
Abstract
Machine learning provides a set of new tools for the analysis, reduction and acceleration of combustion chemistry. The implementation of such tools is not new. However, with the emerging techniques of deep learning, renewed interest in implementing machine learning is fast growing. In this chapter, we illustrate applications of machine learning in understanding chemistry, learning reaction rates and reaction mechanisms and in accelerating chemistry integration.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


1 Introduction and Motivation
Machine-learning (ML), a term associated with a range of data analysis and discovery methods, can provide enabling tools for effective data-based science in the analysis, reduction and acceleration of combustion chemistry. The tools associated with ML can carry out a variety of automated tasks that either serve as effective substitutes for modern data analysis and discovery techniques applied to combustion chemistry or additional tools for its effective integration in CFD codes.
The implementation of ML in combustion chemistry is not new. Several tools have been used for chemistry reduction or chemistry acceleration. Perhaps one of the earliest analysis tools used for combustion chemistry is principal component analysis (PCA) (Vajda et al. 2006). By identifying redundant species in a mechanism and eventually eliminating their reactions, PCA plays a similar role to more recent methods based on directed relations graphs (DRG) (Lu and Law 2005).
Artificial neural networks (ANN) also have been used in combustion chemistry. Since the early work of Christos et al. (1995), ANNs have been used as substitutes for the direct evaluation of the chemical source terms in combustion. Beside their use as generalized function evaluators, ANNs have been used in other contexts, as discussed below. More recent applications of ANNs in combustion chemistry addressed the integration of chemically-stiff systems of equations.
The premise of ML tools in combustion chemistry lies in the availability of an ever-expanding body of data from experiments and computations and the complexity of handling this chemistry in the presence of 10–1000s of chemical species and 100–10,000s chemical reactions. Some of the challenges associated with combustion chemistry and potential applications of ML are highlighted below.
First, chemistry integration represents the ultimate bottleneck in reacting flow simulations. This is partly attributed to the size of chemical systems, involving many species and reactions, and the stiffness of their chemistry. This stiffness is associated with the presence of disparate timescales for the different reactions in a chemical mechanism. Approaches to overcome the presence of such bottlenecks can rely on chemistry reduction, chemistry tabulation and strategies to remove the fast time scales in chemistry integration. This reduction can be implemented offline from detailed chemistry or in situ using adaptive chemistry techniques. Careful chemistry reduction can also achieve a significant reduction of the stiffness of the chemical systems through the elimination of fast reactions and associated species.
Second, another difficult challenge with combustion chemistry is the development of new chemical mechanisms for an expanding range of fuels. Detailed mechanism development is a complex and time-consuming process that usually represents a first step prior to chemistry reduction. Identifying the elementary reactions relevant to a particular fuel oxidation, then determining their rates and relative importance in the mechanisms are integral steps in this process. Such an effort cannot be sustained given the need to develop the important elementary reaction data, especially data critical for the low-temperature oxidation for these fuels. More importantly, practical fuels tend to be complex blends and mixtures of different molecules. Establishing the chemical description of 10 or 100s of molecules is very challenging and must include models for their transport and thermodynamic properties. Until recently, strategies to develop a reduced description of chemistry without access to detailed or skeletal descriptions of chemistry have been limited to ad hoc global chemistry approaches that optimize rate constant and stoichiometric coefficients for the global reactions by matching global observables, such as flame speeds, ignition delay times or extinction strain rates.
However, a growing body of data and detailed mechanisms is now available that can be exploited to develop “rules” for representing the chemistry of complex fuels (Buras et al. 2020; Ilies et al. 2021; Zhang and Sarathy 2021b, c; Zhang et al. 2021). Temporal measurements from shock tubes and rapid compression machines (RCMs), although may be limited to a subset of the chemical species present, which may be subject to experimental uncertainty, can provide important relief to detail mechanism development as discussed below.
The challenges listed above can lend themselves to applications of data science and the implementation of ML tools for combustion chemistry discovery, reduction and acceleration. The various ML methods in combustion chemistry and other applications can generally be classified as either supervised (e.g. classification, regression models) or unsupervised (e.g. clustering and PCA). Supervised models are a class of models in which both input and output are known and prescribed from the training data. This data is called labeled. For example, in a regression ANN for chemical source terms, we attempt to map the thermo-chemical state (i.e. pressure, temperature and composition) to a chemical source terms. During unsupervised learning, the output is not labeled. This approach may include for example identifying principal components (using PCA) from a thermo-chemical state or clustering of states based on the proximity of the thermo-chemical state vector.
Another class of models that have not been extensively used in combustion chemistry are the so-called semi-supervised models. In semi-supervised models, both labeled and unlabeled data are used for the training of these models. These models include for example generative models where available data is trained to generate new similar data. A popular such model is the generative adversarial network (GAN). As expected, ML approaches require data. The quality and quantity of the data is critical as discussed below. These approaches are trained on this data, while a portion can be used for either validation or testing.
In this chapter, we illustrate different implementations of ML tools in combustion chemistry. The goal is not to provide a comprehensive review of these tools or to address all studies involving ML for combustion chemistry. Instead, we attempt to provide an overview of various applications of ML in combustion chemistry. It is important to note that research in ML for combustion chemistry is a very active area of research and more progress is expected in the coming years. The chapter is divided into 3 general topics related to: (1) learning reaction rates, (2) learning reaction mechanisms and (3) chemistry integration and acceleration.
2 Learning Reaction Rates
The law of mass action and the Arrhenius model for the rate constant form the traditional representation of the rate of reaction of chemical species in combustion. This rate can be expressed in terms of a linear combination of rate of progress for each elementary reaction a species is involved in. The integration of chemistry is limited by the cost of this evaluation as well as the inherent stiffness of reaction mechanisms, exhibited by a wide range of timescales involved and the time-step size required to integrate chemistry in combustion simulations.
Artificial neural networks (ANNs) have been proposed as an alternative tool to the direct evaluation of reaction rates based on the law-of-mass-action and the Arrhenius law. Perhaps one of the earliest implementations of ANNs in combustion is through their use in their implementation as regression tools for species and temperature chemical source terms (Blasco et al. 1998, 1999, 2000; Chatzopoulos and Rigopoulos 2013; Chen et al. 2000; Christo et al. 1996; Christos et al. 1995; Flemming et al. 2005; Franke et al. 2017; Ihme 2010; Ihme et al. 2008, 2009; Sen and Menon 2010a, b; Sinaei and Tabejamaat 2017). The primary goal of representing chemical source terms with ANN is to accelerate the evaluation of chemistry. The different demonstrations of ANN for chemistry tabulation have shown that the ANN-based chemistry tabulation method is computationally efficient and accurate.
ANNs are perhaps the most versatile ML tools that have been used for combustion chemistry and other applications. Among these ANNs, one of the most popular ANN architectures are the so-called multi-layer perceptions (MLP). A representative MLP-ANN architecture is shown in Fig. 1. It is designed to construct a functional relation between a prescribed input vector \(\textbf{x} \left( x_1, x_2\right) \) and an output vector \(\textbf{y} \left( y_1, y_2\right) \). Within the context of a regression model, the ANN forms a function for y in terms of x, i.e., \(\textbf{y} = \textbf{f} \left( \textbf{x} \right) \). The input layer in the figure contains the input vector elements, which are represented by “neurons”. A similar arrangement is present for the output layer where each element is represented by a neuron. The neurons carrying values are in the hidden layer, which separate the input and the output layers. In the illustration, there is only one hidden layer with 4 neurons shown. The illustrated MLP here is fully-connected, meaning that starting with the first hidden layer all the way to the output layer, the neurons carrying values are in the hidden layers, which separate the input and output layers. The strength of the connections are represented by “weights” and the value at the neurons at these layers is expressed in terms of the values of the neurons of the previous layers weighted the strength of the connections. Although not shown in the figure, additional “bias” neurons can be added to the input and all hidden layers. The role of the bias neurons is to provide more flexibility to train the model that relates the input to the output vectors.

Illustration of the ANN-based matrix formulation for reaction rates
To illustrate the relation between the input and the output layers, we use the network illustrated in Fig. 1. The output \(y_1\), which corresponds to the value of the first neuron in the output layer, is expressed in terms of the hidden layer:
where the superscript (1) corresponds the first hidden layer with weights \(w_i^{(1)}\) and values \(a_i^{(1)}\) at the ith neuron in the hidden layer. \(b^{(1)}\) is the bias value at the hidden layer and f is the activation function. The bias neuron value serves as an additional parameters to fine-tune the network architecture and potentially reduce its complexity (i.e., less hidden layers or less neurons per hidden layer). The values of the ith neuron, \(a_i^{(1)}\), in the hidden layer can be related to the input variables as follows:
Here \(w_{1i}^{(0)}\) and \(w_{2i}^{(0)}\) correspond to the weights of the connections between the input layer and the ith neuron in the first hidden layer associated with inputs \(x_1\) and \(x_2\), respectively. The network is trained to determine the weights of all connections from input to output layers and the bias values.
In matrix form, the output values for the hidden layer neurons and the output layer neurons can be expressed as follows:
and
where \(\textbf{W}^{(0)}\) and \(\textbf{W}^{(1)}\) are the weight matrices corresponding to the weights of the connections between the input and the first hidden layer and the first hidden layer and the output layer, respectively. \(\textbf{b}^{(0)}\) and \(\textbf{b}^{(1)}\) are the bias vectors for the input and the first hidden layers, respectively, with identical elements in each vector.
The expression above can be generalized to related on hidden layer or an output layer at a level \(n+1\) to the vector of values from the previous layer level n:
MLPs vary in complexity as well as in purpose. Accommodating complexity can be achieved by increasing the number of hidden layers, the number of neurons per hidden layer and the activation functions, which can be varied from one layer to another. Prescribing the loss function can also improve the prediction of the target output. Although, there are usual choices for the activation functions, there is an inherent flexibility in the choice of network parameters, including the activation function to represent systems of equations representing physics, as illustrated below.
2.1 Chemistry Regression via ANNs
In this section, we briefly summarize key considerations for establishing efficient regression for chemical reaction rates using ANNs. Figure 2 illustrates a relatively deep network topology that constructs a regression of the reaction rates for 10 species and the heat release rate for the temperature equation from the work of Wan et al. (2020). This network has 5 fully connected dense layers between the input and output layers. In dense layers, neurons in a given layer are connected through weights to all neurons in the previous layer. As indicated, the number of neurons in the hidden layers is higher towards the input layer and decays towards the output layer. The rectified linear unit (ReLU) activation function is used. The network has approximately 180,000 weights to be optimized during the training stage, which required approximately 2.2 h on an Nvidia GeForce GTX 1089 Ti GPU. Other variants of the topology shown in Fig. 2 have been adopted in the literature (see for example, (Blasco et al. 1998, 1999, 2000; Chatzopoulos and Rigopoulos 2013; Chen et al. 2000; Christo et al. 1996; Christos et al. 1995; Flemming et al. 2005; Franke et al. 2017; Ihme 2010; Ihme et al. 2008, 2009; Sen and Menon 2010a, b; Sinaei and Tabejamaat 2017)).
Determining all these chemical source terms invariably requires more complex neural networks than those specialized to predict only one quantity. Within such complex networks, the weights from the input layer to the layer prior to the last layer are shared among all the input quantities; and the weights relating the last hidden layer to the output layer are the primary differentiators for the individual reaction rates. There are potentially 3 attractive features for the use of ANNs to model chemical source terms. The first feature is the potential acceleration in the evaluation of the chemical source through graphical processing units (GPUs) through integration of neural networks with existing accelerated packages designed to optimize ANN evaluations through mixed hardware frameworks.

Illustration of the ANN-based matrix formulation for reaction rates with multiple inputs and multiple outputs (from Wan et al. (2020))
A second attractive feature is that ANNs can be made simpler by adopting only a subset of the input. This is motivated by the inherent correlation of thermo-chemical scalars in a chemical mechanism, which lends itself to dimensionality reduction methods. Alternatively, low-dimensional manifold parameters, such as principal components (PCs) from PCA, or a choice of representative species, including major reactants, products and intermediates can be used.
A third feature of using ANNs for learning chemical reaction rates, related to the previous one, is that, if a subset of the inputs is used, then, the solution vector may also require only a subset of the thermo-chemical scalars to be transported, which corresponds primarily to the thermo-chemical scalars in the input vector. This can reduce the computational cost. It follows that, if species and associated reactions that represent a bottle-neck in chemistry integration are eliminated, then, the stiffness of the chemical system is significantly reduced, further accelerating chemistry integration.
Implementing the regression for chemical source terms within a single ANN has a number of advantages. First, constraints can be built in the training for the chemical source terms, for example to enforce the conservation of elements, mass or energy. Moreover, a single network with a number of shared weights may be exploited for computational efficiency, since the contributions to the individual source terms occur primarily at the connections between the last hidden layer and the output layer.
However, accommodating all species chemical source terms in a single layer may also require a more complex ANN architecture. Alternative strategies to reduce this complexity have been used. One approach relies on adapting different ANNs for different clusters of data, such as different networks for the reacting and the non-reacting zones in the mixture. This approach has been implemented by Blasco et al. (2000), Chatzopoulos and Rigopoulos (2013) and Franke et al. (2017) using self-organizing maps (SOM) (Kohonen 2013). In these studies, chemistry tabulation was implemented in conjunction with closure models for turbulent combustion and SOM was used as an adaptive tool to cluster similar conditions of the composition space to establish a single ANN regression tables for them.
SOMs are a popular method and an unsupervised ML technique for clustering and model reduction as stated earlier. They are single-layer neural networks that connect inputs, which corresponds to data to be clustered, to a (generally 2D) map of nodes or clusters. The clustering of the input data is based on their weights relative to the different nodes, which are determined iteratively by measuring their “proximity” to the node measures. The outcome of this iterative procedure is a mapping of the original data into a lower-dimensional space represented by the 2D map of nodes. The versatility of SOM in addressing how data is grouped is established through the choice of measures of similarity that are used to identify the mapping. For tabulation, these measures can be related to the proximity in thermo-chemical space (e.g. similar temperatures and compositions); while, for identifying different phases, these measures may rely on the evolution of marker thermo-chemical scalars in time and their correlations with other scalars.
Alternatively, clustering was implemented to group thermo-chemical scalars of similar behavior, such as the construction of an ANN for intermediates and another for reactants and products (Owoyele et al. 2020). This approach attempts to construct a minimum set of neural networks that are also less complex than the ones that accommodate all thermo-chemical scalars.
Additional consideration for constructing ANNs for reaction rate regression is related to the high variability of the input, the thermo-chemical scalars, and the output data, their chemical source terms, resulting in strongly nonlinear regressions, which may require, unnecessarily, complex and deeper ANNs. A potential way of “taming” the data variability is to pre-process the input and the output data. Sharma et al. (2020) used log-normalization to pre-process free radicals, which tend to skew towards zero.
Finally, determining an optimum topology for a chemistry regression network is not a trivial task. A shallow (one hidden layer) to a moderately deep network may not be sufficient to capture the functional complexity of the chemical source terms and may result in “under-fitting”. Meanwhile, a much deeper network with numerous neurons in their hidden layers may achieve better predictions with an increased cost of evaluating the networks and the associated storage needed for the trained weights. It can also result in “over-fitting” when data is sparse or does not represent the true variability of the accessed composition space.
Ihme et al. (2008), Ihme (2010); Ihme et al. (2009) proposed an approach to determine an optimum artificial neural network (OANN) using the generalized pattern search (GPS) method (Torczon 1997). The GPS method is a derivative-free optimization that generates a sequence of iterates with a prescribed objective functions. The optimum network in this method is designed to determine the choice of network parameters (number of hidden layers, number of neurons in hidden layers) that minimize the memory requirements, the computational cost and the approximation error of the network.
Nowadays, other automated tools can be used to help optimize a given network. These include the so-called automated machine learning (or AutoML) tools, such as the Keras Tuner, Auto-PyTorch and the AutoKeras tools (Hutter et al. 2019). However, special attention must be paid to the choice of the measure of convergence of the training schemes.
3 Learning Reaction Mechanisms
Machine learning tools are set to provide greater insight into (1) the discovery of chemical pathways and key reactions in a mechanism, and (2) the reduction and representation of chemical mechanisms. In this section, we review a number of applications in which ML tools have been used for learning reaction mechanisms.
3.1 Learning Observables in Complex Reaction Mechanisms
Although, for many, the ultimate goal of understanding chemical mechanisms is to develop ways to reduce them, developing a qualitative and quantitative understanding of important pathways for reaction and the various stages of oxidation and identifying the main species and reactions important to this oxidation are important crucial steps towards mechanism reduction. ML offers powerful tools to achieve these goals.
Clustering methods have been used in a different context by Blurock and co-workers Blurock (2004), Blurock (2006), Tuner et al. (2005), Blurock et al. (2010) to identify the different mechanistic phases of fuel oxidation, which can be helpful in devising reduced chemistry schemes for these different phases. In Blurock (2004, 2006) clustering based on reaction sensitivity is used to identify the different phases of oxidation of aldehyde combustion and the ignition stages of ethanol, respectively. These studies exploit the presence of “similarity” between chemical states to identify the phases were the associated species are dominant. Identifying such phases can be important in several respects. For example, during the high-temperature oxidation of complex hydrocarbon fuels, identifying the two distinct phases of fuel pyrolysis and subsequent oxidation have enabled pathways to the development of hybrid chemistry approaches (Wang et al. 2018) (see Sect. 3.4). A less obvious distinction between the different phases of the low-temperature oxidation of the same complex fuels, can also reveal similar strategies to construct hybrid chemistry descriptions by identifying representative or marker species for each phase.
Insight to the physics from simulations or experiments can also provide a pathway towards generalizing observations, such as among different fuel functional groups. A recent study by Buras et al. (2020) used convolutional neural networks (CNNs) to construct correlations between the time scales of the low-temperature fuel spontaneous oxidation and chemical species profiles, primarily for OH, HO\(_2\), CH\(_2\)O and CO\(_2\) from plug-flow reactors (PFRs) and the first stage autoignition delay time (IDT). In their study, the authors relied on PFR simulation of 23 baseline fuels (18 pure fuels and 5 fuel blends) spanning a range of functional groups, including alkanes, alkenes/aromatics, oxygenates and fuel blends. They used existing mechanisms and perturbations of the parameters of these mechanisms to construct a wide database of species profiles. Emphasis on OH and HO\(_2\) is motivated by their role during the onset of spontaneous fuel oxidation. These intermediates exhibit different behaviors for two general fuels that show different propensities to form OH and HO\(_2\) during their oxidation cycle resulting in different correlations between the time scales for spontaneous fuel oxidation and the first stage IDT, one showing comparable values between the two quantities and another exhibiting a much slower first stage IDT. These different propensities are exhibited in the temporal profiles of these 2 intermediates as shown by Buras et al. (2020).

Reproduced with permission from Buras et al. (2020)
A schematic of the CNN architecture used by Buras et al. (2020) to construct correlations between profiles of OH, HO\(_2\), CH\(_2\)O and CO\(_2\) from PFR simulations of the low-temperature oxidation of a range of fuels and the first stage ignition delay times (IDTs).
CNNs are a different class of neural networks compared to the fully-connected multi-layer perceptrons shown in Figs. 1 and 2. They are specialized for multi-dimensional inputs, such as 2D images and include intermediate processing layers, convolutional and pooling layers, that are designed to dissect patterns in multi-dimensional and structured input data. Within the context of the work by Buras et al. (2020), the CNN architecture captures the different patterns with the profiles of the intermediates, OH and HO\(_2\).
Figure 3 shows a schematic of the CNN architecture used by Buras et al. (2020) to construct correlations between profiles of OH and HO\(_2\) from PFR simulations of the low-temperature oxidation for a range of fuels and the first stage ignition delay times (IDTs). The input data corresponds to 1D profiles of both OH and HO\(_2\); while the output (or target) is represented by the first stage IDT. By using a CNN, Buras et al. (2020) show that they can generate adequate predictions of the first stage IDT as shown in Fig. 4.

Reproduced with permission from Buras et al. (2020)
Correlations of predictions of first stage IDT from CNN to the simulated values using OH and HO\(_2\) profiles as inputs to the CNN. The inset also shows the histogram of the percent error indicating a relatively narrow distribution.
3.2 Chemical Reaction Neural Networks
One of the more recent developments in ML learning for chemical kinetics is the representation of reaction rates with prescribed inputs as the thermo-chemical state in terms of neural networks (Barwey and Raman 2021; Ji and Deng 2021). Such a representation enables the use of various tools to both accelerate the evaluation of reaction rates and develop skeletal descriptions of detailed mechanisms.
The rate of progress of a global reaction, \(\nu _A \textrm{A} + \nu _B \textrm{B} \rightarrow \nu _C \textrm{C} + \nu _D \textrm{D}\), can be expressed as:
where the rate constant k is expressed in terms of the Arrhenius law:
In this expression, A, b and \(E_a\) correspond to the frequency factor, the pre-exponential temperature power and the activation energy. This expression can be re-written as follows:
This expression can be formulated as an artificial neural network as illustrated in Fig. 5a for a single reaction and Fig. 5b for multi-step reactions. In Fig. 5a, the network emulates the structure of an ANN with no hidden layers. In this network, the input layer corresponds to the natural logs of the concentrations for A, B, C and D. The output layer corresponds to their rate of change, \(-\nu _A \ r\), \(-\nu _B \ r\), \(\nu _C \ r\) and \(\nu _D \ r\), respectively. The activation function is the exponential functions and the bias is \(\ln k\). The stoichiometric coefficients, \(\nu _A\), \(\nu _B\), \(\nu _C\) and \(\nu _D\) correspond to the weights of the network. The bias \(\ln k\), which represents the temperature-dependent rate constant, incorporates to the contributions of the rate parameters, A, b and \(E_a\). The illustrated CRNN can be generalized to accommodate more reactions and more species, as shown in Fig. 5b, thus enabling a neural network description of a set of global reactions to be optimized via ANNs. However, perhaps the main advantages of CRNN beyond the ability to frame reaction mechanisms within a neural network are the potential implications for such network for chemistry reduction and acceleration. Ji and Deng (2021) demonstrated a framework where the CRNN can be learned in the context of neural ODEs as discussed in Sect. 4 below.

An additional advantage of the CRNN is the potential for chemistry reduction via threshold pruning where input and output weights are clipped below a certain threshold. This pruning enhances the sparsity of the CRNN, which in turn can help speed up the evaluation of reaction rates. Ji and Deng (2021) showed that this pruning can still recover accurately the reaction rates in the CRNN by re-balancing the remaining weights.
A similar formulation was proposed by Barwey and Raman (2021). These authors also recast Arrhenius kinetics as a neural network using matrix-based formulations. By this process, the evaluation of the neural network can exploit specially optimized libraries for machine learning that are also optimized for use with graphical processing units (GPUs).
3.3 PCA-Based Chemistry Reduction and Other PCA Applications
As indicated earlier, PCA has been one of the earliest ML tools implemented for combustion chemistry. From the earlier work of Turány and co-workers (see for example (Vajda et al. 2006)) PCA was used to identify the most influential reactions in a mechanism through an eigen decomposition related to the sensitivity matrix. Their analysis is based on identifying the contributions to a “response function”:
which evaluates the cumulative contribution of the normalized deviations of perturbed kinetic model response parameters relative to the original non-perturbed kinetic model. Here, \(f_i\) can correspond to temperature, a measure of species concentrations, or both and other global parameters, such as flame speeds or extinction strain rates. \(\alpha _j\) is a reaction rate kinetic parameter, which is normally adopted as the rate constants for the reaction in a mechanism. Also, l and m in the sum correspond to the total number of analysis point (in space or time) and the number of target functions (e.g. species concentrations, temperatures). \(x_j\) corresponds to positions or times that involve all the samples in the calculation of Q.
PCA is implemented on the matrix \(\textbf{S}^\textbf{T} \textbf{S}\), where \(\textbf{S}\) is the matrix of normalized sensitivity coefficients whose component i, j can be expressed as \(\partial \ln f_i / \partial \ln \alpha _j\). An eigen-decomposition of the matrix yields a set of eigenvalues \(\lambda _i\) (ordered from high to lower magnitudes) and associated eigenvectors (which form an orthonormal set) and principal components (PCs), \(\mathbf{\phi }\), which can be expressed in terms of the kinetic parameters as:
where \(\boldsymbol{\psi }\) is the vector logarithmic parameters \(\psi _j = \ln \alpha _j\). The eigen-decomposition can be used to approximate the response function Q as follows (Vajda et al. 2006):
By ordering the eigenvalues, the PCs corresponding to the largest eigenvalues determine the influential part of the mechanism.

Illustration of the network architecture of an autoencoder
PCA can also be implemented within the context of a neural network using autoencoders. Figure 6 shows the architecture of an autoencoder with an input and an output layer and 3 hidden layers. The hidden layers are implemented with a decreasing number of neurons to a bottleneck layer, then an increasing number of neurons to the output. The dimensional of the output is identical to the input and the values of its neurons is designed to reproduce the corresponding values at the input layer. Therefore, the goal of an autoencoder is to reproduce the original data (at the input) by representing the data through a reduced dimension corresponding to the number of neurons in that hidden layer.
An autoencoder with one hidden layer, the bottleneck layer, a linear activation function and a penalty function that is the mean squared error (MSE) is designed to reproduce the PCA space from a prescribed input dimension to a dimension that corresponds to the number of neurons in the hidden layer. Additional steps are needed to reproduce the PCs from PCA analysis given that PCA also requires an orthonormal set of eigevenvectors for the PCs.
Recently, Zhang et al. (2021) proposed the use of autoencoders as a tool for chemistry reduction. These autoencoders exploit the dimensionality reduction at the bottleneck to construct a reduced description of chemistry. Given the inherent risk of extrapolation when the autoencoder attempts to access out-of-distribution (OOD) regions via extrapolation, Zhang et al. (2021) proposed the coupling of the autoencoder with either a deep ensemble (DE) method (Lakshminarayanan et al. 2017) or the so-called PI3NN method (Zhang et al. 2021). Within an autoencoder structure, the DE method accounts for a predicted mean (the predicted values) as well as the output variance to assess uncertainty (Lakshminarayanan et al. 2017). While in the PI3NN method, two additional neural networks are introduced to estimate the upper and lower bounds of the data reconstruction, again as a measure to assess the uncertainty in the autoencoder performance.

Reproduced with permission from Zhang et al. (2021)
Illustration of two OOD-aware autoencoder architectures with DE (left) and PI3NN (right). The input layer, \(\textbf{x}\) corresponds to the full chemistry description; while the bottleneck \(\textbf{z}\) represents the reduced chemical description. The autoencoder is designed to reproduce the input in the output; and the DE and PI3NN modifications attempt to assess the uncertainty of the predictions, especially when extrapolation is needed.
Figure 7 illustrates the two OOD-aware autoencoder configurations investigated by Zhang et al. (2021). The authors showed that by using these configurations, the number of input species is reduced from 12 to 2 at the bottleneck. This reduction can translate into a reduction in the number of transported scalars.
Finally, another implementation of PCA in combustion chemistry has been proposed by D’Alessio et al. (2020a, b). In their recent studies, they proposed an adaptive reduced chemistry scheme in which the composition space is partitioned into different clusters where appropriate and efficient reduced chemistry models can be implemented. The partitioning is implemented, instead of using a standard clustering approach such as K-Means or SOM, using local PCA (or LPCA) (Kambhatla and Leen 1997). The main difference between the use of LPCA vs K-Means, for example, is in the criteria established to partition the composition space. Instead of minimizing the Euclidean error between data of a given cluster and its centroid, the criteria is to miminize the reconstruction error of the PCA within a given cluster. D’Alessio et al. (2020b) showed that superior performance is established by adopting LPCA as part of the clustering algorithm instead of a hybrid clustering approach based on the coupling of self-organizing maps (SOMs) and K-Means in an unsteady laminar co-flow diffusion flame of methane in air. Within the context of a CFD simulation, LPCA is used as a classifier to determine the cluster to which a given cell state belongs. In each cluster, an a priori chemistry reduction is implemented using the training data, which in the studies of D’Alessio et al. (2020a, b) correspond to a series of unsteady 1D flames or data from 2D simulations of the same configuration, respectively.
3.4 Hybrid Chemistry Models and Implementation of ML Tools
The oxidation chemistry of a typical transportation fuel poses severe computational challenges for multi-dimensional reacting flow simulations. These challenges may be attributed primarily to the sheer size of associated chemical mechanisms when available. However, and oftentimes, the chemical kinetic data may not be available. While chemistry reduction strategies have been reasonably successful in overcoming the challenge of handling chemical complexity (Battin-Leclerc 2008; Turányi and Tomlin 2014), such strategies can only be used when reliable detailed mechanisms for the fuels of interest are available.
Experimental data-based chemistry reduction is one viable strategy for modeling the chemistry of complex fuels. Recently, the hybrid chemistry (HyChem) approach was proposed by Wang and co-workers Wang et al. (2018), Xu et al. (2018), Tao et al. (2018), Wang et al. (2018), Saggese et al. (2020), Xu et al. (2020), Xu and Wang (2021) as a chemistry reduction approach for the high-temperature oxidation of transportation fuels starting from time-series measurements of fuel fragments (and other relevant species) to capture the pyrolysis stage of these fuels. Such measurements can be achieved primarily using shock tubes and a variety of optical diagnostic techniques and sampling methods.
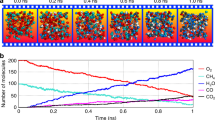
The approach is based on the premise that, at high temperatures, fuel oxidation undergoes: (1) a fast fuel pyrolysis step resulting in the formation of smaller fuel fragments, followed by (2) a longer oxidation step for these fragments. Figure 8 shows experimental observations by Davidson et al. (2011), which illustrate the 2 stages of n-dodecane oxidation through time-history measurements of the fuel, a fuel fragment, C\(_2\)H\(_4\), and oxidation species, OH, H\(_2\)O and CO\(_2\). The figure shows that the fuel is depleted in the first 30\(\upmu \textrm{s}\) and it is replaced by pyrolysis fragments, which eventually oxidize towards simpler hydrocarbons.

(Reproduced with permission from Davidson et al. (2011))
Species time-history measurements for n-dodecane oxidation. Initial mixture conditions: 1410 K, 2.3 atm, 457 ppm n-dodecane/O2/argon, \(\phi = 1\).
In HyChem, a hybrid chemistry model represented by a set of lumped fuel pyrolysis steps is augmented by foundational C\(_0\)–C\(_4\) chemistry for the fragments-oxidation. With experimental measurements of the key fragments, the stoichiometric coefficients and rate constants for the global reactions are determined through an optimization approach. The lumped reactions for the fuel pyrolysis is modeled using the following two reaction steps for a fuel C\(_m\)H\(_n\):
-
Unimolecular decomposition reaction
$$\begin{aligned} \begin{aligned} \textrm{C}_m \textrm{H}_n \rightarrow&e_d \left( \textrm{C}_2 \textrm{H}_4 + \lambda _3 \ \textrm{C}_3 \textrm{H}_6 + \lambda _4 \ \textrm{C}_4 \textrm{H}_8 \right) \\ + \,&b_d \left[ \chi \textrm{C}_6 \textrm{H}_6 + \left( 1-\chi \right) \right] + \alpha \ \textrm{H} + \left( 2 - \alpha \right) \ \textrm{CH}_3 \end{aligned} \end{aligned}$$(13) -
H-atom abstraction and \(\beta \)-scission reactions of fuel radicals
$$\begin{aligned} \begin{aligned} \textrm{C}_m \textrm{H}_n + \textrm{R} \rightarrow \,&\textrm{RH} + \gamma \ \textrm{CH}_4 + e_a \left( \textrm{C}_2 \textrm{H}_4 + \lambda _3 \ \textrm{C}_3\textrm{H}_6 + \lambda _4 \ \textrm{C}_4\textrm{H}_8 \right) \\ +\,&b_a \left[ \chi \textrm{C}_6 \textrm{H}_6 + \left( 1-\chi \right) \right] + \beta \ \textrm{H} + \left( 1 - \beta \right) \ \textrm{CH}_3 \end{aligned} \end{aligned}$$(14)
where R represents the following species: H, CH\(_3\), O, OH, O\(_2\) and HO\(_2\). In these reactions, \(\alpha \), \(\beta \), \(\lambda _3\), \(\lambda _4\) and \(\chi \) are the stoichiometric parameters that need to be determined for each fuel chemistry. More specifically, \(\alpha \) and \(\beta \) correspond to the number of H atoms per C\(_m\)H\(_n\) in the two reactions, respectively. The remaining parameters, \(e_d\), \(e_a\), \(b_d\) and \(b_a\) can be expressed in terms of the stoichiometric parameters using elemental conservation principles across each reaction (Wang et al. 2018).
The HyChem approach relies on the ability to measure some key fuel fragments, CH\(_4\), C\(_2\)H\(_4\) (in shock tubes), C\(_3\)H\(_6\), C\(_4\)H\(_8\) isomers, C\(_6\)H\(_6\) and C\(_7\)H\(_8\) (in flow reactors). Therefore, these fuel fragments represent much less complex species than the original fuel and their oxidation can be modeled using a simpler foundational chemistry model as the subsequent oxidation stage. More importantly, the fragments’ measurements can be used to determine the stoichiometric parameters and the rate constants of the lumped reactions needed to model the pyrolysis stage.
Hybrid chemistry approaches, such as the HyChem ML can play useful roles to formulate robust chemistry descriptions for complex fuels. In two recent studies, Ranade and Echekki (2019a, b) proposed an ANN-based implementation of HyChem. In a first step, a shallow regression ANN is implemented on the temporal species measurements to evaluate directly their rate of change, which directly measures their rate of reaction. In the second step, deep regression ANNs are trained to relate fragments’ concentrations to their rate of reaction. This network, as in the HyChem approach, is used to evaluate the fragments’ chemical source terms during the pyrolysis stage. Ranade and Echekki (2019b) showed that the procedure can be extended beyond the pyrolysis stage to enable the use of a simpler foundational chemistry.
More recently, Echekki and Alqahtani (2021) proposed a data-based hybrid chemistry approach to accelerate chemistry integration during the high-temperature oxidation of complex fuels. The approach is based on the ANN regression of representative species, which may or may not include the pyrolysis fragments, during the pyrolysis stage. These representative C\(_0\)–C\(_4\) species are determined using reactor simulation data and PCA on all species reaction rates. This PCA is used to determine the most important species to represent the evolution of the oxidation process. Beyond the pyrolysis stage, these species can be modeled with a foundational chemistry model like the remaining species.
Since the representative species are not tied to a particular list of fragments, the approach can be extended to the modeling of low-temperature oxidation where some of the initial intermediates are fuel-dependent. The work of Alqahtani (2020) demonstrated the feasibility of this extension to low-temperature fuel oxidation.
The approaches implemented in Ranade and Echekki (2019a, b), Echekki and Alqahtani (2021) or Alqahtani (2020) rely on ANN for the regression of the fragments or representative species in terms of the species concentrations. These studies suggest that the associated architectures of the ANN can be further simplified by using a subset of these species as inputs. This choice is motivated by the inherent correlations of the fragments/representative species and rely on the same motivation for using PCA in combustion modeling. However, ANNs may have limited interpretability unless they are implemented in the context of CRNN, as presented in Sect. 3.2.
The CRNNs (Ji and Deng 2021) offer an alternative optimization of the global reactions of the pyrolysis stage using the law of mass action and the Arrhenius form for the rate constants. Zanders et al. (2021) implemented a stochastic gradient descent (SGD) approach to optimize the lumped global reactions of pyrolysis starting with data of ignition delay times. Their approach was implemented within their Arrhenius.jl open-source software (Ji and Deng 2021) and by implementing the lumped reaction steps of pyrolysis within a CRNN. Their evaluation of the rate parameters of the lumped pyrolysis reactions yielded both an enhanced computational efficiency compared to approaches based on genetic algorithms and an improved predictions of IDT for ranges of temperature and equivalence ratios.
3.5 Extending Functional Groups for Kinetics Modeling
Functional group information has recently been used for the bottom-up development of chemical kinetic models. This approach was developed following the initial insight that AI models can predict combustion properties from several key functional group features of a fuel mixture. Recently, the team led by Zhang et al. advanced lumped fuel chemistry modeling approach using functional groups for mechanism development (FGMech) (Zhang and Sarathy 2021b; Zhang et al. 2021). They created a functional group-based approach, which can account for mixture variability and predict stoichiometric parameters of chemical reactions without the need for any tuning against experiments on the real fuel.
Figure 9 presents an overview of the functional group approach for kinetic model development. The effects of functional groups on the stoichiometric parameters and/or yields of key pyrolysis products were identified and quantified based on previous modeling of pure components (Zhang and Sarathy 2021a; Zhang et al. 2022; Zhang and Sarathy 2021c). A quantitative structure-yield relationship was developed by a multiple linear regression (MLR) model, which was used to predict the stoichiometric parameters and/or yields of key pyrolysis products based on ten input features (eight functional groups, molecular weight, and branching index). The approach was then extended to predict thermodynamic data, lumped reaction rate parameters and transport data based on the functional-group characterization of real fuels. FGMech is fundamentally different in that no parameters need to be tuned to match actual real-fuel pyrolysis/oxidation data, and all the model parameters were derived only from functional group data. It was shown that the FGMech approach can make good predictions on the reactivity of various aviation, gasoline, and diesel fuels (Zhang and Sarathy 2021b; Zhang et al. 2021).

Overview of the functional group approach for kinetic model development
3.6 Fuel Properties’ Prediction Using ML
The properties of fuels are carefully controlled to enable engines to operate at their optimal conditions and to ensure that fuels can be safely handled and stored. Important properties include those that can be easily determined based on simple thermo-physical models and linear blending (e.g., density, viscosity, heating values) to more complex properties that cannot be easily determined from physical modeling (e.g., octane number, cetane number, and sooting tendency). For the latter, ML techniques may be used to predict these fuel properties.
The first requirement for fuel property prediction is a suitable input descriptor for model training. Various molecular 1-3D representations such as SMILES (Simplified Molecular Input Line Entry Specification), InChI (International Chemical Identifier) or connectivity matrices can be used to obtain molecular descriptors for AI-based quantitative structure-property relationships (QSPR). Table 1 illustrates the use of different ML approaches to evaluate fuel properties.
Abdul Jameel et al. have demonstrated significant progress in the use of ANNs to predict various fuel properties including octane numbers (Jameel et al. 2018), derived cetane number (Jameel et al. 2016, 2021), flash point (Aljaman et al. 2022), and sooting indices (Jameel 2021). In general, they used functional groups derived from \(^1\)H NMR spectra of pure hydrocarbons and real fuel mixtures as input descriptors for model training, as illustrated in Fig. 10. The functional groups used include nine structural descriptors (paraffinic primary to tertiary carbons, olefinic, naphthenic, aromatic and ethanolic OH groups, molecular weight and branching index). Ibrahim and Farooq (2020, 2021) utilized the methodology proposed by Abdul Jameel et al. for fuel property (RON, MON, DCN, H/C ratio) prediction based on infrared (IR) absorption spectra rather than NMR shifts.

Conversion of NMR spectra to functional groups followed by training for ML model for property prediction
3.7 Transfer Learning for Reaction Chemistry
Chemical kinetic modelling is an indispensable tool for our understanding of the formation and composition of complex mixtures. These models are routinely used to study pollution, air quality, and combustion systems. Recommendations from kinetic models often help shape and guide environmental policies and future research directions. There are two essential data feeds for such models: species thermochemistry and rate coefficients of elementary reactions. Uncertainties in these feeds directly affect the predictive accuracy of chemical kinetic models. Historically, these data were measured experimentally and/or estimated from simple rules, such as group-additivity and structure-activity-relations. Ab-initio quantum chemistry based theoretical models have been developed over the years to calculate thermochemistry and reaction rate coefficients, and the accuracies of these calculations have been increasing steadily. These methods, however, require significant computational power and are challenging to apply to large molecular systems. In recent times, machine-learning based methods have attracted significant attention for the prediction of thermochemistry and reaction rate coefficients. In particular, inspired by the success of transfer learning approach in image processing, researchers have applied it in the domain of reaction chemistry. Transfer learning applies the knowledge (or model) learned in one task to another task. One of the benefits of transfer learning is that it can overcome the lack of large datasets, which are generally needed for machine learning algorithms.

Reproduced with permission from Grambow et al. (2019)
Transfer learning model architecture to learn molecular embedding and neural network parameter initialization for application to small datasets.
Grambow et al. (2019) trained three base models, one each for enthalpy of formation, entropy and heat capacity, on a large dataset (\(\approx \)130,000) generated from low-level (high uncertainty) theoretical calculations. These based models were then used as the starting models for the prediction of more accurate values of those thermochemistry properties by using a much smaller (<10,000) dataset of experimental values and high-accuracy theoretical calculations (see Fig. 11). Bhattacharjee and Vlachos (2020) implemented a ‘data fusion’ methodology to map thermo-chemical quantities, calculated at various levels of theory, to a higher level of theory. Zhong et al. (2022) overcame the challenge of small datasets by transferring knowledge among them for predictions with higher accuracy (see Fig. 12). The authors also compared their results with two other similar approaches, namely multitask learning and image-based transfer learning. Likewise, Han and Choi (2021) presented a framework of leveraging the learning from a large simulated database (with high uncertainty) to a small experimental database (with small uncertainty) for reliably predicting NMR (nuclear magnetic resonance) chemical shifts over a wide range of chemical space.
More recently, Ibrahim and Farooq (2022) showcased a temperature-dependent multi-target model with a custom-made Arrhenius loss applied to the AtmVOCkin reaction rate dataset. The Arrhenius loss dictates physically sound temperature-dependence which reduces overfitting, makes use of all available data in literature, and it outputs the three Arrhenius parameters which are compatible with modern automated chemical mechanism generator inputs. The graph-based D-MPNN was used for transfer learning from the publicly available QM9 dataset which stretches the applicability domain and supplements fixed molecular descriptors. Multi-target predictions were also implemented to enable cross-reaction learning which can enhance predictive capability for reactions with small datasets. Tuning was done using Bayesian optimization which gives robust/automatic predictions and a fair comparison among various models. The model was used to predict the three modified-Arrhenius parameters for the temperature-dependent reactions of OH, O\(_3\), NO\(_3\) and Cl with a wide range of hydrocarbons (see Fig. 13).

Reproduced with permission from Zhong et al. (2022)
Transfer learning approach for combining small datasets.

(Courtesey of Ibrahim and Farooq (2022))
Reaction rate prediction scheme (with toluene shown as a representative molecule).
4 Chemistry Integration and Acceleration
Chemistry integration represents a true bottleneck in combustion simulations involving both transport and chemistry. Measures to accelerate chemistry have adopted different strategies that are often combined with an initial step of chemistry reduction to global or skeletal mechanisms. Such strategies include chemistry tabulation, such as the use of in situ adaptive tabulation (ISAT) (Pope 1997), regression (such as ANN-based regression discussed in Sect. 2) and the piecewise reusable implementation of solution mapping (PRISM) (Tonse et al. 2003), adaptive chemistry, including dynamic approaches (see for example Liang et al. (2009), Continuo et al. (2011), Sun and Ju (2017) and D’Alessio et al. (2020a)), manifold-based methods, such as intrinsic low-dimensional manifolds (ILDM) (Maas and Pope 1992) and computational singular perturbation (CSP) methods (Lam and Goussis 1994). Chemistry acceleration primarily relies on operator splitting of the chemical source terms resulting in the solution for ordinary differential equations (ODEs).
In the last few years, there has been a growing excitement about the potential of neural ODE (NODE) solutions (Chen et al. 2018; Rackauckas et al. 2020). NODEs construct solutions for ODEs using neural networks and ODE solvers where model parameters (i.e. weights) are evaluated by a backward solution of the adjoint state. Implementing NODEs for combustion reaction presents numerous challenges associated with the inherent stiffness of the ODEs and the requirement for the simultaneous solutions of multiple ODEs for species and energy (Kim et al. 2021).
However, there have been several attempts in recent years to implement chemistry integration with neural networks. Owoyele and Pal (2022) proposed the so-called ChemNODE approach. The implementation of ChemNODE is summarized in Fig. 14. In ChemNODE, a stiff ODE solver is used to advance the solution of a thermo-chemical state at different time increments. These solutions constitute the observations that are used to train for the reaction rates implemented on the right column of the figure. These ANN-based reaction rates are integrated as well using the same ODE solver. The loss function to be minimized is the mean squared error comparing the solutions at the various observation points based on integration with the Arrhenius law and integration with the ANN-based reaction rates. Recognizing the difficulty of learning chemical sources within the proposed ChemNODE approach, Owoyele and Pal (2022) used a progressive approach for training these terms where each species is trained sequentially while the remaining species’ source terms are modeled with the solution from the ODE solver based on the Arrhenius law. Moreover, the optimization process involves the evaluation of derivatives of the neural network solution with respect to the network parameters, Owoyele and Pal (2022) adopted a forward-mode continuous sensitivity analysis using packages available for the Julia language.

Reproduced with permission from Owoyele and Pal (2022)
Illustration of the ChemNODE algorithm.
An alternative procedure for accelerating chemistry integration is proposed by Galassi et al. (2022). Their acceleration strategy is built on the use of CSP to remove the fast time scales from the chemistry integration. CSP usually requires the evaluation of a Jacobian matrix for the local chemical source terms and its eigen-decomposition. This decomposition is needed to identify the fast and slow timescales of the chemical system. By projecting the fast time scales out of the chemistry integration, the inherent stiffness of this chemical system is significantly reduced. However, there is an inherent cost to the evaluation of the Jacobian and the process of its eigen-decomposition, which scales strongly with the size of the chemical mechanism. Galassi et al. (2022) proposed the use of ANN regression as a cheaper surrogate to the local projection basis. Otherwise the CSP procedure shown in Fig. 15 is adopted. Figure 15 shows the general algorithm used to integrate chemistry within the proposed CSP-ANN framework. Given a current chemical state, the CSP basis is retrieved using ANN. The training for this basis is implemented offline, which was carried out in the Galassi et al. (2022) study using 0D ignition data for hydrogen-air mixtures. The procedure, then, involves an implementation of “radical correction” to account for the fast time scales, an explicit integration using the projection into the slow invariant manifold, then another radical correction. For the 9-species mechanism, 7 neural networks are trained in the Galassi et al. (2022) study. They each feature 2 hidden layers with 128 neurons each in each layer.

Reproduced with permission from Galassi et al. (2022)
Illustration of the CSP-ANN algorithm.
Zhang et al. (2021) proposed a different scheme for chemistry integration, which is based on training a deep neural network (DNN) to project a solution of the thermo-chemical state vector at a given time (i.e. the input) to the corresponding solution after a small time increment (i.e. the output). Figure 16 illustrates the structure of the DNN, which was implemented for a dimethyl ether (DME) mechanism with 54 species. The input solution at a given time includes 56 neurons for the species, pressure and temperature. The DNN features two independent, fully-connected branches for the low- and high-temperature oxidation for DME. Each branch has 3 hidden layers with 1600, 400 and 400 neurons. The output corresponds to the projection of the solution at a later time with 56 neurons in the output layer. The approach adopted by Zhang et al. (2021) is very reminiscent of the ISAT approach (Pope 1997), except for relying on DNNs to project solutions instead of a tree-based storage and tabulation.

Reproduced with permission from Zhang et al. (2021)
Illustration of the deep neural network for DLODE.
5 Conclusions
In this chapter, we have illustrated a number of applications of ML tools in combustion chemistry. These applications span the scopes of understanding, the reduction and the acceleration of chemistry in combustion applications. Based on the material presented, we anticipate important advances in the following areas:
-
The development of experiment-based HyChem-style mechanisms for a broad range of fuels that also rely on rules extracted for fuels of the same functional group.
-
The implementation of novel ML tools for chemistry reduction either by developing skeletal mechanisms, global mechanisms or hybrid chemistry models combining empirical global steps coupled with foundational chemistry.
-
The development of chemistry acceleration schemes that exploit dimensionality reduction of the composition space and features of stiffness removal.
References
Al Ibrahim E, Farooq A (2020) Octane prediction from infrared spectroscopic data. Energy Fuels 34(1):817–826
Al Ibrahim E, Farooq A (2021) Prediction of the derived cetane number and carbon/hydrogen ratio from infrared spectroscopic data. Energy Fuels 35:8141–8152
Al Ibrahim E, Farooq A (2022) A transfer learning approach to multi-target temperature-dependent reaction rate prediction. Submitted
Aljaman B, Ahmed U, Zahid U, Reddye VM, Sarathy SM, Jameel AGA (2022) A comprehensive neural network model for predicting flash point of oxygenated fuels using a functional group approach. Fuel 317:123428
Alqahtani SSH (2020) Machine learning methods for chemistry reduction in combustion. PhD thesis, North Carolina State University
Barwey S, Raman V (2021) A neural network-inspired matrix formulation of chemical kinetics for acceleration on gpus. Energies 14(9)
Battin-Leclerc F (2008) Detailed chemical kinetic models for the low-temperature combustion of hydrocarbons with application to gasoline and diesel fuel surrogates. Prog Energy Combust Sci 34:40–498
Bhattacharjee H, Vlachos DG (2020) Thermochemical data fusion using graph representation learning. J Chem Info Model 60:4673–4683
Blasco JA, Fueyo N, Dopazo C, Ballester J (1998) Modelling the temporal evolution of a reduced combustion chemical system with an artificial neural network. Combust Flame 113:38–52
Blasco JA, Fueyo N, Larroya JC, Dopazo C, Chen J-Y (1999) A single-step time-integrator of a methane-air chemical system using artificial neural networks. Comput Chem Eng 23:1127–1133
Blasco JA, Fueyo N, Dopazo C, Chen J-Y (2000) A self-organizing-map approach to chemistry representation in combustion applications. Combust Theo Model 4:61–76
Blurock ES (2004) Characterizing complex reaction mechanisms using machine learning clustering techniques. Int J Chem Kin 36:107–118
Blurock ES (2006) Automatic characterization of ignition processes with machine learning clustering techniques. Int J Chem Kin 38:621–633
Blurock ES, Tuner M, Mauss F (2010) Phase optimized skeletal mechanisms for engine simulations. Combust Theo Model 14:295–313
Buras ZJ, Safta C, Zádor J, Sheps L (2020) Simulated production of OH, HO$_2$, CH$_2$O, and CO$_2$ during dilute fuel oxidation can predict 1st-stage ignition delays. Combust Flame 216:472–484
Chatzopoulos AK, Rigopoulos S (2013) A chemistry tabulation approach via rate-controlled constrained equilibrium (RCCE) and artificial neural networks (ANNs), with application to turbulent non-premixed CH$_4$/H$_2$/N$_2$ flames. Proc Combust Inst 34:1465–1473
Chen RT, Rubanova Y, Bettencourt J, Duvenaud DK (2018) Neural ordinary differential equations. Adv Neural Info Sys 6571–6583
Chen J-Y, Blasco JA, Fueyo N, Dopazo C (2000) An economical strategy for storage of chemical kinetics: fitting in situ adaptive tabulation with artificial neural networks. Proc Combust Inst 28:115–121
Christo FC, Masri AR, Nebot EM (1996) Artificial neural network implementation of chemistry with PDF simulation of H$_2$/CO$_2$ flames. Combust Flame 106:406–427
Christos FC, Masri AR, Nebot EM, Turanyi T (1995) Utilising artificial neural network and repro-modelling in turbulent combustion. In: 1995 IEEE international conference on neural networks proceedings, pp 911–916
Continuo F, Jeanmart H, Lucchini T, D’Errico G (2011) Coupling of in situ adaptive tabulation and dynamic adaptive chemistry: an effective method for solving combustion in engine simulations. Proc Combust Inst 33:3057–3064
D’Alessio G, Cuoci A, Aversano G, Bracconi M, Stagni A, Parente A (2020a) Impact of the partitioning method on multidimensional adaptive-chemistry simulations. Energies 13
D’Alessio G, Parente A, Stagni A, Cuoci A (2020b) Adaptive chemistry via pre-partitioning of composition space and mechanism reduction. Combust Flame 211:68–82
Davidson DF, Hong Z, Pilla G, Farooq A, Cook R, Hanson RK (2011) Multi-species time-history measurements during n-dodecane oxidation behind reflected shock waves. Proc Combust Inst 33:151–157
Echekki T, Alqahtani S (2021) A data-based hybrid model for complex fuel chemistry acceleration at high temperatures. Combust Flame 223:142–152
Flemming F, Sadiki A, Janicka J (2005) LES using artificial neural networks for chemistry representation. Prog Comput Fluid Dyn 5:375–385
Franke LLC, Chatzopoulos AK, Rigopoulos S (2017) Tabulation of combustion chemistry via artificial neural networks (ANNs): methodology and application to LES-PDF simulation of Sydney flame L. Combust Flame 185:245–260
Galassi RM, Ciottoli PP, Valorani M, Im HG (2022) An adaptive time-integration scheme for stiff chemistry based on computational singular perturbation and artificial neural networks. J Comput Phys
Grambow CA, Li Y-P, Green WH (2019) Accurate thermochemistry with small data sets: a bond additivity correction and transfer learning approach. J Phys Chem A 123(27):5826–5835
Han H, Choi S (2021) Transfer learning from simulation to experimental data: NMR chemical shift predictions. J Phys Chem Lett 12:3662–3668
Hutter F, Kotthoff L, Vanschoren J (2019) Automated machine learning methods, systems, challenges. In: Hutter F, Kotthoff L, Vanschoren J (eds) Automated machine learning methods, systems, challenges. Springer Series on Challenges in Machine Learning
Ihme M (2010) Topological optimization of artificial neural networks using a pattern search method. NOVA Science Inc., USA, pp 323–343
Ihme M, Marsden AL, Pitsch H (2008) Generation of optimal artificial neural networks using a pattern search algorithm: application to approximation of chemical systems. Neural Comput 20:573–601
Ihme M, Schmidt C, Pitsch H (2009) Optimal artificial neural networks and tabulation methods for chemistry representation in LES of a bluff-body swirl-stabilized flame. Proc Combust Inst 32:1527
Ilies BD, Khandavilli M, Li Y, Kukkadapu G, Wagnon SW, Jameel AGA, Sarathy SM (2021) Probing the chemical kinetics of minimalist functional group gasoline surrogates. Energy Fuels 35(4):3315–3332
Jameel AGA (2021) Predicting sooting propensity of oxygenated fuels using artificial neural networks. Proc 9
Jameel AGA, van Oudenhoven VCO, Naser N, Emwas AH, Gao X, Sarathy SM (2021) Predicting ignition quality of oxygenated fuels using nuclear magnetic resonance spectroscopy and artificial neural networks. SAE Int J Fuels Lubr
Jameel AGA, Naser N, Emwas A-H, Dooley S, Sarathy SM (2016) Predicting fuel ignition quality using h-1 NMR spectroscopy and multiple linear regression. Energy Fuels 30(11):9819–9835
Jameel AGA, Van Oudenhoven VCO, Emwas A-H, Sarathy SM (2018) Predicting octane number using nuclear magnetic resonance spectroscopy and artificial neural networks. Energy Fuels 32(5):6309–6329
Ji W, Deng S (2021) Autonomous discovery of unknown reaction pathways from data by chemical reaction neural networks. J Phys Chem A 125:1082–1092
Ji W, Qiu W, Shi Z, Pan S, Deng S (2021) Stiff-pinn: physics-informed neural network for stiff chemical kinetics. J Phys Chem A 125:8098–8106
Ji W, Deng S (2021) Arrhenius.jl: a differentiable combustion simulation package
Ji W, Zanders J, Park J-W, Deng S (2021) Machine learning approaches to learn HyChem models. In: Proceedings of the ASME 2021 international combustion conference, number paper ICEF2021/69657
Kambhatla N, Leen TK (1997) Dimension reduction by local principal component analysis. Neural Comput 9:1493–1516
Kim S, Ji W, Deng S, Ma Y, Rackauckas C (2021) Stiff neural ordinary differential equations. Chaos 31(093122)
Kohonen T (2013) Essentials of the self-organizing map. Neural Netw 37:52–65
Lakshminarayanan B, Pritzelnd A, Blundell C (2017) Simple and scalable predictive uncertainty estimation using deep ensembles. Proc Adv Neural Inf Process Syst 6402–6413
Lam SH, Goussis DA (1994) The CSP method for simplifying kinetics. Int J Chem Kin 26:461–486
Liang L, Stevens JG, Raman S, Farrell JT (2009) The use of dynamic adaptive chemistry in combustion simulation of gasoline surrogate fuels. Combust Flame 156:1493–1502
Lu TF, Law CK (2005) A directed relation graph method for mechanism reduction. Proc Combust Inst 30:1333–1341
Maas U, Pope SB (1992) Simplifying chemical kinetics: intrinsic low-dimensional manifolds in composition space. Combust Flame 88:239–264
Owoyele O, Kundu P, Ameen MM, Echekki T, Som S (2020) Application of deep artificial neural networks to multi-dimensional flamelet libraries and spray flames. Int J Engine Res 21(1, SI):151–168
Owoyele O, Pal P (2022) ChemNODE: a neural ordinary differential equations framework for efficient chemical kinetic solvers. Energy AI 7
Pope SB (1997) Computationally efficient implementation of combustion chemistry using in situ adaptive tabulation. Combust Sci Tech 1:41–63
Rackauckas C, Ma Y, Martensen J, Warner C, Zubov K, Supekar R, Skinner D, Ramadhan A, Edelman A (2020) Universal differential equations for scientific machine learning. arXiv:2001.04385
Ranade R, Echekki T (2019a) A framework for data-based turbulent combustion closure: a priori validation. Combust Flame 206:490–505
Ranade R, Echekki T (2019b) A framework for data-based turbulent combustion closure: a posteriori validation. Combust Flame 210:279–291
Saggese C, Wan K, Xu R, Tao Y, Bowman CT, Park JW, Lu T, Wang H (2020) A physics-based approach to modeling real-fuel combustion chemistry—V. NO$_x$ formation from a typical jet A. Combust Flame 212:270–278
Sen BA, Menon S (2010a) Linear eddy mixing based tabulation and artificial neural networks for large eddy simulations of turbulent flames. Combust Flame 157:62–74
Sen BA, Menon S (2010b) Large eddy simulation of extinction and reignition with artificial neural networks based chemical kinetics. Combust Flame 157:566–578
Sharma AJ, Johnson RF, Kessler DA, Moses A (2020) Deep learning for scalable chemical kinetics. In: AIAA scitech 2020 forum, number AIAA paper 2020-0181
Sinaei P, Tabejamaat S (2017) Large eddy simulation of methane diffusion jet flame with representation of chemical kinetics using artificial neural network. Proc Inst Mech Eng Part E: J Process Mech Eng 231:147–163
Sun W, Ju Y (2017) TA multi-timescale and correlated dynamic adaptive chemistry and transport (CO-DACT) method for computationally efficient modeling of jet fuel combustion with detailed chemistry and transport. Combust Flame 184:297–311
Tao Y, Xu R, Wang K, Shao J, Johnson SE, Movaghar A, Han X, Park JW, Lu T, Brezinsky K, Egolfopoulos FN, Davidson DF, Hanson RK, Bowman CT, Wang H (2018) A physics-based approach to modeling real-fuel combustion chemistry—III: reaction kinetic model of JP10. Combust Flame 198:466–476
Tonse SR, Moriarty NW, Frenklach M, Brown NJ (2003) Computational economy improvements in PRISM. Int J Chem Kin 35:438–452
Torczon V (1997) On the convergence of pattern search algorithms. SIAM J Opt 7(1):1–25
Tuner M, Blurock ES, Mauss F (2005) Phase optimized skeletal mechanisms in a stochastic reactor model for engine simulation. SAE, USA
Turányi T, Tomlin AS (2014) Reduction of reaction mechanisms. Springer, pp 183–312
Vajda S, Valko P, Turányi T (2006) Principal component analysis of kinetic models. Int J Chem Kin 17:55–81
Wan K, Barnaud C, Vervisch L, Domingo P (2020) Chemistry reduction using machine learning trained from non-premixed micro-mixing modeling: application to DNS of a syngas turbulent oxy-flame with side-wall effects. Combust Flame 220:119–129
Wang K, Xu R, Parise T, Shao J, Movaghar A, Lee DJ, Park JW, Gao Y, Lu T, Egolfopoulos FN, Davidson DF, Hanson RK, Bowman CT, Wang H (2018) A physics based approach to modeling real-fuel combustion chemistry—IV: HyChem modeling of combustion kinetics of a bio-derived jet fuel and its blends with a conventional jet A. Combust Flame 198:477–489
Wang H, Xu R, Wang K, Bowman CT, Hanson RK, Davidson DF, Brezinsky K, Egolfopoulos FN (2018) A physics-based approach to modeling real-fuel combustion chemistry—I: evidence from experiments, and thermodynamic, chemical kinetic and statistical considerations. Combust Flame 193:502–519
Xu R, Saggese C, Lawson R, Movaghar A, Parise T, Shao J, Choudhary R, Park JW, Lu T, Hanson RK, Davidson DF, Egolfopoulos FN, Aradi A, Prakash A, Raja V, Mohan R, Cracknell R, Wang H (2020) A physics-based approach to modeling real-fuel combustion chemistry—VI: predictive kinetic models of gasoline fuels. Combust Flame 220:475–487
Xu R, Wang H (2021) A physics-based approach to modeling real-fuel combustion chemistry—VII: relationship between speciation measurement and reaction model accuracy. Combust Flame 224(SI):126–135
Xu K, Wang R, Banerjee S, Shao J, Parise T, Zhu Y, Wang S, Movaghar A, Lee DJ, Zhao R, Han X, Gao Y, Lu T, Brezinsky K, Egolfopoulos FN, Davidson DF, Hanson RK, Bowman CT, Wang H (2018) A physics-based approach to modeling real-fuel combustion chemistry—II: reaction kinetic models of jet and rocket fuels. Combust Flame 193:520–537
Zhang X, Sarathy SM (2021a) High-temperature pyrolysis and combustion of C$_5$–C$_{19}$ fatty acid methyl esters (FAMEs): a lumped kinetic modeling study. Energy Fuels 35(23):19553–19567
Zhang X, Sarathy SM (2021b) A functional-group-based approach to modeling real-fuel combustion chemistry—II: kinetic model construction and validation. Combust Flame 227:510–525
Zhang X, Sarathy SM (2021c) A lumped kinetic model for high-temperature pyrolysis and combustion of 50 surrogate fuel components and their mixtures. Fuel 286
Zhang X, Yalamanchi KK, Sarathy SM (2021) A functional-group-based approach to modeling real-fuel combustion chemistry—I: prediction of stoichiometric parameters for lumped pyrolysis reactions. Combust Flame 227:497–509
Zhang P, Liu S, Lu D, Sankaran R, Zhang G (2021) An out-of-distribution-aware autoencoder model for reduced chemical kinetics. Disc Contin Dyn Syst—Series S
Zhang P, Liu S, Lu D, Zhang G, Sankaran R (2021) A prediction interval method for uncertainty quantification of regression models. In: Conference: ninth international conference on learning representations (ICLR), Virtual, Austria—5/7/2021
Zhang X, Li W, Xu Q, Zhang Y, Jing Y, Wang Z, Sarathy SM (2022) A decoupled modeling approach and experimental measurements for pyrolysis of C$_6$–C$_{10}$ saturated fatty acid methyl esters (FAMEs). Combust Flame, page in press
Zhang T, Zhang Y, E W, Ju Y (2021) DLODE: a deep learning-based ode solver for chemical kinetics (AIAA paper 2021-1139)
Zhong S, Zhang Y, Zhang H (2022) Machine learning-assisted QSAR models on contaminant reactivity toward four oxidants: combining small data sets and knowledge transfer. Env Sci Tech 56:681–692
Acknowledgements
The authors would like to acknowledge the support of King Abdullah University of Science and Technology under grant: 4351-CRG9.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2023 The Author(s)
About this chapter

Cite this chapter
Echekki, T., Farooq, A., Ihme, M., Sarathy, S.M. (2023). Machine Learning for Combustion Chemistry. In: Swaminathan, N., Parente, A. (eds) Machine Learning and Its Application to Reacting Flows. Lecture Notes in Energy, vol 44. Springer, Cham. https://doi.org/10.1007/978-3-031-16248-0_5
Download citation
DOI: https://doi.org/10.1007/978-3-031-16248-0_5
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-16247-3
Online ISBN: 978-3-031-16248-0
eBook Packages: EnergyEnergy (R0)