
Abstract
With industrialization, the production of chemicals and their introduction into the environment have increased massively. These new agents included many chemical classes and comprise an integral part of the world economy and commerce [1]. Nevertheless, several of the chemicals used today are called endocrine-disrupting compounds (EDCs).
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
2.1 Endocrine Disruptors: Impact on Health
With industrialization, the production of chemicals and their introduction into the environment have increased massively. These new agents included many chemical classes and comprise an integral part of the world economy and commerce [1]. Nevertheless, several of the chemicals used today are called endocrine-disrupting chemicals (EDCs).
A chemical agent is classified as an endocrine-disrupting chemical when it can interfere with the synthesis, metabolism, and action of endogenous hormones.
These substances have been even more tested systematically for endocrine-disrupting effects in organisms as the production of large amounts of synthetic industrial and biomedical chemicals, as well as unwanted pollutants, poses destructive consequences to our ecosystem and imposes negative health effects to wildlife and humans [2, 3].
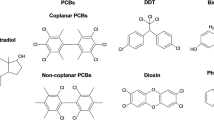
Several compounds have been identified as potential EDCs due to their actions of estrogenic and androgenic gene regulation, altering the synthesis, metabolism, and action of endogenous hormones interfering with different human mechanisms of male and female reproductive systems. The main chemical and natural compounds that are currently classified and approved as EDCs that deserve a special attention are summarized in Table 2.1.
Epidemiological studies suggest an association between the increasing exposure to chemicals and the development of some of the main ailments of the industrialized world (e.g., disturbances in reproduction, hormone-related cancers, and metabolic disorders like obesity and type 2 diabetes) [44]. In particular, some of these chemicals act as endocrine disruptors as they disturb endogenous hormone signaling pathways.
The consequences of endocrine disruption in human health are discussed. In particular, the possible involvement of EDCs’ exposure on the development of metabolic diseases and their possible responsibility in health disorders are the basic reasons for understanding the mechanisms of action of EDCs. This research field is, however, the subject of scientific and public controversy due to the lack of knowledge about the possible molecular mechanisms underlying endocrine disruption and disease development [45].
A recent review reports that about 40% of human death (62 million per year) is attributed to the effect of exposure to chemical pollutants [46]. Moreover, a study from the US Center for Disease Control (CDC) reported that in the bodies of Americans of all ages have been found over 116 extraneous chemicals [47] and in the cord blood of American infants have been detected over 358 industrial chemicals and pesticides [48].
In some cases, it has been suggested that precise diseases (cancer, neurological disorders, allergies, and reproductive disorders) may be associated with exposure to chemical agents, like EDCs [49]. Focusing on the reproductive disorders, according to the World Health Organization (WHO)’s data, about 80 million people worldwide were estimated to be affected by infertility [50] because of the interference with external agents.
According to these evidences, these compounds raise serious concerns about their potential health impact. These chemicals are prevalently synthetic molecules from an industrial origin [51, 52], but also include some natural molecules [53, 54]. In the latter case, the presence of these compounds ubiquitously in the environment makes really difficult to avoid exposure to them.
The endocrine system of vertebrates is an intricate web of stimulatory and inhibitory hormone signals that control basic functions of body such as metabolism, growth, digestion, and cardiovascular function, as well as more specialized processes such as behavior, sexual differentiation (during embryogenesis), sexual maturation (during puberty), and adult reproduction [55]. EDCs affect human health by disturbing normal endocrine activity through interaction with different receptors involved in key metabolic interaction strategies. This detrimental effect is due to the ability of EDCs to interfere with or mimic endogenous hormones and other signaling molecules of the endocrine system [56, 57].
Due to the huge impact of EDCs on hormonal system, these substances can cause different clinical conditions, including infertility, alterations in sperm quality, abnormalities in sex organs and growth, endometriosis, early puberty [58, 59], altered nervous system function, neurological and learning disabilities [60, 61], immune function [62], certain cancers [63, 64], respiratory problems, metabolic issues, diabetes, obesity, cardiovascular problems [44], thyroid function alterations [65], immune diseases [62], and more (Table 2.2). This aspect highlights the importance of the evaluation of EDC exposure on a developing organism in order to prevent the development of diseases or dysfunctions later in life [74].
2.1.1 Effects of EDCs on Metabolism
Due to the ability of EDCs to interfere with hormone signaling and metabolism, exposure to these substances could lead to the development of metabolic diseases.
In particular, these chemicals cause consequences on the metabolic system by interacting with the hormone receptors (HRs) of the nuclear receptor (NR) family [75] that represents a family of structurally related transcription factors that are involved in various essential biological functions (e.g., fetal development, homeostasis, reproduction, metabolism, and response to xenobiotic substances).
Some EDCs can bind directly to these receptors either as agonists or antagonists, thus enhancing or inhibiting the effect of hormones [44]. In this way, EDCs interfere with the process of hormone biosynthesis, transport to the target tissue, levels of hormone-binding proteins, and hormone catabolism and deregulate hormone availability [76, 77]. One of the main alterations involved in the development of metabolic diseases after EDC exposure is represented by the onset of metabolic syndrome (MetS), due to alteration in fat metabolism and glucose uptake because of the engagement of NRs by EDCs [78] (Table 2.2) that lead to obesity [68, 69] (Table 2.2). For this reason, EDCs involved in MetS onset are called “obesogens,” and it was assumed that already in utero and onward exposure may play a role in the development of obesity and related diseases during life [78,79,80] (Table 2.2).
In addition to EDCs–NRs interaction, the involvement of other receptors in the modulation of the metabolic system has been reported, for example, the aryl hydrocarbon receptor (AhR) [81, 82], estrogen receptors (ERs), androgen receptors (ARs), retinoid X receptor (RXR), and peroxisome proliferator-activated receptors (PPARs) [83].
According to the data reported in the literature, there are specific molecules that interact with the aforementioned receptors, causing a negative effect on human metabolism: For example, dioxin exposure has been reported to increase the risk for type 2 diabetes [84] (Table 2.2), while persistent organic pollutants (POPs), a group of AhR ligands, have been found to be associated with both diabetes and MetS in an epidemiological study of human serum samples [70, 85]. Other authors conclude that low-dose exposure to polychlorinated biphenyls-77 (PCB-77), which could be accumulated in the adipose tissue, may contribute to the development of obesity and atherosclerosis [67] (Table 2.2). In particular, it is known that estrogen and its receptors play an important role in adipogenesis, and exogenous estrogens have an impact on adipose metabolism; for example, octylphenol, a chemical widely used as a surfactant and frequently found in wastewater, decreases adipocyte differentiation [86] (Table 2.2).
Again, bisphenol A (BPA), a monomer with strong estrogenic properties, largely used in a lining of food and beverage containers, medical tubing, and dental fillings, has been associated with cardiovascular disease, liver abnormalities, and diabetes [71] (Table 2.2).
2.1.2 EDCs and Cancer
As mentioned earlier, EDCs are substances that interfere with the endocrine system and therefore with hormones, which are involved in the evolution of cancer.
Exposure to EDCs, in particular to estrogen- or androgen-mimicking EDCs, can promote tumor formation, especially prostate and breast cancers, the latter in particular during the prenatal period, whereas the exposure to some EDCs may affect mammary gland development and increase breast cancer risk later in life [72].
Moreover, EDC exposure could also interfere with hormonal cancer therapy (Table 2.2).
Regard the exposure to EDCs in the prenatal period, there are some indications confirming that exposure to endocrine-disrupting substances in utero can confer an increased risk of cancer in humans (Table 2.2). A clear example is the diethylstilbestrol, reported to be associated with increased incidence of clear-cell carcinoma of the vagina when the exposure occurred during fetal life [87].
Moreover, perinatal exposure to low doses of BPA was reported to result in altered mammary gland morphogenesis and carcinoma in situ [88]. Similar to what is reported referring to the mammary gland, the fetal development of the prostate is affected by the exposure to BPA, which increases the adult prostate size, promoting prostate cancer development [89] (Table 2.2).
One of the main mechanisms responsible for tumorigenesis due to EDC exposure involves epigenetic changes on chromatin and DNA [90, 91]. These kinds of modifications, even if are not affecting DNA sequence, are stable over rounds of cell division and could also be heritable. Epigenetic modifications, such as chromatin methylation or histone modifications, lead to alteration in gene expression that may have repercussions at a phenotypical level, inducing syndromes or tumors, during both prenatal period and adult life.
2.1.3 Effects of EDCs on Reproduction, Growth, and Development
The impairment of the endocrine system regulation may raise abnormal function and development of the reproductive systems [59] (Table 2.2).
In particular, early-life exposures to high level of EDCs have been linked to developmental abnormalities and may increase the risk for a variety of diseases later in life [92]. In fact, EDC exposures during fetal development and childhood can cause long-lasting health effects, as during this developmental period, hormones regulate both formation and maturation of organs [57].
In particular, these hormones are subjected to hypothalamic control during fetal and early postnatal life and are crucial to enable a correct sexual differentiation and a successful reproduction in adulthood [93, 94]. Vilahur et al. [95] showed that prenatal exposure to endocrine-disrupting chemicals causes changes in DNA methylation that were manifested as the latent development of male infertility, reproductive cancers, and other dysfunctions (Table 2.2).
Some classes of EDCs (dichlorodiphenyltrichloroethanes [DDTs], BPA, phthalates, PCBs, and others) can mimic or block the effects of male and female sex hormones, exhibiting a diverse effect on the reproductive and development sphere in men and women, leading to various hormonal changes [59]. In the case of women, EDCs are implicated in the development of some gynecologic pathologies and fertility problems: de Cock et al. (1994) found that reduced fecundability ratio and longer time-to-pregnancy are associated with the application of pesticides fecundability [96] (Table 2.2). Moreover, a positive association between EDCs, especially estrogen-like BPA [97], and gynecological problems highlighting recurrent miscarriages was observed.
On the other hand, men are known to be more susceptible to steroidogenesis, the process for steroid hormone production. Several evidences supported the role of EDCs in interfering with steroidogenesis (particularly through interaction with NRs), modulating the release of endogenous steroid hormones that may cause subsequent reproductive dysfunction [58] (Table 2.2).
The effects of EDCs on fetal testis seem to be more striking as the disruption of steroidogenesis at this early developmental stage can also affect the proliferation of germ cells and Sertoli cells [98,99,100] (Table 2.2), which supports the maximum number of sperms that can be produced in adulthood [101, 102].
2.2 Targets of EDCs: Genomic and Nongenomic Modulation
Insecticides, plasticizers, and detergents can be classified as potential EDCs, and the exposure to these chemicals in the environment in the food chain, or occupational exposures, may affect human health inducing developmental effects and birth defects [103].
EDCs act on human health through various mechanisms, which can be classified as genomic and nongenomic (Fig. 2.1). EDCs can affect every possible cellular hormonal pathway: For example, some of these compounds can bind directly to hormone receptors either as agonists or antagonists, thus enhancing or inhibiting the effect of a hormone regulating gene expression [45]. Despite this, nongenomic modulation was observed, referring to epigenetic modifications, as a system of interaction on human health [104] (Fig. 2.1).

Schematic representation of the types of modulation of the EDCs, genomic and nongenomic
2.2.1 EDCs and Genomic Modulation
A wide range of EDCs exert their effects using a hormone-type mimicking mechanism. These synthetic molecules can be present in rather high quantities and therefore can compete with endogenous hormones despite their lower affinity toward hormone receptors (HRs).
EDCs exhibit structures that are similar to hormones [105]; thus, they can interfere with their binding site as a agonist, activating the receptor, or as an antagonist, inhibiting HRs (Fig. 2.1). This mechanism is defined as “genomic” due to the fact that hormone receptors, like androgen receptors (ARs), estrogen receptors (ERs), and aryl hydrocarbon receptors (AhRs), are nuclear receptors (NRs) that have a direct effect into DNA as a transcription factor [75]. In the specific case of estrogen receptors, p-nonylphenol and bisphenol A act as agonists for ERs, and like estradiol [103], vinclozolin [106], DDT [107], atrazine, and lindane [108] are AR antagonists and have antiandrogenic effects, reducing the expression of ARs [56].
In addition to this mechanism, EDCs are able to interact directly with hormonal regulation by acting on the components of the hormonal signaling pathway (Fig. 2.1). The interaction between EDCs and these components involves a modification of the hormone biosynthesis, positively or negatively, or their degradation [45]. Indeed, xenobiotics and many EDCs have an indirectly effect on hormone regulation through the activation/inhibition of receptors that induced the expression of enzymes involved in activation, conjugation, and elimination of endogenous hormone [56] (Fig. 2.1). The most striking example is in the interference of these compounds with enzyme cytochrome P450, modifying the hormone synthesis as in the case of steroid synthesis [109]. Despite this, also many other metabolic enzymes involved in the synthesis, elimination, and conversion of steroid hormones, such as testosterone to 17β-estradiol (E2) and progesterone to testosterone [110], could be affected by EDCs.
EDCs could also disturb the balance of circulating and local tissue concentration of hormones disturbing the normal functions of the endocrine system. In particular, steroid hormones are hydrophobic and require to be bound by blood proteins to be transported. This characteristic is shared also by EDCs, which have this characteristic of hydrophobicity [111]. In this mechanism, EDCs do not compete with hormones at the receptor level, but at the level of their circulating binding proteins; in fact, these endocrine disruptors are susceptible to compete with hydrophobic hormone-binding transport proteins [112]; otherwise, other EDCs can affect the biosynthesis or degradation of hormone-binding transport protein [113] (Fig. 2.1).
Finally, a further mechanism responsible for EDCs alteration with the endocrine system is represented by the inhibition of receptor expression, modifying endogenous hormone receptor turnover [114] (Fig. 2.1).
Each single mechanism of interaction does not exclude the other; EDCs represent a category of compounds with various and complex activities in the endogenous system.
2.2.2 EDCs and Nongenomic Modulation
The conjugation of the term “epigenetics” [115] gives importance to the effects of the environment on the human gene apparatus. Epigenetics relates the changes that occur in gene expression and phenotype with the inheritance of these, not altering the DNA sequences and identifying external agents from the environment as the cause. The environment can be perturbed by EDCs, and endocrine disruptors affect the epigenetic processes such as DNA methylation [116, 117] and histone modification [118]; this can be considered the nongenomic modulation by EDCs.
Nongenomic modulation by EDCs has importance during development, particularly in three windows of exposure when the epigenome is susceptible to reprogramming by EDCs during gamete maturation [119], implantation of fetus [120], and differentiation of pluripotent cells [121].
The reprogram of the epigenome caused by exposure to EDCs is supported by several evidences; in fact, chemical disruptors such as DES [122], BPA, genistein [123], and vinclozolin [124] all have been shown to alter patterns of DNA methylation. Moreover, Newbold [125] and Tang [126] explored DNA methylation changes induced by developmental exposure to EDCs, specifically to DES.
Finally, Anway et al. [31] demonstrated the transgenerational effects of vinclozolin on spermatogenesis and fertility in males after gestational exposure to these EDCs. This supports the fact that EDCs induce transgenerational inheritance by assuming transmission via germ line alterations [127].
2.3 Hormone Mimicry and Disruption by EDCs: Modulation of Hormone Activity
The name “EDCs” itself refers to the ability of a specific group of environmental endocrine-active chemicals to interfere with the endocrine system, as well as other systems (mainly the reproductive system), disturbing endogenous hormone signaling pathways [44]. As already mentioned, EDCs can influence and modulate the hormonal system, by acting as endogenous hormones and other signaling molecules of the endocrine system, or even mimicking them. This particular property represents a great potential risk for human health, and therefore, the increasing release of contaminants in the environment should be a concern of a global interest.
The principal molecular mechanism of action exhibited by EDCs is represented by their interaction with hormone receptors (HRs), influencing hormonal activity directly (Table 2.3). Receptors have evolved to be protected against binding with endogenous molecules other than hormones; however, the growing environmental contamination of synthetic toxicants having the shape and size of the actual hormone could not have been encountered before during receptor evolution. For this reason, even though EDCs have lower receptor affinity compared with physiological hormones, because of their abundance in the environment, these chemicals can compete with endogenous hormones [105].
Endocrine disruptors are notably able to bind to the family of nuclear receptors (NRs), including the estrogen receptors (ERs) and androgen receptors (ARs) [44], but they can also be associated with some membrane receptors. Nevertheless, thyroid hormone receptors, retinoid X receptor (RXR), and peroxisome proliferator-activated receptors (PPARs) have been recently identified as additional binding targets too [44]. This is the case of estradiol and bisphenol A that can bind both the nuclear receptors ERα and ERβ and a transmembrane receptor called G protein-coupled estrogen receptor 1 (GPER) (GPR30) [128, 129].
Importantly, depending on the binding of EDCs with NRs or membrane receptors, it is possible to observe different kinds of effects. In fact, in the case of NR engagement by EDCs, we observe a modulation of gene expression that exerts a long-term effect on target cells’ phenotype [5, 130] due to their transcriptional factor function, while in the case of binding with membrane receptors the result is a short-term and more acute effect [5, 131].
On the other hand, behaving like agonists, EDCs can also hinder endogenous hormones, by occupying HRs and antagonizing the proper ligand–hormone interaction (Table 2.3) [132]. Many of these EDCs showing antiestrogenic, antiandrogenic, antiprogesteronic, and anti-ER activities have been detected in wastewater [133].
The antagonism exerted by EDCs could be explained by the ability of some chemical compounds to block receptor conformation in their inactive state, resulting in the inhibition of their signaling pathways [45]. For example, polychlorinated biphenyls (PCBs) can prevent the association between triiodothyronine (T3) and thyroid hormone receptor (THR), with the consequent dissociation of the transcriptionally active THR/retinoid X receptor heterodimer complex from the thyroid response element (TRE) [113].
Besides the hormonal activity exhibited by EDCs via receptor binding, toxicants can influence hormonal system activating other signaling pathways, that is, interacting with components of hormone signaling pathways downstream of receptor activation (Table 2.3) [45]. Since this phenomenon does not involve any binding with hormone receptors, such EDCs may present different structures from endogenous hormones [45]. An example is represented by fluoxetine (FLX), a selective serotonin reuptake inhibitor (SSRI) active substance present in antidepressant drugs, which has the potential to alter many intracellular signaling pathways in different cellular types, without HR association [134,135,136]. In addition, some bisphenols can interact with Ras small G proteins (e.g., K-Ras4B) and activate the Ras signaling cascade, causing the increase of pERK and pAKT [137]. Finally, the herbicide atrazine can inhibit cAMP-specific phosphodiesterase 4 (PDE4) [130], leading to cAMP intracellular accumulation, and tolylfluanid is able to reduce insulin receptor substrate-1 (IRS-1) levels, downstream from the insulin receptor, in human adipocytes [131].
Another way by which exogenous molecules exert endocrine disruption is by directly affecting the endogenous hormone biosynthesis or degradation (Table 2.3). Again, in this specific case, there is no interaction with hormone receptors; therefore, chemicals can exhibit different structures than physiological hormones [45]. Evidences of altered hormone biosynthesis have been shown after exposition to different EDCs; for example, low dose of BPA inhibits adiponectin secretion in vitro in human adipocytes [114, 138, 139], 4-nonyphenol (4-NP) inhibits the synthesis of testosterone by Leydig cells following the stimulation by human chorionic gonadotropin [140], or triclosan stimulates vascular endothelial growth factor (VEGF) secretion by human prostate cancer cells [141]. Concerning EDCs’ influence on hormone degradation, polybrominated diphenyl ethers (PBDEs) have been described to potentially increase thyroxine (T4) elimination, lowering its concentration level in blood [113], while parabens inhibit estrogen degradation [142], increasing the hormone concentration in blood.
Since the majority of the hormones are hydrophobic, like steroids and thyroid ones, they necessarily must be transported in association with specific binding proteins through the bloodstream. Thus, since EDCs present structural similarity to hormones, they are hydrophobic and can bind the same hormone-binding transport proteins, competing with endogenous hormones (Table 2.3) [143,144,145]. The result of the association between binding proteins and toxicants is the decrease in hormone transport that decreased their concentration in blood. Examples of transport proteins that are usually subjected to be bound by chemicals instead of hormones include steroid hormone-binding protein (SHBG) or α-fetoprotein (AFP) [112, 146].
Furthermore, EDCs can modulate endogenous free active hormone concentration not only by taking their place in binding transport protein, but also via the direct modification of these protein levels in the bloodstream, affecting their biosynthesis or degradation (Table 2.3) [45]. In fact, a lower availability of transport proteins means at the same time a lower concentration of hormones. EDCs responsible for this mechanism are binding-independent, so they can exhibit different structures from those of endogenous hormones. The advantage of such mechanism of action is that chemicals mainly target liver because this is the classic degrading organ, and at the same time, liver is the place where binding transport proteins are synthetized and degraded [45]. PBDEs, for example, may downregulate transport protein transthyretin (TTR) level [113] and consequently lower T4 amount in blood.
Among the several mechanisms affecting hormonal system to mention, there is also the ability of EDCs to regulate HR turnover through the stimulation or inhibition of their expression (Table 2.3). The absence of the binding with hormone receptors in this specific mechanism allows such chemicals to exhibit structural differences from hormones [45]. BPA, for example, has been shown to induce leptin receptor expression in ovarian cancer cells in vitro [31], cadmium increased estrogen receptor beta (ERβ) and Cyp19a1 enzymes in endothelial human umbilical vein endothelial cells (HUVECs) in vitro, and a dose-dependent decrease of androgen receptor (AR) expression levels was observed after 24 h of exposure [38]. Conversely, the inhibition of receptors has been shown after the administration of a low oral dose of BPA to rats, which decreased estrogen receptor expression in their hypothalamic cells [39]. The inhibition of androgen receptors by BPA has also been observed in vivo [29] and in vitro cells of patients with breast or prostate cancer [40].
Nowadays, it is well known that EDCs may also influence human epigenome by acting through various mechanisms, such as DNA methylation and histone code alteration [1]. Epigenetic modifications consist of changes in gene expression and resulting phenotype, without any alteration in nucleotide sequence of DNA. The alteration of gene expression can act on differential physiological pathways and can affect normal hormonal activity, for example, by alternating hormone signaling pathway downstream of receptor or provoking aberrant receptor turnover, therefore constituting another nontraditional mechanism of action of EDCs on hormonal system (Table 2.3). Waalkes et al. [147] demonstrated that inorganic arsenic exposure in utero induced a significantly decreased promoter methylation of ER in the liver, resulting in increased ER expression, which also correlated with the increased incidence of hepatocellular carcinoma in exposed animals. The soybean isoflavone genistein (GE) has been shown to prevent breast cancer and to induce epigenetic reactivation of estrogen receptors [148]. Recent studies have suggested that GE, besides its ability to enhance the anticancer capacity of the estrogen antagonist tamoxifen (TAM) in ERα-positive breast cancer cells, can also reactivate ERα expression in ERα-negative breast cancer cells. This positive influence on estrogen receptor expression was enhanced when combined with a histone deacetylase (HDAC) inhibitor, named trichostatin A (TSA). GE treatment also resensitized ERα-dependent cellular responses to the activator 17β-estradiol (E2) and the antagonist TAM. The following research revealed that GE can remodel chromatin structure and consequently reactivate ERα gene expression.
Although the spectrum of chemical compounds to which we may be exposed is broad and can influence human health by multiple mechanisms of action, EDCs can be classified according to their endocrine effect in the main classes of estrogenic, antiestrogenic, androgenic, and antiandrogenic EDCs. The name of the class they belong to suggests the type of endogenous hormonal pathway that is disrupted by those specific chemicals.
Among the endocrine-disrupting chemicals, environmental estrogens were the first to cause concern. Lately, it was discovered that also other hormonal systems are susceptible to disruption, like androgen signaling pathway. At last, thyroid hormone was identified as a target for endocrine disruption too [149], as well as peroxisome proliferator-activated receptor (PPAR)/retinoid X receptor (RXR) system [150, 151].
2.4 Mechanism of Action of EDCs and Estrogens
17-beta-Estradiol (E2) is a key hormone involved in many biological processes in humans, like development and maintenance of the female reproductive tract, brain, bone, and cardiovascular system. Moreover, E2 is a key component in male development too [152]. In female adulthood, estrogen takes part in metabolism and coordinates the morphological alterations occurring during physiological menstrual cycle and pregnancy, together with differentiation and proliferation of target tissues [152].
The classical and conventional estrogenic function is notably mediated by the interaction of E2 with specific estrogen receptors ERα and ERβ, which are steroid receptors belonging to the superfamily of nuclear receptors, in particular type I NRs [152, 153]. ERα and ERβ are tissue-specific; therefore, they are thought to possess distinct physiological roles [154]. ERα is expressed in breast, uterus, pituitary, testis, and kidney, while ERβ is found in cardiovascular system, prostate, hypothalamus, gastrointestinal tract, ovary, kidney, lungs [155,156,157,158,159], and breast [160,161,162]. The tissue distribution pattern of ERβ suggests its role both in male and female reproduction and development and in central neuroendocrine regulation. Even if most tissues show a predominance of either ERα or ERβ, many others coexpress both [163]. To further support the different regulatory roles that distinguish the two types of ERs, ERα contains dissimilar domains compared with ERβ, suggesting a potential for different ligand specificity that results in different effects [103].
As already mentioned before, all nuclear receptors directly participate in the cellular response to hormone, affecting gene expression by acting as transcription factors. Ligand-activated ER generally induces the increase of target gene expression in target tissues, even if in some other cases it can decrease specific gene transcription [164]. However, the mechanism of negative regulation is still less characterized.
Thus, estrogens mainly work through the binding to ERs in order to transactivate the expression of estrogen-responsive genes. The latter contain an estrogen-responsive element (ERE) in their promoters/enhancers, which represents a recognition sequence to which ER binds. Before activation by association with E2 ligand, ERs exhibit inactivation due to the binding with the heat shock protein-90 (HSP90) [152] and display a diffuse nuclear localization [163]. After ER binding with the E2, ER dimerizes and becomes an active transcription factor acting on the expression of several important genes such as progesterone receptor (another nuclear hormone receptors), vascular endothelial growth factor, c-Fos/c-Jun (proto-oncogenes), and cyclin D1 (cell cycle regulators) [152], stimulating proliferation or differentiation at the cellular level. Several coactivators are also recruited to the complex and, together with ER, participate in remodeling of chromatin structure in order to provide access for transcription machinery to the target gene promoter and allow the transcription to begin [165].
Finally, an additional form of nuclear regulation has been discovered via ERs named “composite regulation,” which is based on protein–protein interaction with other transcription factor, requiring the association between activated steroid receptor and members of the activator protein 1 (AP-1) complex, Fos and Jun [103, 166,167,168,169,170], resulting in either gene activation or repression [166]. Nevertheless, it is not still clear how negative, positive, and composite regulations participate together in the complex cellular functions related to the action of estrogens, like differentiation, organ development, and growth.
Among the EDCs, a huge variety of compounds have been identified as estrogenic chemicals, also called “xenoestrogens” [171, 172]. EDCs with the ability to interfere with estrogenic signaling are, for example, genistein, octylphenol, nonylphenol, bisphenol A (BPA), and diethylstilbestrol (DES), and many of these xenoestrogens are ligands for ERs and therefore can act either as agonists (estrogenic) or as antagonists (antiestrogenic) toward endogenous estrogens.
Several EDCs (e.g., DES, BPA, methoxychlor [173], and genistein) show ER-mediated effects on gene expression, competing with E2 for binding to the same ERs. However, xenoestrogens exhibit a different affinity for the two types of ERs: While DES and methoxychlor have a higher affinity for ERα, genistein and BPA mostly bind ERβ [174]. Interestingly, recent studies have identified a natural compound proposed to be an ERβ-specific ligand [175], which may play a role in preventing progression to prostate cancer by activating prostatic ERβ [176]. Differential ERα- or ERβ-mediated effects may partially account for parallel differential effects of EDCs on different target tissues. This is confirmed in the reproductive tract, where the deleterious effects of EDCs are xenoestrogen-specific [177]. Thus, xenoestrogens can disrupt the endocrine system, both showing differential effects on ERα or ERβ or via differential binding affinity.
Similar to the physiological estrogenic pathway, the association between xenoestrogens and ERs leads to the expression of estrogen-responsive genes (Fig. 2.2), as observed following DES, 7-methylbenz[a]anthracene-3,9-diol (MBA), coumestrol, and genistein (GE) [178] exposure. On the contrary, other xenoestrogens can act as hormonal antagonists (Fig. 2.2) employing multiple mechanisms, for example, by preventing the binding of ER to DNA (e.g., BPA) [179] or inhibiting the binding of ER coactivators [174] to avoid transactivation of gene expression.

Summary of the main mechanisms of action of EDCs involved in estrogenic pathway
However, in addition to the classical ER binding, a possible parallel pathway of estrogenic regulation may involve the existence and binding of alternative “estrogenic receptors,” that is, orphan receptors called ER-related receptors (ERRs), -1 and -2 (Fig. 2.2), which share a similar sequence to ER [180]. Several researchers have focused on the nature (constitutive or liganded) of their transcriptional activities. Moreover, it has been showed that ERRs can interfere with estrogen signaling in various ways, either positively or negatively [181]. Therefore, the identification of possible modulators (positive or negative) of ERR activities could be highly useful in understanding some estrogen-related pathologies.
Besides mimicking and antagonizing endogenous estrogens, EDCs may also mediate estrogenic biological effects by inducing enzymes that accelerate the metabolism of estradiol (Fig. 2.2) [103]. Dioxins, for example, have provided additional evidence for their antiestrogenic effects inducing an increased metabolism of E2 [182, 183].
An additional endocrine-disrupting effect of EDCs on estrogenic responses consists in their ability to increase or decrease the amount of available ERs (Fig. 2.2) [184,185,186]. Many members of NR superfamily are degraded by the ubiquitin-proteasome pathway in a ligand-dependent manner, in order to prevent cells from overstimulation by endogenous hormones or other activating signals [187]. Consequently, EDCs might act on proteasome-mediated degradation of nuclear receptors, altering physiological estrogenic pathway. Masuyama et al. [188] compared the effects of BPA and estradiol treatments on ER-mediated transcription, to try to explain the observations relating to differential effects of BPA treatment on ER levels [189]. In the presence of estradiol, both ERα and ERβ interacted directly with suppressor for Gal 1 (SUG1) of the proteasome. In contrast, BPA activated ER-mediated transcription, without enhancing the interaction between ERβ and SUG1. In the presence of BPA, ubiquitination and degradation of ERβ were also slower than those in the presence of estradiol or phthalic acid, suggesting that BPA may affect the ERβ-mediated transcription of target genes by inhibiting ERβ degradation [190].
Lastly, an attractive hypothesis contemplates the conversion of endocrine chemicals from inactive to active ER-binding estrogens, due to the action of a biotransformation happening in vivo (Fig. 2.2) [103]. A phenomenon of biotransformation has been reported for tamoxifen [191], after the isolation of its metabolite 4-OH-tamoxifen from animals and humans treated with the compound. 4-OH-tamoxifen induces antiestrogenic effects, and it has an even higher affinity for ERs compared with its parent compound. Additional EDCs that are metabolically converted into active agents include ethylene glycol monomethyl ether (EGME) [192] and methoxychlor [193].
Endocrine compounds can also exert apparent estrogenic growth effects by interacting with different cellular factors that could occur downstream from ER of the estrogenic regulatory cascade in target cells (Fig. 2.2) [103]. Tamoxifen, for example, affects calmodulin regulation without ER mediation and directly inhibits protein kinase C through non-ER mechanisms [194, 195]. Other xenoestrogens may rapidly modify other signaling pathways, such as DES, nonylphenol (NP), BPA, and polychlorinated biphenyl (PCB), which are able to alter phosphorylation state of proteins belonging to the large family of mitogen-activated protein kinases (MAPK) in mussel hemocytes [196, 197]. Finally, phytoestrogens possess a variety of nonhormonal properties; for example, dietary phytoestrogens are capable of inhibiting tyrosine kinase activity, which are involved in various growth factor signaling pathways implicated in control of cell growth and differentiation [163]. Anyway, some phytoestrogens, like isoflavones in soy and resveratrol in grapes, have been identified as active agents responsible for benefits to human health exerting hormonal mimicry or antagonism through endocrine pathways or endocrine disruption. Recently, the Food and Drug Administration recognized that the Asian diet, high in soy consumption [198], can be associated with lowered cholesterol and reduction in cardiac risk [199], and the same was sustained by the “French Paradox” that associates red wine consumption to decreased cardiac risk [200]. New scientific data would link the dietary assumption of phytoestrogens with the epigenetic mechanism of histone acetylation (Fig. 2.2) through the phenomenon of gene superinduction. The term “superinduction” means the increased expression of nuclear receptor-activated genes to higher levels than those observed with the established ligand, for example, estradiol for ERs [103]. These observations could explain how soy phytoestrogens and grape may act as nontraditional molecular mechanism, leading to health benefits and anticancer effects. In fact, some of these beneficial effects are now associated with a particular family of histone deacetylases (HDACs), including the human SirT1. Again, the action of fiber assumption in lowering colon cancer incidence has been explained by the presence of butyrate and through the effects on histone acetylation status [201, 202]. Lately, several investigations have asserted that the soy isoflavone phytoestrogens, genistein and daidzein, can be linked to resveratrol, butyrate, and histone acetylation state. Both the isoflavone phytoestrogens act as superinducers of estrogen signaling pathways [203]. This evidence could be the proof that histone acetylation status can also be affected by these compounds. Furthermore, similar superinduction properties are also seen with grape phytoestrogen, resveratrol [103, 204], which has been linked to the effects on HDAC Sir2/SirT1 and, thus, also histone acetylation status [205, 206].
2.5 Mechanism of Action of EDCs and Androgens
Testosterone constitutes the key hormone of androgen hormonal pathway in human, and male testis already produces it around gestational day 65 [207], in order to guarantee the proper establishment of sexual behaviors, male reproductive tract development, and masculinization of other organs. In fact, androgens mediate a wide range of biological responses, such as testicular and accessory sex gland development and function, pubertal sexual maturation, maintenance of spermatogenesis and maturation of sperm, male gonadotropin regulation through feedback loops, and various male secondary characteristics like bone mass, musculature, fat distribution, and hair patterning [208]. Moreover, testosterone becomes critical for brain development too, thanks to its aromatization in 17β-estradiol (E2) by the action of the aromatase CYP19 [209].
Similar to estrogens, both testosterone and its metabolite dihydrotestosterone (DHT) can bind type I NRs, called androgen receptors (ARs). ARs, as well as all the members included in the family of nuclear receptors, are a class of ligand-activated proteins that can enter the nucleus functioning as transcription factors and regulating specific gene expression. ARs can be found in multiple organs, such as hypothalamus [210], pituitary, kidney, prostate, and adrenals (and ovary) [211, 212].
During the perinatal period of programming of the endocrine axis, the hormonal feedback from the gonads to the hypothalamus and pituitary gland represents an event of extreme importance and sensitivity toward endogenous and exogenous stimuli [152]. While female sexual differentiation is considered as a default developmental pathway since it is independent of estrogens and androgens, male sexual differentiation is driven by the fetal testes and it is entirely androgen-dependent [213]. For this reason, male sexual differentiation is highly susceptible to androgen disruptors that could affect developmental programming and reproductive tract maturation [214]. In fact, the eventual lack of testosterone in male fetus due to antiandrogenic exposure, to a genetic mutation in AR or to a blocked metabolism of the hormone, can induce the development of phenotypic female with testes.
The exposure to EDCs during male reproductive tract development may alter testosterone–AR association or the endogenous hormone metabolism, resulting in permanent reprogramming of male reproductive tract and its hormonal communication with the entire hypothalamic–pituitary–gonadal (HPG) axis. In adulthood, HPG axis is already established, and antiandrogen compounds can cause aberration in sperm production and libido in males [215, 216].
The mechanisms of action exploited by endocrine compounds to disrupt androgen pathway are multiple. Among these, we can mention the influence on receptor turnover by decreasing AR levels (Fig. 2.3), the alteration of luteinizing hormone (LH) stimulation (Fig. 2.3), the interference with androgen synthesis, metabolism and clearance (Fig. 2.3), and the alteration of proper folding of the ligand-binding domain (LBD) in ARs after EDC binding [107, 214, 217,218,219,220,221]. Aberrant LBD misfolding means AR inactivity, due to its inability to recruit coactivators and to initiate transcription.

Summary of the main mechanisms of action of EDCs involved in the androgenic pathway
Although there are many sites of action for chemicals to interfere with androgen signaling, endocrine chemicals are classified into two main categories: those that interfere with androgen biosynthesis or metabolism (non-receptor-mediated disruptors) (Fig. 2.3) and those that interact with ARs to interfere with the ligand-dependent transcriptional function (receptor-mediated disruptors) (Fig. 2.3). Despite these two principal classes, some chemicals such as PCBs [219], DES [222], cyproterone acetate (CPA), and hydroxyflutamide (OHF) [217] affect AR activity through the reduction of its expression and level. In addition, another group of EDCs disrupts androgen pathway by inhibiting AR ligand, binding, dimerization, and DNA binding or by silencing expression of AR target genes affecting downstream cellular response.
Androgen receptor–mediated disruptors can be further divided into agonists and antagonists. Agonists bind to androgen receptors and activate a response mimicking the action of endogenous androgens; on the contrary, antagonists block AR transactivation.
A pilot study by Araki et al. [223] reported that some industrial or environmental chemicals show AR agonist activity, and in particular, the compound 1,2-dibromo-4-(1,2-dibromoethyl) cyclohexane (DBE-DBCH) was recently identified as the first potent environmental activator of the human AR [224, 225]. DBE-DBCH can exist in four diastereoisomeric forms: α and β that can be converted into γ and δ at specific conditions [226]. Several analyses showed that diastereomers γ and δ are more potent activators of human AR than α and β, but all the DBE-DBCH diastereomers induced the expression of the downstream target prostate-specific antigen (PSA) in vitro [227].
Contrariwise, EDCs with antiandrogenic action are, for example, dichlorodiphenyltrichloroethanes (DDTs), whose isomers [228] and metabolite [41] were shown to reduce the association between DHT and AR in vivo and to inhibit DHT-induced transcriptional activation in vitro [107]. In addition to AR antagonistic effects of DDT, high concentrations of its metabolite have been shown to function as inhibitors of 5α-reductase that converts testosterone to DHT [229], providing a clear example of how chemical compounds can affect androgen signaling at multiple sites of action. Fetal and neonatal DDT exposure in male produced demasculinizing effects with a high incidence of epididymal and testicular lesions [107, 230] and reduced prostate growth and inflammation [231]. Methoxychlor has a similar structure to DDT and, beyond its well-known estrogenic activity, also shows affinity to the AR at comparable or even higher levels than DDTs [41]. In addition to methoxychlor, BPA is first believed to act in estrogen signaling pathway; however, many scientific evidences have shown its association with AR [41] and its antagonist activity [232]. Lastly, vinclozolin also exerts an endocrine-disrupting potential as an AR antagonist through its primary metabolites [233]. Its mechanism of action consists of inhibiting AR transactivation and androgen-dependent gene expression. In vivo administration of vinclozolin at different doses, routes, and periods (gestation, lactation, puberty, and adulthood) is closely related to a different kind of effect on the male reproductive tract [214]. In addition, vinclozolin exposure during sex determination in developing male germ cells (fetal days 8–14) may lead to transgenerational effect. This phenomenon is based on epigenetic modification on male germ cells that consist of perturbations in DNA methylation patterns, underlining the presence of a relationship between this antiandrogenic compound and epigenetics [38].
2.6 Conclusions
Today, several compounds are classified as endocrine disruptor compounds (EDCs), intended as chemical agents that can interfere with the synthesis, metabolism, and action of endogenous hormones [56]. The EDCs are present in the environment ubiquitously and include both natural molecules [53, 54] and synthetic molecules [51, 52]. Several of these compounds used today have been tested systematically for endocrine-disrupting effects in organisms, as demonstrated by epidemiological studies that suggest an association between the exposure to chemicals and the development of some of the main ailments (e.g., metabolic disorders like obesity and type 2 diabetes) [44] (Table 2.2). This action is due to the ability of EDCs to interfere, with different strategies, with endogenous hormones and other signaling molecules of the endocrine system [56, 57].
The effect of these compounds on the endocrine system contributes to the emergence of several problems in the metabolism and systems of the human body. The first system to be negatively affected by these compounds is the metabolic system, through the interaction of EDCs with the hormone receptors (HRs) of the nuclear receptor (NR) family [75], mainly estrogen receptors (ERs) and androgen receptors (ARs) [103]. The direct receptor interaction of EDCs as agonists or antagonists enhances or inhibits the hormones’ action [44], leading to the development of conditions such as the metabolic syndrome (MetS) [78, 79, 125] (Table 2.2). Another negative manifestation of the effect of EDCs on health is the development of cancer [72], caused by both the induction due to these substances of epigenetic changes [54] and the interference of these with the endocrine system and hormones, which are involved in the evolution of cancer (Table 2.2).
EDCs, in addition to the evolution of cancer and problems related to metabolism, affect most of all reproduction, growth, and development. In particular, early-life exposures to high level of EDCs have been associated with developmental abnormalities and may increase the risk for a variety of diseases later in life [23]. In particular, some classes of EDCs can mimic or block the effects of male and female sex hormones, reducing fecundability and alternating reproductive development in men and women [59] (Table 2.2).
Due to the importance and the impact on human health of EDCs, even more studies have begun to focus on potentially harmful compounds.
As illustrated in Table 2.3, endocrine-active compounds can exert their disrupting potential toward hormonal signaling through a huge variety of mechanisms acting at different levels.
According to their endocrine effect, EDCs can be classified as estrogenic, antiestrogenic, androgenic, and antiandrogenic based on their effects on the hormone system.
Of course, the main classification of EDCs is represented by division between estrogenic and androgenic compounds, intended as products that bind estrogen receptors (ERs) and androgen receptors (ARs) with an activating or inhibiting function (Table 2.1). The EDCs–hormone receptor interaction causes various effects, especially on the reproductive system [56]. Within this classification of EDCs, there are several compounds that are included in the classes of pesticides [49], phytoestrogens [26], plastics or associated chemicals (such as phthalates) [23], and drugs (especially anticancer) [234] (Table 2.1). These molecules have effects on human health mainly through two mechanisms: genomic and nongenomic (Fig. 2.1). The genomic modulation induces the regulation of hormone gene expression [45] due to the fact that hormone receptors bound by EDCs are nuclear receptors (NRs) and have a direct effect on DNA as transcription factors [75]. Despite this, EDCs also show nongenomic modulation, referring to epigenetic modifications [127] (Fig. 2.1).
EDCs have the ability to modulate endocrine system and hormonal activity by hormone mimicry and disruption as the type of genomic modulation mechanism [44]. In fact, EDCs may act as endogenous hormones and other signaling molecules of the endocrine system or even mimic them.
EDCs that compete with endogenous hormones as agonists bind HRs inducing their activation and the initiation of the hormonal signaling pathway [105]. On the contrary, EDCs can also act as antagonists of endogenous hormones (Table 2.3), by binding the same hormone receptors but occupying receptor binding site and antagonizing the proper ligand–hormone interaction [132].
Besides the hormonal activity exhibited by EDCs via receptor binding, toxicants can influence hormonal system activating other signaling pathways, that is, interacting with components of hormone signaling pathways downstream of receptor activation (Table 2.3) [45]. A parallel way to modulate hormonal system consists of altering endogenous hormone biosynthesis or degradation (Table 2.3). Again, EDCs can affect hormonal activity by regulating HR turnover through the stimulation or inhibition of their expression (Table 2.3) [45]. Finally, some EDCs exert endocrine disruption on hormonal system via epigenetic modifications as nongenomic modulation (Table 2.3), via DNA methylation and histone code alteration [1] that occurs during the development, when the epigenome is more susceptible to reprogramming by EDCs [119, 120].
This chapter introduced the general characteristics of endocrine-disrupting chemical compounds and summarized their main molecular mechanisms of action, which involve both genomic and nongenomic modulations. Endocrine disruption is associated with inappropriate regulation of hormone activity, underlining the complexity of physiological hormonal signaling and suggesting a large number of potential targets for EDC disruption. Increased studies concerning EDCs’ action in hormone effects, signaling, and transcriptional regulation could provide a better understanding of the danger and their potential consequences of endocrine-active substances in human health.
References
Guerrero-Bosagna C, Valladares L. Endocrine disruptors, epigenetically induced changes, and transgenerational transmission of characters and epigenetic states. In: Endocrine-disrupting chemicals. Cham: Springer; 2007. p. 175–89.
Jablonka E, et al. The genome in context: biologists and philosophers on epigenetics. BioEssays. 2002;24(4):392–4.
Singal R, Ginder GD. DNA methylation. Blood. 1999;93(12):4059–70.
Cooper RL, et al. Atrazine disrupts the hypothalamic control of pituitary-ovarian function. Toxicol Sci. 2000;53(2):297–307.
MH Yu, H.T., M Tsunoda, Environmental toxicology: biological and health effects of pollutants. 2011.
Waller SA, et al. Agricultural-related chemical exposures, season of conception, and risk of gastroschisis in Washington state. Am J Obstet Gynecol. 2010;202(3):241 e1–6.
Victor-Costa AB, et al. Changes in testicular morphology and steroidogenesis in adult rats exposed to atrazine. Reprod Toxicol. 2010;29(3):323–31.
Maffini MV, et al. Endocrine disruptors and reproductive health: the case of bisphenol-a. Mol Cell Endocrinol. 2006;254–255:179–86.
Kandaraki E, et al. Endocrine disruptors and polycystic ovary syndrome (PCOS): elevated serum levels of bisphenol A in women with PCOS. J Clin Endocrinol Metab. 2011;96(3):E480–4.
Caserta D, et al. Bisphenol A and the female reproductive tract: an overview of recent laboratory evidence and epidemiological studies. Reprod Biol Endocrinol. 2014;12(1):37.
Souter I, et al. The association of bisphenol-a urinary concentrations with antral follicle counts and other measures of ovarian reserve in women undergoing infertility treatments. Reprod Toxicol. 2013;42:224–31.
Bauer SM, et al. The effects of maternal exposure to bisphenol a on allergic lung inflammation into adulthood. Toxicol Sci. 2012;130(1):82–93.
Connor TH, et al. Reproductive health risks associated with occupational exposures to antineoplastic drugs in health care settings: a review of the evidence. J Occup Environ Med. 2014;56(9):901–10.
Richter P, Calamera JC, Morgenfeld MC, Kierszenbaum AL, Lavieri JC, Mancini RE. Effect of chlorambucil on spermatogenesis in the human with malignant lymphoma. Cancer. 1970;25(5):1026–30.
Russell LB, et al. Chlorambucil effectively induces deletion mutations in mouse germ cells. Proc Natl Acad Sci U S A. 1989;86(10):3704–8.
Russell LB, Hunsicker PR, Shelby MD. Melphalan, a second chemical for which specific-locus mutation induction in the mouse is maximum in early spermatids. Mutat Res Lett. 1992;282(3):151–8.
Ning Y, et al. 5-Aza-2′-deoxycytidine inhibited PDGF-induced rat airway smooth muscle cell phenotypic switching. Arch Toxicol. 2013;87(5):871–81.
Stenzig J, et al. DNA methylation in an engineered heart tissue model of cardiac hypertrophy: common signatures and effects of DNA methylation inhibitors. Basic Res Cardiol. 2016;111(1):9.
Klaver R, et al. Direct but no transgenerational effects of decitabine and vorinostat on male fertility. PLoS One. 2015;10(2):e0117839.
Newbold RR, et al. Proliferative lesions and reproductive tract tumors in male descendants of mice exposed developmentally to diethylstilbestrol. Carcinogenesis. 2000;21(7):1355–63.
Li S, et al. Environmental exposure, DNA methylation, and gene regulation: lessons from diethylstilbesterol-induced cancers. Ann N Y Acad Sci. 2003;983(1):161–9.
Axelsson J, Rylander L, Rignell-Hydbom A, Lindh CH, Jönsson BA, Giwercman A. Prenatal phthalate exposure and reproductive function in young men. Environ Res. 2015;138:264–70.
Kim SH, Park MJ. Phthalate exposure and childhood obesity. Ann Pediatr Endocrinol Metab. 2014;19(2):69–75.
Rusyn I, Corton JC. Mechanistic considerations for human relevance of cancer hazard of di(2-ethylhexyl) phthalate. Mutat Res. 2012;750(2):141–58.
Jung T, et al. Effects of the protein phosphorylation inhibitor genistein on maturation of pig oocytes in vitro. J Reprod Fertil. 1993;98(2):529–35.
Helferich WG, Andrade JE, Hoagland MS. Phytoestrogens and breast cancer: a complex story. Inflammopharmacology. 2008;16(5):219–26.
Rawlings NC, Cook SJ, Waldbillig D. Effects of the pesticides carbofuran, chlorpyrifos, dimethoate, lindane, triallate, trifluralin, 2,4-D, and pentachlorophenol on the metabolic endocrine and reproductive endocrine system in ewes. J Toxicol Environ Health A. 1998;54(1):21–36.
Beard AP, Rawlings NC. Thyroid function and effects on reproduction in ewes exposed to the organochlorine pesticides lindane or pentachlorophenol (PCP) from conception. J Toxicol Environ Health A. 1999;58(8):509–30.
Witt KL, Bishop JB. Mutagenicity of anticancer drugs in mammalian germ cells. Mutat Res-Fund Mol Mech Mutagen. 1996;355(1–2):209–34.
Sifakis S, et al. Human exposure to endocrine disrupting chemicals: effects on the male and female reproductive systems. Environ Toxicol Pharmacol. 2017;51:56–70.
Anway MD, et al. Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science. 2005;308(5727):1466–9.
Cupp AS, et al. Effect of transient embryonic in vivo exposure to the endocrine disruptor methoxychlor on embryonic and postnatal testis development. J Androl. 2003;24(5):736–45.
Jager C, Bornman MS, Horst G, I. The effect of p-nonylphenol, an environmental toxicant with oestrogenic properties, on fertility potential in adult male rats. Andrologia. 2009;31(2):99–106.
Zamkowska D, et al. Environmental exposure to non-persistent endocrine disrupting chemicals and semen quality: an overview of the current epidemiological evidence. Int J Occup Med Environ Health. 2018;31(4):377–414.
Miyagawa S, Sato T, Iguchi T. Nonylphenol. Handbook of hormones comparative endocrinology for basic and clinical research. Academic Press. 2016; Subchapter 101A5: 73–574.
Gilbert SF, Epel D. Ecological developmental biology: integrating epigenetics, Medicine, and evolution. Sunderland, MA: Sinauer Associates Inc; 2009.
Brieno-Enriquez MA, et al. Exposure to endocrine disruptor induces transgenerational epigenetic deregulation of microRNAs in primordial germ cells. PLoS One. 2015;10(4):e0124296.
Guerrero-Bosagna C, et al. Epigenetic transgenerational actions of vinclozolin on promoter regions of the sperm epigenome. PLoS One. 2010;5(9):e13100.
Andersen HR, et al. Effects of currently used pesticides in assays for estrogenicity, androgenicity, and aromatase activity in vitro. Toxicol Appl Pharmacol. 2002;179(1):1–12.
Mikamo E, et al. Endocrine disruptors induce cytochrome P450 by affecting transcriptional regulation via pregnane X receptor. Toxicol Appl Pharmacol. 2003;193(1):66–72.
Fang H, et al. Study of 202 natural, synthetic, and environmental chemicals for binding to the androgen receptor. Chem Res Toxicol. 2003;16(10):1338–58.
Anway MD, Leathers C, Skinner MK. Endocrine disruptor vinclozolin induced epigenetic transgenerational adult-onset disease. Endocrinology. 2006;147(12):5515–23.
Skinner MK, et al. Transgenerational epigenetic programming of the brain transcriptome and anxiety behavior. PLoS One. 2008;3(11):e3745.
Swedenborg E, et al. Endocrine disruptive chemicals: mechanisms of action and involvement in metabolic disorders. J Mol Endocrinol. 2009;43(1):1–10.
Combarnous Y, Nguyen TMD. Comparative overview of the mechanisms of action of hormones and endocrine disruptor compounds. Toxics. 2019;7(1):5.
Pimentel D, et al. Ecology of increasing diseases: population growth and environmental degradation. Hum Ecol Interdiscip J. 2007;35(6):653–68.
National Center for Environmental Health (U.S.). Division of Laboratory Sciences.; National Health and Nutrition Examination Survey (U.S.). Third national report on human exposure to environmental chemicals. 2005:1–467.
Environmental Working Group. Pollution in people: Cord blood contaminants in minority newborns. 2009;6(2011):3–60.
Mnif W, et al. Effect of endocrine disruptor pesticides: a review. Int J Environ Res Public Health. 2011;8(6):2265–303.
Jarow JP, et al. Best practice policies for male infertility. J Urol. 2002;167(5):2138–44.
Westerhoff P, et al. Fate of endocrine-disruptor, pharmaceutical, and personal care product chemicals during simulated drinking water treatment processes. Environ Sci Technol. 2005;39(17):6649–63.
Zoeller RTJM. Environmental chemicals as thyroid hormone analogues: new studies indicate that thyroid hormone receptors are targets of industrial chemicals? Mol Cell Endocrinol. 2005;242(1–2):10–5.
Lecomte S, et al. Phytochemicals targeting estrogen receptors: Beneficial rather than adverse effects? Int J Mol Sci. 2017;18(7):1381.
Wynne-Edwards KE. Evolutionary biology of plant defenses against herbivory and their predictive implications for endocrine disruptor susceptibility in vertebrates. Environ Health Perspect. 2001;109(5):443–8.
Lopez-Rodriguez D, et al. Cellular and molecular features of EDC exposure: consequences for the GnRH network. Nat Rev Endocrinol. 2021;17:1–14.
Greathouse KL, Walker CL. Environmental impacts on reproductive health and fertility. mechanisms of endocrine disruption. Cambridge University Press. 2010:72.
Gore AC. Developmental programming and endocrine disruptor effects on reproductive neuroendocrine systems. Front Neuroendocrinol. 2008;29(3):358–74.
Yeung BH, et al. Endocrine disrupting chemicals: multiple effects on testicular signaling and spermatogenesis. Spermatogenesis. 2011;1(3):231–9.
Caserta D, et al. Impact of endocrine disruptor chemicals in gynaecology. Hum Reprod Update. 2008;14(1):59–72.
Kajta M, Wojtowicz AK. Impact of endocrine-disrupting chemicals on neural development and the onset of neurological disorders. Pharmacol Rep. 2013;65(6):1632–9.
Kajta M, Wójtowicz AK. Impact of endocrine-disrupting chemicals on neural development and the onset of neurological disorders. Pharmacol Rep. 2013;65(6):1632–9.
Segovia-Mendoza M, et al. How microplastic components influence the immune system and impact on children health: focus on cancer. Birth Defects Res. 2020;112(17):1341–61.
Brisken C. Endocrine disruptors and breast cancer. Chimia Int J Chem. 2008;62(5):406–9.
Soto AM, Sonnenschein C. Environmental causes of cancer: endocrine disruptors as carcinogens. Nat Rev Endocrinol. 2010;6(7):364–71.
Moriyama K, et al. Thyroid hormone action is disrupted by bisphenol a as an antagonist. J Clin Endocrinol Metab. 2002;87(11):5185–90.
Diamanti-Kandarakis E, et al. The impact of endocrine disruptors on endocrine targets. Horm Metab Res. 2010;42(8):543–52.
Arsenescu V, et al. Polychlorinated biphenyl-77 induces adipocyte differentiation and proinflammatory adipokines and promotes obesity and atherosclerosis. Environ Health Perspect. 2008;116(6):761–8.
Després J-P, et al. Abdominal obesity and the metabolic syndrome: contribution to global cardiometabolic risk. Arterioscler Thromb Vasc Biol. 2008;28(6):1039–49.
Phillips LK, Prins JB. The link between abdominal obesity and the metabolic syndrome. Curr Hypertens Rep. 2008;10(2):156–64.
Lee D-H, et al. A strong dose-response relation between serum concentrations of persistent organic pollutants and diabetes: results from the National Health and Examination Survey 1999–2002. Diabetes Care. 2006;29(7):1638–44.
Lang IA, et al. Association of urinary bisphenol a concentration with medical disorders and laboratory abnormalities in adults. JAMA. 2008;300(11):1303–10.
Brisken C. Endocrine disruptors and breast cancer. Chimia. 2008;62(5):406–9.
Anway MD, Skinner MK. Epigenetic programming of the germ line: effects of endocrine disruptors on the development of transgenerational disease. Reprod BioMed Online. 2008;16(1):23–5.
Dickerson SM, Gore AC. Estrogenic environmental endocrine-disrupting chemical effects on reproductive neuroendocrine function and dysfunction across the life cycle. Rev Endocr Metab Disord. 2007;8(2):143–59.
Gronemeyer H, Gustafsson JA, Laudet V. Principles for modulation of the nuclear receptor superfamily. Nat Rev Drug Discov. 2004;3(11):950–64.
Baker ME, Medlock KL, Sheehan DM. Flavonoids inhibit estrogen binding to rat alpha-fetoprotein. Proc Soc Exp Biol Med. 1998;217(3):317–21.
Boas M, et al. Environmental chemicals and thyroid function. Eur J Endocrinol. 2006;154(5):599–611.
Baillie-Hamilton PF. Chemical toxins: a hypothesis to explain the global obesity epidemic. J Altern Complement Med. 2002;8(2):185–92.
Newbold RR, et al. Developmental exposure to endocrine disruptors and the obesity epidemic. Reprod Toxicol. 2007;23(3):290–6.
Newbold RR, et al. Effects of endocrine disruptors on obesity. Int J Androl. 2008;31(2):201–8.
McMillan BJ, Bradfield CA. The aryl hydrocarbon receptor sans xenobiotics: endogenous function in genetic model systems. Mol Pharmacol. 2007;72(3):487–98.
Beischlag TV, et al. The aryl hydrocarbon receptor complex and the control of gene expression. Crit Rev Eukaryot Gene Expr. 2008;18(3):207–50.
Gore AC. Endocrine-disrupting chemicals: from basic research to clinical practice. Totowa, NJ: Humana Press; 2007.
Warner M, et al. Diabetes, metabolic syndrome, and obesity in relation to serum dioxin concentrations: the Seveso women’s health study. Environ Health Perspect. 2013;121(8):906–11.
Wang CX, et al. Exposure to persistent organic pollutants as potential risk factors for developing diabetes. Sci Chin-Chem. 2010;53(5):980–94.
Bonefeld-Jørgensen EC, et al. Endocrine-disrupting potential of bisphenol A, bisphenol A dimethacrylate, 4-n-nonylphenol, and 4-n-octylphenol in vitro: new data and a brief review. Environ Health Perspect. 2007;115(Suppl 1):69–76.
Baird DD, Newbold R. Prenatal diethylstilbestrol (DES) exposure is associated with uterine leiomyoma development. Reprod Toxicol. 2005;20(1):81–4.
Mustieles Miralles V. Maternal and paternal preconception exposure to phenols and preterm birth. Environ Int. 2020;137:105523.
Taylor JA, et al. Interactive effects of perinatal BPA or DES and adult testosterone and estradiol exposure on adult urethral obstruction and bladder, kidney, and prostate pathology in male mice. Int J Mol Sci. 2020;21(11):3902.
Anway MD, Skinner MK. Epigenetic transgenerational actions of endocrine disruptors. Endocrinology. 2006;147(6 Suppl):S43–9.
Chang HS, et al. Transgenerational epigenetic imprinting of the male germline by endocrine disruptor exposure during gonadal sex determination. Endocrinology. 2006;147(12):5524–41.
Watkins DJ, et al. Impact of phthalate and BPA exposure during in utero windows of susceptibility on reproductive hormones and sexual maturation in peripubertal males. Environ Health. 2017;16(1):69.
Dumesic DA, Abbott DH, Padmanabhan V. Polycystic ovary syndrome and its developmental origins. Rev Endocr Metab Disord. 2007;8(2):127–41.
Padula AM. The freemartin syndrome: an update. Anim Reprod Sci. 2005;87(1–2):93–109.
Vilahur N, et al. Prenatal exposure to mixtures of xenoestrogens and repetitive element DNA methylation changes in human placenta. Environ Int. 2014;71:81–7.
de Cock J, et al. Time to pregnancy and occupational exposure to pesticides in fruit growers in The Netherlands. Occup Environ Med. 1994;51(10):693–9.
Sugiura-Ogasawara M. Reply to: ‘limitations of a case–control study on bisphenol a (BPA) serum levels and recurrent miscarriage’. Hum Reprod. 2006;21(2):566–7.
Scott HM, Mason JI, Sharpe RM. Steroidogenesis in the fetal testis and its susceptibility to disruption by exogenous compounds. Endocr Rev. 2009;30(7):883–925.
Sharpe RM. Pathways of endocrine disruption during male sexual differentiation and masculinization. Best Pract Res Clin Endocrinol Metab. 2006;20(1):91–110.
Sharpe RM. Environmental/lifestyle effects on spermatogenesis. Philos Trans R Soc Lond Ser B Biol Sci. 2010;365(1546):1697–712.
Orth JM, Gunsalus GL, Lamperti AA. Evidence from Sertoli cell-depleted rats indicates that spermatid number in adults depends on numbers of Sertoli cells produced during perinatal development. Endocrinology. 1988;122(3):787–94.
Orth JM, et al. Gonocyte-Sertoli cell interactions during development of the neonatal rodent testis. Curr Top Dev Biol. 2000;50(50):103–24.
Adler SR. Cellular mechanisms of endocrine disruption. In: Endocrine-disrupting chemicals. Cham: Springer; 2007. p. 135–74.
Martini M, Corces VG, Rissman EF. Mini-review: epigenetic mechanisms that promote transgenerational actions of endocrine disrupting chemicals: applications to behavioral neuroendocrinology. Horm Behav. 2020;119:104677.
Montes-Grajales D, Olivero-Verbel J. EDCs DataBank: 3D-structure database of endocrine disrupting chemicals. Toxicology. 2015;327:87–94.
Wong C, et al. Androgen receptor antagonist versus agonist activities of the fungicide vinclozolin relative to hydroxyflutamide. J Biol Chem. 1995;270(34):19998–20003.
Kelce WR, et al. Persistent DDT metabolite p,p'-DDE is a potent androgen receptor antagonist. Nature. 1995;375(6532):581–5.
Daxenberger A. Pollutants with androgen-disrupting potency. Eur J Lipid Sci Technol. 2002;104(2):124–30.
Klaassen CD. Casarett and Doull's toxicology: the basic science of poisons. Ann Intern Med. 1992;117(5)
Norman AW, Henry HL. Hormones. Amsterdam: Elsevier; 1997.
Wan Q, et al. Research progress on the relationship between sex hormone-binding globulin and male reproductive system diseases. Andrologia. 2021;53:e13893.
Sheikh IA, et al. Endocrine disruption: computational perspectives on human sex hormone-binding globulin and phthalate plasticizers. PLoS One. 2016;11(3):e0151444.
Boas M, Feldt-Rasmussen U, Main KM. Thyroid effects of endocrine disrupting chemicals. Mol Cell Endocrinol. 2012;355(2):240–8.
Qiu LL, et al. Decreased androgen receptor expression may contribute to spermatogenesis failure in rats exposed to low concentration of bisphenol a. Toxicol Lett. 2013;219(2):116–24.
Waddington CH. The epigenotype. 1942. Int J Epidemiol. 2012;41(1):10–3.
Holliday R, Pugh JE. DNA modification mechanisms and gene activity during development. Science. 1975;187(4173):226–32.
Riggs AD. X inactivation, differentiation, and DNA methylation. Cytogenet Cell Genet. 1975;14(1):9–25.
Quivy V, et al. Gene activation and gene silencing: a subtle equilibrium. Cloning Stem Cells. 2004;6(2):140–9.
Schaefer CB, et al. Epigenetic decisions in mammalian germ cells. Science. 2007;316(5823):398–9.
Santos F, et al. Dynamic reprogramming of DNA methylation in the early mouse embryo. Dev Biol. 2002;241(1):172–82.
Sanz LA, et al. A mono-allelic bivalent chromatin domain controls tissue-specific imprinting at Grb10. EMBO J. 2008;27(19):2523–32.
Alworth LC, et al. Uterine responsiveness to estradiol and DNA methylation are altered by fetal exposure to diethylstilbestrol and methoxychlor in CD-1 mice: effects of low versus high doses. Toxicol Appl Pharmacol. 2002;183(1):10–22.
Dolinoy DC, Huang D, Jirtle RL. Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proc Natl Acad Sci U S A. 2007;104(32):13056–61.
Anway MD, Rekow SS, Skinner MK. Transgenerational epigenetic programming of the embryonic testis transcriptome. Genomics. 2008;91(1):30–40.
Newbold RR, et al. Developmental exposure to diethylstilbestrol alters uterine gene expression that may be associated with uterine neoplasia later in life. Mol Carcinog. 2007;46(9):783–96.
Tang WY, et al. Persistent hypomethylation in the promoter of nucleosomal binding protein 1 (Nsbp1) correlates with overexpression of Nsbp1 in mouse uteri neonatally exposed to diethylstilbestrol or genistein. Endocrinology. 2008;149(12):5922–31.
Champagne FA. Epigenetic mechanisms and the transgenerational effects of maternal care. Front Neuroendocrinol. 2008;29(3):386–97.
Thomas P, Dong J. Binding and activation of the seven-transmembrane estrogen receptor GPR30 by environmental estrogens: a potential novel mechanism of endocrine disruption. J Steroid Biochem Mol Biol. 2006;102(1–5):175–9.
Hiroi H, et al. Differential interactions of bisphenol a and 17beta-estradiol with estrogen receptor alpha (ERalpha) and ERbeta. Endocr J. 1999;46(6):773–8.
Kucka M, et al. Atrazine acts as an endocrine disrupter by inhibiting cAMP-specific phosphodiesterase-4. Toxicol Appl Pharmacol. 2012;265(1):19–26.
Sargis RM, et al. The novel endocrine disruptor tolylfluanid impairs insulin signaling in primary rodent and human adipocytes through a reduction in insulin receptor substrate-1 levels. Biochim Biophys Acta. 2012;1822(6):952–60.
Tabb MM, Blumberg B. New modes of action for endocrine-disrupting chemicals. Mol Endocrinol. 2006;20(3):475–82.
Rao K, et al. In vitro agonistic and antagonistic endocrine disrupting effects of organic extracts from waste water of different treatment processes. Front Environ Sci Eng. 2013;8(1):69–78.
Stepulak A, et al. Fluoxetine inhibits the extracellular signal regulated kinase pathway and suppresses growth of cancer cells. Cancer Biol Ther. 2008;7(10):1685–93.
Liu XL, et al. Fluoxetine regulates mTOR signalling in a region-dependent manner in depression-like mice. Sci Rep. 2015;5(1):16024.
Ofek K, et al. Fluoxetine induces vasodilatation of cerebral arterioles by co-modulating NO/muscarinic signalling. J Cell Mol Med. 2012;16(11):2736–44.
Schopel M, et al. Allosteric activation of GDP-bound Ras isoforms by Bisphenol derivative plasticisers. Int J Mol Sci. 2018;19(4):1133.
Ptak A, Gregoraszczuk EL. Bisphenol a induces leptin receptor expression, creating more binding sites for leptin, and activates the JAK/stat, MAPK/ERK and PI3K/Akt signalling pathways in human ovarian cancer cell. Toxicol Lett. 2012;210(3):332–7.
Hugo ER, et al. Bisphenol A at environmentally relevant doses inhibits adiponectin release from human adipose tissue explants and adipocytes. Environ Health Perspect. 2008;116(12):1642–7.
Jambor T, et al. In vitro effect of 4-nonylphenol on human chorionic gonadotropin (hCG) stimulated hormone secretion, cell viability and reactive oxygen species generation in mice Leydig cells. Environ Pollut. 2017;222:219–25.
Derouiche S, et al. Activation of TRPA1 channel by antibacterial agent Triclosan induces VEGF secretion in human prostate cancer stromal cells. Cancer Prev Res (Phila). 2017;10(3):177–87.
Engeli RT, et al. Interference of paraben compounds with estrogen metabolism by inhibition of 17beta-Hydroxysteroid dehydrogenases. Int J Mol Sci. 2017;18(9):2007.
Ishihara A, Sawatsubashi S, Yamauchi K. Endocrine disrupting chemicals: interference of thyroid hormone binding to transthyretins and to thyroid hormone receptors. Mol Cell Endocrinol. 2003;199(1–2):105–17.
Déchaud H, et al. Xenoestrogen interaction with human sex hormone-binding globulin (hSHBG)1. Steroids. 1999;64(5):328–34.
Yamauchi K, Ishihara A. Thyroid system-disrupting chemicals: interference with thyroid hormone binding to plasma proteins and the cellular thyroid hormone signaling pathway. Rev Environ Health. 2006;21(4):229–51.
Hong H, et al. Human sex hormone-binding globulin binding affinities of 125 structurally diverse chemicals and comparison with their binding to androgen receptor, estrogen receptor, and alpha-fetoprotein. Toxicol Sci. 2015;143(2):333–48.
Waalkes MP, et al. Estrogen signaling in livers of male mice with hepatocellular carcinoma induced by exposure to arsenic in utero. J Natl Cancer Inst. 2004;96(6):466–74.
Li Y, et al. Epigenetic reactivation of estrogen receptor-alpha (ERalpha) by genistein enhances hormonal therapy sensitivity in ERalpha-negative breast cancer. Mol Cancer. 2013;12(1):9.
Schmutzler C, et al. Thyroid hormone biosynthesis is a sensitive target for the action of endocrine disrupting chemicals (EDC). Exp Clin Endocrinol Diabetes. 2006;114(S 1):OR8_44.
Grun F, Blumberg B. Environmental obesogens: organotins and endocrine disruption via nuclear receptor signaling. Endocrinology. 2006;147(6 Suppl):S50–5.
Kanayama T, et al. Organotin compounds promote adipocyte differentiation as agonists of the peroxisome proliferator-activated receptor gamma/retinoid X receptor pathway. Mol Pharmacol. 2005;67(3):766–74.
Woodruff TJ, et al. Environmental impacts on reproductive health and fertility. Cambridge University Press; 2010.
Beato M. Gene regulation by steroid hormones. In: Gene Expression. Springer; 1993. p. 43–75.
Gustafsson JA. Estrogen receptor beta--a new dimension in estrogen mechanism of action. J Endocrinol. 1999;163(3):379–83.
Couse JF, et al. Tissue distribution and quantitative analysis of estrogen receptor-alpha (ERalpha) and estrogen receptor-beta (ERbeta) messenger ribonucleic acid in the wild-type and ERalpha-knockout mouse. Endocrinology. 1997;138(11):4613–21.
Mitchner NA, Garlick C, Ben-Jonathan N. Cellular distribution and gene regulation of estrogen receptors alpha and beta in the rat pituitary gland. Endocrinology. 1998;139(9):3976–83.
Sar M, Welsch F. Differential expression of estrogen receptor-beta and estrogen receptor-alpha in the rat ovary. Endocrinology. 1999;140(2):963–71.
Shughrue PJ, Komm B, Merchenthaler I. The distribution of estrogen receptor-beta mRNA in the rat hypothalamus. Steroids. 1996;61(12):678–81.
Shughrue PJ, et al. Comparative distribution of estrogen receptor-alpha (ER-alpha) and beta (ER-beta) mRNA in the rat pituitary, gonad, and reproductive tract. Steroids. 1998;63(10):498–504.
Dotzlaw H, et al. Expression of estrogen receptor-beta in human breast tumors. J Clin Endocrinol Metab. 1997;82(7):2371–4.
Ferguson AT, Lapidus RG, Davidson NE. The regulation of estrogen receptor expression and function in human breast cancer. In: Biological and hormonal therapies of cancer. Cham: Springer; 1998. p. 255–78.
Vladusic EA, et al. Expression of estrogen receptor beta messenger RNA variant in breast cancer. Cancer Res. 1998;58(2):210–4.
Yoon K, et al. Estrogenic endocrine-disrupting chemicals: molecular mechanisms of actions on putative human diseases. J Toxicol Environ Health B Crit Rev. 2014;17(3):127–74.
Beato M. Transcriptional control by nuclear receptors. FASEB J. 1991;5(7):2044–51.
Klinge CM. Estrogen receptor interaction with co-activators and co-repressors. Steroids. 2000;65(5):227–51.
Diamond MI, et al. Transcription factor interactions: selectors of positive or negative regulation from a single DNA element. Science. 1990;249(4974):1266–72.
Tzukerman M, Zhang XK, Pfahl M. Inhibition of estrogen receptor activity by the tumor promoter 12-O-tetradeconylphorbol-13-acetate: a molecular analysis. Mol Endocrinol. 1991;5(12):1983–92.
Jonat C, et al. Antitumor promotion and antiinflammation: down-modulation of AP-1 (Fos/Jun) activity by glucocorticoid hormone. Cell. 1990;62(6):1189–204.
Lucibello FC, et al. Mutual transrepression of Fos and the glucocorticoid receptor: involvement of a functional domain in Fos which is absent in FosB. EMBO J. 1990;9(9):2827–34.
Yang-Yen H-F, et al. Transcriptional interference between c-Jun and the glucocorticoid receptor: mutual inhibition of DNA binding due to direct protein-protein interaction. Cell. 1990;62(6):1205–15.
Vondracek J, Kozubik A, Machala M. Modulation of estrogen receptor-dependent reporter construct activation and G0/G1-S-phase transition by polycyclic aromatic hydrocarbons in human breast carcinoma MCF-7 cells. Toxicol Sci. 2002;70(2):193–201.
Coldham NG, et al. Evaluation of a recombinant yeast cell estrogen screening assay. Environ Health Perspect. 1997;105(7):734–42.
Eroschenko VP, Rourke AW, Sims WF. Estradiol or methoxychlor stimulates estrogen receptor (ER) expression in uteri. Reprod Toxicol. 1996;10(4):265–71.
Routledge EJ, et al. Differential effects of xenoestrogens on coactivator recruitment by estrogen receptor (ER) alpha and ERbeta. J Biol Chem. 2000;275(46):35986–93.
Pak TR, et al. The androgen metabolite, 5α-Androstane-3β, 17β-diol, is a potent modulator of estrogen receptor-β1-mediated gene transcription in neuronal cells. Endocrinology. 2005;146(1):147–55.
Imamov O, Lopatkin NA, Gustafsson JA. Estrogen receptor beta in prostate cancer. N Engl J Med. 2004;351(26):2773–4.
Welshons WV, et al. Low-dose bioactivity of xenoestrogens in animals: fetal exposure to low doses of methoxychlor and other xenoestrogens increases adult prostate size in mice. Toxicol Ind Health. 1999;15(1–2):12–25.
Oostenbrink C, van Gunsteren WF. Free energies of ligand binding for structurally diverse compounds. Proc Natl Acad Sci U S A. 2005;102(19):6750–4.
Gould JC, et al. Bisphenol a interacts with the estrogen receptor alpha in a distinct manner from estradiol. Mol Cell Endocrinol. 1998;142(1–2):203–14.
Evans RM. The steroid and thyroid hormone receptor superfamily. Science. 1988;240(4854):889–95.
Horard B, Vanacker JM. Estrogen receptor-related receptors: orphan receptors desperately seeking a ligand. J Mol Endocrinol. 2003;31(3):349–57.
Spink DC, et al. Stimulation of 17 beta-estradiol metabolism in MCF-7 cells by bromochloro- and chloromethyl-substituted dibenzo-p-dioxins and dibenzofurans: correlations with antiestrogenic activity. J Toxicol Environ Health. 1994;41(4):451–66.
Spink DC, et al. 17β-estradiol hydroxylation catalyzed by human cytochrome P450 1A1: a comparison of the activities induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin in MCF-7 cells with those from heterologous expression of the cDNA. Arch Biochem Biophys. 1992;293(2):342–8.
La Rosa P, et al. Xenoestrogens alter estrogen receptor (ER) alpha intracellular levels. PLoS One. 2014;9(2):e88961.
Sheeler CQ, Dudley MW, Khan SA. Environmental estrogens induce transcriptionally active estrogen receptor dimers in yeast: activity potentiated by the coactivator RIP140. Environ Health Perspect. 2000;108(2):97–103.
Khurana S, Ranmal S, Ben-Jonathan N. Exposure of newborn male and female rats to environmental estrogens: delayed and sustained hyperprolactinemia and alterations in estrogen receptor expression. Endocrinology. 2000;141(12):4512–7.
Dennis AP, Haq RU, Nawaz Z. Importance of the regulation of nuclear receptor degradation. Front Biosci. 2001;6:D954–9.
Masuyama H, et al. Endocrine disrupting chemicals, phthalic acid and nonylphenol, activate Pregnane X receptor-mediated transcription. Mol Endocrinol. 2000;14(3):421–8.
Inoshita H, Masuyama H, Hiramatsu Y. The different effects of endocrine-disrupting chemicals on estrogen receptor-mediated transcription through interaction with coactivator TRAP220 in uterine tissue. J Mol Endocrinol. 2003;31(3):551–61.
Masuyama H, Hiramatsu Y. Involvement of suppressor for Gal 1 in the ubiquitin/proteasome-mediated degradation of estrogen receptors. J Biol Chem. 2004;279(13):12020–6.
Jordan VC, et al. A monohydroxylated metabolite of tamoxifen with potent antioestrogenic activity. J Endocrinol. 1977;75(2):305–16.
Johanson G. Toxicity review of ethylene glycol monomethyl ether and its acetate ester. Crit Rev Toxicol. 2000;30(3):307–45.
Cummings AM. Methoxychlor as a model for environmental estrogens. Crit Rev Toxicol. 1997;27(4):367–79.
O'Brian CA, et al. Inhibition of protein kinase C by tamoxifen. Cancer Res. 1985;45(6):2462–5.
Issandou M, et al. Opposite effects of tamoxifen on in vitro protein kinase C activity and endogenous protein phosphorylation in intact MCF-7 cells. Cancer Res. 1990;50(18):5845–50.
Canesi L, et al. Effects of PCB congeners on the immune function of Mytilus hemocytes: alterations of tyrosine kinase-mediated cell signaling. Aquat Toxicol. 2003;63(3):293–306.
Canesi L, et al. Environmental estrogens can affect the function of mussel hemocytes through rapid modulation of kinase pathways. Gen Comp Endocrinol. 2004;138(1):58–69.
Rose DP, Boyar AP, Wynder EL. International comparisons of mortality rates for cancer of the breast, ovary, prostate, and colon, and per capita food consumption. Cancer. 1986;58(11):2363–71.
Food and A Drug. Food labeling health claims; soy protein and coronary heart disease. Fed Regist. 1999;64:57699–733.
Wu JM, et al. Mechanism of cardioprotection by resveratrol, a phenolic antioxidant present in red wine (review). Int J Mol Med. 2001;8(1):3–17.
Marlett JA, et al. Position of the American dietetic association: health implications of dietary fiber. J Am Diet Assoc. 2002;102(7):993–1000.
Hinnebusch BF, et al. The effects of short-chain fatty acids on human colon cancer cell phenotype are associated with histone hyperacetylation. J Nutr. 2002;132(5):1012–7.
Villar-Garea A, Esteller M. Histone deacetylase inhibitors: understanding a new wave of anticancer agents. Int J Cancer. 2004;112(2):171–8.
Gehm BD, et al. Resveratrol, a polyphenolic compound found in grapes and wine, is an agonist for the estrogen receptor. Proc Natl Acad Sci U S A. 1997;94(25):14138–43.
Howitz KT, et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003;425(6954):191–6.
Wood JG, et al. Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature. 2004;430(7000):686–9.
Kelce WR, Wilson EM. Antiandrogenic effects of environmental endocrine disruptors. In: Endocrine disruptors—part I. cham: Springer; 2001. p. 39–61.
Matsumoto T, et al. Androgen receptor functions in male and female physiology. J Steroid Biochem Mol Biol. 2008;109(3–5):236–41.
Holloway CC, Clayton DF. Estrogen synthesis in the male brain triggers development of the avian song control pathway in vitro. Nat Neurosci. 2001;4(2):170–5.
Beyer C, Green SJ, Hutchison JB. Androgens influence sexual differentiation of embryonic mouse hypothalamic aromatase neurons in vitro. Endocrinology. 1994;135(3):1220–6.
Burek M, et al. Tissue-specific distribution of the androgen receptor (AR) in the porcine fetus. Acta Histochem. 2007;109(5):358–65.
Davison SL, Davis SR. Androgens in women. J Steroid Biochem Mol Biol. 2003;85(2–5):363–6.
Nef S, Parada LF. Hormones in male sexual development. Genes Dev. 2000;14(24):3075–86.
Luccio-Camelo DC, Prins GS. Disruption of androgen receptor signaling in males by environmental chemicals. J Steroid Biochem Mol Biol. 2011;127(1–2):74–82.
Chapin RE, et al. Endocrine modulation of reproduction. Fundam Appl Toxicol. 1996;29(1):1–17.
Welsch F. How can chemical compounds alter human fertility? Eur J Obstet Gynecol Reprod Biol. 2003;106(1):88–91.
List HJ, et al. Effects of antiandrogens on chromatin remodeling and transcription of the integrated mouse mammary tumor virus promoter. Exp Cell Res. 2000;260(1):160–5.
Massaad C, et al. How can chemical compounds alter human fertility? Eur J Obstet Gynecol Reprod Biol. 2002;100(2):127–37.
Portigal CL, et al. Polychlorinated biphenyls interfere with androgen-induced transcriptional activation and hormone binding. Toxicol Appl Pharmacol. 2002;179(3):185–94.
Bonefeld-Jorgensen EC, et al. Endocrine-disrupting potential of bisphenol A, bisphenol A dimethacrylate, 4-n-nonylphenol, and 4-n-octylphenol in vitro: new data and a brief review. Environ Health Perspect. 2007;115(Suppl 1):69–76.
Ostby J, et al. The fungicide procymidone alters sexual differentiation in the male rat by acting as an androgen-receptor antagonist in vivo and in vitro. Toxicol Ind Health. 1999;15(1–2):80–93.
McKinnell C, et al. Suppression of androgen action and the induction of gross abnormalities of the reproductive tract in male rats treated neonatally with diethylstilbestrol. J Androl. 2001;22(2):323–38.
Araki N, et al. Screening for androgen receptor activities in 253 industrial chemicals by in vitro reporter gene assays using AR-EcoScreen cells. Toxicol in Vitro. 2005;19(6):831–42.
Larsson A, et al. Identification of the brominated flame retardant 1,2-dibromo-4-(1,2-dibromoethyl)cyclohexane as an androgen agonist. J Med Chem. 2006;49(25):7366–72.
Nyholm JR, et al. Maternal transfer of brominated flame retardants in zebrafish (Danio rerio). Chemosphere. 2008;73(2):203–8.
Arsenault G, et al. Structure characterization and thermal stabilities of the isomers of the brominated flame retardant 1,2-dibromo-4-(1,2-dibromoethyl)cyclohexane. Chemosphere. 2008;72(8):1163–70.
Khalaf H, et al. Diastereomers of the brominated flame retardant 1,2-dibromo-4-(1,2 dibromoethyl)cyclohexane induce androgen receptor activation in the hepg2 hepatocellular carcinoma cell line and the lncap prostate cancer cell line. Environ Health Perspect. 2009;117(12):1853–9.
Danzo BJ. Environmental xenobiotics may disrupt normal endocrine function by interfering with the binding of physiological ligands to steroid receptors and binding proteins. Environ Health Perspect. 1997;105(3):294–301.
Lo S, et al. Effects of various pesticides on human 5alpha-reductase activity in prostate and LNCaP cells. Toxicol in Vitro. 2007;21(3):502–8.
Wolf C Jr, et al. Administration of potentially antiandrogenic pesticides (procymidone, linuron, iprodione, chlozolinate, p,p'-DDE, and ketoconazole) and toxic substances (dibutyl- and diethylhexyl phthalate, PCB 169, and ethane dimethane sulphonate) during sexual differentiation produces diverse profiles of reproductive malformations in the male rat. Toxicol Ind Health. 1999;15(1–2):94–118.
You L, Brenneman KA, Heck H. In utero exposure to antiandrogens alters the responsiveness of the prostate to p,p'-DDE in adult rats and may induce prostatic inflammation. Toxicol Appl Pharmacol. 1999;161(3):258–66.
Xu LC, et al. Evaluation of androgen receptor transcriptional activities of bisphenol A, octylphenol and nonylphenol in vitro. Toxicology. 2005;216(2–3):197–203.
Kelce WR, et al. Environmental hormone disruptors: evidence that vinclozolin developmental toxicity is mediated by antiandrogenic metabolites. Toxicol Appl Pharmacol. 1994;126(2):276–85.
Mittendorf R. Teratogen update: carcinogenesis and teratogenesis associated with exposure to diethylstilbestrol (DES) in utero. Teratology. 1995;51(6):435–45.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2023 The Author(s)
About this chapter

Cite this chapter
Rizzo, R., Bortolotti, D., Rizzo, S., Schiuma, G. (2023). Cellular Mechanisms of Endocrine Disruption. In: Marci, R. (eds) Environment Impact on Reproductive Health. Springer, Cham. https://doi.org/10.1007/978-3-031-36494-5_2
Download citation
DOI: https://doi.org/10.1007/978-3-031-36494-5_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-36493-8
Online ISBN: 978-3-031-36494-5
eBook Packages: MedicineMedicine (R0)