
Abstract
Clastic-dominated lead–zinc (CD Pb–Zn) deposits are an important source of the world’s Pb and Zn supply. Their genesis is contentious due to uncertainties regarding the time of ore formation relative to the deposition of the fine-grained carbonaceous strata that host CD Pb–Zn mineralization. Sulfur-isotopic studies are playing an important role in determining if ore minerals precipitated when hydrothermal fluids exhaled into the water column from which the host strata were being deposited, or when hydrothermal fluids entered the host strata during diagenesis or even later after lithification. Older conventional S-isotopic studies, based on analyses of bulk mineral-separate samples obtained by either physical or chemical separation methods, provided data that has been widely used to support a syngenetic-exhalative origin for CD Pb–Zn mineralization. However, with the advent in the late 1980’s of in situ S-isotopic studies using micro-analytical methods, it soon became apparent that detailed S-isotopic variations of genetic importance are blurred in conventional analytical data sets because of averaging during sample preparation. Clastic-dominated Pb–Zn mineralization in the North Australian Proterozoic metallogenic province and the North American Paleozoic Cordilleran province has been the subject of many stable isotope studies based on both bulk and in situ analytical methods. Together with detailed mineral texture observations, the studies have revealed a similar sulfide mineral paragenesis in both provinces. The earliest sulfide phase in the paragenesis is fine-grained pyrite that sometimes has a framboidal texture. This pyrite typically has a wide range of δ34S values that are more than 15‰ lower than the value of coeval seawater sulfate. These features are typical of, and very strong evidence for, pyrite formation by bacterial sulfate reduction (BSR) either syngenetically in an anoxic water column or during early diagenesis in anoxic muds. The formation of this early pyrite is followed by one or more later generations of pyrite that often occur as overgrowths around the early pyrite generation. The later pyrite generations have δ34S values that are much higher than the early pyrite, often approaching the value of coeval seawater sulfate. Later pyrite formation has been variously attributed to BSR in a more restricted diagenetic environment, to sulfate driven-anaerobic oxidation of methane (SD-AOM) and to abiotic thermal sulfate reduction (TSR), with all three mechanisms again involving coeval seawater sulfate. The main sulfide ore minerals, galena and sphalerite, either overlap with or postdate later pyrite generations and are most often attributed to TSR of seawater sulfate. However, in comparison with pyrite, there is a dearth of in situ δ34S data for galena and sphalerite that needs to be rectified to better understand ore forming processes. Importantly, the available data do not support a simple sedimentary-exhalative model for the formation of all but part of one of the Northern American and Australian deposits. The exception is the giant Red Dog deposit group in Alaska where various lines of evidence, including stable isotopic data, indicate that ore formation was protracted, ranging from early syn-sedimentary to early diagenetic sulfide formation through to late sulfide deposition in veins and breccias. The Red Dog deposits are the only example with early sphalerite with extremely low negative δ34S values typical of a BSR-driven precipitation mechanism. By contrast, later stages of pyrite, sphalerite and galena have higher positive δ34S values indicative of a TSR-driven precipitation mechanism. In CD Pb–Zn deposits in carbonate-bearing strata, carbon and oxygen isotope studies of the carbonates provide evidence that the dominant carbonate species in the ore-forming hydrothermal fluids was H2CO3, and that the fluids were initially warm (≥ 150 °C) and neutral to acid. The δ18O values of the hydrothermal fluids are ≥ 6‰, suggesting these fluids were basinal fluids that evolved through exchange with the basinal sedimentary rocks. Known CD Pb–Zn deposits all occur at or near current land surfaces and their discovery involved traditional prospecting, geophysical and geochemical exploration techniques. Light stable isotopes are unlikely to play a significant role in the future search for new CD Pb–Zn deposits deep beneath current land surfaces, but are likely to prove useful in identifying ore-forming hydrothermal fluid pathways in buried CD Pb–Zn systems and be a vector to new mineralization.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others

1 Introduction
Clastic-dominated lead–zinc (CD Pb–Zn) mineral deposits are the main source of the world’s zinc and lead supply. They are therefore an attractive exploration target but their genesis, a critical element in exploration strategies, remains contentious (Huston et al. 2006; Magnall et al. 2021; Spinks et al. 2021). Reviews of the CD Pb–Zn deposit type include Gustafson and Williams (1981), Leach et al. (2005), Leach et al. (2010) and Wilkinson (2014). Common features of the deposit type are: (1) their occurrence in fine-grained carbonaceous and pyritic sedimentary rocks; (2) ores that are sedimentary in appearance with sulfide banding that parallels bedding in adjacent inter-ore strata; (3) a stratiform and stratabound relationship with their host sedimentary rock sequences; (4) stacked layers of mineralization; (5) high sulfide sulfur contents (typically ≥ 20 wt% S); (6) a simple sulfide mineralogy dominated by pyrite, sphalerite and galena and (7) low Cu (≤ ~0.4 wt% Cu) and variable Ag (≤ ~150 g/t Ag) contents. Deposits have an irregular age distribution. They first appeared ~1850 Ma but there are few in rocks aged between 1350 and 760 Ma. Barite is often associated with Paleozoic-age deposits but less so with Proterozoic examples.
This chapter discusses the genetic and exploration implications of the light-element (carbon, oxygen and sulfur) isotope geochemistry of CD Pb–Zn mineralization in the two most comprehensively studied CD Pb–Zn metallogenic provinces. These are the North Australian Proterozoic province (Huston et al. 2006) and the North American Paleozoic Cordilleran province (Leach et al. 2010). General information on light stable isotopes, including their chemistry, fractionation, analysis and applications can be found in Huston et al. (2023). Stable isotope applications of particular relevance to CD Pb–Zn deposits are highlighted in the following summary of CD Pb–Zn mineral systems.
2 Clastic-Dominated Pb–Zn Mineral Systems
The mineral system concept is growing in prominence in ore genesis studies and mineral exploration (Hagemann et al. 2016). The main components of a mineral system are the source of ore- and gangue-mineral constituents; the transport of these constituents to sites of ore formation; the trap where ore forms and the energy that drives the system.
The generic CD Pb–Zn mineral systems in Fig. 1 are based on the work of Goodfellow (1987), Hoggard et al. (2020), Huston et al. (2006, 2016), Manning and Embso (2018) and Sangster (2002, 2018). The sedimentary rock sequences in the system occur in sedimentary basins formed in extensional tectonic settings. Basin fill includes both shallow- and deeper-water facies material. During burial and compaction seawater-sulfate bearing basinal waters are thought to react with the basin fill and scavenge metals, generating the mineralizing hydrothermal fluids. Possible energy sources that drive systems include heat flow from the basement, gravity, compaction, seismic pumping, and compressional tectonism (Cooke et al. 1998), and the release of pressure in over-pressured fluid reservoirs triggered by extension (Vearncombe et al. 1996). The main channelways along which the hydrothermal fluids move from deep within host basins to trap sites are extensional growth faults.

Generic clastic-dominated Pb–Zn mineral systems showing a syngenetic ore formation, and b syndiagenetic to epigenetic ore formation. Pathways (1 & 2) in a are discussed in the text
The trap for CD Pb–Zn mineralization is the environment associated with the deposition and lithification of the fine-grained carbonaceous strata that host the mineralization. These host rocks are typically deposited in half-graben tectonic settings adjacent to the same extensional growth faults that may have been hydrothermal fluid conduits. Wall rocks adjacent to feeder faults are often hydrothermally altered and spatially associated with feeder-zone breccia- and vein-hosted mineralization.
Ore deposition occurs when the chemistry of the hydrothermal fluid changes upon arrival at the trap site. One possible trigger for change is the exhalation of the fluid into the host-sediment depositional environment (syngenetic ore formation), a scenario illustrated in Fig. 1a. If the hydrothermal fluid has a low salinity, ore-forming reactions would most likely be triggered by mixing between the hydrothermal fluid and seawater (Pathway 1, Fig. 1a). On the other hand, if the exhaling hydrothermal fluid has a high salinity, it would mostly like be denser than seawater and would pond in sea-floor depressions and form brine pools in which sulfide formation is triggered by changes associated with the development and evolution of the brine pool chemistry (Pathway 2, Fig. 1a). Figure 1b illustrates subsurface ore forming scenarios that are triggered by reactions between the hydrothermal fluid and components of the host strata during either diagenesis (syndiagenetic ore formation) or after lithification (epigenetic ore formation). The timing of CD Pb–Zn ore formation relative to the deposition of the host rocks has been debated for decades and remains a critical uncertainty in CD Pb–Zn mineral systems. In the case of syndiagenetic and epigenetic ore formation (Fig. 1b) an outflow zone for the hydrothermal fluids may occur in rocks downstream from the trap environment.
2.1 Applications of Stable Isotopes in Clastic-Dominated Pb–Zn Systems
Many of the important chemical reactions in CD Pb–Zn systems fractionate stable isotopes and carbon, oxygen and sulfur isotopic studies of systems have done much to improve our understanding of how ore forms within the systems.
2.1.1 Metal Source Identification
It is difficult to elucidate metal sources for CD Pb–Zn deposits using stable isotope evidence. However, some insights can be gained indirectly through stable isotope studies of possible source rocks. For example, Cooke et al. (1998) found that altered volcanic horizons stratigraphically below CD Pb–Zn mineralization in the North Australian CD Pb–Zn province (Fig. 2) are almost 100% depleted in Pb, Zn and Cu and are therefore a possible metal source. Cooke et al. (1998) used fluid inclusion and bulk C- and O-isotope data from the altered volcanic rocks to conclude that the fluids which caused the alteration were warm (≈ 100 °C) and saline (> 20 wt% NaCl equivalent) and had a δ18OSMOW value of ≈ −1‰ and a δ13CPDB value of ≈ −7‰. These values are consistent with the idea that the metal-depleting fluids were originally meteoric waters ± seawater and that their salinity and 18O content increased as they moved through overlying basinal carbonates and evaporites and deeper into underlying volcanic rocks. The question of whether or not the isotopic composition of these fluids prove that volcanic rocks were the source of metals in CD Pb–Zn mineralization higher in the stratigraphic succession is discussed later in this Chapter.

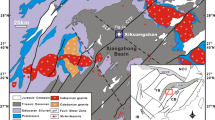
Location of the significant clastic-dominated Pb–Zn deposits and prospects in the Carpentaria Zinc Belt of Northern Australia
2.1.2 Sulfide-Sulfur Source Identification
In contrast to metal sources, sulfur isotope studies have proved very useful in identifying the source of sulfide-S in CD Pb–Zn mineralization. Sulfur isotope data, along with other geological evidence, suggest strongly that the sulfide-S source was seawater sulfate, and came either directly from seawater or indirectly via sulfate minerals such as gypsum, anhydrite or barite. The value of δ34Sseawater sulfate has varied significantly through geological time, and the secular δ34Sseawater sulfate curve was initially constructed using worldwide sulfur isotopic data from marine evaporites (Claypool et al. 1980). Evaporites are best preserved in Phanerozoic strata and the secular δ34Sseawater sulfate curve is therefore best documented in the Phanerozoic, but less so in the Precambrian where evaporites are rare. During the Phanerozoic the curve varies between ~12 and 30‰ and the processes controlling δ34Sseawater sulfate values are well understood (Bottrell and Newton 2006; Halevy et al. 2012; Canfield 2013). Although the Precambrian part of the curve is not so well documented, it continues to be improved using not only scant evaporite data, but also carbonate associated sulfate (CAS) data (Kah et al. 2004; Gellatly and Lyons 2005; Chu et al. 2007; Luo et al. 2015; Crockford et al. 2019; Turchyn and DePaolo 2019). In this Chapter the best estimates of the value of δ34Sseawater sulfate at the time of deposition of the strata hosting CD Pb–Zn deposits are presented under the Source heading, whereas the applicability of this value to ore formation is discussed under the Trap heading because, as will become apparent later in this Chapter, one of the main issues relating to CD Pb–Zn ore genesis is how and where in the mineral system sulfate is reduced to sulfide.
2.1.3 Transport Mechanisms
In a seminal study of hydrothermal fluids capable of forming CD Pb–Zn mineralization, Cooke et al. (2000) used geochemical modelling to show that both cool (≤ 150 ℃) oxidized hydrothermal fluids (dominated by SO42− ± HSO4−) and warmer (~250 ℃) reduced acidic fluids (dominated by H2S or HS−) can transport and deposit sufficient quantities of Pb and Zn to form CD Pb–Zn deposits. Stable isotopic studies can help determine which fluid type was involved in the formation of a particular deposit and examples are discussed in this Chapter.
2.1.4 Trap Mechanisms
The S-isotopic composition of galena, sphalerite and pyrite are important determinants of the processes by which the sulfides form in CD Pb–Zn deposits, the relative timing of sulfide mineral formation (paragenesis), and temperatures of ore formation.
A low-temperature (< 100 °C) process frequently mentioned in discussions of CD Pb–Zn ore formation is bacterial sulfate reduction (BSR). BSR is the main process by which pyrite forms in modern carbonaceous muds (Berner 1970), sediments that may be modern analogues of the pyritic carbonaceous fine-grained strata that host CD Pb–Zn mineralization. Critical components of the modern process are organic matter, sulfate, a source of iron (typically hydrous iron oxide coatings on detrital minerals) and sulfate reducing bacteria. In modern carbonaceous muds pyrite formation by BSR most often occurs during early diagenesis, within centimeters of the sediment–water interface. Where the rate of organic matter accumulation exceeds the rate of oxidation of the organic matter, anoxic conditions develop and sulfate reducing bacteria become active. The bacteria metabolize the organic matter and reduce sulfate that diffuses into the sediments from the overlying water column. The basic reaction in the process is:
The H2S produced by BSR reacts with hydrous iron oxides in the muds to form either mackinawite (FeS) and/or greigite (Fe3S4). Some H2S is bacterially oxidized to elemental sulfur which in turn reacts with the mackinawite and greigite to form pyrite. Pyrite formation by BSR is a low temperature process as most sulfate reducing bacteria live within a temperature range of 0° to ~48 °C with a few, the thermophiles, able to live in a range of ~30° to ~100 °C (Ohmoto and Goldhaber 1997). In detail, pyrite formation by BSR is a complex process and further information can be found in Berner (1970, 1984), Goldhaber and Kaplan (1974) and Rickard and Luther (2007). In modern environments pyrite sometimes forms by BSR in stratified water bodies where stagnant anoxic layers underlie oxic surface waters. In the anoxic layer, provided there are sufficient concentrations of SO42−, suitable oxidants, reactive organic matter and iron, pyrite can form by the same BSR reaction pathway to that just described. The main locus of such pyrite formation is close to the oxic-anoxic interface, and after settling into the underlying muds pyrite grain growth may continue by BSR in the early diagenetic environment (Wilkin and Barnes 1997).
Pyrite can also form diagenetically in anoxic sediments deeper (meters to ~10 m) beneath the sediment–water interface by a different biogenic process (Borowski et al. 2013). This second low-temperature process operates in the sulfate-methane transition zone (SMT) where methane and sulfate-reducing bacteria form a diagenetic environment characterized by the anoxic oxidation of methane. Here methane rather than solid organic matter is involved in sulfate reduction by way of reactions of the type:
Following Rieger et al. (2020) this sulfate driven-anaerobic oxidation of methane process is referred to in this Chapter as SD-AOM.
A third pathway for pyrite formation involves the inorganic reduction of sulfate, a reaction requiring temperatures higher than those for BSR and SD-AOM, and either organic carbon or another reductant. Such reduction is referred to as thermal sulfate reduction (TSR), a process that appears to become important at temperatures of 100–140 °C and greater (Huston et al. 2023).
The formation of pyrite, as well as galena and sphalerite, in CD Pb–Zn deposits has been variously attributed to BSR, SD-AOM and TSR, and sulfur isotope studies are one of the main ways distinguishing between these pathways. Details about the S-isotopic fractionations associated with BSR and TSR are discussed by Huston et al. (2023). Typically, Ssulfide − Ssulfate fractionation by BSR ranges from ~15 to 60‰ while the kinetic isotope fractionation for TSR has a smaller range of ~10–20‰ (Kiyosu and Krouse 1990). As the δ34Sseawater sulfate value through geological time is ≥ ~10‰ (Bottrell and Newton 2006), the δ34S values of sulfides formed by the BSR pathway are typically low and negative, whereas those formed by the TSR pathway are high and positive. This difference is used often to distinguish between the two reduction pathways (Machel et al. 1995).
However, the SD-AOM study of Borowski et al. (2013) has complicated interpretations of δ34Ssulfide ranges and values. Whereas in modern sediments pyrite formed by BSR is characterized by low negative δ34S values, pyrite formed by SD-AOM is characterized by high δ34S positive values, making it difficult to distinguish between TSR and SD-AOM using sulfur isotopic data alone. The interpretation of high positive δ34S values is further complicated by two other processes that have been shown to produce high positive δ34S values. One is when sulfides form by BSR in euxinic sulfate-limited conditions (Gomes and Hurtgen 2015) and the other is when sulfides form by BSR in a restricted or closed system in which Rayleigh fractionation becomes important (Ohmoto and Goldhaber 1997; Huston et al. 2023). Choosing between these possible causes of high positive δ34S values requires other evidence besides sulfur isotope data and is a topic examined in many of the studies reviewed in this chapter.
Huston et al. (2023) used the exchange of 32S and 34S between galena and sphalerite (their Eq. 2) to illustrate their discussion of stable isotope fractionation. The magnitude of Δ34Ssphalerite-galena is temperature dependent and can be used as a CD Pb–Zn ore-forming geothermometer provided the two sulfides coprecipitated in isotopic equilibrium or formed at different times under the same physicochemical conditions and assuming, in both instances, that the samples analysed are pure and are not contaminated with other sulfides. Throughout this chapter, for the sake of consistency, all temperatures derived using on the Δ34Ssphalerite-galena geothermometer are based on the experimental calibration equation of Ding et al. (2003) which is almost identical to the preferred equation of Ohmoto and Rye (1979). The cited temperatures also assume the above-mentioned conditions have been met for accurate temperature determinations.
2.1.5 Isotopic Analytical Methods and Reporting Nomenclature
Stable isotopic analytical techniques have evolved through time (Huston et al. 2023). The older techniques required ~10 mg of sample for analysis and bulked mineral separates were typically obtained either by physical (e.g. hand picking) or selective chemical dissolution methods. By contrast, starting in the late 1980s, bulk techniques began giving way to in situ microanalytical techniques. To maintain consistency with Huston et al. (2023), isotopic values determined by the bulk analytical techniques are identified in this Chapter by the subscript “bulk” (e.g. δ34Sbulk pyrite), whereas values determined by in situ microanalytical techniques are identified by the subscript “in situ” (e.g. δ34Sin situ pyrite).
Both analytical techniques pose analytical challenges (Huston et al. 2023). Nevertheless, data acquired using both techniques have helped advance our understanding of CD Pb–Zn mineral systems. Because of the small grain sizes of minerals in many CD Pb–Zn deposits (diameters < 1 mm) bulk analytical techniques produce values that can be averages of many tens of millions of individual grains (Eldridge et al. 1993). As shown by Eldridge et al. (1993), the averaging aspect of bulk analyses has been found to have obscured genetically important isotopic complexities and Ohmoto and Goldhaber (1997) warned that interpretations developed using bulk data will not always be accurate. Therefore, where possible, this Chapter emphasizes conclusions reached using in situ data, with the implications of bulk data only discussed where they are important to the interpretation of in situ data or have had a major impact on our understanding of particular CD Pb–Zn mineral systems.
3 The Northern Australian Proterozoic Clastic-Dominated Pb–Zn Province
The sedimentary host-rock sequences of this province are Palaeo- to Mesoproterozoic in age (Southgate et al. 2000) and occur in the McArthur Basin in the north, and in the Mt Isa Inlier in the south (Fig. 2). The province hosts five of the world’s ten largest CD Pb–Zn deposits based on Pb + Zn content (Leach et al. 2005). These, in order of decreasing Pb–Zn content, are HYC (3rd), Hilton (6th), Mt Isa (7th), George Fisher (9th) and Century (10th). All five deposits have supported significant mining operations. All but one of the five deposits occur in strata deposited between ~1665 and ~1635 Ma. The exception is Century which occurs in strata deposited ~1595 Ma (Page and Sweet 1998). The province also contains a number of smaller CD Pb–Zn type deposits, including Dugald River, Lady Loretta, Walford Creek, and Teena (Rohrlach et al. 1998; Williams 1998; Large et al. 2005; Hayward et al. 2021).
3.1 Clastic-Dominated Pb–Zn Deposits in the McArthur Basin
The largest CD Pb–Zn deposit in the McArthur Basin is the HYC deposit, sometimes referred to as the McArthur River deposit (Fig. 3a). The HYC deposit has been described by Croxford and Jephcott (1972), Murray (1975), Lambert (1976), Eldridge et al. (1993), Large et al. (1998), Ireland et al. (2004a,b) and Spinks et al. (2021). The HYC deposit is located in the Batten Fault Zone portion of the McArthur Basin, as are several smaller CD Pb–Zn deposits and occurrences (Fig. 3a), including Emu Plains (Ahmad et al. 2013; Walker et al. 1977), Mitchell Yard and Myrtle (Ahmad et al. 2013), the stratiform part of Ridge II (Williams 1978a), Teena (Hayward et al. 2021) and W-Fold (Murray 1975; Walker et al. 1977).

a Location of the HYC deposit and associated deposits and prospects, McArthur Basin, Northern Territory, Australia. b McArthur Group stratigraphy in the HYC area. Stratigraphic units discussed in this Chapter are highlighted in color
Almost all the CD Pb–Zn mineralization in the McArthur Basin occurs in carbonaceous and dolomitic mudstones of the 1640 ± 3 Ma HYC Pyritic Shale Member of the Barney Creek Formation in the McArthur Group (Fig. 3b). Away from CD Pb–Zn mineralization pyrite is common in the Barney Creek Formation, and sometimes in the overlying Reward Dolostone (Fig. 3b). The most pyritic and organic-rich portions of the two formations occur in McArthur Basin sub-basin depocenters.
A number of small discordant vein- and breccia-hosted Pb–Zn and Cu deposits also occur in the Batten Fault Zone in dolostone formations older than the Barney Creek Formation. Two of these, Cooley and Ridge (Fig. 3a), lie immediately east of, and appear to be a feeder-zone component of the HYC mineral system (Williams 1978a; Rye and Williams 1981).
3.2 Stable Isotope Studies in the McArthur Basin
3.2.1 Source
As noted earlier, Cooke et al. (1998) used fluid inclusion and bulk C–O (carbonate) and oxygen (silicate) isotope measurements of altered volcanic rocks lying stratigraphically deep beneath the Barney Creek Formation to determine if they were the source of the metals in the McArthur CD Pb–Zn mineralization. The volcanic rocks studied by Cooke et al. (1998) are exposed in the Mallapunyah Dome region, ~80 km SSW of the HYC deposit. Bulk whole rock δ18OSMOW data from altered mafic volcanic rocks in the Dome region range from 15.7 to 19.7‰, a range used with other evidence to conclude that alteration occurred at ≈ 100 °C and involved saline (> 20 wt% NaCl equivalent) fluids with a δ18OSMOW value of ≈ −1‰ and a δ13CPDB value of ≈ −7‰. In contrast bulk whole rock δ18OSMOW data for similarly-altered mafic rocks between ~80 and 200 km east of the Mallapunyah Dome range from 6.0 to 10.4‰ (Champion et al. 2020), a difference attributed either to alteration at ≈ 250 °C by similar fluids (with a δ18OSMOW value of 1.7‰) to those involved in the Mallapunyah Dome alteration or to alteration by very different fluids. Although neither study proves conclusively that the metals in the McArthur Basin CD Pb–Zn mineralization came from the altered volcanic rocks, both postulate the stable isotopic composition of the hydrothermal fluids that might have formed the CD Pb–Zn mineralization.
The value of δ34Sseawater sulfate at the time of deposition of the strata hosting McArthur CD Pb–Zn deposits was taken by Magnall et al. (2020b) to be, at a minimum, 25‰ based on the global comparisons of Li et al. (2015). This is in good agreement with the median δ34S evaporative gypsum value of 26.8‰ for the Myrtle Shale Member (Fig. 3b) of the McArthur Group (Crockford et al. 2019). However, the secular δ34Sseawater sulfate curve of Chu et al. (2007), based on CAS data through the Jixian section, northern China, gives a δ34Sseawater sulfate value of 20‰ for sedimentary rocks of the same age as the Barney Creek Formation. The sulfide-forming processes discussed in the McArthur Trap section below considers the genetic implications of both a 20‰ and a 26.8‰ value for McArthur δ34Sseawater sulfate.
3.2.2 Transport
Rye and Williams (1981) studied the bulk isotopic composition of ore-stage vein-fill dolomite in the Cooley and Ridge mineralization and the results are shown in Fig. 4. The δ13C and δ18O values correlate strongly, and dolomite from the Emu Fault zone (Smith and Croxford 1975) falls at the isotopically light end of the trend. Moreover, δ13C and δ18O values increase westward from the Emu Fault. Two explanations were proposed for the correlation using temperature-dependent fractionation modelling. The first is that the correlation reflects vein dolomite deposition from a hydrothermal fluid with δ13CH2CO3 ~−2.75‰ and δ18OSMOW ~12.5‰ that cooled from ~325 to 100 °C as it flowed westwards from the Emu Fault. The isotopic composition of this fluid suggests it was a highly evolved basinal brine that had exchanged with a large marine carbonate reservoir (Taylor 1979). If the explanation is correct then the Emu Fault dolomite formed at ~325 °C, the Cooley dolomites formed between ~275 and ~185 °C, and the Ridge I dolomite formed between ~190 and ~175 °C. The second explanation is that the correlation again reflects a cooling trend, this time of a hydrothermal fluid that had a constant δ18OSMOW but a δ13C that became progressively lower due to local oxidation of organic matter during hydrothermal alteration of the host rocks. This second explanation yields a cooling from ~220 to ~125 °C of a fluid that had initial δ13CH2CO3 and δ18OSMOW values respectively of −4.5‰ and 8.1‰. By the time the fluid cooled to 125 °C the δ13CH2CO3 value of the fluid would have decreased to −5.25‰. Rye and Williams (1981) noted that there were no unique fits of model curves to the dolomite isotope trend, and simply concluded that the data are consistent with the formation of ore-stage dolomite from a single hydrothermal fluid that cooled, and possibly changed in isotopic composition, as it moved westwards from the Emu Fault zone and deposited the Cooley and Ridge mineralization.

δ18O versus δ13C values of dolomite in the discordant and concordant Ridge –Cooley mineralization and HYC deposit (after Rye and Williams 1981; Smith and Croxford 1975). The data field for the unmineralized McArthur Basin rocks represents, respectively, the 1st and 3rd quartiles of the δ18O and δ13C values determined by Kunzmann et al. (2019) for 485 drill-core samples remote from mineralization in the McArthur Basin
Importantly, Rye and Williams (1981) found that the C- and O-isotopic composition of dolomites in well-mineralized HYC samples (Smith and Croxford 1975) also correlate strongly and that the lines of best fit for the HYC and Cooley-Ridge data sets are almost indistinguishable (Fig. 4). The similarity is strong evidence that the Cooley and Ridge prospects and HYC deposit are genetically related and are parts of the one mineral system in which the discordant Cooley-Ridge mineralization is feeder-zone style mineralization. Dolomite vein-fill in the discordant Cooley-Ridge mineralization precipitated at warmer temperatures (~170–310 °C, 1st model or ~170–210 °C, 2nd model) from the same hydrothermal fluids that at lower temperatures (~170–240 °C, 1st model or ~125–175 °C, 2nd model) completely exchanged and isotopically equilibrated with preexisting sedimentary dolomite in the HYC deposit and/or deposited hydrothermal dolomite in the HYC mineralization.
Large et al. (2001) studied the isotopic composition of carbonates in an interpreted hydrothermal alteration halo around the HYC deposit. The halo is characterized by anomalous whole-rock Zn, Pb and Tl values and, locally, Mn-rich ferroan dolomite-ankerite carbonates (Large et al. 2000). Carbonate C- and O-isotopic values in the halo define a field that is bordered by the HYC and Cooley-Ridge lines (Fig. 5) and extends away from the lines, and from the unaltered McArthur Basin dolomite field of Kunzmann et al. (2019), towards higher δ18Odolomite values and lower δ13Cdolomite values. Using a temperature-dependent fluid-rock interaction modelling approach Large et al. (2001) found that the hydrothermal alteration was caused either by a HCO3−dominant neutral to alkaline fluid with a δ18OSMOW value of 5 ± 5‰ and a δ13C value of −6‰ ± 1‰ over a temperature range of 50–120 °C, or a H2CO3-dominant neutral to acidic fluid with a δ18O value of ~15‰ and a δ13C value of −4 to −6‰ over a temperature of greater than 200 °C. Based on mineralogical features, interpreted to indicate alteration was a low-temperature event, Large et al (2001) concluded that a HCO3−dominant fluid caused the alteration. The conclusion is puzzling because the modelling of Cooke et al. (1998) showed that the isotopic composition of carbonates define a δ13C − δ18O curve with a low positive slope like that shown in Fig. 4 when in equilibrium over a range of temperatures (25–200 °C) with H2CO3-dominant fluids, but not with HCO3−-dominant fluids which define a U-shaped curve over the same temperature range (Cooke et al. 1998: Fig. 19). Therefore the weight of evidence favors the formation of the HYC mineralization and associated hydrothermal alternation by a hot (initial temperature > 200 °C) neutral to acidic H2CO3-dominant hydrothermal fluid.

δ18O versus δ13C values of carbonates in the alteration halo associated with the HYC deposit, together with carbonates from the HYC and Cooley-Ridge mineralization and unmineralized dolomitic rocks in the McArthur Basin and Lady Loretta areas (after Kunzmann et al. 2019; Large et al. 2001; McGoldrick et al. 1998). The data field for the unmineralized McArthur Basin rocks is from Fig. 4
A comparison of the various C- and O-isotopic compositions postulated for the HYC ore-forming hydrothermal fluids with those postulated for the metal-leaching fluids by Cooke et al. (1998) is shown in Fig. 6a. The metal-leaching fluid is close in composition to the low δ13Cfluid and δ18Ofluid extremes of the wide isotopic compositional field calculated by Large et al. (2001) for their lower-temperature HCO3−dominant HYC fluid, but is distinctly lower isotopically to the isotopic compositions calculated by Large et al. (2001) and Rye and Williams (1981) for the three higher-temperature H2CO3dominant HYC hydrothermal fluids. As the HYC fluids were most likely H2CO3-dominant, it is clear from Fig. 6a that the metal-leaching fluids discussed earlier were not the immediate ore-forming fluids. However, if the metal-leaching fluids had continued to interact with McArthur Basin strata after leaching metal from volcanic material, they may have evolved to become the ore-forming fluids. As illustrated in Fig. 6b this evolution would require increasing temperatures and an increase in δ18Ofluid values of between ~9 and ~16‰ and in δ13Cfluid values of between ~2 and ~4‰. Further work is clearly needed to better understand the origin of the metals and hydrothermal fluids in the HYC mineral system.

δ18O versus δ13C values of the fluids postulated to have leached base metals volcanic rocks in the lower McArthur Basin (after Cooke et al. 1998) and to be possible hydrothermal fluids involved in the formation of the HYC and Lady Loretta clastic-dominated Pb–Zn deposits (after Large et al. 2001; McGoldrick et al. 1998; Rye and Williams 1981)
3.2.3 Trap
As well as C- and O-isotopic studies, S-isotopic studies have also added to an understanding of CD Pb–Zn mineral systems in the McArthur Basin. Pyrite has been the studied isotopically in detail in the HYC and Teena deposits (Fig. 3a). In the HYC deposit there are two generations of pyrite (Williams 1978a; Eldridge et al. 1993; Large et al. 1998; Ireland et al. 2004b). The earlier generation, Py1, comprises small (typically 5–10 µm diameter) euhedral to subhedral crystals that occur either in bedding parallel laminae or in spherical to ellipsoidal aggregates with diameters of up to ~100 µm. The second generation, Py2, occurs either as overgrowths on individual Py1 grains or as interstitial cement in Py1 aggregates. Py2 is optically duller and slightly browner and softer than Py1. Chemically, the main chemical distinction between the pyrite generations appears to be an enrichment of Tl in Py2 relative to Py1 (Spinks et al. 2021).
In situ sulfur isotope analyses show that Py1 and Py2 are isotopically different (Eldridge et al. 1993). Py2 has a broader spread of δ34Sin situ pyrite values than Py1 and a median δ34Sin situ pyrite value ~16‰ higher (Fig. 7). If the value of δ34Sseawater sulfate during the deposition of the Barney Creek Formation was 26.8‰, based on the Myrtle Shale Member median δ34Sevaporative gypsum value discussed above, then Δ34Sseawater sulfate-pyrite 1 is ≤ 40‰ and that for Py2 is ≤ 30% (Fig. 7). Both fractionations are in the range of pyrite formation by BSR. Eldridge et al. (1993) concluded that Py1 formed by BSR during early diagenesis when the system was open to sulfate, whereas Py2 was formed during later diagenesis when the system was closed to sulfate, with δ34Ssulfate increasing due to closed-system Rayleigh fractionation.

Box and whisker diagrams of sulfur isotopic data for the HYC, Ridge and Cooley mineralization (after Smith and Croxford 1973; Rye and Williams 1981; Ireland et al. 2004b). Principal features of these and subsequent box and whisker plots are illustrated in the insert. The box shows the 25th (Q1) to 75th (Q3) percentile range of isotopic values in each data set. This range is the interquartile range (IQR). The vertical line within the box marks the median value of the data set and the cross marks the average value. The whiskers extend respectively each side of the box to the smallest data point ≥ Q1 − 1.5*IQR (the minimum) and to the largest data point ≤ Q3 + 1.5*IQR (the maximum). Outliers are those data point beyond the minimum and maximum limits and are shown at dots The dashed blue and orange vertical lines show the value of δ34Ssulfide formed by thermochemical sulfate reduction at various temperatures. These lines have been determined using the fractionation equation of Kiyosu and Krouse (1990) assuming a seawater sulfate with a value of 26.8‰, and b seawater sulfate with a value of 20‰
Magnall et al. (2020b) studied pyrite in and around the Teena deposit from three drill holes aligned in a roughly east–west direction over a distance of ~1.75 km across the Teena sub-basin (Hayward et al. 2021). Two holes intersect the mineralized Lower Pyritic Shale Member of the HYC Pyritic Shale and the third intersects the poorly-mineralized (< 6 wt% Zn) Lower Pyritic Shale Member ~115 m west of Teena. The study also included pyrite from the Middle HYC Pyritic Shale Member above the deposit. Two pyrite generations were identified at Teena, Py-1 and Py-2, each with two sub-types (a and b). Py-1a comprises < 5 µm grains and framboids, whereas Py-1b comprises > 5 µm idiomorphic euhedral pyrite that typically occurs on the margins of nodular carbonate. Py-2a comprises spherical and concentrically zoned crystals containing abundant host rock inclusions, and Py-2b comprises irregular, anhedral overgrowths on earlier Py-1 and contains interstitial sphalerite and galena. A minor, later pyrite type, Py-3, comprises coarse-grained euhedral crystals in late-stage sulfide-carbonate-quartz veins. The Teena Py-1 and Py-2 generations appear to be the same as the two HYC pyrite generations.
Py-1a is present throughout the stratigraphic interval studied at Teena and is abundant within correlative carbonaceous intervals in the three drill holes. Py-1a is interpreted to also predate the growth of diagenetic dolomite nodules. The restriction of Py-1b to the margins of the nodules is interpreted to indicate that Py-1b formed late during nodule growth. Py-2 is limited to regions of Pb–Zn mineralization. It is enriched in Pb and As and is interpreted to be hydrothermal in origin.
The Teena δ34Sin situ pyrite data of Magnall et al. (2020b) are summarized in Fig. 8. The data reveal that there are two classes of Py-1a at Teena, and these are shown separately in Fig. 8. The class with the lower median value has a large offset from coeval seawater sulfate (assumed here to be 26.8‰) of ~30‰, consistent with formation by BSR under relatively open system conditions. Py-1a is widespread both within, adjacent to, and stratigraphically above the Teena mineralization. In contrast the Py1a class with the higher median value of 33.3‰ is comparable in value to the assumed value of coeval seawater sulfate. This Py1a class is restricted to an interval of high pyrite abundance in the Middle HYC Pyritic Shale Member and it occurs in all three drill holes. Magnall et al. (2020b) concluded that the formation of this high-median-value Py-1a was not a localized process, but rather was a sub-basin wide process. Highly positive δ34Spyrite values are typical of sulfate reduction in a sulfate-limited environment that is widely interpreted to reflect water-mass restriction (e.g. Lyons et al. 2000). However, because the high positive Py-1a occurs in a part of the HYC Pyritic Shale that was laid down at a time of high relative sea level during a marine transgression (Kunzmann et al. 2019), Magnall et al. (2020b), concluded that sulfate limitation was caused by a high organic carbon flux that promoted BSR in an euxinic water column.

Box and whisker diagram of sulfur isotopic data for the Teena mineralization (after Magnall et al. 2020b)
Py-1b generally has higher δ34S values than those of the low-median value Py-1a class and is interpreted by Magnall et al. (2020b) as indicating that Py-1b formed in pore fluids during later diagenesis, either by BSR or SD-AOM. However, the range of Py-1b spans the entire range of all the pyrite in the Lower HYC Pyritic Shale Member, suggesting the possibility that some of the pyrite classified as Py-1b on textural criteria may be Py-2, a possibility that requires further investigation. The δ34Sin situ Py2 values are intermediate between the py-1a end members, but showed no systematic trends within individual zoned aggregate samples. Because Py-2 only occurs within Pb–Zn mineralized rocks, and Py-2b sometimes has interstitial galena and sphalerite, Magnall et al. (2020b) concluded that Py-2 is part of the Teena hydrothermal event that postdates the early diagenetic Py-1 event. Unfortunately, Magnall et al. (2020b) did not measure the S-isotopic compositions of galena and sphalerite at Teena to determine the isotopic relationship of Py-2 to the two ore sulfides and further test their paragenetic sequence conclusion that Py-2, galena and sphalerite are cogenetic and formed during later diagenesis at Teena.
The S-isotope geochemistry of galena and sphalerite, however, has been studied in the HYC, Cooley and Ridge deposits. In a bulk isotope study Smith and Croxford (1973) reported δ34Sbulk values for coexisting pyrite, galena and sphalerite in a vertical section through the HYC mineralization (Fig. 9). The Δ34Sbulk sphalerite-bulk galena values are relatively uniform, suggesting the two ore minerals were in, or have approached, S-isotopic equilibrium. The fractionations give equilibration temperatures ranging from ~90 to ~240 °C. The δ34Sbulk pyrite values of Smith and Croxford (1973), as well as being shown in Fig. 9, are also included in Fig. 7. They reflect mixtures of Py1 and Py2 and it is therefore not surprising that the median δ34Sbulk pyrite value falls between those of Py1 and Py2. The δ34Sbulk pyrite values increase progressively upwards from −3.9‰ at the base of the deposit to 14.3‰ at the top, whereas galena and sphalerite values do not (Fig. 9). The pattern is strong evidence that galena and sphalerite are not in isotopic equilibrium with either Py1 or Py2. The pattern further suggests that the ratio Py1/Py2 is highest at the base of the HYC deposit and decreases systematically up through the deposit. Smith and Croxford’s (1973) preferred genetic interpretation is that the HYC mineralization formed syngenetically by a dual S-source process in which Zn and Pb, either as sulfides or sulfide complexes, were introduced intermittently into a restricted local basin by exhaled basinal brines whereas Fe was contributed continuously as iron oxides or hydroxides that were converted to pyrite by BSR in a restricted environment in which the δ34Sseawater sulfate increased upward due to closed system Rayleigh fractionation.

Stratigraphic variations in bulk δ34S values of pyrite, sphalerite and galena though the HYC deposit intersected in the V121 Shaft (after Smith and Croxford 1973)
In situ HYC δ34Sgalena and δ34Ssphalerite data, reported by Eldridge et al. (1993), are included in Fig. 7. Unlike the bulk and in situ pyrite values, the bulk sphalerite and galena values are comparable to their in situ values. Eldridge et al. (1993) concluded that sphalerite and galena could not have formed from the same H2S pool as the pyrite if all three sulfide minerals had had a common hydrothermal origin, and also that sphalerite and galena could not have formed from the same BSR generated H2S pool as pyrite even if all three sulfides had a biogenic origin. To explain these conclusions Eldridge et al (1993) proposed a syndiagenetic model for the HYC mineralization in which the two pyrite generations formed at different stages of diagenesis by BSR whereas sphalerite and galena, as well as some gangue minerals, precipitated later from hydrothermal fluids that travelled parallel to bedding in the host sedimentary sequence before it was totally lithified. In this genetic model the two pyrite generations formed diagenetically by BSR from relatively cool fluids dominated by Fe, SO42− and CO2, whereas the base-metal sulfides formed later from a warmer Pb–Zn bearing hydrothermal fluid. The in situ data could not be used to differentiate between galena and sphalerite formation involving a sulfate-dominant oxidized hydrothermal fluid and inorganic sulfate reduction (i.e. TSR) as proposed by Williams (1978b), and precipitation from a H2S-dominant reduced hydrothermal fluid. The genetic model is similar to the Teena model of Magnall et al. (2020b), the main difference being that Eldridge et al. (1993) regarded Py2 to be an extension of the BSR diagenetic event that formed Py1 rather than a part of the hydrothermal ore-forming event.
The possibility that galena and sphalerite formed by TSR is problematic if the sulfate being reduced is coeval seawater sulfate with the δ34S value of ~26.8‰ discussed above. Figure 7a shows the δ34S values of sulfides that would form by TSR from this sulfate at various temperatures, based on the kinetic fractionation equation of Kiyosu and Krouse (1990). The majority of the galena and sphalerite δ34S values plot below 100 °C and below the temperature range for TSR, and below the temperature range suggested by the HYC Δ34Sbulk sphalerite-bulk galena values. If the two ore minerals did form in the trap environment by TSR, then the sulfate involved was not coeval seawater sulfate with a δ34S value of ~26.8‰, but rather was formed from sulfate with a lower δ34S value. One possibility is that the sulfate had a δ34S value of ~20‰, consistent with the secular δ34Sseawater sulfate curve of Chu et al. (2007). This possibility is illustrated in Fig. 7b. The resulting temperatures of galena and sphalerite formation are closer to those indicated by the galena-sphalerite sulfur isotope fractionation geothermometer. If the coeval seawater sulfate indeed had a δ34S value of ~26.8‰ and was the source of reduced sulfur in the pyrite, then it is possible that galena and sphalerite formed by TSR from an oxidized hydrothermal fluid that transported sulfate with a δ34S value of ~20‰, or from a reduced hydrothermal fluid in which the reduced sulfur had a value between ~0 and ~10‰. The possibility that the sulfide sulfur in the two ore minerals was not coeval seawater sulfate, and the oxidation state of the HYC hydrothermal fluid, are subjects that need further investigation.
The few bulk δ34S values of coexisting sphalerite and galena in the Cooley and Ridge prospects (Rye and Williams 1981) are similar to those in the HYC, but Δ34Sbulk sphalerite-bulk galena values are smaller and give a temperature range 130–300 °C. The temperatures fall westwards away from the Emu Fault, consistent with the temperatures indicated by the isotopic composition of the associated ore-stage dolomite.
Ireland et al. (2004b) measured the in situ sulfur isotopic composition of two grain-size variants of sphalerite in the HYC deposit, named Sp1 and Sp2 (Fig. 7). Sp1 is the dominant type. It is very fine grained (1–10 µm) and occurs as irregular elongate aggregates (up to 200 µm long) that combine to form sphalerite laminae up to ~1 mm thick (Ireland et al. 2004b). The less abundant second type, Sp2, occurs as partial to almost complete replacement of the nodular dolomite found predominately towards the fringes of the HYC deposit. Ireland et al. (2004b) only analysed Sp1 and Sp2 in the lower HYC ore lenses but sampling was widespread and included material from both the center and fringes of the deposit. Sp1 δ34Sin situ values are similar to the in situ sphalerite values reported by Eldridge et al. (1993), indicating that most of the sphalerite analysed in the previous study was Sp1. The median values of δ34Sin situ Sp1, δ34Sin situ Sp 2 and δ34S bulk sphalerite (Fig. 7) suggest that Sp1 represents ~60% of all the HYC sphalerite, rather than > 80% as estimated by Ireland et al. (2004b). The difference raises the possibility of there being a strong positively-skewed continuum of δ34Ssphalerite values across the whole of the HYC and that all the sphalerite formed during a single event, a possibility also requiring further investigation.
Ireland et al. (2004b) concluded that Sp1 formed when pulsed distal exhalations of warm (100–150 °C) hypersaline sulfate-predominant metalliferous brines ponded in a local depression, producing a stratified brine pool with each stratum having different chemistries and temperatures. It was argued the brine pool was overlain by anoxic seawater and, above that layer, oxic seawater. To explain the lack of isotopic equilibrium between pyrite and sphalerite, Ireland et al. (2004b) postulated that Sp1 formed in, and settled from, a layer near the base of the brine pool by reactions involving H2S produced by BSR and minor amounts of H2S generated by thermal sulfate reduction (TSR). In contrast, Py1 formed at the same time as Sp1 in a higher layer of the brine pool utilizing H2S formed by BSR and more oxidized sulfur species (SO42−, S0 and S2O3−1) produced by the cyclical sulfur oxidation and disproportionation in the oxic zone overlying the brine pool. By contrast Sp2 and Py2 are postulated to have formed diagenetically beneath the brine pool in a metalliferous brine environment that was partially closed and dominated by BSR-generated H2S that produced heavier δ34S values for Sp2 and Py2 than those for Sp1 and Py1 due to closed-system Rayleigh fractionation. No attempt is made to explain the disequilibrium isotopic relationship between Sp2 and Py2. The genetic model of Ireland et al (2004b) is complex and additional detail to that covered in the above summary can be found in the original reference.
Two more recent studies of the McArthur CD Pb–Zn mineralization, neither of which involve new stable isotope data, further supports the diagenetic/epigenetic ore-forming conclusions of Eldridge et al. (1993) and Magnall et al. (2020b) rather than the syngenetic-exhalative conclusions of Ireland et al. (2004b). Spinks et al. (2021) used Maia Mapper, a µXRF mapping system, and SEM and electron backscatter diffraction observations to show that galena and sphalerite in the HYC deposit formed by the hydrothermal replacement of dolomite. Similarly, Magnall et al. (2021), building on their earlier S-isotopic study at Teena, used other mineralogical and geochemical evidence to conclude that the Teena galena and sphalerite also formed by carbonate replacement.
3.3 Clastic-Dominated Pb–Zn Mineralization in the Southern Mt Isa Inlier
The most economically important CD Pb–Zn deposits in the Mt Isa Inlier are the Mt Isa, Hilton and George Fisher deposits (Fig. 2). All occur in the ca 1655 ± 7 Ma dolomitic Urquhart Shale (Page and Sweet 1998). The Urquhart Shale also hosts the large Mt Isa Cu orebodies that postdate the Pb–Zn orebodies (Kawasaki and Symons, 2011; Large et al. 2005; Perkins 1984). Unlike the McArthur Basin CD Pb–Zn mineralization, that in the Mt Isa district is complexly altered by faulting, folding, metamorphism and by hydrothermal alteration related to Cu mineralization. Much of the Mt Isa district CD Pb–Zn mineralization is banded like the McArthur ores but the banding and grain sizes are coarser. The main sulfide minerals are sphalerite, galena, pyrite and pyrrhotite. Deformed mineralization displays textures indicative of extensive sulfide remobilization, but some breccia and vein-hosted mineralization appears to have been introduced after the CD Pb–Zn mineralization (Chapman 2004).
3.3.1 Source
The value of δ34Sseawater sulfate at the time of deposition of the strata hosting CD Pb–Zn deposits of the Mt Isa district is uncertain. The secular δ34Sseawater sulfate curve of Chu et al. (2007) suggests the value was ~28‰, whereas in their study of the George Fisher deposit Rieger et al. (2020) assume a value between 18 and 25‰.
3.3.2 Transport
Carbon and oxygen isotope studies in the Mt Isa CD Pb–Zn district include Smith et al. (1978), Heinrich et al. (1989) and Waring et al. (1998). However, these studies are of little use in elucidating the nature of the fluids that formed the Mt Isa CD Pb–Zn deposits because, as was shown by Waring et al. (1998), the isotopic geochemistry of the Mt Isa carbonates reflects hydrothermal alteration associated with the later Mt Isa Cu mineralization, an event that overprinted C- and O-isotopic evidence of the earlier Pb–Zn mineralizing event.
3.3.3 Trap
Pyrite in the Urquhart Shale along a ~17 km interval of the formation stretching from ~4 km south of the Mt Isa deposit to a point ~14 km north of the deposit and ~5 km south of the George Fisher deposit has been studied by Painter et al (1999). The study included 104 δ34Sbulk measurements of fine-grained (< 30 µm) pyrite, the earliest recognized pyrite generation in the Mt Isa region). Although the most pyritic portion of the studied interval is in the Mt Isa deposit itself, Painter et al. (1999) found no obvious difference in δ34Sbulk pyrite values between mineralized and unmineralized Urquhart Shale.
The δ34Sbulk pyrite values have a positively skewed distribution (Fig. 10), interpreted by Painter et al. (1999) as indicating the pyrite formed in a restricted environment during late diagenesis from sulfate-bearing fluids that flowed parallel to bedding. Petrographic and spatial arguments were used to conclude that the pyrite formed by TSR rather than BSR. Painter et al (1999) used geological and textural arguments and a statistical analysis of all previous bulk sulfur isotope results from the Mt Isa deposit (Fig. 10) to conclude that fine-grained pyrite predated the Pb–Zn mineralization and that the mineralizing hydrothermal fluids were dominated by reduced sulfur rather than by sulfate. This two stage syndiagenetic model is similar to that proposed for the HYC mineralization by Eldridge et al. (1993).

Further insights into the formation of pyrite in the Urquhart Shale and its CD Pb–Zn mineralization are provided by Rieger et al. (2020) in an in situ sulfur isotope study of pyrite in and near the George Fisher CD Pb–Zn deposit. The study includes data from unmineralized Urquhart Shale at Shovel Flats at the northern end of the study area of Painter et al. (1999). Four significant stages of sulfide deposition are recognized by Rieger et al. (2020). These are an early pre-ore pyrite stage, followed by three stages of hydrothermal sulfide deposition comprising pyrite, sphalerite, galena, pyrrhotite and chalcopyrite. The first hydrothermal stage is the dominant one and is the CD Pb–Zn stage. Five pyrite types are identified by Rieger et al. (2020). The earliest is Py-0, the fine-grained pyrite also studied by Painter et al. (1999). Rieger et al. (2020) divided Py-0 into two sub-types: Py-0a that occurs as the bright cores of zoned Py-0 crystals and Py-0b that occurs as duller overgrowths around Py-0a. The first hydrothermal pyrite phase, Py-1, is stratabound and coarser (≤ several 100 µm) than Py-0. Py-1 is sometimes replaced by sphalerite or galena to give the pyrite atoll texture first described in the Mt Isa deposit by Grondijs and Schouten (1937). The second hydrothermal pyrite phase, Py-2, occurs in veins and breccias that cut and replace the host sediments and previous sulfide phases. The third hydrothermal pyrite stage, Py3, comprises stratabound, massive units or veins and breccias that cut all previous stages and host-rock bedding. Py-3 is often accompanied by pyrrhotite. Another pyrite type, euhedral pyrite (Py-euh), occurs as overgrowths on Py-0 and is the coarsest (up to several mm in grain size) pyrite type. It occurs at Shovel Flats as well as in the George Fisher deposit. Although Py-euh postdates Py-0, its relationship, if any, to either one or more of the hydrothermal pyrite phases is unclear. Further and very detailed descriptions of the textural relationship between the various sulfides in the George Fisher mineralization and enclosing sediments can be found in Rieger et al. (2020).
The in situ sulfur isotopic composition of the George Fisher pyrite types are shown in Fig. 11. Py-0 has the lowest isotopic values. In the George Fisher mineralization the median δ34S value of Py-0a is −1.1‰ and that of Py-0b is 0.7‰, whereas at Shovel Flat the median Py-0a value is 4‰. The in situ Shovel Flats values are consistent with the bulk values of samples from the same area reported by Painter et al. (1999). The order of median δ34S values is Py-0 < Py2 < Py-1 < Py-3. Py-euh at both George Fisher and Shovel Flats have similar wide distributions of δ34S of values.

Box and whisker diagram of sulfur isotope data for sulfides in the George Fisher (G.F.) deposit and unmineralized Urquhart Shale in the Shovel Flats (S.F.) area south the George Fisher (after Rieger et al. 2020)
Because δ34Sin situ Py-0 values are offset from coeval seawater sulfate values (assumed to be between 18 and 25‰) by ≤ 25–32.5‰, Rieger et al. (2020) concluded Py-0 formed during early diagenesis by BSR under open-system conditions rather than during late diagenesis by TSR as argued by Painter et al. (1999). As Py-0b δ34S values are slightly higher than those of Py-0a Rieger et al. (2020) also concluded that Py-0b formed under increasingly sulfate-limited conditions as diagenetic pore fluids became more restricted.
Rieger et al. (2020) noted that the variability of Py-1 δ34S values is at the hand-specimen scale, suggesting Py-1 formed a transport-limited environment that was not linked to the closing environment in which Py-0 formed because of the separation of the δ34S values of the two pyrite types (Fig. 11). Given the high positive δ34S values of Py-1 it was concluded it formed at a temperature of > 100 °C by TSR involving the oxidation of organic matter in the host Urquhart Shale. During deformation following the Stage 1 hydrothermal event it is argued that the earlier sulfides were recycled and replaced to form the Stage 2 mineralization, resulting in Py-2 being isotopically intermediate in values between Py-0 and Py-1. Py-3 is postulated to have formed by TSR during a later deformational event associated with the Cu mineralization event. Because Py-euh occurs in both mineralized and unmineralized samples Rieger et al. (2020) concluded that this pyrite type is unlikely to have had a hydrothermal origin. Py-euh could have formed by either TSR under conditions of variable rates of sulfate replenishment relative to reduction, or by sulfate reduction involving SD-AOM. Rieger et al. (2020) did not measure the isotopic composition of the sphalerite and galena in the George Fisher deposit to test their conclusion that the hydrothermal pyrite-forming mechanisms also extends to the formation of galena and sphalerite.
However, bulk S-isotopic values for galena and sphalerite, as well as for pyrite and pyrrhotite, have been reported from the Mt Isa CD Pb–Zn deposit by Solomon (1965) and Smith et al. (1978) and their data are shown in Fig. 10. The distributions of δ34S values for the four minerals are similar in both studies, but those from Smith et al. (1978) are more useful from a genetic viewpoint as they include 14 measurements of coexisting galena, sphalerite, pyrrhotite and pyrite. Their δ34S values, with one exception, decrease in the order pyrite > sphalerite > pyrrhotite > galena, suggesting sulfur isotope equilibrium was reached or approached between the four minerals. However, because of uncertainties regarding the purity of their mineral separates, and discrepancies in equilibration temperatures determined from Δ34S values of the various sulfide pairs, Smith et al (1978) concluded that isotopic equilibrium was never fully established. The median values of pyrite measured by Solomon (1965) and Smith et al. (1978) are respectively 17.0 and 18.1‰ and are clearly different to the fine-grained pyrite studied by Painter et al. (1999). Rather the pyrite is similar to Py-1 at the George Fisher deposit which has a median value of 16.9‰. The similarity is further support for the conclusion of Rieger et al. (2020) that Py-1 formation is a part of the CD Pb–Zn ore-forming event.
The S-isotopic data of Smith et al. (1978) also show that the Py-1 type pyrite is more similar isotopically to coexisting galena and sphalerite than is the case with the pyrite coexisting with galena and sphalerite in the HYC ores. The difference suggests strongly that unlike the Mt Isa ores, the HYC ores do not contain significant quantities of hydrothermal pyrite, suggesting that the hydrothermal fluids that formed the Mt Isa mineralization had a higher abundance of Fe than the HYC hydrothermal fluids.
3.4 Clastic-Dominated Pb–Zn Mineralization in the Central Mt Isa Inlier
A small but interesting CD Pb–Zn deposit, Lady Loretta, occurs in the central Mt Isa Inlier (Fig. 2). The host is the 1653 ± 7 Ma Lady Loretta Formation which lies at the top McNamara Group (Page and Sweet 1998). The mineralization and its geological setting has been described by Carr (1984), Carr and Smith (1977) and Duffett (1998). Unlike the other North Australian CD Pb–Zn deposits, Lady Loretta has thick lenses of layered barite with sphalerite, pyrite and chert at the top of the mineralized sequence. The strata hosting Lady Loretta may have been formed in a lacustrine, rather than marine environment (Large and McGoldrick 1998). The strata are dolomitic, but the carbonate directly associated with the mineralization is siderite (Large and McGoldrick 1998). Lady Loretta is less metamorphosed than the ore deposits of the Mt Isa district, but more metamorphosed than those in the McArthur Basin (Carr and Smith 1977).
3.4.1 Source
A strong indication of the value of δ34Swater column sulfate at the time of deposition of the strata hosting the Lady Loretta CD Pb–Zn deposit has been provided by Gellatly and Lyons (2005) who reported 23 CAS analyses from the Paradise Creek Formation that lies ~500 m beneath the Lady Loretta Formation in the McNamara Group (Southgate et al. 2000). The CAS δ34S values range from 14.1 to 37.3‰ and have a median of 30.6‰ which is taken here to be the value of δ34Swater column sulfate during Lady Loretta times.
3.4.2 Transport
Dolomite outside the Lady Loretta mineralization has bulk δ18OSMOW and δ13C values respectively of 16.5–18.2‰ and −1.5 to −0.9‰ (McGoldrick et al. 1998). The δ13C values fall in the range of unaltered McArthur Basin dolomites whereas the δ18OSMOW values are lower (Fig. 5), a difference attributed by Large et al. (2001) to a marine depositional environment in the McArthur Basin and a lacustrine one at Lady Loretta. Siderite within the Lady Loretta mineralization has bulk δ18O and δ13C values respectively of 23.2–28.3‰ and −3.2 to −1.9‰. McGoldrick et al. (1998) calculated that the siderite formed at temperatures near 100 °C from a fluid with a δ18OSMOW value of 6‰ and a δ13C value of −6‰. Such a fluid falls in the low-temperature HYC hydrothermal fluid (Fig. 6) modelled by Large et al. (2001). The values are also consistent with the ore-forming hydrothermal fluid being an evolved basinal brine.
3.4.3 Trap
Bulk δ34S values of pyrite, sphalerite, galena and barite at Lady Loretta are reported by Carr and Smith (1977). The median δ34S values for the three sulfides are similar to those at Mt Isa, while barite has a median δ34S value of 39.4‰ (Fig. 12). In the seven sets of δ34S of coexisiting pyrite-sphalerite-galena measurements made by Carr and Smith (1977) only one set has the relationship δ34Sbulk pyrite > δ34Sbulk sphalerite > δ34Sbulk galena, suggesting the three sulfides are not in, or approached, isotopic equilibrium. By contrast, in all seven sets δ34Sbulk sphalerite > δ34Sbulk galena, indicating the two ore minerals are in, or have approached, sulfur isotope equilibrium and that they are both in isotopic disequilibrium with pyrite. Bulk Δ34Ssphalerite-galena values range from +2.3 to +4.7‰, corresponding to a temperatures range of ~125–300 °C. However, this range should be treated with caution as Carr and Smith (1977) had difficulties obtaining pure mineral separates for analysis. Had galena and sphalerite formed by TSR of the ambient water column sulfate with a value of 30.6‰, then the range of ore-forming temperature, based on the fractionation equation of Kiyosu and Krouse (1990) would be ~110–160 °C (Fig. 12).

Box and whisker diagram of sulfur isotopic data for sulfides and barite in the Lady Loretta deposit (after Carr and Smith 1977). The dashed blue, vertical lines show the value of δ34Ssulfide formed by thermochemical sulfate reduction at various temperatures assuming a seawater sulfate with a value of 30.6‰
Carr and Smith (1977) noted a subtle increase in δ34Sbulk pyrite values up section at Lady Loretta and suggested the trend reflected pyrite formation by BSR in a restricted environment with an increasingly limited supply of sulfate, as suggested by Smith and Croxford (1973) to explain a similar increase in δ34Sbulk pyrite values up section in the HYC deposit. The suggestion is supported by the δ34Sbulk barite values from the barite lenses at the top of the deposit which are ~9‰ higher than the assumed ambient water column sulfate value (Fig. 7). As sphalerite and galena appear isotopically unrelated to pyrite, Carr and Smith (1977) proposed a two S-source model for the formation of the Lady Loretta deposit in which Pb and Zn sulfides, or sulfide complexes of Pb and Zn, were exhaled into the restricted reservoir of seawater from which the pyrite formed independently by BSR, the same genetic model proposed for the HYC by Smith and Croxford (1973).
3.5 Clastic-Dominatd Pb–Zn Mineralization in the Northern Mt Isa Inlier
The large CD Pb–Zn Century deposit in the northern Mt Isa Inlier (Fig. 2) is hosted by the siliciclastic Lawn Hill Formation that was deposited at 1595 ± 6 Ma (Page and Sweet 1998). Although some pyrite occurs in a halo around the deposit, the mineralization mostly comprises sphalerite with only minor galena (Broadbent et al. 1998). Some 80–90% of the Century ores are finely banded and are interlayered with sideritic siltstone and black shale, with the remainder comprising later coarser-grained vein- and breccia-style mineralization.
There are two types of sphalerite in the banded mineralization, one with a porous texture and a high pyrobitumen content, and the other with a non-porous texture and a low pyrobitumen content (Broadbent et al. 1998). A number of minor vein and breccia-style occurrences of Pb–Zn mineralization occur in a 10 by 20 km area to the north, northwest and south of Century, and these, together with Century, comprise the Burketown mineral field (Polito et al. 2006).
3.5.1 Source
The value of δ34Sseawater sulfate at the time of deposition of the strata hosting the Century deposit is uncertain. The secular δ34Sseawater sulfate curve of Chu et al. (2007) indicates that the value changed suddenly from ~16 to ~22‰ around the Century host strata were being deposited.
3.5.2 Transport
Five textural types of siderite occur in the Century deposit. Broadbent et al. (1998) report δ18Obulk and δ13Cbulk values for siderite mixtures that range respectively from 13.9 to 25.3‰ and −8.3 to 2.5‰ (Broadbent et al. 1998: Fig. 17). Because the analyses are of mixtures of siderite types, they could not be used to make quantitative estimates of the C- and O-isotopic composition of the hydrothermal fluids that formed the Century deposit. Broadbent et al. (1998) simply concluded that the siderites were deposited from an 18O-enriched fluid, the source possibly being evaporative carbonate sequences deep beneath the Lawn Hill Formation. A subsequent fluid inclusion study (Polito et al. 2006) found that the porous sphalerite was deposited at temperatures between 120 and 160 °C from a saline basinal brine (~21% NaCl equivalent) with a δDfluid range of −89 to −83‰. It was concluded that the fluids were highly evolved meteoric waters that came from siliciclastic strata some 8–10 km beneath the Century deposit site.
3.5.3 Trap
The bulk sulfur isotope values of sphalerite, galena and pyrite at Century measured by Broadbent et al. (1998) and Polito et al. (2006) are illustrated in Fig. 13. The median values of the pyrite, porous and non-porous sphalerite and galena in the stratiform mineralization reported by Broadbent et al. (1998) are similar (respectively 11.0‰, 10.2‰, 9.2‰ and 11.1‰) which, along with textural evidence, led Broadbent et al (1998) to conclude they are cogenetic. However, the sphalerite in the stratiform mineralization measured by Polito et al. (2006) has a median value of 13.2‰ indicating that the spread of δ34Ssulfide values in the CD Pb–Zn mineralization at Century is greater than that found by Broadbent et al. (1998) and a good understanding of the formation of the Century mineralization requires a comprehensive in-situ S-isotope study of the deposit.

Sulfides in the Century vein and breccia mineralization have higher bulk values than those in the stratiform mineralization, while those in the nearby Burketown mineral field are even higher (Fig. 12). To explain this observation Broadbent et al (1998) argued that that the Century and Burketown deposits are part of a single mineralizing system involving a large closed-system sulfate-rich fluid reservoir that became progressively enriched in 34S as sulfides formed by TSR over a long period. These authors also postulated that the Century CD Pb–Zn mineralization formed by the subsurface replacement of silica and infill of pore space in the host sediments during late diagenesis, with porous sphalerite forming by oil-mediated TSR reactions and the non-porous sphalerite by gas-mediated TSR reactions. However, Polito et al. (2006) concluded that the fluids that formed Century did not form the vein and breccia mineralization elsewhere in the Burketown mineral field.
4 The North American Paleozoic Cordillera Clastic-Dominated Pb–Zn Province—Canada
Several well-studied CD Pb–Zn deposits occur in the Selwyn Basin in the Canadian portion of the North American Cordillera (Fig. 14), including the Howards Pass deposit which is the second largest of the world’s ten largest CD Pb–Zn deposits based on Pb + Zn content (Leach et al. 2005). The Selwyn Basin CD Pb–Zn deposits are hosted by strata ranging in age from Cambrian to Devonian. The Basin is well suited to sulfur isotope studies to elucidate the role seawater sulfate might have been played in the formation of these deposits because the secular δ34S seawater sulfate curve for the Paleozoic is well documented (Claypool et al. 1980); there are numerous horizons of sedimentary/early diagenetic pyrite within the basin, as well as horizons rich in barite, and there are three significant groups of CD Pb–Zn deposits of different ages in the basin. They are the Cambrian Anvil Range group, the Ordovician–Silurian Howards Pass group and the Late Devonian MacMillan Pass group. (Fig. 13). The Anvil Range deposits have not been studied using in situ techniques, unlike those MacMillan and Howards Pass deposits, and are therefore not considered further in this Chapter. However, information about their stable isotope characteristics can be found in Campbell and Ethier (1974) and Shanks et al. (1987).

Location of the most significant CD Pb–Zn deposit districts in the North American Cordillera CD Pb–Zn Province. The term Realm in the geological map legend refers to the various components of the pre-accretionary terranes that today comprise tectonic elements of the North American Cordillera
In the first of two related papers that greatly influenced ideas on the formation of CD Pb–Zn mineralization worldwide, Goodfellow and Jonasson (1984) presented secular S-isotopic curves for pyrite and barite in the Selwyn Basin. The barite curve is similar in shape to the global seawater sulfate curve of Claypool et al. (1980), but is consistently 5–10‰ higher than the seawater sulfate curve. By contrast, the pyrite curve of Goodfellow and Jonasson (1984) is different and exceeds the δ34S seawater sulfate curve in three periods: the Late Ordovician, the Early Devonian and the Late Devonian. These three periods were interpreted to be times of almost complete reduction of seawater sulfate by BSR in restricted and stagnant deep euxinic layers of a stratified ocean. In the second paper Goodfellow (1987) noted that the interpreted periods of stagnation appear to coincide with periods of CD Pb–Zn mineralization and linked ore formation to stagnation. Because the isotopic curves for sphalerite and galena generally track the pyrite curve in the Selwyn Basin, Goodfellow (1987) further argued that the sulfide-S in the ore minerals also formed in the stagnant layer and concluded that CD Pb–Zn ores formed in the Selwyn Basin upon the exhalation of base-metal rich, but S-poor, hydrothermal fluids into stagnant euxinic ocean water (Fig. 1a, Pathway 1). This sedimentary-exhalative model is based on bulk sulfur isotopic measurements, but with the advent of in situ isotopic analytical technologies and their application to the MacMillan Pass and Howards Pass CD Pb–Zn deposits, a very different history of ore-forming processes has emerged.
4.1 Clastic-Dominated Pb–Zn Mineralization in the MacMillan Pass District
The MacMillan Pass CD Pb–Zn mineralization occurs in the Late Devonian Earn Group. Although the host rocks have been slightly metamorphosed (to sub-greenschist facies) and deformed into tight folds, mineralization displaying well-preserved sedimentary and hydrothermal features occurs in the limbs of folds (Magnall et al. 2016b). There are two significant deposits at MacMillan Pass, Tom (Andsell et al. 1989) and Jason (Gardner and Hutcheon 1985). Tom is located ~6 km east of Jason. Both deposits have several zones of stratiform mineralization in a sequence of chert conglomerates and biosiliceous mudstones that accumulated in a submarine fan on the eastern edge of the Selwyn Basin. Both deposits have well-developed feeder zones.
4.1.1 Source
The value of δ34Sseawater sulfate at the time of deposition of the Late Devonian strata hosting the Tom and Jason CD Pb–Zn deposits is ~24‰ (Magnall et al. 2016a).
4.1.2 Transport
A study of the Tom and Jason feeder zones by Magnall et al. (2016a) provides important insights into the nature of the fluids involved in the formation of the deposits. Two stages of feeder-zone development are recognized. Stage 1 comprises pervasive ankerite alteration of the organic-rich mudstone host rock and associated crosscutting ankerite stockwork veining (±pyrobitumen, pyrite and quartz). Stage 2 involves the deposition of massive sulfide (galena, pyrrhotite and pyrite ± chalcopyrite and sphalerite) and siderite (±quartz and barytocalcite).
δ13Cbulk ankerite and δ18Obulk ankerite values for Jason feeder complex correlate positively and strongly but those for Tom do not (Fig. 15). The Tom δ13C bulk ankerite values are similar to those at Jason, but Tom δ18Obulk ankerite values are on average ~2.5‰ higher. Temperature-dependent fluid-rock interaction modelling by Magnall et al. (2016a) indicates that the earliest ankerite-forming event involved a fluid with an initial isotopic value of −3‰ for δ13C and + 9.5‰ for δ18O at Jason and −2.5‰ for δ13C and + 10.0‰ for δ18O at Tom (Fig. 6). Fluid inclusion data indicate that the hydrothermal fluids entering the feeder zones were initially hot (270–300 °C) but cooled during the evolution of the Stage 1 ankerite event. It is concluded that the hydrothermal fluids had a modest salinity (6 wt% NaCl), were initially hot (270–300 °C) with a pH (≤ 4.5) and were metal rich (≫ 100 ppm Pb, Zn). As there are no evaporites in the Selwyn Basin, the modest salinity of the hydrothermal fluid is interpreted to have been derived originally from seawater trapped in Selwyn basinal rocks. Their high δ18O values of ~10.0‰ suggest these basinal fluids evolved as they interacted with the basinal sedimentary lithologies (Magnall et al. 2016a).

4.1.3 Trap
Stage 2 fluid inclusion data of Magnall et al. (2016a) indicate that during sulfide deposition the hydrothermal fluids in the feeder zones to the Tom and Jason deposits cooled to between 215 and 108 °C. Stage 2 bulk pyrite, galena and sphalerite δ34S values from feeder complexes are illustrated in Fig. 16. The Jason feeder zone bulk galena δ34S values have limited range compared to those at Tom and are also significantly lower. Because the feeder-zone sulfide minerals appear to have formed close to the sediment–water interface, Magnall et al. (2016a) assumed that Late Devonian seawater sulfate with δ34Ssulfate ~24‰ was the source of the sulfide S. As the fluid inclusion data indicate that the temperature of sulfide formation was ≥ 108 °C, Magnall et al. (2016a) argued that the sulfide sulfur was generated by the TSR of seawater sulfate. Based on their sulfur isotope measurements and the kinetic isotope fractionation equation of Kiyosu and Krouse (1990) for TSR, Magnall et al. (2016a) found that the sulfides at Jason formed at temperatures broadly consistent with the temperatures derived from their fluid inclusion measurements. Given the contrasting S-isotopic composition of the Tom sulfides it was further suggested that the sulfide sulfur at Tom might have formed at cooler temperatures by BSR.

Box and whisker diagram of sulfur isotopic data for sulfides in the feeder zones of the Tom and Jason deposits (after Gardner and Hutcheon 1985; Magnall et al. 2016a). The dashed blue, vertical lines show the value of δ34Ssulfide formed by thermochemical sulfate reduction at various temperatures assuming a seawater sulfate with a value of 24‰

Box and whisker diagram of sulfur isotopic data for sulfides and barite in the stratiform portions of the Tom and Jason deposits (after Gardner and Hutcheon 1985; Magnall et al. 2016b). The dashed blue, vertical lines show the value of δ34Ssulfide formed by thermochemical sulfate reduction at various temperatures assuming a seawater sulfate with a value of 24‰
Turning to the stratiform CD Pb–Zn mineralization adjacent to the Tom and Jason feeder complexes, Magnall et al. (2015) observed that the ore-bearing sequences at Tom and Jason contain abundant preserved radiolarians, including beds up to 10-cm-thick with > 50% radiolarians that are partially preserved by an early generation of pyrite that is overprinted by base-metal sulfides. The radiolarian-rich lithologies are interpreted to be a favorable host for mineralization due to high porosities during early diagenesis. Because the lithology also contains abundant organic matter it has a high capacity for the generation of sulfide via in situ sulfate reduction.
The stable isotope geochemistry of the Tom and Jason CD Pb–Zn mineralization was studied by Magnall et al. (2016b). The study includes in situ δ34S data for barite and pyrite from relatively undeformed drill core samples, as well as bulk δ34S values for pyrite extracted from barren mudstones between the two deposits and above and below the Tom deposit. Magnall et al. (2016b) identified three phases of mineralization. Stage 1 includes > 7 µm framboidal pyrite (Py–I) and small (< 25 µm) anhedral crystals of barite (brt-I) that replace quartz in the host mudstones. Stage 2 is characterized by euhedral pyrite that occurs as stratiform accumulations (Py–IIa) or solitary crystals (Py–IIb), and by coarser-grained (> 25 µm) and equant barite (brt–II) that is often associated with Py-IIa. Stage 2 also has barite in veins (brt–III) as well as euhedral celsian. The main mineralization event is Stage 3. It includes pyrite, sphalerite, galena and witherite. Stage 3 pyrite (Py–III), occurs as large sub- to anhedral replacements of earlier barite, and witherite occurs as an accessory phase to sphalerite. Galena is the final phase in the Stage 3 event, occurring as anhedral, interstitial crystals that replace earlier barite and pyrite along stratiform horizons.
The in situ barite δ34S values from the three stages are similar (Fig. 17) and Magnall et al. (2016b) concluded that the barite sulfate originated from modified Late Devonian seawater. They also observed that radiolarian tests in the host sediments have high Ba contents and concluded the enrichment reflected high levels of biological activity that predated diagenetic base-metal mineralization.
The various pyrite generations have distinctive sulfur isotopic compositions (Fig. 17). Py-I has the lowest δ34S values, Py-IIa has the highest, and Py-IIb and Py-III have values between Py-I and Py-IIa. Py-I values are offset from the ambient seawater SO42− value by between 44 and 54‰ and are interpreted to reflect formation by BSR, consistent with the framboidal texture of Py-I. Because the majority of framboids are > 7 µm in diameter it is further argued that Py-I formed diagenetically below the sediment–water interface rather than in a euxinic water column because framboids that form above the sediment–water interface typically have smaller diameters. Py–II is the heaviest isotopically and Magnall et al. (2016b) concluded from its isotopic composition and relationship with brt-II barite that it formed diagenetically below the sediment–water interface by SD-AOM. Py-III has a broad distribution of δ34S values that overlap Py-II, but which extends to lower values. The formation of sphalerite and galena is not discussed in detail in Magnall et al. (2016b) other than a brief comment that barite replacement by these sulfides might be an important mineralizing pathway in those CD Pb–Zn deposits rich in barite. Bulk pyrite δ34S values from barren mudstones range from −15.6 to +8.7‰ (n = 37). Because the range falls between the distributions of Py–I and II the bulk pyrite is interpreted to be mixtures of these two pyrite types.
Magnall et al. (2020c) presented a new subseafloor replacement model for the MacMillan Pass deposits using the stable isotope data and arguments discussed above, as well as new geological, geochemical and mineralogical information from Magnall et al. (2020a). The new model has ore formation occurring entirely in the subsurface from hot (300 °C) mineralizing fluids that thermally degraded organic matter, generating CO2 that promoted barite dissolution and not only triggered sulfide precipitation by TSR but also increased porosity and permeability that further facilitated ore formation. Gardner and Hutcheon (1985) report bulk (physical separates) S-isotopic values of galena and sphalerite from both the feeder zone and the CD Pb–Zn mineralization at Jason (Figs. 16 and 17). The values are consistent with the derivation of their sulfide-S by TSR of Late Devonian seawater sulfate at temperatures ≥ ~150 °C, with feeder zone having the higher temperatures (Fig. 16).
4.2 Clastic-Dominated Pb–Zn Mineralization in the Howards Pass District
CD Pb–Zn mineralization in the Howards Pass district occurs in the Ordovician–Silurian Duo Lake Formation. There are fourteen known deposits at Howards Pass, the largest being XY, Don, and Anniv. The main sulfides are sphalerite and galena, with pyrite a minor though ubiquitous component (Goodfellow and Jonasson 1986). The host sediments comprise carbonaceous and calcareous mudstones and some siliceous mudstones (Gadd et al. 2016). Descriptions of the geological and geochemical characteristics of the Howards Pass mineralization can be found in Gadd et al. (2016), Kelley et al. (2017), Martel (2017) and Slack et al. (2017). Following the bulk sulfur isotopic Howards Pass studies of Goodfellow and Jonasson (1984) and Goodfellow (1987) discussed above, Gadd et al. (2017) presented a more detailed study of the Howards Pass mineralization that included both in situ and bulk isotopic isotopic measurements. Johnson et al. (2018) subsequently published a new and very comprehensive bulk pyrite δ34S stratigraphic profile for an unmineralized Duo Lake Formation section in the region of the XY, Central, Don and Don East mineralization.
4.2.1 Source
The value of δ34Sseawater sulfate at the time of deposition of the Silurian strata hosting the Howards Pass CD Pb–Zn deposits is ~24‰ (Gadd et al. 2017).
4.2.2 Trap
In a detailed textural study Gadd et al. (2016) showed that pyrite of the Howards Pass district had a protracted growth history. Py1 is the earliest generation. It is framboidal and formed either in the water column or during very early diagenesis. During later diagenesis two younger pyrite generations (Py2a and Py2b) formed concurrently with sphalerite and galena. An even younger generation, Py3, occurs as porphyroblasts in strain shadows in deformed rocks and is therefore related to later deformation rather than to the mineralizing event.
Gadd et al. (2017) measured the in situ sulfur isotopic composition of the four pyrite types and galena from across the Howards Pass district, as well as the bulk sulfur isotopic compositions of coexisting sphalerite and galena in the XY deposit. The data are summarized in Fig. 18. The δ34S values of the framboidal Py1 are all negative and Δ34Sseawater sulfate-pyrite 1 values range from ~34 to 54‰. The range is strong evidence that Py1 formed by BSR either syngenetically in the water column or during early diagenesis from coeval seawater sulfate with a δ34S of ~28‰. The δ34S values of Py2a and Py2b are much higher than those of Py1 and are interpreted to reflect formation during later diagenesis by TSR. Gadd et al. (2017) note that the Py2 values mimic the secular Selwyn Basin pyrite curve that Goodfellow and Jonasson (1984) used to argue that the Howards Pass host rocks were deposited during a period of basin-wide stagnation when the oceanic environment was restricted and euxinic. However, as pointed out by Gadd et al. (2017), it is the isotopic composition of Py1 that reflects conditions in the oceanic water column and the Py1 data do not support the idea that the Howards Pass host rocks were deposited in a restricted euxinic environment.

Box and whisker diagram of sulfur isotopic data for sulfides in the Howards Pass deposits (after Gadd et al. 2017). The dashed blue, vertical lines show the value of δ34Ssulfide formed by thermochemical sulfate reduction at various temperatures assuming a seawater sulfate with a value of 24‰

Stratigraphic variations in δ34Sbulk pyrite values in the five members of Duo Lake Formation, Selwyn Basin; the Pyritic Siliceous Mudstone (PSM), the Calcareous Mudstone (CCM), the Lower Cherty Mudstone (LCM), the Active Member (AM and host to the Howards Pass mineralization) and the Upper Siliceous Mudstone (USM) (adapted from Johnson et al. 2018)
The δ34Sin situ galena values are significantly lighter than the δ34Sbulk galena values (Fig. 18), but the reason for this difference remains unclear. Amongst the bulk sulfur isotope measurements there are 12 coexisting sphalerite-galena pairs, all with δ34Sbulk sphalerite > δ34Sbulk galena. Gadd et al. (2017) concluded that the two phases formed concurrently in isotopic equilibrium, with Δ34Ssphalerite-galena yielding a median temperature of ~160 °C. It was further concluded galena and sphalerite formed by TSR involving coveal seawater sulfate with a δ34S value of ~28‰, a conclusion further supported by the sulfide δ34S values predicted by the kinetic fractionation equation of Kiyosu and Krouse (1990) for TSR involving sulfate with a δ34S value of ~28‰ (Fig. 18).
Gadd et al. (2017) concluded that Howards Pass mineralization formed diagenetically beneath a brine pool in a depression floored (Fig. 1a Pathway 2) by water saturated, organic-rich hemipelagic sediments. Initial BSR of seawater sulfate triggered Py1 precipitation at the top of the sediment pile or just above in the overlying water column. Subsequently exhaled dense warm (> 100 °C) hydrothermal fluids entered the system and ponded above the pyritic muds. These fluids then seeped down into the porous muds where TSR, initially catalyzed by BSR-produced H2S, precipitated both Py2 generations, sphalerite and galena deeper in the sediment pile. Py3 is postulated to have formed later in response to metamorphism associated with Jurassic to Cretaceous orogenesis. In support of their argument that Py2a and 2b, sphalerite and galena formed diagenetically in the sediment pile, Gadd et al. (2017) noted that calcite associated with the mineralization has δ13C values ranging from −1 to −6‰. Much of the calcite is nodular, suggesting it formed diagenetically, with its low δ13C values coming from the oxidation of organic carbon (with δ13C values of ~−28‰) associated with the TSR event that formed Py2, galena and sphalerite.
In an attempt to reconcile the older stagnant euxinic basin ore-forming model at Howards Pass with the new contradictory findings just discussed, Johnson et al. (2018) constructed a new bulk pyrite δ34S stratigraphic profile through an unmineralized section of the Duo Lake Formation at Howards Pass. The new profile is shown in Fig. 19. The δ34S values of pyrite range from ~−15‰ at the base of the section to 30‰ at the top, which is twice the range found by Goodfellow and Jonasson (1984). Johnson et al. (2018) argued that if the high positive values had been due to stagnation in a restricted anoxic oceanic basin, then the anoxic environment must have remained uninterrupted for some 20 Myr. The improbability of this situation, together with other geochemical evidence that the sequence was at least intermittently oxygenated (Slack et al. 2017), led Johnson et al. (2018) to propose that the base of the Duo Lake Formation was dominated by isotopically low Py1, and that pyrite with highly positive δ34S values became increasingly abundant up section. It is unclear from the textural descriptions of the latter pyrite generation (predominantly subhedral to euhedral crystals) if it is Py2 and/or Py3 as defined by Gadd et al. (2016, 2017). Johnson et al. (2018) also found that δ13Cbulk carbonate values decrease up section from ~−2.5 to ~−12.2‰, a trend that correlates with an increase in TOC. The trends are interpreted as indicating that beneath the Howards Pass mineralization pyrite formed by BSR, whereas starting immediately below the mineralized horizon, pyrite with high positive δ34S values began forming by SD-AOM that also generated oxidized carbon complexes with low δ13C values that concurrently formed low-δ13C authigenic carbonate. Johnson et al. (2018) suggested the high positive isotopic composition of galena and sphalerite in the Howards Pass mineralization might also have formed by SD-AOM, rather than by TSR as proposed by Gadd et al. (2017). However, the various temperature estimates of galena and sphalerite at Howards Pass are inconsistent with SD-AOM being an important ore-forming process.
5 The North American Paleozoic Cordillera Clastic-Dominated Pb–Zn Province—United States
The Alaskan part of the Northern American Cordillera hosts the world’s largest CD Pb–Zn deposit based on Pb + Zn content (Leach et al. 2005). It is the Red Dog deposit complex located in the Western Brooks Range in northern Alaska (Fig. 14). The complex comprises of four large CD Pb–Zn deposits (Qanaiyaq, Main, Aqqaluk and Paalaaq). They occur in organic-rich siliceous mudstones, shales, cherts and carbonates of the Carboniferous Kuna Formation (Leach et al. 2004). The deposits were originally one single deposit but were dismembered during the Mesozoic Brookian orogeny ~100 Ma after the deposition of the Kuna Formation.
The stratigraphic top of the deposits is marked by a sulfide-poor barite facies, beneath which, in descending order, lies a sulfide-bearing barite facies, a silica rock facies, a massive sulfide facies and, at the base, a sulfide vein facies (Leach et al. 2004). About 20% of the Red Dog production has come from the sulfide-bearing barite facies, 60% from the massive sulfide facies, and 20% from the sulfide vein facies (Kelley et al. 2004).
The sulfide-bearing barite facies comprises two textural types of barite, a fine-grained (10–50 µm) equigranular type and a coarse-grained (up to 3 cm) type (Kelley et al. 2004). Both types are intergrown with sulfide minerals, with sulfides locally interlayered with barite. The silica rock facies is characterized by strongly silicified barite dominated by medium-grained zoned euhedral quartz. The massive sulfide facies (> 40% sulfides) comprises semi-massive and massive, unbedded sulfide mineralization, with the sulfides disseminated in a dense silica matrix. Within this facies there are rare zones of banded and fragmental sulfides. Although sulfide veins occur throughout the mineralization, they are most abundant at the base and periphery of the mineralization. Veins vary in width from ~1 mm to 1 m and in places the vein density is sufficient to constitute ore.
The main sulfides in the Red Dog deposits are sphalerite, galena, pyrite and marcasite. Kelley et al. (2004) divided the mineralization into 4 temporal stages, each characterized by different colors of sphalerite. The earliest stage, Stage 1, is characterized by early brown sphalerite, Stage 2 by yellow–brown sphalerite, Stage 3 by red-brown sphalerite and the final stage, Stage 4, by tan sphalerite.
5.1 Source
The value of δ34Sseawater sulfate at the time of deposition of the Carboniferous strata hosting the Red Dog is 17.5 ± 1‰ (Johnson et al. 2004).
5.2 Transport
To date there have been no stable isotope studies directly relevant to the elucidation of the origin of the hydrothermal fluids that formed the Red Dog deposits. However, Leach et al. (2004) found that the Stage 4 red-brown sphalerite at Red Dog contains fluid inclusions suitable for study. They revealed that red-brown sphalerite formed between 100 and 180 °C from a fluid with a salinity of 14–19 wt% NaCl equivalent, consistent with the fluid being an evolved basin brine that obtained its salinity through the evaporation of seawater. No fluid inclusions data could be obtained from the earlier three sphalerite stages.
5.3 Trap
In situ sulfur isotopic values of sphalerite, pyrite and galena reported by Kelley et al. (2004) are shown in Fig. 20. As the value of δ34Sseawater sulfate at the time of deposition of the Red Dog host rocks was ~17.5‰. Δ34S sulfide-sulfate values for Stage 1 sphalerite and pyrite range from ~40 to 55‰ providing good evidence that the two sulfides formed by BSR. However, given the possibility of barite-inclusion contamination in some samples, Kelley et al. (2004) noted that the fractionation could be considerably smaller—in the range 15–25‰. In either case, the Stage 1 sphalerite and pyrite data are consistent with BSR involvement in their formation. The rare Stage 1 galena has low positive δ34S values, unlike the low negative values for sphalerite and pyrite, indicating that the ore-forming sulfides did not approach or achieve S-isotopic equilibrium. This isotopic relationship is interpreted as indicating rapid low temperature sulfide precipitation.

Box and whisker diagram of in situ sulfur isotopic data for sulfides in the Red Dog deposits (after Kelley et al. 2004). The dashed blue, vertical lines show the value of δ34Ssulfide formed by thermochemical sulfate reduction at various temperatures assuming a seawater sulfate with a value of 17.5‰
In contrast, Kelley et al. (2004) concluded that the formation of the Stage 2 and 3 mineralization was dominated by TSR based on Δ34S sulfide-sulfate values of ~15‰ for Stage 2 and 3 sulfides and Red Dog barite. Few available pyrite and galena sulfur isotope measurements from the Stage 2 and 3 mineralization lie within the ranges of the sphalerite measurements, suggesting that their formation also involved TSR. Sulfides in the Stage 4 mineralization are characterized by isotopic values similar to those in Stage 1 mineralization, suggesting BSR was also involved in their formation (Kelley et al. 2004). A comparison of the δ34S values of sphalerite and galena with the sulfide δ34S values predicted by the kinetic fractionation equation of Kiyosu and Krouse (1990) for TSR involving sulfate with a δ34S value of ~17.5 (Fig. 20) are consistent with the Stage 1 colloform and dendritic galena, and Stages 2 and 3 galena and sphalerite all forming by TSR from coeval seawater sulfate.
In summary, Kelley et al (2004) concluded from geological, mineralogical, textural, geochemical and isotope data that the formation of the Red Dog mineralization was protracted and complex. During the early Stage 1 event barite, brown sphalerite, pyrite and galena formed by BSR in organic-rich muds at or below the seafloor at temperatures from cool (< 100 °C) hydrothermal fluids rich in Ba, Zn and Pb. Warmer (100–200 °C) hydrothermal fluids recrystallized barite and deposited Stage 2 yellow–brown sphalerite, pyrite and galena in partly-lithified organic-rich muds below the seafloor, with H2S produced by TSR of sulfate sourced from either barite or seawater. During Stage 3 the continued alteration of mudstones produced methane, hydrofracturing and veins in which barite, red-brown sphalerite, pyrite or marcasite and galena were deposited from the evolving hydrothermal fluids. Red-brown sphalerite and pyrite also formed from the fluids by the replacement of earlier-deposited barite. Finally, Stage 4 event appears to be related to Brookian-age thrusting; it is thus not part of the main hydrothermal event that formed the original mineralization.
6 Implications for Clastic-Dominated Pb–Zn Mineral Systems
6.1 Source
The consensus view that Pb and Zn in CD Pb–Zn deposits is leached from host-basin sedimentary and/or volcanic rocks lying stratigraphy beneath the deposits is difficult to test using stable isotopes. At best, as shown by Cooke et al (1998) and Champion et al. (2020) in their source-related studies in the McArthur Basin, stable isotopes can be used to infer the C- and O-isotopic composition of fluids that might have leached metals from potential source rocks, but only in those rare cases where potential source rocks can be accessed and analysed. As both studies showed, the approach does not produce definitive outcomes. So far little attention has been paid to the source of the significant amounts of iron contained in the early pyrite attributed to BSR in pyrite-rich CD Pb–Zn deposits such as those in the McArthur Basin and Mt Isa district. Possibilities include an ordinary oceanic environment, a ferruginous ocean, and/or hydrothermal fluids. Which, if any, of these sources are important is a subject requiring further research.
The consensus view that contemporaneous seawater sulfate is the source of sulfide-S in CD Pb–Zn deposits has been clearly demonstrated in the case of the North American Paleozoic deposits. The involvement of seawater sulfate in the formation of the Proterozoic age Australian CD Pb–Zn deposits cannot be demonstrated so well, because the secular δ34S seawater sulfate curve for the Proterozoic is less well understood. Nevertheless the S-isotopic geochemistry of sulfides in the older Australian CD Pb–Zn deposits is consistent with their derivation from sulfate with large positive δ34S values which in all probability was seawater sulfate. However, in the case of the HYC deposit, it is possible that the ore-forming reduced sulfur was generated by TSR at depth and transported to the trap in a reduced fluid. While this reduced sulfur was formed from seawater sulfate, there is some evidence to suggest it had a different δ34S value to that of the sulfate in the water column from which the host strata of the HYC deposit were deposited.
6.2 Transport
Insights into the nature of the transporting hydrothermal fluids that form CD Pb–Zn deposits have come from carbon and oxygen isotopic composition of carbonates in and surrounding CD Pb–Zn mineralization. Fluid-mineral modelling of carbon and oxygen isotopic data typically yields results suggesting that the fluids had high δ18O values (≥ 6‰), typical of basinal-generated fluids that evolved geochemically through exchange with the basinal sedimentary rock pile (Fig. 6). In the HYC and Jason deposits where carbonate was either deposited from the hydrothermal fluids or fully exchanged with the fluids, δ13Ccarbonate and δ18Ocarbonate values correlate and have low positive slopes (Figs. 4 and 15) suggesting that the dominant carbonate species in the fluids was H2CO3 rather than HCO3− (Cooke et al. 1998: Fig. 19). The hydrothermal fluids that formed both the HYC and MacMillan Pass mineralization were therefore neutral to acidic.
6.3 Trap
The preferred trap-related ore-precipitating processes presented in the more comprehensive in situ studies discussed above are summarized in Table 1. The studies do not support the simple sedimentary-exhalative process favoured in the era preceding in situ sulfur isotope studies. Rather they indicate trap processes involving a protracted period of sulfide precipitation. In most of the recent studies it is argued that hydrothermal fluids did not exhale into the water column from which the host strata were deposited, but rather infiltrated the host strata after sedimentation. Only two of the recent studies (Ireland et al. 2004b; Gadd et al. 2017) retain an exhalative genetic component, and in both of these the critical trap element is a brine pool in which the exhalative fluids pond. It is argued by Ireland et al. (2004b) that sulfide precipitation is syngenetic and occurs mainly in the brine pool, whereas Gadd et al. (2017) argue that sulfides precipitate within the sedimentary pile beneath the pool.
In situ studies show that both the Northern Australian and Selwyn Basin CD Pb–Zn deposits have remarkably similar histories of sulfide deposition, commencing with pre-ore diagenetic pyrite formation involving BSR in a system open to sulfate and followed in most deposits by additional pyrite formation in more restricted environments as diagenesis and lithification progressed. This restriction leads to Rayleigh fractionation of sulfur isotopes and to higher δ34S values in the later pyrite compared to the earlier pyrite. The process of later pyrite formation has been variously attributed to BSR (HYC: Eldridge et al. 1993), BSR or SD-AOM (Teena: Magnall et al. 2020b), SD-AOM (Tom and Jason: Magnall et al. 2020c) and to TSR (George Fisher: Rieger et al. 2020; Howards Pass: Gadd et al. 2017).
By comparison with pyrite, there is a lack of in situ sulfur isotope data available for galena and sphalerite. The main impact to date of in situ sulfur isotope studies on the formation of the ore minerals has been the detailed sulfide mineral textural observations required for good in situ analyses. These observations show that galena and sphalerite mostly form after early diagenetic pyrite and therefore do not have a syngenetic to earlier diagenetic origin. Processes by which the ore minerals form have been mostly attributed to TSR-generated reduced S, exceptions being BSR for Sp-1 at HYC as suggested by Ireland et al. (2004b) and the possibility of SD-AOM at Howards Pass (Johnson et al. 2018). However, as noted earlier, ore-forming temperatures at Howards Pass appear too high for SD-AOM to have been a significant process in ore formation. Similarly, in the case of the HYC mineralization, Δ34Ssphalerite-galena values and dolomite C-O isotopic modelling indicate that mineralization formed at temperatures ≥ 100 °C, consistent with the production of sulfide by TSR rather than BSR. This conclusion is further strengthened by a study, unrelated to stable isotopes, of the temperature of alteration of organic matter in and around the HYC mineralization by Vinnichenko et al. (2021).
The Red Dog district is the only example discussed in this Chapter that does not follow the same ore-forming mechanism of early pyrite formation involving BSR and the latter formation of galena and sphalerite involving TSR. For the Red Dog district there is good sulfur isotopic evidence that Stage 1 brown sphalerite formed by BSR in unconsolidated organic-rich muds. The subsequent main stages of base metal mineralization involved TSR, mostly likely associated with barite replacement, as is also postulated by Magnall et al. (2020c) for some of the Tom and Jason mineralization. In a much later stage of ore formation in the Red Dog district, some ~100 my younger than the main ore-forming event, sulfur isotopic data suggests sphalerite formation again involved BSR (Kelley et al. 2004).
6.3.1 Trap-Related Questions Still to be Answered
Cool-oxidized or warm-reduced hydrothermal fluids? It was noted at the beginning of this Chapter that Cooke et al. (2000) used geochemical modelling to show that both cool (≤ 150 °C) oxidized hydrothermal fluids (dominated by SO42− ± HSO4−) and warmer (~250 °C) reduced acidic fluids (dominated by H2S or HS−) can transport and deposit sufficient quantities of Pb and Zn to form CD Pb–Zn deposits. The cool oxidized fluids were termed McArthur-type and Cooke et al. (2000) argued these fluids formed deposits such as HYC, Mt Isa and Hilton by in situ sulfate reduction and/or by low-temperature fluid mixing or interaction with earlier-formed pyrite. In contrast the warm reduced fluids were termed Selwyn-type and it was argued they formed the Selwyn Basin deposits at higher temperatures by processes like cooling, addition of H2S and pH increases.
In the in situ isotopic studies reviewed above, the trap-related paragenetic sequences and their interpretation in the North Australian and Selwyn Basin CD Pb–Zn deposits are strikingly similar. The similarity suggests that the hydrothermal fluids in both metallogenic provinces were warm and very likely had similar chemistries, contrary to the conclusions of Cooke et al. (2000). However, in deposits such as Tom and Jason, where barite-replacement appears to be a significant ore-forming process, it is likely that TSR occurred within the trap itself, whereas in other deposits, such as HYC, the site of TSR is less obvious. Eldridge et al. (1993) suggested H2S required for ore formation was introduced into the HYC trap by the hydrothermal fluid, implying that TSR occurred deeper in the mineral system whereas Williams (1978b), in a study not involving stable isotopes, favoured in situ TSR and a SO42-dominant hydrothermal fluid. The site of TSR for CD Pb–Zn ore formation and the oxidation state of the hydrothermal fluids has not been discussed specifically in many of the studies reviewed in this Chapter, and is another subject worthy of further investigation to improve understanding of trap processes in the CD Pb–Zn mineral systems.
What is the temperature of clastic-dominated Pb–Zn ore-formation? A significant uncertainty in our understanding of CD Pb–Zn mineral systems, and one related to uncertainty about the oxidation state of the ore-forming hydrothermal fluids, is the temperature of ore formation. In the McArthur Basin, for example, Ireland et al. (2004b) argued that galena and sphalerite formed by BSR at temperatures < 100 °C whereas others (e.g. Eldridge et al.1993; Rye and Williams 1981; Magnall et al. 2020b) presented a range of evidence indicating temperatures were > 100 °C and therefore involved TSR. One line of evidence used to support temperatures of > 100 °C, Δ34Ssphalerite-galena values, is permissive of warmer fluid temperatures but, as mentioned earlier, this geothermometer is based on assumptions that require further testing. There is a paucity of in situ sulfur isotopic data for galena and sphalerite in CD Pb–Zn deposits and more such data might help improve temperature estimates using the Δ34Ssphalerite-galena fractionation geothermometer. However, irrespective of whether or not they do, new in situ galena and sphalerite sulfur isotope studies will almost certainly produce fresh and unexpected genetic insights as has already happened with in situ S-isotopic studies of pyrite in CD Pb–Zn deposits.
What is the correct explanation for the formation of sulfides with high δ34S values? In many of the studies discussed in this Chapter sulfides with high δ34S values have been variously attributed to sulfate-restricted conditions caused by BSR in a stagnant and restricted basin, BSR in an euxinic water column triggered by a high flux of organic carbon, BSR in a restricted diagenetic environment, SD-AOM during later diagenesis and TSR. All these processes have been shown elsewhere to produce sulfides with high δ34S values, so high δ34S values in and of themselves are not indicative of a particular process. The only S-isotopic signature that appears to be diagnostic of a particular process is a Δ34Ssulfide − sulfate range of ~15–60‰, with much of the range having negative values. This S-isotope signature appears only to be displayed by sulfides formed by BSR in a system open to sulfate. Other processes are difficult to differentiate, although if the temperatures of sulfide reduction could be ascertained accurately, then choosing between biogenic and abiogenic processes would be possible. Clearly new and/or better diagnostic criteria would improve our understanding of the formation of sulfides with high δ34S values in CD Pb–Zn deposits and help to remove uncertainties about the position of sulfides in the paragenetic sequence in those instances where mineral textures are inconclusive.
Where has all the oxidized organic carbon gone? Sedimentary organic matter is characterized by low negative δ13C values (~−25‰), an isotopic signature that transfers to oxidized organic carbon species (Ohmoto and Goldhaber 1997). Low negative δ13C values in carbonates are therefore a good indication that oxidized organic carbon is a significant component of those carbonates. Authigenic carbonates associated with both SD-AOM (Borowski et al. 2013) and TSR (Machel et al. 1995; Machel 2001) typically have low value δ13C and is one line of evidence used to conclude that the respective sulfate-reduction mechanisms involved the oxidation of organic matter. Ireland et al. (2004b) used a lack of low δ13C values in nodular dolomites in the HYC deposit to argue that their formation did not involve the oxidation of organic matter during ore formation by either BSR or TSR beneath the nodular dolomite-bearing layers as suggested by Logan et al. (2001). Similarly, Rye and Williams (1981) found no C-isotopic evidence for significant amounts of oxidized organic carbonate in the dolomites in and surrounding the HYC deposit. This isotopic evidence may be used to suggest that sulfide formation in the HYC deposit did not involve in situ sulfate reduction and concomitant oxidation of organic matter. This would be consistent with the model of Eldridge et al. (1993) in which reduced sulfur was transported into the HYC trap by the hydrothermal fluids and the model of Ireland et al. (2004b) in which the HYC ores formed syngenetically by BSR in a brine pool from which oxidized organic carbon was able to escape into the overlying water column. Alternatively, if ore formation did involve in situ sulfate reduction during diagenesis or later, then the large amounts of oxidized of organic carbonate that would have been generated during sulfate reduction (Williams 1978b) must have been removed through the outflow zone of the HYC CD Pb–Zn mineral system. In the other CD Pb–Zn deposits reviewed in this chapter, in which ore-formation is postulated to involve in situ TSR, little attention has been paid to the quantities and fate of the oxidized organic carbon that would have been generated as galena and sphalerite were precipitated. The fate of this carbon is another subject worthy of further investigation to improve our understanding of trap processes in the CD Pb–Zn mineral systems.
7 Implications for Future Exploration
The CD Pb–Zn deposits discussed in this chapter are located at, or close to, current land surfaces where traditional prospecting, geophysical and geochemical exploration methods have been effective. However, the search for new CD Pb–Zn deposits will become increasingly focused on buried deposits that have no near-surface expression. New exploration methods will be needed in the future and the stable isotope studies summarized in this chapter provide some clues as to how light stable isotopes might assist in the discovery of CD Pb–Zn mineralization at depth.
In the case of an undiscovered CD Pb–Zn deposit located deep beneath current land surfaces, the parts of the CD Pb–Zn mineral system most likely to have a surface or near-surface expression are extensions of the feeder faults above buried deposits and the discharge zones for the hydrothermal fluids, downstream from buried deposits (Fig. 1). If exploration is taking place in a metallogenic province containing previously discovered and well-studied CD Pb–Zn deposits, knowledge of the isotopic geochemistry of the hydrothermal fluids that formed the known deposits could help identify extensions of feeder faults above buried deposits. Such extensions might have hydrothermal alternation minerals and/or vein- and breccia-fill minerals with isotopic signatures consistent with alteration and formation from fluids matching those that formed the known deposits in the province. If an exploration strategy is based on a genetic ore-forming model in which galena and sphalerite formed in the trap environment by in situ TSR and the concomitant oxidation of organic matter, then authigenic or vein-fill carbonates with low negative δ13C values in prospective terrains may help to identify the discharge zone of a CD Pb–Zn deposit and thus be a vector to mineralization at depth.
Both potential exploration applications of stable isotope geochemistry would require large numbers of analyses and, as discussed by Huston et al. (2023), the approach would require reductions in costs and the time required for analyses, and a significant increase in the number of analytical laboratories required to provide the analyses. These conditions would not be such a problem were the two applications used more selectively to analyse appropriate minerals in drill core from drill holes sited using other exploration tools.
8 Future Directions
Stable isotopes, particularly where integrated with other geochemical, mineralogical and geological techniques, are playing an increasing role in the study of CD Pb–Zn deposits and the trend will continue as stable isotope studies and technologies continue to evolve. As shown in this chapter, in situ stable isotope studies are proving to be particularly useful in genetic studies of CD Pb–Zn mineralization, but future in situ investigations would benefit from more integration with bulk isotopic and other whole-rock geochemical studies that would facilitate the quantification of ore-forming process. At present it is very difficult to estimate from existing data the relative importance of BSR, SD-AOM and TSR in ore formation. Also, integrated studies at a variety of sampling sizes would provide important new insights into the scales at which processes such as sulfate reduction operate during CD Pb–Zn ore formation.
Galena and sphalerite have been neglected relative to pyrite in in situ isotopic studies and the imbalance needs to be addressed given the uncertainties about galena and sphalerite formation in CD Pb–Zn deposits. Studies of the abundances of 33S and 36S, in addition to 32S and 34S, are providing new information about the formation of pyrite in sedimentary rocks unrelated to CD Pb–Zn mineralization as shown, for example, by Johnston et al. (2008) in their McArthur Basin pyrite study. Abundance data for all four sulfur isotopes in pyrite, galena and sphalerite in CD Pb–Zn mineralization should similarly refine our understanding of the ore-forming processes.
Huston et al. (2023) note that the development of analytical techniques to measure fractionations of “clumped isotopes” has the potential to become an important new ore genesis tool. Clumped isotope studies of carbonates in CD Pb–Zn deposits show promise as a much-needed and improved geothermometer that could help improve knowledge of the temperature of ore formation.
Stable isotopes will very likely play a greater role in CD Pb–Zn mineral exploration but, as is currently the case, that role is likely to remain subsidiary to the various geophysical and other geochemical exploration tools which are widely used today in the mineral exploration industry and which are being continually adapted to meet the challenges of locating ore at ever-increasing depths beneath the current land surface.
References
Ahmad M, Dunster JN, Munson TJ (2013) Chapter 15: McArthur basin. Northern Territory Geol Surv Spec Publ 5:15:1–15:72
Andsell KA, Nesbitt BE, Longstaffe FJ (1989) A fluid inclusion and stable isotope study of the Tom Ba-Pb-Zn deposit, Yukon Territory, Canada. Econ Geol 84:841–856
Berner RA (1970) Sedimentary pyrite formation. Am J Sci 268:1–23
Berner RA (1984) Sedimentary pyrite formation: an update. Geochim Et Cosochim Acta 48:605–615
Borowski WS, Rodriguez NM, Paull CK, Ussler W III (2013) Are 34S-enriched authigenic sulfide minerals a proxy for elevated methane flux and gas hydrates in the geologic record? Mar Pet Geol 43:381–395
Bottrell SH, Newton RJ (2006) Reconstruction of changes in global sulfur cycling from marine sulfate isotopes. Earth Sci Rev 75:59–83
Broadbent GC, Myers RF, Wright JV (1998) Geology and origin of shale-hosted Zn-Pb-Ag mineralization at the Century deposit, northwest Queensland, Australia. Econ Geol 93:1264–1294
Campbell FA, Ethier VG (1974) Sulfur isotopes, iron content of sphalerite, and ore textures in the Anvil ore body, Canada. Econ Geol 69:482–493
Canfield DE (2013) Sulfur isotopes in coal constrain the evolution of the Phanerozoic sulfur cycle. Proc Natl Acad Sci 110:8443–8446
Carpenter SJ, Lohmann KC, Holden P, Walter LM, Huston TJ, Halliday AN (1991) δ18O, 87Sr/86Sr and Sr/Mg ratios of Late Devonian abiotic marine calcite: implications for the composition of ancient seawater. Geochim Et Cosochim Acta 55:1991–2010
Carr GR (1984) Primary geochemical and mineralogical dispersion in the vicinity of the Lady Loretta Zn–Pb–Ag deposit, North Queensland. J Geochem Expl 22:217–238
Carr GR, Smith JW (1977) A comparative isotopic study of the Lady Loretta zinc-lead-silver deposit. Mineral Deposita 12:105–110
Champion DC, Huston Dl, Bastrakov E, Siegel C, Thorne J, Gibson GM, Houser J (2020) Alteration of mafic igneous rocks of the southern McArthur Basin: comparison with the Mount Isa region and implications for basin-hosted base metal deposits. In: Exploring for the future: extended abstracts. Geoscience Australia, Canberra. https://doi.org/10.11636/134206
Chapman LH (2004) Geology and mineralization styles of the George Fisher Zn–Pb–Ag deposit, Mount Isa, Australia. Econ Geol 99:233–254
Chu X, Zhang T, Zhang Q, Lyons TW (2007) Sulfur and carbon isotope records from 1700 to 800 Ma carbonates of the Jixian section, northern China: implications for secular isotope variations in Proterozoic seawater and relationships to global supercontinental events. Geochim Cosochim Acta 71:4668–4692
Claypool GE, Holser WT, Kaplan IR, Sakai H, Zak I (1980) The age curves of sulfur and oxygen isotopes in marine sulfate and their mutual interpretation. Chem Geol 28:199–260
Cooke DR, Bull SW, Donovan S, Rogers JR (1998) K-metasomatism and base metal depletion in volcanic rocks from the McArthur Basin, Northern Territory—implications for base metal mineralization. Econ Geol 93:1237–1263
Cooke DR, Bull SW, Large RR, McGoldrick PJ (2000) The importance of oxidized brines for the formation of Australian Proterozoic stratiform sediment-hosted Pb–Zn (Sedex) deposits. Econ Geol 95:1–18
Crockford PW, Kunzmann M, Bekker A, Hayles J, Huiming B, Halverson GP, Peng Y, Bui TH, Cox GM, Gibson TM, Worndle S, Rainbird R, Lepland A, Swanson-Hysell NL, Master S, Sreenivas B, Kuznetsov A, Krupenik V, Wing BA (2019) Claypool continued: extending the isotopic record of sedimentary sulfate. Chem Geol 513:200–225
Croxford NJW, Jephcott S (1972) The McArthur lead-zinc-silver deposit, Northern Territory, Australia. Proc Austr Inst Mining Metall. 243:1–26
Ding T, Zhang C, Wan D, Liu Z, Zhang G (2003) An experimental calibration on the sphalerite-galena sulfur isotope geothermometers. Acta Geol Sin 77:519–521
Duffett ML (1998) Gravity, magnetic and radiometric evidence for the geological setting of the Lady Loretta Pb–Zn–Ag deposit—a qualitative appraisal. Econ Geol 93:1295–1306
Eldridge CS, Williams N, Walshe JL (1993) Sulfur isotope variability in sediment-hosted massive sulfide deposits as determined using the ion microprobe SHRIMP: II a study of the H.Y.C. deposit at McArthur River, Northern Territory, Australia. Econ Geol 88:1–26
Gadd MG, Layton-Matthews D, Peter JM, Paradis SJ (2016) The world-class Howard’s Pass SEDEX Zn-Pb district, Selwyn Basin, Yukon. Part I: trace element compositions of pyrite record input of hydrothermal, diagenetic, and metamorphic fluids to mineralization. Mineral Deposita 51:319–342
Gadd MG, Layton-Matthews D, Peter JM, Paradis SJ, Jonasson IR (2017) The world-class Howard’s Pass SEDEX Zn-Pb district, Selwyn Basin, Yukon. Part II: the roles of thermochemical and bacterial sulfate reduction in metal fixation. Mineral Deposita 52:405–419
Gardner HD, Hutcheon I (1985) Geochemistry, mineralogy and geology of the Jason Pb–Zn deposits, MacMillan Pass, Yukon, Canada. Econ Geol 80:1257–1276
Gellatly AM, Lyons TW (2005) Trace sulfate in mid-Proterozoic carbonates and the sulfur isotope record of biospheric evolution. Geochim Et Cosochim Acta 69:3813–3829
Goldhaber MB, Kaplan IR (1974) The sulfur cycle. In: Goldberg ED (ed) The sea: 5 marine chemistry. Wiley, New York, pp 569–655
Gomes ML, Hurtgen MT (2015) Sulfur isotope fractionation in modern euxinic systems: implication for paleoenvironmental reconstructions of paired sulfate-sulfide isotope records. Geochim Cosmochim Acta 157:39–55
Goodfellow WD (1987) Anoxic stratified oceans as a source of sulphur in sediment-hosted stratiform Pb-Zn deposits (Selwyn Basin, Yukon, Canada). Chem Geol 65:359–382
Goodfellow WD, Jonasson IR (1984) Ocean stagnation and ventilation defined by δ34S secular trends in pyrite and barite. Geology 12:583–586
Goodfellow WD, Jonasson IR (1986) Environment of formation of the Howards Pass (XY) Zn–Pb deposit, Selwyn Basin, Yukon. Can Inst Mining Metall Spec 37:19–50
Grondijs HF, Schouten C (1937) A study of the Mount Isa ores. Econ Geol 32:407–450
Gustafson LB, Williams N (1981) Sediment-hosted stratiform deposits of copper, lead and zinc. Econ Geol 75th Anniv Vol 139–178
Hagemann SG, Lisitsin VA, Huston DL (2016) Mineral system analysis: quo vadis. Ore Geol Rev 76:504–522
Halevy I, Peters SE, Fischer WW (2012) Sulfate burial constraints on the Phanerozoic sulfur cycle. Science 337:331–334
Hayward N, Magnall JM, Taylor M, King R, McMillan N, Gleeson SA, (2021) The Teena Zn–Pb deposit (McArthur Basin, Australia). Part 1: syndiagenetic base metal sulfide mineralization related to dynamic subbasin evolution. Econ Geol 116:1743–1768
Heinrich CA, Andrew AS, Wilkins RWT, Patterson DJ (1989) A fluid inclusion and stable isotope study of synmetamorphic copper formation at Mount Isa, Australia. Econ Geol 84:529–550
Hoggard MJ, Czarnota K, Richards FD, Huston DL, Jaques LA, Ghelichkhan S (2020) Global distribution of sediment-hosted metals controlled by craton edge stability. Nat Geosci 13:504–510
Huston DL, Stevens B, Southgate PN, Muhling P, Wyborn L (2006) Australian Zn–Pb–Ag ore-forming systems: a review and analysis. Econ Geol 101:1117–1157
Huston DL, Mernagh TP, Hagemann SG, Doublier MP, Fiorentini M, Champion DC, Jaques AL, Czarnota K, Cayley R, Skirrow R, Bastrakov E (2016) Tectono-metallogenic systems—the place of mineral systems within tectonic evolution, with an emphasis on Australian examples. Ore Geol Rev 76:168–210
Huston DL, Trumbull RB, Beaudoin G, Ireland T (2023) Light stable isotopes (H,B,C, O and S) in ore studies—methods, theory, applications and uncertainties. In: Huston DL, Gutzmer J (eds) Isotopes in economic geology, metallogensis and exploration. Springer, Berlin (this volume)
Ireland T, Bull SR, Large RR (2004a) Mass flow sedimentology within the HYC Zn–Pb–Ag deposit, Northern Territory, Australia: evidence for syn-sedimentary ore genesis. Mineral Deposita 39:143–158
Ireland T, Large RR, McGoldrick P, Blake M (2004b) Spatial distribution patterns of sulfur isotopes, nodular carbonate, and ore textures in the McArthur River (HYC) Zn–Pb–Ag deposit, Northern Territory, Australia. Econ Geol 99:1687–1709
Johnson CA, Kelley KD, Leach DL (2004) Sulfur and oxygen isotopes in barite deposits of the western Brooks Range, Alaska, and implications for the origin of the Red Dog massive sulfide deposits. Econ Geol 99:1435–1448
Johnson CA, Slack JF, Dumoulin JA, Kelley KD, Falck H (2018) Sulfur isotopes of host strata for Howards Pass (Yukon—Northwest Territories) Zn–Pb deposits implicate anaerobic oxidation of methane not basin stagnation. Geology 46:619–622
Johnston DT, Farquhar J, Summons RE, Shen Y, Kaufman AJ, Masterson AL, Canfield DE (2008) Sulfur isotope biogeochemistry of the Proterozoic McArthur Basin. Geochim Cosmochim Acta 72:4278–4290
Kah LC, Lyons TW, Frank TD (2004) Low marine sulphate and protracted oxygenation of the Proterozoic biosphere. Nature 431:834–838
Kawasaki K, Symons DTA (2011) Paleomagnetism of the Mt Isa Zn–Pb–Cu–Ag and George Fisher Zn–Pb–Ag deposits, Australia. Aust J Earth Sci 58:335–345
Kelley KD, Leach DL, Johnson CA, Clark JL, Fayek M, Slack JF, Anderson VM, Ayuso RA, Ridley WI (2004) Textural, compositional, and sulfur isotope variations of sulfide minerals in the Red Dog Zn–Pb–Ag Deposits, Brooks Range, Alaska: implications for ore formation. Econ Geol 99:1509–1532
Kelley KD, Selby D, Falck H, Slack JF (2017) Re-Os systematics and age of pyrite associated with stratiform Zn–Pb mineralization in the Howards Pass district, Yukon and Northwest Territories, Canada. Mineral Deposita 52:317–335
Kiyosu Y, Krouse RH (1990) The role of organic and acid in the sulfur abiogenic isotope reduction effect. Geochem J 24:21–27
Kunzmann M, Schmid S, Blaikie TN, Halverson GP (2019) Facies analysis sequence stratigraphy, and carbon isotope chemostratigraphy of a classic Zn-Pb host succession: the Proterozoic middle McArthur Group, McArthur Basin, Australia. Ore Geol Rev 106:150–175
Lambert IB (1976) The McArthur zinc-lead-silver deposit: features, metallogenesis and comparisons with some other stratiform ores. In: Wolf KH (ed) Handbook of stratabound and stratiform ore deposits, vol 6, pp 535–585
Large RR, McGoldrick PJ (1998) Lithogeochemical halos and geochemical vectors to stratiform sediment-hosted Zn-Pb-Ag deposits 1. Lady Loretta deposit, Queensland. J Geochem Expl 63:37–56
Large RR, Bull SW, Cooke DR, McGoldrick PJ (1998) A genetic model for the HYC deposit, Australia: based on regional sedimentology, geochemistry, and sulfide-sediment relationships. Econ Geol 93:1345–1368
Large RR, Bull SW, McGoldrick PJ (2000) Lithogeochemical halos and geochemical vectors to stratiform sediment hosted Zn–Pb–Ag deposits. Part 2: HYC deposit, Northern territory. J Geochem Exploration 64:105–126
Large RR, Bull SW, Winefield PR (2001) Carbon and oxygen isotope halo in carbonates related to the McArthur River (HYC) Zn–Pb–Ag deposit. Northern Australia: implications for sedimentation, ore genesis, and mineral exploration. Econ Geol 96:1567–1593
Large RR, Bull SW, McGoldrick PJ, Walters W, Derrick GM, Carr GR (2005) Stratiform and strata-bound Zn–Pb–Ag deposits in Proterozoic sedimentary basins, Northern Australia. Econ Geol 100th Anniv Vol 931–963
Leach DL, Marsh E, Emsbo P, Rombach CS, Kelley KD, Anthony M (2004) Nature of hydrothermal fluids at the shale-hosted Red Dog Zn–Pb–Ag deposits, Brooks Range, Alaska. Econ Geol 99:1449–1480
Leach DL, Bradley DC, Huston DL, Pisarevsky SA, Taylor RD, Gardoll SJ (2010) Sediment-hosted lead-zinc deposits in earth history. Econ Geol 105:593–625
Leach DL, Sangster DF, Kelley KD, Large RR, Garven G, Allen CR, Gutzmer J, Walters S (2005) Sediment-hosted lead-zinc deposits: a global perspective. Econ Geol 100th Anniv Vol 561–607
Li C, Planavsky NJ, Love GD, Reinhard CT, Hardisty D, Feng L, Bates SM, Huang J, Zhang Q, Chu X, Lyons, TW (2015) Marine redox conditions in the middle Proterozoic ocean and isotopic constraints on authigenic carbonate formation: Insights from the Chuanlinggou Formation, Yanshan Basin, North China. Geochim Cosmochim Acta 150:90–105
Logan GA, Hinman MC, Walter MR, Summons RE (2001) Biogeochemistry of the 1640 Ma McArthur River (HYC) lead-zinc ore and host-sediments, Northern Territory, Australia. Geochim Cosmochim Acta 65:2317–2336
Luo G, Ono S, Huang J, Algeo TJ, Li C, Zhou L, Robinson A, Lyons TW, Xie S (2015) Decline in oceanic sulfate levels during the early Mesoproterozoic. Precambrian Res 258:36–47
Lyons TW, Luepke JJ, Schreiber ME, Zieg GA (2000) Sulfur geochemical constraints on Mesoproterozoic restricted marine deposition: Lower Belt Supergroup, northwestern United States. Geochim Cosmochim Acta 64:427–437
Machel HG (2001) Bacterial and thermochemical sulfate reduction in diagenetic settings—old and new insights. Sediment Geol 140:143–175
Machel HG, Krouse HR, Sassen R (1995) Products and distinguishing criteria of bacterial and thermochemical sulfate reduction. Appl Geochem 10:373–389
Magnall JM, Gleeson SA, Paradis S (2015) The importance of siliceous radiolarian-bearing mudstones in the formation of sediment-hosted Zn-Pb±Ba mineralization in the Selwyn Basin, Yukon, Canada. Econ Geol 110:2139–2146
Magnall JM, Gleeson SA, Blamey NJF, Paradis S, Luo Y (2016a) The thermal and chemical evolution of hydrothermal vent fluids in shale hosted massive sulphide (SHMS) systems from the MacMillan Pass district (Yukon, Canada). Geochim Cosmochim Acta 193:251–273
Magnall JM, Gleeson SA, Stern RA, Newton RJ, Poulton SW, Paradis S (2016b) Open system sulphate reduction in a diagenetic environment – isotopic analysis of barite (δ34S and δ18O)and pyrite (δ34S) from the Tom and Jason Late Devonian Zn–Pb–Ba deposits, Selwyn Basin, Canada. Geochim Cosmochim Acta 180:146–163
Magnall JM, Gleeson SA, Creaser RA, Paradis S, Glodny J, Kyle JR (2020a) The mineralogical evolution of the clastic dominant-type Zn–Pb±Ba deposits at Macmillan Pass (Yukon, Canada)—tracing subseafloor barite replacement in the layered mineralization. Econ Geol 115:961–979
Magnall JM, Gleeson SA, Hayward N, Rocholl A (2020b) Massive sulfide Zn deposits in the Proterozoic did not require euxinia. Geochem Persp Lett 13:19–24
Magnall JM, Gleeson SA, Paradis S (2020c) A new subseafloor replacement model for the Macmillan Pass clastic-dominant Zn-Pb-±Ba deposits (Yukon Canada). Econ Geol 115:935–959
Magnall JM, Hayward N, Gleeson SA, Schleicher A, Dalrymple I, King R, Mahlstadt N (2021) The Teena Zn-Pb deposit (McArthur Basin, Australia) Part II: carbonate replacement sulfide mineralization during burial diagenesis—implications for mineral exploration. Econ Geol 116:1769–1801
Manning AH, Embso P (2018) Testing the potential of brine reflux in the formation of sedimentary exhalative (sedex) ore deposits. Ore Geol Rev 102:862–874
Martel E (2017) The importance of structural mapping in ore deposits—a new perspective on the Howard’s Pass Zn–Pb district, Northwest Territories, Canada. Econ Geol 112:1285–1304
McGoldrick PJ, Kitto PA, Large RR (1998) Variation of carbon and oxygen isotopes in the alteration halo to the Lady Loretta deposit—implications for exploration and ore genesis. In: Water-rock interaction: Proceeding of the 9th international symposium on water-rock interaction, 30 March–3 April 1998, Taupo, New Zealand, pp 561–564
Murray WJ (1975) McArthur River H.Y.C. lead-zinc and related deposits. Austr Inst Mining Metall Mon 5:329–339
Ohmoto H, Goldhaber MB (1997) Sulfur and carbon isotopes. In: Barnes HL (ed) Geochemistry of hydrothermal ore deposits, 3rd edn. Wiley, New York, pp 517–611
Ohmoto H, Rye RO (1979) Isotopes of sulfur and carbon. In: Barnes HL (ed) Geochemistry of hydrothermal ore deposits, 2nd edn. Wiley, New York, pp 509–567
Page RW, Sweet IP (1998) Geochronology of basin phases in the western Mt Isa Inlier and correlation with the McArthur Basin. Austr J Earth Sci 45:201–232
Painter MGM, Golding SD, Hannan KW, Neudert MK (1999) Sedimentologic, petrographic, and sulfur isotope constraints on fine-grained pyrite formation at Mt Isa Mine and environs, northwest Queensland, Australia. Econ Geol 94:883–912
Perkins WG (1984) Mount Isa silica-dolomite and copper orebodies. Econ Geol 79:601–637
Polito PA, Kyser TK, Golding SD, Southgate PN (2006) Zinc deposits and related mineralization of the Burketown mineral field, including the world class Century deposit, northern Australia: fluid inclusion and stable isotope evidence for basin fluid sources. Econ Geol 101:1251–1273
Rickard D, Luther GW III (2007) Chemistry of iron sulfides. Chem Rev 107:514–562
Rieger P, Magnall JM, Gleeson SA, Lilly R, Rocholl A, Kusebauch C, (2020) Sulfur isotope constraints on the conditions of pyrite formation in the Paleoproterozoic Urquhart Shale Formation and George Fisher Zn–Pb–Ag Deposit, northern Australia. Econ Geol 115:1003–1020
Rohrlach BD, Fu M, Clarke JDA (1998) Geological setting, paragenesis and fluid history of the Walford Creek Zn–Pb–Cu–Ag prospect, Mt Isa Basin, Australia. Austr J Earth Sci 45:68–81
Rye DM, Williams N (1981) Studies of the base metal sulfide deposits at McArthur River, Northern Territory, Australia: III. The stable isotope geochemistry of the H.Y.C., Ridge, and Cooley deposits. Econ Geol 76:1–26
Sangster DF (2002) The role of dense brines in the formation of vent-distal sedimentary-exhalative (SEDEX) lead-zinc deposits: field and laboratory evidence. Mineral Deposita 37:149–157
Sangster DF (2018) Toward an integrated genetic model for vent-distal SEDEX deposits. Mineral Deposita 53:509–527
Shanks WC III, Woodruff LG, Jilson GA, Jennings DS, Modene JS, Ryan BD (1987) Sulfur and lead isotope studies of stratiform Zn–Pb–Ag deposits, Anvil Range, Yukon: basinal brine exhalation and anoxic bottom-water mixing. Econ Geol 82:600–634
Slack JF, Falck H, Kelley KD, Xue GG (2017) Geochemistry of host rocks in the Howards Pass district, Yukon-Northwest Territories, Canada: implications for sedimentary environments of Zn–Pb and phosphate mineralization. Mineral Deposita 52:565–593
Smith JW, Croxford NJW (1973) Sulphur-isotope ratios in the McArthur lead-zinc-silver deposit. Nature 245:10–12
Smith JW, Croxford NJW (1975) An isotopic investigation of the environment of deposition of the McArthur mineralization. Mineral Deposita 10:269–276
Smith JW, Burns MS, Croxford NJW (1978) Stable isotope studies of the origins of mineralization at Mount Isa I. Mineral Deposita 13:369–381
Solomon PJ (1965) Investigations into sulfide mineralization at Mount Isa, Queensland Part 1: textural studies of the ores. Econ Geol 60:737–765
Southgate PN, Bradshaw BE, Domagala J, Jackson MJ, Idnurm M, Krasay AA, Page RW, Sami TT, Scott DF, Lindsay JF, McConachie BA, Tarlowski C (2000) Chronostratigraphic basin framework for Palaeoproterozoic rocks (1730–1575 Ma) in northern Australia and implications for base-metal mineralisation. Austr J Earth Sci 47:461–483
Spinks S, Pearce MA, Weihua L, Kunzmann M, Ryan CG, Moorhead GF, Kirkham R, Blaikie T, Sheldon HA, Schaubs PM, Rickard WDA (2021) Carbonate replacement as the principal ore formation process in the Proterozoic McArthur River (HYC) sediment-hosted Zn–Pb deposit, Australia. Econ Geol 116:693–718
Taylor HP (1979) Oxygen and hydrogen isotope relationships in hydrothermal mineral deposits. In: Barnes HL (ed) Geochemistry of hydrothermal ore deposits, 2nd edn. Wiley, New York, pp 236–277
Turchyn AV, DePaolo DJ (2019) Seawater chemistry through Phanerozoic time. Annu Rev Earth Planet Sci 47:197–224
Vearncombe JR, Chisnall AW, Dentith M, Dorling S, Rayner MN, Holyland PW (1996) Structural controls on Mississippi Valley-type mineralization, the southeast Lennard Shelf, Western Australia. Econ Geol Spec Publ 4:74–95
Vinnichenko G, Hope JM, Jarrett AJM, Williams N, Brocks JJ (2021) Reassessment of thermal preservation of organic matter in the Paleoproterozoic McArthur River (HYC) Zn-Pb ore deposit, Australia. Ore Geol Rev 133:104–129
Walker RN, Logan RG, Binnekamp JG (1977) Recent advances concerning the H.Y.C. and associated deposits, McArthur River, N.T. J Geol Soc Austr 24:365–380
Waring CL, Andrew AS, Ewers GR (1998) Use of O, C and S stable isotopes in regional mineral exploration. AGSO J Geol Geophys 17:301–313
Wilkin RT, Barnes HL (1997) Formation processes of framboidal pyrite. Geochim Cosmochim Acta 61:323–339
Wilkinson JJ (2014) Sediment-hosted zinc–lead mineralization. Treatise on geochemistry, 2nd edn, vol 3, pp 219–249
Williams N (1978a) Studies of the base metal sulfide deposits at McArthur River, Northern Territory, Australia: I. the Cooley and Ridge deposits. Econ Geol 73:1005–1035
Williams N (1978b) Studies of the base metal sulfide deposits at McArthur River, Northern Territory, Australia: II. The sulfide-S and organic-C relationships of the concordant deposits and their significance. Econ Geol 73:1036–1056
Williams PJ (1998) An introduction to the metallogeny of the McArthur River-Mount Isa-Cloncurry minerals province. Econ Geol 93:1120–1131
Acknowledgements
I thank David Huston for his invitation to write this chapter and his support and advice as the chapter evolved, Jens Gutzmer for his wise counsel and guidance during the preparation of the chapter, and reviewers Joe Magnall and Raphael Baumgartner for their constructive criticism that helped greatly in the development of the final manuscript. Finally, I owe a great debt of gratitude to my erstwhile colleagues in the Mt Isa Mines group of companies, particularly Ross Logan and the late Bill Croxford, the late Bill Murray and late Tim Bennett who introduced me to the HYC, Teena, Lady Loretta and Mt Isa district Pb–Zn CD deposits and the many genetic, developmental and exploration challenges posed by these deposits.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2023 The Author(s)
About this chapter

Cite this chapter
Williams, N. (2023). Light-Element Stable Isotope Studies of the Clastic-Dominated Lead–Zinc Mineral Systems of Northern Australia and the North American Cordillera: Implications for Ore Genesis and Exploration. In: Huston, D., Gutzmer, J. (eds) Isotopes in Economic Geology, Metallogenesis and Exploration. Mineral Resource Reviews. Springer, Cham. https://doi.org/10.1007/978-3-031-27897-6_11
Download citation
DOI: https://doi.org/10.1007/978-3-031-27897-6_11
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-27896-9
Online ISBN: 978-3-031-27897-6
eBook Packages: Earth and Environmental ScienceEarth and Environmental Science (R0)