
Abstract
Metabolic cardiomyopathies represent a large heterogeneous group of diseases caused by inborn errors of metabolism (IEM). So far, more than 1000 distinct disorders of metabolism have been described. Cardiac involvement is present in about 5% of these metabolic defects. While in some diseases, the cardiac involvement is representing the dominant and potentially fatal condition, in the others it may be only part of a complex multisystemic disease manifestation.
Cardiac involvement has been described in various types of metabolic defects including lysosomal storage diseases, different types of glycogenoses and other inborn errors of metabolism such as disorders of amino acid metabolism, organic acid metabolism, and fatty acid metabolism. In broader term of the definition, metabolic cardiomyopathies include also oxidative phosphorylation defects (OXPHOS) usually listed among mitochondrial diseases.
Most IEM are inherited as autosomal recessive traits, however, some highly prevalent diseases with cardiac involvement are X-linked (Anderson Fabry disease, Hunter syndrome—type II mucopolysaccharidosis, Danon disease).
Cardiomyopathies caused by IEM are phenotypically heterogeneous, most of them characterized by cardiac hypertrophy and/or by different degrees of progressive dilatation and systolic dysfunction resulting in dilated cardiomyopathy phenotype. However, several diseases may appear as restrictive cardiomyopathy phenotype. The tableau of the disease may include different electrophysiological changes (short PR, preexcitation pattern, AV conduction abnormalities) and/or valvular involvement.
Access this chapter
Tax calculation will be finalised at checkout
Purchases are for personal use only
Similar content being viewed by others
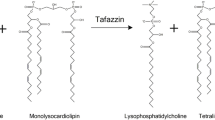
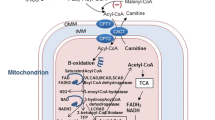

References
Sweet ME, Mestroni L, Taylor MRG. Genetic infiltrative cardiomyopathies. Heart Fail Clin. 2018;14(2):215–24.
Cox GF. Diagnostic approaches to pediatric cardiomyopathy of metabolic genetic etiologies and their relation to therapy. Prog Pediatr Cardiol. 2007;24(1):15–25.
Fukao T, Sasai H, Aoyama Y, Otsuka H, Ago Y, Matsumoto H, Abdelkreem E. Recent advances in understanding beta-ketothiolase (mitochondrial acetoacetyl-CoA thiolase, T2) deficiency. J Hum Genet. 2019;64(2):99–111.
Arora N, Stumper O, Wright J, Kelly DA, McKiernan PJ. Cardiomyopathy in tyrosinaemia type I is common but usually benign. J Inherit Metab Dis. 2006;29(1):54–7.
Lorenzo V, Torres A, Salido E. Primary hyperoxaluria. Nefrologia. 2014;34(3):398–412.
Merritt JL 2nd, Norris M, Kanungo S. Fatty acid oxidation disorders. Ann Transl Med. 2018;6(24):473.
Nair V, Belanger EC, Veinot JP. Lysosomal storage disorders affecting the heart: a review. Cardiovasc Pathol. 2019;39:12–24.
Linhart A, Cecchi F. Common presentation of rare diseases: left ventricular hypertrophy and diastolic dysfunction. Int J Cardiol. 2018;257:344–50.
Arbustini E, Di Toro A, Giuliani L, Favalli V, Narula N, Grasso M. Cardiac phenotypes in hereditary muscle disorders: JACC state-of-the-art review. J Am Coll Cardiol. 2018;72(20):2485–506.
Akhtar M, Elliott P. The genetics of hypertrophic cardiomyopathy. Glob Cardiol Sci Pract. 2018;2018(3):36.
Lipshultz SE, Sleeper LA, Towbin JA, Lowe AM, Orav EJ, Cox GF, Lurie PR, McCoy KL, McDonald MA, Messere JE, Colan SD. The incidence of pediatric cardiomyopathy in two regions of the United States. N Engl J Med. 2003;348(17):1647–55.
Doheny D, Srinivasan R, Pagant S, Chen B, Yasuda M, Desnick RJ. Fabry Disease: prevalence of affected males and heterozygotes with pathogenic GLA mutations identified by screening renal, cardiac and stroke clinics, 1995-2017. J Med Genet. 2018;55(4):261–8.
Towbin JA, Lowe AM, Colan SD, Sleeper LA, Orav EJ, Clunie S, Messere J, Cox GF, Lurie PR, Hsu D, Canter C, Wilkinson JD, Lipshultz SE. Incidence, causes, and outcomes of dilated cardiomyopathy in children. JAMA. 2006;296(15):1867–76.
Thevenon J, Laurent G, Ader F, Laforêt P, Klug D, Duva Pentiah A, Gouya L, Maurage CA, Kacet S, Eicher JC, Albuisson J, Desnos M, Bieth E, Duboc D, Martin L, Réant P, Picard F, Bonithon-Kopp C, Gautier E, Binquet C, Thauvin-Robinet C, Faivre L, Bouvagnet P, Charron P, Richard P. High prevalence of arrhythmic and myocardial complications in patients with cardiac glycogenosis due to PRKAG2 mutations. Europace. 2017;19(4):651–9.
Platt FM, Boland B, van der Spoel AC. The cell biology of disease: lysosomal storage disorders: the cellular impact of lysosomal dysfunction. J Cell Biol. 2012;199(5):723–34.
Burke MA, Cook SA, Seidman JG, Seidman CE. Clinical and Mechanistic Insights Into the Genetics of Cardiomyopathy. J Am Coll Cardiol. 2016;68(25):2871–86.
Vasilescu C, Ojala TH, Brilhante V, Ojanen S, Hinterding HM, Palin E, Alastalo TP, Koskenvuo J, Hiippala A, Jokinen E, Jahnukainen T, Lohi J, Pihkala J, Tyni TA, Carroll CJ, Suomalainen A. Genetic basis of severe childhood-onset cardiomyopathies. J Am Coll Cardiol. 2018;72(19):2324–38.
Lipshultz SE, Cochran TR, Briston DA, Brown SR, Sambatakos PJ, Miller TL, Carrillo AA, Corcia L, Sanchez JE, Diamond MB, Freundlich M, Harake D, Gayle T, Harmon WG, Rusconi PG, Sandhu SK, Wilkinson JD. Pediatric cardiomyopathies: causes, epidemiology, clinical course, preventive strategies and therapies. Future Cardiol. 2013;9(6):817–48.
Linhart A, Palecek T, Bultas J, Ferguson JJ, Hrudová J, Karetová D, Zeman J, Ledvinová J, Poupetová H, Elleder M, Aschermann M. New insights in cardiac structural changes in patients with Fabry’s disease. Am Heart J. 2000;139(6):1101–8.
Lieske JC, Monico CG, Holmes WS, Bergstralh EJ, Slezak JM, Rohlinger AL, Olson JB, Milliner DS. International registry for primary hyperoxaluria. Am J Nephrol. 2005;25(3):290–6.
Salido E, Pey AL, Rodriguez R, Lorenzo V. Primary hyperoxalurias: disorders of glyoxylate detoxification. Biochim Biophys Acta. 2012;1822(9):1453–64.
Wilcken B, Wiley V, Sim KG, Carpenter K. Carnitine transporter defect diagnosed by newborn screening with electrospray tandem mass spectrometry. J Pediatr. 2001;138(4):581–4.
Fu L, Huang M, Chen S. Primary carnitine deficiency and cardiomyopathy.Korean. Circ J. 2013;43(12):785–92.
Frigeni M, Balakrishnan B, Yin X, Calderon FRO, Mao R, Pasquali M, Longo N. Functional and molecular studies in primary carnitine deficiency. Hum Mutat. 2017;38(12):1684–99.
Guertl B, Noehammer C, Hoefler G. Metabolic cardiomyopathies. Int J Exp Pathol. 2000;81(6):349–72.
Vishwanath VA. Fatty acid beta-oxidation disorders: a brief review. Ann Neurosci. 2016;23(1):51–5.
Kang E, Kim YM, Kang M, Heo SH, Kim GH, Choi IH, Choi JH, Yoo HW, Lee BH. Clinical and genetic characteristics of patients with fatty acid oxidation disorders identified by newborn screening. BMC Pediatr. 2018;18(1):103.
Mehta A, Ricci R, Widmer U, Dehout F, Garcia de Lorenzo A, Kampmann C, Linhart A, Sunder-Plassmann G, Ries M, Beck M. Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry Outcome Survey. Eur J Clin Invest. 2004;34(3):236–242.
Echevarria L, Benistan K, Toussaint A, Dubourg O, Hagege AA, Eladari D, Jabbour F, Beldjord C, De Mazancourt P, Germain DP. X-chromosome inactivation in female patients with Fabry disease. Clin Genet. 2016;89(1):44–54.
Linhart A, Elliott PM. The heart in Anderson-Fabry disease and other lysosomal storage disorders. Heart. 2007;93(4):528–35.
Linhart A, Kampmann C, Zamorano JL, Sunder-Plassmann G, Beck M, Mehta A, Elliott PM, European FOS Investigators. Cardiac manifestations of Anderson-Fabry disease: results from the international Fabry outcome survey. Eur Heart J. 2007;28(10):1228–35.
Militaru S, Ginghina C, Popescu BA, Saftoiu A, Linhart A, Jurcut R. Multimodality imaging in Fabry cardiomyopathy: from early diagnosis to therapeutic targets. Eur Heart J Cardiovasc Imaging. 2018;19(12):1313–22.
Germain DP, Brand E, Burlina A, Cecchi F, Garman SC, Kempf J, Laney DA, Linhart A, Maródi L, Nicholls K, Ortiz A, Pieruzzi F, Shankar SP, Waldek S, Wanner C, Jovanovic A. Phenotypic characteristics of the p.Asn215Ser (p.N215S) GLA mutation in male and female patients with Fabry disease: a multicenter Fabry Registry study. Mol Genet Genomic Med. 2018. https://doi.org/10.1002/mgg3.389.
Nowak A, Mechtler T, Kasper DC, Desnick RJ. Correlation of Lyso-Gb3 levels in dried blood spots and sera from patients with classic and Later-Onset Fabry disease. Mol Genet Metab. 2017;121(4):320–4.
Kramer J, Weidemann F. Biomarkers for diagnosing and staging of Fabry disease. Curr Med Chem. 2018;25(13):1530–7.
Smid BE, van der Tol L, Cecchi F, Elliott PM, Hughes DA, Linthorst GE, Timmermans J, Weidemann F, West ML, Biegstraaten M, Lekanne Deprez RH, Florquin S, Postema PG, Tomberli B, van der Wal AC, van den Bergh Weerman MA, Hollak CE. Uncertain diagnosis of Fabry disease: consensus recommendation on diagnosis in adults with left ventricular hypertrophy and genetic variants of unknown significance. Int J Cardiol. 2014;177(2):400–8.
Germain DP, Elliott PM, Falissard B, Fomin VV, Hilz MJ, Jovanovic A, Kantola I, Linhart A, Mignani R, Namdar M, Nowak A, Oliveira JP, Pieroni M, Viana-Baptista M, Wanner C, Spada M. The effect of enzyme replacement therapy on clinical outcomes in male patients with Fabry disease: a systematic literature review by a European panel of experts. Mol Genet Metab Rep. 2019;19:100454.
Wanner C, Arad M, Baron R, Burlina A, Elliott PM, Feldt-Rasmussen U, Fomin VV, Germain DP, Hughes DA, Jovanovic A, Kantola I, Linhart A, Mignani R, Monserrat L, Namdar M, Nowak A, Oliveira JP, Ortiz A, Pieroni M, Spada M, Tylki-Szymańska A, Tøndel C, Viana-Baptista M, Weidemann F, Hilz MJ. European expert consensus statement on therapeutic goals in Fabry disease. Mol Genet Metab. 2018;124(3):189–203.
Ortiz A, Germain DP, Desnick RJ, Politei J, Mauer M, Burlina A, Eng C, Hopkin RJ, Laney D, Linhart A, Waldek S, Wallace E, Weidemann F, Wilcox WR. Fabry disease revisited: management and treatment recommendations for adult patients. Mol Genet Metab. 2018;123(4):416–27.
Hughes DA, Nicholls K, Shankar SP, Sunder-Plassmann G, Koeller D, Nedd K, Vockley G, Hamazaki T, Lachmann R, Ohashi T, Olivotto I, Sakai N, Deegan P, Dimmock D, Eyskens F, Germain DP, Goker-Alpan O, Hachulla E, Jovanovic A, Lourenco CM, Narita I, Thomas M, Wilcox WR, Bichet DG, Schiffmann R, Ludington E, Viereck C, Kirk J, Yu J, Johnson F, Boudes P, Benjamin ER, Lockhart DJ, Barlow C, Skuban N, Castelli JP, Barth J, Feldt-Rasmussen U. Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomised phase III ATTRACT study. J Med Genet. 2017;54(4):288–96.
Welford RWD, Mühlemann A, Garzotti M, Rickert V, Groenen PMA, Morand O, Üçeyler N, Probst MR. Glucosylceramide synthase inhibition with lucerastat lowers globotriaosylceramide and lysosome staining in cultured fibroblasts from Fabry patients with different mutation types. Hum Mol Genet. 2018;27(19):3392–403.
Guérard N, Morand O, Dingemanse J. Lucerastat, an iminosugar with potential as substrate reduction therapy for glycolipid storage disorders: safety, tolerability, and pharmacokinetics in healthy subjects. Orphanet J Rare Dis. 2017;12(1):9.
Germain DP, Hughes DA, Nicholls K, Bichet DG, Giugliani R, Wilcox WR, Feliciani C, Shankar SP, Ezgu F, Amartino H, Bratkovic D, Feldt-Rasmussen U, Nedd K, Sharaf El Din U, Lourenco CM, Banikazemi M, Charrow J, Dasouki M, Finegold D, Giraldo P, Goker-Alpan O, Longo N, Scott CR, Torra R, Tuffaha A, Jovanovic A, Waldek S, Packman S, Ludington E, Viereck C, Kirk J, Yu J, Benjamin ER, Johnson F, Lockhart DJ, Skuban N, Castelli J, Barth J, Barlow C, Schiffmann R. Treatment of Fabry’s disease with the pharmacologic chaperone migalastat. N Engl J Med. 2016; 375(6):545–55.
Khan SA, Peracha H, Ballhausen D, Wiesbauer A, Rohrbach M, Gautschi M, Mason RW, Giugliani R, Suzuki Y, Orii KE, Orii T, Tomatsu S. Epidemiology of mucopolysaccharidoses. Mol Genet Metab. 2017;121(3):227–40.
Muenzer J. Overview of the mucopolysaccharidoses. Rheumatology (Oxford). 2011;50 Suppl 5:v4–12.
Braunlin E, Wang R. Cardiac issues in adults with the mucopolysaccharidoses: current knowledge and emerging needs. Heart. 2016;102(16):1257–62.
Soliman OI, Timmermans RG, Nemes A, Vletter WB, Wilson JH, ten Cate FJ, Geleijnse ML. Cardiac abnormalities in adults with the attenuated form of mucopolysaccharidosis type I. J Inherit Metab Dis. 2007;30(5):750–7.
Wraith JE, Jones S. Mucopolysaccharidosis type I. Pediatr Endocrinol Rev. 2014;12(Suppl 1):102–6.
Hendriksz CJ, Berger KI, Giugliani R, Harmatz P, Kampmann C, Mackenzie WG, Raiman J, Villarreal MS, Savarirayan R. International guidelines for the management and treatment of Morquio A syndrome. Am J Med Genet A. 2015;167A(1):11–25.
Lin HY, Chuang CK, Chen MR, Lin SM, Hung CL, Chang CY, Chiu PC, Tsai WH, Niu DM, Tsai FJ, Lin SJ, Hwu WL, Lin JL, Lin SP. Cardiac structure and function and effects of enzyme replacement therapy in patients with mucopolysaccharidoses I, II, IVA and VI. Mol Genet Metab. 2016;117(4):431–7.
Kampmann C, Lampe C, Whybra-Trümpler C, Wiethoff CM, Mengel E, Arash L, Beck M, Miebach E. Mucopolysaccharidosis VI: cardiac involvement and the impact of enzyme replacement therapy. J Inherit Metab Dis. 2014;37(2):269–76.
Braunlin E, Rosenfeld H, Kampmann C, Johnson J, Beck M, Giugliani R, Guffon N, Ketteridge D, Sá Miranda CM, Scarpa M, Schwartz IV, Leão Teles E, Wraith JE, Barrios P, Dias da Silva E, Kurio G, Richardson M, Gildengorin G, Hopwood JJ, Imperiale M, Schatz A, Decker C, Harmatz P, MPS VI Study Group. Enzyme replacement therapy for mucopolysaccharidosis VI: long-term cardiac effects of galsulfase (Naglazyme®) therapy. J Inherit Metab Dis. 2013;36(2):385–94.
Montaño AM, Lock-Hock N, Steiner RD, Graham BH, Szlago M, Greenstein R, Pineda M, Gonzalez-Meneses A, Çoker M, Bartholomew D, Sands MS, Wang R, Giugliani R, Macaya A, Pastores G, Ketko AK, Ezgü F, Tanaka A, Arash L, Beck M, Falk RE, Bhattacharya K, Franco J, White KK, Mitchell GA, Cimbalistiene L, Holtz M, Sly WS. Clinical course of sly syndrome (mucopolysaccharidosis type VII). J Med Genet. 2016;53(6):403–18.
McCafferty EH, Scott LJ. Vestronidase alfa: a review in mucopolysaccharidosis VII. BioDrugs. 2019;33(2):233–40.
Hack HA, Walker R, Gardiner P. Anaesthetic implications of the changing management of patients with mucopolysaccharidosis. Anaesth Intensive Care. 2016;44(6):660–8.
Walker R, Belani KG, Braunlin EA, Bruce IA, Hack H, Harmatz PR, Jones S, Rowe R, Solanki GA, Valdemarsson B. Anaesthesia and airway management in mucopolysaccharidosis. J Inherit Metab Dis. 2013;36(2):211–9.
Braunlin EA, Harmatz PR, Scarpa M, Furlanetto B, Kampmann C, Loehr JP, Ponder KP, Roberts WC, Rosenfeld HM, Giugliani R. Cardiac disease in patients with mucopolysaccharidosis: presentation, diagnosis and management. J Inherit Metab Dis. 2011;34(6):1183–97.
Braunlin E, Orchard PJ, Whitley CB, Schroeder L, Reed RC, Manivel JC. Unexpected coronary artery findings in mucopolysaccharidosis. Report of four cases and literature review. Cardiovasc Pathol. 2014;23(3):145–51.
Boffi L, Russo P, Limongelli G. Early diagnosis and management of cardiac manifestations in mucopolysaccharidoses: a practical guide for paediatric and adult cardiologists. Ital J Pediatr. 2018;44(Suppl 2):122.
Muenzer J, Wraith JE. Clarke LA; International Consensus Panel on Management and Treatment of Mucopolysaccharidosis I. Mucopolysaccharidosis I: management and treatment guidelines. Pediatrics. 2009;123(1):19–29.
Aldenhoven M, Wynn RF, Orchard PJ, O’Meara A, Veys P, Fischer A, Valayannopoulos V, Neven B, Rovelli A, Prasad VK, Tolar J, Allewelt H, Jones SA, Parini R, Renard M, Bordon V, Wulffraat NM, de Koning TJ, Shapiro EG, Kurtzberg J, Boelens JJ. Long-term outcome of Hurler syndrome patients after hematopoietic cell transplantation: an international multicenter study. Blood. 2015;125(13):2164–72.
Laraway S, Mercer J, Jameson E, Ashworth J, Hensman P, Jones SA. Outcomes of long-term treatment with laronidase in patients with mucopolysaccharidosis type I. J Pediatr. 2016;178:219–26.e1.
Concolino D, Deodato F, Parini R. Enzyme replacement therapy: efficacy and limitations. Ital J Pediatr. 2018;44(Suppl 2):120
Muenzer J. Early initiation of enzyme replacement therapy for the mucopolysaccharidoses. Mol Genet Metab. 2014;111(2):63–72.
Hicks J, Wartchow E, Mierau G. Glycogen storage diseases: a brief review and update on clinical features, genetic abnormalities, pathologic features, and treatment. Ultrastruct Pathol. 2011;35(5):183–96.
Kishnani PS, Steiner RD, Bali D, Berger K, Byrne BJ, Case LE, Crowley JF, Downs S, Howell RR, Kravitz RM, Mackey J, Marsden D, Martins AM, Millington DS, Nicolino M, O’Grady G, Patterson MC, Rapoport DM, Slonim A, Spencer CT, Tifft CJ, Watson MS. Pompe disease diagnosis and management guideline. Genet Med. 2006;8(5):267–88.
Bembi B, Cerini E, Danesino C, Donati MA, Gasperini S, Morandi L, Musumeci O, Parenti G, Ravaglia S, Seidita F, Toscano A, Vianello A. Diagnosis of glycogenosis type II. Neurology. 2008;71(23 Suppl 2):S4–11.
Chan J, Desai AK, Kazi ZB, Corey K, Austin S, Hobson-Webb LD, Case LE, Jones HN, Kishnani PS. The emerging phenotype of late-onset Pompe disease: a systematic literature review. Mol Genet Metab. 2017;120(3):163–72.
Boentert M, Florian A, Dräger B, Young P, Yilmaz A. Pattern and prognostic value of cardiac involvement in patients with late-onset Pompe disease: a comprehensive cardiovascular magnetic resonance approach. J Cardiovasc Magn Reson. 2016;18(1):91.
Fayssoil A. Cardiomyopathy in Pompe’s disease. Eur J Intern Med. 2008;19(1):57–9.
Schoser B, Stewart A, Kanters S, Hamed A, Jansen J, Chan K, Karamouzian M, Toscano A. Survival and long-term outcomes in late-onset Pompe disease following alglucosidase alfa treatment: a systematic review and meta-analysis. J Neurol. 2017;264(4):621–30.
Joanne M, Skye N, Tracy M. The effectiveness of enzyme replacement therapy for juvenile-onset Pompe disease: a systematic review. J Inherit Metab Dis. 2019;42(1):57–65.
Vita G, Vita GL, Musumeci O, Rodolico C, Messina S. Genetic neuromuscular disorders: living the era of a therapeutic revolution. Part 2: diseases of motor neuron and skeletal muscle. Neurol Sci. 2019;40(4):671–81.
D’souza RS, Levandowski C, Slavov D, Graw SL, Allen LA, Adler E, Mestroni L, Taylor MR. Danon disease: clinical features, evaluation, and management. Circ Heart Fail. 2014;7(5):843–9.
Nascimbeni AC, Fanin M, Angelini C, Sandri M. Autophagy dysregulation in Danon disease. Cell Death Dis. 2017;8(1):e2565.
Konrad T, Sonnenschein S, Schmidt FP, Mollnau H, Bock K, Ocete BQ, Münzel T, Theis C, Rostock T. Cardiac arrhythmias in patients with Danon disease. Europace. 2017;19(7):1204–10.
Sternick EB, Oliva A, Magalhães LP, Gerken LM, Hong K, Santana O, Brugada P, Brugada J, Brugada R. Familial pseudo-Wolff-Parkinson-White syndrome. J Cardiovasc Electrophysiol. 2006;17(7):724–32.
Arad M. Cardiac Danon disease: insights and challenges. Int J Cardiol. 2017;245:211–2.
Rowland TJ, Sweet ME, Mestroni L, Taylor MR. Danon disease - dysregulation of autophagy in a multisystem disorder with cardiomyopathy. J Cell Sci. 2016;129(11):2135–43.
Brambatti M, Caspi O, Maolo A, Koshi E, Greenberg B, Taylor MRG, Adler ED. Danon disease: gender differences in presentation and outcomes. Int J Cardiol. 2019;286:92–8.
Konrad T, Sonnenschein S, Schmidt FP, Mollnau H, Bock K, Ocete BQ, Münzel T, Theis C, Rostock T. Cardiac arrhythmias in patients with Danon disease. Europace. 2017;19(7):1204–10.
Boucek D, Jirikowic J, Taylor M. Natural history of Danon disease. Genet Med. 2011;13:563–8.
Stokes MB, Taylor AJ, McLean CA, D’Arcy CE, Mariani JA. Severe left ventricular hypertrophy and marked cardiac fibrosis in Danon disease. Int J Cardiol. 2016;221:14–6.
Ng KM, Mok PY, Butler AW, Ho JC, Choi SW, Lee YK, Lai WH, Au KW, Lau YM, Wong LY, Esteban MA, Siu CW, Sham PC, Colman A, Tse HF. Amelioration of X-linked related autophagy failure in Danon disease with DNA methylation inhibitor. Circulation. 2016;134(18):1373–89.
Finsterer J, Stöllberger C, Maeztu C. Sudden cardiac death in neuromuscular disorders. Int J Cardiol. 2016;203:508–15.
Zaha VYL. Amp-activated protein kinase regulation and biological actions in the heart. Circ Res. 2012;111:800–14.
Hedberg-Oldfors C, Oldfors A. Polyglucosan storage myopathies. Mol Aspects Med. 2015;46:85–100.
Murphy RT, Mogensen J, McGarry K, Bahl A, Evans A, Osman E, Syrris P, Gorman G, Farrell M, Holton JL, Hanna MG, Hughes S, Elliott PM, Macrae CA, McKenna WJ. Adenosine monophosphate-activated protein kinase disease mimicks hypertrophic cardiomyopathy and Wolff-Parkinson-White syndrome: natural history. J Am Coll Cardiol. 2005;45(6):922–30.
Gollob MH, Green MS, Tang AS, Gollob T, Karibe A, Ali Hassan AS, Ahmad F, Lozado R, Shah G, Fananapazir L, Bachinski LL, Roberts R. Identification of a gene responsible for familial Wolff-Parkinson-White syndrome. N Engl J Med. 2001;344(24):1823–31.
Porto AG, Brun F, Severini GM, Losurdo P, Fabris E, Taylor MRG, Mestroni L, Sinagra G. Clinical spectrum of PRKAG2 syndrome. Circ Arrhythm Electrophysiol. 2016;9(1):e003121.
Arad M, Maron BJ, Gorham JM, Johnson WH Jr, Saul JP, Perez-Atayde AR, Spirito P, Wright GB, Kanter RJ, Seidman CE, Seidman JG. Glycogen storage diseases presenting as hypertrophic cardiomyopathy. N Engl J Med. 2005;352(4):362–72.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Linhart, A. (2020). Metabolic Cardiomyopathy. In: Baars, H.F., Doevendans, P.A.F.M., Houweling, A.C., van Tintelen, J.P. (eds) Clinical Cardiogenetics. Springer, Cham. https://doi.org/10.1007/978-3-030-45457-9_9
Download citation
DOI: https://doi.org/10.1007/978-3-030-45457-9_9
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-45456-2
Online ISBN: 978-3-030-45457-9
eBook Packages: MedicineMedicine (R0)