
Abstract
According to data from the American Cancer Society, cancer is one of the deadliest health problems globally. Annually, renal cell carcinoma (RCC) causes more than 100,000 deaths worldwide [1–4], posing an urgent need to develop effective treatments to increase patient survival outcomes. New therapies are expected to address a major factor contributing to cancer’s resistance to standard therapies: oncogenic heterogeneity. Gene expression can vary tremendously among different types of cancers, different patients of the same tumor type, and even within individual tumors; various metabolic phenotypes can emerge, making singletherapy approaches insufficient. Novel strategies targeting the diverse metabolism of cancers aim to overcome this obstacle. Though some have yielded positive results, it remains a challenge to uncover all of the distinct metabolic profiles of RCC. In the quest to overcome this obstacle, the metabolic oriented research focusing on these cancers has offered freshly new perspectives, which are expected to contribute heavily to the development of new treatments.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others

Keywords
- Renal cell carcinoma
- Metabolic phenotypes
- Glucose metabolism
- Glutamine metabolism
- Oncogenic heterogeneity—intratumoral heterogeneity
-
Different oncogenic mutations lead to various metabolic phenotypes in renal cell carcinomas (RCC).
-
Loss of the von Hippel-Lindau (VHL) tumor-suppressor gene results in metabolic alterations, including shifts toward aerobic glycolysis in RCC.
-
Fumarate hydratase mutations result in an increase in aerobic glycolysis in RCC.
-
The upregulation of glycolysis enzymes is correlated with high-grade RCC.
-
An increase in glutathione (GSH)/glutathione disulfide (GSSG) ratio protects RCC against oxidative stress in a grade-dependent manner.
-
Metabolic spatial heterogeneity can be induced by both gene-dependent and independent processes.
-
Metabolic intratumoral heterogeneity explains the failure of monotherapy and necessitates the need for personalized and/or combination therapies for RCC.
1 Introduction
According to data from the American Cancer Society, cancer is one of the deadliest health problems globally. Annually, renal cell carcinoma (RCC) causes more than 100,000 deaths worldwide [1,2,3,4], posing an urgent need to develop effective treatments to increase patient survival outcomes. New therapies are expected to address a major factor contributing to cancer’s resistance to standard therapies: oncogenic heterogeneity. Gene expression can vary tremendously among different types of cancers, different patients of the same tumor type, and even within individual tumors; various metabolic phenotypes can emerge, making single-therapy approaches insufficient. Novel strategies targeting the diverse metabolism of cancers aim to overcome this obstacle. Though some have yielded positive results, it remains a challenge to uncover all of the distinct metabolic profiles of RCC. In the quest to overcome this obstacle, the metabolic oriented research focusing on these cancers has offered freshly new perspectives, which are expected to contribute heavily to the development of new treatments.
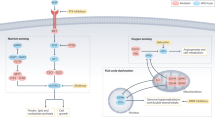
2 Different Oncogenic Mutations Lead to Different Metabolic Phenotypes in RCC (Fig. 1)
Renal cell carcinoma (RCC), or hypernephroma, is the most common type of kidney cancer in adults, responsible for approximately 90–95% of all cases. RCC originates from the network of convoluted tubules of the nephron [5] and consists of diverse histological subtypes, each with unique sets of metabolic rearrangements that can be traced back to gene alterations [6, 7]. These genomic abnormalities provide cancer cells with the advantageous abilities to adapt to the limitations of their microenvironments and meet the demands of rapid and deleterious cell division.

Heterogeneous metabolic phenotypes in renal cell carcinoma as a result of different oncogenic mutations. AKT protein kinase B, AMPK AMP-activated protein kinase, ePC ether-type phosphatidylcholine, ePE ether type, FH fumarate hydratase, G6P glucose-6-phosphate, GLUT 2 glucose transporter 2, GSH glutathione, HIF-1α hypoxia-inducible factor 1-alpha, mTOR mammalian target of rapamycin, PEP phosphoenolpyruvic acid, ROS reactive oxygen species, SM sphingomyelin, TCA cycle tricarboxylic acid cycle
2.1 Loss of the von Hippel-Lindau Tumor-Suppressor Gene Results in Metabolic Alterations Including Shifts Toward Aerobic Glycolysis in RCC
Loss of the von Hippel-Lindau (VHL) tumor-suppressor gene is the most prominent genetic alteration in RCC, commonly associated with over 80% of clear-cell renal cell carcinoma (ccRCC) tumors [8, 9]. The protein product of VHL facilitates the degradation of hypoxia-inducible factor (HIF) [10]. Thus, the loss of VHL leads to the accumulation of HIF-1α and the constitutive activation of hypoxia-inducible genes, even in oxygenated conditions. This includes the enhanced expressions of glucose transporter 2 (GLUT2), hexokinase 2 (HK2), and lactate dehydrogenase A (LDHA), which are keys to the metabolic shift towards aerobic glycolysis in tumors with this genotype [11, 12]. In fact, enhanced HIF-1α activity is thought to mediate the Warburg effect in RCC [13, 14]. Moreover, a study by Gill et al. found that HIF upregulation led to increases in glucose and amino acid uptake, lipogenesis, and augmentation of glycolysis through upregulation of MET protein expression and subsequent phosphatidylinositol 3-kinase (PI3K)/Akt signaling pathway activation [14,15,16].
In addition, pentose phosphate shunt dependence, increases in glutamine transport, and fatty acid production all have been documented as VHL-associated metabolic alterations in ccRCC [17]. The increased activity of the pentose phosphate pathway (PPP) plays a significant role in protecting cancer cells from oxidative stress as this pathway generates NADPH and enables the maintenance of glutathione levels [13]. The Consortia of The Cancer Genome Atlas Research Network revealed that upregulation of PPP genes (G6PH, PGLS, TALDO, and TKT), fatty acid synthesis genes (ACC and FASN), and PI3K pathway-enhancing genes (MIR21) correlated with worse survival. In contrast, upregulation of AMP-activated protein kinase (AM PK) complex genes, multiple Krebs cycle genes, and PI3K pathway inhibitors (phosphatase and tensin homolog deleted in chromosome 10 (PTEN), tuberous sclerosis 2 (TSC2)) correlated with better survival [17].
2.2 Fumarate Hydratase Mutations Result in an Increase in Aerobic Glycolysis in RCC
Studies by Tong et al. indicated that RCC cells carrying inactivated fumarate hydratase (FH), a tricarboxylic acid (TCA) cycle enzyme, demonstrated metabolic changes that were distinct from other genetically defined RCC, such as an increase in aerobic glycolysis and advanced tumorigenicity. Thus, fumarate hydratase-deficient kidney cancer has low oxygen consumption rates as well as low mitochondrial complex I activities [18, 19]. In addition to having a glycolytic shift, FH-deficient kidney tumors and cell lines from patients with hereditary leiomyomatosis and RCC also exhibited decreased levels of AMPK, a key metabolic regulator. Glycolytic upregulation allows the cells to adapt to growth demands by generating NADPH, acetyl-CoA, and precursors for ribose, protein, and fatty acid biosynthesis through reduced AMPK signaling. Furthermore, another study by Massari et al. demonstrated that intracellular augmentation of fumarate has an inhibitory effect on HIF prolyl hydroxylase (PHD) and consequently inhibited the activity of the VHL ubiquitination complex, which, in turn, led to the upregulation of HIF-target genes (VEGF and GLUT1) in a VHL-independent manner by stabilizing HIF1a [20]. On the other hand, intracellular reactive oxygen species (ROS) accumulation under glycolysis can also result in HIF1a stabilization [21].

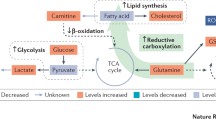
Spatial and temporal intratumoral heterogeneity in renal cell carcinoma. Gal galactose, GSH glutathione, GSSG glutathione disulfide, Man mannose, MCFAs medium-chain fatty acids
LDHA inhibition has shown promise against FH-deficient RCC cells in vitro and in vivo [22] and other cancers [23,24,25,26]. Metformin, an antidiabetic medication, was reported to activate AMPK and inhibit RCC growth in vitro and in vivo [27].
3 Metabolic Signatures of RCC
3.1 Metabolic Differences Between Normal Renal Cells and RCC
To compensate for the high demands of energy and biosynthetic macromolecules for proliferation, RCC cells change their metabolic phenotypes from that of normal renal cells to satisfy the demands. This leads to the different metabolic signatures of RCC cells as compared to normal renal cells. One of the metabolic signatures presenting in RCC cells found by Saito et al. is the decrease in the level of phosphatidylethanolamine (PE) in RCC cells as compared to normal cells [28]. PE is one of the most abundant glycerophospholipids in eukaryotes and plays a crucial role in autophagy, cell division, and protein folding and acts as a precursor for synthesizing phosphatidylcholine (PC), phosphatidylserine (PS) N-acylethanolamine (NAE) [29,30,31,32]. Furthermore, the study by Saito et al. shows that increased PE, which is induced by ethanolamine, inhibits RCC cell proliferation [28]. Low levels of PE inhibit cell apoptosis, which explains why the downregulation of PE benefits RCC cells. The study also found that increases in ether-type PE (ePE) and ether-type phosphatidylcholine (ePC) are associated with ccRCC metastasis [28].
Interestingly, other studies showed that there is an upregulation of the fetal isoform of pyruvate kinase, isoform M2 (PKM2), across all grades of RCC, suggesting that tumor cells utilize different enzymes from those found in normal tissue to enhance their growth [33, 34].
Moreover, another metabolic feature of RCC is the decrease of sphingomyelin (SM), an essential component of the plasma membrane that regulates the formation of lipid microdomains through interacting with cholesterol and glycerophospholipids [28, 35]. A high level of SM was reported to make the cells more vulnerable to apoptosis [36].
In addition to the metabolic signatures of RCC reported by Saito et al., the study by Catchpole et al. found that the upregulation of fatty acid levels was potentially linked to the metastatic stage of the malignancy [37]. The high fatty acid levels were the results of an increase in de novo fatty acid synthesis and/or a decrease in fatty acid oxidation, which often occurred during the invasive and metastatic stage of RCC [37, 38]. Another compound with high concentration, α-tocopherol, was found in RCC and ovarian cancer to protect tumor cells against oxidative stress [37, 39].
Another interesting metabolic signature in RCC cells is related to amino acid metabolism. Specifically, while arginosuccinate synthase 1 (ASS1) is known to play an important role in the urea cycle via ammonia detoxification through the conversion of citrulline to arginine [40], the study by Yoon et al. revealed an absence or decrease of ASS1 in ccRCC; therefore, arginine was identified as an auxotrophic marker for ccRCC [41]. With the use of proteomics, another study by Perroud et al. similarly demonstrated the downregulation of ASS1 across all the grades of ccRCC [34]. Metabolic profiling using metabolomics technologies [42] has revealed different key metabolic phenotypes of RCC and identified potential targets for new treatments for RCC patients.
3.2 Temporal Impact of RCC Metabolism (Fig. 2)
Using the Fuhrman grading system, Wettersten et al. demonstrated that different grades of RCC were associated with different biochemical processes that have distinctive and prominent metabolic reprogramming, which is supported by the observation of clinically distinguishable features within various tumor grades [43]. In addition, Kang et al. confirmed the metabolic reprogramming in ccRCC by demonstrating the stage-dependent alteration in the expression of solute carriers (SLCs) [44]. SLCs have an important role in transmembrane transportation of specific metabolic substrates, including inorganic ions, nucleotides, amino acids, fatty acids, and sugars [45].
Using a combination of proteomics and metabolomics analysis, Wettersten et al. correlated increasing amounts of metabolites in the aerobic glycolysis pathway, including glucose, glucose-6-phosphate, and fructose-6-phosphate, with higher tumor grade. Likewise, an indication that glucose metabolism is reprogrammed to the lactate fermentation pathway, especially in advanced disease, is supported by the grade-dependent upregulation of lactate [43]. Consistently, a study by Hakimi et al. reported a more than twofold downregulation of citrate concentration in high-stage tumors [46] that may contribute to a reduction in oxidative phosphorylation capacity. However, in a study conducted by Perroud et al., the correlation of the upregulation of phosphoglycerate kinase 1 (PGK1) and grade 1 and 2 tumors is debatable [34]. On the other hand, Hakimi et al. showed that high levels of galactose and mannose were associated with an advanced stage of tumor progression [46].
A study by Horiguchi et al. first revealed the correlation between fatty acid synthase expression and high grade and metastasis of RCC [47]. On the other hand, Wettersten et al. found a broad reduction in shorter chain free fatty acid (FFA) oxidation in high-grade tumors, whereas there was an increase in carnitine and acylcarnitine levels in all grades of RCC. In accordance with this finding, Hakimi et al. discovered in high-stage tumors a reduction in cis-aconitate and medium-chain fatty acids [43, 46]. Furthermore, Xiao et al. showed that genes related to fatty acid β-oxidation enzymes, EHHADH (3-hydroxyacyl CoA dehydrogenase and enoyl-CoA hydratase) and ACADM (medium-chain acyl-CoA dehydrogenase), were simultaneously downregulated at the mRNA and protein levels in opposite proportion to stages of ccRCC [48].
Glutathione (GSH)/glutathione disulfide (GSSG) ratio is an important indicator of oxidative stress. Wettersten et al. revealed that this ratio is grade dependent and consistent with the downregulation of the catabolic enzymes responsible for utilizing glutamine and glutamate in the TCA and urea cycles, indicating a protective role of glutamine reprogramming against oxidative stress in high-grade tumors [43, 49]. Similarly, upregulation of the majority of metabolites involved in the glutathione synthesis such as cysteine, γ-glutamyl cysteine (GLU-CYS), and GSH was observed by Hakimi et al. in a stage-dependent manner [46]. Furthermore, they demonstrated that methionine augmentation at higher tumor stages could be related to the upregulation of glutathione metabolism. In fact, the glutathione regeneration pathway is carried out by homocysteine being converted to cysteine via cystathionine [50].
Interestingly, Hakimi et al. found a grade-dependent increase in the level of methylthioadenosine (MTA). MTA is a downstream product of the polyamine biosynthetic pathway, implying that alterations in the polyamine pathway are correlated to tumor progression [46]. MTA is also part of the methionine and adenine salvage pathway, where phosphorylation of MTA via methylthioadenosine phosphorylase (MTAP) leads to the regeneration of adenine and methionine [51]. Using metabolomic approaches, Xu et al. recently identified a stage-dependent decrease in the expression of MTAP that consequently resulted in the accumulation of MTA in RCC cells [52].
3.3 Intratumoral Heterogeneity of RCC (Fig. 2)
3.3.1 Gene Independence
While intratumoral genetic heterogeneity has been extensively investigated in ccRCC [53, 54], only a small number of recent studies has documented the regional variations in metabolic patterns [55,56,57]. Specifically, mutations of SET domain containing 2 (SETD2), PTEN, and lysine-specific histone demethylase (KDM5C) were found within the same tumor, suggesting that convergent phenotypic evolution and mutations of VHL were ubiquitously detected by multi-region sequencing in all analyzed regions [54]. Using global metabolomics analyses, Okegawa et al. investigated the heterogeneity of intratumoral metabolic profile and gene mutations in multiple regions of a single primary RCC tumor. Two major metabolically different tumor clusters, metabolic cluster 1 (MC1) and metabolic cluster 2 (MC2), were identified among 32 spatially separated sections of the tumor tissue. They observed an upregulation of the glycolysis-PPP, glutathione, and amino acids in the MC1 region as opposed to the downregulation of glycolysis-PPP metabolites and glutathione (reduced) in MC2 region. They also showed that the MC2 region had elevated levels of pyruvate, cystine, and 2-oxobutyric acid. This suggested that tumor cells are dependent on the presence of pyruvate in MC2 regions for growth stimulation, which may make pyruvate metabolism an ideal therapeutic target [55]. Importantly, they showed that the Warburg-like effect is present in all tumor sites without any correlation to the status of the VHL gene. Ultimately, the clear intratumoral metabolic heterogeneity identified within a single tumor may contribute to treatment resistance [56, 57]. In contrast, an almost uniform pattern of lipid profile was observed across the samples [55, 58]. Interestingly, the study also showed no correlation between the mutational status of genes and metabolic patterns, suggesting that other factors, such as noncanonical metabolic flux, tumor microenvironment, and epigenetics, may regulate the metabolic phenotype [55].
Accumulating evidence from various cancer studies suggests the important role of long noncoding RNAs (lncRNAs) in different cellular processes, including metabolism [59,60,61] through regulation of gene expression at the epigenetic level [62, 63]. Specifically, a study by Li et al. found intertumoral heterogeneity of lncRNAs in ccRCC that can contribute to the regulation of the HIF-1 signaling pathway [64].
Using hyperpolarized carbon-13 (13C) magnetic resonance imaging (HP-MRI) technique, Tran et al. noted the intertumoral heterogeneity of pyruvate delivery, pyruvate-to-lactate metabolic conversion, and ratio of lactate to pyruvate across the RCC but did not investigate whether this heterogeneity is gene dependent or not [65].
Using Dixon-based MRI technique, Zhang et al. demonstrated the correlation between fat fraction and intratumoral heterogeneity of metabolites; however, the heterogeneity was more prominent between different tumor types [66].
3.3.2 Gene Dependence
Using [18F] fluorodeoxyglucose (FDG) and positron-emission tomography (PET)/MR imaging, Brooks et al. also demonstrated that the presence of glycolytic enzyme FBP1, higher levels of glucose transporter GLUT1, and lower expression of FBP1 and HIF-1α transcription factor led to regional variation in metabolic heterogeneity. This study revealed an association between [18F]FDG avidity and distinct patterns of metabolic genes and protein expression. The [18F] FDG-avid region had a lower expression of FBP1 and HIF1a and higher expression of glucose transporter GLUT1, which could be related to a higher metabolic activity supported by boosting glucose uptake [22].
Polybromo-1 (PBRM1) gene is one of the major subunits of PBAF (polybromo-associated BRG1 or BRM-associated factors). Loss of its expression is considered as another common mutation in tumor-suppressor gene in ccRCC, besides VHL [67, 68]. Interestingly, Chowdhury et al. demonstrated that re-expression of PBAF in a PBAF-deficient ccRCC could lead to the reversion of several important metabolic signatures in RCC, including glucose and cholesterol metabolism [69]. Using tissue microarray (TMA)-based immunohistochemistry from each tumor, a study by Jiang et al. revealed different expressions of PBRM1, AT-rich interaction domain 1A (ARID1A), SETD2, brahma-related gene 1 (BRG1), and brahma gene (BRM) among 160 ccRCCs, indicating high intratumoral heterogeneity of PBRM1 expression and its associated proteins of the same tumor [70].
4 RCC Therapy
The current standard frontline therapies for metastatic RCC are largely VEGFR inhibitors, such as sunitinib and sorafenib. However, about 20–30% of patients do not respond to these therapies, and among those, nearly all the patients become resistant to these drugs [71].
Activation of the mammalian target of rapamycin (mTOR), a member of PI3K-related kinases, through VEGF signaling pathways, can lead to protein synthesis and energy production in RCC [72]. Although a variety of agents have been investigated to target the mTOR pathway, only recently three mTOR inhibitors have been approved as second-line therapy in patients with RCC: temsirolimus (Torisel®–CCI-779, 4), everolimus or rad001 (Afinitor®, 4a), and sirolimus (Rapamycin®, 4b) [73, 74]. Interestingly, a study by Li et al. revealed that mTOR inhibitor (everolimus) treatment could overcome the Warburg effect via mTOR pathway blocking and HIF1α expression inhibition [75]. Recent work by Gameiro et al. found that loss of VHL rendered RCC cells sensitive to glutamine deprivation [76]. In line with this finding, they found that systematic treatment with glutaminase inhibitors suppressed ccRCC growth both in vitro and in vivo. Other metabolic targeting therapies for RCC include mitochondrial inhibition by auraptene [77] and GLUT1 inhibition by STF-3 [78].
5 Conclusion
Metabolic spatial and temporal heterogeneity represents one of the main mechanisms behind the high therapy resistance of RCC. Recent findings suggest that initial monotherapy failure can be associated with subclonal variation in metabolic heterogeneity. Targeting several metabolic pathways is the key strategy for effective therapy [79, 80]. It is also important to mention that metabolic phenotypic heterogeneity is regulated through both gene-dependent and gene-independent manners. Taken together, RCC treatment has a variety of challenges posed by intratumoral metabolic heterogeneity, highlighting the need for the further development of novel approaches to identify clonally dominant metabolic targets for the future development of effective therapies for RCC.
Abbreviations
- ACADM:
-
Medium-chain acyl-CoA dehydrogenase
- ACC:
-
Acetyl-CoA carboxylase
- AMPK:
-
AMP-activated protein kinase
- ARID1A:
-
AT-rich interaction domain 1A
- ASS1:
-
Arginosuccinate synthase
- ATP:
-
Adenosine triphosphate
- BRG1:
-
Brahma-related gene 1
- BRM:
-
Brahma gene
- ccRCC:
-
Clear-cell renal cell carcinoma
- ePC:
-
Ether-type PC
- ePE:
-
Ether-type PE
- ERRα:
-
Estrogen-related receptor Α
- FASN:
-
Fatty acid synthase
- FDG:
-
Fluorodeoxyglucose
- FFAs:
-
Free fatty acids
- FH:
-
Fumarate hydratase
- G6PH:
-
Glucose-6-phosphate dehydrogenase
- GLS2:
-
Glutaminase 2
- Glu:
-
Glutamine
- GLU-CYS:
-
Cysteine, γ-glutamyl cysteine
- GLUT1:
-
Glucose transporter 1
- GLUT2:
-
Glucose transporter 2
- GSH:
-
Glutathione
- GSSG:
-
Glutathione disulfide
- HCC:
-
Hepatocellular carcinoma
- HIF:
-
Hypoxia-inducible factor
- HIF-1α:
-
Hypoxia-inducible factor 1-alpha
- HK2:
-
Hexokinase 2
- KDM5C:
-
Lysine-specific histone demethylase 5C
- LDHA:
-
Lactate dehydrogenase A
- lncRNAs:
-
Long noncoding RNAs
- MC1:
-
Metabolic cluster 1
- MC2:
-
Metabolic cluster 2
- MIR21:
-
MicroRNA 21
- MRI:
-
Magnetic resonance imaging
- mTOR:
-
Mammalian target of rapamycin
- mTORC1:
-
Mechanistic target of rapamycin complex 1
- NADPH:
-
Nicotinamide adenine dinucleotide phosphate
- NAE:
-
N-acylethanolamine
- PBRM1:
-
Polybromo-1
- PC:
-
Phosphatidylcholine
- PE:
-
Phosphatidylethanolamine
- PET:
-
Positron-emission tomography
- PGK1:
-
Phosphoglycerate kinase 1
- PGLS:
-
6-Phosphogluconolactonase
- PHD:
-
Prolyl hydroxylase
- PI3K:
-
Phosphatidylinositol-3 kinases
- PKM2:
-
Pyruvate kinase isoform M2
- PPP:
-
Pentose phosphate pathway
- PTEN:
-
Phosphatase and tensin homolog deleted in chromosome 10
- PS:
-
Phosphatidylserine
- RCC:
-
Renal cell carcinoma
- ROS:
-
Reactive oxygen species
- SETD2:
-
SET domain containing 2
- SLCs:
-
Solute carriers
- SM:
-
Sphingomyelin
- TALDO:
-
Transaldolase
- TCA:
-
Tricarboxylic acid
- TKT:
-
Transketolase
- TSC2:
-
Tuberous sclerosis 2
- VEGFR:
-
Vascular endothelial growth factor receptor
- VHL:
-
Von Hippel-Lindau tumor-suppressor gene
- α-KG:
-
α-Ketoglutarate
References
Jemal, A., et al. (2011). Global cancer statistics. CA: A Cancer Journal for Clinicians, 61(2), 69–90.
Global Burden of Disease Study. (2016). Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet, 388(10053), 1459–1544.
Siegel, R. L., Miller, K. D., & Jemal, A. (2017). Cancer statistics, 2017. CA: A Cancer Journal for Clinicians, 67(1), 7–30.
Akinyemiju, T., et al. (2017). The burden of primary liver cancer and underlying etiologies from 1990 to 2015 at the global, regional, and national level: Results from the Global Burden of Disease Study 2015. JAMA Oncology, 3(12), 1683–1691.
Gu, F. L., et al. (1991). Cellular origin of renal cell carcinoma--an immunohistological study on monoclonal antibodies. Scandinavian Journal of Urology and Nephrology. Supplementum, 138, 203–206.
Rini, B. I., Campbell, S. C., & Escudier, B. (2009). Renal cell carcinoma. Lancet, 373(9669), 1119–1132.
Sudarshan, S., et al. (2013). Metabolism of kidney cancer: From the lab to clinical practice. European Urology, 63(2), 244–251.
Sato, Y., et al. (2013). Integrated molecular analysis of clear-cell renal cell carcinoma. Nature Genetics, 45(8), 860–867.
Nickerson, M. L., et al. (2008). Improved identification of von Hippel-Lindau gene alterations in clear cell renal tumors. Clinical Cancer Research, 14(15), 4726–4734.
Czyzyk-Krzeska, M. F., & Meller, J. (2004). von Hippel-Lindau tumor suppressor: Not only HIF’s executioner. Trends in Molecular Medicine, 10(4), 146–149.
Stubbs, M., & Griffiths, J. R. (2010). The altered metabolism of tumors: HIF-1 and its role in the Warburg effect. Advances in Enzyme Regulation, 50(1), 44–55.
Gordan, J. D., Thompson, C. B., & Simon, M. C. (2007). HIF and c-Myc: Sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell, 12(2), 108–113.
Pinthus, J. H., et al. (2011). Metabolic features of clear-cell renal cell carcinoma: Mechanisms and clinical implications. Canadian Urological Association Journal, 5(4), 274–282.
Semenza, G. L. (2007). HIF-1 mediates the Warburg effect in clear cell renal carcinoma. Journal of Bioenergetics and Biomembranes, 39(3), 231–234.
Gill, A. J., et al. (2014). Succinate dehydrogenase (SDH)-deficient renal carcinoma: A morphologically distinct entity: A clinicopathologic series of 36 tumors from 27 patients. The American Journal of Surgical Pathology, 38(12), 1588–1602.
Sulpice, E., et al. (2009). Cross-talk between the VEGF-A and HGF signalling pathways in endothelial cells. Biology of the Cell, 101(9), 525–539.
The Cancer Genome Atlas Research Network. (2013). Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature, 499(7456), 43–49.
Yang, Y., et al. (2010). UOK 262 cell line, fumarate hydratase deficient (FH-/FH-) hereditary leiomyomatosis renal cell carcinoma: In vitro and in vivo model of an aberrant energy metabolic pathway in human cancer. Cancer Genetics and Cytogenetics, 196(1), 45–55.
Tong, W. H., et al. (2011). The glycolytic shift in fumarate-hydratase-deficient kidney cancer lowers AMPK levels, increases anabolic propensities and lowers cellular iron levels. Cancer Cell, 20(3), 315–327.
Massari, F., et al. (2015). Metabolic alterations in renal cell carcinoma. Cancer Treatment Reviews, 41(9), 767–776.
Ishikawa, K., et al. (2008). ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science, 320(5876), 661–664.
Brooks, S. A., et al. (2016). Alternate metabolic programs define regional variation of relevant biological features in renal cell carcinoma progression. Clinical Cancer Research, 22(12), 2950–2959.
Le, A., et al. (2010). Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proceedings of the National Academy of Sciences of the United States of America, 107(5), 2037–2042.
Rajeshkumar, N. V., et al. (2015). Therapeutic targeting of the Warburg effect in pancreatic cancer relies on an absence of p53 function. Cancer Research, 75(16), 3355–3364.
Dutta, P., et al. (2013). Evaluation of LDH-A and glutaminase inhibition in vivo by hyperpolarized 13C-pyruvate magnetic resonance spectroscopy of tumors. Cancer Research, 73(14), 4190–4195.
Bose, S., Zhang, C., & Le, A. (2021). Glucose metabolism in cancer: The Warburg effect and beyond. Advances in Experimental Medicine and Biology, 1311, https://doi.org/10.1007/978-3-030-65768-0_1
Liu, J., et al. (2013). Metformin inhibits renal cell carcinoma in vitro and in vivo xenograft. Urologic Oncology, 31(2), 264–270.
Saito, K., et al. (2016). Lipidomic signatures and associated transcriptomic profiles of clear cell renal cell carcinoma. Scientific Reports, 6, 28932.
Fagone, P., & Jackowski, S. (2013). Phosphatidylcholine and the CDP-choline cycle. Biochimica et Biophysica Acta, 1831(3), 523–532.
Vance, J. E., & Tasseva, G. (2013). Formation and function of phosphatidylserine and phosphatidylethanolamine in mammalian cells. Biochimica et Biophysica Acta, 1831(3), 543–554.
Vance, J. E. (2008). Phosphatidylserine and phosphatidylethanolamine in mammalian cells: Two metabolically related aminophospholipids. Journal of Lipid Research, 49(7), 1377–1387.
Farine, L., & Bütikofer, P. (2013). The ins and outs of phosphatidylethanolamine synthesis in Trypanosoma brucei. Biochimica et Biophysica Acta, 1831(3), 533–542.
Christofk, H. R., et al. (2008). Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature, 452(7184), 181–186.
Perroud, B., et al. (2009). Grade-dependent proteomics characterization of kidney cancer. Molecular & Cellular Proteomics, 8(5), 971–985.
Barceló-Coblijn, G., et al. (2011). Sphingomyelin and sphingomyelin synthase (SMS) in the malignant transformation of glioma cells and in 2-hydroxyoleic acid therapy. Proceedings of the National Academy of Sciences of the United States of America, 108(49), 19569–19574.
Ding, T., et al. (2008). SMS overexpression and knockdown: Impact on cellular sphingomyelin and diacylglycerol metabolism, and cell apoptosis. Journal of Lipid Research, 49(2), 376–385.
Catchpole, G., et al. (2011). Metabolic profiling reveals key metabolic features of renal cell carcinoma. Journal of Cellular and Molecular Medicine, 15(1), 109–118.
Menendez, J. A., & Lupu, R. (2007). Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nature Reviews. Cancer, 7(10), 763–777.
Ham, A. J., & Liebler, D. C. (1997). Antioxidant reactions of vitamin E in the perfused rat liver: Product distribution and effect of dietary vitamin E supplementation. Archives of Biochemistry and Biophysics, 339(1), 157–164.
Haines, R. J., Pendleton, L. C., & Eichler, D. C. (2011). Argininosuccinate synthase: At the center of arginine metabolism. International Journal of Biochemistry and Molecular Biology, 2(1), 8–23.
Yoon, C. Y., et al. (2007). Renal cell carcinoma does not express argininosuccinate synthetase and is highly sensitive to arginine deprivation via arginine deiminase. International Journal of Cancer, 120(4), 897–905.
Hoang, G., Udupa, S., & Le, A. (2019). Application of metabolomics technologies toward cancer prognosis and therapy. International Review of Cell and Molecular Biology, 347, 191–223.
Wettersten, H. I., et al. (2015). Grade-dependent metabolic reprogramming in kidney cancer revealed by combined proteomics and metabolomics analysis. Cancer Research, 75(12), 2541–2552.
Kang, W., et al. (2020). The SLC family are candidate diagnostic and prognostic biomarkers in clear cell renal cell carcinoma. BioMed Research International, 2020, 1932948.
Hediger, M. A., et al. (2004). The ABCs of solute carriers: Physiological, pathological and therapeutic implications of human membrane transport proteins: Introduction. Pflügers Archiv, 447(5), 465–468.
Hakimi, A. A., et al. (2016). An integrated metabolic atlas of clear cell renal cell carcinoma. Cancer Cell, 29(1), 104–116.
Horiguchi, A., et al. (2008). Fatty acid synthase over expression is an indicator of tumor aggressiveness and poor prognosis in renal cell carcinoma. The Journal of Urology, 180(3), 1137–1140.
Xiao, H., et al. (2019). Three novel hub genes and their clinical significance in clear cell renal cell carcinoma. Journal of Cancer, 10(27), 6779–6791.
Li, T., Copeland, C., & Le, A. (2021). Glutamine metabolism in cancer. Advances in Experimental Medicine and Biology, 1311, https://doi.org/10.1007/978-3-030-65768-0_2
Lu, S. C. (1999). Regulation of hepatic glutathione synthesis: current concepts and controversies. The FASEB Journal, 13(10), 1169–1183.
Kirovski, G., et al. (2011). Down-regulation of methylthioadenosine phosphorylase (MTAP) induces progression of hepatocellular carcinoma via accumulation of 5’-deoxy-5’-methylthioadenosine (MTA). The American Journal of Pathology, 178(3), 1145–1152.
Xu, J., et al. (2019). Targeting the insulin-like growth factor-1 receptor in MTAP-deficient renal cell carcinoma. Signal Transduction and Targeted Therapy, 4, 2.
Gerlinger, M., et al. (2014). Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nature Genetics, 46(3), 225–233.
Gerlinger, M., et al. (2012). Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. The New England Journal of Medicine, 366(10), 883–892.
Okegawa, T., et al. (2017). Intratumor heterogeneity in primary kidney cancer revealed by metabolic profiling of multiple spatially separated samples within tumors. eBioMedicine, 19, 31–38.
Nabi, K., & Le, A. (2021). The intratumoral heterogeneity of cancer metabolism. Advances in Experimental Medicine and Biology, 1311, https://doi.org/10.1007/978-3-030-65768-0_11
Antonio, M. J., Zhang, C., & Le, A. (2021). Different tumor microenvironments lead to different metabolic phenotypes. Advances in Experimental Medicine and Biology, 1311, https://doi.org/10.1007/978-3-030-65768-0_10
Park, J. K., et al. (2021). The heterogeneity of lipid metabolism in cancer. Advances in Experimental Medicine and Biology, 1311, https://doi.org/10.1007/978-3-030-65768-0_3
Xiong, H., et al. (2017). LncRNA HULC triggers autophagy via stabilizing Sirt1 and attenuates the chemosensitivity of HCC cells. Oncogene, 36(25), 3528–3540.
Rupaimoole, R., et al. (2015). Long noncoding RNA ceruloplasmin promotes cancer growth by altering glycolysis. Cell Reports, 13(11), 2395–2402.
Zheng, J., et al. (2016). Pancreatic cancer risk variant in LINC00673 creates a miR-1231 binding site and interferes with PTPN11 degradation. Nature Genetics, 48(7), 747–757.
Schmitt, A. M., & Chang, H. Y. (2016). Long noncoding RNAs in cancer pathways. Cancer Cell, 29(4), 452–463.
Huarte, M. (2015). The emerging role of lncRNAs in cancer. Nature Medicine, 21(11), 1253–1261.
Li, X., et al. (2018). Dissecting LncRNA roles in renal cell carcinoma metastasis and characterizing genomic heterogeneity by single-cell RNA-seq. Molecular Cancer Research, 16(12), 1879–1888.
Tran, M., et al. (2019). First-in-human in vivo non-invasive assessment of intra-tumoral metabolic heterogeneity in renal cell carcinoma. BJR Case Report, 5, 3.
Zhang, Y., et al. (2017). Addressing metabolic heterogeneity in clear cell renal cell carcinoma with quantitative Dixon MRI. JCI Insight, 2, 15.
Peña-Llopis, S., et al. (2012). BAP1 loss defines a new class of renal cell carcinoma. Nature Genetics, 44(7), 751–759.
Varela, I., et al. (2011). Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature, 469(7331), 539–542.
Chowdhury, B., et al. (2016). PBRM1 regulates the expression of genes involved in metabolism and cell adhesion in renal clear cell carcinoma. PLoS One, 11(4), e0153718.
Jiang, W., et al. (2016). Immunohistochemistry successfully uncovers intratumoral heterogeneity and widespread co-losses of chromatin regulators in clear cell renal cell carcinoma. PLoS One, 11(10), e0164554.
Rini, B. I., & Atkins, M. B. (2009). Resistance to targeted therapy in renal-cell carcinoma. The Lancet Oncology, 10(10), 992–1000.
Saxton, R. A., & Sabatini, D. M. (2017). mTOR signaling in growth, metabolism, and disease. Cell, 169(2), 361–371.
Masoud, G. N., & Li, W. (2015). HIF-1α pathway: Role, regulation and intervention for cancer therapy. Acta Pharmaceutica Sinica B, 5(5), 378–389.
Kamli, H., Li, L., & Gobe, G. C. (2019). Limitations to the therapeutic potential of tyrosine kinase inhibitors and alternative therapies for kidney cancer. The Ochsner Journal, 19(2), 138–151.
Li, X., et al. (2020). The tumor suppressor NDRG2 cooperates with an mTORC1 inhibitor to suppress the Warburg effect in renal cell carcinoma. Investigational New Drugs, 38(4), 956–966.
Gameiro, P. A., et al. (2013). In vivo HIF-mediated reductive carboxylation is regulated by citrate levels and sensitizes VHL-deficient cells to glutamine deprivation. Cell Metabolism, 17(3), 372–385.
Jang, Y., et al. (2015). Suppression of mitochondrial respiration with auraptene inhibits the progression of renal cell carcinoma: Involvement of HIF-1α degradation. Oncotarget, 6(35), 38127–38138.
Chan, D. A., et al. (2011). Targeting GLUT1 and the Warburg effect in renal cell carcinoma by chemical synthetic lethality. Science Translational Medicine, 3(94), 94ra70.
Dang, C. V., et al. (2011). Therapeutic targeting of cancer cell metabolism. Journal of Molecular Medicine (Berlin), 89(3), 205–212.
Hirschey, M. D., et al. (2015). Dysregulated metabolism contributes to oncogenesis. Seminars in Cancer Biology, 35(Suppl), S129–S150.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2021 The Author(s)
About this chapter

Cite this chapter
Zarisfi, M., Nguyen, T., Nedrow, J.R., Le, A. (2021). The Heterogeneity Metabolism of Renal Cell Carcinomas. In: Le, A. (eds) The Heterogeneity of Cancer Metabolism. Advances in Experimental Medicine and Biology, vol 1311. Springer, Cham. https://doi.org/10.1007/978-3-030-65768-0_8
Download citation
DOI: https://doi.org/10.1007/978-3-030-65768-0_8
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-65767-3
Online ISBN: 978-3-030-65768-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)