
Abstract
Primary liver cancer is the fourth leading cause of cancer death around the world. Histologically, it can be divided into two major groups, hepatocellular carcinoma (75% of all liver cancer) and intrahepatic cholangiocarcinoma (15% of all liver cancer) [1, 2]. Primary liver cancer usually happens in liver disease or cirrhosis patients [1], and the risk factors for developing HCC depend on the etiology [3] and the country of provenance [1]. There is an urgent need for an accurate diagnostic test given the high proportion of false positives and false negatives for alpha-fetoprotein (AFP), a common HCC biomarker [4]. Due to often being diagnosed in advanced stages, HCCrelated deaths per year have doubled since 1999 [3]. With the use of metabolomics technologies [5], the aberrant metabolism characteristics of cancer tissues can be discovered and exploited for the new biomarkers and new therapies to treat HCC [6, 7].
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
- Primary liver cancer
- Metabolic phenotypes
- Glucose metabolism
- Glutamine metabolism
- Oncogenic heterogeneity
- Lipid metabolism
- Redox metabolism
- Sorafenib
-
Different oncogenic mutations lead to different metabolic phenotypes in primary liver cancer.
-
MYC and MET mutations regulate glucose and glutamine metabolism differently in primary liver cancer.
-
Glucose metabolism increased by acetylated phosphoglycerate kinase 1 (PGK1) leads to the promotion of cancer cell proliferation and tumorigenesis in the liver.
-
There exist metabolic differences between hepatocellular carcinoma (HCC) and normal liver tissue or other liver diseases.
1 Introduction
Primary liver cancer is the fourth leading cause of cancer death around the world. Histologically, it can be divided into two major groups, hepatocellular carcinoma (75% of all liver cancer) and intrahepatic cholangiocarcinoma (15% of all liver cancer) [1, 2]. Primary liver cancer usually happens in liver disease or cirrhosis patients [1], and the risk factors for developing HCC depend on the etiology [3] and the country of provenance [1]. There is an urgent need for an accurate diagnostic test given the high proportion of false positives and false negatives for alpha-fetoprotein (AFP), a common HCC biomarker [4]. Due to often being diagnosed in advanced stages, HCC-related deaths per year have doubled since 1999 [3]. With the use of metabolomics technologies [5], the aberrant metabolism characteristics of cancer tissues can be discovered and exploited for the new biomarkers and new therapies to treat HCC [6, 7].

The heterogeneity of liver cell carcinoma metabolism and its associated oncogenic mutations
2 Different Oncogenic Mutations Lead to Different Metabolic Phenotypes in Primary Liver Cancer
Most patients with HCC are diagnosed at advanced stages, and the current effective treatments for these patients are limited. Nevertheless, if HCC patients are diagnosed at an early stage, the tumors can be resected or ablated. However, these patients often experience recurrence after resection/ablation [8]. Strategies to improve patient survival outcomes involve therapies exploiting the metabolic vulnerabilities of cancer cells. However, within the tumor microenvironments, the alterations in metabolic pathways [9, 10], resulting from the combinational effect of genetic, epigenetic, and transcriptomic variations [11, 12], occur frequently to accommodate the high energy demands of tumor growth. Consequently, the complexity of the heterogeneity of altered cancer metabolism leads to resistance in therapeutic cancer treatments [13, 14]. Additionally, different patients also exhibit different forms of liver cancer that correspond to genetic differences [11, 15,16,17,18,19]. Given the genetic and metabolic complexities of HCC, identifying core metabolic pathways utilized by the tumors to drive metabolic phenotypic plasticity of this neoplasm will substantially contribute to the development of effective metabolic therapies.
2.1 MYC and MET Mutations Regulate Glucose and Glutamine Metabolism Differently in Primary Liver Cancer
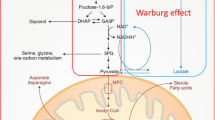
MYC is a well-known regulator of metabolism in cancer [20, 21]. Yuneva et al. found that the reprogramming of glucose and glutamine metabolism was different depending on the activation of the MYC oncogene or the MET proto-oncogene even within a specific liver cancer type [22]. They found an increased uptake and catabolism of glucose in primary liver cancer as compared to the normal liver in both MET- and MYC-induced liver tumors. However, MYC-induced liver tumors exhibited the Warburg effect [23] in which these tumors produced significantly high levels of lactate, a phenotype not observed in MET-induced tumors [22]. This study suggests that within the same cancer type, cells exhibit diverse genetic abnormalities that result in diverging and distinct metabolic manifestations. These numerous and remarkably pliant alterations appear to be essential for meeting the variety of demands of cell proliferation, which include ATP production, biosynthesis of cellular building blocks, reactive oxygen species (ROS) detoxification, and degradation of the extracellular matrix scaffolding to allow for angiogenesis and thus tumorigenesis.

The alteration of glucose metabolism involves upregulation of glycolysis enzymes and downregulation of gluconeogenic enzymes and mitochondrial aerobic activity. GLUT glucose transporter, MCT1 protein monocarboxylate transporter isoform 1, HK2 hexokinase 2, PFK1 phosphofructokinase-1, FBP1 fructose-1,6-bisphosphatase, PCK phosphoenolpyruvate carboxykinase, GCK glucokinase, TCA cycle tricarboxylic acid cycle, miR-383 microRNA-383
Tumor cell metabolism of the same tissue type has been shown to depend on the identities of the genetic mutations. While MYC-induced mouse liver tumors exhibit enhanced glutamine [24] and glucose [23] metabolism, accompanied by an increase in lactate production and Krebs cycle intermediates, MET-induced mouse liver tumors are found to consume glucose as a means of synthesizing glutamine [22] (Fig. 1). Thus, it is reasonable to conclude that these two genes dictate radically opposite roles for glutamine, a central player in cancer metabolism. This fact illustrates, once again, that cancer metabolism can be determined by the nature of the genetic alterations and that how the metabolism is altered across different tumors can be extremely substantial. This helps to explain how the same tissue of origin in different patients, in this case, can have different genetic alterations and metabolic phenotypes, thus substantiating the potential role of heterogeneity, even in a single tumor of the same patient.
2.2 Liver Receptor Homolog 1 (LRH-1) Regulates Mitochondrial Glutamine Metabolism
The study led by Xu et al. has revealed the crucial role of liver receptor homolog 1 (LRH-1) in regulating mitochondrial glutamine metabolism, which eventually leads to the production of NADPH through a noncanonical glutamine pathway. Specifically, the study found that the regulation of malic enzyme 1 (Me1), an enzyme that catalyzes the conversion of malate to pyruvate to produce NADPH through a noncanonical glutamine pathway [25], is dependent on LRH-1 [26].
Furthermore, using 13C5-labeled glutamine, the study highlighted the essential role of LRH-1 in promoting the production of glutamate from glutamine via controlling mitochondrial glutaminase 2 (GLS2) [26]. Consequently, the production of α-ketoglutarate (α-KG) from glutamate activates the mechanistic target of the rapamycin complex 1 (mTORC1) signaling pathway [26], a regulator of cell growth metabolism including proteins, lipids, and nucleotides [27]. Due to its pivotal role in the production of the reductive biosynthetic product NADPH and the activation of the mTORC1 signaling pathway through glutamine metabolism, LRH-1 promotes cell proliferation. Thus, loss of LRH-1 prevents diethylnitrosamine (DEN)-induced liver carcinogenesis [26]. Targeting glutamine metabolism as a strategy for the treatment has been studied in several cancers [24, 28,29,30,31,32,33,34] (Fig. 1).
2.3 Glucose Metabolism Increased by Acetylated Phosphoglycerate Kinase 1 (PGK1) Leads to the Promotion of Cancer Cell Proliferation and Tumorigenesis in Liver
Compared to normal liver cells, cancerous liver cells, and cancer cells, in general, need a much greater amount of energy to fuel their proliferation. One of the ways to satisfy these high demands of energy is to adjust the energy-yielding pathways accordingly to produce energy in the most efficient manner. Deciphering different energy production-enhancing mechanisms in cancers has attracted a lot of attention because having a better understanding of these mechanisms provides strategies to advance treatments for cancers. Similarly, the study led by Hu et al. elucidated the role of phosphoglycerate kinase 1 (PGK1), an enzyme catalyzing the conversion of 1,3-bisphosphoglycerate to 3-phosphoglycerate yielding one ATP equivalent, in glycolysis, cell proliferation, and tumorigenesis. The formation of acetylated PGK1 at position K323 is required to activate PGK1 [35]. Activated PGK1, in turn, regulates cancer cell metabolism. Specifically, acetylated PGK1 enhances the production of energy in the form of ATP [35]. Ultimately, the acetylation of PGK1 at K323 promotes liver tumorigenesis. With this understanding of how PGK1 K323 acetylation functions in liver cancer, the emergence of new effective treatments using PGK1 as a therapeutic target for patients with liver cancer is promising (Fig. 1).

The alteration of lipid metabolism involves upregulation of fatty acid biosynthesis enzymes and downregulation of fatty acid oxidation enzymes. FASN fatty acid synthase, ACC acetyl-CoA carboxylase, ACLY ATP citrate lyase, ACSS1 acyl-CoA synthetase short-chain family member 1, ACADSB acyl-CoA dehydrogenase family, HADH hydroxyacyl-CoA dehydrogenases, CPT carnitine palmitoyltransferase 2, ACS acetyl-coA synthase
3 Metabolic Differences Between Liver Cancer Stem Cells (LCSCs) and Non-liver Cancer Stem Cells (Non-LCSCs)
Given their metabolic heterogeneity, LCSCs are able to adapt to many different environments, which causes therapeutic resistance to many available treatments for HCC. Understanding the metabolism of LCSCs is crucial not only for the improvement of currently available therapies but also for paving a new path for developing other therapies targeting the revealed metabolic pathways. In an effort to elaborate on the understanding of the metabolism of LCSCs, Hur et al. found the following differences in the metabolism of LCSCs as compared to non-LCSCs. The increased proliferation of LCSCs can be explained based on the metabolomics analysis, which reveals the higher presence of essential metabolites that are either resulting from highly activated catabolism or acting as substrates to promote other energy-yielding processes. Specifically, the study found a higher concentration of lactate, the final product from glycolysis, citrate, succinate, and several amino acids such as aspartate, glutamate, isoleucine, leucine, phenylalanine, tyrosine, and valine in LCSCs as compared to non-LCSCs. Moreover, they showed that MYC, a known regulator of glycolytic metabolism [20, 36], is highly expressed in LCSCs as compared to non-LCSCs, and this resulted in an increased amount of energy available for the rapid proliferation of the cancer cells [37]. This study also found that fatty acid oxidation in LCSCs is less active than that in non-LCSCs [37]. Consequently, the production of NADPH from fatty acid oxidation, contributing to oxidative phosphorylation to generate ATP, is less for LCSCs. Nevertheless, the assessment of three genes—cytochrome C oxidase subunit 5B (COX5B), adenosine triphosphate-5-alpha (ATP5α), and estrogen-related receptor Α (ERRα)—involved in oxidative phosphorylation showed no differences between LCSCs and non-LCSCs [37]. This means that LCSCs must have utilized more glycolysis to produce ATP to satisfy the demands of energy for their rapid proliferation (Fig. 1).
4 Metabolic Signatures of Liver Cancer
4.1 Metabolism of HCC Is Different from that of Normal Liver Tissue
This section presents evidence of changes in the concentrations of specific metabolites in liver cancer as compared to normal hepatocytes and how this knowledge allows the development of new therapeutic models.
From recent studies, it is known that glucose metabolism is reprogrammed in HCC with the aim of allowing its growth, proliferation, and metastasis. One of the most outstanding characteristics of cancer cell metabolism is the high demand for glucose [38]. Cassim et al. found that GLUT1 expression is increased in HCC tumorigenic cells as opposed to normal liver cells, which have high GLUT2 expression [39]. Furthermore, Kim et al. found that the expression of GLUT1 (SLC2A1) is increased in advanced stages of HCC [38] (Fig. 2). Moreover, there is a switch in the expression of hexokinase (HK) enzyme from low-affinity glucokinase (GCK) isoenzyme in normal hepatocytes to high-affinity hexokinase 2 (HK2) isoenzyme. DeWaal et al. showed that HCC cells predominantly use HK2, unlike noncancerous liver cells that use GCK (or HK4) [40]. In addition, they showed that there is an increased expression of HK2 in HCC cells and cells with evidence of dysplasia [40].
On the other hand, Fang et al. studied the influence of miR-383 on the expression of lactate dehydrogenase A (LDHA), which plays a key role in the progression of many types of cancer [41]. An overexpression of miR-383, an endogenous non-coding microRNA, has been reported to markedly inhibit HCC cells’ glycolysis, proliferation, and invasion, through its binding to the LDHA-expressing gene [41] (Fig. 2). The LDHA inhibitor, FX11, has also shown promising results in several studies [42,43,44]. While it is known that there is an increase in the activity of aerobic glycolysis in HCC, Wang et al. showed that there is also a decrease in gluconeogenesis activity in HCC [45]. Björnson et al. discovered downregulation of phosphoenolpyruvate carboxykinase (PCK1 and PCK2), the first gluconeogenesis enzyme in HCC [46]. Hirata et al. also discovered that in HCC samples there is low expression of fructose-1,6-bisphosphatase (FBP1), a gene that encodes the gluconeogenic enzyme FBP1 [45].
Furthermore, they also discovered that FBP1 overexpression is associated with decreased expression of hexokinase-2 (HK2) and phosphofructokinase-1 (PFK1), key enzymes of aerobic glycolysis in cancer cells [45]. It was also shown that low FBP1 levels were associated with tumor progression in HCC samples [45] (Fig. 2).
Regarding the altered lipid metabolism [47], Björnson et al. found evidence of a significant increase in the expression of key genes related to fatty acid biosynthesis (FAB) and pentose phosphate pathway (PPP) in HCC samples compared to noncancerous liver samples [46] (Fig. 3). The upregulation of these genes and the corresponding enzymes thus provide the necessary supplies for the synthesis of fatty acids, nucleotides, and NADPH, essential for tumor growth and proliferation [46]. Acetyl-CoA is required for fatty acid biosynthesis, and it is commonly produced by the oxidation of fatty acids [46]. However, Björnson et al. found that fatty acid oxidation is decreased in HCC and the main supply for the generation of fatty acids is exogenously acquired acetate [46]. According to this study, fatty acid oxidation (FAO) enzymes such as carnitine palmitoyltransferase 2 (CPT2), acyl-CoA dehydrogenase family (ACADSB), and hydroxyacyl-CoA dehydrogenases (HADH) are significantly downregulated in HCC tumors [46]. Acetate of exogenous origin enters the cell and, through acetyl-CoA synthases, is transformed into acetyl-CoA, a necessary substrate for the biosynthesis of fatty acids [46]. In addition, the expression of the acyl-CoA synthetase short-chain family member 1 (ACSS1) subtype, which resides in the mitochondria, ATP-citrate lyase (ACLY), acetyl-CoA carboxylase (ACC), and fatty acid synthase (FASN), are significantly increased in patients with HCC compared to noncancerous liver samples [46]. Furthermore, in the same study, two groups with highly differentiated ACSS1 expression levels were observed within the HCC patient samples [46] (Fig. 3). Further analysis revealed that the group with high levels of ACSS1 had a greater suppression of the enzymes related to FAO as compared to the group with low levels of ACSS1 [46]. Furthermore, it was also observed that the group with high ACSS1 expression had four times the expression of pyruvate kinase (PKM) compared to the group with low levels of ACSS1 [46] (Fig. 3). Increased expression of PKM is related to a greater capacity for metabolic adaptation to different concentrations of nutrients and oxygen [46].
4.2 Oxidative Stress Signature in HCC
Oxidative stress , which is determined by the balance of oxidant and antioxidant species, also plays an important role in HCC. Among the antioxidant species, glutathione (GSH) is the major nonenzymatic regulator of intracellular redox homeostasis [48], and it is the most abundant antioxidant in hepatocytes [49]. In the serum of patients with HCC, a significant increase in amino acids related to the synthesis of GSH, the reduced form of glutathione, and glucose 6-phosphate (G6P), a compound necessary to form NADPH, has been evidenced [50]. On the other hand, the quantification of oxidative damage is made possible through measuring oxidative products, and in HCC, 8-hydroxydeoxy guanosine and 4-hydroxynonenal (oxidative products of DNA and lipids, respectively) were found [49]. De Matteis et al. also found that there were elevated levels of 8-hydroxydeoxy-guanosine in the serum of patients with chronic hepatitis, which can lead to an increased risk of HCC [49].
It is important to note that Wang et al. found that a large elevation of canavaninosuccinate (CSA) in HCC patients from the first affiliated hospital of the medical school of Zhejiang University, as opposed to samples from cirrhosis patients, is a signature of oxidized stress [51]. CSA produces fumarate, which is a key metabolite of the tricarboxylic acid (TCA) cycle, and elevated CSA levels indicate that its conversion to fumarate is impaired and thus negatively impacting the subsequent TCA cycle intermediate formation while promoting Warburg’s glycolysis [52]. They also discovered the sensitivity and specificity of alpha-fetoprotein (AFP) and CSA to distinguish HCC patients from cirrhosis controls [51]. It is important to note that these authors do not discuss dietary factors due to the plant origin of canavanine.
5 New Therapeutic Investigations Based on Metabolism Studies
Currently, the multikinase inhibitor sorafenib is the main drug for the treatment of advanced HCC that delays tumur progression and improves overall survival. However, its ability to shrink the tumor in patients is very modest [53]. Therefore, other treatments that may potentiate or replace sorafenib are being studied. DeWaal et al. studied the synergism between HK2 silencing and sorafenib because the first one can induce cell death, and the second one can inhibit growth factor receptors. It was observed that HK2 silencing made HCC cells more sensitive to sorafenib. Therefore, the combination therapy using sorafenib and HK2 silencing decreased tumor growth substantially more than either treatment alone [40].
Another protein that has garnered attention because it is expressed in high amounts in many types of malignant cells is a transmembrane protein known as CD147 (Fig. 2). It is associated with the lactate transporter monocarboxylate transporter isoform 1 (MCT1), a key protein that exports lactate to the extracellular environment from HCC cells and is necessary for the proliferation of HCC [49]. Huang et al. found that CD147 overexpression significantly promoted glycolysis via p53/TIGAR/PFK and inhibited mitochondrial activity via p53 in HCC cells [54]. Furthermore, the silencing of CD147 was associated with a relative increase in mtDNA content in HCC cells compared to control cells [54]. Additionally, this silencing was associated with a high oxygen consumption rate, a decrease in the intracellular concentration of glucose and pyruvate, a high intracellular lactate concentration, and a significant decrease in cell growth [54]. Inhibition of CD147 phosphorylation was found in HCC samples from patients with distant metastases and was associated with elevated levels of AFP. This phosphorylation requires further studies as a potential prognosis biomarker, as well as a potential strategy for the development of treatments against HCC metastasis [55].
6 Conclusion
Given the complex heterogeneity and intricate evolutionary characteristics of liver cancers, the increase in resistance rate to current therapies has emerged as the main obstacle that many studies focusing on HCC have been trying to overcome. Among the different methods available to tackle the problem, metabolomics-based approaches serve as powerful strategies—allowing researchers to uncover metabolic profiles of different cancers. In addition, using metabolomics technologies to track a variety of metabolites in cancers offers researchers a better picture of the interactions that occur within the tumor microenvironments. Understanding the heterogeneity of cancer metabolism will pave a new path for the development of metabolism-based therapies to improve the outcome of cancer therapy. As has been shown, genes such as MYC, MET, or LRH-1 activate various metabolic pathways for cell growth and survival. On the other hand, the combination of AFP with CSA has shown good results as a diagnostic test, which compels us to continue studying the metabolic pathway of CSA within HCC cells. However, more research is required to understand the progression of liver cancer and how to evaluate it at different stages for a more favorable prognosis, as well as less invasive and more effective treatments. The study of the metabolome is yielding good results, but it is still necessary to discover new relationships between the different metabolic pathways.
Abbreviations
- ACC:
-
Acetyl-CoA carboxylase
- ACSS1:
-
Acyl-CoA synthetase short-chain family member 1
- AFP:
-
Alpha-fetoprotein
- ATP:
-
Adenosine triphosphate
- COX5B:
-
Cytochrome C oxidase subunit 5B
- CSA:
-
Canavaninosuccinate
- DEN:
-
Diethylnitrosamine
- ERRα:
-
Estrogen-related receptor Α
- FAO:
-
Fatty acid oxidation
- FASN:
-
Fatty acid synthase
- FBP1:
-
Fructose-1,6-bisphosphatase
- G6P:
-
Glucose 6-phosphate
- G6PD:
-
Glucose-6-phosphate dehydrogenase
- GCK:
-
Glucokinase
- GLS2:
-
Glutaminase 2
- Glu:
-
Glutamine
- GLUT1:
-
Glucose transporter 1
- GSH:
-
Glutathione
- GLUT2:
-
Glucose transporter 2
- HCC:
-
Hepatocellular carcinoma
- HK2:
-
Hexokinase 2
- LCSCs:
-
Liver cancer stem cells
- LDHA:
-
Lactate dehydrogenase A
- LRH-1:
-
Liver receptor homolog 1
- MCT1:
-
Protein monocarboxylate transporter isoform 1
- Me1:
-
Malic enzyme 1
- miR-383:
-
MicroRNA-383
- mTORC1:
-
Mechanistic target of rapamycin complex 1
- NADPH:
-
Nicotinamide adenine dinucleotide phosphate
- Non-LCSCs:
-
Non-liver cancer stem cells
- PCK:
-
Phosphoenolpyruvate car-boxykinase
- PFK1:
-
Phosphofructokinase-1
- PGK1:
-
Phosphoglycerate kinase 1
- PKM:
-
Pyruvate kinase
- ROS:
-
Reactive oxygen species
- α-KG:
-
α-Ketoglutarate
References
Dasgupta, P., et al. (2020). Global trends in incidence rates of primary adult liver cancers: A systematic review and meta-analysis. Frontiers in Oncology, 10, 171.
Lin, H. S., et al. (2019). Identification of novel anti-liver cancer small molecules with better therapeutic index than sorafenib via zebrafish drug screening platform. Cancers (Basel), 11, 6.
Kim, H. S., & El-Serag, H. B. (2019). The epidemiology of hepatocellular carcinoma in the USA. Current Gastroenterology Reports, 21(4), 17.
Gao, R., et al. (2015). Serum metabolomics to identify the liver disease-specific biomarkers for the progression of hepatitis to hepatocellular carcinoma. Scientific Reports, 5, 18175.
Hoang, G., Udupa, S., & Le, A. (2019). Application of metabolomics technologies toward cancer prognosis and therapy. International Review of Cell and Molecular Biology, 347, 191–223.
Dang, C. V., et al. (2011). Therapeutic targeting of cancer cell metabolism. Journal of Molecular Medicine (Berlin), 89(3), 205–212.
Hirschey, M. D., et al. (2015). Dysregulated metabolism contributes to oncogenesis. Seminars in Cancer Biology, 35(Suppl), S129–S150.
Llovet, J. M., et al. (2016). Hepatocellular carcinoma. Nature Reviews. Disease Primers, 2, 16018.
Nabi, K., & Le, A. (2021). The intratumoral heterogeneity of cancer metabolism. Advances in Experimental Medicine and Biology, 1311, https://doi.org/10.1007/978-3-030-65768-0_11
Antonio, M. J., Zhang, C., & Le, A. (2021). Different tumor microenvironments lead to different metabolic phenotypes. Advances in Experimental Medicine and Biology, 1311, https://doi.org/10.1007/978-3-030-65768-0_10
Forner, A., Llovet, J. M., & Bruix, J. (2012). Hepatocellular carcinoma. Lancet, 379(9822), 1245–1255.
Zucman-Rossi, J., et al. (2015). Genetic landscape and biomarkers of hepatocellular carcinoma. Gastroenterology, 149(5), 1226–1239. e4.
Schulze, K., Nault, J. C., & Villanueva, A. (2016). Genetic profiling of hepatocellular carcinoma using next-generation sequencing. Journal of Hepatology, 65(5), 1031–1042.
Bobrovnikova-Marjon, E., & Hurov, J. B. (2014). Targeting metabolic changes in cancer: Novel therapeutic approaches. Annual Review of Medicine, 65, 157–170.
Wolpaw, A. J., & Dang, C. V. (2018). Exploiting metabolic vulnerabilities of cancer with precision and accuracy. Trends in Cell Biology, 28(3), 201–212.
Cancer Genome Atlas Research Network. (2017). Electronic address, w.b.e. and N. Cancer genome atlas research, comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell, 169(7), 1327–1341.e23.
Calderaro, J., et al. (2017). Histological subtypes of hepatocellular carcinoma are related to gene mutations and molecular tumour classification. Journal of Hepatology, 67(4), 727–738.
Zheng, H., et al. (2018). Single-cell analysis reveals cancer stem cell heterogeneity in hepatocellular carcinoma. Hepatology, 68(1), 127–140.
Xue, R., et al. (2016). Variable Intra-tumor genomic heterogeneity of multiple lesions in patients with hepatocellular carcinoma. Gastroenterology, 150(4), 998–1008.
Dang, C. V., Le, A., & Gao, P. (2009). MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clinical Cancer Research, 15(21), 6479–6483.
Le, A., & Dang, C. V. (2013). Studying Myc’s role in metabolism regulation. Methods in Molecular Biology, 1012, 213–219.
Yuneva, M. O., et al. (2012). The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metabolism, 15(2), 157–170.
Bose, S., Zhang, C., & Le, A. (2021). Glucose metabolism in cancer: The Warburg effect and beyond. Advances in Experimental Medicine and Biology, 1311, https://doi.org/10.1007/978-3-030-65768-0_1
Li, T., Copeland, C., & Le, A. (2021). Glutamine metabolism in cancer. Advances in Experimental Medicine and Biology, 1311, https://doi.org/10.1007/978-3-030-65768-0_2
Wise, D. R., et al. (2008). Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proceedings of the National Academy of Sciences of the United States of America, 105(48), 18782–18787.
Xu, P., et al. (2016). LRH-1-dependent programming of mitochondrial glutamine processing drives liver cancer. Genes & Development, 30(11), 1255–1260.
Laplante, M., & Sabatini, D. M. (2012). mTOR signaling in growth control and disease. Cell, 149(2), 274–293.
Zimmermann, S. C., et al. (2016). Allosteric glutaminase inhibitors based on a 1,4-di(5-amino-1,3,4-thiadiazol-2-yl)butane scaffold. ACS Medicinal Chemistry Letters, 7(5), 520–524.
Rais, R., et al. (2016). Discovery of 6-diazo-5-oxo-l-norleucine (DON) prodrugs with enhanced CSF delivery in monkeys: A potential treatment for glioblastoma. Journal of Medicinal Chemistry, 59(18), 8621–8633.
Xiang, Y., et al. (2015). Targeted inhibition of tumor-specific glutaminase diminishes cell-autonomous tumorigenesis. The Journal of Clinical Investigation, 125(6), 2293–2306.
Le, A., et al. (2012). Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metabolism, 15(1), 110–121.
Elgogary, A., et al. (2016). Combination therapy with BPTES nanoparticles and metformin targets the metabolic heterogeneity of pancreatic cancer. Proceedings of the National Academy of Sciences of the United States of America, 113(36), E5328–E5336.
Udupa, S., et al. (2019). Upregulation of the glutaminase II pathway contributes to glutamate production upon glutaminase 1 inhibition in pancreatic cancer. Proteomics, 19(21–22), e1800451.
Nguyen, T., et al. (2019). Uncovering the role of N-acetyl-aspartyl-glutamate as a glutamate reservoir in cancer. Cell Reports, 27(2), 491–501. e6.
Hu, H., et al. (2017). Acetylation of PGK1 promotes liver cancer cell proliferation and tumorigenesis. Hepatology, 65(2), 515–528.
Dang, C. V. (2010). Rethinking the Warburg effect with Myc micromanaging glutamine metabolism. Cancer Research, 70(3), 859–862.
Hur, W., et al. (2017). Systems approach to characterize the metabolism of liver cancer stem cells expressing CD133. Scientific Reports, 7, 45557.
Kim, Y. H., et al. (2017). SLC2A2 (GLUT2) as a novel prognostic factor for hepatocellular carcinoma. Oncotarget, 8(40), 68381–68392.
Cassim, S., et al. (2018). Metabolite profiling identifies a signature of tumorigenicity in hepatocellular carcinoma. Oncotarget, 9(42), 26868–26883.
DeWaal, D., et al. (2018). Hexokinase-2 depletion inhibits glycolysis and induces oxidative phosphorylation in hepatocellular carcinoma and sensitizes to metformin. Nature Communications, 9(1), 446.
Fang, Z., et al. (2017). The miR-383-LDHA axis regulates cell proliferation, invasion and glycolysis in hepatocellular cancer. Iranian Journal of Basic Medical Sciences, 20(2), 187–192.
Le, A., et al. (2010). Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proceedings of the National Academy of Sciences of the United States of America, 107(5), 2037–2042.
Rajeshkumar, N. V., et al. (2015). Therapeutic targeting of the Warburg effect in pancreatic cancer relies on an absence of p53 function. Cancer Research, 75(16), 3355–3364.
Dutta, P., et al. (2013). Evaluation of LDH-A and glutaminase inhibition in vivo by hyperpolarized 13C-pyruvate magnetic resonance spectroscopy of tumors. Cancer Research, 73(14), 4190–4195.
Hirata, H., et al. (2016). Decreased expression of fructose-1,6-bisphosphatase associates with glucose metabolism and tumor progression in hepatocellular carcinoma. Cancer Research, 76(11), 3265–3276.
Bjornson, E., et al. (2015). Stratification of hepatocellular carcinoma patients based on acetate utilization. Cell Reports, 13(9), 2014–2026.
Park, J. K., et al. (2021). The heterogeneity of lipid metabolism in cancer. Advances in Experimental Medicine and Biology, 1311, https://doi.org/10.1007/978-3-030-65768-0_3
Arauz, J., Ramos-Tovar, E., & Muriel, P. (2016). Redox state and methods to evaluate oxidative stress in liver damage: From bench to bedside. Annals of Hepatology, 15(2), 160–173.
De Matteis, S., et al. (2018). Aberrant metabolism in hepatocellular carcinoma provides diagnostic and therapeutic opportunities. Oxidative Medicine and Cellular Longevity, 2018, 7512159.
Andrisic, L., et al. (2018). Short overview on metabolomics approach to study pathophysiology of oxidative stress in cancer. Redox Biology, 14, 47–58.
Wang, B., et al. (2012). Metabonomic profiles discriminate hepatocellular carcinoma from liver cirrhosis by ultraperformance liquid chromatography-mass spectrometry. Journal of Proteome Research, 11(2), 1217–1227.
Fitian, A. I., & Cabrera, R. (2017). Disease monitoring of hepatocellular carcinoma through metabolomics. World Journal of Hepatology, 9(1), 1–17.
Assenat, E., et al. (2019). Sorafenib alone vs. sorafenib plus GEMOX as 1(st)-line treatment for advanced HCC: The phase II randomised PRODIGE 10 trial. British Journal of Cancer, 120(9), 896–902.
Huang, Q., et al. (2014). CD147 promotes reprogramming of glucose metabolism and cell proliferation in HCC cells by inhibiting the p53-dependent signaling pathway. Journal of Hepatology, 61(4), 859–866.
Jin, J., et al. (2019). Hypo-phosphorylated CD147 promotes migration and invasion of hepatocellular carcinoma cells and predicts a poor prognosis. Cellular Oncology (Dordrecht), 42(4), 537–554.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2021 The Author(s)
About this chapter

Cite this chapter
Salazar, J., Le, A. (2021). The Heterogeneity of Liver Cancer Metabolism. In: Le, A. (eds) The Heterogeneity of Cancer Metabolism. Advances in Experimental Medicine and Biology, vol 1311. Springer, Cham. https://doi.org/10.1007/978-3-030-65768-0_9
Download citation
DOI: https://doi.org/10.1007/978-3-030-65768-0_9
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-65767-3
Online ISBN: 978-3-030-65768-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)