for the CoU. Note that the CoU can be modified, and the credibility assessment repeated if the model fails the credibility assessment. Alternatively, the model itself, or the credibility activities, can be revisited and improved to reach the required level of model credibility. A comprehensive summary of the computational model, model results and conclusions must be documented and archived upon conclusion of the modelling project.
4.9 Essential Good Simulation Practice Recommendations
-
The credibility of in silico methodologies based on predominantly mechanistic models can be effectively demonstrated following the risk-based approach to model verification, validation and uncertainty quantification as detailed in the ASME VV-40:2018 technical standard. The credibility of methodologies based on predominantly data-driven models should follow a Predetermined Change Control Plan, where the model's credibility is periodically retested using new test data.
-
Where applicable, the validation of predominantly mechanistic models should be performed separately for the physiological modelling layer, the disease modelling layer, and the treatment modelling layers.
-
Regulators qualifying in silico methodologies to be used as drug-development tools expect that prior knowledge is generally scarcely informative.
-
Regulators currently require that in silico methodologies used as drug-development tools are qualified following the same regulatory framework used for experimental methodologies. In particular, the technical validation is expected to be separated from the clinical validation.
-
In analogy to what is proposed to evaluate the applicability of the analytical validation activities for biomarkers, we recommend describing and assessing the collection/acquisition, preparation/processing, and storage of the comparator data used to validate in silico methodologies.
Notes
- 1.
EMA letter “Letter of support for Mobilise-D digital mobility outcomes as monitoring biomarkers”, Apr 2020. https://www.ema.europa.eu/en/documents/other/letter-support-mobilise-D-digital-mobility-outcomes-monitoring-biomarkers_en.pdf.
- 2.
FDA guidance “Exposure-Response Relationships—Study Design, Data Analysis, and Regulatory Applications”, Apr 2003. https://www.fda.gov/media/71277/download.
- 3.
EMA guideline “Guideline on the Reporting of Physiologically Based Pharmacokinetic (PBPK) Modelling and Simulation”, Dec 2018. https://www.ema.europa.eu/documents/scientific-guideline/guideline-reporting-physiologically-based-pharmacokinetic-pbpk-modelling-simulation_en.pdf.
- 4.
FDA guidance “Physiologically Based Pharmacokinetic Analyses—Format and Content”, Dec 2018. https://www.fda.gov/media/101469/download.
- 5.
Please note that the term ‘verify’ is used in place of validate in the guidance document cited in Footnote.
- 6.
FDA guidance “Reporting of Computational Modeling Studies in Medical Device Submissions”, Sep 2016. https://www.fda.gov/media/87586/download.
- 7.
ASME standard “Standard for Verification and Validation in Computational Solid Mechanics V V 10 - 2019”, 2020. https://www.asme.org/codes-standards/find-codes-standards/v-V-10-standard-verification-validation-computational-solid-mechanics.
- 8.
ASME standard “Standard for Verification and Validation in Computational Fluid Dynamics and Heat Transfer V V 20 - 2009 (R2021)”, 2009. https://www.asme.org/codes-standards/find-codes-standards/v-V-20-standard-verification-validation-computational-fluid-dynamics-heat-transfer.
- 9.
ASME standard “Assessing Credibility of Computational Modeling through Verification and Validation: Application to Medical Devices”, 2018. https://www.asme.org/codes-standards/find-codes-standards/v-v-40-assessing-credibility-computational-modeling-verification-validation-application-medical-devices.
- 10.
FDA guidance “Assessing the Credibility of Computational Modeling and Simulation in Medical Device Submissions”, Nov 2023. https://www.fda.gov/media/154985/download.
- 11.
ASME Standard “Standard for Verification and Validation in Computational Solid Mechanics V V 10 - 2019”, 2020. https://www.asme.org/codes-standards/find-codes-standards/v-V-10-standard-verification-validation-computational-solid-mechanics.
- 12.
- 13.
U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), and Center for Biologics Evaluation and Research (CBER), “Biomarker Qualification: Evidentiary Framework Guidance for Industry and FDA Staff DRAFT GUIDANCE,” 2018. https://www.fda.gov/media/119271/download.
- 14.
FDA white paper “Artificial Intelligence/Machine (AI/ML)-Based Software as a Medical Device (SaMD) Action Plan”, Jan 2021. https://www.fda.gov/media/145022/download.
References
Berg, P., Saalfeld, S., Voß, S., Beuing, O., & Janiga, G. (2019). A review on the reliability of hemodynamic modeling in intracranial aneurysms: Why computational fluid dynamics alone cannot solve the equation. Neurosurgical Focus, 47, E15. https://doi.org/10.3171/2019.4.FOCUS19181
Cartailler, T., Guaus, A., Janon, A., Monod, H., Prieur, C., & Saint-Geours, N. (2014). Sensitivity analysis and uncertainty quantification for environmental models. ESAIM: Proceedings, 44, 300–321. https://doi.org/10.1051/proc/201444019
Curreli, C., Pappalardo, F., Russo, G., Pennisi, M., Kiagias, D., Juarez, M., & Viceconti, M. (2021). Verification of an agent-based disease model of human Mycobacterium tuberculosis infection. International Journal of Numerical Method Biomedical Engineering, 37, e3470. https://doi.org/10.1002/cnm.3470
Farmer, C. L. (2017). Uncertainty quantification and optimal decisions. Proceedings of the Royal Society A: Mathematical, Physical and Engineering Sciences, 473, 20170115. https://doi.org/10.1098/rspa.2017.0115
Farrar, D., Allen, B., Crump, K., & Shipp, A. (1989). Evaluation of uncertainty in input parameters to pharmacokinetic models and the resulting uncertainty in output. Toxicology Letters, 49, 371–385. https://doi.org/10.1016/0378-4274(89)90044-1
Horner, M. (2021). The method of rotated solutions: A highly efficient procedure for code verification. Journal of Verification, Validation and Uncertainty Quantification, 6. https://doi.org/10.1115/1.4049322
Killewo, J., Heggenhougen, K., & Quah, S. R. (2010). Epidemiology and demography in public health. Academic Press.
Kuemmel, C., Yang, Y., Zhang, X., Florian, J., Zhu, H., Tegenge, M., Huang, S.-M., Wang, Y., Morrison, T., & Zineh, I. (2020). Consideration of a credibility assessment framework in model-informed drug development: potential application to physiologically-based pharmacokinetic modeling and simulation. CPT: Pharmacometrics & Systems Pharmacology 9, 21–28. https://doi.org/10.1002/psp4.12479
Mirams, G. R., Pathmanathan, P., Gray, R. A., Challenor, P., & Clayton, R. H. (2016). Uncertainty and variability in computational and mathematical models of cardiac physiology. Journal of Physiology, 594, 6833–6847. https://doi.org/10.1113/JP271671
Musuamba, F. T., Bursi, R., Manolis, E., Karlsson, K., Kulesza, A., Courcelles, E., Boissel, J., Lesage, R., Crozatier, C., Voisin, E. M., Rousseau, C. F., Marchal, T., Alessandrello, R., & Geris, L. (2020). Verifying and validating quantitative systems pharmacology and in silico models in drug development: Current needs, gaps, and challenges. CPT Pharmacometrics & Systems Pharmacology, 9, 195–197. https://doi.org/10.1002/psp4.12504
Musuamba, F. T., Skottheim Rusten, I., Lesage, R., Russo, G., Bursi, R., Emili, L., Wangorsch, G., Manolis, E., Karlsson, K. E., Kulesza, A., Courcelles, E., Boissel, J.-P., Rousseau, C. F., Voisin, E. M., Alessandrello, R., Curado, N., Dall’ara, E., Rodriguez, B., Pappalardo, F., & Geris, L. (2021). Scientific and regulatory evaluation of mechanistic in silico drug and disease models in drug development: Building model credibility. CPT: Pharmacometrics & Systems Pharmacology, 10, 804–825. https://doi.org/10.1002/psp4.12669
Nikishova, A., Veen, L., Zun, P., & Hoekstra, A. G. (2019). Semi-intrusive multiscale metamodelling uncertainty quantification with application to a model of in-stent restenosis. Philosophical Transaction: A Mathematical Physical Engineering Science, 377, 20180154. https://doi.org/10.1098/rsta.2018.0154
Parvinian, B., Pathmanathan, P., Daluwatte, C., Yaghouby, F., Gray, R. A., Weininger, S., Morrison, T. M., & Scully, C. G. (2019). Credibility evidence for computational patient models used in the development of physiological closed-loop controlled devices for critical care medicine. Frontiers in Physiology, 10, 220. https://doi.org/10.3389/fphys.2019.00220
Pathmanathan, P., & Gray, R. A. (2014). Verification of computational models of cardiac electro-physiology. International Journal of Numerical Method Biomedical Engineering, 30, 525–544. https://doi.org/10.1002/cnm.2615
Pathmanathan, P., Gray, R. A., Romero, V. J., & Morrison, T. M. (2017). Applicability analysis of validation evidence for biomedical computational models. Journal of Verification, Validation and Uncertainty Quantification, 2. https://doi.org/10.1115/1.4037671
Pathmanathan, P., Shotwell, M. S., Gavaghan, D. J., Cordeiro, J. M., & Gray, R. A. (2015). Uncertainty quantification of fast sodium current steady-state inactivation for multi-scale models of cardiac electrophysiology. Progress in Biophysics and Molecular Biology, 117, 4–18. https://doi.org/10.1016/j.pbiomolbio.2015.01.008
Roache, P. J. (2019). The method of manufactured solutions for code verification. In Beisbart, C. & Saam, N. J. (Eds.), Computer simulation validation: Fundamental concepts, methodological frameworks, and philosophical perspectives, simulation foundations, methods and applications (pp. 295–318). Springer International Publishing, Cham. https://doi.org/10.1007/978-3-319-70766-2_12
Roy, C. J., & Oberkampf, W. L. (2011). A comprehensive framework for verification, validation, and uncertainty quantification in scientific computing. Computer Methods in Applied Mechanics and Engineering, 200, 2131–2144. https://doi.org/10.1016/j.cma.2011.03.016
Sarrami-Foroushani, A., Lassila, T., Gooya, A., Geers, A. J., & Frangi, A. F. (2016). Uncertainty quantification of wall shear stress in intracranial aneurysms using a data-driven statistical model of systemic blood flow variability. Journal of Biomechanics, 49, 3815–3823. https://doi.org/10.1016/j.jbiomech.2016.10.005
Schaefer, J. A., Romero, V. J., Schafer, S. R., Leyde, B., Denham, C. L. (2020). Approaches for quantifying uncertainties in computational modeling for aerospace applications. In AIAA Scitech 2020 Forum, AIAA SciTech Forum. American Institute of Aeronautics and Astronautics. https://doi.org/10.2514/6.2020-1520
Smith, R. C. (2013). Uncertainty quantification: theory, implementation, and applications. SIAM - Society for Industrial and Applied Mathematics.
Tsakalozou, E., Babiskin, A., & Zhao, L. (2021). Physiologically-based pharmacokinetic modeling to support bioequivalence and approval of generic products: A case for diclofenac sodium topical gel, 1. CPT: Pharmacometrics & Systems Pharmacology, 10, 399–411. https://doi.org/10.1002/psp4.12600
Viceconti, M., Emili, L., Afshari, P., Courcelles, E., Curreli, C., Famaey, N., Geris, L., Horner, M., Jori, M. C., Kulesza, A., Loewe, A., Neidlin, M., Reiterer, M., Rousseau, C. F., Russo, G., Sonntag, S. J., Voisin, E. M., & Pappalardo, F. (2021b). Possible contexts of use for in silico trials methodologies: A consensus-based review. IEEE Journal of Biomedical and Health Informatics, 25, 3977–3982. https://doi.org/10.1109/JBHI.2021.3090469
Viceconti, M., Hernandez Penna, S., Dartee, W., Mazzà, C., Caulfield, B., Becker, C., Maetzler, W., Garcia-Aymerich, J., Davico, G., & Rochester, L. (2020). Toward a regulatory qualification of real-world mobility performance biomarkers in Parkinson’s patients using digital mobility outcomes. Sensors (basel), 20, 5920. https://doi.org/10.3390/s20205920
Viceconti, M., Pappalardo, F., Rodriguez, B., Horner, M., Bischoff, J., & Musuamba Tshinanu, F. (2021a). In silico trials: Verification, validation and uncertainty quantification of predictive models used in the regulatory evaluation of biomedical products. Methods, Methods on Simulation in Biomedicine, 185, 120–127. https://doi.org/10.1016/j.ymeth.2020.01.011
Villaverde, A. F., Raimundez, E., Hasenauer, J., & Banga, J. R. (2022). Assessment of prediction uncertainty quantification methods in systems biology. IEEE/ACM Transaction on Computational Biology and Bioinformatics. https://doi.org/10.1109/TCBB.2022.3213914
Viceconti, M., Tome, M., Dartee, W., Knezevic, I., Hernandez Penna, S., Mazzà, C., Caulfield, B., Garcia-Aymerich, J., Becker, C., Maetzler, W., Troosters, T., Sharrack, B., Davico, G., Corriol-Rohou, S., Rochester, L., & the Mobilise-D Consortium. (2022). On the use of wearable sensors as mobility biomarkers in the marketing authorization of new drugs: A regulatory perspective. Frontiers in Medicine, 9.
Xin, Y., & McIntosh, E. (2017). Assessment of the construct validity and responsiveness of preference-based quality of life measures in people with Parkinson’s: A systematic review. Quality of Life Research, 26, 1–23. https://doi.org/10.1007/s11136-016-1428-x
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2024 The Author(s)
About this chapter

Cite this chapter
Courcelles, E. et al. (2024). Model Credibility. In: Viceconti, M., Emili, L. (eds) Toward Good Simulation Practice. Synthesis Lectures on Biomedical Engineering. Springer, Cham. https://doi.org/10.1007/978-3-031-48284-7_4
Download citation
DOI: https://doi.org/10.1007/978-3-031-48284-7_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-48283-0
Online ISBN: 978-3-031-48284-7
eBook Packages: Synthesis Collection of Technology (R0)