
Abstract
A Clinical Trials Centre (CTC) is an institution that unites experts and expertise for successful designing, conducting and publishing of clinical trials. It provides competence and the infrastructure required for conducting clinical trials in compliance with international and national regulations, guidelines and standards, ICH GCP (good clinical practice) and the Declaration of Helsinki. Mostly, the CTCs are institutions of the Medical Faculty of the University or the University Hospitals, sometimes both. In Germany, after passing an audit providing reassurance of compliance with required standards, a Clinical Trials Centre is invited to join the Network of Coordinating Centers for Clinical Trials (KKS-Network). Challenges in establishing these standards are diverse and start at the underlying IT (information technology) structure that might not be suitable for providing validated data systems and continue in generating awareness for the need of centralised structures for being compliant with regulations and guidance in conducting high-quality clinical trials.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
- Clinical trial
- Clinical Trials Centre
- Investigator-initiated trial
- Good clinical practice
- Quality management
- Pharmacovigilance
- Sponsor
Challenge: IT Systems
One of the main challenges of the CTC Ulm as joint institution of the Medical Faculty and the University Hospital Ulm is the qualitative and quantitative development of competencies with regard to planning, organisation and conduct of clinical trials to improve translational, patient-oriented research and thus increase study activities according to applicable laws and regulations.

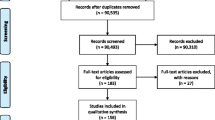
Review process and numbers of EDC systems screened for suitability for being used within an academic CTC. The figure illustrates the stepwise selection and exclusion process for the identification of an EDC system meeting the requirements (a) to be validated, (b) providing hosting in the EU according to the GDPR and (c) providing eCRFs
Keys in performing clinical trials are validated, clinical databases that capture, transfer and store data correctly and should comply with current regulatory standards EMA (2020).
Due to the fact that patient’s data within a hospital is stored in a secure environment, the data generated in clinical trials also should be stored within this central, protected system. However, the common patient-centred hospital IT system is neither equipped nor prepared for hosting clinical trial data and centrally based data management structures. A dedicated IT team who is trained in clinical trial IT structure requirements and allocated time resource is usually not available. Therefore, the CTC chose relevant clinical trial-related database structures to be hosted externally, which is favourable with regard to the pharmacovigilance database. However, external hosting is not the best option for other relevant data capture systems as the clinical study database (EDC, electronic data capture) due to the high costs of external hosting services and limited flexibility. Therefore, a solution might be to choose a ready-made system including the service of free system installation and minimal regular update and maintenance challenges to the existing IT system.
After identifying about 17 EDC systems, a short review revealed four systems that were not supported anymore and therefore could be excluded. From the remaining 13 systems, 6 were not validated and therefore could be excluded as not providing the needed validation requirements for investigational medicinal product clinical trials. The remaining seven systems were validated EDC systems, but only six of those offered servers within the European Union (EU), which was considered a prerequisite for complying with the legal requirements of the EU General Data Protection Regulation (GDPR). In addition, only four of those six EDC systems offered the possibility of in-house programming of eCRF (electronic case report form) (Fig. 1). The companies providing those remaining EDC systems were invited to present the systems to the university clinical research teams within the Clinical Trial Management Meeting (see section “Governance”). Discussions on costs, hosting systems, flexibility, national user support and availability, as well as familiarity within the academic community, finally lead to a decision for one of these systems.
Establishing a Central Quality Management System
Quality (according to ISO 9000, https://www.iso.org/) is a set of characteristics that a product must have to satisfy needs and expectations of the customer. Indeed, the output (product) from a clinical trial is an information that provides an answer to a scientific question and thus underlies the same standards as any other product.
According to international law and guidelines, one of those being the ICH GCP E6 (R2) (good clinical practice) guideline (https://ichgcp.net/), the sponsor should implement a system to manage quality throughout all stages of the trial process to ensure subject protection and reliability of trial results.
A quality management system (QMS) therefore comprises the design of efficient clinical trial tools and procedures for data collection and processing as well as the control thereof within a quality assurance system.
Quality management consists of quality assurance (QA), comprising all those planned and systematic actions that are established to ensure that the trial is performed and the data are generated, documented, recorded and reported in compliance with GCP and regulatory requirements as SOPs (standard operating procedures) and quality control (QC). QC comprises the operational techniques and activities undertaken within the quality assurance system to verify that the requirements for quality of the trial-related activities have been fulfilled as, for example, monitoring. As within an academic institution as a university and university hospital, the individual clinical trial facilities within the diverse medical specialities generate diverse QMS or parts of it to best comply with required regulation and guidelines. Those decentralised, individual systems provide different quality guidance within SOPs, working instructions as well as QC measures. Therefore, clinical trials, originating from the same sponsor, the University Hospital of Ulm, might range from poor to fair overall quality. To overcome the differing qualities and to guarantee a high-quality standard according to applicable laws and regulations, a centralised QMS for all trials sponsored by the University Hospital Ulm should be implemented, trained and controlled. With a centralised system, insufficient quality within clinical trials and consequential potential harm to patients is more likely ensured to be avoided, and study data integrity is maintained.
The first step within this process is to define and draft overarching, general, central SOPs for clinical trials. This happens with the provision that the major steps are agreed upon by stakeholders, and processes and templates are generated. These SOPs have to be thoroughly taught and made available either through a central platform for clinical trials or a document management system. To assure compliance with these SOPs and general guidance and regulation, and thus adequate quality, major documents of each clinical trial have to undergo a compliance check process within the CTC before sponsor acceptance and signature. In implementing a compliance check that is independent from the clinical trial facility planning the trial, central quality aspects can be implemented for all clinical trials, and GCP compliance can be confirmed. Importantly, the sponsor signature will not be provided without recommendation for signature from the CTC compliance check process, and therefore the compliance check is mandatory for all clinical trials performed at the University Hospital Ulm. Finally, this process leads to a reasonably well-adjusted good quality of all clinical trials performed at the University Hospital Ulm as sponsor of clinical trials. Additionally, the mandatory compliance check task provides a sponsor oversight over existing studies. This oversight is not only requested according to regulation but also needed within other specialities of the Clinical Trials Centre as pharmacovigilance to comply with, for example, cross-reporting obligations.
However, the implementation of a compliance check has to be considered as a first step within the scope of a central QMS for clinical trials. Further quality measures have to be implemented centrally, and compliance should be audited to further improve and control the quality management of clinical trials.
Establishing a Central Pharmacovigilance System
All clinical trials and medical device studies taking place at the University Hospital Ulm (UKU) are subject to the statutory or other provisions to collect, evaluate and report adverse events that occur during the study. SUSARs (suspected unexpected serious adverse reactions) are to be reported electronically to the European EudraVigilance Clinical Trial Module, to ethics committee and to investigators; additionally, a continuous benefit-risk evaluation is mandatory in clinical trials.
As pharmacovigilance requires prompt action and demanding processes in short time frames as well as specially trained pharmacovigilance experts with a proof of work experience in their field, this task was found to be outsourced to CROs (clinical research organisations) at the UKU. A “responsible person for EudraVigilance” must be announced to the EMA (European Medicines Agency), who as a named person represents the sponsor at the European agency and is the point of contact for all safety-related issues for regulatory bodies.
Outsourcing implies several disadvantages in general such as high costs, obscure and complicated processes, the need of controlling and auditing, allocating resources and thus generating redundant processes.
As the Clinical Trials Centre Ulm was faced with the fact that the CRO providing the responsible person function and SUSAR reporting for all IITs (investigator-initiated trials) of the University Hospital Ulm was insolvent, immediate action was necessary. The CTC was notified that services would be stopped within 4 weeks of notification.
Due to this short timeline, a transfer to another CRO including audit, contract negotiations, database and process transfer was not feasible. To guarantee continued safety management for the clinical trials concerned, the CTC decided to assume the responsible person function and SUSAR reporting responsibilities from the CRO. Fortunately, the CTC staff was already trained and listed within the EMA database, and necessary certificates were available.
To guarantee a smooth transition, a central SUSAR reporting email address within UKU was established. Important for choosing this email address was the need to guarantee that external emails, including relevant attachments, will be received in a secure environment (encrypted connection) without being blocked or refuted. In addition, it was indispensable that the CTC staff will be able to access this mailbox 24/7 also from external locations to assure timely processing in accordance with the business continuity plan in case of emergency situations. Additionally, it was guaranteed that this safety email address stored all email conversations and that there is no possibility of emails being deleted or moved by any CTC staff. A regular backup procedure was confirmed to be in place. After successful testing of the email address as well as fax, a process of regular mailbox check was established: The responsible person, deputy or delegates will make sure to regularly check the mailbox for SUSARs according to a predefined schedule, indicating responsibility and backup. It is expected that SUSAR notifications will be sent and received mainly via email, and fax is expected as a fallback solution. Fax will be checked on a daily basis during usual working hours.
After establishing the communication structure at the CTC for SUSAR cases, all stakeholders performing clinical trials had to be identified and notified, and relevant documents of studies concerned had to be amended. All relevant documents (safety management plans, protocols and others) were reviewed for the changes to be implemented due to the pharmacovigilance responsibility transfer from the CRO to the CTC Ulm. Stakeholders were notified by telephone and by email within predefined timelines, and the relevant documents were identified, adapted and signed. Notification dates and acknowledgement dates were tracked within a “CRO-PhV Transfer UKU” table.
To take action according to a predefined strategy and schedule, the transfer modalities and timelines were all documented within a “Transfer Plan.” After successful transfer, this “Transfer Plan” was integrated within a “Transfer Document”, documenting the transfer and closing the process by signature.
Following this structured approach of process and data transfer, delays or inconsistencies were not noted after the day the system was switched from the CRO to the UKU process, and thus a smooth and successful transition was performed.
By establishing a pharmacovigilance system on-site at the CTC Ulm, streamlined and transparent processes with an adequate price-performance ratio can be set up centrally. Lengthy negotiations with third-party providers are no longer necessary, as are the sometimes complex processes of control and interaction. Pharmacovigilance processes are audit- and inspection-relevant and can be simplified, made transparent and optimised for internal procedures through central SOPs, so that compliance with regulatory requirements is made easier for all those involved. Another advantage is the continuous availability of the expertise on-site, as well as the access to and overview of the safety data at any time, thus guaranteeing the regular risk-benefit assessments required by law.
The sponsorship obligations with regard to pharmacovigilance in IITs are regulated by law. As a sponsor of IITs, the UKU is legally obliged to put in place pharmacovigilance arrangements as sending safety-related information (SUSARs) to the (European/responsible) authority (electronic notification to the EMA (European Medicines Agency)), ethics committee and participating investigators and, if necessary, to data safety monitoring boards (DSMBs)) and marketing authorisation holders (MAHs). The SUSAR reporting to the EMA has to take place centrally via the EudraVigilance database. Pharmacovigilance can be considered a central task per se as the legislation clarifies the ultimate responsibility being with the sponsor of the clinical trial. Therefore, outsourcing to a CRO has different implications as on the one hand being costly, and cost considerations are always high priority within the public sector. On the other hand, due to the sponsor obligations as outlined above, extensive oversight mechanism of third-party providers would have to be established, which would lead to resource allocation at the sponsor site. After review of existing processes with CROs, we found extensive, intransparent, time-consuming and error-prone processes, resulting in potential incompliance with pharmacovigilance legal and regulatory requirements. In accordance with Dinnett et al. (2013), we concluded that without a centralised pharmacovigilance system, the pharmacovigilance responsibilities of the sponsor are hardly to be adequately fulfilled.
Therefore, in order to meet the legal requirements for drug safety in the studies initiated at the UKU (IIT), as well as to provide the qualitative requirements to guarantee a high level of patient safety and regulatory compliance, streamlined processes and development of know-how and cross-departmental specialist expertise in pharmacovigilance locally, it was decided to implement a centralised pharmacovigilance system within the UKU. To meet these requirements, a GCP-compliant pharmacovigilance database (PhV-DB), and a pharmacovigilance quality management system (PhV QMS), including training of its management has to be implemented.
The refinancing of the pharmacovigilance system to be established at the CTC takes place through an internally defined scale of fees and represents a cost-adapted solution to the situation of self-initiated medical research in university medicine.
The goal of providing a pharmacovigilance system with a high-quality pharmacovigilance database that meets current regulatory requirements, embedded in a pharmacovigilance QMS for self-initiated studies at the UKU, represents a major local advantage for medical research at the University of Ulm (Table 1).
With regard to IT (information technology) resource limitations as well as limited knowledge in validation procedures, an in-house solution with a self-developed pharmacovigilance DB was found to be unfeasible due to the extensive regulatory requirements of the pharmacovigilance database. Therefore, it was decided to approach PhV database vendors for feasible solutions.
Before screening PhV database vendors, intensive communications and discussion with the stakeholders of the individual study centres were performed. Information about the current pharmacovigilance solutions (status quo) and processes was obtained. A needs analysis as well as a query of needed capacities regarding the provision of study-specific pharmacovigilance services at the UKU was performed. Analysis of the findings revealed the volume of cases and required processes so that vendors of two PhV databases were approached and compared with regard to price, support services, follow-up costs and expenses (e.g. required in-house validation), server location, validation (GAMP-5), MedDRA implementation, guaranteed availability time, gateway function to EMA, etc. Other CTCs using these databases were interviewed and pros and cons opposed. Finally, a decision for one of the systems was made during a CTC board meeting.
Subsequently, the CTC Ulm began with the creation and implementation of a PhV QMS (SOPs, manuals, conventions) and subsequent training of the employees of the study centres as well as information of the respective clinics, institutes and project managers.
The pharmacovigilance system consisting of the pharmacovigilance database and the PhV QMS, which in addition to SOPs, manuals, conventions and other documents also includes the necessary training for internal employees, project managers and collaborators as well as study participants, is being developed and continuously adapted by the CTC Ulm.
Objectives of the implementation of a central pharmacovigilance system at the CTC are the following:
-
1.
Support of medical research and relief of the study centres by creating ICSRs (individual case safety reports) and other necessary reports (including DSURs/SAE listings) on adverse events from clinical trials.
-
2.
Provision of a high-quality pharmacovigilance standard through a pharmacovigilance system for the documentation and tracking of serious adverse events (e.g. for DSMBs) as well as reporting of SUSARs to the EMA and respective competent authorities nationally and internationally according to legal requirements (mandatory electronically via EMA portal from CTC Ulm). This also includes the creation of data sets that are consistent and complete in terms of content, which can be sent to all institutions/affected persons within the narrow legally prescribed time window, thus promoting and ensuring the contemporary and required quality of clinical trials in the field of pharmacovigilance.
-
3.
Promotion and implementation of a uniform and high-quality procedure within the UKU with regard to SAE processing and data entry through the provision of central SOPs, data entry and coding conventions, as well as regular training (PhV QMS).
Central Training
As part of a university medicine medical faculty and central structure of clinical trial research, a CTC is responsible for training and further educating employees involved in clinical studies for their special requirements. Training is a cornerstone to enable investigators and study staff to conduct clinical trials safely and ensure the implementation of clinical studies according to applicable laws and guidelines. Training and advanced training are therefore essential aspects of a CTC. In order to meet these requirements, the CTCs, which are part of the CTC network (KKS-Netzwerk e. V. n.d.), have established their own departments, which guarantee high-quality training and further education of qualified study staff for the implementation of clinical studies as well as for the further training of medical professionals in the field of study design and coordination (Stellungnahme der Arbeitsgruppe “Klinische Studien” der DFG-Senatskommission für Grundsatzfragen in der Klinischen Forschung 2018; Wissenschaftsrat 2018).
The training courses include ethical, regulatory, qualitative, safety, operational and other scientific requirements for clinical studies in order to ensure the implementation according to global quality and safety standards. The qualification and training of study staff ensure the safety of the study participants on-site and the validity and robustness of the study data.
Depending on the course programme, the CTC organises the certification of the courses by the German Medical Association and approval by the ethics committee. The medical participants receive appropriate training points (CME) from the medical association after successfully passing a knowledge test. The courses are continuously evaluated by the participants as well as the training management and adjusted accordingly.
The challenges of implementing such a central training environment are organisational as well as content related. With regard to organisational aspects, the following issues need to be considered: A lecture room or an auditorium for up to 150 participants needs to be reserved. As some trainings are whole-day trainings or even comprise several days, catering should be offered. Upfront, invitations, agenda and further information and communication should be disseminated. The target audience has to be defined, and the means of communication such as email, publication within print media or within intra- and/or Internet page has to be determined. For drafting a participation list, registration should be organised by a predefined central email address and contact, and each registration should be followed by registration approval message. If the training is held by live webinar, the system and webinar platform needs to be tested upfront; in general, IT support should be organised before and during the whole meeting to resolve upcoming issues with login and connection. In general, automatic functions as training registration approval message should be considered. After the training, tests need to be collected and reviewed. A process for failed participants and the possibility of re-testing should be defined upfront. Confirmation of participation and training certificates need to be printed, signed and forwarded to the participants. Finally, the evaluation sheets need to be reviewed and possible actions taken.
With regard to organisational aspects, an early start should be envisaged, and the efforts should not be underestimated. Therefore, enough time should be reserved for the preparation as well as follow-up activities.
With regard to content-related aspects, experts within their fields need to be identified and asked for their willingness to prepare and hold a lecture as part of the training. Backup solutions should be in place for individuals, and the presentations need to be collected upfront and reviewed by the study team for content and format.
The challenge of training and transfer of study-related knowledge as, for example, GCP to all stakeholders in all parts of the academic research facilities is a key part of quality assurance in clinical studies.
Governance
It is key to implement a central, superordinate institution for implementing overarching standards for clinical trials according to applicable laws, regulations and guidelines, GCP as well as local processes. As the sponsor oversight needs to be guaranteed by law, it was decided that stakeholders at the individual study centres should be identified and invited to regular meetings for information exchange. This meeting was named Clinical Trial Management (CTM) meeting and invites all stakeholders not only to be informed about the current status and standards of clinical studies at the UKU but also to be actively involved in decisions and upcoming actions. To be invited to the CTM meeting, the CTC asks within the introductory visit at the clinics and institutes for a representative and deputy to be invited to regular CTM meetings. These stakeholders are the links to the individual study centres in the different departments and responsible for disseminating the information provided within the CTM meetings. Also, dedicated project groups, which concentrate on specific solutions, are recruited from the CTM members and report their solutions to the CTM team. Based on these meetings, governance was implemented and is executed by the CTC.
References
Dinnett et al (2013) Implementing a centralised pharmacovigilance service in a non-commercial setting in the United Kingdom Trials. 14:171.
EMA (2020) Notice to sponsors on validation and qualification of computerised systems used in clinical trials. EU Regulation 536/2014, FDA Title 21 CRF Part 11, ICH E6.
KKS-Netzwerk e. V. (n.d.). https://www.kks-netzwerk.de/index.php
Stellungnahme der Arbeitsgruppe “Klinische Studien” der DFG-Senatskommission für Grundsatzfragen in der Klinischen Forschung. Bonn, 2018.
Wissenschaftsrat. Empfehlungen zu Klinischen Studien. Drs. 7301–18 Hannover 19 10 2018.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2022 The Author(s)
About this chapter

Cite this chapter
Lang, N. (2022). Challenges in Establishing the Clinical Trials Centre at the University of Ulm. In: Schmidt-Straßburger, U. (eds) Improving Oncology Worldwide. Sustainable Development Goals Series. Springer, Cham. https://doi.org/10.1007/978-3-030-96053-7_11
Download citation
DOI: https://doi.org/10.1007/978-3-030-96053-7_11
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-96052-0
Online ISBN: 978-3-030-96053-7
eBook Packages: MedicineMedicine (R0)