
Abstract
Wheat is challenged by rapidly evolving pathogen populations, resulting in yield losses. Plants use innate immune systems involving the recognition of pathogen effectors and subsequent activation of defense responses to respond to pathogen infections. Understanding the genes, genetic networks, and mechanisms governing plant-pathogen interactions is key to the development of varieties with robust resistance whether through conventional breeding techniques coupled with marker selection, gene editing, or other novel strategies. With regards to plant-pathogen interactions, the most useful targets for crop improvement are the plant genes responsible for pathogen effector recognition, referred to as resistance (R) or susceptibility (S) genes, because they govern the plant’s defense response. Historically, the molecular identification of R/S genes in wheat has been extremely difficult due to the large and repetitive nature of the wheat genome. However, recent advances in gene cloning methods that exploit reduced representation sequencing methods to reduce genome complexity have greatly expedited R/S gene cloning in wheat. Such rapid cloning methods referred to as MutRenSeq, AgRenSeq, k-mer GWAS, and MutChromSeq allow the identification of candidate genes without the development and screening of high-resolution mapping populations, which is a highly laborious step often required in traditional positional cloning methods. These new cloning methods can now be coupled with a wide range of wheat genome assemblies, additional genomic resources such as TILLING populations, and advances in bioinformatics and data analysis, to revolutionize the gene cloning landscape for wheat. Today, 58 R/S genes have been identified with 42 of them having been identified in the past six years alone. Thus, wheat researchers now have the means to enhance global food security through the discovery of R/S genes, paving the way for rapid R gene deployment or S gene elimination, manipulation through gene editing, and understanding wheat-pathogen interactions at the molecular level to guard against crop losses due to pathogens.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others

Keywords
10.1 Introduction
Pathogens and pests pose a significant threat to global food security, affecting not just primary yields, but also the stability and distribution of production and the quality of food (Savary et al. 2017). An estimated 21.47% of global wheat yields are lost annually due to pathogens and pests (Savary et al. 2019), equating to ~210 million metric tons of grain per year, enough to bake 290 billion loaves of bread (Wulff and Krattinger 2022). Combining agronomic practices that reduce the initial disease inoculum and infection rate with selection of genetically resistant varieties is an effective crop disease management strategy, and to develop genetically resistant wheat, resistance (R) genes need to be identified, characterized, and deployed. In some diseases, for example tan spot or septoria nodorum blotch, susceptibility is conferred by dominant genes. In these cases, the priority is to remove or disrupt susceptibility (S) genes rather than deploy novel R genes. Gene cloning is crucial to the efficient deployment of R genes and removal of S genes, requiring the identification of the nucleotide sequence of a target gene and validating its function. Diversity and functional studies can assess the effects of genetic variation within an R or S gene on their respective resistance/susceptibility, allowing researchers to develop molecular markers targeting the variants, which can then be used to select breeding lines with the most beneficial alleles. Cloned R genes can also be introduced into modern cultivars via gene complementation or cross-hybridization, and S genes can be removed through marker-assisted elimination or gene editing. The methods and resources used to clone R and S genes are shared, and as such R and/or S genes will be referred to as “R/S genes” in this chapter.
Although over 460 R/S genes in wheat have been described (Hafeez et al. 2021), only 58 have been cloned (Table 10.1). The genome of hexaploid bread wheat is large and repetitive due, in part, to its evolutionary history, making it challenging to clone R/S genes. The basic seven-chromosome Triticeae progenitor split into the Triticum and Aegilops branches about 3 million years ago (MYA) (reviewed by Faris 2014). Modern-day bread wheat (Triticum aestivum ssp. aestivum L., 2n = 6x = 42, AABBDD) is an allohexaploid that evolved as a result of two amphiploidization events involving the hybridization of two different species followed by spontaneous chromosome doubling through meiotic restitution division, several mutations, and interspecific gene flow. Around 0.5 MYA the wild diploid species T. urartu Tumanian ex Gandylian (2n = 2x = 14, AA) hybridized with a species similar to Aegilops speltoides Tausch (2n = 2x = 14, SS) to form tetraploid wheat Triticum turgidum ssp. dicoccoides Thell (2n = 4x = 28, AABB), also known as wild emmer. T. turgidum ssp. durum (2n = 4x = 28, AABB), durum wheat, is a free-threshing derivative of T. turgidum ssp. dicoccoides, and it is today widely cultivated and used to make pasta and other semolina-based products. The second amphiploidization event occurred around 8000 years ago. A T. turgidum ssp. and the diploid wild goat grass Aegilops tauschii Coss. (2n = 2x = 14, DD) hybridized to form hexaploid (common or bread) wheat T. aestivum (2n = 6x = 42, AABBDD). Due to the differential presence of Ae. tauschii lineage specific sequences in modern cultivars, it is possible that more than one hybridization even occurred between T. turgidum spp. and Ae. tauschii (Gaurav et al. 2022). Together, bread and durum wheat provide about 18% of the caloric intake of humans worldwide, but in some regions of the world, wheat accounts for over a third of the caloric and protein intake (Erenstein et al. 2022).
Despite their polyploid nature, bread and durum wheat behave like diploid plants genetically, with homologous chromosomes pairing and segregating during meiosis. The pairing of homoeologous chromosomes is prevented by genes Ph1 and Ph2 (Riley and Chapman 1958; Sears and Okamoto 1958; Mello-Sampayo and Lorente 1968) with the resulting diploid-like pairing of wheat chromosomes in meiosis simplifying segregation studies and genetic mapping of traits. Due to their formation through amphiploidization, hexaploid and tetraploid wheats often have three or two copies of each gene, respectively, called homoeologous genes. Homoeologous genes are often highly conserved, with ~97% identity across their coding regions (Schreiber et al. 2012), and this high sequence conservation among homoeologous genes hinders the development of homoeolog-specific molecular markers. Additionally, approximately 85% of the wheat genome is comprised repetitive elements (Wicker et al. 2018), making it difficult to design molecular markers that only target one locus for use in molecular mapping or marker-assisted selection.
Bread and durum wheat genomes are relatively large at 12 and 17 Gb, respectively (Bennett and Smith 1976). The sequencing and assembly of such large genomes are computationally challenging and further complicated by the highly repetitive nature of wheat genomes and interchromosomal gene duplications (IWGSC et al. 2014). The complexity of the wheat genome has hampered the generation of genomic data and bioinformatic analysis. Despite the challenges, multiple high-quality genome assemblies have been constructed (Table 10.2). Genome assemblies are used to design molecular markers and bait libraries, assess candidate genes, and evaluate structural variation as well as acting as a foundation for developing genomic resources and tools that aid in the cloning of R/S genes.
The first cloned S and R genes in wheat, TaMlo-B1 and Lr10, were published in 2002 (Elliott et al. 2002) and 2003 (Feuillet et al. 2003), respectively. Since then, 48 more R/S genes have been cloned from Triticum or Aegilops species, and an additional eight R/S genes have been cloned from related species and shown to be functional in wheat (Table 10.1, current as of 8/1/2022). In just the last two years, more R/S genes were cloned than were cloned in the first decade of R/S gene cloning. Here, we review the surge of genomic resources and gene cloning methods that have contributed to the acceleration of R/S gene cloning in wheat.
10.2 Advances in Wheat Genome Sequencing
High-quality genomic sequences and assemblies act as the basis for gene cloning efforts in wheat, and the recognition of this requirement led to the formation of the International Wheat Genome Sequencing Consortium (IWGSC) in 2005. Several hexaploid, tetraploid, and diploid Triticum full genome assemblies have been released in the last five years (Table 10.2). The bread wheat variety CHINESE SPRING was selected for sequencing due to the extensive genetic and molecular resources developed using this variety (Gill et al. 2004), including aneuploid stocks developed by Ernie Sears that could be used to physically map genes and markers to specific chromosomes (Sears 1954, 1966; Sears and Sears 1978). Segmental deletion lines (Endo and Gill 1996) further specified physical regions within chromosomal arms and were used to map 16,000 expressed sequence tag (EST) loci (Qi et al. 2004).
Hexaploid wheat was estimated to be 17 Gb and included families of DNA sequences that were highly repetitive (Bennett and Smith 1976). A reduced-representation sequencing approach was used to reduce the genome complexity and size (IWGSC et al. 2014), making use of CHINESE SPRING ditelosomic stocks developed by Sears and Sears (1978) to isolate each chromosome arm by flow cytometry, and BAC libraries were subsequently constructed from the DNA of individual arms. The bin-mapped ESTs were used to assess the purity of the sorted chromosome fractions (Qi et al. 2004). Short read paired-end sequences of each BAC library were assembled resulting in a 10.2 Gb draft assembly referred to as the Chinese Spring Survey Sequences (CSS) and represented 61% of the genome sequence (IWGSC et al. 2014).
A pseudomolecule level assembly of chromosome 3B was produced separately using a minimum tiling path of 8,452 BACs sequenced with Roche/454 paired-end reads (Choulet et al. 2014). After scaffold assembly, Illumina reads from flow sorted chromosome 3B were used to fill gaps. A detailed SNP-based genetic map from the CHINESE SPRING × RENAN population was used to orient and order scaffolds. Ultimately, the pseudomolecule level assembly represented 93% of chromosome 3B. A total of 124,201 high-confidence gene loci were annotated in the CSS and chromosome 3B assembly (IWGSC et al. 2014).
Whole genome shotgun (WGS) assemblies of the Triticum turgidum ssp. durum cultivars CAPPELLI and STRONGFIELD were released in 2014 alongside an assembly of Ae. speltoides accession ERX391140 (SS) (IWGSC et al. 2014). Although these assemblies consisted of numerous small contigs with unknown order, orientation, and space between contigs, partly due to the piling of repetitive elements, they offer a draft assembly of low-copy DNA and therefore can be used to identify alleles, design gene-specific markers, or compare genes and gene families among assemblies. Chapman et al. (2015) integrated WGS and genetic mapping to assemble and order contigs of the synthetic hexaploid W7984. Despite the WGS method and lack of chromosome isolation via flow sorting, the assembly was 9.1 Gb, just 1.1 Gb smaller than the CSS assembly.
With the growth of sequencing and assembly methods, more wheat scaffold and pseudomolecule level assemblies became available (Figs. 10.1 and 10.2). As of August 2022, 46 unique accessions have scaffold and/or pseudomolecule level assemblies (Table 10.2). In 2020, there was a significant increase in the number of hexaploid accessions with pseudomolecule or scaffold level assemblies. Through a large international collaborative effort, Walkowiak et al. (2020) published the 10+ Wheat Genomes’ paper, which included pseudomolecule assemblies of nine bread wheat lines and one T. aestivum ssp. spelta accession plus the scaffold level assemblies of five additional bread wheat lines. Prior to this, CHINESE SPRING and the synthetic hexaploid W7984 were the only hexaploids with either a pseudomolecule or scaffold level assembly. Principal component analysis of exome sequence capture alleles in ~1200 hexaploid accessions revealed that CHINESE SPRING was genetically distant from other hexaploid wheats (Walkowiak et al. 2020). The accessions included in the Walkowiak et al. (2020) paper were selected to more accurately represent the full diversity of hexaploid wheat allowing analysis of intergenome variability. The genome of the Tibetan semi-wild wheat (T. aestivum ssp. tibetanum Shao) accession ZANG1817 was also published the same year (Guo et al. 2020).

Cumulative accessions with pseudomolecule level assemblies. Color corresponds to the subgenome of the accession

Cumulative accessions with scaffold level assemblies. Color corresponds to the subgenome of the accession
Most of the Triticum and Aegilops assemblies and genome browsers are hosted on websites. Not all assemblies are hosted on a single website and different assembly and annotation versions are available on different websites, so care should be taken when comparing assemblies or annotations from different sources. Many of these websites host additional resources that may be useful in the gene cloning and characterization process, such as molecular markers, exome capture data, varietal SNPs, and TILLING mutants.
The following are useful websites for accessing the genome assemblies:
-
GrainGenes (Yao et al. 2022)—https://wheat.pw.usda.gov.
-
Ensembl Plants—http://plants.ensembl.org/Triticum_aestivum.
-
Grassroots Infrastructure—https://grassroots.tools/service/blast-blastnCerealsDB.
-
https://www.cerealsdb.uk.net/cerealgenomics/CerealsDB/blast_WGS.php.
10.3 Map-based Cloning
Map-based cloning was used to clone the first wheat R gene, Lr10 (Feuillet et al. 2003). Since then, map-based cloning has been the most frequently used method to clone R/S genes in wheat (around 50%, Table 10.1). Map-based cloning uses the genetic relationship between a gene and molecular markers to place a gene on a genetic map. Originally, an iterative approach termed chromosome walking was used to define the candidate gene region. The two closest molecular markers were then used to screen large-insert libraries of cloned fragments of DNA (yeast artificial chromosomes or bacterial artificial chromosomes, YACs or BACs) to identify flanking clones, and new markers developed from the ends of the clones were used to rescreen the library and “walk” closer to the gene of interest until a clone containing the gene was identified. Sequencing of the clone(s) spanning markers defined by flanking genetic recombinants would reveal the nucleotide sequence of the R/S gene. While we still use the term “cloning,” the development and screening of large-insert genomic clones are seldom still necessary to clone a gene. The development of molecular markers and subsequent high-density, or saturation, mapping of target R/S genes in segregating populations is a critical step in the map-based cloning process. Historically, high-density mapping was conducted on a low-throughput basis using markers such as restriction fragment length polymorphisms (RFLPs), amplified fragment length polymorphisms (AFLPs), or simple-sequence repeats (SSRs, or microsatellites). Recent advances in high-throughput genotyping technologies such as Diversity Arrays Technology (DArT), DNA SNP arrays, custom Kompetitive allele-specific PCR (KASP) arrays, or genotyping by sequencing offer high-density genotyping at affordable costs. These genotyping technologies can also be used in combination with a bulked segregant analysis (BSA) approach to quickly find markers associated with a phenotype without having to genotype a large mapping population (see also Chap. 9).
The size of the candidate gene region, as defined by the genetic region between the closest markers flanking the R/S gene, is dependent on both the marker density and the recombination rate. In a population of fixed size, such as a recombinant inbred or doubled haploid population, there is a finite number of recombination events. Sometimes, there are not enough recombination events in a population to reduce the candidate gene region to a reasonable size. If the marker density is too low, recombination events can go undetected, resulting in a larger candidate gene region. Additional molecular markers in a region cosegregating with the gene will not increase resolution. Even in cases where marker density and recombination rate are high, a candidate gene region may be gene-rich, making it difficult to identify the trait-associated gene. Map-based cloning also requires access to the DNA sequence between the flanking markers. This need is often met by the multiple sequenced wheat genomes. It is important to remember that even if the sequenced wheat genotypes do not carry a functional allele of a target R/S gene, they may carry a nonfunctional allele. As such, it may be useful to identify candidate genes even in genotypes that do not display the desired resistant or susceptible phenotype. If the phenotypes of the sequenced wheat genomes are known, candidate genes may be eliminated based on a comparison of gene content between lines with and without the trait of interest (Running and Faris, unpublished).
If the sequenced wheat genotypes do not carry an allele of the R/S gene, or when the R/S gene is in an area of low recombination, such as an introgressed segment from a wild relative or near a centromere, alternate gene cloning methods may be more appropriate. Map-based cloning can be slow, dependent on the generation of the mapping population, and requires screening of 1000’s of recombinant gametes.
10.4 Reduced-Representation Sequencing Methods
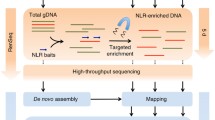
Reduced-representation sequencing (RRS) is a key step in the rapid cloning methods that are used in wheat (described below), and it can be combined with traditional map-based cloning methods to quickly identify candidate genes. RRS reduces genome complexity and therefore the cost and time of sequencing and analysis. The three main methods of RRS are transcriptome or RNA sequencing, exome capture, and chromosome flow sorting (Fig. 10.3). These methods allow preferential sequencing of more relevant spaces, either genic regions or promoters, or the specific chromosome containing an R/S gene. In some cases, RRS methods are incorporated into rapid cloning methods.

Reduced sequencing methods. a Transcriptome sequencing. RNA is isolated from tissue and reverse transcribed into cDNA, which is sequenced and mapped to a reference assembly. b Exome sequencing. DNA is isolated from tissue and a DNA sequencing library is prepared. Short biotinylated baits complementary to the targets hybridize to the DNA, bind to magnetic beads, and are captured by a magnet, yielding a target-enriched sequencing library. Exome sequencing can target the whole exome or a particular gene family such as NLRs as is done in the RenSeq method. c Chromosome flow sorting. Liquid suspensions of mitotic chromosomes collected from dividing root cells are fluorescently labeled and separated using flow cytometry based on the fluorochrome signal and relative DNA content
10.4.1 Exome Capture
In exome capture, the baits, or capture probes, hybridize to the targets and then are bound by streptavidin-coated magnetic beads. The magnetic beads are “captured” by a magnet, unbound DNA is washed away, and the remaining target-enriched library is amplified and sequenced. Capture probes’ assays can target genes, promoters, and even specific types of genes like nucleotide-binding domain leucine-rich repeat containing genes (NLRs). Exome capture assays targeting the genic regions of wheat have been designed from the sequenced wheat genomes, each using an increasing design space size as additional wheat genome sequences became available.
Jordan et al. (2015) designed an exome capture probe assay called the “wheat exome capture” (WEC) using a design space of 110 Mb from a 3.8 Gb low-copy number genome assembly of CHINESE SPRING (Brenchley et al. 2012). To identify genic regions, they aligned reported wheat cDNA and EST sequences and conducted a BLASTn search using Brachypodium exon sequences. Krasileva et al. (2017) designed T. turgidum and T. aestivum exome capture probes to target gene annotations from the CSS assembly, transcripts from transcriptome studies, and unannotated homologs of barley in wheat. The exome capture probes targeted 85 Mb. Following the publication of high-quality reference wheat genome assemblies and annotations in 2017 and 2018, Gardiner et al. (2019) discovered that the existing exome capture assay only targeted 32.6% of the high-confidence gene set of wheat. Using the high-confidence annotated genes in the CHINESE SPRING-TGACv1 and RefSeq.v1 genome assemblies, Ae. tauschii assembly Aet v4.0, and the T. turgidum ssp. dicoccoides WEWSeq v1.0 assembly, they designed exome capture probe sets targeting genes and putative promoters. Probes of ~75 bp were designed approximately every 120 bp across 786 Mb of design space, of which 509 Mb was gene space, and 277 Mb was putative promoter sequences. The exome capture and promoter capture probe sets designed by Jordan et al. (2015), Krasileva et al. (2017), and Gardiner et al. (2019) were available through NimbleGen (Roche) but have since been discontinued. The most recent exome capture assay, the myBaits® Expert Wheat Exome capture, designed using the CHINESE SPRING-RefSeq v1.0 assembly, captures over 250 Mb of coding sequence (Daicel Arbor Biosciences).
To further reduce genome complexity, capture probes assays can be developed to target a particular gene class such as NLRs. NLRs are the most common class of cloned R/S gene in wheat (Table 10.1), and the wheat pangenome is estimated to contain 6–8 thousand NLR genes (Walkowiak et al. 2020). Exome capture of NLR genes and subsequent sequencing is termed Resistance gene enrichment Sequencing (RenSeq). The first R genes cloned using RenSeq were Rpi-ber2 and Rpi-rzc1, which confer resistance against Phytophthora infestans infections in potato (Jupe et al. 2013). Since then, RenSeq has been incorporated into rapid cloning methods AgRenSeq and MutChromSeq (discussed below). RenSeq was also recently combined with BSA in a method termed BSR-Seq (Lin et al. 2022). RenSeq was applied to DNA pools of resistant and susceptible plants allowing the identification of SNPs in NLRs linked to resistance. RenSeq is a key method in multiple rapid cloning strategies, efficiently enriching NLR genes. Kale et al. (2022) found the Triticeae RenSeq Baits V3 probe set (Zhang et al. 2021a) resulted in target enrichment of 220-fold of 18 Mb of NLR genes annotated in CHINESE SPRING-RefSeq v1.0. However, because probes were designed to target previously annotated NLR genes, RenSeq captures are biased and may not capture unannotated NLRs, i.e., NLRs not present in the sequences and annotated genome assemblies. RenSeq also relies on the assumption that the target R/S gene is a member of the NLR class. If it is suspected that the target gene might belong to a different class, then other methods should probably be considered.
10.4.2 Transcriptome Sequencing
Transcriptome sequencing, or RNA-Seq, is a less biased RRS method as it is not limited to previously annotated genes and/or a gene family. RNA-Seq combined with BSA (BSR-Seq) was applied to two Ae. tauschii populations to map leaf rust resistance gene Lr42, yielding just three candidate genes (Lin et al. 2022). RNA-Seq is limited to detecting genes that are expressed at the time of RNA collection in sufficient levels, and assembly of transcripts can be challenged by the co-expression of homoeologs. Lin et al. (2022) avoided the latter challenge by conducting RNA-Seq on a diploid.
10.4.3 Chromosome Flow Sorting
Chromosome flow sorting separates an individual chromosome via flow cytometry based on the chromosome size and base-pair composition (Doležel et al. 2011). Following separation, the individual chromosome can be sequenced and assembled as was done to complete the CSS assembly (IWGSC et al. 2014). Chromosome flow sorting is a highly specialized skill requiring unique equipment available in few labs. Also, not all chromosomes are able to be sorted from all others at sufficient efficiency to obtain a sample with adequate purity, and the time and labor needed to develop cytogenetic stocks such as the ditelosomics developed by Sears and Sears (1978) in CHINESE SPRING preclude that from being a viable option. Therefore, it is important to first determine whether a target chromosome can be efficiently sorted using flow cytometry before embarking on a project that relies on it to be successful.
10.5 Rapid Cloning Methods
10.5.1 MutRenSeq
RenSeq is coupled with mutational genomics in the MutRenSeq rapid cloning strategy (Steuernagel et al. 2016). In the MutRenSeq method, a mutant population is screened to identify the expected mutant phenotype and then RenSeq is conducted on confirmed mutants (Fig. 10.4). Independent mutation events within a single NLR associated with the mutant phenotype reveal the candidate gene(s). Sr22 and Sr45 were the first wheat R/S genes cloned in wheat using MutRenSeq. Sr22, which provides stem rust resistance, resides in introgressions from T. boeoticum and T. monococcum that had poor agronomic performance due to linkage drag (Olson et al. 2010). Additionally, mapping efforts were hampered by reduced recombination in the Sr22 region (Steuernagel et al. 2016). To clone stem rust resistance genes Sr22 and Sr45, Steuernagel et al. (2016) developed EMS-mutant populations for each R gene and applied RenSeq to six mutants/population and the wild type. In each mutant population, comparative sequence analysis of the NLRs in the mutants and wild type revealed one gene with mutations in all six mutants. MutRenSeq effectively eliminated the need for high-resolution mapping, which is particularly difficult when the R/S gene of interest resides in a low recombination region. MutRenSeq has since been used to clone stem rust resistance genes Sr26, Sr27, and Sr61, stripe rust resistance genes Yr5 and Yr7, leaf rust resistance gene Lr13/Ne2, and powdery mildew resistance gene Pm21 (Xing et al. 2018; Marchal et al. 2018; Zhang et al. 2021a; Hewitt et al. 2021a, b; Yan et al. 2021; Upadhyaya et al. 2021).

Overview of R/S gene rapid cloning methods in wheat. The paths of MutChromSeq (orange), MutRenSeq (red), AgRenSeq (teal), and k-mer GWAS (seafoam) are shown with stops at particular methods numbered and connected with solid lines. a Source of phenotypic variation. Rapid cloning methods use one of two forms of phenotypic variation, induced phenotypic variation through mutagenesis (left) or natural variation in a diversity panel (right). b Genome complexity reduction. After phenotyping, the next step is genome complexity reduction through either chromosome flow sorting or R gene enrichment through gene family capture. Note, the k-mer GWAS path moves directly to sequencing. c Sequencing. Next, the flow sorted chromosome, captured genes, or diversity panel is/are sequenced. Depending on the target, personal preferences, and resources available, different sequencing methods may be used. d R/S gene identification. The final step involves identifying candidate genes. Left, candidate genes are identified through comparison of mutant (light teal) and wild-type sequences to identify regions with mutation overlap. Right, associations between particular NLRs or k-mers are identified with the highest associations being candidate genes or near candidate genes. Association genetics yields candidate genes that require functional validation while methods using induced variation through mutagenesis already include a functional validation step
MutRenSeq is a powerful tool to quickly clone NLR resistance genes and is particularly advantageous when trying to clone a gene in an area of low recombination. However, it is limited to genotypes that can be easily mutagenized and R genes in the NLR family. In general, higher ploidy levels tend to tolerate higher EMS levels. The lower tolerance of mutagen dose results in lower mutation density, increasing the number of mutants that must be generated and phenotypically evaluated to identify independent lines with mutant alleles. In some cases, mutagenesis of diploids can result in sterile plants making the MutRenSeq method a less attractive option.
10.5.2 AgRenSeq
To address the limitations of MutRenSeq, Association Genetics RenSeq (AgRenSeq) was developed (Arora et al. 2019) by combining association genetics and RenSeq. A diversity panel is phenotyped for disease reactions and RenSeq is conducted on the panel. K-mers within the sequenced NLR are identified and mapped to a reference assembly. Associations between k-mers and phenotypes are then calculated and plotted, similar to a Manhattan plot. Significant k-mers map to contigs that represent candidate genes. To test AgRenSeq, a panel of 174 Ae. tauschii ssp. strangulata accessions was genotyped and evaluated for reaction to six races of wheat stem rust pathogen Puccinia graminis f. sp. tritici (PGT). Two previously cloned genes, Sr33 and Sr45, served as positive controls (Periyannan et al. 2013; Steuernagel et al. 2016). K-mers associated with resistance to PGT race RKQQC, which is avirulent to Sr33, resided on the contig containing the previously cloned Sr33. Sr45, which was previously identified using MutRenSeq (Steuernagel et al. 2016), was also identified via AgRenSeq. Candidate genes for Sr46 and SrTA1662 were also identified in this study, and the Sr46 candidate was functionally validated by mutagenesis and gene complementation. Thus, Arora et al. (2019) demonstrated the ability of AgRenSeq to directly identify candidate genes. However, as with other RenSeq-based cloning methods, AgRenSeq is limited to cloning NLR genes.
10.5.3 K-mer GWAS
K-mer-based association mapping, or k-mer GWAS, is an extension of AgRenSeq, but it excludes the RenSeq step and is therefore not limited to the detection of only NLRs. Instead, k-mers are identified from whole-genome shotgun sequencing reads and projected onto a reference assembly. The analysis is similar to AgRenSeq, but because k-mers can be anywhere and not just within candidate genes, one must analyze the genes near the k-mers that were in linkage disequilibrium with the phenotype. Gaurav et al. (2022) conducted whole-genome shotgun sequencing on 242 Ae. tauschii accessions and used k-mer GWAS to identify a 50-kb linkage disequilibrium block containing two candidate genes for the stem rust resistance gene SrTA1662. Subsequent functional validation via gene complementation confirmed that SrTA1662 is an NLR. The panel sequenced in Gaurav et al. (2022) is publicly available and can be used to rapidly clone R/S genes from Ae. tauschii accessions.
In 2020, Voichek and Wiegel published a reference-free k-mer GWAS method. In this method, the associations between k-mers and the phenotype were calculated prior to mapping the k-mers to a reference, allowing the identification of k-mers significantly associated with the trait, including those absent in a reference. In a case study in Arabidopsis, the authors identified k-mers significantly associated with two traits—growth in the presence of a flg22 variant and germination in darkness under low nutrient supply—neither of which mapped to their reference genome. Assembly and subsequent analysis of the short reads used to identify the significant k-mers revealed alternate structural variants of genes associated with the two traits. Although reference-free k-mer GWAS has not yet been used to clone R/S genes in wheat, it has been applied to map resistance to yellow rust and leaf rust (Kale et al. 2022). R/S genes display abundant presence/absence and copy number variation (Van de Weyer et al. 2019; Walkowiak et al. 2020), so the potential to detect structural variants not in a reference assembly via reference-free k-mer GWAS is appealing.
Both AgRenSeq and k-mer-GWAS require shot gun sequencing of an entire diversity panel, which can initially be expensive and laborious. However, once this has been completed, the same panel can be used to clone multiple R/S genes. Additionally, AgRenSeq and k-mer GWAS can be limited by the population structure of the diversity panel (Yu et al. 2006) and choice of the reference sequence can influence which associations are detected (Voichek and Weigel 2020; Kale et al. 2022).
10.5.4 MutChromSeq
In 2016, Sánchez-Martín et al. published the rapid cloning method MutChromSeq and used it to clone the powdery mildew resistance gene Pm2a, which had previously mapped to chromosome 6A (Huang and Röder 2004). Using the MutChromSeq method, which applied the RRS method chromosome flow sorting, chromosome 6A was sorted from six confirmed EMS-derived powdery mildew susceptible mutants and wild-type genotypes. The separated chromosomes were sequenced and assembled followed by sequence analysis to identify mutation overlap. Contigs with mutations in all or most of the mutant lines are most likely to contain the candidate gene. Two contigs were identified, although one was later discarded due to an abnormal SNV frequency, leaving just one contig with a NLR gene. MutChromSeq is similar to MutRenSeq, but it is not limited to NLR genes. MutChromSeq was also used to clone leaf rust resistance gene Lr14a with ankyrin transmembrane protein domains and Pm4b, which contains kinase, C2, and transmembrane domains (Kolodziej et al. 2021; Sánchez-Martín et al. 2021).
10.6 Validating Candidate Genes
Validating candidate genes is a critical step in proving a gene confers a particular phenotype. Forward and reverse genetics approaches can be used to identify and validate candidate genes. Forward genetics approaches start from a phenotype and identify the gene that confers the phenotype (Fig. 10.5). Many of the rapid cloning methods are considered forward genetics approaches as they start with variation in a phenotype, either natural or induced through mutagenesis. However, not all forward genetics approaches serve as functional validation methods. For example, map-based cloning and association genetics approaches often yield multiple candidate genes and must be followed up with functional validation to determine which candidate gene is the gene of interest. Because rapid cloning methods MutRenSeq and MutChromSeq use mutagenized populations, these methods both identify and validate candidate genes.

Commonly used forward and reverse genetics methods to identify and/or validate R/S genes in wheat. Arrows indicate the direction of the genetic approaches with forward genetics approaches starting with a known phenotype and identifying the gene underlying the phenotype, while reverse genetics starts with a known gene sequence and identifies the phenotypic effects of genic or transcriptomic alternations. Common methods used to identify and/or validate R/S genes in wheat are listed under their approach type
Reverse genetics approaches start with the gene sequence and identify the phenotypic effects of particular gene states (Fig. 10.5). Functional validation methods that use a reverse genetics approach include methods like RNA interference, gene complementation, or CRISPR/CAS9 gene editing, which alter the genetic or transcriptomic makeup of an individual to identify the phenotypic effect of the alteration. The Targeting Induced Local Lesions in Genomes (TILLING) resource can also be used to functionally validate genes in a reverse genetics manner. Krasileva et al. (2017) sequenced the exomes of 1200 CADENZA and 1535 KRONOS EMS mutants and characterized and cataloged the mutations relative to the CSS assembly. When the CHINESE SPRING-RefSeqv1.0 assembly was published, the TILLING raw reads were realigned to the new assembly. The TILLING resources expedite functional validation of genes as researchers do not need to create the genetic or transcriptomic alteration. Instead, mutant lines with known alterations in candidate genes can be selected on Ensembl Plants and ordered from SeedStor (https://www.seedstor.ac.uk/). However, the TILLING resource is limited to functionally validating genes present in CADENZA or KRONOS and annotated in the CHINESE SPRING-RefSeqv1.1 gene models.
Often both forward and reverse genetics approaches are applied to functionally validate R/S genes. The two most commonly used functional validation methods are mutagenesis and gene complementation, both of which have been used to validate around two-thirds of the cloned R/S genes. About 43% of the cloned R/S genes have been validated using both mutagenesis and gene complementation. Gene silencing, transient expression, gene editing, and the TILLING populations are less frequently used methods of functional validation, with each being used to validate 15 or fewer R/S genes.
Clustered regularly interspaced short palindromic repeats (CRISPR) and its associated protein (Cas) can be used to produce site-specific double-stranded breaks, often resulting in gene knockout. Wang et al. (2014) used CRISPR-Cas9 and transcription activator-like effector nuclease (TALEN) technologies to knock out three homoeoalleles of the powdery mildew susceptibility gene Mlo in the cultivar Bobwhite, resulting in reduced susceptibility to Blumeria graminis f. sp. tritici. While CRISPR-Cas mediated gene knockout is a highly specific and targeted functional validation method, unlike random mutagenesis, it is somewhat limited to functionally validating genes present in easily transformable cultivars such as FIELDER or BOBWHITE. However, advancements in gene editing and transformation methods are expanding the definition of “transformable cultivars.”
10.7 Conclusions and Future Outlook
The expansion of wheat genomic resources, genomic complexity reduction methods coupled with advanced sequencing technologies, and rapid cloning methods has enabled the accelerated cloning of R/S genes in wheat. In 2020 and 2021, sixteen cloned R/S genes were published, a feat that in the earlier years of R/S gene cloning took thirteen years to accomplish; it was not unheard of for cloning an R/S gene to take 10 years. Now, cloning an R/S gene is possible in less than a year. Undoubtedly, R/S gene cloning will continue to accelerate as more reference genomes are published, sequencing costs decrease, and cloning methods advance. The multiple sequenced wheat genomes that are currently available are a tremendous resource and make it relatively easy to assess gene content in a given R/S gene candidate gene region. However, given the common presence/absence and copy number variation displayed by R/S genes (Van de Weyer et al. 2019; Walkowiak et al. 2020), it is still possible for a gene of interest to be absent in all the wheat genomes currently available. We have not yet reached a true wheat pangenome, but costs for sequencing and assembly of entire wheat genomes continue to decline, and the data can be obtained in a matter of months. Therefore, it is now becoming more feasible to sequence and assemble the entire genome of a wheat line with the primary goal of cloning a single R/S gene, a feat that was nearly unthinkable when wheat genomics researchers met in 2003 to carve a path forward to sequence the first wheat genome (Gill et al. 2004).
Wheat’s wild relatives offer a greater pool of R genes, as they have not undergone the genetic bottleneck characteristic of domestication. Association genetics methods, like k-mer GWAS and AgRenSeq, address some of the limitations of traditional map-based cloning, exploiting greater genetic diversity and ancestral recombination to identify unique disease resistance loci. Additionally, these diversity panels often allow the isolation of more than one R gene as they segregate for resistance to multiple isolates and/or races of multiple pathogens, whereas biparental mapping populations are often designed to segregate for only one R/S locus for ease of genetic mapping. The advances in sequencing technologies, cloning methods, and gene editing technologies will likely soon reshape the way R genes from wild relatives are deployed in adapted germplasm. Historically, chromosome engineering strategies involving cytogenetic methods to achieve chromosome substitutions, translocations, and ultimately introgressions of smaller segments containing target genes, were extremely laborious and time-consuming, and the end product usually suffered from deleterious linkage drag. The modern sequencing and cloning technologies discussed in this chapter may make it more feasible to clone the target gene in the wild relative accession itself. Although genetic transformation (GMO wheat) is currently not accepted, the acceptance of gene editing appears more promising. Thus, once a target R gene is cloned from a wild relative, it is conceivable that a homologous gene could be identified in wheat and edited to acquire the desired function.
With the availability of multiple reference wheat genomes, in some cases, the bottleneck of cloning R/S genes has shifted from candidate gene identification to functional validation. The use of the CADENZA- and KRONOS-TILLING populations offers rapid functional validation. However, a single bread wheat and durum wheat cultivar cannot feasibly represent the R/S gene content of all bread and durum wheat. Still, due to ease of use and affordability, the TILLING populations are an excellent resource worth considering.
Cloning and deploying R genes and removing S genes is a constant highly coordinated race to keep up with evolving pathogen populations. We suspect that as more R/S genes are cloned, more research will focus on identifying unique durable combinations of R/S genes. For example, Luo et al. (2021) transformed a five-gene cassette of stem rust resistance genes into bread wheat cultivar FIELDER, resulting in broad-spectrum resistance. Another benefit of cloning multiple R/S genes in a given system is that the cumulative knowledge acquired can begin to shed light on the essential components, which can lead to the development of designer genes that could operate to govern broad-spectrum resistance and perhaps resistance less prone to being overcome due to natural mutations occurring in the pathogen. The use of R gene cassettes, disruption of S genes, or the development and deployment of designer genes made possible through advancements in tissue culture, transformation methods, and gene editing technologies are promising directions to ensure stable wheat production enhancing global food security.
References
Acevedo-Garcia J, Spencer D, Thieron H, Reinstädler A, Hammond-Kosack K, Phillips AL, Panstruga R (2017) mlo-based powdery mildew resistance in hexaploid bread wheat generated by a non-transgenic TILLING approach. Plant Biotechnol J 15:367–378
Akpinar BA, Leroy P, Watson-Haigh NS et al (2022) The complete genome sequence of elite bread wheat cultivar, “Sonmez”. F1000Res 11:614. https://doi.org/10.12688/f1000research.121637.1
Arora S, Steuernagel B, Gaurav K et al (2019) Resistance gene cloning from a wild crop relative by sequence capture and association genetics. Nat Biotechnol 37:139–143. https://doi.org/10.1038/s41587-018-0007-9
Athiyannan N, Abrouk M, Boshoff WHP et al (2022) Long-read genome sequencing of bread wheat facilitates disease resistance gene cloning. Nat Genet 54:227–231. https://doi.org/10.1038/s41588-022-01022-1
Aury J-M, Engelen S, Istace B et al (2022) Long-read and chromosome-scale assembly of the hexaploid wheat genome achieves high resolution for research and breeding. GigaScience 11:giac034. https://doi.org/10.1093/gigascience/giac034
Avni R, Nave M, Barad O et al (2017) Wild emmer genome architecture and diversity elucidate wheat evolution and domestication. Science 357:93–97. https://doi.org/10.1126/science.aan0032
Avni R, Lux T, Minz-Dub A et al (2022) Genome sequences of three Aegilops species of the section Sitopsis reveal phylogenetic relationships and provide resources for wheat improvement. Plant J 110:179–192. https://doi.org/10.1111/tpj.15664
Bennett MD, Smith J (1976) Nuclear DNA amounts in angiosperms. Phil Trans R Soc B 274:227–274
Brenchley R, Spannagl M, Pfeifer M et al (2012) Analysis of the bread wheat genome using whole-genome shotgun sequencing. Nature 491:705–710. https://doi.org/10.1038/nature11650
Cao A, Xing L, Wang X et al (2011) Serine/threonine kinase gene Stpk-V, a key member of powdery mildew resistance gene Pm21, confers powdery mildew resistance in wheat. Proc Natl Acad Sci USA 108:7727–7732. https://doi.org/10.1073/pnas.1016981108
Chapman JA, Mascher M, Buluç A et al (2015) A whole-genome shotgun approach for assembling and anchoring the hexaploid bread wheat genome. Genome Biol 16:26. https://doi.org/10.1186/s13059-015-0582-8
Chen S, Zhang W, Bolus S et al (2018) Identification and characterization of wheat stem rust resistance gene Sr21 effective against the Ug99 race group at high temperature. PLoS Genet 14:e1007287. https://doi.org/10.1371/journal.pgen.1007287
Chen S, Rouse MN, Zhang W et al (2020) Wheat gene Sr60 encodes a protein with two putative kinase domains that confers resistance to stem rust. New Phytol 225:948–959. https://doi.org/10.1111/nph.16169
Choulet F, Alberti A, Theil S et al (2014) Structural and functional partitioning of bread wheat chromosome 3B. Science 345:1249721. https://doi.org/10.1126/science.1249721
Clavijo BJ, Venturini L, Schudoma C et al (2017) An improved assembly and annotation of the allohexaploid wheat genome identifies complete families of agronomic genes and provides genomic evidence for chromosomal translocations. Genome Res 27:885–896. https://doi.org/10.1101/gr.217117.116
Cloutier S, McCallum BD, Loutre C et al (2007) Leaf rust resistance gene Lr1, isolated from bread wheat (Triticum aestivum L.) is a member of the large psr567 gene family. Plant Mol Biol 65:93–106. https://doi.org/10.1007/s11103-007-9201-8
Doležel J, Kubaláková M, íhalíková J et al (2011) Chromosome analysis and sorting using flow cytometry. In: Birchler JA (ed) Plant chromosome engineering. Humana Press, Totowa, NJ, pp 221–238
Elliott C, Zhou F, Spielmeyer W, Panstruga R, Schulze-Lefert P (2002) Functional conservation of wheat and rice Mlo orthologs in defense modulation to the powdery mildew fungus. Mol Plant Microbe Interact 15:1069–1077
Endo TR, Gill BS (1996) The deletion stocks of common wheat. J Hered 87:295–307
Erenstein O, Jaleta M, Mottaleb KA et al (2022) Global trends in wheat production, consumption and trade. In: Reynolds MP, BraunHJ (eds) Wheat Improvement. Springer, Cham. https://doi.org/10.1007/978-3-030-90673-3_4
Faris JD (2014) Wheat domestication: key to agricultural revolutions past and future. In: Tuberosa R, Graner A, Frison E (eds) Genomics of plant genetic resources. Springer, Netherlands, Dordrecht, pp 439–464
Faris JD, Zhang Z, Lu H et al (2010) A unique wheat disease resistance-like gene governs effector-triggered susceptibility to necrotrophic pathogens. Proc Natl Acad Sci USA 107:13544–13549. https://doi.org/10.1073/pnas.1004090107
Feuillet C, Travella S, Stein N et al (2003) Map-based isolation of the leaf rust disease resistance gene Lr10 from the hexaploid wheat (Triticum aestivum L.) genome. Proc Natl Acad Sci USA 100:15253–15258. https://doi.org/10.1073/pnas.2435133100
Fu D, Uauy C, Distelfeld A et al (2009) A kinase-START gene confers temperature-dependent resistance to wheat stripe rust. Science 323:1357–1360. https://doi.org/10.1126/science.1166289
Gardiner L-J, Brabbs T, Akhunov A et al (2019) Integrating genomic resources to present full gene and putative promoter capture probe sets for bread wheat. GigaScience 8:1–13. https://doi.org/10.1093/gigascience/giz018
Gaurav K, Arora S, Silva P et al (2022) Population genomic analysis of Aegilops tauschii identifies targets for bread wheat improvement. Nat Biotechnol 40:422–431. https://doi.org/10.1038/s41587-021-01058-4
Gill BS, Appels R, Botha-Oberholster A-M et al (2004) A workshop report on wheat genome sequencing. Genetics 168:1087–1096. https://doi.org/10.1534/genetics.104.034769
Guo W, Xin M, Wang Z et al (2020) Origin and adaptation to high altitude of Tibetan semi-wild wheat. Nat Commun 11:5085. https://doi.org/10.1038/s41467-020-18738-5
Hafeez AN, Arora S, Ghosh S et al (2021) Creation and judicious application of a wheat resistance gene atlas. Mol Plant 14:1053–1070. https://doi.org/10.1016/j.molp.2021.05.014
He H, Zhu S, Ji Y et al (2017) Map-based cloning of the gene Pm21 that confers broad spectrum resistance to wheat powdery mildew. https://doi.org/10.1101/177857
Hewitt T, Müller MC, Molnár I et al (2021a) A highly differentiated region of wheat chromosome 7AL encodes a Pm1a immune receptor that recognizes its corresponding AvrPm1a effector from Blumeria graminis. New Phytol 229:2812–2826. https://doi.org/10.1111/nph.17075
Hewitt T, Zhang J, Huang L et al (2021b) Wheat leaf rust resistance gene Lr13 is a specific Ne2 allele for hybrid necrosis. Mol Plant 14:1025–1028. https://doi.org/10.1016/j.molp.2021.05.010
Huai B, Yuan P, Ma X et al (2022) Sugar transporter TaSTP3 activation by TaWRKY19/61/82 enhances stripe rust susceptibility in wheat. New Phytol. https://doi.org/10.1111/nph.18331
Huang X-Q, Röder MS (2004) Molecular mapping of powdery mildew resistance genes in wheat: a review. Euphytica 137:203–223. https://doi.org/10.1023/B:EUPH.0000041576.74566.d7
Huang L, Brooks SA, Li W et al (2003) Map-based cloning of leaf rust resistance gene Lr21 from the large and polyploid genome of bread wheat. Genetics 164:655–664. https://doi.org/10.1093/genetics/164.2.655
Hurni S, Brunner S, Buchmann G et al (2013) Rye Pm8 and wheat Pm3 are orthologous genes and show evolutionary conservation of resistance function against powdery mildew. Plant J 76:957–969. https://doi.org/10.1111/tpj.12345
Ingvardsen CR, Massange-Sánchez JA, Borum F et al (2019) Development of mlo-based resistance in tetraploid wheat against wheat powdery mildew. Theor Appl Genet 132:3009–3022. https://doi.org/10.1007/s00122-019-03402-4
International Wheat Genome Sequencing Consortium (IWGSC) (2014) A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science 355:1251788. https://doi.org/10.1126/science.1251788
Jia J, Zhao S, Kong X et al (2013) Aegilops tauschii draft genome sequence reveals a gene repertoire for wheat adaptation. Nature 496:91–95. https://doi.org/10.1038/nature12028
Jordan KW, Wang S, Lun Y et al (2015) A haplotype map of allohexaploid wheat reveals distinct patterns of selection on homoeologous genomes. Genome Biol 16:48. https://doi.org/10.1186/s13059-015-0606-4
Jupe F, Witek K, Verweij W et al (2013) Resistance gene enrichment sequencing (RenSeq) enables reannotation of the NB-LRR gene family from sequenced plant genomes and rapid mapping of resistance loci in segregating populations. Plant J 76:530–544. https://doi.org/10.1111/tpj.12307
Kale SM, Schulthess AW, Padmarasu S et al (2022) A catalogue of resistance gene homologs and a chromosome‐scale reference sequence support resistance gene mapping in winter wheat. Plant Biotechnol J pbi.13843. https://doi.org/10.1111/pbi.13843
Kan J, Cai Y, Cheng C et al (2022) Simultaneous editing of host factor gene TaPDIL5-1 homoeoalleles confers wheat yellow mosaic virus resistance in hexaploid wheat. New Phytol 234:340–344. https://doi.org/10.1111/nph.18002
Klymiuk V, Yaniv E, Huang L et al (2018) Cloning of the wheat Yr15 resistance gene sheds light on the plant tandem kinase-pseudokinase family. Nat Commun 9:3735. https://doi.org/10.1038/s41467-018-06138-9
Kolodziej MC, Singla J, Sánchez-Martín J et al (2021) A membrane-bound ankyrin repeat protein confers race-specific leaf rust disease resistance in wheat. Nat Commun 12:956. https://doi.org/10.1038/s41467-020-20777-x
Krasileva KV, Vasquez-Gross HA, Howell T et al (2017) Uncovering hidden variation in polyploid wheat. Proc Natl Acad Sci USA 114. https://doi.org/10.1073/pnas.1619268114
Krattinger SG, Lagudah ES, Spielmeyer W et al (2009) A putative ABC transporter confers durable resistance to multiple fungal pathogens in wheat. Science 323:1360–1363. https://doi.org/10.1126/science.1166453
Li G, Zhou J, Jia H et al (2019) Mutation of a histidine-rich calcium-binding-protein gene in wheat confers resistance to Fusarium head blight. Nat Genet 51:1106–1112. https://doi.org/10.1038/s41588-019-0426-7
Li M, Dong L, Li B et al (2020) A CNL protein in wild emmer wheat confers powdery mildew resistance. New Phytol 228:1027–1037. https://doi.org/10.1111/nph.16761
Li L-F, Zhang Z-B, Wang Z-H et al (2022) Genome sequences of five Sitopsis species of Aegilops and the origin of polyploid wheat B subgenome. Mol Plant 15:488–503. https://doi.org/10.1016/j.molp.2021.12.019
Lin G, Chen H, Tian B et al (2022) Cloning of the broadly effective wheat leaf rust resistance gene Lr42 transferred from Aegilops tauschii. Nat Commun 13:3044. https://doi.org/10.1038/s41467-022-30784-9
Ling H-Q, Zhao S, Liu D et al (2013) Draft genome of the wheat A-genome progenitor Triticum urartu. Nature 496:87–90. https://doi.org/10.1038/nature11997
Ling H-Q, Ma B, Shi X et al (2018) Genome sequence of the progenitor of wheat A subgenome Triticum urartu. Nature 557:424–428. https://doi.org/10.1038/s41586-018-0108-0
Liu W, Frick M, Huel R et al (2014) The stripe rust resistance gene Yr10 encodes an evolutionary-conserved and unique CC–NBS–LRR Sequence in wheat. Mol Plant 7:1740–1755. https://doi.org/10.1093/mp/ssu112
Lu P, Guo L, Wang Z et al (2020) A rare gain of function mutation in a wheat tandem kinase confers resistance to powdery mildew. Nat Commun 11:680. https://doi.org/10.1038/s41467-020-14294-0
Luo M-C, Gu YQ, Puiu D et al (2017) Genome sequence of the progenitor of the wheat D genome Aegilops tauschii. Nature 551:498–502. https://doi.org/10.1038/nature24486
Luo M, Xie L, Chakraborty S et al (2021) A five-transgene cassette confers broad-spectrum resistance to a fungal rust pathogen in wheat. Nat Biotechnol 39:561–566. https://doi.org/10.1038/s41587-020-00770-x
Maccaferri M, Harris NS, Twardziok SO et al (2019) Durum wheat genome highlights past domestication signatures and future improvement targets. Nat Genet 51:885–895. https://doi.org/10.1038/s41588-019-0381-3
Mago R, Zhang P, Vautrin S et al (2015) The wheat Sr50 gene reveals rich diversity at a cereal disease resistance locus. Nat Plants 1:15186. https://doi.org/10.1038/nplants.2015.186
Marchal C, Zhang J, Zhang P et al (2018) BED-domain-containing immune receptors confer diverse resistance spectra to yellow rust. Nat Plants 4:662–668. https://doi.org/10.1038/s41477-018-0236-4
Mello-Sampayo T, Lorente R (1968) The role of chromosome 3D in the regulation of meiotic pairing in hexaploid wheat. EWAC Newslett 2:16–24
Moore JW, Herrera-Foessel S, Lan C et al (2015) A recently evolved hexose transporter variant confers resistance to multiple pathogens in wheat. Nat Genet 47:1494–1498. https://doi.org/10.1038/ng.3439
Olson EL, Brown-Guedira G, Marshall D et al (2010) Development of wheat lines having a small introgressed segment carrying stem rust resistance gene Sr22. Crop Sci 50:1823–1830. https://doi.org/10.2135/cropsci2009.11.0652
Periyannan S, Moore J, Ayliffe M et al (2013) The gene Sr33, an ortholog of barley Mla genes, encodes resistance to wheat stem rust race Ug99. Science 341:786–788. https://doi.org/10.1126/science.1239028
Poddar S, Tanaka J, Running KLD, Kariyawasam GK, Faris JD, Friesen TL, Cho M-J, Cate JHD, Staskawicz B (2022) bioRxiv. https://doi.org/10.1101/2022.04.05.487229
Poddar S, Tanaka J, Running KLD et al (2023) Optimization of highly efficient exogenous-DNA-free Cas9-ribonucleoprotein mediated gene editing in disease susceptibility loci in wheat (Triticum aestivum L.) Front Plant Sci 13:1084700. https://doi.org/10.3389/fpls.2022.1084700
Qi LL, Echalier B, Chao S et al (2004) A chromosome bin map of 16,000 expressed sequence tag loci and distribution of genes among the three genomes of polyploid wheat. Genetics 168:701–712. https://doi.org/10.1534/genetics.104.034868
Rawat N, Pumphrey MO, Liu S et al (2016) Wheat Fhb1 encodes a chimeric lectin with agglutinin domains and a pore-forming toxin-like domain conferring resistance to Fusarium head blight. Nat Genet 48:1576–1580. https://doi.org/10.1038/ng.3706
Riley R, Chapman V (1958) Genetic control of the cytologically diploid behaviour of hexploid wheat. Nature 182:713–715
Saintenac C, Zhang W, Salcedo A et al (2013) Identification of wheat gene Sr35 that confers resistance to Ug99 stem rust race group. Science 341:783–786. https://doi.org/10.1126/science.1239022
Saintenac C, Lee W-S, Cambon F et al (2018) Wheat receptor-kinase-like protein Stb6 controls gene-for-gene resistance to fungal pathogen Zymoseptoria tritici. Nat Genet 50:368–374. https://doi.org/10.1038/s41588-018-0051-x
Saintenac C, Cambon F, Aouini L et al (2021) A wheat cysteine-rich receptor-like kinase confers broad-spectrum resistance against Septoria tritici blotch. Nat Commun 12:433. https://doi.org/10.1038/s41467-020-20685-0
Sánchez-Martín J, Steuernagel B, Ghosh S et al (2016) Rapid gene isolation in barley and wheat by mutant chromosome sequencing. Genome Biol 17:221. https://doi.org/10.1186/s13059-016-1082-1
Sánchez-Martín J, Widrig V, Herren G et al (2021) Wheat Pm4 resistance to powdery mildew is controlled by alternative splice variants encoding chimeric proteins. Nat Plants 7:327–341. https://doi.org/10.1038/s41477-021-00869-2
Sato K, Abe F, Mascher M et al (2021) Chromosome-scale genome assembly of the transformation-amenable common wheat cultivar ‘Fielder.’ DNA Res 28:dsab008. https://doi.org/10.1093/dnares/dsab008
Savary S, Bregaglio S, Willocquet L et al (2017) Crop health and its global impacts on the components of food security. Food Sec 9:311–327. https://doi.org/10.1007/s12571-017-0659-1
Savary S, Willocquet L, Pethybridge SJ et al (2019) The global burden of pathogens and pests on major food crops. Nat Ecol Evol 3:430–439. https://doi.org/10.1038/s41559-018-0793-y
Schreiber AW, Hayden MJ, Forrest KL et al (2012) Transcriptome-scale homoeolog-specific transcript assemblies of bread wheat. BMC Genomics 13:492. https://doi.org/10.1186/1471-2164-13-492
Sears ER (1954) The aneuploids of common wheat. Mo Agr Exp Sta Res Bull 572:1–59
Sears ER (1966) Nullisomic-tetrasomic combinations in hexaploid wheat. In: Riley R, Lewis KR (eds) Chromosome manipulation and plant genetics. Oliver & Boyd, Edinburgh, pp 29–45
Sears ER, Okamoto M (1958) Intergenomic chromosome relationships in hexaploid wheat. Proc Int Congr Genet 2:258–259
Sears ER, Sears LMS (1978) The telocentric chromosomes of common wheat In: Ramanujam S (ed) Proceedings of the 5th international wheat genetics symposium. Indian Society of Genetics and Plant Breeding, New Delhi, pp 389–407
Shi G, Zhang Z, Friesen TL et al (2016) The hijacking of a receptor kinase–driven pathway by a wheat fungal pathogen leads to disease. Sci Adv 2:e1600822. https://doi.org/10.1126/sciadv.1600822
Singh SP, Hurni S, Ruinelli M et al (2018) Evolutionary divergence of the rye Pm17 and Pm8 resistance genes reveals ancient diversity. Plant Mol Biol 98:249–260. https://doi.org/10.1007/s11103-018-0780-3
Srichumpa P, Brunner S, Keller B, Yahiaoui N (2005) Allelic series of four powdery mildew resistance genes at the Pm3 locus in hexaploid bread wheat. Plant Physiol 139:885–895. https://doi.org/10.1104/pp.105.062406
Steuernagel B, Periyannan SK, Hernández-Pinzón I et al (2016) Rapid cloning of disease-resistance genes in plants using mutagenesis and sequence capture. Nat Biotechnol 34:652–655. https://doi.org/10.1038/nbt.3543
Su Z, Bernardo A, Tian B et al (2019) A deletion mutation in TaHRC confers Fhb1 resistance to Fusarium head blight in wheat. Nat Genet 51:1099–1105. https://doi.org/10.1038/s41588-019-0425-8
The International Wheat Genome Sequencing Consortium (IWGSC), Mayer KFX, Rogers J et al (2014) A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science 345:1251788. https://doi.org/10.1126/science.1251788
The International Wheat Genome Sequencing Consortium (IWGSC), Appels R, Eversole K et al. (2018) Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 361:eaar7191. https://doi.org/10.1126/science.aar7191
Thind AK, Wicker T, Šimková H et al (2017) Rapid cloning of genes in hexaploid wheat using cultivar-specific long-range chromosome assembly. Nat Biotechnol 35:793–796. https://doi.org/10.1038/nbt.3877
Upadhyaya NM, Mago R, Panwar V et al (2021) Genomics accelerated isolation of a new stem rust avirulence gene–wheat resistance gene pair. Nat Plants 7:1220–1228. https://doi.org/10.1038/s41477-021-00971-5
Van de Weyer AL, Monteiro F, Furzer OJ, Nishimura MT, Cevik V, Witek K, Jones JDG, Dangl JL, Weigel D, Bemm F (2019) A species-wide inventory of NLR genes and alleles in Arabidopsis thaliana. Cell 178(5):1260-1272.e14. https://doi.org/10.1016/j.cell.2019.07.038
Várallyay É, Giczey G, Burgyán J (2012) Virus-induced gene silencing of Mlo genes induces powdery mildew resistance in Triticum aestivum. Arch Virol 157:1345–1350
Voichek Y, Weigel D (2020) Identifying genetic variants underlying phenotypic variation in plants without complete genomes. Nat Genet 52(5):534–540. https://doi.org/10.1038/s41588-020-0612-7
Walkowiak S, Gao L, Monat C et al (2020) Multiple wheat genomes reveal global variation in modern breeding. Nature 588:277–283. https://doi.org/10.1038/s41586-020-2961-x
Wang Y, Cheng X, Shan Q, Zhang Y, Liu J, Gao C, Qiu J-L (2014) Simultaneous editing of three homoeoalleles in hexaploid bread wheat confers heritable resistance to powdery mildew. Nat Biotechnol 32:947
Wang H, Sun S, Ge W et al (2020a) Horizontal gene transfer of Fhb7 from fungus underlies Fusarium head blight resistance in wheat. Science 368:eaba5435. https://doi.org/10.1126/science.aba5435
Wang H, Zou S, Li Y et al (2020b) An ankyrin-repeat and WRKY-domain-containing immune receptor confers stripe rust resistance in wheat. Nat Commun 11:1353. https://doi.org/10.1038/s41467-020-15139-6
Wang L, Zhu T, Rodriguez JC et al (2021) Aegilops tauschii genome assembly Aet v5.0 features greater sequence contiguity and improved annotation. Genes Genom Genet 11:jkab325. https://doi.org/10.1093/g3journal/jkab325
Wicker T, Gundlach H, Spannagl M et al (2018) Impact of transposable elements on genome structure and evolution in bread wheat. Genome Biol 19:103. https://doi.org/10.1186/s13059-018-1479-0
Wulff BB, Krattinger SG (2022) The long road to engineering durable disease resistance in wheat. Curr Opin Biotechnol 73:270–275. https://doi.org/10.1016/j.copbio.2021.09.002
Xie J, Guo G, Wang Y et al (2020) A rare single nucleotide variant in Pm5e confers powdery mildew resistance in common wheat. New Phytol 228:1011–1026. https://doi.org/10.1111/nph.16762
Xing L, Hu P, Liu J et al (2018) Pm21 from Haynaldia villosa encodes a CC-NBS-LRR protein conferring powdery mildew resistance in wheat. Mol Plant 11:874–878. https://doi.org/10.1016/j.molp.2018.02.013
Yahiaoui N, Srichumpa P, Dudler R, Keller B (2004) Genome analysis at different ploidy levels allows cloning of the powdery mildew resistance gene Pm3b from hexaploid wheat: Positional cloning of Pm3 from hexaploid wheat. Plant J 37:528–538. https://doi.org/10.1046/j.1365-313X.2003.01977.x
Yan X, Li M, Zhang P et al (2021) High-temperature wheat leaf rust resistance gene Lr13 exhibits pleiotropic effects on hybrid necrosis. Mol Plant 14:1029–1032. https://doi.org/10.1016/j.molp.2021.05.009
Yao E, Blake VC, Cooper L et al (2022) GrainGenes: a data-rich repository for small grains genetics and genomics. Database 2022:baac034. https://doi.org/10.1093/database/baac034
Yu J, Pressoir G, Briggs W et al (2006) A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet 38:203–208. https://doi.org/10.1038/ng1702
Yu G, Matny O, Champouret N et al (2022) Aegilops sharonensis genome-assisted identification of stem rust resistance gene Sr62. Nat Commun 13:1607. https://doi.org/10.1038/s41467-022-29132-8
Yuan C, Wu J, Yan B et al (2018) Remapping of the stripe rust resistance gene Yr10 in common wheat. Theor Appl Genet 131:1253–1262. https://doi.org/10.1007/s00122-018-3075-9
Zhang W, Chen S, Abate Z et al (2017) Identification and characterization of Sr13, a tetraploid wheat gene that confers resistance to the Ug99 stem rust race group. Proc Natl Acad Sci USA 114. https://doi.org/10.1073/pnas.1706277114
Zhang C, Huang L, Zhang H et al (2019) An ancestral NB-LRR with duplicated 3′UTRs confers stripe rust resistance in wheat and barley. Nat Commun 10:4023. https://doi.org/10.1038/s41467-019-11872-9
Zhang J, Hewitt TC, Boshoff WHP et al (2021a) A recombined Sr26 and Sr61 disease resistance gene stack in wheat encodes unrelated NLR genes. Nat Commun 12:3378. https://doi.org/10.1038/s41467-021-23738-0
Zhang Z, Running KLD, Seneviratne S et al (2021b) A protein kinase–major sperm protein gene hijacked by a necrotrophic fungal pathogen triggers disease susceptibility in wheat. Plant J 106:720–732. https://doi.org/10.1111/tpj.15194
Zhang L, Liu Y, Wang Q et al (2022a) An alternative splicing isoform of wheat TaYRG1 resistance protein activates immunity by interacting with dynamin-related proteins. J Exp Bot erac245 https://doi.org/10.1093/jxb/erac245
Zhang X, Wang G, Qu X et al (2022b) A truncated CC-NB-ARC gene TaRPP13L1-3D positively regulates powdery mildew resistance in wheat via the RanGAP-WPP complex-mediated nucleocytoplasmic shuttle. Planta 255:60. https://doi.org/10.1007/s00425-022-03843-0
Zhao G, Zou C, Li K et al (2017) The Aegilops tauschii genome reveals multiple impacts of transposons. Nat Plants 3:946–955. https://doi.org/10.1038/s41477-017-0067-8
Zhou Y, Bai S, Li H et al (2021) Introgressing the Aegilops tauschii genome into wheat as a basis for cereal improvement. Nat Plants 7:774–786. https://doi.org/10.1038/s41477-021-00934-w
Zhu T, Wang L, Rodriguez JC et al (2019) Improved genome sequence of wild emmer wheat Zavitan with the aid of optical maps. Genes Genom Genet 9:619–624. https://doi.org/10.1534/g3.118.200902
Zhu T, Wang L, Rimbert H et al (2021) Optical maps refine the bread wheat Triticum aestivum cv. Chinese Spring genome assembly. Plant J 37:528–538. https://doi.org/10.1046/j.1365-313X.2003.01977.x
Zimin AV, Puiu D, Hall R et al (2017a) The first near-complete assembly of the hexaploid bread wheat genome, Triticum aestivum. GigaScience 6. https://doi.org/10.1093/gigascience/gix097
Zimin AV, Puiu D, Luo M-C et al (2017b) Hybrid assembly of the large and highly repetitive genome of Aegilops tauschii, a progenitor of bread wheat, with the MaSuRCA mega-reads algorithm. Genome Res 27:787–792. https://doi.org/10.1101/gr.213405.116
Zou S, Wang H, Li Y et al (2018) The NB-LRR gene Pm60 confers powdery mildew resistance in wheat. New Phytol 218:298–309. https://doi.org/10.1111/nph.14964
Acknowledgements
Figures were created with Biorender.com.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2024 The Author(s)
About this chapter

Cite this chapter
Running, K.L.D., Faris, J.D. (2024). Rapid Cloning of Disease Resistance Genes in Wheat. In: Appels, R., Eversole, K., Feuillet, C., Gallagher, D. (eds) The Wheat Genome. Compendium of Plant Genomes. Springer, Cham. https://doi.org/10.1007/978-3-031-38294-9_10
Download citation
DOI: https://doi.org/10.1007/978-3-031-38294-9_10
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-38292-5
Online ISBN: 978-3-031-38294-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)