
Abstract
Management of patients with advanced cancer includes individualized treatment recommendations guided by molecular profiles. Refined complex molecular and immunological diagnostics are developed in parallel to the rapidly growing number of targeted therapies for defined genetic alterations and novel immunotherapies. For adequate counseling, patients are presented to Molecular Tumor Boards within the framework of precision oncology programs established at virtually all large cancer centers worldwide. The annotation and clinical interpretation of molecular pathology results are carried out by a multiprofessional team of experts formulating individualized treatment recommendations, taking also into account clinical characteristics. The process of annotation and clinical interpretation of molecular events in tumors also considers predictive factors defined in randomized studies as well as clinical judgement. All steps described above are not standardized, resulting in relevant heterogeneity in treatment recommendations among MTBs in different institutions.
In this chapter, contemporary challenges will be discussed, including intratumoral heterogeneity, use of diverse molecular diagnostic systems with inherent differences in sensitivity and specificity of detecting genetic alterations; the yet insufficiently addressed need for harmonizing variant annotation and interpretation; and the currently rather intuitive inclusion of multiple further “soft” parameters; all of which may significantly contribute to the current heterogeneity of recommendations.
You have full access to this open access chapter, Download conference paper PDF
Similar content being viewed by others


Keywords
- Precision medicine
- Clinical next generation sequencing
- Molecular diagnostics
- Targeted therapy
- Tumor heterogeneity
Introduction
Precision oncology is a rapidly evolving approach of tailoring therapeutic interventions to the individual molecular features of patients and/or their disease that moves beyond the conventional approach of stratifying patients into treatment groups based on tumor stage and phenotypic biomarkers [1]. Central to precision oncology is the ability to characterize precisely the molecular and cellular features of a tumor and its microenvironment, to determine which treatments are likely to confer the greatest benefit. For adequate counseling, patients are presented to Molecular Tumor Boards (MTBs) established at virtually all large cancer centers worldwide within the framework of precision oncology programs. The aim of MTBs is to identify and discuss all potential therapeutic strategies, based on genetic analysis, for patients who are not responding to standard-of-care systemic therapies. The individualized treatment recommendations by the MTB should be derived from a multidisciplinary discussion, including not only specific molecular alterations but also features concerning the patient (e.g., performance status, comorbidities).
Major scientific advances, in particular high-throughput sequencing technologies, animal and (increasingly also) ex vivo organoid models, play a pivotal role in the translational research that reinforces the current practice of medical oncology. However, the simply stated goal–tailoring oncological treatment to individual characteristics of a cancer patient–hides much complexity, and there are considerable challenges to be addressed. For a patient with cancer, major factors to consider include technical feasibility and validity of biomarkers, inter- and intratumoral heterogeneity, the challenging task of integrating and interpreting the ever-increasing volume of omics data, and the changing perspectives on value and cost-effectiveness in personalized cancer medicine.
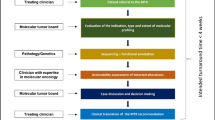
The analysis of molecular profiles of tumors with the primary goal of detecting targets for molecular therapies relies on the collection of an appropriate biological specimen, optimal sample handling and processing, and accurate data acquisition and analysis (Fig. 4.1). Several efforts have led to technological improvements in terms of biospecimen acquisition and processing, parallel with assay procedure homogenization. Professional bodies have been providing guidance for the validation of clinical next-generation sequencing (NGS) tests [2], and rigorous validation programs [3, 4] have helped to successfully implement and continuously improve the usage of NGS tests in a routine clinical setting [5]. In the following sections, we will discuss relevant aspects to be considered in order to obtain high-quality samples and to standardize molecular analysis. We will review current challenges and potential solutions to maximize the clinical utility of a molecular profiling program.

Overview of the precision oncology workflow. Relevant steps are listed in the left and important considerations in the right boxes
Relevant Aspects for Sample Collection and Processing
Though sample collection and processing can differ depending on the type of molecular analysis, there are general quality aspects to be considered for any type of NGS-based molecular profiling (Table 4.1). Low tumor cellularity and thus insufficient tumor DNA present in a sample is one of the most frequent reasons of molecular testing failure. According to our experience from molecular profiling programs at the Charité Comprehensive Cancer Center, tumor specimens selected for sequencing do not pass quality control in approximately 10% of patients referred to the MTB [6], stressing the importance of pathologic assessment in sample selection. The neoplastic cell, stromal and necrotic content can be highly variable within a sample, and even expert pathologists can judge the sample purity and suitability for testing very differently. Training programs for pathologists [7] that cover the principles and pitfalls of tumor cellularity scoring on sections [8] might help in reducing the reported wide variation in cellularity scoring amongst pathologists [8]. Latest technological advancement in digital pathology, including tissue scanners capable of scanning whole slides at high resolution also opens the possibility to leverage artificial intelligence-based image analysis techniques to further improve scoring accuracy [9].
Intratumoral heterogeneity (ITH) has been described as a further potential confounding factor in molecular profiling of tumors [10]. Regionally separated driver mutations, as reported among others for patients with non-small cell lung cancer [11] and renal cancer [12], are likely to be missed by single biopsy-based routine mutational analysis. Considerable ITH has also been reported for head neck squamous cell carcinoma [13,14,15,16,17,18], further increasing evidence that a single biopsy might not be enough for capturing the entire mutational landscape of individual tumors. A recent systematic review of studies on genetic mutation testing revealed that formal guidelines on how to avoid sampling bias due to ITH are lacking [19]. Of note, only 58% of the 40 genetic / biomarker studies included in this review reported on tumor purity thresholds, widely ranging from 10 to 100% [19]. As potential strategies to reduce sample bias due to ITH, Pongor and colleagues tested the effect of sample size, pooling as well as sequencing depth on the results of multiregional sequencing in ovarian cancer [20]. They observed similar genetic compositions from spatially neighboring regions, with only few private mutations [20]. Pooling samples from multiple distinct regions of the primary tumor did not increase the overall number of identified mutations. They further showed that pooling of multiregional biopsies was especially not suitable for hypermutated tumors since it diluted subclonal private mutations below detection thresholds [20]. In view of the limitations of present technologies, they recommended only one sequencing run per sample combined with high coverage (100–300x) sequencing, regardless of the number of samples taken from the same patient, as affordable and practical approach [20]. Another potential solution in tumor entities with known high ITH might be a complementary approach, in which mutational profiling of a single tumor biopsy is combined with the analysis of cell-free circulating tumor DNA [21], representing the cumulative reservoir of regionally separated mutant variants in the tumor.
Despite increasing numbers of studies focusing on the prevalence of ITH, its impact on clinical management of patients remains largely unknown. Evidence from breast cancer research has shown that molecular profiling of multiple tumor foci rather than the single largest tumor can lead to a change in treatment in 12.5% of cases [22]. In neuroblastoma, high degree of spatial heterogeneity was observed for genetic alterations in the druggable target genes ALK and BRAF [23]. Temporal ITH reflected by considerable differences in the genetic profiles of primary HNSCC tumors and their local relapses [24] stresses the importance of considering ITH in molecularly guided treatment selection in HNSCC as well. This preliminary evidence strongly supports further studies of ITH in precision oncology.
Clinical Next-Generation Sequencing
Clinical NGS has experienced rapid uptake in recent years, with a large number of academic and commercial certified laboratories offering NGS testing of tumor specimens [25]. Targeted NGS panels have come into widespread use for solid cancer patients including also patients with head neck cancer [26, 27]. Large-scale sequencing platforms, such as whole-genome and whole-exome sequencing (WGS and WES, respectively), are already frequently used for research purposes, revealing prognostic or predictive profiles that may ultimately guide therapy [28]. In the prospective observational DKTK-MASTER study by the German Cancer Consortium, it was shown that WGS/WES and RNA sequencing enables molecularly informed treatments that lead to clinical benefit in a substantial proportion of patients with advanced rare cancers [28]. Further proof that WGS can meet high-quality diagnostics standards in clinical routine was recently provided by Roepman and colleagues who, after optimizing sample and data processing procedures, reported a technical success rate of 95.6% for WGS analysis of fresh-frozen tumor samples [29]. It is thus expected that in the near-future, WES and WGS will be applied in standard care.
However, since formalin fixation and paraffin embedding (FFPE) will remain the most widely used method for tissue fixation in the diagnostic setting, targeted NGS will likely remain the predominant application in molecular pathology [30]. This is due to the fact that: (1) WES and WGS are not yet ready for clinical FFPE-based NGS; (2) low sequencing coverage remains an issue especially for samples with low tumor cell content; (3) turn-around times are still too long for the routine diagnostic setting; (4) the costs are much too high; and last but not least, (5) the majority of sequence information generated by WES and WGS cannot directly be translated into clinical intervention. By contrast, targeted NGS strategies can overcome most of these disadvantages. Since the first reports showing that it is feasible to use small amounts of genomic DNA derived from FFPE biopsies for NGS-based analyses [31], several protocols have been established that adapt DNA extraction methods as well as target region capture to the specific requirements of FFPE tissue.
Targeted Gene Panel Sequencing
The clinical demand for mutation detection within multiple genes from a single tumor sample requires molecular diagnostic laboratories to develop rapid, highly sensitive, accurate and high-throughput testing within tight budget constraints. To meet this demand, most clinical laboratories including the Molecular Pathology at our institution use NGS panels which interrogate a specific set of clinically relevant cancer-related genes (Table 4.2). The targeted sequences of interest may be enriched by either amplification or hybrid capture. While amplification-based sequencing offers certain advantages including a generally lower input requirement and faster turn-around times, hybrid capture often gives greater library complexity and uniformity [32]. Validated assays incorporating both approaches have demonstrated strong performance characteristics on all major variant classes, including single nucleotide variants, insertion/deletions, copy number variants and structural rearrangements such as gene fusions.
The list of gene alterations targeted by clinical NGS panels varies largely. It may be focused on one or a few histologies, such as lung or colon cancer, that have a high prevalence of clinically actionable mutations (e.g. the 52-gene Oncomine focus assay, ThermoFisher Scientific). More typically however, the panel includes several hundred pan-cancer genes, such as the MH IVD 600+ gene test (a customized test from Molecular Health GmbH, Heidelberg, Germany), the 505-gene MSK-IMPACT test [33] or the 324-gene FoundationOne CDx® test [34]. The overlap between these comprehensive gene panels is moderate (Fig. 4.2), with only 208 (22.8%) of the 911 genes captured by all of them. The impact of this heterogeneity in available NGS tests on the clinical benefit rate of molecular profiling programs remains unclear. Also unresolved remains the question whether focused panels targeting only a limited number of molecular alterations are sufficient for routine patient care since they cover all alterations for which molecular drugs are currently available. For a preliminary analysis, we determined the portion of molecular alterations in HNSCC classified as main actionable targets based on the European Society for Medical Oncology Scale for Clinical Actionability of Molecular Targets (ESCAT) [35] that would be captured by the small 52-gene Oncomine Focus assay or the more comprehensive 324-gene FoundationOne CDx® test. As shown in Fig. 4.3, alterations in 14 of the 34 (41%) genes could be captured by both tests, and 4 of 34 (12%) genes by none of them. Of note, alterations in 16 of the 34 genes (47%) would be missed by the small NGS test, including genes of the DNA repair (BRCA2, PALB2, POLE), oxidative stress (KEAP1, CUL3, NFE2L2) and PI3K pathways (PTEN). Alterations in these genes mostly classify to the ESCAT tier III category [35] which is defined by a clinical benefit demonstrated in other tumor types or for similar molecular targets.

Venn diagram of unique and common gene targets among three clinical next generation sequencing tests (blue: Molecular Health IVD 600+, red: MSK-IMPACT, green: FoundationOne CDx®)

Venn diagram of genes affected by selected potentially targetable molecular alterations in head neck squamous cell carcinoma (red), captured by a large (blue: FoundationOne CDx®) or small next generation sequencing panel (green: Oncomine™ Focus Assay). Genes captured by both tests (n=14), only the large test (n=16) or none of the tests (n=4) are listed in the boxes
Clinical Interpretation of Molecular Alterations
Personalized treatment requires the identification of predictive biomarkers. In an evidence-based medicine sense, these are biomarkers that are associated with response to a particular treatment, irrespective of the mechanism. In this sense, any molecular alteration that provides information on the probability of response to a therapy is a predictive biomarker [36]. At the same time, the inter- and intratumoral heterogeneity of tumors and the number of different alterations make it important to keep biological mechanisms in mind, to adequately interpret molecular alterations that might be similar but not identical to previously described ones [37]. Thus, the identification of molecular alterations and their clinical interpretation requires interdisciplinary analyses by bioinformaticians, biologists and physicians. Identified molecular alterations in a tumor need to be interpreted and annotated in an interdisciplinary setting in a third step to assess their value for guiding personalized therapy (Fig. 4.1). Furthermore, predictive biomarkers are not limited to mutations (e.g. amplification, methylation, gene expression changes) and change rapidly with new data and drugs. These challenges furthermore stress the need for the timeliness of data as well as workflow flexibility.
Usually, these workflows consist of the identification of published data for a given alteration, their annotation with an evidence level, rating the quality and applicability of the data and the interdisciplinary discussion of annotated molecular alterations in an interdisciplinary molecular tumor board [6]. Several databases have been established to allow for up-to-date searches of predictive biomarker data, of which part of the OncoKB database has recently gained FDA-recognition [38, 39]. Due to the wealth of clinical and preclinical data, most databases contain non-overlapping information [40], which has sparked the development of a meta-database [41]. Identified clinical and preclinical studies then need to be evaluated and ranked, for which several evidence level systems have been created [42, 43]. These evidence levels consider type and quality of underlying clinical trials, tumor histologies or even preclinical data. Negative predictive biomarkers indicating resistance to a specific therapy should also be integrated [6].
The individual annotation of molecular alterations, especially in the context of co-occurring alterations, is largely manual work and has been coined the bottleneck of personalized oncology [44]. This manual work remains unstandardized and substantial heterogeneity in the interpretation of molecular alterations, especially in patients with complex tumors and not well-described alterations, was shown, whereas fewer better-described alterations led to more concordant treatment recommendations [45, 46]. Following the identification of potential predictive biomarkers, supporting data and the annotation of molecular alterations with evidence levels, an interpreted molecular patient profile is usually presented in an MTB. These MTBs have been established at many institutions but standards, guidelines, or quality requirements for MTBs are currently absent. All are oriented to molecular tumor profiling and its relevance to treatment decisions, and all consist of a multidisciplinary team. Clinicians (mostly medical oncologists) and pathologists form the core of virtually every MTB, and medical biologists and bioinformaticians take part in most MTBs, but other than that, composition can vary widely [6, 45, 47, 48]. The MTB critically appraises molecular alterations, identified predictive biomarkers and patient factors to ultimately identify treatment options.
Treatment and Follow-up
Though individualized treatment recommendations provided by the MTB based on molecular aberrations, relevant patient characteristics, drug and clinical trial availability can guide therapy selection, the final recommendation to the patients remains at the discretion of the treating physician. In the above mentioned DKTK-MASTER study by the German Cancer Consortium in patients with rare cancers, the recommended therapies were administered in 32% of cases [28]. A lack of drug and trial availability, low evidence levels for identified treatment options and deteriorating performance status of patients remain the major causes for this relatively small percentage. Prioritization of recommendations by the MTB was also identified as an important factor in clinical decision-making, as the highest-ranked recommendations could be implemented in 84% of cases [28]. Another important observation of the DKTK-MASTER trial was also that 25% of recommendations were based on ESCAT tier III molecular alterations, for which potential clinical benefit may be predicted because they represent specific alteration (as tiers I and II) but the molecular alteration-drug efficacy relationship was established in a different tumor type.
About one third of patients receive molecularly targeted treatment in unstratified trials [28, 49]. To extend the potential benefit to more patients, overarching precision oncology trials, an access program for drugs and an early integration of precision oncology are required. Available data suggest a greater benefit for patients that receive treatment that is better matched to their individual tumor’s molecular profile [49]. Therefore, the integration of novel drugs and customized drug combinations need specific attention. The resulting complexity makes the development of prospective clinical trials to answer specific questions relating to these highly heterogeneous patient cohorts extremely difficult. Precision oncology programs are therefore also required to collect evidence from treated patients through a structured follow-up program. These data should ultimately be standardized and shared between centers to allow for an optimal use of available evidence.
Conclusions
Molecular profiling programs are highly complex and still largely unstandardized. Many of the technical aspects such as sample acquisition, sequencing and variant reporting as well as the clinical interpretation of the results are already performed in a routine setting at many cancer centers, even though many aspects such as tumor heterogeneity require specific attention. Given that the knowledge of specific biomarker actionability and the armamentarium of molecularly targeted drugs are rapidly evolving, it can be envisioned that the fraction of patients who will benefit from genomically agnostic precision oncology will significantly increase in the next years. Therefore, continuously revisiting the whole MTB workflow from NGS platform selection, data acquisition, annotation and interpretation within molecular profiling programs is warranted to capture the actionable cancer genome and transcriptome as completely as possible.
The translation of findings into clinical care will continue to depend on additional factors like patient performance status and the availability of drugs and clinical trials. The integration of novel drugs and drug combinations as well as increasing evidence from dedicated follow-up programs is expected to improve outcome of precision oncology programs.
References
Yates LR, Seoane J, Le Tourneau C, Siu LL, Marais R, Michiels S, et al. The european society for medical oncology (ESMO) precision medicine glossary. Ann Oncol. 2018;29(1):30–5.
Jennings LJ, Arcila ME, Corless C, Kamel-Reid S, Lubin IM, Pfeifer J, et al. Guidelines for validation of next-generation sequencing-based oncology panels: a joint consensus recommendation of the association for molecular pathology and college of american pathologists. J Mol Diagn. 2017;19(3):341–65.
Hirsch B, Endris V, Lassmann S, Weichert W, Pfarr N, Schirmacher P, et al. Multicenter validation of cancer gene panel-based next-generation sequencing for translational research and molecular diagnostics. Virchows Arch. 2018;472(4):557–65.
Lier A, Penzel R, Heining C, Horak P, Frohlich M, Uhrig S, et al. Validating comprehensive next-generation sequencing results for precision oncology: the NCT/DKTK molecularly aided stratification for tumor eradication research experience. JCO Precis Oncol. 2018;2:1–13.
Mosele F, Remon J, Mateo J, Westphalen CB, Barlesi F, Lolkema MP, et al. Recommendations for the use of next-generation sequencing (NGS) for patients with metastatic cancers: a report from the ESMO precision medicine working group. Ann Oncol. 2020;31(11):1491–505.
Lamping M, Benary M, Leyvraz S, Messerschmidt C, Blanc E, Kessler T, et al. Support of a molecular tumour board by an evidence-based decision management system for precision oncology. Eur J Cancer. 2020;127:41–51.
Genomics Education Programme: Tumour Assessment in the Genomic Era: NHS Health Education England. https://www.genomicseducation.hee.nhs.uk/education/online-courses/tumour-assessment-in-the-genomic-era/.
Sloan P, Robinson M. Quality assessment across disciplines in head and neck cancer treatment diagnostic pathology in HNSCC. Front Oncol. 2020;10:364.
Akbar S, Peikari M, Salama S, Panah AY, Nofech-Mozes S, Martel AL. Automated and manual quantification of tumour cellularity in digital slides for tumour burden assessment. Sci Rep. 2019;9(1):14099.
Bosman FT. Tumor heterogeneity: will it change what pathologists do. Pathobiology. 2018;85(1–2):18–22.
de Bruin EC, McGranahan N, Mitter R, Salm M, Wedge DC, Yates L, et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science. 2014;346(6206):251–6.
Gerlinger M, Rowan AJ, Horswell S, Math M, Larkin J, Endesfelder D, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366(10):883–92.
Zhang XC, Xu C, Mitchell RM, Zhang B, Zhao D, Li Y, et al. Tumor evolution and intratumor heterogeneity of an oropharyngeal squamous cell carcinoma revealed by whole-genome sequencing. Neoplasia. 2013;15(12):1371–8.
Ledgerwood LG, Kumar D, Eterovic AK, Wick J, Chen K, Zhao H, et al. The degree of intratumor mutational heterogeneity varies by primary tumor sub-site. Oncotarget. 2016;7(19):27185–98.
Jie W, Bai J, Yan J, Chi Y, Li BB. Multi-site tumour sampling improves the detection of intra-tumour heterogeneity in oral and oropharyngeal squamous cell carcinoma. Front Med (Lausanne). 2021;8: 670305.
Zandberg DP, Tallon LJ, Nagaraj S, Sadzewicz LK, Zhang Y, Strome MB, et al. Intratumor genetic heterogeneity in squamous cell carcinoma of the oral cavity. Head Neck. 2019;41(8):2514–24.
Mroz EA, Tward AD, Pickering CR, Myers JN, Ferris RL, Rocco JW. High intratumor genetic heterogeneity is related to worse outcome in patients with head and neck squamous cell carcinoma. Cancer. 2013;119(16):3034–42.
Mroz EA, Patel KB, Rocco JW. Intratumor heterogeneity could inform the use and type of postoperative adjuvant therapy in patients with head and neck squamous cell carcinoma. Cancer. 2020;126(9):1895–904.
Swift SL, Duffy S, Lang SH. Impact of tumor heterogeneity and tissue sampling for genetic mutation testing: a systematic review and post hoc analysis. J Clin Epidemiol. 2020;126:45–55.
Pongor LS, Munkacsy G, Vereczkey I, Pete I, Gyorffy B. Currently favored sampling practices for tumor sequencing can produce optimal results in the clinical setting. Sci Rep. 2020;10(1):14403.
Heitzer E, van den Broek D, Denis MG, Hofman P, Hubank M, Mouliere F, et al. Recommendations for a practical implementation of circulating tumor DNA mutation testing in metastatic non-small-cell lung cancer. ESMO Open. 2022;7(2):100399.
Boros M, Ilyes A, Nechifor Boila A, Moldovan C, Eniu A, Stolnicu S. Morphologic and molecular subtype status of individual tumor foci in multiple breast carcinoma. A study of 155 cases with analysis of 463 tumor foci. Hum Pathol. 2014; 45(2):409–16.
Schmelz K, Toedling J, Huska M, Cwikla MC, Kruetzfeldt LM, Proba J, et al. Spatial and temporal intratumour heterogeneity has potential consequences for single biopsy-based neuroblastoma treatment decisions. Nat Commun. 2021;12(1):6804.
de Roest RH, Mes SW, Poell JB, Brink A, van de Wiel MA, Bloemena E, et al. Molecular characterization of locally relapsed head and neck cancer after concomitant chemoradiotherapy. Clin Cancer Res. 2019;25(23):7256–65.
Karlovich CA, Williams PM. Clinical applications of next-generation sequencing in precision oncology. Cancer J. 2019;25(4):264–71.
The Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015; 517(7536): 576−82.
Su SC, Lin CW, Liu YF, Fan WL, Chen MK, Yu CP, et al. Exome sequencing of oral squamous cell carcinoma reveals molecular subgroups and novel therapeutic opportunities. Theranostics. 2017;7(5):1088–99.
Horak P, Heining C, Kreutzfeldt S, Hutter B, Mock A, Hullein J, et al. Comprehensive genomic and transcriptomic analysis for guiding therapeutic decisions in patients with rare cancers. Cancer Discov. 2021;11(11):2780–95.
Roepman P, de Bruijn E, van Lieshout S, Schoenmaker L, Boelens MC, Dubbink HJ, et al. Clinical validation of whole genome sequencing for cancer diagnostics. J Mol Diagn. 2021;23(7):816–33.
Dietel M. Molecular pathology: a requirement for precision medicine in cancer. Oncol Res Treat. 2016;39(12):804–10.
Kerick M, Isau M, Timmermann B, Sultmann H, Herwig R, Krobitsch S, et al. Targeted high throughput sequencing in clinical cancer settings: formaldehyde fixed-paraffin embedded (FFPE) tumor tissues, input amount and tumor heterogeneity. BMC Med Genomics. 2011;4:68.
Samorodnitsky E, Jewell BM, Hagopian R, Miya J, Wing MR, Lyon E, et al. Evaluation of hybridization capture versus amplicon-based methods for whole-exome sequencing. Hum Mutat. 2015;36(9):903–14.
Memorial Sloan Kettering Cancer Center: MSK-IMPACT: A Targeted Test for Mutations in Both Rare and Common Cancers. https://www.mskcc.org/msk-impact.
Foundation Medicine: FoundationOne®CDx. https://www.foundationmedicine.com/test/foundationone-cdx.
Marret G, Bieche I, Dupain C, Borcoman E, du Rusquec P, Ricci F, et al. Genomic alterations in head and neck squamous cell carcinoma: level of evidence according to ESMO scale for clinical actionability of molecular targets (ESCAT). JCO Precis Oncol. 2021;5:215–26.
Pezo RC, Bedard PL. Definition: translational and personalised medicine, biomarkers, pharmacodynamics. ESMO Handbook of Translational Research;2015.
Horak P, Griffith M, Danos AM, Pitel BA, Madhavan S, Liu X, et al. Standards for the classification of pathogenicity of somatic variants in cancer (oncogenicity): joint recommendations of clinical genome resource (ClinGen), cancer genomics consortium (CGC), and variant interpretation for cancer consortium (VICC). Genet Med. 2022;24(5):986–98.
Chakravarty D, Gao J, Phillips SM, Kundra R, Zhang H, Wang J, et al. OncoKB: a precision oncology knowledge base. JCO Precis Oncol. 2017;2017.
Griffith M, Spies NC, Krysiak K, McMichael JF, Coffman AC, Danos AM, et al. CIViC is a community knowledgebase for expert crowdsourcing the clinical interpretation of variants in cancer. Nat Genet. 2017;49(2):170–4.
Pallarz S, Benary M, Lamping M, Rieke D, Starlinger J, Sers C, et al. Comparative analysis of public knowledge bases for precision oncology. JCO Precis Oncol. 2019;3.
Wagner AH, Walsh B, Mayfield G, Tamborero D, Sonkin D, Krysiak K, et al. A harmonized meta-knowledgebase of clinical interpretations of somatic genomic variants in cancer. Nat Genet. 2020;52(4):448–57.
Horak P, Leichsenring J, Goldschmid H, Kreutzfeldt S, Kazdal D, Teleanu V, et al. Assigning evidence to actionability: an introduction to variant interpretation in precision cancer medicine. Genes Chromosomes Cancer. 2021.
Mateo J, Chakravarty D, Dienstmann R, Jezdic S, Gonzalez-Perez A, Lopez-Bigas N, et al. A framework to rank genomic alterations as targets for cancer precision medicine: the ESMO scale for clinical actionability of molecular targets (ESCAT). Ann Oncol. 2018;29(9):1895–902.
Good BM, Ainscough BJ, McMichael JF, Su AI, Griffith OL. Organizing knowledge to enable personalization of medicine in cancer. Genome Biol. 2014;15(8):438.
Rieke DT, Lamping M, Schuh M, Le Tourneau C, Baste N, Burkard ME, et al. Comparison of treatment recommendations by molecular tumor boards worldwide. JCO Precis Oncol. 2018;2:1–14.
Koopman B, Groen HJM, Ligtenberg MJL, Grunberg K, Monkhorst K, de Langen AJ, et al. Multicenter comparison of molecular tumor boards in the netherlands: definition, composition, methods, and targeted therapy recommendations. Oncologist. 2021;26(8):e1347–58.
Schwaederle M, Parker BA, Schwab RB, Fanta PT, Boles SG, Daniels GA, et al. Molecular tumor board: the university of california-san diego moores cancer center experience. Oncologist. 2014;19(6):631–6.
van der Velden DL, van Herpen CML, van Laarhoven HWM, Smit EF, Groen HJM, Willems SM, et al. Molecular tumor boards: current practice and future needs. Ann Oncol. 2017;28(12):3070–5.
Rodon J, Soria JC, Berger R, Miller WH, Rubin E, Kugel A, et al. Genomic and transcriptomic profiling expands precision cancer medicine: the WINTHER trial. Nat Med. 2019;25(5):751–8.
National Netwerk Genomic Medicine Lung Cancer. Molecular pathological Diagnostics. https://ngm-cancer.com/en/diagnostics/.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2023 The Author(s)
About this paper

Cite this paper
Tinhofer, I., Keilholz, U., Rieke, D. (2023). How to Standardize Molecular Profiling Programs for Routine Patient Care. In: Vermorken, J.B., Budach, V., Leemans, C.R., Machiels, JP., Nicolai, P., O'Sullivan, B. (eds) Critical Issues in Head and Neck Oncology. Springer, Cham. https://doi.org/10.1007/978-3-031-23175-9_4
Download citation
DOI: https://doi.org/10.1007/978-3-031-23175-9_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-23174-2
Online ISBN: 978-3-031-23175-9
eBook Packages: MedicineMedicine (R0)